
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization and magnetic properties of undoped and copper-doped cobalt ferrite nanoparticles prepared by the octanoate coprecipitation route at very low dopant concentrations
Hypolite Mathias Kamta Tedjieukeng *a,
Patrice Kenfack Tsobnangb,
Roussin Lontio Fomekonga,
Ekane Peter Etapec,
Pattayil A. Joyd,
Arnaud Delcortee and
John Ngolui Lambia
*a,
Patrice Kenfack Tsobnangb,
Roussin Lontio Fomekonga,
Ekane Peter Etapec,
Pattayil A. Joyd,
Arnaud Delcortee and
John Ngolui Lambia
aMaterials Chemistry Laboratory, Department of Chemistry, Higher Teacher Training College, University of Yaoundé I, P. O. Box 47, Yaoundé, Cameroon
bDepartment of Chemistry, University of Dschang, P. O. Box 67, Dschang, Cameroon
cDepartment of Chemistry, Faulty of Science, University of Buea, P. O. Box 63, Buea, Cameroon
dPhysical Chemistry Division, National Chemical Laboratory, Pune 411008, India
ePole Bio & Soft Mater, Institut de la Matière Condensée et des Nanosciences, Université Catholique de Louvain, Croix du Sud 1, 1348, Louvain-La-Neuve, Belgium
First published on 16th November 2018
Abstract
Nanoparticles of undoped and copper-doped cobalt ferrite Co1−xCuxFe2O4 at very low dopant concentrations (x = 0; 0.02; 0.04; 0.06; 0.08) were successfully synthesized by pyrolysis of the corresponding hetero metal octanoate precursors obtained via coprecipitation using the octanoate ligand as precipitating agent. The precursors were then characterized by FTIR, ICP-AES and TG-DTA analyses and the results reveal the formation of a copper-cobalt-iron hydroxooctanoate represented by the formula [Co1−xCuxFe2(C8H15O2)6(OH)2·2H2O]. The decomposition products obtained upon pyrolysis in air at 400 °C for 3 h were characterized by FTIR, XRD, SEM, TEM, XPS and VSM analyses. FTIR and XRD analyses showed the formation of a single phase mixed spinel ferrite while TEM analysis showed that the particles have a spherical shape with a mean size of 20 nm and form spherical agglomerates with sizes reaching 500 nm in some cases as the SEM images show. The chemical states of the metallic species in the samples were revealed by XPS to be Cu2+, Co2+ and Fe3+. These results combined with XRD confirmed the mixed spinel structure, Co1−xCuxFe2O4 in which Cu2+ ions substitute Co2+ ions in tetrahedral sites for x lower than 0.06 and in octahedral sites for x between 0.06 and 0.08. Magnetic parameters such as saturation magnetization (Ms), coercivity (Hc), remanent magnetization (Mr), magnetocrystalline anisotropy constant (K) and reduced magnetization (Mr/Ms), obtained from magnetic hysteresis loops measured at room temperature, are in agreement with this mixed spinel structure and also indicate that these materials are ferromagnetic and could be good candidates for applications in biomedicine and in microwave devices.
1. Introduction
Magnetic spinel ferrite nanomaterials have nowadays attracted a lot of interest because of the important roles they play in enhancing knowledge and understanding of the functioning of magnetic materials in general and ferrites in particular.1–4 This is more so given the fact that ferrites have several technological applications such as in high density magnetic storage, sensors, biomedical drug delivery, broadband transformers, computer circuitry, carrier telephony and highly efficient catalysts.5–11 Most of these applications depend on the magnetic properties of these materials which could be ascribed to a complex interplay of several factors where cationic distribution and spin canting are fundamental and predominant.12The general formula for spinel ferrites is MFe2O4, where M is a divalent metal ion and Fe is present in the +3 oxidation state.13–16 Generally, there are two types of ferrite spinel structures: the normal and the inverse, even though most spinel ferrites are inverse.17,18 In the normal spinel, the tetrahedral sites occupied contain the M2+ ions while the Fe3+ ions are located in the octahedral sites occupied.19–21 In the inverse spinel, however, all the M2+ ions and half of the Fe3+ ions are located in the octahedral sites occupied, with the other half of the Fe3+ ions in the tetrahedral sites occupied. This implies that the cation distribution between the two interstitial sites could influence the magnetic properties of the spinel ferrites.18
The cobalt ferrite spinel (CoFe2O4), in particular, is a hard magnetic material with well-known properties such as mechanical hardness, remarkable chemical stability, high anisotropy (Ka = 3 × 105 J m−3)8 and a relatively high saturation magnetization (Ms) and coercivity (Hc). As a result, it has many applications such as in ferrofluids technology, as biocompatible magnetic nanoparticles for cancer treatment, for magnetic resonance imaging and in microwave devices.5,22–25 The substitution of cobalt by copper in CoFe2O4 is known to modify the structural and magnetic properties of the material making it a suitable candidate for some applications like in biomedical drug delivery and in microwave, switching and memory devices.10,26
Most often, the magnetic properties of cobalt ferrite spinels depend on the preparation method27–29 and the nature of the dopant used.30,31 Ma et al.28 synthesized the cobalt ferrite (CoFe2O4) by the solvothermal method at 160 °C and obtained a saturation magnetization of 73 emu g−1 while Gopalan et al.29 obtained a saturation magnetization of 48 emu g−1 for the same material synthesized by sol–gel combustion method. Prathapani et al.30 synthesized CoFe2−xErxO4 ferrite nanoparticles (with x = 0, 0.01, 0.02, 0.03 and 0.04) by the sol–gel assisted auto combustion method. For x = 0, they obtained a magnetization (M) of 72.1 emu g−1 at room temperature. As x increased from 0.01 to 0.02 this value increased to 75.3 emu g−1 while as x increased from 0.03 to 0.04 the value decreased to 73.2 emu g−1. Tholkappiyan et al.31 synthesized MgFe2−xErxO4 ferrite nanoparticles (with x = 0, 0.02, 0.04 and 0.06) by combustion method and showed that at room temperature, the saturation magnetization (Ms) increased from 13.2 emu g−1 for x = 0 to 44.1 emu g−1 for x = 0.06.
Several methods6,7,22–24,26,32,33 have been used to synthesize nano-structured mixed copper-cobalt ferrites. These methods include the microwave assisted sol–gel,6 co-precipitation,7,24 sol–gel,22,23 ceramic,26 microemulsion32 and soft chemistry methods.33 Among all these methods, coprecipitation has been preferred. It involves precipitating simultaneously two or more metal salts in solution by using a precipitating agent (or ligand) and subsequently thermally decomposing the precipitate.34 Relative to the other methods, it offers several advantages such as better distribution of the constituents in the resulting ferrites, low operating temperature, low cost and good control of stoichiometry.7,35 Many studies10,24,36 have reported on high amounts of substitution, where an increase in Cu content has resulted in a decrease in the magnetization. For example, Jnaneshwara et al.10 synthesized Co1−xCuxFe2O4 (with x = 0, 0.1, 0.2, 0.3, 0.4, 0.5) by the solution combustion method and showed that the saturation magnetization decreased from 38.5 to 26.7 emu g−1 as the concentration of Cu2+ increased. Balavijayalakshmi et al.,24 on the other hand, synthesized Co1−xCuxFe2O4 nanoparticles (x = 0, 0.2, 0.4, 0.6, 0.8, 1) by coprecipitation method followed by sintering at 900 °C while Samavati et al.36 obtained the same material (x = 0, 0.3, 0.5, 0.7, 1) by the same method with sintering at 800 °C. Their results showed that when the copper concentrations increased, the saturation magnetization (Ms) decreased from 72.06 emu g−1 to 21.11 emu g−1 for the former and from 61.06 to 39.52 emu g−1 for the latter. From all what precedes, it is evident that the preparation method and sometimes the sintering temperature influence either the size or the morphology of the spinel ferrite nanomaterial, which in turn, influences the magnetic properties.
