
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTripterine alleviates lipopolysaccharide-induced airway epithelial barrier dysfunction through suppressing the Hippo pathway
Jie Gao * and
Wenying Wang
* and
Wenying Wang
Department of Pediatrics, Huaihe Hospital of Henan University, No. 115 Ximen Street, Kaifeng 475000, Henan, P. R. China. E-mail: jie_gao11@163.com
First published on 27th November 2018
Abstract
Recent studies show that airway epithelial barrier dysfunction is closely associated with allergic inflammation and asthma pathogenesis. Tripterine, a pentacyclic triterpenoid isolated from the plant family Celastraceae, possesses anti-inflammatory and anti-oxidant properties. Our study aimed to investigate the effects of tripterine on lipopolysaccharide (LPS)-induced airway epithelial barrier dysfunction and the molecular mechanism involved. Cell viability and apoptosis were evaluated by CCK-8 assay and flow cytometry, respectively. The mRNA expressions and secretion of interleukin (IL)-6, IL-8, IL-1β, and mucin 5AC (MUC5AC) were detected by qRT-PCR and ELISA, respectively. The changes of the Hippo pathway were examined by western blot analyses of phosphorylated yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). Results showed that LPS treatment induced viability inhibition and apoptosis in lung bronchial epithelial cell line (16HBE) cells. Exposure to LPS increased the mRNA expression and concentrations of IL-6, IL-8, IL-1β, and MUC5AC in 16HBE cells. However, pretreatment with tripterine attenuated the effects of LPS on 16HBE cells. Tripterine inhibited LPS-induced activation of the Hippo pathway in 16HBE cells. Moreover, knockdown of YAP attenuated LPS-induced airway epithelial barrier dysfunction in 16HBE cells. In conclusion, tripterine attenuated LPS-induced airway epithelial barrier dysfunction through suppressing the Hippo pathway, providing new insight into the mechanism responsible for the effects of tripterine in asthma.
1. Introduction
Asthma, caused by both genetic and environmental factors, is a chronic inflammatory disorder in the respiratory tract that affects approximately 300 million individuals all over the world and causes at least 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths each year.1,2 Its incidence and prevalence is still constantly increasing year by year, which results in a substantial burden for health care systems worldwide.3 Asthma is characterized by reversible airflow obstruction and airway hyperresponsiveness (AHR), which are attributed to chronic airway inflammation in the airway wall.4 The airway epithelium serves as the first structural barrier against the inhaled environmental insults, and plays a pivotal role in the initiation of allergic airway inflammation. A number of observations have suggested that a variety of stimuli such as allergens usually cause asthma by disrupting airway epithelial barrier integrity.5,6 As a common feature of asthma, airway epithelial barrier dysfunction is closely associated with the pathogenesis of asthma.7 Accordingly, it is urgently needed to explore novel effective therapeutic strategies to attenuate airway epithelial barrier dysfunction.
000 deaths each year.1,2 Its incidence and prevalence is still constantly increasing year by year, which results in a substantial burden for health care systems worldwide.3 Asthma is characterized by reversible airflow obstruction and airway hyperresponsiveness (AHR), which are attributed to chronic airway inflammation in the airway wall.4 The airway epithelium serves as the first structural barrier against the inhaled environmental insults, and plays a pivotal role in the initiation of allergic airway inflammation. A number of observations have suggested that a variety of stimuli such as allergens usually cause asthma by disrupting airway epithelial barrier integrity.5,6 As a common feature of asthma, airway epithelial barrier dysfunction is closely associated with the pathogenesis of asthma.7 Accordingly, it is urgently needed to explore novel effective therapeutic strategies to attenuate airway epithelial barrier dysfunction.
Tripterine (C29H38O4), also known as celastrol, is a pentacyclic triterpenoid derived from the root extracts of the traditional Chinese medicinal herb Tripterygium wilfordii Hook F. According to the previous studies, tripterine has been shown to display potent anti-inflammatory, anti-oxidant, immunosuppressive and anti-cancer activities in multiple experimental models.8–10 Due to its remarkable anti-inflammatory activity, tripterine has been widely used as a traditional Chinese medicine for a long history to treat many inflammatory diseases including asthma.11,12 However, the effect of tripterine on airway epithelial barrier dysfunction in asthma remains largely unknown.
In the present study, we assessed the effects of tripterine on lipopolysaccharide (LPS)-induced human airway epithelial barrier dysfunction and found that tripterine could alleviate LPS-induced cell viability inhibition, apoptosis, release of inflammatory cytokines and mucin 5AC (MUC5AC) expression in human lung bronchial epithelial cell line (16HBE) cells. The ameliorative effects of tripterine on LPS-induced human airway epithelial barrier dysfunction were mediated by suppressing the Hippo pathway.
