
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving the inhibitory effect of CXCR4 peptide antagonist in tumor metastasis with an acetylated PAMAM dendrimer†
Changliang Liu a,
Hongyang Duanab,
Zijian Zhaoa,
Wenzhe Liab,
Lilusi Maa,
Xiaocui Fang*a,
Chen Wang*a and
Yanlian Yang*a
a,
Hongyang Duanab,
Zijian Zhaoa,
Wenzhe Liab,
Lilusi Maa,
Xiaocui Fang*a,
Chen Wang*a and
Yanlian Yang*a
aCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Key Laboratory of Biological Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: fangxc@nanoctr.cn; wangch@nanoctr.cn; yangyl@nanoctr.cn; Tel: +86-10-82545561 Tel: +86-10-82545559
bAcademy for Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China
First published on 30th November 2018
Abstract
The metastasis of breast cancer is one of the main factors resulting in the high fatality of patients. Although many antagonists have been developed to inhibit the metastasis of breast cancer, their practical application has been limited because of the poor solubility of many chemotherapeutic drugs in the physiological environment. Herein, a complex of E5 peptide antagonist and acetylated PAMAM G5 (PAC80) has been constructed to enhance the solubility of the peptide antagonist. The E5 peptide antagonist has been designed and it was confirmed that it could specifically bind to CXCR4, which is a chemokine receptor involved in the metastasis of several types of cancers, in our previous work. The results demonstrated that PAC80 could significantly increase the solubility of the E5 peptide in the physiological environment, as well as the affinity of the E5 peptide to CXCR4-positive cell lines, and the inhibitory effect of the E5 peptide for cell migration in vitro. Meanwhile, the passive lung metastasis model of breast cancer was established and the anti-tumor metastasis of the PAC80–E5 complex was evaluated in vivo. The results show that the PAC80–E5 complex demonstrated excellent inhibition for the tumor metastasis at an E5 dosage of 10 and 20 mg kg−1. These effects indicate a feasible strategy to apply the PAC80–peptide complex in cancer therapies to improve the solubility and bioavailability.
1. Introduction
Breast cancer is one of the most common malignancies and lethal diseases in women.1 The high fatality rate of breast cancer patients results mainly from metastasis, which consists of multiple steps including the escape of tumor cells from the primary site, reaching systemic circulation and surviving in the circulation, adhering to the capillaries and extravasating into the targeting organs.2,3 One of the major reasons for the distance organ specific metastasis is the high chemokine expression in target organs and the presence of their cognate receptor in breast cancer cells.4The C-X-C chemokine receptor-4 (CXCR4) has been found to be relevant to the specific distant metastasis of various types of human cancers, including breast cancers,5,6 prostate cancers,7,8 colon cancers9,10 and multiple myeloma.11 High expression levels of CXCR4 in cancer cells enable them to migrate to their target tissues, such as lung, liver, brain and bone, in which there is an abundant source of the natural ligand CXCL12. CXCL12 can guide the directional migration of cancer cells via a CXCL12 chemotactic gradient. For instance, more and more published literature has reported that, myeloid12,13 and lymphoid14,15 leukemia cells have a high CXCR4 expression level, which induces leukemia cells migration towards stromal cells secreting CXCL12, and thereby reside in the tumor microenvironment that protects the leukemia cells from chemotherapy.16,17
Nowadays, in order to inhibit breast cancer metastasis, great efforts have been made to design antagonists, such as small molecules,18,19 antibodies20 and peptides,21,22 to inhibit the interaction between the chemokines and their receptors. Compared to small molecules and antibodies, peptides can be readily synthesized, metabolically cleaved and rapidly cleared from the body without introducing serious immune responses and toxic side effects, which shows promising potential in cancer therapies. However, poor solubility and the lack of serum stability limit the in vivo efficacy of the peptide drugs. To improve the solubility and bioavailability, various drug delivery systems have been developed, such as polymers, liposomes, micelles and other nanoparticles.23,24 Polyamidoamine (PAMAM) dendrimers have attracted considerable research interest as drug carriers owing to their unique properties. The hyperbranched structure and abundant surface amino groups mean that the dendrimer can be easily modified by acetylation25 or PEG26 to improve their biocompatibility.27 In addition, their monodispersed tree-like structure with many empty interior cavities enables the dendrimers to encapsulate drug molecules that have poor solubility.27 Partial acetylation has been reported to be used to neutralize the cationic surface of the dendrimers, which can reduce their cytotoxicity and nonspecific binding and also increase their solubility in the physiological environment.25 The remaining non-acetylated primary amino groups can also be used for covalent and non-covalent interaction with various functional molecules.28
According to previous work conducted in our lab, the designed E5 peptide targeting CXCR4 has been shown to inhibit CXCL12 induced migration of multiple leukemia cells in vitro and to prolong the survival of mice transplanted with acute myeloid leukemia cell in vivo.29 However, the E5 peptide has poor solubility in the physiological environment owing to its sensitivity to salt ions, which leads to its inefficiency in the inhibition of cancer cell migration. Thus, in this present work, an acetylated PAMAM G5 (PAC80) has been constructed to enhance the solubility of the E5 peptide (Scheme 1). The results demonstrated that the introduction of PAC80 not only improved the solubility of the E5 peptide, but also enhanced the binding affinity of the E5 peptide to CXCR4-positive cancer cells. Therefore, compared with free E5 peptide, the E5 peptide has a better inhibitory effect on CXCR4/CXCL12 mediated cancer cell migration in the presence of PAC80 both in vivo and in vitro. This method sheds light on the development of dendrimer-based drug delivery systems and provides a possible avenue towards cancer therapy.