In this paper, we therefore report for the first time and to the best of our knowledge, a controlled synthesis of Co1−xCuxFe2O4 ferrite nanoparticles at very low dopant concentrations (x = 0, 0.02, 0.04, 0.06, 0.08) by a simple but versatile method which involves the pyrolysis of the precursors obtained by a coprecipitation route using aqueous solutions of the octanoate ligand as precipitating agent. The use of octanoate as precipitating agent and the interval of the dopant applied in this synthesis could certainly have an influence on the magnetic properties of the as-synthesized materials. The precursors were characterized by elemental (C, H, N) analyses, ICP-AES, FTIR and TGA. The decomposition products, obtained from thermal decomposition of the precursors, were characterized by FTIR, XRD, TEM, SEM, XPS and VSM.
2. Experimental section
2.1. Synthesis procedure
Copper chloride (CuCl2·2H2O), cobalt nitrate [Co(NO3)2·6H2O], ferric nitrate [Fe(NO3)3·9H2O], lithium hydroxide (LiOH·H2O) and octanoic acid, all obtained from Aldrich, were used without further purification.The undoped and copper-doped cobalt ferrite nanoparticles, Co1−xCuxFe2O4 (x = 0, 0.02, 0.04, 0.06, 0.08) were obtained via two experimental steps: the precursors were first synthesized by coprecipitation in aqueous solution containing iron, copper and cobalt ions in the right ratio using the octanoate ligand as precipitating agent then followed by thermal decomposition of the precursors obtained. For example, for the synthesis of cobalt iron octanoate precursor: lithium octanoate was first prepared in aqueous solution by adding lithium hydroxide (13 mmol) to octanoic acid (13 mmol). After a few minutes, an aqueous solution containing cobalt(II) nitrate (1.6 mmol) and iron(III) nitrate (3.2 mmol) was poured drop by drop into the previously prepared solution of lithium octanoate with continuous stirring. The resulting brown solution was then stirred for another 1 h at room temperature (30 °C). The resulting precipitate (cobalt-iron octanoate) was then filtered (to remove the LiNO3), washed with ethanol and dried in a desiccator for two days at room temperature to obtain a light brown powder. The as-prepared precursor powder was decomposed in air at 400 °C in a tubular furnace (heating rate 15 °C min−1) for 3 h. The scheme of the synthesis is shown in Fig. 1. The as-prepared powder samples of Co1−xCuxFe2O4 for x = 0, 0.02, 0.04, 0.06, and 0.08 were labelled, respectively, as S1, S2, S3, S4 and S5 while the corresponding decomposed products were labelled as S1P, S2P, S3P, S4P, and S5P.
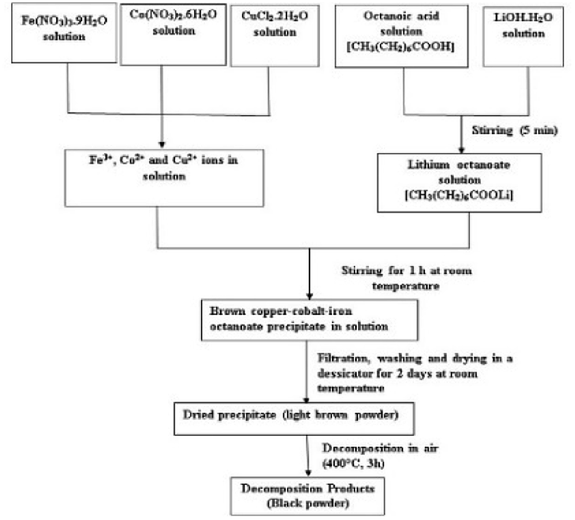 | ||
| Fig. 1 Flow chart for synthesis of Co1−xCuxFe2O4 nanoparticles using co-precipitation method. | ||
2.2. Characterization methods
The metal contents of the precursors were estimated by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES) using a Thermo Jarrell Ash Iris Advantage apparatus. The solution used for measurements was obtained as follows. A few milligrams (∼80 mg) of the sample were digested in 4 mL of mixed acid (3 mL of concentrated nitric acid + 1 mL of concentrated hydrochloric acid) and the resulting solution was diluted to 1000 mL using distilled water. A LECO CHN 900 Elemental Analyzer was used for the C and H analyses in the precursors. The functional groups of the octanoate ligand were determined using Fourier Transform Infra-Red (FTIR) spectroscopy using a Quick Lock Single Reflection Horizontal ATR Accessory from Bruker. The thermal decomposition behaviour of the precursors was studied by Thermogravimetric Analysis (TGA) on a METTLER TOLEDO Thermal Analyzer in air at a flow rate of 100 mL min−1, between 25–900 °C and at a heating rate of 10 °C min−1. Powder X-Ray Diffraction (XRD) data of the decomposed products were collected at room temperature on a D5000 Siemens Kristalloflex θ–2θ powder diffractometer which has a Bragg–Brentano geometry, is equipped with a graphite-monochromated Cu-Kα (λ = 1.54056 Å) radiation. For the experiment, the samples were spread out on a flat silicon plate in such a manner as to avoid preferred orientations. The patterns were