2. Materials and methods
2.1. Cell culture and treatment
The human airway epithelial cell line 16HBE was purchased from American type culture collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco's Modified Eagle Medium (DMEM) medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS, Hyclone, Logan, UT, USA), and 1% penicillin/streptomycin (Gibco) at 37 °C in a humidified incubator containing 5% CO2/95% air. Tripterine (purity > 98%; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in dimethyl sulphoxide (DMSO, Sigma-Aldrich) to prepare stock solution and stored at −20 °C. For some experiments, 16HBE cells were treated with a series of concentrations of tripterine (0, 1, 2, or 4 μM) or LPS (Sigma-Aldrich) (0, 1, 5, 10 μg ml−1) for 24 h, or pretreated with 1 or 2 μM tripterine for 30 min and then stimulated with 10 μg ml−1 LPS for 24 h. siRNA targeting yes-associated protein (YAP) (si-YAP) and siRNA negative control (si-NC) were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). 16HBE cells were transfected with si-YAP or si-NC using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions.2.2. Cell viability assay
16HBE cells were seeded into 96-well plates at 2 × 103 cells per well and treated as described above. After treatment, 10 μl of cell counting kit-8 reagent (Dojindo, Kumamoto, Japan) was added into each well and incubated for 2 h in the dark at 37 °C. Finally, the absorbance at 450 nm was recorded using a microplate reader (Bio-Tek Instruments Inc., Winooski, VT, USA).2.3. Flow cytometry analysis
Following treatment as above, 16HBE cells were harvested, digested with trypsin, and washed twice with ice-cold PBS. The cell apoptosis was then examined using Annexin V-FITC/PI Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA). Finally, the apoptotic rate was measured with FACSCanto II Flow Cytometer (BD Biosciences).2.4. Enzyme-linked immunosorbent assay (ELISA)
After different treatments, the supernatants of 16HBE cells were collected for ELISA analysis. The levels of interleukin (IL)-6, IL-8, IL-1β, and MUC5AC in the supernatants were measured using respective commercial available ELISA kits (BlueGene Biotech, Shanghai, China) according to the manufacturer's instructions.2.5. Quantitative real-time PCR (qRT-PCR)
Total RNA were extracted from cultured 16HBE cells using TRIzol (Qiagen, Valencia, CA, USA) and purified using an RNeasy kit (Qiagen). Approximately 1 μg of total RNA was reverse transcribed into cDNA using PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Kusatsu, Japan). For the detection of mRNA expressions of IL-6, IL-8, IL-1β, and MUC5AC, qPCR was performed using SYBR premix Ex Taq II (Takara) on the