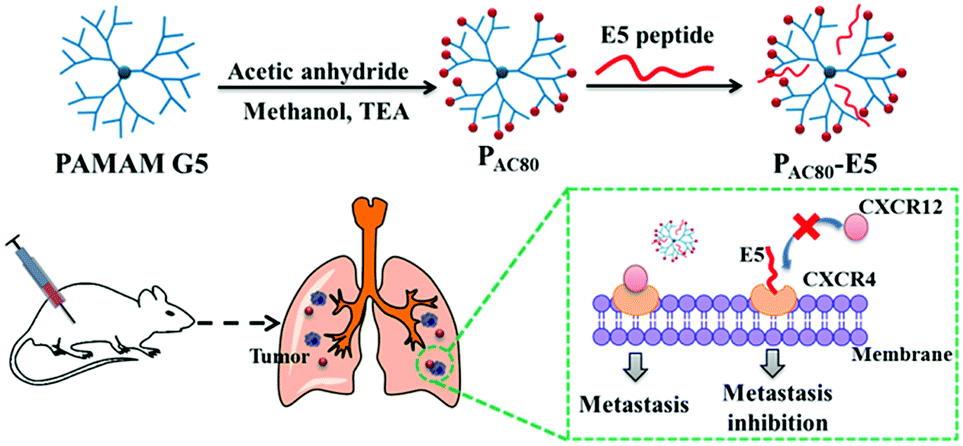 | ||
| Scheme 1 Schematic illustration of the preparation of the PAC80–E5 complex and the process of anti-tumor metastasis of the E5 peptide in the presence of PAC80. | ||
2. Experimental
2.1 Materials
PAMAM G5 dendrimer with an ethylenediamine core, acetic anhydride and 2,5-dihydroxybenzoic acid (DHB) were purchased from Sigma-Aldrich (USA). Triethylamine, deuteroxide, anhydrous methanol, glutamine, sodium pyruvate, Hepes, glucose and acetonitrile were purchased from J&K Chemical (Beijing, China). The Cell Counting Kit-8 (CCK-8) was purchased from Fanbo Biochemicals (Beijing, China). RPMI-1640, DMEM, opti-MEM medium, penicillin and streptomycin were purchased from Gibco Inc. (Thermo Fisher, USA). E5 peptide (GGRSFFLLRRIQGCRFRNTVDD) was synthesized by Guoping Pharmaceutical Co., Ltd. (Anhui, China). CXCL12 was purchased from R&D Systems (USA), and lysis buffer was purchased from Pierce (USA).2.2 Synthesis of partially acetylated PAMAM (PAC80)
PAC80 was synthesized by the reaction of the periphery amino group of the PAMAM dendrimer with acetic anhydride. Briefly, 26 mg (0.9 μmol) PAMAM G5 was dissolved in 2 mL anhydrous methanol, and then 6.8 μL (72 μmol) acetic anhydride and 12.48 μL (90 μmol, 25% molar excess) triethylamine were added. The mixture was stirred at room temperature for 4 h. The excess solvent and reagents in the reaction system were removed using rotary evaporation followed by dialyzing against deionized water for three cycles. The pure acetylated dendrimer was lyophilized to yield the product. PAC60, PAC100 were synthesized following a similar procedure with different molar equivalents of acetic anhydride.2.3 Characterization of PAC80
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Next, 2 μL of the samples was spotted onto the target plate (Bruker Daltonics, USA), and the solvent was allowed to evaporate under vacuum conditions. Analysis was performed in the linear mode. The PAMAM dendrimer that did not undergo acetylation was used as the control.
:1, v/v). Next, 2 μL of the samples was spotted onto the target plate (Bruker Daltonics, USA), and the solvent was allowed to evaporate under vacuum conditions. Analysis was performed in the linear mode. The PAMAM dendrimer that did not undergo acetylation was used as the control.![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ); δ 2.68–2.74 (–CH2CH2CO–); δ 2.95–3.20 (–CONHCH2CH2– and –CH2CH2NH2); δ 1.89 (–COCH3).
); δ 2.68–2.74 (–CH2CH2CO–); δ 2.95–3.20 (–CONHCH2CH2– and –CH2CH2NH2); δ 1.89 (–COCH3).2.4 Cell lines and animals
Murine breast cancer 4T1 cells and murine stromal MS-5 cells were kindly provided by Professor Haiyan Xu, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College (Beijing, China). Human breast cancer MCF-7 and MDA-MB-231 cells were gifted by Professor Chunying Chen, National Center for Nanoscience and Technology (Beijing, China). MS-5 cells were cultured in RPMI-1640 culture medium supplemented with high glucose, 10% fetal bovine serum (FBS), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. MCF-7 and MDA-MB-231 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with high glucose, 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. 4T1 cells were cultured in RPMI-1640 culture medium supplemented with 2 mmol L−1 glutamine, 10 mmol L−1 Hepes, 1 mmol L−1 sodium pyruvate, 4.5 g L−1 glucose, 10% FBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. All of the cells were maintained at conditions of 37 °C and 5% CO2.Female BALB/C mice (6–8 weeks, 18–20 g) were purchased from Charles River Laboratory Animal Technology Co., Ltd. (Beijing, China). All animal procedures were performed in strict accordance with the Guidelines for Care and Use of Laboratory Animals of Peking University and approved by the Animal Ethics Committee of National Center for Nanoscience and Technology, Chinese Academy of Sciences.
2.5 Cytotoxicity of PAC80
A Cell Counting Kit-8 (CCK-8) assay was used to measure the cytotoxicity of the PAMAM and acetylated PAMAM on MDA-MB-231 and 4T1 cell lines. Briefly, MDA-MB-231 and 4T1 cells were seeded into 96-well plates at a density of 4000/well in 200 μL medium and cultured for 24 h at conditions of 37 °C and 5% CO2. Then, different acetylated PAMAM was added into the cells over a range of concentrations (0, 5, 10, 15, 20 μM). Cells without treatment were used as the control. After incubating for 48 h, 10 μL of the CCK-8 reagents were added into each well and followed by incubation for another 2 h. The absorbance at 450 nm was determined using a plate reader (Infinite M200, Tecan, Switzerland). The cell viability was calculated using the following formulas: cell viability% = Atreated/Acontrol × 100%, in which Atreated was the absorbance value of the cells with samples, Acontrol was the absorbance value of the cells without samples. Each assay was repeated five times.2.6 Preparation of PAC80–E5 complex
E5 peptide and PAC80 were dissolved in sterile water to the concentration of 10 mg mL−1 and 200 mg mL−1, respectively. A certain amount of the PAC80 aqueous solution was mixed into 10 mg mL−1 of the E5 aqueous solution. After it was fully mixed, the aqueous solution was then added into an appropriate amount of 10× PBS buffer and deionized water to prepare a solution of PAC80–E5 complex with a final concentration of then E5 peptide of 1 mg mL−1 and the PBS at 1×. In the subsequent experiments, the solution could be diluted to the concentration needed by 1× PBS or culture medium.2.7 Semi-quantitative fluorescence assay
To investigate the solubility of the E5 peptide in the absence and presence of PAC80, a semi-quantitative fluorescence assay was performed in PBS buffer. PAC80 bound with FITC labelled E5 peptide with different molar ratios of 2:1, 1:1, 1:2, 1:3 and 1:4 were prepared according to the above mentioned procedure. At the initial moment, all of samples were centrifuged, and 100 μL supernatant per sample was transferred to a 96-well plate. Then, the fluorescence intensities of the FITC labelled E5 peptide were measured at an excitation wavelength of 488 nm with an emission wavelength of 525 nm using a plate reader. Then the samples were stored at 4 °C in the dark and tested as above after incubating for 24 h. The fluorescent intensity of the free FITC labelled E5 peptide was set as the control. The relative solubility of the FITC labelled E5 peptide in each group was calculated by comparison with the control group.
2.8 Flow cytometry assay
The affinity of the E5 peptide towards CXCR4 was evaluated in MS-5, MCF-7, MDA-MB-231 and 4T1 cell lines using flow cytometry (FCM). Briefly, the cells were collected and washed once with washing buffer (PBS containing 2% FBS) and then resuspended with complete medium to 5 × 106/mL. 50 μL of the cell suspension was added into the tube and centrifuged at 800 g for 3 min. Then, the supernatant was removed carefully and 60 μL of the FITC labelled E5 peptide, or PAC80–E5 complex (the molar ratio of PAC80:E5 = 1:3) at different concentrations in complete medium was added into the tube. The cells were incubated at room temperature for 1 h and then washed twice with 1 mL washing buffer. The supernatant was then removed and the cells were resuspended with 500 μL washing buffer. 1 × 104 cells were analyzed using a C6 Accuri flow cytometer (BD Biosciences, USA).