recorded in the range of 5–90° with a scan step of 0.02° (2θ) and a 2 s acquisition interval. The XRD patterns were compared to those of the CoFe2O4 patterns of the ICCD using the High-Score Plus software for phase identification. The recorded patterns were also indexed and the unit cell refined by using the FULLPROF suite program. The morphology of the powder was determined by Scanning Electron Microscopy (SEM) using a high resolution FEG JEOL7600F with an accelerating voltage of 1 kV. The morphologies of the samples were confirmed by Transmission Electron Microscopy (TEM, Leo922 Model from Zeiss) at an accelerating voltage of 120 kV. The TEM samples were prepared by dropping a sonicated water dispersion of the powder samples on a carbon-coated copper grid. The composition, the chemical state of the various constituents and the stoichiometry of each sample were determined by X-ray Photoelectron Spectroscopy (XPS) using a Kratos Axis Ultra spectrometer (Kratos Analytical, Manchester, UK) equipped with a monochromatized aluminum X-ray source (powered at 10 mA and 15 kV) and an eight-channeltron detector. The magnetic measurements were carried out at room temperature a SQUID-Vibrating Sample Magnetometer (Quantum Design) with an applied field in the range ± 60 kOe.3. Results and discussions
3.1. Characterization of precursors
| Samples | Elemental contents (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Fe | Co | Cu | C | H | ||||||
| Theo* | Exp** | Theo | Exp | Theo | Exp | Theo | Exp | Theo | Exp | |
| a *Theo = theoretical; **Exp = experimental. | ||||||||||
| S1 | 10.2 | 9.6 | 5.4 | 3.4 | 0 | 0 | 52.41 | 53.12 | 8.80 | 8.58 |
| S2 | 10.2 | 9.4 | 5.3 | 3.6 | 0.12 | 0.13 | — | — | — | — |
| S3 | 10.2 | 9.6 | 5.1 | 3.4 | 0.23 | 0.20 | — | — | — | — |
| S4 | 10.2 | 9.7 | 5.0 | 3.3 | 0.35 | 0.34 | — | — | — | — |
| S5 | 10.2 | 9.9 | 4.9 | 3.2 | 0.46 | 0.43 | 52.40 | 52.83 | 8.80 | 8.46 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)] vibration around 1700 cm−1 is an indication that the entire carboxylate group is engaged in the coordination bond. Lambi et al.37 and Mesubi et al.38 have shown that when the value of the separation between the bands, Δν (=νCOOasym − νCOOsym) is above 200 cm−1, a monodentate mode of bonding between the carboxylate group (COO−) of the ligand and the metal atoms is indicated, a value of Δν below 110 cm−1 is attributable to the chelating bidentate mode while Δν between 140 cm−1 and 200 cm−1 is attributed to the bridged bidentate bonding mode.39 All the samples investigated (S1 to S5) showed Δν values of 150 cm−1 corresponding to the bridged bidentate bonding mode of the octanoic acid to the metals These FTIR results are in good agreement with those reported on iron carboxylate complexes.40,41
O)] vibration around 1700 cm−1 is an indication that the entire carboxylate group is engaged in the coordination bond. Lambi et al.37 and Mesubi et al.38 have shown that when the value of the separation between the bands, Δν (=νCOOasym − νCOOsym) is above 200 cm−1, a monodentate mode of bonding between the carboxylate group (COO−) of the ligand and the metal atoms is indicated, a value of Δν below 110 cm−1 is attributable to the chelating bidentate mode while Δν between 140 cm−1 and 200 cm−1 is attributed to the bridged bidentate bonding mode.39 All the samples investigated (S1 to S5) showed Δν values of 150 cm−1 corresponding to the bridged bidentate bonding mode of the octanoic acid to the metals These FTIR results are in good agreement with those reported on iron carboxylate complexes.40,41
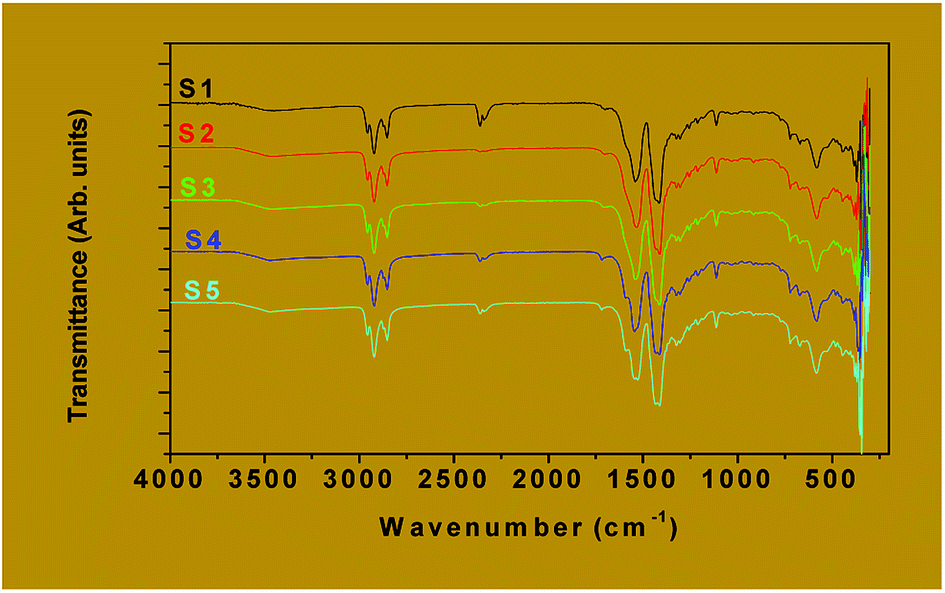 | ||
| Fig. 2 FTIR spectra of the as-prepared samples S1 to S5. | ||
| Functional groups | Vibration mode | Wave number (cm−1) |
|---|---|---|
| a ν: stretching (as: asymmetric; s: symmetric); δ: deformation in the plane; π: deformation outside the plane and ρ: wagging. | ||
| O–H/H2O | ν | 3420 |
| δ | 1536 | |
| O–H/Fe–OH | ν | 3420 |
| CH/CH3 | νas | 2957 |
| νs | 2872 | |
| CH/CH2 | νas | 2923 |
| νs | 2853 | |
| C–C | νas | 920 |
| OCO | νas | 1581 |
| νs | 1431 | |
| δ | 674 | |
| π | 641 | |
| (CH2)6 | δ | 1321 |