StepOnePlus™ Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). The sequences of primers were as follows: IL-6 (forward, 5′-TCC ACA AGC GCC TTC GGT CC-3′; reverse, 5′-GTG GCT GTC TGT GTG GGG CG-3′), IL-8 (forward, 5′-CTC TTG GCA GCC TTC CTG ATT-3′; reverse, 5′-TAT GCA CTG ACA TCT AAG TTC TTT AGC A-3′), IL-1β (forward, 5′-GCC CTA AAC AGA TGA AGT GCT C-3′; reverse, 5′-GAA CCA GCA TCT TCC TCA-3′), MUC5AC (forward, 5′-TGA TCA TCC AGC AGC AGG GCT-3′; reverse, 5′-CCG AGC TCA GAG GAC ATA TGG G-3′), GAPDH (forward, 5′-GAC CTG ACC TGC CGT CTA-3′; reverse, 5′-GTT GCT GTA GCC AAA TTC GTT-3′). The relative gene expression levels were calculated using the comparative 2−ΔΔCt method, with GAPDH as an internal standard.2.6. Western blot analysis
The treated 16HBE cells were collected and lysed in RIPA lysis buffer (Beyotime, Shanghai, China) and protein concentrations were quantified using the BCA method (Thermo Fisher Scientific). Protein samples were denatured by boiling and equivalent amounts of proteins were loaded on 12% SDS-PAGE and electro-transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA). The membranes were then blocked for 2 h with 5% non-fat milk at room temperature, followed by overnight incubation at 4 °C with the appropriate primary antibodies against phosphorylated-YAP (p-YAP), YAP, phosphorylated transcriptional co-activator with PDZ-binding motif (TAZ) (p-TAZ), TAZ, and β-actin (all from Cell Signaling Technology, Beverly, MA, USA). After washing for three times, the membrane was then incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) for 1 h at room temperature. Finally, the protein bands were visualized using the Pierce ECL Western Blotting kit (Pierce, Rockford, IL, USA) and band intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD).2.7. Statistical analysis
All data are shown as mean ± standard deviation (SD). Statistical analysis was carried out using the SPSS17.0 version software (SPSS, Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) was used to analyze the significance among groups. Results were considered significantly different at P values less than 0.05.3. Results
3.1. Tripterine alleviated LPS-induced viability inhibition in 16HBE cells
To assess the cytotoxicity of tripterine and LPS on 16HBE cells, 16HBE cells were incubated in different concentrations of tripterine (0, 1, 2, or 4 μM) or LPS (0, 1, 5, or 10 μg ml−1) for 24 h. Subsequent CCK-8 assay showed that low dose of tripterine at 1 or 2 μM exhibited no effect on viability of 16HBE cells, but tripterine at 4 μM suppressed the viability of 16HBE cells (Fig. 1A). LPS at 5 and 10 μg ml−1 impeded the viability of 16HBE cells (Fig. 1B). Therefore, 1 and 2 μM tripterine with no cytotoxicity