2.9 Laser scanning confocal microscope assay
The affinity of the E5 peptide towards CXCR4 was also investigated using MDA-MB-231 cells as a representative cell model and examining using laser scanning confocal microscopy (LSCM). 1 × 105 MDA-MB-231 cells were seeded in glass bottom dishes and incubated for 24 h. Then the cells were washed three times and 1 mL of complete medium containing FITC labelled E5 peptide, or PAC80–E5 (the molar ratio of PAC80:E5 = 1:3), was added. The final concentration of the FITC labelled E5 peptide was 1 μM. After incubating for 1 h, the cells were washed and mounted with complete medium containing Hoechst 33258. The cells were then washed with PBS after incubating for 15 min. Images were photographed using a laser scanning confocal microscope (LSM760, Zeiss, Germany).
2.10 Transwell assay
The transwell assay was applied using millicell hanging cell culture inserts for MCF-7, MDA-MB-231 and 4T1 cell lines (8 μm, Millipore, Switzerland). The MCF-7 (1 × 105 cells per chamber), MDA-MB-231 (1 × 105 cells per chamber) and 4T1 (5 × 104 cells per chamber) cells were added into the upper chambers of the inserts in opti-MEM medium containing different concentrations of E5 or PAC80–E5 (the molar ratio of PAC80:E5 = 1:3). Then, the inserts were placed into a well in a 24-well plate containing 800 μL culture medium in the presence or absence of CXCL12. The concentration of CXCL12 was 100 ng mL−1. After incubation for 24 h at 37 °C, the top surface of the insert was scraped using a cotton swab and the cells on the lower surface of the membrane were fixed and stained with crystal violet. Cells that had migrated to the bottom of the membrane were visualized and counted using a digital microscope (DMI3000B, Leica, Germany).
2.11 Western blot analysis
MCF-7 and MDA-MB-231 cells were maintained overnight in a medium containing 0.5% FBS at 1 × 106 cells per well in a 6-well plate. After incubating for 24 h, CXCL12 was added into the medium to a final concentration of 100 ng mL−1. Then, the E5 peptide and PAC80–E5 was added into the medium to a final concentration of E5 peptide of 1 μM. After incubating for 15 min at 37 °C, the cells were washed with PBS and lysed in RIPA lysis buffer supplemented with protease inhibitors and a phosphatase inhibitors mixture on ice for 1 h. The lysates were clarified by centrifugation at 14000 g for 10 min. Equal protein (10 μg) was loaded and separated on 10% bis–tris gels (Invitrogen, USA), and transferred to PVDF membranes (0.45 μm, Millipore, USA). Phosphorylated and total proteins were probed with specific primary antibodies (Akt, phosphorylated-Akt, Erk, phosphorylated-Erk, p38, phosphorylated-p38, GAPDH; Cell Signaling Technology, USA) overnight at 4 °C and the secondary antibodies were labelled with HRP (Cell Signaling Technology, USA) for 1 h at room temperature. The immunocomplex on the membrane was visualized using ChemiDoc® Touch Imaging System (Bio-Rad, USA) with SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Scientific, USA).
2.12 Tumor metastatic inhibition assay in vivo
4T1 cells were injected into the mice at 5 × 104 cells per mouse via the tail vein to develop a lung metastatic model of breast cancer.29 The mice were randomly divided into five groups (five mice per group) and maintained for 24 h. The E5 and PAC80–E5 (the molar ratio of PAC80:E5 = 1:3) were injected into the celiac plexus at an E5 dose of 10 mg kg−1 and 20 mg kg−1 every two days (on day 1, 3, 5, 7, 9, 11, 13, 15). Sterile water was included as a control. The body weight of each mouse was measured every two days. At day 16, the mice were sacrificed and the lung tissues from each group were collected and photographed. The number of macroscopic metastatic nodules per lung was recorded carefully to calculate the inhibitory effects on lung metastasis of breast cancer. Furthermore, histological examinations of the lung tissues were performed using hematoxylin and eosin (H&E) staining to detect the metastatic foci.
2.13 Histological analysis
Histological analysis was also performed to evaluate the safety and tissue toxicity of E5 and PAC80–E5. At the end of the dose, all of the mice were sacrificed and the main organs (heart, liver, spleen, and kidney) were excised and fixed in 4% paraformaldehyde solution followed by embedding in paraffin. The sliced organ tissues were mounted on glass slides and were stained using H&E and observed using a digital microscope.2.14 Statistical analysis
All of the experiments were carried out in triplicate and a Student's t-test was performed to assess the statistical significance of the results (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).3. Result and discussion
3.1 Synthesis and characterization of acetylated PAMAM G5
The partially acetylated PAMAM dendrimer was synthesized by the reaction of the surface amino and acetic anhydride in the presence of triethylamine. The degree of acetylation was analyzed using MALDI-TOF MS and 1H NMR. As shown in Fig. 1a, MALDI-TOF MS data demonstrated that the molecular weight of PAMAM increased as the number of surface acetyl groups increased [MW (PAMAM) = 28825, MW (PAC80) = 31998], suggesting the formation of a covalent bond. The average number of acetyl group on the PAMAM periphery was calculated, with an average value of 73.8 [MW (–COCH3) = 43].
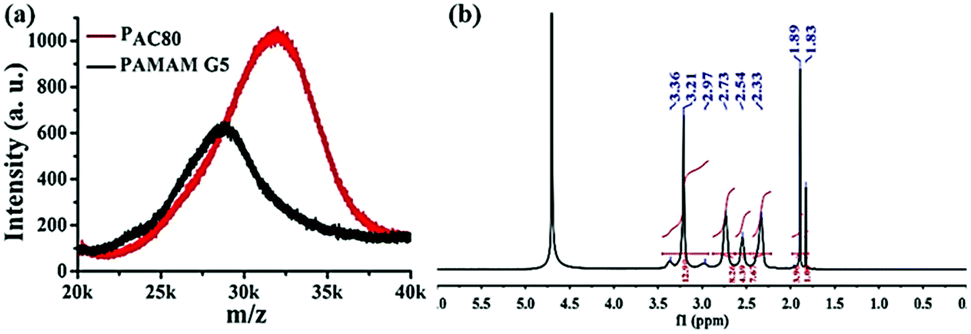 | ||
| Fig. 1 (a) MALDF-TOF MS spectra of PAMAM G5 and acetylated PAMAM G5 (PAC80). (b) 1H NMR spectra of PAC80 (D2O as solvent). | ||
In addition, 1H NMR measurements were also used to verify the chemical structure of the PAC80 (Fig. 1b). A new peak at 1.89 ppm corresponding to the proton of the methyl next to the acylamino group (–NHCOCH3) appeared, indicating the successful modification of the PAMAM by the acetyl group.28 As a result of the integral of the proton signal of the methyl group (a sharp peak at 1.89 ppm) and the protons of the methylene next to the amido group (–CONHCH2CH2 and –CH2CH2NH2, 2.95–3.20 ppm) in the PAMAM dendrimer, the grafting ratio of the acetyl group on the PAMAM periphery was calculated with an average value of 73, which was consistent with the results of MALDI-TOF MS.
The particle sizes of the PAMAM dendrimer and the PAC80 conjugates were determined using DLS. The PAC80 conjugates (6.81 ± 0.96 nm) showed an increased particle size compared with the unmodified PAMAM dendrimer (5.85 ± 0.83 nm). This result was in agreement with the MALDI-TOF MS and 1H NMR data, which indicated the successful acetylation of PAMAM. Moreover, the zeta potential of the unmodified PAMAM was 29.7 ± 5.68 mV and 14.1 ± 1.15 mV in deionized and incubation medium, respectively, while the zeta potential was decreased to 5.95 ± 0.2 mV and 2.85 ± 0.66 mV respectively after acetylation. These results reveal that the cationic groups of PAMAM were neutralized as the newly introduced acetyl group can block the surface amines in the dendrimer and further shield the positive charge. As a result, the acetyl groups were successfully conjugated to the PAMAM by anhydride and the amine interaction. The almost neutralized nature of the acetylated dendrimer could then improve the biocompatibility and safety of the carrier.