| ρ | 723 | |
| Fe–O | ν | 580 |
| Cu/Co–O | ν | 370 |
 | ||
| Fig. 3 TGA curves of the as-prepared samples S2 (a) and S1 to S5 (b). | ||
3.2. Analysis of the decomposition products
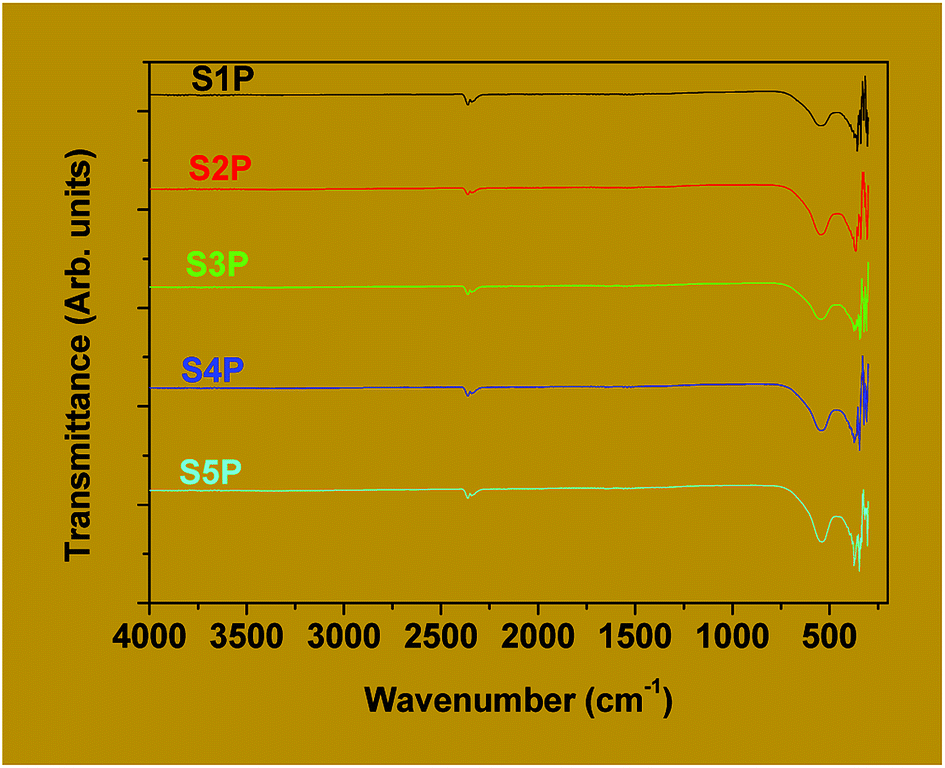 | ||
| Fig. 4 FTIR spectra of the decomposition products S1P to S5P. | ||
For spinel ferrites, the band appearing at the higher wave number (500–600 cm−1) is usually assigned to tetrahedral coordination while that at the lower wave number (385–450 cm−1) is often ascribed to octahedral coordination.46,47 Thus, it can be concluded that the products, S1P to S5P, correspond to the cobalt ferrite spinel. Avazpour et al.45 have obtained similar results with the absorption bands at 540 cm−1 and 331 cm−1 corresponding, respectively, to the tetrahedral and octahedral groups in cobalt ferrite.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group).24 The lattice parameters “a” of all these samples are presented in Table 3. The parameter decreases from 8.352 Å to 8.344 Å as the doping percentage (x) increases from 0 to 0.04 but increases from 8.344 to 8.366 Å as x increases from 0.06 to 0.08. These variations are probably related to the structure of the compounds. The parent material (CoFe2O4) obtained through the coprecipitation method used in this work could be a mixed spinel structure where a part of Co(II) and Fe(III) ions are found in the tetrahedral and octahedral interstices occupied. For a copper content (x) less than 0.06, only the cobalt(II) ions of the tetrahedral sites are substituted. The unit cell parameter of the compound slightly decreases since the ionic radii of copper(II) and cobalt(II) ions are, respectively, 0.57 Å and 0.58 Å in a tetrahedral coordination.48 But for a copper content greater than or equal to 0.06, all the copper ions of the tetrahedral site are substituted and a part of the cobalt(II) ions located in the octahedral sites are also replaced. The unit cell parameter of the compound, therefore, increases since the ionic radius of the copper(II) ion (0.73 Å) is greater than that of cobalt(II) (0.65 Å) in an octahedral coordination.48
m space group).24 The lattice parameters “a” of all these samples are presented in Table 3. The parameter decreases from 8.352 Å to 8.344 Å as the doping percentage (x) increases from 0 to 0.04 but increases from 8.344 to 8.366 Å as x increases from 0.06 to 0.08. These variations are probably related to the structure of the compounds. The parent material (CoFe2O4) obtained through the coprecipitation method used in this work could be a mixed spinel structure where a part of Co(II) and Fe(III) ions are found in the tetrahedral and octahedral interstices occupied. For a copper content (x) less than 0.06, only the cobalt(II) ions of the tetrahedral sites are substituted. The unit cell parameter of the compound slightly decreases since the ionic radii of copper(II) and cobalt(II) ions are, respectively, 0.57 Å and 0.58 Å in a tetrahedral coordination.48 But for a copper content greater than or equal to 0.06, all the copper ions of the tetrahedral site are substituted and a part of the cobalt(II) ions located in the octahedral sites are also replaced. The unit cell parameter of the compound, therefore, increases since the ionic radius of the copper(II) ion (0.73 Å) is greater than that of cobalt(II) (0.65 Å) in an octahedral coordination.48
 | ||
| Fig. 5 Powder XRD patterns of the decomposition products S1P to S5P. | ||
| Samples | S1P | S2P | S3P | S4P | S5P |
|---|---|---|---|---|---|
| a (Å) | 8.352 | 8.350 | 8.338 | 8.344 | 8.366 |
| D (nm) | 16.8 | 13.8 | 13.7 | 13.5 | 13.6 |