and 10 μg ml−1 LPS were used for subsequent experiments. To clarify the effect of tripterine on LPS-induced viability inhibition in 16HBE cells, 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then stimulated with 10 μg ml−1 LPS for 24 h. CCK-8 assay demonstrated that 10 μg ml−1 LPS repressed the viability of 16HBE cells compared with control group, which was restored by pretreatment with 1 or 2 μM tripterine, suggesting that tripterine alleviated LPS-induced viability inhibition in 16HBE cells.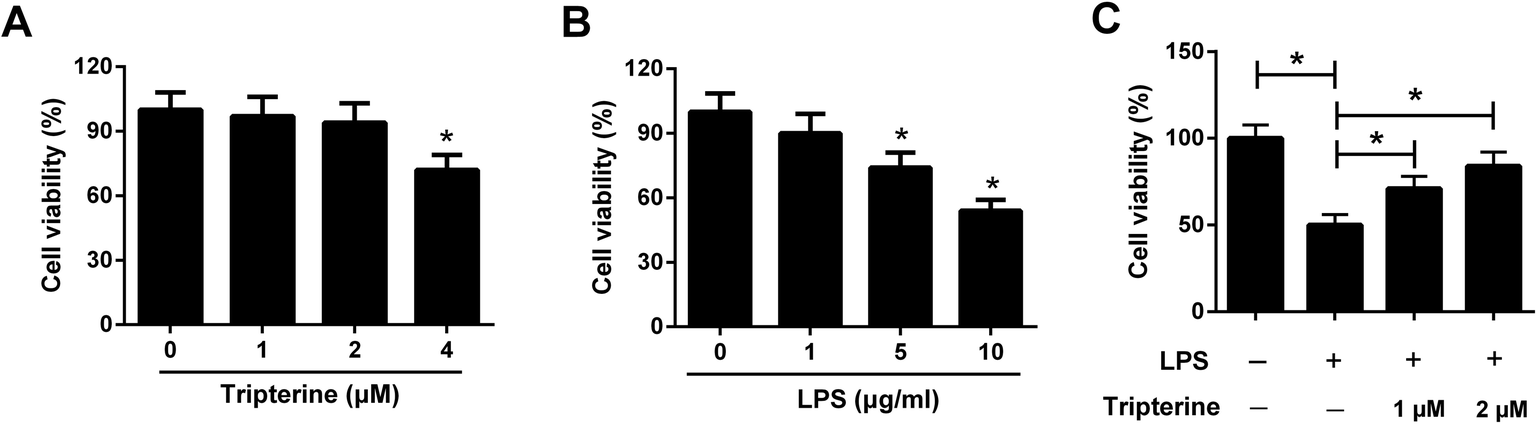 | ||
| Fig. 1 Tripterine alleviated LPS-induced viability inhibition in 16HBE cells. (A) 16HBE cells were incubated in different concentrations of tripterine (0, 1, 2, or 4 μM) for 24 h, followed by evaluation of cytotoxicity by CCK-8 assay. (B) 16HBE cells were treated with a series of concentration of LPS (0, 1, 5, or 10 μg ml−1) for 24 h, followed by assessment of cytotoxicity by CCK-8 assay. (C) 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then stimulated with 10 μg ml−1 LPS for 24 h. Cell viability was examined by CCK-8 assay. *P < 0.05. | ||
3.2. Tripterine attenuated LPS-induced apoptosis in 16HBE cells
We further determined the effects of tripterine on LPS-induced apoptosis in 16HBE cells by flow cytometry. As shown in Fig. 2, exposure to 10 μg ml−1 LPS induced apoptosis in 16HBE cells, while cotreatment with 10 μg ml−1 LPS and 1 or 2 μM tripterine counteracted LPS-induced apoptosis, indicating that tripterine attenuated LPS-induced apoptosis in 16HBE cells.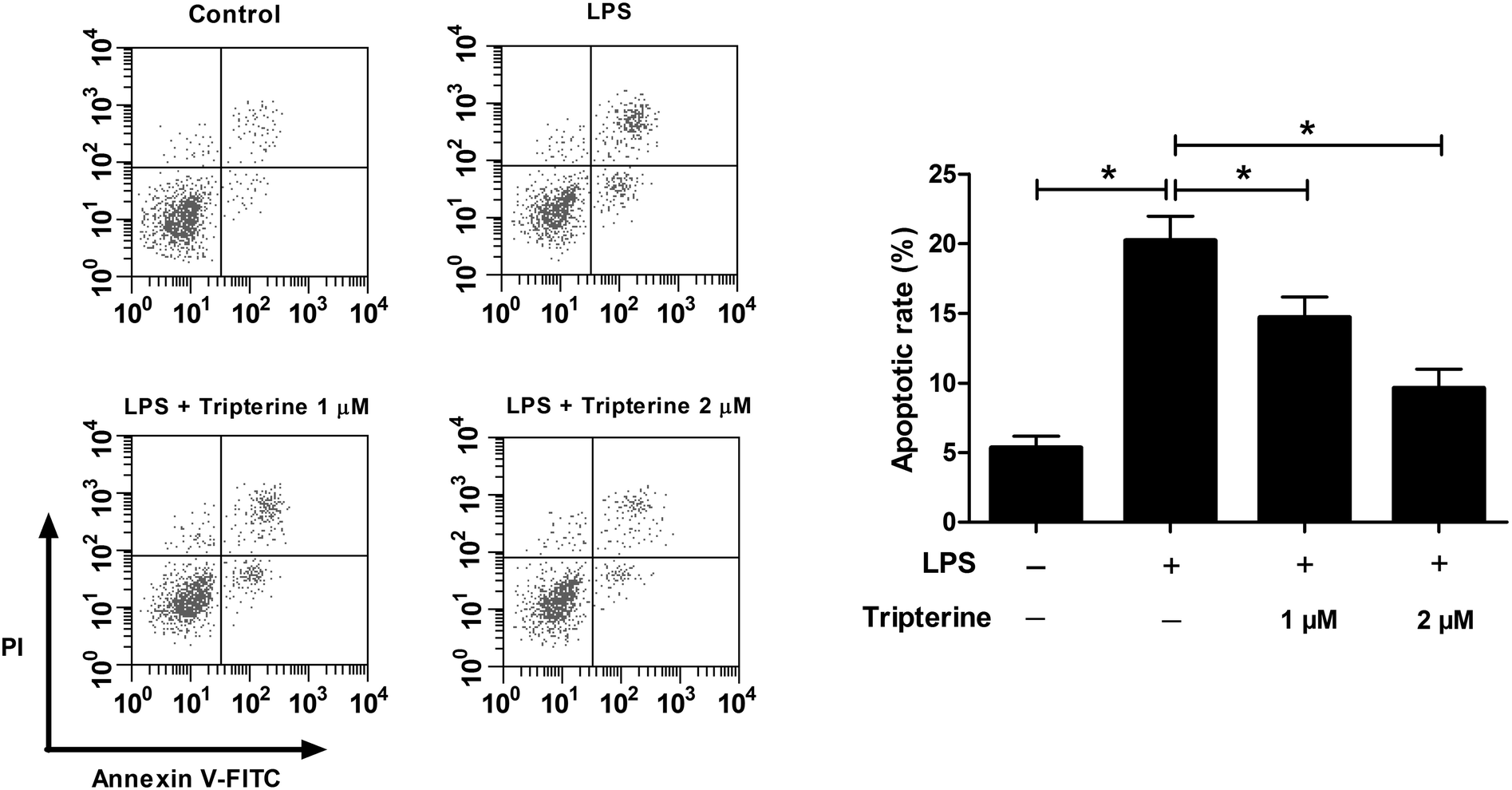 | ||
| Fig. 2 Tripterine attenuated LPS-induced apoptosis in 16HBE cells. 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then exposed to 10 μg ml−1 LPS for 24 h. Flow cytometry analysis was performed to analyze apoptosis. | ||
3.3. Tripterine attenuated LPS-induced IL-6, IL-8, and IL-1β expressions in 16HBE cells
To explore the anti-inflammatory effect of tripterine on LPS-stimulated 16HBE cells, the levels of the inflammatory cytokines including IL-6, IL-8, and IL-1β were detected by qRT-PCR and ELISA assays. The results revealed that the mRNA expressions and secretion of IL-6 (Fig. 3A and D), IL-8 (Fig. 3B and E), and IL-1β (Fig. 3C and F) in 16HBE cells were elevated after treatment with 10 μg ml−1 LPS. However, pretreatment with tripterine antagonized LPS-induced increase in IL-6, IL-8, and IL-1β expressions in 16HBE cells.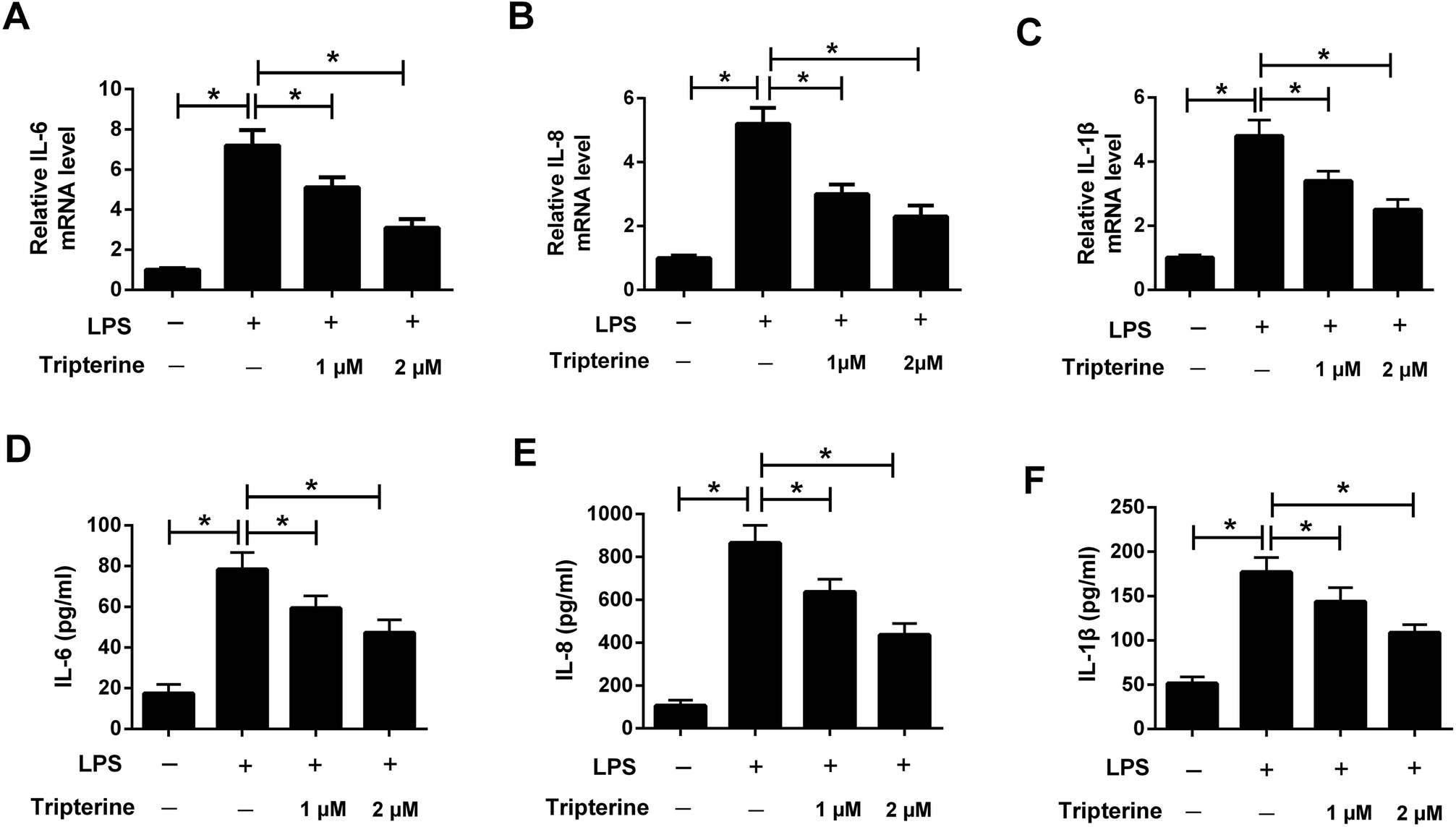 | ||
| Fig. 3 Tripterine attenuated LPS-induced IL-6, IL-8, and IL-1β expressions in 16HBE cells. 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then exposed to 10 μg ml−1 LPS for 24 h. (A–C) qRT-PCR was conducted to detect the mRNA expressions of IL-6, IL-8, and IL-1β in treated 16HBE cells. (D–F) ELISA assay was performed to measure the concentrations of IL-6, IL-8, and IL-1β in the supernatant of treated 16HBE cells. *P < 0.05. | ||