3.2 Cytotoxicity of PAC80 in vitro
The cytotoxicity of the acetylated PAMAM was investigated in MDA-MB-231 and 4T1 breast cancer cells. The results indicated that the dendrimer that had different degrees of surface amine acetylation displayed a different cytotoxicity in a concentration dependent manner (Fig. 2). Compared with the unmodified dendrimers, the cytotoxicity of the acetylated PAMAM was reduced significantly, the decrease of cationic groups through the neutralization of the acetyl groups contributed to this. Meanwhile, the cytotoxicity for both of the two kinds of cancers cell lines exhibited a correlative reduction as the acetylation degree increased. In general, the cell viability increased with the reduction of the surface amines, which is probably caused by the intrinsic toxicity associated with the cationic density. Compared with PAC80 and PAC100, PAC60 exhibited the highest cytotoxicity for both the MDA-MB-231 and 4T1 cancer cells. The cell viabilities reduced to 70% after PAC60 treatment at a concentration of 20 μM for the MDA-MB-231 cancer cells (Fig. 2a) and even dropped to below 40% after the same treatment in the 4T1 cancer cells (Fig. 2b). However, the cell viabilities of the two kinds of cancer cell lines were still above 70% after incubation with PAC80 and PAC100 for 48 h at the concentration of 20 μM, which revealed the increased safety of the dendrimers with a higher degree of acetylation (≥80 acetyl groups). These results suggest that the neutralization of the cationic groups of the PAMAM dendrimer could reduce the intrinsic toxicity. This could be useful for predicting the appropriate surface modifications required to minimize the toxicity of PAMAM dendrimers in the size range studied.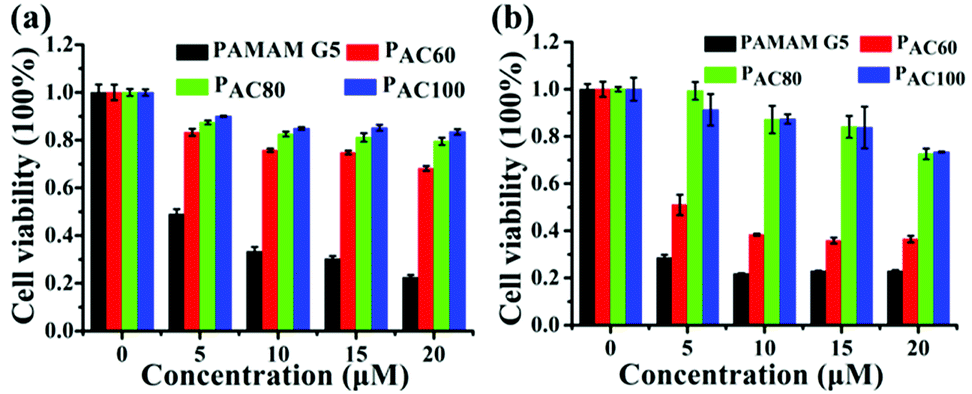 | ||
| Fig. 2 Cell viability of: (a) MDA-MB-231 cells; and (b) 4T1 cells after incubating with PAMAM G5, PAC60, PAC80 and PAC100. | ||
3.3 Increased solubility and stability of E5 peptide by PAC80
To investigate the solubility of the E5 peptide in the absence and presence of PAC80, we performed a semi-quantitative fluorescence assay to obtain the relative concentration of dissolved E5 in the PBS buffer. The final concentration of the FITC labelled E5 was 0.1 mM in the experiment. The PAC80 was added into the E5 peptide solution with a molar ratio of PAC80:E5 of 2:1, 1:1, 1:2, 1:3 and 1:4 to obtain the PAC80–E5 complex. The quantification of the dissolved E5 in the absence and presence of different amounts of PAC80 was performed using the relative fluorescent intensity (Fig. 3b). We found that when E5 was dissolved in PBS, the actual concentration of the E5 peptide was much lower at 0 h compared with the PAC80–E5 complex, and only one third of the fluorescent intensity was recorded compared with the PAC80–E5 complex at a 2:1 molar ratio (PAC80:E5), which indicated that the PAC80 can improve the solubility of E5 in the PBS buffer. When the molar ratios of PAC80 and E5 were ranged from 2:1 to 1:3, the concentration of dissolved E5 in the supernatant was maintained at a high level. The dissolved E5 was decreased obviously when the molar ratio of PAC80 and E5 was 1:4, which could result from the saturation of the interaction between PAC80 and E5. After 24 h, the concentration of the dissolved E5 significantly dropped to half of the initial time (0 h). This result demonstrated that the E5 peptide had an aggregation tendency in PBS as time went on. After binding with PAC80, the concentration of the dissolved E5 changed slightly during 24 h, which revealed that PAC80 inhibited E5 aggregation in the PBS buffer.
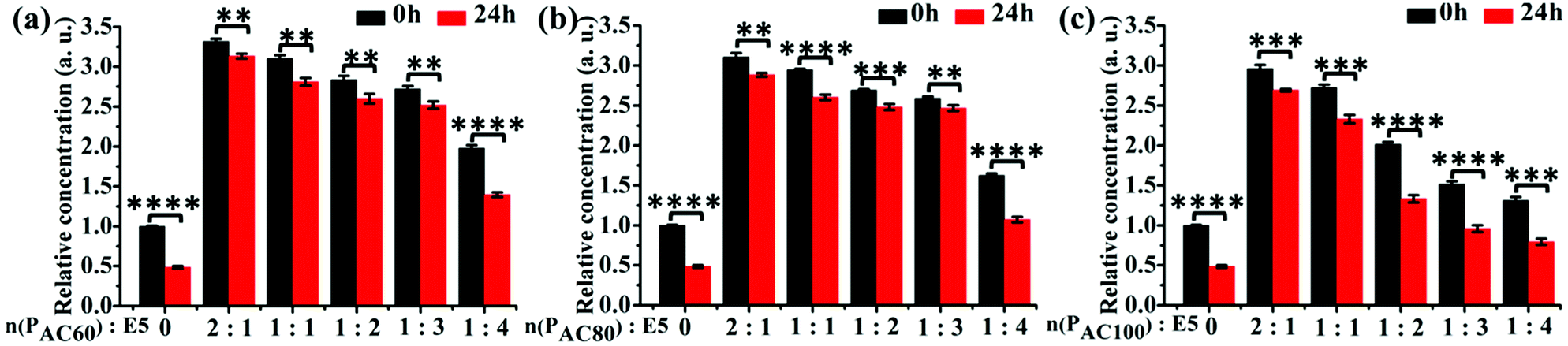 | ||
| Fig. 3 The relative solubility of the E5 peptide in the presence of: (a) PAC60; (b) PAC80; and (c) PAC100 detected using a semi-quantitative fluorescence assay over 24 h. The acetylated PAMAM G5 bound with the FITC labeled-E5 peptide with different molar ratios of 2:1, 1:1, 1:2, 1:3 and 1:4. The concentration of E5 was 0.1 mM in the experiment. Error bars represent the standard deviation (n = 3). | ||
Furthermore, the solubility of E5 in the presence of PAC60 and PAC100 were also investigated under the same experimental conditions (Fig. 3a and c). The results indicated that PAC60 improved the solubility of E5 and inhibited its aggregation when the molar ratios of PAC60:E5 ranged from 2:1 to 1:3. In the presence of PAC100, we found that the dissolved E5 in the supernatant was maintained at a high concentration level only when the molar ratio of PAC100:E5 was 2:1 and 1:1. On the whole, the remaining surface amine groups on the partially acetylated dendrimers could affect the drug loading through hydrogen bonds and electrostatic interactions, and affect the cytotoxicity of the carriers in parallel. Thus, the observations from the current study indicate that PAC80 is an optimized strategy that could be utilized for the delivery of the E5 peptide at a molar ratio of 1:3 to form a complex of PAC80–E5.