From the (311) diffraction line, the average crystallite diameters (D) of the samples (Table 3) were estimated using the Scherrer equation5,49 and were found to decrease from 16.8 nm in the pure cobalt ferrite with increasing the copper content (x), to a value of 13.5 nm for x = 0.06. Between x = 0.06 and x = 0.08, however, the crystallite sizes increased to 13.6 nm. The observed decrease in crystallite size with doping up to x = 0.06 can be attributed to the substitution of cobalt(II) ions by the relatively smaller copper(II) ions in the tetrahedral site. The increase in size between x = 0.06 and x = 0.08 could be attributed to the fact that the substitution of the cobalt(II) ions by the relatively larger copper(II) ions rather takes place in the octahedral site, which is larger, thus, provoking an increase in the particle size. This phenomenon has been observed by other researchers for values of x higher than 0.08.10,50 Furthermore, the crystallite sizes of the samples vary only slightly with increasing doping. This is normal since the doping levels are very low and the differences in doping concentrations are also very small. The closeness in these values is an indication that nano-sized particles of these materials have been obtained via the coprecipitation method employed.
The SEM images showing the morphology and microstructure of the decomposition products are as presented in Fig. 6 for the three (03) samples S1P (Fig. 6a), S3P (Fig. 6b) and S5P (Fig. 6c) while a further investigate on the morphology carried out by TEM is shown in Fig. 7 (S1P (Fig. 7a) and S2P (Fig. 7b)). The SEM images indicate that there is agglomeration and that the crystals are homogeneous, regular and yeast-like in shape. The particle sizes of about 20 nm estimated from TEM analysis are in good agreement with those (14 nm) deduced from XRD data. The phenomenon of agglomeration observed with these samples can be explained on the basis of their small particle sizes and the absence of steric hindrance both of which facilitate the particles coming together to form agglomerates. The very low agglomeration depicted by TEM analysis suggests that the critical coagulation concentration of the cobalt spinel ferrite samples correlates positively with the specific surface area. In other words, samples with higher specific surface areas are indicative of the fact that it is the surface chemistry rather than the bulk properties that are dominant in spinel ferrite aggregation.51 This agglomeration phenomenon in the cobalt ferrites has also been reported in the literature.28,31,36,52,53
 | ||
| Fig. 6 SEM images of the decomposition products S1P (a), S3P (b) and S5P(c). | ||
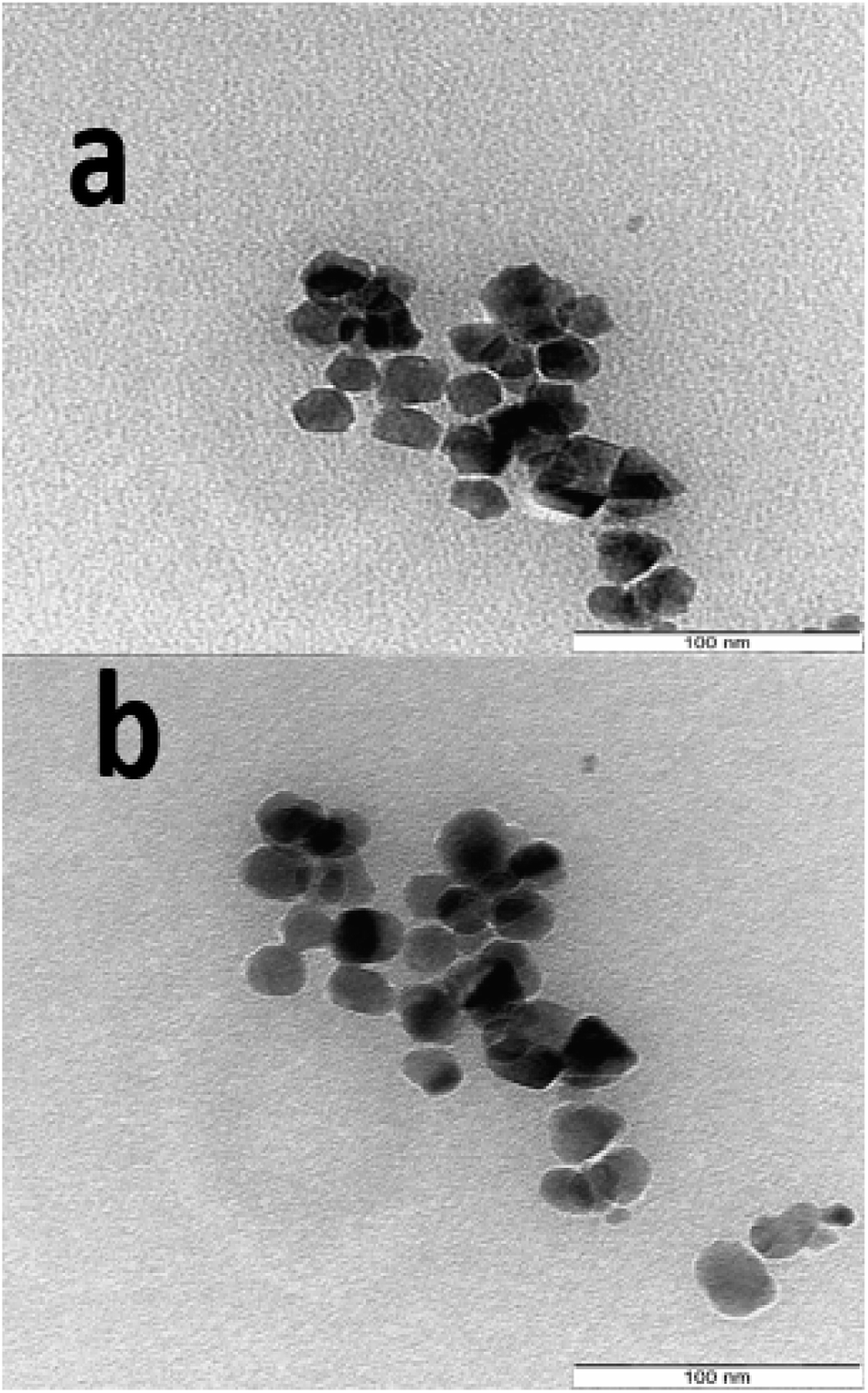 | ||
| Fig. 7 TEM images of the decomposition products S1P (a) and S5P (b). | ||
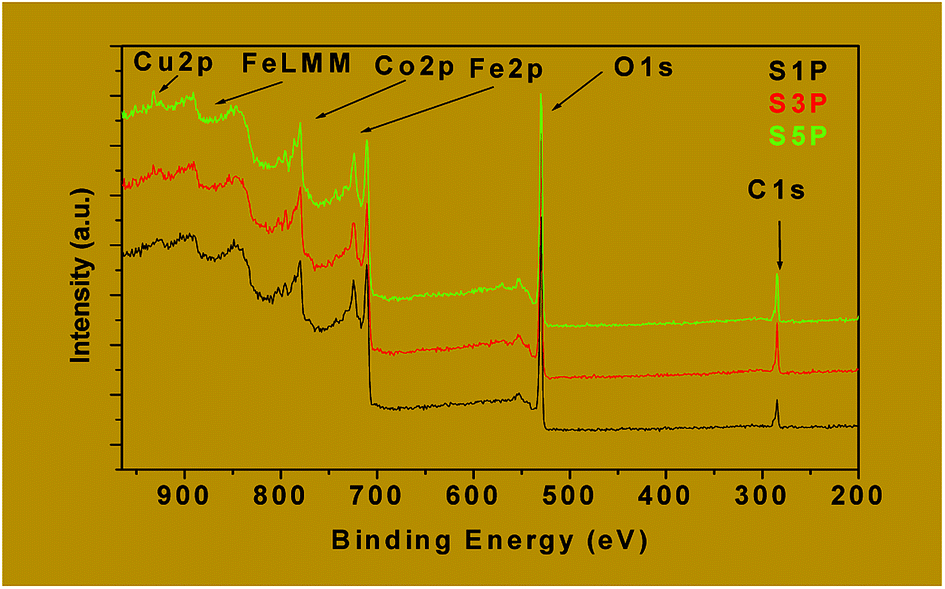 | ||
| Fig. 8 Wide scan XPS spectra of the decomposition products S1P, S3P and S5P. | ||
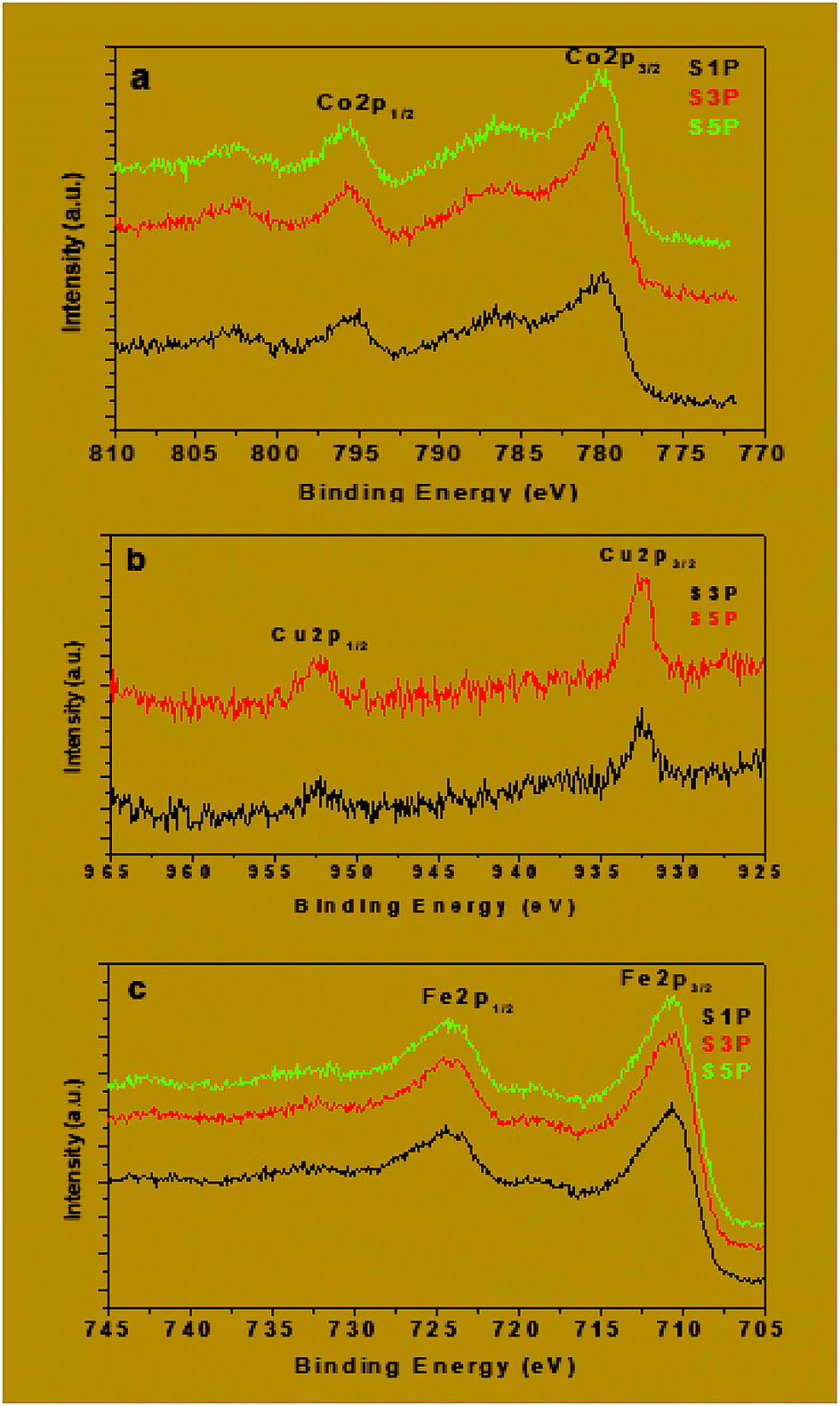 | ||
| Fig. 9 Co2p (a), Cu2p (b) and Fe2p (c) XPS spectra of the decomposition products S1P, S3P and S5P. | ||
The binding energies of Fe2p, Co2p and Cu2p are presented in Table 4. The binding energies of Cu 2p3/2 and Cu 2p1/2 for the samples S3P were obtained, respectively, as 932.6 eV and 952.4 eV while for S5P the values were, respectively, 932.7 eV and 952.4 eV (Fig. 9b and Table 4). This suggests the presence of Cu2+ as reported elsewhere.55,56 Table 4 and Fig. 9a show that the average binding energy of Co 2p3/2 for S1P, S3P and S5P is about 780.4 eV while that of Co 2p1/2 is 795.4 eV for the same samples. Fig. 9c gives an average value of 710.9 eV for Fe 2p3/2 and 719 eV for Fe 2p1/2 for samples S1P, S3P and S5P. The results obtained in this work for Co and Fe have been ascribed to Co2+ and Fe3+ by other researchers.57–60 The slight differences observed in the binding energies of Co and Fe in the three (03) samples, S1P, S2P and S3P, are most likely due to the change in their chemical environment provoked by the presence of the dopant (Cu). These differences could also be as a result of the formation of a mixed spinel as revealed by XRD. The fact that the characteristic peaks of Fe, Cu, Co, Cu+, Co3+ and Fe2+ species are absent from the spectra, indicates that only Fe3+, Co2+ and Cu2+ ions are present on the surface of the compounds and, thus, confirms the proposed formulation Co1−xCuxFe2O4. The XPS results above are consistent with those of XRD for the decomposition products S1P, S2P and S5P, thus, confirming the formation of undoped and copper-doped cobalt ferrite structures and excluding the presence of any mixed phases of CuO, CoO and Fe2O3.