3.4. Tripterine inhibited LPS-induced MUC5AC expression in 16HBE cells
MUC5AC is a well-known major mucus protein secreted from the airway surface epithelium and has been demonstrated to be highly expressed in asthma.13 To determine the effect of tripterine on MUC5AC expression, 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then treated with 10 μg ml−1 LPS for 24 h. The expression of MUC5AC was detected by qRT-PCR and ELISA. The results implied that LPS exposure promoted the mRNA expression of MUC5AC (Fig. 4A) and enhanced the concentration of MUC5AC (Fig. 4B) in the supernatant of 16HBE cells, which were undermined by 1 and 2 μM tripterine pretreatment. These data demonstrated that tripterine inhibited LPS-induced MUC5AC expression in 16HBE cells.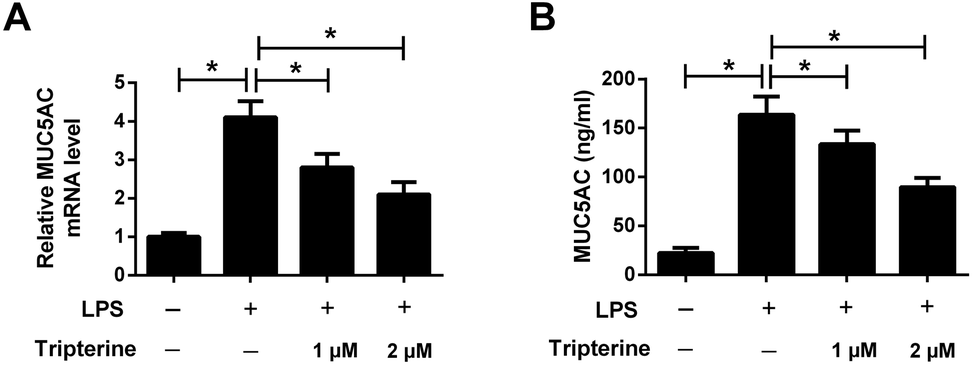 | ||
| Fig. 4 Tripterine inhibited LPS-induced MUC5AC expression in 16HBE cells. 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then treated with 10 μg ml−1 LPS for 24 h. (A) qRT-PCR was conducted to detect the mRNA expressions of MUC5AC in treated 16HBE cells. (B) ELISA assay was performed to measure the concentrations of MUC5AC in the supernatant of treated 16HBE cells. *P < 0.05. | ||
3.5. Tripterine inhibited LPS-induced activation of the Hippo pathway in 16HBE cells
Recent studies reported that the Hippo pathway plays an important role in the pathogenesis of human asthma.14 To investigate the effect of tripterine on the Hippo pathway in LPS-stimulated 16HBE cells, western blot analysis was performed to detect the protein levels of YAP and TAZ, which are two key downstream targets of the Hippo pathway. As shown in Fig. 5, LPS increased the protein levels of YAP and TAZ, but decreased the protein levels of p-YAP and p-TAZ in 16HBE cells, while pretreatment with tripterine weakened the effect of LPS on the expression levels of YAP, p-YAP, TAZ and p-TAZ. Thus, we concluded that tripterine inhibited LPS-induced activation of the Hippo pathway in 16HBE cells.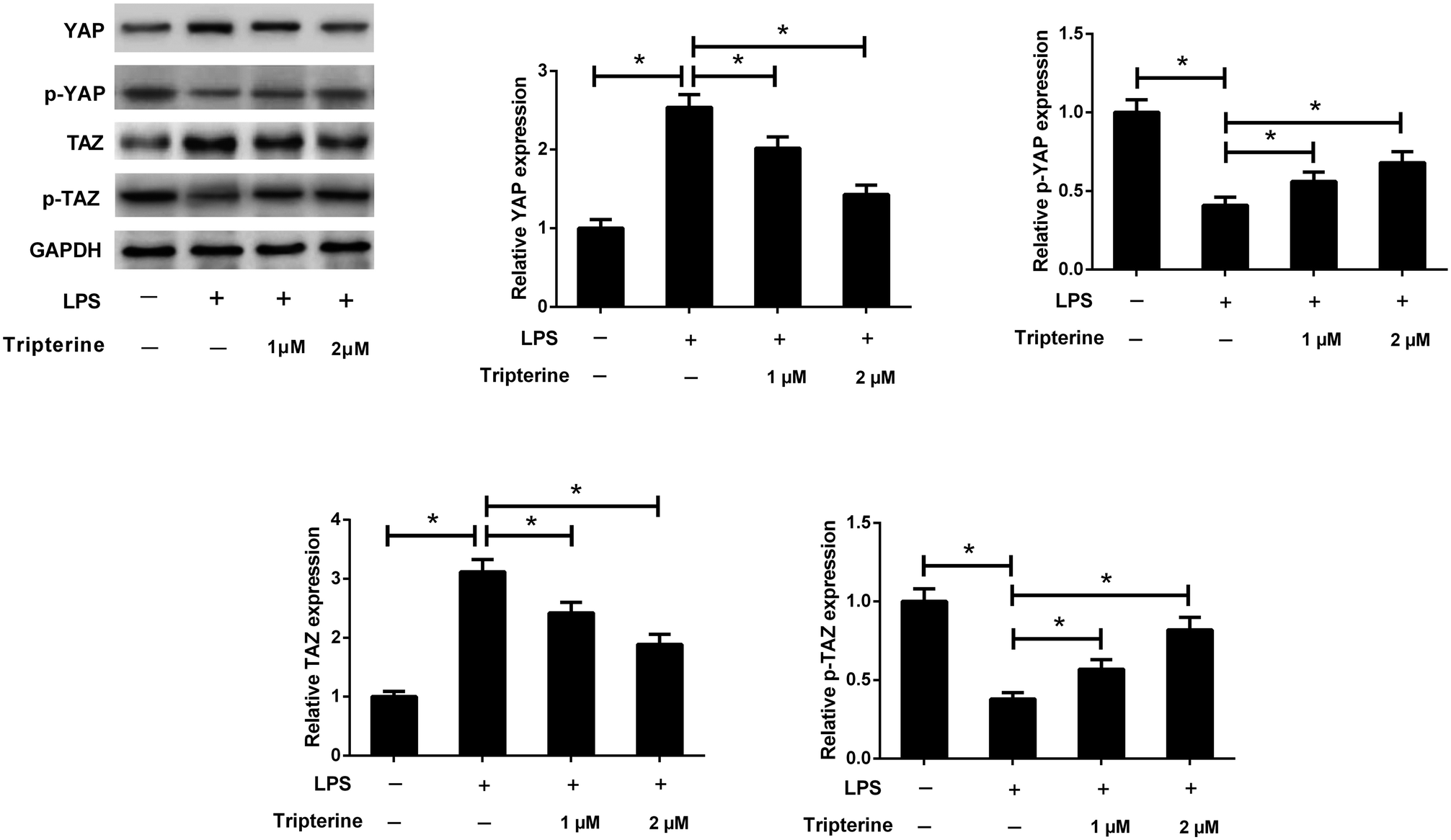 | ||
| Fig. 5 Tripterine inhibited LPS-induced activation of the Hippo pathway in 16HBE cells. 16HBE cells were pretreated with 1 or 2 μM tripterine for 30 min and then treated with 10 μg ml−1 LPS for 24 h. Western blot analysis was performed to detect the protein levels of YAP, p-YAP, TAZ, and p-TAZ. *P < 0.05. | ||
3.6. Knockdown of YAP attenuated LPS-induced airway epithelial barrier dysfunction in 16HBE cells