The stability of the PAC80–E5 complex was then detected using the DLS assay in deionized water and PBS buffer, as shown in Fig. S3.† The results revealed that the free E5 peptide had a tendency to aggregate in both water and PBS buffer. Larger aggregates were observed in the PBS buffer and these resulted from the ionic sensitivity of the E5 peptide. However, the mean diameter of the PAC80–E5 complex was small and hardly changed within 24 h in deionized water and PBS buffer, which suggested that the complex had a good stability over time.
3.4 Enhanced affinity of E5 peptide to cancer cells by PAC80
To investigate the binding affinity of the E5 peptide and PAC80–E5 complex to CXCR4 in cells, four cell lines with different CXCR4 expression levels were chosen, including MS-5, MCF-7, MDA-MB-231 and 4T1 cell lines. As shown in Fig. S1,† the CXCR4 expression levels in MCF-7, MDA-MB-231, 4T1 and MS-5 were determined to be 32.0%, 51.9%, 52.6% and 1.1%, respectively. Then, the four cell lines were incubated with different concentrations of E5 peptide in the absence and presence of PAC80. After incubation for 1 h, the cells were analyzed using FCM. As shown in Fig. 4, the percentage of fluorescent cells increased gradually with the increase of the concentration of E5. However, the amount of the fluorescent cells after incubation with free E5 was much lower than that observed after incubation with PAC80–E5 in MDA-MB-231, 4T1 and MCF-7 cell lines under the same conditions. The percentage of fluorescent cells was 42.3% and 35.3% for MDA-MB-231 and 4T1 cells, respectively, after incubation with free E5 at the concentration of 10 μM, while the percentage reached 84.1% and 68.0% for the MDA-MB-231 and 4T1 cancer cells, respectively, after incubation with PAC80–E5 at the same concentration as the E5 peptide. Meanwhile, the results also showed that the binding amounts of the PAC80–E5 to three kinds of CXCR4-overexpressing cancer cell lines were increased more rapidly compared to those of the free E5. These results indicated the PAC80 improved the binding affinity of the E5 peptide towards CXCR4. In addition, the binding amount of E5 on the MS-5 cells was negligible for both the free E5 and PAC80–E5, which revealed the high binding specificity of the E5 peptide towards CXCR4.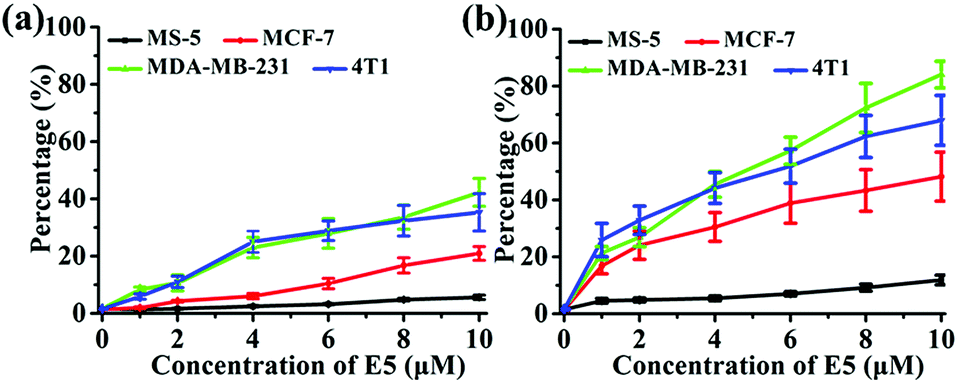 | ||
| Fig. 4 Affinity of: (a) free E5 peptide and (b) PAC80–E5 complex to cell lines with different expression levels of CXCR4. | ||
The binding affinity was also observed using LSCM using MDA-MB-231 as a representative cell model. The E5 peptide exhibited a limited interaction with MDA-MB-231 owing to the low solubility in the cell culture medium. However, the binding amount of the E5 peptide in the presence of PAC80 was increased significantly under the same concentration of the E5 peptide (Fig. 5). This conclusion is in agreement with the FCM results that indicate that PAC80 could improve the binding affinity of the E5 peptide by improving the solubility of the E5 peptide.
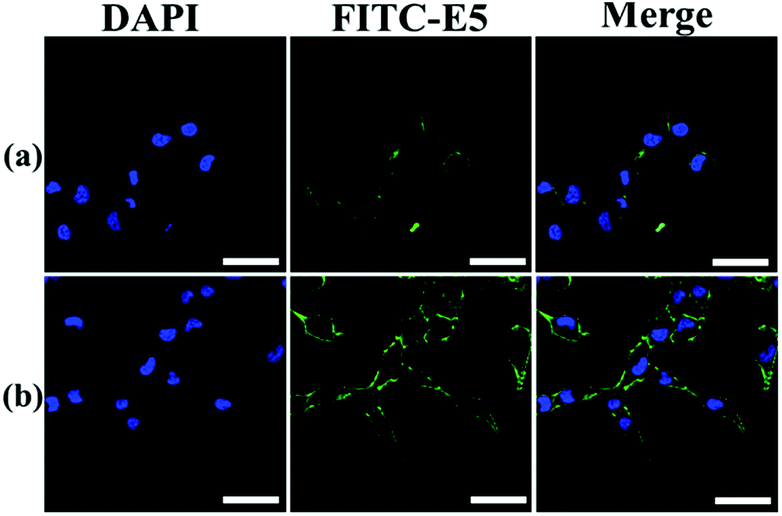 | ||
| Fig. 5 The affinity of: (a) free E5 peptide; and (b) PAC80–E5 bound to MDA-MB-231 cells were detected using LSCM using FITC as the label for E5. Scale bar = 50 μm. | ||
3.5 Improved anti-tumor metastasis effect of E5 peptide in vitro by PAC80
It has been reported that the E5 peptide can inhibit CXCL12-induced cell migration in human acute myelocytic leukemia cell lines and prolong the survival of leukemia mice.29 Overexpression of CXCR4 was also observed in breast cancer cells, and its activation by CXCL12 could induce chemotaxis in metastatic breast cancer cells, which consequently allows cells to migrate to the target organs and survive in the distance organs.4 Herein, the transwell assay was performed on the MCF-7, MDA-MB-231 and 4T1 cell lines to determine whether PAC80 could improve the inhibitory effect of E5 on cancer cell migration. The results showed that CXCL12 could enhance the migration of cancer cells compared with the untreated cells. Thus, the migration rate of cancer cells after CXCL12 treatment was set as 100% as a control (Fig. 6a–c and S2†). When the cancer cells were treated with free E5 peptide at 0.01, 0.1 and 1 μM, the cancer cell migration was inhibited slightly for the MCF-7, MDA-MB-231 and 4T1 cancer cell lines. The migration rate was even