| S1P | S3P | S5P | |
|---|---|---|---|
| Fe 2p1/2 | 724.4 | 724.4 | 724.4 |
| Fe 2p3/2 | 710.9 | 710.8 | 711 |
| Co 2p1/2 | 795.3 | 795.4 | 795.5 |
| Co 2p3/2 | 780.3 | 780.4 | 780.5 |
| Cu 2p1/2 | 0.0 | 952.4 | 952.4 |
| Cu 2p3/2 | 0.0 | 932.6 | 932.7 |
 | ||
| Fig. 10 Magnetic hysteresis loops of the ferrite samples S1P to S5P. | ||
| Samples | Hc (Oe) | Ms (emu g−1) | Mr (emu g−1) | Mr/Ms | K (J m−3) |
|---|---|---|---|---|---|
| S1P | 1284.9 | 67.1 | 21.2 | 0.32 | 0.023 |
| S2P | 1362.8 | 69.5 | 23.8 | 0.34 | 0.029 |
| S3P | 1377.9 | 71.1 | 23.5 | 0.33 | 0.026 |
| S4P | 1632.3 | 73.5 | 26.4 | 0.36 | 0.032 |
| S5P | 1544.5 | 68.5 | 25.2 | 0.37 | 0.028 |
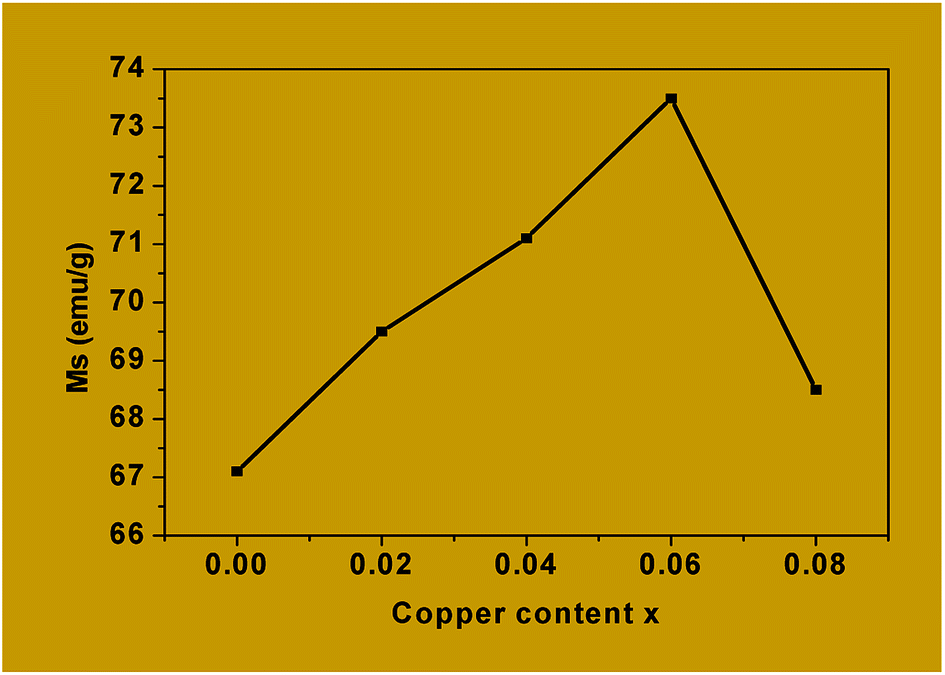 | ||
| Fig. 11 Saturation magnetization Ms versus copper content x in Co1−xCuxFe2O4. | ||
The values of Ms obtained for Co1−xCuxFe2O4 are lower than for the bulk cobalt ferrite and this could be attributed to the very small size of the nanoparticles in the former. For example, the saturation magnetization, Ms, obtained for x = 0 is 67.1 emu g−1 which is much lower than that reported for the bulk cobalt ferrite (≈90 emu g−1).65,66 Canting of spins at the surface of the particles and/or the magnetically dead layer at the surface of the particles is responsible for the observed lower value of magnetization.66–69
Table 5 also shows that the coercivity (Hc) increases from 1284.9 Oe to 1632.3 Oe as x is increased from 0 to 0.06 and then decreases to 1544.5 Oe for x = 0.08. The decrease in coercivity for x = 0.08 is being attributed to the replacement of Co2+ by Cu2+ from the octahedral sites. This, in turn, is responsible for the high magnetocrystalline anisotropy of cobalt ferrite, where the coercivity is related to the magnetocrystalline anisotropy constant through the relation Hc = 2K/μ0Ms.25 The small magnetic anisotropy of Cu2+ ions as compared to that of Co2+ ions reduces the magnetocrystalline anisotropy constant at higher levels of substitution.70,71
The values of Mr/Ms for copper-doped cobalt ferrites which vary from 0.32 to 0.37 are shown in Table 5. These values are less than the typical value of “1.0” expected for single domain isolated ferrimagnetic samples. The observed deviation from unity (1) could be attributed to the interactions between the grains which, in turn, are affected by the grain size distribution in the material.29,72
For CoFe2O4 (x = 0) obtained by calcining the precursor at 400 °C, the coercivity Hc is 1284.9 Oe (Table 5). This value is higher than that of 1100.72 Oe obtained by Samavati et al.,36 heating at 800 °C and that of 1060.24 Oe obtained by Balavijayalakshmi et al.,24 heating at 900 °C. These results clearly indicate that, for the same synthesis route, the processing temperature has an influence on the magnetic properties of the CoFe2O4 ferrite nanoparticles. This can be explained by the fact that by heating above 400 °C, the thermal energy is sufficient to allow the migration of Co2+ ions from the octahedral to the tetrahedral sites and the number of such Co2+ ions migrating increases with the processing temperature. This migration leads to a decrease in the magnetocrystalline anisotropy which, in turn, is responsible in the decrease of coercivity.73 Another possible explanation is that the increase in the size of the particles when calcined at higher temperatures could cause a decrease in the coercivity.
All the results obtained in this work from the magnetic measurements for the materials (Co1−xCuxFe2O4) indicate that they are ferromagnetic owing to the fact that, as can be observed from the hysteresis loops, the samples have a spontaneous magnetization even in the absence of an applied magnetic field. This, therefore, makes them potential candidates for various applications such as in biomedicine and in microwave devices. As far as applications in biomedicine are concerned, the samples obtained in this work have smaller sizes which are the predominant factors in the killing bacteria very fast.23,36 In the same light, Samavati et al.36 and Noppakun et al.23 have synthesized Co1−xCuxFe2O4 (x = 0, 0.3, 0.5, 0.7, 1) by the coprecipitation method using hydroxide ion as ligand, for the former and by the sol–gel method using citric acid as chelating agent, for the latter. The groups obtained particle sizes varying from 30 nm to 20 nm and 42 nm to 38 nm, respectively, as the copper concentration was increased. They are all concluded that the smallest particles have the highest killing rate on the E. coli bacteria. With regards to microwave applications, G. P. Rodrigue74 has showed that the ferrites are used in general in microwave devices. More precisely, they are used as signal amplitude limiters75 and frequency multipliers76 in microwave devices.