To explore the role of the Hippo pathway in LPS-induced airway epithelial barrier dysfunction in 16HBE cells, 16HBE cells were transfected with si-YAP or si-NC for 24 h, followed by stimulation with 10 μg ml−1 LPS for another 24 h. Western blot analysis manifested that YAP knockdown conspicuously abolished the protein levels of YAP in 16HBE cells (Fig. 6A). CCK-8 assay demonstrated that LPS-induced viability inhibition in 16HBE cells was ameliorated by transfection with si-YAP (Fig. 6B). Meanwhile, we discovered that silencing of YAP offset LPS-induced apoptosis in 16HBE cells (Fig. 6C). YAP knockdown also restrained the concentrations of IL-6 (Fig. 6D), IL-8 (Fig. 6E), and IL-1β (Fig. 6F) in 16HBE cells in the presence of LPS. Furthermore, LPS-induced MUC5AC secretion in the supernatant of 16HBE cells was mitigated by YAP knockdown (Fig. 6G). Collectively, we concluded that inactivation of the Hippo pathway attenuated LPS-induced airway epithelial barrier dysfunction in 16HBE cells.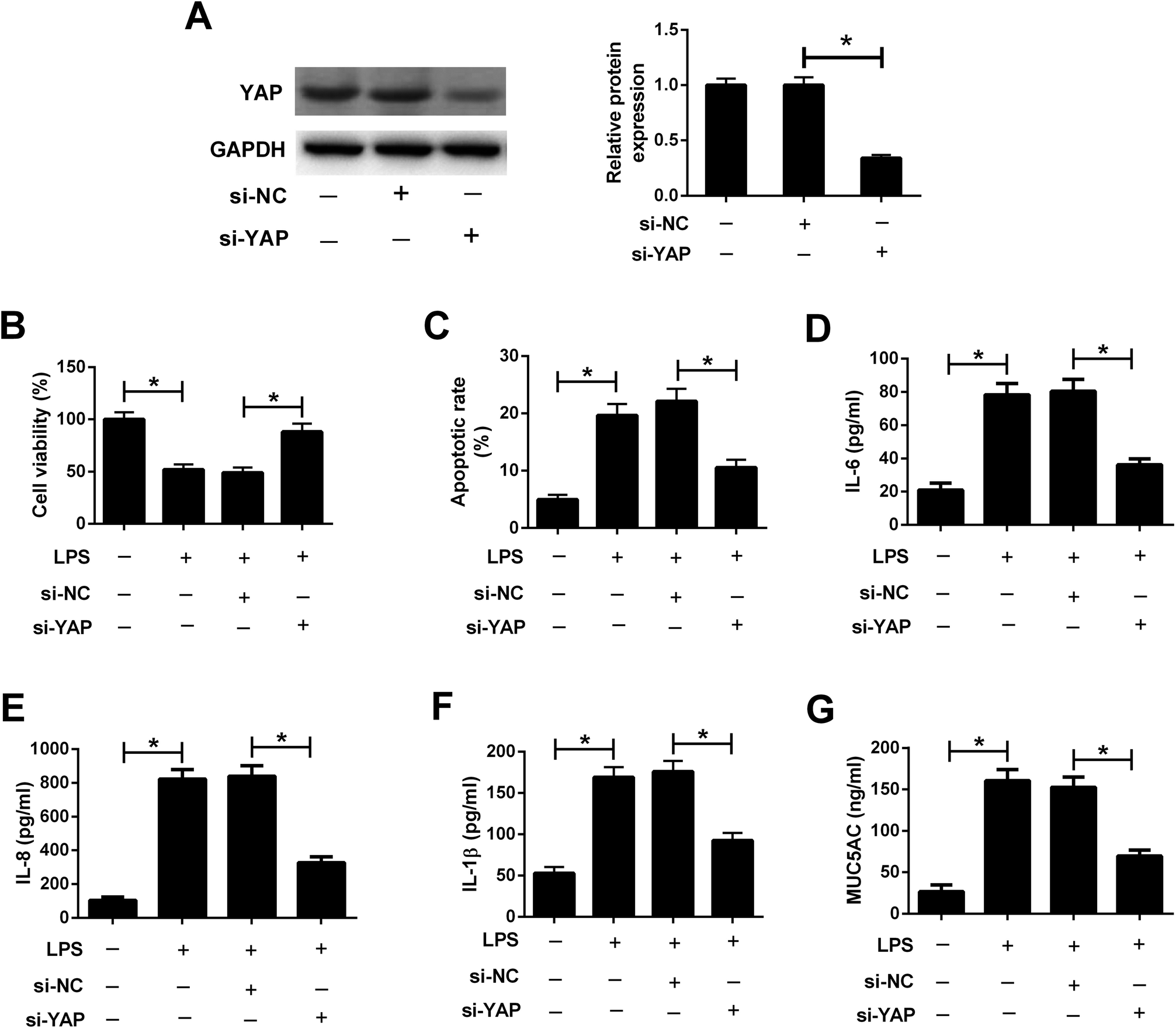 | ||
| Fig. 6 Knockdown of YAP attenuated LPS-induced airway epithelial barrier dysfunction in 16HBE cells. 16HBE cells were transfected with si-YAP or si-NC for 24 h, followed by stimulation with 10 μg ml−1 LPS for another 24 h. (A) Western blot analysis was conducted to detect the protein levels of YAP in treated 16HBE cells. (B) Cell viability in treated 16HBE cells was evaluated by CCK-8 assay. (C) Apoptosis of treated 16HBE cells was examined by flow cytometry analysis. The secretion of IL-6 (D), IL-8 (E), IL-1β (F) and MUC5AC (G) in the supernatant of treated 16HBE cells were determined by ELISA. *P < 0.05. | ||
4. Discussion
Airway epithelial cells form the first line of defense against inhaled environmental insults including allergens, environmental pollutants and respiratory viruses, and are often disrupted in asthma. It has been widely recognized that airway epithelial barrier dysfunction is considered as a common physiological feature of asthma and contribute to asthma pathogenesis.15 Therefore, identifying therapeutic agents to prevent disruption of the airway epithelial barrier function may serve as promising therapeutic strategies for alleviating asthma symptoms. To the best of our knowledge, our study is the first time to demonstrate that tripterine could attenuate LPS-induced airway epithelial barrier dysfunction via inactivation of the Hippo-YAP/TAZ pathway.Tripterine, a naturally occurring quinone methide triterpene isolated from the plant family Celastraceae, possesses a broad range of biological activities including anti-inflammatory and anti-oxidant properties. Tripterine has been shown to exert therapeutic potential on the treatment of autoimmune and inflammatory diseases,16 such