greater than 60% when the concentration of E5 reached 1 μM in these three cancer cell lines. However, PAC80–E5 was able to markedly inhibit the migration of all cancer cell lines at the same concentration of E5 peptide. By visualizing the cells that had migrated to the bottom of the culture inserts, the migration percentage was substantially reduced to 37.4% for MCF-7, 23.2% for MDA-MB-231 and 34.6% for the 4T1 cancer cells. There were significant differences between the free E5 and PAC80–E5. Overall, these results indicated that PAC80–E5 could effectively inhibit CXCL12-induced cell migration in CXCR4-overexpressing breast cell lines compared with the free E5 peptide.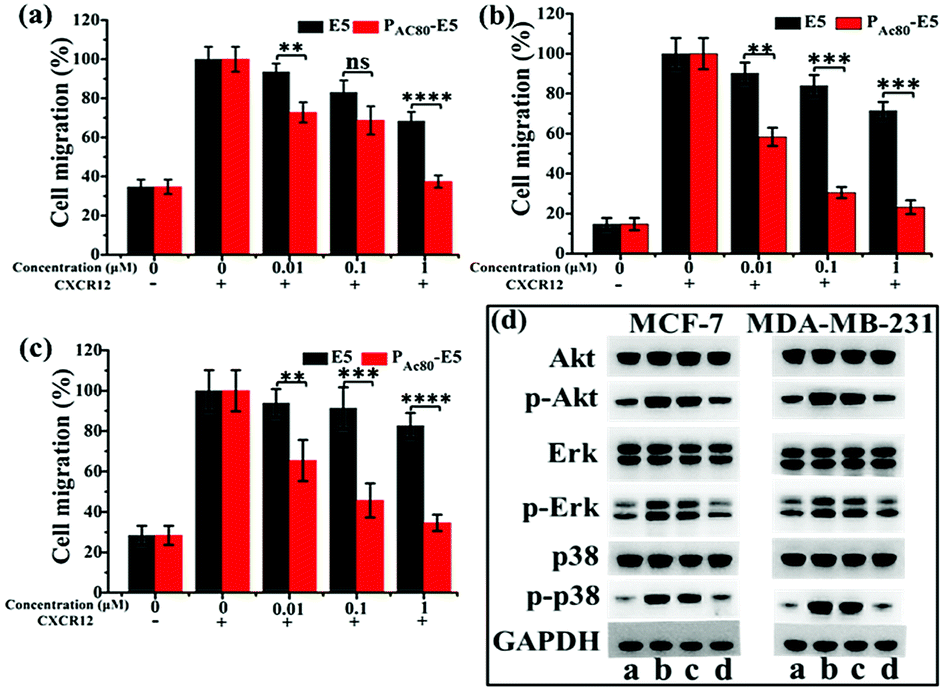 | ||
| Fig. 6 (a) The inhibitory effect of E5 and PAC80–E5 on: (a) MCF-7; (b) MDA-MB-231; and (c) 4T1 cells detected by transwell assay. The CXCR12 supplemented sample without E5 or PAC80–E5 was set as 100% as the control. Error bars represent the standard deviation (n = 3). (d) Effects of E5 and PAC80–E5 on CXCL12-induced Akt, Erk and p38 activation of cancer cells using western blot analysis. (a) Control; (b) treated with CXCL12 only; (c) treated with CXCL12 and E5; and (d) treated with CXCL12 and PAC80–E5. The concentration of E5 peptide was 1 mM. | ||
3.6 Enhanced down-regulation effect of E5 peptide towards CXCL12-induced Akt, Erk and p38 phosphorylation by PAC80
It has been confirmed that the E5 peptide could bind to CXCR4 and inhibit the phosphorylation levels of the Akt, Erk and p38 proteins, which account for the migration of cells stimulated by CXCL12.8 Therefore, next we performed western blot experiments to detect the expression levels of Akt (phospho-Akt), Erk (phospho-Erk) and p38 (phospho-p38) in MDA-MB-231 and MCF-7 cell lines to further clarify whether PAC80 could enhance the down-regulation effect of the E5 peptide, which could interrupt the intracellular signaling pathway of CXCL12/CXCR4. The results displayed in Fig. 6d showed that the CXCL12 treatment enhanced the phosphorylation protein expression levels of Akt, Erk and p38 (lane b) in reference to those of the control (lane a), while PAC80–E5 dramatically inhibited the robust phosphorylation protein expression of Akt, Erk and p38 in cancer cells stimulated by CXCL12 (lane d) compared with free E5 at 1 μM (lane c). These results clearly indicated that PAC80 could enhance the interaction of the E5 peptide and the CXCR4 protein on the CXCR4-positive cells, which would inhibit the phosphorylation levels of p-Akt, p-Erk and p-p38 proteins stimulated by CXCL12.3.7 Enhanced anti-tumor metastasis effect of E5 peptide in vivo by PAC80
Finally, in order to contrast the anti-tumor metastasis effect of E5 peptide and PAC80–E5 complex in vivo, the breast cancer passive lung metastasis model was established by inoculating the 4T1 cells via the caudal vein. Before treatment, the lifetime of the PAC80–E5 complex was evaluated in vitro using DLS. The results indicated that the complex could maintain its structure for 24 h, and that the complex would disrupt gradually with time after 24 h (Fig. S4†). Thus, the E5 peptide and PAC80–E5 were administrated every two days with a E5 peptide dosage of 10 mg kg−1 and 20 mg kg−1 via celiac plexus injection. The mice treated with sterile water were used as a control. The results demonstrated that the body weight of the mice from each group was not significantly changed during the treatment (Fig. 8a), which revealed the low side effects of the E5 peptide and the good biocompatibility of PAC80. After 15 days, all of the mice were sacrificed and the lung tissues were carefully collected and photographed (Fig. 7). The number of macroscopic metastatic nodules in the lung tissues was recorded to evaluate the lung metastasis of breast cancer (Fig. 8b and 7). Compared with the control group, the formulation of lung metastasis was slightly decreased after E5 peptide treatment, but there were no significant differences between the E5-treated groups with different doses (10 and 20 mg kg−1). However, in the PAC80–E5 treated groups, the average numbers of metastatic nodules per lung were 32 and 19 for the dosage of 10 mg kg−1 and 20 mg kg−1, respectively, compared with average numbers of metastasis nodules of 50 and 48 for the free E5 at 10 and 20 mg kg−1, respectively. The anti-tumor metastasis effect of PAC80–E5 was 64.3% and 39.2% for the free E5 groups at the same dosages and 59.8% and 34.6% for the control group. However, the average number treated with the free E5 peptide was 92.9% and 88.5% of the control group, which indicated a slight inhibition of metastasis for the free E5 treatment. Accordingly, PAC80–E5 with a dose of 10 and 20 mg kg−1 could produce a 40.2% and 65.4% metastasis inhibition rate on the lung metastasis of breast cancer compared with the free E5 at the same dosage and the control, which was 5.66 and 5.68 fold higher than that of the free E5 at the same dosage, respectively. These results indicated that PAC80 enhanced the inhibitory effect of the E5 peptide on the tumor metastasis in vivo by improving the solubility of E5 in the physiological environment. | ||
| Fig. 7 Photographs of lungs excised from mice, the arrows refer to the tumor nodes in the lungs. | ||
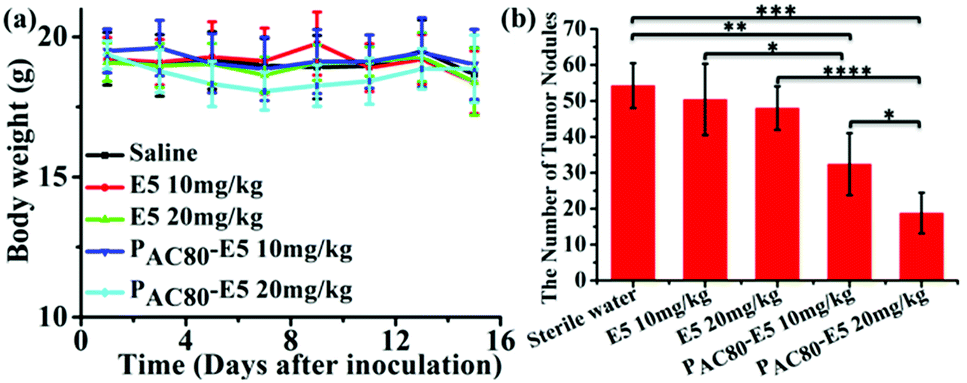 | ||
| Fig. 8 (a) The body weights of the mice were measured every two days. (b) The number of tumor nodes in the lungs of the mice were counted. | ||
Histological examination was also achieved using H&E staining to detect the metastatic lesions and the toxicity of PAC80. The metastatic lesions were shown as cell clusters with dark stained nuclei (Fig. 9, lung). Compared with the groups treated with both sterile water and the free E5 peptide, the obvious decrease of the metastatic foci in the PAC80–E5 groups suggested the efficient anti-metastatic efficacy of PAC80–E5. As a result, the lung metastasis of breast cancer was notably inhibited by the PAC80–E5 complex, whereas the inhibitory effect was significantly difference compared with the negative control and the free E5 groups. In addition, H&E analysis of other tissues, including the heart, liver, spleen, lung and kidney, is shown in Fig. 9. No visualized tissue damage was observed from these results, which revealed that the acetylated PAMAM dendrimer has an excellent biocompatibility as an auxiliary agent to improve the solubility of hydrophobic drugs.
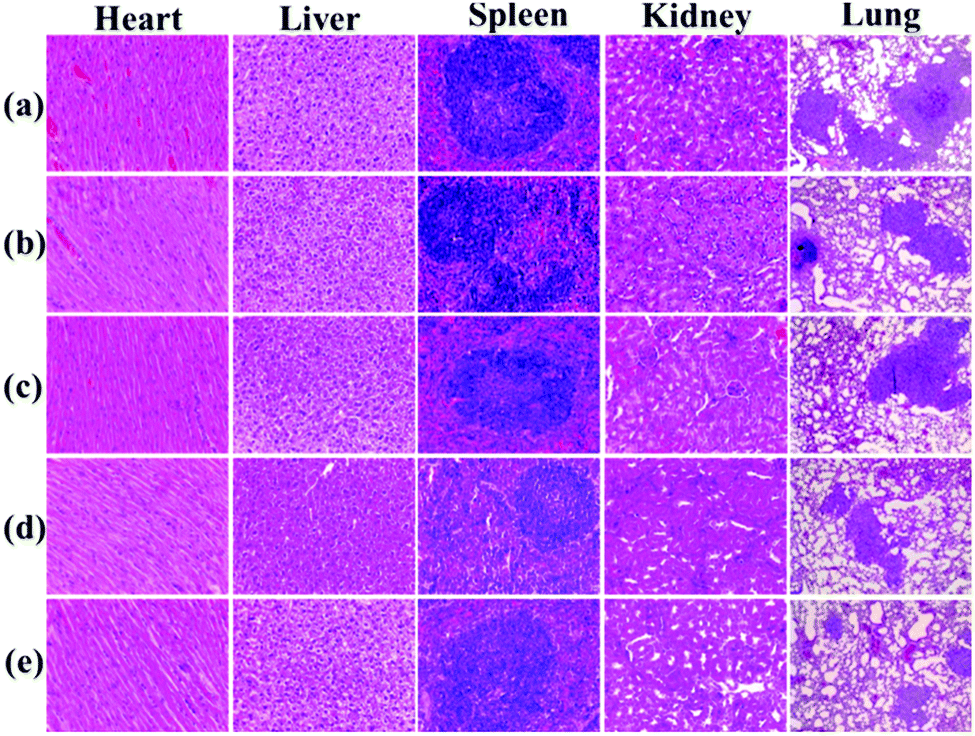 | ||
| Fig. 9 Histological analysis of tissues from major organs of female BALB/C mice after treatment with: (a) sterile water; (b) E5 with a dosage of 10 mg kg−1; (c) E5 with a dosage of 20 mg kg−1; (d) PAC80–E5 with a dosage of 10 mg kg−1; and (e) PAC80–E5 with a dosage of 20 mg kg−1. | ||
The poor solubility and low dispersion performance of the E5 peptide in the physiological environment owing to its sensitivity to salt ions have restricted its clinical application. In this present work, acetylated PAMAM (PAC80) was synthesized to improve the solubility of the E5 peptide. As expected, the PAC80 could enhance the solubility and suppress the aggregation of the E5 peptide effectively through electrostatic interaction and hydrophobic interactions (Fig. S3†). Additionally, PAC80 could also improve the affinity of the E5 peptide to the CXCR4-positive cells and enhance the inhibitory effect on cell migration. As detailed in the previous report, PAMAM dendrimers could penetrate through cell membranes when the periphery amine groups remain on the surface.25 Therefore, the enhanced activity of the E5 peptide in cell levels was contributed to by the sufficient interaction of the E5 peptide with its target CXCR4 in the presence of PAC80. This phenomenon mainly resulted from the increased solubility of the E5 peptide in the presence of PAC80, which could maintain the effective concentration and long-term stability of the E5 peptide in the physiological environment. On the whole, PAC80 could improve the affinity of the E5 peptide to cancer cells overexpressing CXCR4, as well as having inhibitory effects on the cell migration. This enhancement efficacy of PAC80 was also observed in vivo. The addition of PAC80 improved the binding affinity of the E5 peptide towards CXCR4, while free E5 peptide displayed a negligible antitumor activity in vivo owing to the low solubility. Furthermore, the low tissue toxicity means that this superbranched polymer could be a promising adjuvant in diagnosis, imaging in vivo and disease therapy by improving the solubility of hydrophobic probes and therapeutic agents. The enhanced metastasis inhibitory effect of the PAC80–E5 compounds means it could be a promising adjuvant in cancer therapy.
4. Conclusions
This work demonstrated that, compared with the free E5 peptide, the encapsulation/complexation of the E5 peptide into/with dendrimers led to the excellent solubility of this anti-metastatic inhibitor and enhanced its anti-tumor metastasis effect both in vitro and in vivo. In the presence of PAC80, the E5 peptide showed a higher binding affinity to CXCR4-overexpressing breast cancer cells, and exhibited a better inhibitory effect on the CXCL12-induced migration of breast cancer cells. Furthermore, the use of PAC80 enhanced the anti-metastatic efficacy of the E5 peptide significantly in the lung metastatic tumor animal model, without causing tissue damage. These results indicate that PAC80 could be used as a wide range adjuvant in cancer therapy and for other applications in clinical treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21773042), the Beijing Natural Science Foundation (No. 2162044), and the National Key Research and Development Project (2016YFF0203803).References
- W. Chen, A. D. Hoffmann, H. Liu and X. Liu, NPJ Precis. Oncol., 2018, 2, 4–12 CrossRef PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed.
- H. Sun, Q. Meng, S. Tang, J. Su, Q. Yin, L. Chen, W. Gu, H. Yu, Z. Zhang, S. Wang and Y. Li, Mol. Pharm., 2015, 12, 3323–3331 CrossRef CAS PubMed.
- A. Martinez-Ordoñez, S. Seoane, P. Cabezas, N. Eiro, J. Sendon-Lago, M. Macia, T. Garcia-Caballero, L. O. Gonzalez, L. Sanchez, F. Vizoso and R. Perez-Fernandez, Oncogene, 2018, 37, 1430–1444 CrossRef PubMed.
- A. Müller, B. Homey, H. Soto, N. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verástegui and A. Zlotnik, Nature, 2001, 410, 50–56 CrossRef PubMed.
- M. C. P. Smith, K. E. Luker, J. R. Garbow, J. L. Prior, E. Jackson, D. Piwnica-Worms and G. D. Luker, Cancer Res., 2004, 64, 8604–8612 CrossRef CAS PubMed.
- R. S. Taichman, C. Cooper, E. T. Keller, K. J. Pienta, N. S. Taichman and L. K. McCauley, Cancer Res., 2002, 62, 1832–1837 CAS.
- S. Singh, U. P. Singh, W. E. Grizzle and J. W. Lillard Jr, Lab. Invest., 2004, 84, 1666–1676 CrossRef CAS PubMed.
- S. Brand, J. Dambacher, F. Beigel, T. Olszak, J. Diebold, J. M. Otte, B. Goke and S. T. Eichhorst, Exp. Cell Res., 2005, 310, 117–130 CrossRef CAS PubMed.
- J. Kim, H. Takeuchi, S. T. Lam, R. R. Turner, H.-J. Wang, C. Kuo, L. Foshag, A. J. Bilchik and D. S. B. Hoon, J. Clin. Oncol., 2005, 23, 2744–2753 CrossRef CAS PubMed.
- Y. Alsayed, H. Ngo, J. Runnels, X. Leleu, U. K. Singha, C. M. Pitsillides, J. A. Spencer, T. Kimlinger, J. M. Ghobrial, X. Jia, G. Lu, M. Timm, A. Kumar, D. Côté, I. Veilleux, K. E. Hedin, G. D. Roodman, T. E. Witzig, A. L. Kung, T. Hideshima, K. C. Anderson, C. P. Lin and I. M. Ghobrial, Blood, 2007, 109, 2708–2717 CAS.
- Z. Zeng, Y. Xi Shi, I. J. Samudio, R.-Y. Wang, X. Ling, O. Frolova, M. Levis, J. B. Rubin, R. R. Negrin, E. H. Estey, S. Konoplev, M. Andreeff and M. Konopleva, Blood, 2009, 113, 6215–6224 CrossRef CAS PubMed.
- S. Tavor, I. Petit, S. Porozov, A. Avigdor, A. Dar, L. Leider-Trejo, N. Shemtov, V. Deutsch, E. Naparstek, A. Nagler and T. Lapidot, Cancer Res., 2004, 64, 2817–2824 CrossRef CAS PubMed.
- J. A. Burger, M. Burger and T. J. Kipps, Blood, 1999, 94, 3658–3667 CAS.
- D. A. Sipkins, X. Wei, J. W. Wu, J. M. Runnels, D. Côté, T. K. Means, A. D. Luster, D. T. Scadden and C. P. Lin, Nature, 2005, 435, 969–973 CrossRef CAS PubMed.
- J. A. Burger and A. Bürkle, Br. J. Haematol., 2007, 137, 288–296 CrossRef CAS PubMed.
- A. Zepeda-Moreno, R. Saffrich, T. Walenda, V. T. Hoang, P. Wuchter, S. Sanchez-Enriquez, A. Corona-Rivera, W. Wagner and A. D. Ho, Exp. Hematol., 2012, 40, 666–674 CrossRef CAS PubMed.
- W. Zhan, Z. Liang, A. Zhu, S. Kurtkaya, H. Shim, J. P. Snyder and D. C. Liotta, J. Med. Chem., 2007, 50, 5655–5664 CrossRef CAS PubMed.
- V. M. Truax, H. Zhao, B. M. Katzman, A. R. Prosser, A. A. Alcaraz, M. T. Saindane, R. B. Howard, D. Culver, R. F. Arrendale, P. R. Gruddanti, T. J. Evers, M. G. Natchus, J. P. Snyder, D. C. Liotta and L. J. Wilson, ACS Med. Chem. Lett., 2013, 4, 1025–1030 CrossRef CAS PubMed.
- P. A. Mayes, K. W. Hance and A. Hoos, Nat. Rev. Drug Discovery, 2018, 17, 509–527 CrossRef CAS PubMed.
- S. Santagata, L. Portella, M. Napolitano, A. Greco, C. D'Alterio, M. V. Barone, A. Luciano, M. Gramanzini, L. Auletta, C. Arra, A. Zannetti and S. Scala, Sci. Rep., 2017, 7, 2554–2562 CrossRef PubMed.
- M. Lefrançois, M.-R. Lefebvre, G. Saint-Onge, P. E. Boulais, S. Lamothe, R. Leduc, P. Lavigne, N. Heveker and E. Escher, ACS Med. Chem. Lett., 2011, 2, 597–602 CrossRef PubMed.
- M. L. Tan, P. F. Choong and C. R. Dass, Peptides, 2010, 31, 184–193 CrossRef CAS PubMed.
- S. Bae, K. Ma, T. H. Kim, E. S. Lee, K. T. Oh, E. S. Park, K. C. Lee and Y. S. Youn, Biomaterials, 2012, 33, 1536–1546 CrossRef CAS PubMed.
- R. B. Kolhatkar, K. M. Kitchens, P. W. Swaan and H. Ghandehari, Bioconjugate Chem., 2007, 18, 2054–2060 CrossRef CAS PubMed.
- S. Zhu, M. Hong, L. Zhang, G. Tang, Y. Jiang and Y. Pei, Pharm. Res., 2010, 27, 161–174 CrossRef CAS PubMed.
- J. Liu, J. Liu, L. Chu, Y. Wang, Y. Duan, L. Feng, C. Yang, L. Wang and D. Kong, Int. J. Nanomed., 2010, 6, 59–69 Search PubMed.
- I. J. Majoros, T. P. Thomas, C. B. Mehta and J. R. Baker, J. Med. Chem., 2005, 48, 5892–5899 CrossRef CAS PubMed.
- H. Cao, Z. Dan, X. He, Z. Zhang, H. Yu, Q. Yin and Y. Li, ACS Nano, 2016, 10, 7738–7748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08526a |
| This journal is © The Royal Society of Chemistry 2018 |