Table 6 shows a comparison of results from this work with those of other researchers with regards to the variation of particle sizes with the synthesis method and temperature.
| Powder obtained | Synthesis method | Decomposition temperature (°C) | Particle sizes (nm) | Saturation magnetization Ms (emu g−1) | Ref. |
|---|---|---|---|---|---|
| CoFe2O4 nanospheres | Coprecipitation (HO− ligand) | 800 | 32 | 61.06 | 36 |
| CoFe2O4 nanoangles | Sol–gel | 800 | 42.54 | — | 23 |
| CoFe2O4 nanoplates | Coprecipitation (HO− ligand) | 900 | 37 | 72.06 | 24 |
| CoFe2O4 nanospheres | Hydrothermal | 140 | 40 | 86 | 44 |
| CoFe2O4 nanospheres | Solvothermal | 160 | 30 | 73 | 28 |
| CoFe2O4 nanospheres | Coprecipitation (octanoate ion ligand) | 400 | 20 | 67.1 | This work |
4. Conclusion
We have successfully synthesized Co1−xCuxFe2O4 nanoparticles via the pyrolysis of copper-cobalt-iron hydroxooctanoate prepared by coprecipitation at a relatively low temperature (∼380 °C) and, for the first time, at very low dopant concentrations (x = 0, 0.02, 0.04, 0.06, 0.08). The particles obtained are crystalline and are of nano-size based on calculations using the Scherrer equation which gave crystallite sizes ranging between 14 and 17 nm and also from TEM analysis which showed a particle size of ∼20 nm. Direct evidence for the presence of Cu in the cobalt ferrite crystal structure is provided by XPS which, combined with XRD, showed the formation of a single cubic mixed spinel structure in which a maximum value of x = 0.06 of copper is found in the tetrahedral sites and the remainder in the octahedral sites. VSM measurements confirm the above results obtained by XPS combined with XRD. VSM results also showed that the final materials are ferromagnetic and could be good candidates for various applications including in biomedicine and in microwave devices.On the whole, the present results demonstrate that the coprecipitation synthesis technique used in this work is not only simple, low cost, controllable, reproducible and versatile but can also be applied to prepare a wide variety of target compounds with a good control of phase purity and stoichiometry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely thank Sophie Hermans, Pierre Eloy and Christelle Dzesse for the TGA, XPS and C, H analyses, respectively.Notes and references
- A. Godlyn Abraham, A. Manikandan, E. Manikandan, S. Vadivel, S. K. Jaganathan, A. Baykal and P. S. Renganathan, J. Magn. Magn. Mater., 2018, 452, 380–388 CrossRef.
- A. Manikandan, A. Saravanan, S. Arul and M. Bououdina, J. Nanosci. Nanotechnol., 2015, 15, 4358–4366 CrossRef CAS PubMed.
- A. Manikandan and S. Arul Antony, J. Supercond. Novel Magn., 2014, 27, 2725–2733 CrossRef CAS.
- G. Padmapriya, A. Manikandan, V. Krishnasamy, S. K. Jaganathan and S. Arul Antony, J. Supercond. Novel Magn., 2016, 29, 2141–2149 CrossRef CAS.
- A. M. Ibrahim, M. M. A. El-latif and M. M. Mahmoud, J. Alloys Compd., 2010, 506, 201–204 CrossRef CAS.
- A. Azam, J. Alloys Compd., 2012, 540, 145–153 CrossRef CAS.
- J. Balavijayalakshmi, N. Suriyanarayanan, R. Jayaprakash and V. Gopalakrishnan, Phys. Procedia, 2013, 49, 49–57 CrossRef CAS.
- I. H. Gul, A. Maqsood, M. Naeem and M. N. Ashiq, J. Alloys Compd., 2010, 507, 201–206 CrossRef CAS.
- S. Chakrabarty, A. Dutta and M. Pal, J. Alloys Compd., 2014, 625, 216–223 CrossRef.
- D. M. Jnaneshwara, D. N. Avadhani, B. Daruka Prasad, H. Nagabhushana, B. M. Nagabhushana, S. C. Sharma, S. C. Prashantha and C. Shivakumara, Spectrochim. Acta, Part A, 2014, 132, 256–262 CrossRef CAS PubMed.
- M. Sugimoto, J. Am. Ceram. Soc., 1999, 82, 269–280 CrossRef CAS.
- K. N. Trohidou, Magnetic Nanoparticle Assemblies, Pan Stanford, 1st edn, 2014 Search PubMed.
- A. Manikandan, J. Judith Vijaya and L. John Kennedy, Phys. E, 2013, 49, 117–123 CrossRef CAS.
- D. Maruthamani, S. Vadivel, M. Kumaravel, B. Saravanakumar, P. Bappi, S. Sankar Dhar, A. Habibi-Yangjeh, A. Manikandan and G. Ramadoss, J. Colloid Interface Sci., 2017, 498, 449–459 CrossRef CAS PubMed.
- S. Asiri, M. Sertkol, S. Guner, H. Gungunes, K. M. Batoo, T. A. Saleh, H. Sozeri, M. A. Almessiere, A. Manikandan and A. Baykal, Ceram. Int., 2018, 44, 5751–5759 CrossRef CAS.
- G. Mathubala, A. Manikandan, S. Arul Antony and P. Ramar, Nanosci. Nanotechnol. Lett., 2016, 8, 375–381 CrossRef.
- W. Bayoumi, J. Mater. Sci., 2007, 42, 8254–8261 CrossRef CAS.
- N. M. Deraz and A. Alarifi, J. Anal. Appl. Pyrolysis, 2012, 94, 41–47 CrossRef CAS.
- A. Silambarasu, A. Manikandan and K. Balakrishman, J. Supercond. Novel Magn., 2017, 30, 2631–2640 CrossRef CAS.
- A. T. Ravichandran, J. Srinivas, R. Karthick, A. Manikandan and A. Baykal, Ceram. Int., 2018, 44, 13247–13252 CrossRef CAS.
- S. Rajmohan, V. Jeseentharani, A. Manikandan and J. Pragasam, Nanosci. Nanotechnol. Lett., 2016, 8, 393–398 CrossRef.
- M. Hashim, Alimuddin, S. Kumar, B. H. Koo, S. E. Shirsath, E. M. Mohammed, J. Shah, R. K. Kotnala, H. K. Choi, H. Chung and R. Kumar, J. Alloys Compd., 2012, 518, 11–18 CrossRef CAS.
- N. Sanpo, J. Wang and C. C. Berndt, J. Nano Res., 2013, 22, 95–106 Search PubMed.
- J. Balavijayalakshmi, N. Suriyanarayanan and R. Jayapraksah, Mater. Lett., 2012, 81, 52–54 CrossRef CAS.
- K. Maaz, S. Karim, A. Mashiatullah, J. Liu, M. D. Hou, Y. M. Sun, J. L. Duan, H. J. Yao, D. Mo and Y. F. Chen, Phys. B, 2009, 404, 3947–3951 CrossRef CAS.
- B. K. Bammannavar, L. R. Naik and R. B. Pujar, Prog. Electromagnet. Res. Lett., 2008, 4, 121–129 CrossRef.
- D. Makovec and A. Kodre, J. Nano Res., 2010, 12, 1263–1273 CrossRef.
- J. Ma, J. Zhao, W. Li, S. Zhang, Z. Tian and S. Basov, Mater. Res. Bull., 2013, 48, 214–217 CrossRef CAS.