as arthritis,17 atherosclerosis18 and Alzheimer's disease.19 Interestingly, it was reported that celastrol imparted protective effects against LPS-induced acute respiratory distress syndrome in rats through inhibition of the nuclear factor-kappa B (NF-κB) signaling pathway.20 Celastrol was shown to effectively suppress airway inflammation, airway hyperresponsiveness and tissue remodeling in an ovalbumin (OVA)-induced allergic asthma in mice.21 Celastrol was also found to suppress AHR through inhibiting Th17 response in OVA-induced obesity asthmatic mice.22 A previous study showed that tripterine mitigated cigarette smoke extract-induced DNA damage through the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway in human bronchial epithelial cells.23 In our study, we further showed that tripterine dose-dependently attenuated LPS-induced viability inhibition and apoptosis in 16HBE cells. Moreover, tripterine suppressed LPS-induced expressions of inflammatory cytokines including IL-6, IL-8, and IL-1β in 16HBE cells, suggesting that tripterine repressed LPS-induced inflammatory response in 16HBE cells. Mucus hypersecretion, a common pathophysiological feature of asthma, is an important contributor to asthma exacerbation.24 MUC5AC is a major component of mucin expressed by lung goblet cells in asthma, and upregulated production of MUC5AC contributes to airflow limitation and AHR in asthma patients.25 Herein, our study demonstrated that tripterine inhibited LPS-induced MUC5AC expression in 16HBE cells. Collectively, we concluded that tripterine attenuated LPS-induced airway epithelial barrier dysfunction.
The Hippo pathway has emerged as an evolutionarily conserved pathway that plays a key role in controlling organ size, proliferation and apoptosis of immune cells.26 The activation of the Hippo pathway is mediated through phosphorylation of the transcriptional co-activator YAP and its paralogue TAZ, two effectors of the Hippo pathway, in mammal.27 Studies reported that YAP protein was significantly upregulated in the bronchial airway tissues of OVA-induced chronic asthma mouse model,28 suggesting that the Hippo pathway might play a crucial in the pathological process of asthma.14 In the present study, we demonstrated that LPS induced activation of the Hippo pathway in 16HBE cells, which was suppressed by pretreatment with tripterine. Moreover, inhibition of the Hippo pathway by si-YAP significantly attenuated LPS-induced viability inhibition, apoptosis, secretion of inflammatory cytokines and MUC5AC in 16HBE cells. Therefore, it is reasonable to infer that tripterine attenuated LPS-induced airway epithelial barrier dysfunction through suppressing Hippo pathway.
5. Conclusion
In summary, we provided the first evidence that tripterine attenuated LPS-induced airway epithelial barrier dysfunction through suppressing Hippo pathway, providing new insight into the mechanism responsible for the effects of tripterine in asthma.Conflicts of interest
The authors declare that they have no competing interests.References
- F. D. Martinez, Eur. Respir. J., 2007, 29, 179–184 CrossRef CAS PubMed.
- A. B. Becker and E. M. Abrams, Curr. Opin. Allergy Clin. Immunol., 2017, 17, 99–103 CrossRef PubMed.
- M. Masoli, D. Fabian, S. Holt and R. Beasley, Allergy, 2004, 59, 469–478 CrossRef PubMed.
- Y. Bosse, P. D. Pare and C. Y. Seow, Curr. Allergy Asthma Rep., 2008, 8, 357–366 CrossRef CAS PubMed.
- I. H. Heijink, A. van Oosterhout and A. Kapus, Eur. Respir. J., 2010, 36, 1016–1026 CrossRef CAS PubMed.
- S. Post, M. C. Nawijn, M. R. Jonker, N. Kliphuis, M. van den Berge, A. J. van Oosterhout and I. H. Heijink, Allergy, 2013, 68, 1117–1125 CAS.
- S. T. Holgate, Curr. Opin. Pulm. Med., 2009, 15, 63–71 CrossRef CAS PubMed.
- C. Wang, C. Shi, X. Yang, M. Yang, H. Sun and C. Wang, Eur. J. Pharmacol., 2014, 744, 52–58 CrossRef CAS PubMed.
- R. Kannaiyan, M. K. Shanmugam and G. Sethi, Cancer Lett., 2011, 303, 9–20 CrossRef CAS PubMed.
- S. H. Venkatesha, S. Dudics, B. Astry and K. D. Moudgil, Pathog. Dis., 2016, 74, ftw059 CrossRef PubMed.
- A. R. Setty and L. H. Sigal, Semin. Arthritis Rheum., 2005, 34, 773–784 CrossRef PubMed.
- T. Morita, Am. J. Hypertens., 2010, 23, 821 CrossRef PubMed.
- M. Chen, Z. Lv, W. Zhang, L. Huang, X. Lin, J. Shi, W. Zhang, R. Liang and S. Jiang, Mol. Immunol., 2015, 64, 99–105 CrossRef CAS PubMed.
- L. E. Fodor, A. Gezsi, L. Ungvari, A. F. Semsei, Z. Gal, A. Nagy, G. Galffy, L. Tamasi, A. Kiss, P. Antal and C. Szalai, Allergy, Asthma Immunol. Res., 2017, 9, 247–256 CrossRef CAS PubMed.
- I. H. Heijink, M. C. Nawijn and T. L. Hackett, Clin. Exp. Allergy, 2014, 44, 620–630 CrossRef CAS PubMed.
- H. Z. Jin, B. Y. Hwang, H. S. Kim, J. H. Lee, Y. H. Kim and J. J. Lee, J. Nat. Prod., 2002, 65, 89–91 CrossRef CAS.
- S. M. Nanjundaiah, S. H. Venkatesha, H. Yu, L. Tong, J. P. Stains and K. D. Moudgil, J. Biol. Chem., 2012, 287, 22216–22226 CrossRef CAS PubMed.
- L. Gu, W. Bai, S. Li, Y. Zhang, Y. Han, Y. Gu, G. Meng, L. Xie, J. Wang, Y. Xiao, L. Shan, S. Zhou, L. Wei, A. Ferro and Y. Ji, PLoS One, 2013, 8, e65477 CrossRef CAS PubMed.
- A. C. Allison, R. Cacabelos, V. R. Lombardi, X. A. Alvarez and C. Vigo, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2001, 25, 1341–1357 CrossRef CAS PubMed.
- Y. Wei and Y. Wang, J. Immunotoxicol., 2017, 14, 228–234 CrossRef CAS PubMed.
- D. Y. Kim, J. W. Park, D. Jeoung and J. Y. Ro, Eur. J. Pharmacol., 2009, 612, 98–105 CrossRef CAS PubMed.
- Z. Zeng, X. Lin, R. Zheng, H. Zhang and W. Zhang, Front. Pharmacol., 2018, 9, 49 CrossRef PubMed.
- W. Liu, X. Tan, L. Shu, H. Sun, J. Song, P. Jin, S. Yu, M. Sun and X. Jia, Molecules, 2012, 17, 9104–9115 CrossRef CAS PubMed.
- H. Lai and D. F. Rogers, J. Aerosol Med. Pulm. Drug Delivery, 2010, 23, 219–231 CrossRef CAS PubMed.
- C. M. Evans, D. S. Raclawska, F. Ttofali, D. R. Liptzin, A. A. Fletcher, D. N. Harper, M. A. McGing, M. M. McElwee, O. W. Williams, E. Sanchez, M. G. Roy, K. N. Kindrachuk, T. A. Wynn, H. K. Eltzschig, M. R. Blackburn, M. J. Tuvim, W. J. Janssen, D. A. Schwartz and B. F. Dickey, Nat. Commun., 2015, 6, 6281 CrossRef CAS PubMed.
- J. Huang, S. Wu, J. Barrera, K. Matthews and D. Pan, Cell, 2005, 122, 421–434 CrossRef CAS PubMed.
- A. Ramos and F. D. Camargo, Trends Cell Biol., 2012, 22, 339–346 CrossRef CAS PubMed.
- J. Zhou, F. Xu, J. J. Yu and W. Zhang, Int. J. Clin. Exp. Pathol., 2015, 8, 11132–11139 CAS.
| This journal is © The Royal Society of Chemistry 2018 |