- E. V. Gopalan, P. A. Joy, I. A. Al-omari, D. S. Kumar, Y. Yoshida and M. R. Anantharaman, J. Alloys Compd., 2009, 485, 711–717 CrossRef CAS.
- S. Prathapani, M. Vinitha, T. V Jayaraman and D. Das, J. Appl. Phys., 2014, 115, 17A502 CrossRef.
- R. Tholkappiyan and K. Vishista, Mater. Sci. Semicond. Process., 2015, 40, 631–642 CrossRef CAS.
- C. Singh, S. Bansal, V. Kumar, K. B. Tikoo and S. Singhal, RSC Adv., 2015, 5, 39052–39061 RSC.
- A. R. Carole Villette and P. Tailhades, J. Solid State Chem., 1995, 117, 64–72 CrossRef.
- R. L. Fomekong, University catholic of Louvain Belgium/University of Yaounde I Cameroon, Doctorate/PhD thesis, 2016.
- R. Lontio, P. Kenfack, D. Magnin, S. Hermans, A. Delcorte and J. Lambi, J. Solid State Chem., 2015, 230, 381–389 CrossRef.
- A. Samavati, M. K. Mustafa, A. F. Ismail and M. H. D. Othman, Mater. Express, 2016, 6, 473–482 CrossRef CAS.
- J. Lambi, A. Nsehyuka, N. Egbewatt, L. Cafferata and A. Arvia, Thermochim. Acta, 2003, 398, 145–151 CrossRef CAS.
- M. Adediran Mesubi, J. Mol. Struct., 1982, 81, 61–71 CrossRef.
- R. Lenin and P. A. Joy, J. Phys. Chem. C, 2016, 120, 11640–11651 CrossRef CAS.
- M. Popescu, C. Turta, V. Meriacre, V. Zubareva, T. Gutberlet and H. Bradaczek, Thin Solid Films, 1996, 274, 143–146 CrossRef CAS.
- S. Mauchauffee, Université Paul Verlaine-Metz, Doctorate/PhD thesis, 2007.
- R. M. Mohamed, M. M. Rashad, F. A. Haraz and W. Sigmund, J. Magn. Magn. Mater., 2010, 322, 2058–2064 CrossRef CAS.
- H. A. Hamad, M. M. A. El-latif, A. B. Kashyout, W. A. Sadik and M. Y. Feteha, Russ. J. Gen. Chem., 2014, 84, 2205–2210 CrossRef CAS.
- L. Zhao, H. Zhang, Y. Xing, S. Song, S. Yu and W. Shi, J. Solid State Chem., 2008, 181, 245–252 CrossRef CAS.
- L. Avazpour, M. A. Zandi, M. R. Toroghinejad and H. Shokrollahi, J. Alloys Compd., 2015, 637, 497–503 CrossRef CAS.
- R. Fareghi-Alamdari, F. Zandi and M. H. Keshavarz, RSC Adv., 2015, 5, 71911–71921 RSC.
- H. Kumar, J. P. Singh, R. C. Srivastava, P. Negi, H. M. Agrawal and K. Asokan, J. Nanosci., 2014, 2014, 1–10 CrossRef.
- R. D. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- L. Yao, Y. Xi, G. Xi and Y. Feng, J. Alloys Compd., 2016, 680, 73–79 CrossRef CAS.
- M. Hashim, S. E. Shirsath, S. S. Meena, R. K. Kotnala, S. Kumar, P. Bhatt, R. B. Jotania and R. Kumar, Mater. Chem. Phys., 2013, 141, 406–415 CrossRef CAS.
- D. Zhou, Z. Ji, X. Jiang, D. R. Dunphy, J. BrinkerA and A. Keller, PLoS One, 2013, 8, e81239 CrossRef PubMed.
- Z. Gu, X. Xiang, G. Fan and F. Li, J. Phys. Chem. C, 2008, 112, 18459–18466 CrossRef CAS.
- R. Lontio, J. Ngolui, G. Roland, P. Kenfack, H. M. T. Kamta, C. Ngnintedem and A. Delcorte, J. Solid State Chem., 2016, 241, 137–142 CrossRef.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Minesota, United States of America, 1992 Search PubMed.
- Y. B. Yaolin Xu, J. Sherwood, Y. qin and R. A. Holler, Nanoscale, 2015, 7, 12641–12649 RSC.
- R. Jamatia, A. Gupta and A. Kumar, ChemistrySelect, 2016, 4, 852–860 CrossRef.
- Z. Gu, X. Xiang, G. Fan and F. Li, J. Phys. Chem. C, 2008, 112, 18459–18466 CrossRef CAS.
- X. Yang, X. Wang and Z. Zhang, J. Cryst. Growth, 2005, 277, 467–470 CrossRef CAS.
- P. M. Kouotou and Z. Tian, Phys. Status Solidi A, 2015, 212, 1508–1513 CrossRef CAS.
- W. P. Wang, H. Yang, T. Xian and J. L. Jiang, Mater. Trans., 2012, 53, 1586–1589 CrossRef CAS.
- S. U. Bhasker and M. V. R. Reddy, J. Sol-Gel Sci. Technol., 2015, 396–402 CrossRef.
- S. Bhukal, T. Namgyal, S. Mor, S. Bansal and S. Singhal, J. Mol. Struct., 2012, 1012, 162–167 CrossRef CAS.
- T. Tatarchuk, M. Bououdina, W. Macyk, O. Shyichuk, N. Paliychuk, I. Yaremiy, B. Al-Najar and M. Pacia, Nanoscale Res. Lett., 2017, 12, 141 CrossRef PubMed.
- S. Briceño, H. Del Castillo, V. Sagredo, W. Bramer-Escamilla and P. Silva, Appl. Surf. Sci., 2012, 263, 100–103 CrossRef.
- F. Bensebaa, F. Zavaliche, P. L. Ecuyer, R. W. Cochrane and T. Veres, J. Colloid Interface Sci., 2004, 277, 104–110 CrossRef CAS PubMed.
- P. Jeppson, R. Sailer, E. Jarabek, J. Sandstrom, B. Anderson, M. Bremer and D. G. Grier, J. Appl. Phys., 2006, 100, 114324 CrossRef.
- R. H. Kodama, J. Magn. Magn. Mater., 1999, 200, 359–372 CrossRef CAS.
- A. Beitollahi, R. Sani, Y. V Maksimov and I. P. Suzdalev, NSTI-Nanotech., 2006, 1, 431–434 CAS.
- L. Ajroudi, N. Mliki, L. Bessa, V. Madigou and S. Villain, Mater. Res. Bull., 2014, 59, 1–49 CrossRef.
- G. Baldi, D. Bonacchi, C. Innocenti, G. Lorenzi and C. Sangregorio, J. Magn. Magn. Mater., 2007, 311, 10–16 CrossRef CAS.
- S. Ammar, A. Helfen, N. Jouini, F. Fiévet, I. Rosenman, F. Villain, P. Molinie and M. Danot, J. Mater. Chem., 2001, 111, 86–192 Search PubMed.
- S. Ayyappan, S. Mahadevan, P. Chandramohan, M. P. Srinivasan, J. Philip and B. Raj, J. Phys. Chem. C, 2010, 114, 6334–6341 CrossRef CAS.
- P. Tailhades, C. Villette and A. Rousset, J. Solid State Chem., 1998, 141, 56–63 CrossRef CAS.
- G. P. Rodrigue, J. Appl. Phys., 1969, 40, 929–937 CrossRef.
- R. W. Degrasse, J. Appl. Phys., 1959, 30, 1555 CrossRef.
- W. P. Ayres, P. H. Vartanian and J. L. Melchor, J. Appl. Phys., 1956, 27, 188–189 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |