DOI:
10.1039/C8RA08486F
(Paper)
RSC Adv., 2018,
8, 39929-39936
PbII-catalyzed transformation of aromatic nitriles to heptanitrogen anions via sodium azide: a combined experimental and theoretical study†
Received
13th October 2018
, Accepted 26th November 2018
First published on 29th November 2018
Abstract
Under hydrothermal conditions, an open-chain N73− anion stabilized in a metal–organic framework (MOF) was achieved for the first time via the in situ reaction of 4-fluorobenzonitrile and sodium azide with Pb2+ ion as catalyst. The anion with C2h symmetry in the MOF was studied by FT-IR, single-crystal XRD and theoretical calculations. Thermal analysis results demonstrated the stability of the anion in the MOF below 430 °C and a high energy content of 8.61 kJ g−1. The anion is also a good reducing agent. It can easily react with basic KMnO4 solution. Moreover, the present study indicates that the Pb2+ ion activates the azide rather than nitrile in the in situ reaction of nitriles with azides to form polynitrogen and this mechanism is a distinct contradiction with the previous results in which the nitrile reacts with azide in the presence of transition metal ions. Our findings may open a new avenue towards the synthesis and capture of polynitrogen compounds.
Introduction
Metal–organic frameworks (MOFs) have attracted enormous attention for not only fundamental interest but also applied research, such as gas separation, gas storage, catalysis, chemical sensing and drug delivery,1–10 and this is due to the unique nature of the porosity.11 Several surprising studies have recently underscored the potential important application of nitrogen-rich MOFs in the area of energetic materials.12–15 In particular, the metal ions, via coordinating with nitrogen-rich ligands, can be easily constructed as energetic MOFs and most of them possess lower sensitivities.16 To date, albeit that many MOFs based on energetic ligands, including hydrazines, triazoles and tetrazoles, have been designed and synthesized as interesting candidates for primary or secondary explosives,15–24 no significant progress in MOFs involving polynitrogen ligands (Nn, with n more than three) has been achieved due to the assembly difficulties and intrinsic instability of polynitrogen species. In this context, it is a significant challenge to obtain more stable polynitrogen ligands and reveal the reaction mechanism to chemists, since nitrogen atoms do not tend to generate stable multi-atom rings or long chains and most of the reaction intermediates are too active to be captured. Single-crystal XRD is a powerful tool for exploring a reaction mechanism and directly provides information on the nature of reaction intermediates.25–27 However, the precondition is based on the fact that the active intermediates must be captured in their crystalline state. Recently, using single-crystal XRD analysis, Lu and co-workers have demonstrated that metal ion catalysis plays a critical role in the in situ reaction of tetrazole formation28 and it has been found that the common [2 + 3] cycloaddition reaction of nitrile with azide to form tetrazole was easily achieved in water using metal ions as a Lewis acid catalyst29–32 and density-functional theory (DFT) calculations disclosed that the activation of nitrile using Zn2+ ions is the critical step for in situ tetrazole formation,33 such a mechanism was further supported via the capture of a crystalline reaction intermediate during the experiment.28
Currently, a large number of studies have been focused on the in situ formation of metal–tetrazole complexes using transition metal ions, e.g. Zn2+, Co2+, Cd2+ and Mn2+, as the Lewis acid catalyst.29–32 In this study, we noted that the halides or pseudohalides of transition metal ions and the main group Pb2+ ion show different solubility in water. This fact indicates that there exists a distinct coordination interaction between the metal ion and halide or pseudohalide anions between the transition metal ions and main group Pb2+ ion. Consequently, it is possible to obtain different products if the transition metal ions are replaced by the main group Pb2+ ion during the in situ reaction of nitrile with azide to form tetrazole. We attempted to explore the reaction of nitrile and azide in the presence of the main group Pb2+ ion. Surprisingly, a new open-chain polynitrogen anion was isolated in the water instead of the in situ formation of tetrazole. We further designed a reaction system, by which we successfully captured three interrelated intermediate products, H3O[Pb8(μ6-N7)(μ4-O)2(μ6-ox)(μ7-CHDA)4]·4H2O (MOF-N7), Pb3(N3)6 (α-Pb(N3)2) and [Pb2(μ4-O) (FBA)2]·2H2O (Pb-FBA) (HFBA = 4-fluorobenzoic acid; H2CHDA = 1,3-cyclohexane-dicarboxylic acid; H2ox = oxalate acid), the crystal structures of which were characterized. Our results reveal that the azide ion, rather than the nitrile was activated by the Pb2+ ion and this was distinct from that previously reported reaction catalyzed by transition metal ions.28,33
Experimental and computational sections
Caution
Azides and MOF-N7 are a highly energetic. They should be handled only on a very small scale and must be handled with special caution.
Materials and general methods
All chemicals used were obtained commercially and used without further purification. C, H and N microanalyses were performed on a Vario EL III elementary analyzer. FT-IR spectra (KBr pellet) were recorded in the range 400–4000 cm−1 on an iS50 FT-IR (Nicolet) spectrometer. Powder X-ray diffraction pattern (PXRD) was performed on a Rigaku D/max-RA rotating anode X-ray diffractometer at room temperature. N2 sorption isotherm was obtained on the Belsorp MAX volumetric adsorption equipment. Thermogravimetric analysis and differential scanning calorimetry (TGA/DSC) were determined on a Netzsch STA-409PC analyzer in flowing N2 with a linear heating rate of 10 °C min−1.
Synthesis of MOF-N7, α-Pb(N3)2 and Pb-FBA
H3O[Pb8(μ6-N7)(μ4-O)2(μ6-ox)(μ7-CHDA)4]·4H2O (MOF-N7). A mixture of Pb(NO3)2, (0.0662 g, 0.2 mmol), H2CHDA (0.0172 g, 0.1 mmol), 4-FBN (0.0121 g, 0.1 mmol), NaN3 (0.0130 g, 0.2 mmol), NaOH (0.0080 g, 0.2 mmol) and H2O (14 mL) was sealed in a 25 mL Teflon-lined stainless steel container. The reactor was heated to 160 °C for 96 h, and then slowly cooled down to room temperature. Colorless block crystals of MOF-N7 were isolated via filtration and rinsed with distilled water several times in 35% yield (based on Pb(NO3)2). C34H51N7O27Pb8 (2647.41): calc. C, 15.43; H, 1.94 N, 3.70; found C, 15.38; H, 1.99; N, 3.65%. FT-IR (KBr pellet, cm−1): 3446(w), 2915(m), 2849(m), 2061(m), 1585(s), 1544(s), 1506(s), 1399(s), 1354(m), 1341(m), 1320(m), 1294(m), 1268(m), 1250(m), 1226(m), 1185(m), 1140(m), 1027(m), 959(m), 916(m), 766(m), 714(m), 685(m), 655(m), 566(m), 520(m), 487(m).
Pb3(N3)6 (α-Pb(N3)2). The preparation of α-Pb(N3)2 was carried out using the procedure for preparing MOF-N7 with the addition of 4-FBN not included. Yellow rod crystals of α-Pb(N3)2 were isolated via filtration and rinsed with distilled water several times in 70% yield (based on NaN3). N18Pb3 (873.75): calc. N, 28.86; found N, 28.78%. FT-IR (KBr pellet, cm−1): 2036(s), 1384(m), 1328(w), 1128(w), 1043(w), 669(w), 632(w), 606(w), 434(w).
[Pb2(μ4-O)(FBA)2]·2H2O (Pb-FBA). Pb-FBA was obtained via the reaction of Pb(NO3)2 with 4-FBN and NaN3 under the same reaction conditions used for the preparation of MOF-N7. Colorless rod crystals of Pb-FBA were isolated via filtration and rinsed with distilled water several times in 65% yield (based on 4-FBN). C14H12F2O7Pb2 (744.64): calc. C, 22.58; H, 1.62; found C, 22.52; H, 1.75%. FT-IR (KBr pellet, cm−1): 3448(m), 3086(w), 2926(m), 1606(s), 1552(s), 1525(s), 1509(m), 1492(m), 1298(w), 1239(m), 1227(m), 1215(m), 1151(m), 1088(w), 1013(w), 857(m), 782(s), 688(w), 669(w), 610(s), 547(w), 528(w), 492(w), 431(m).
X-ray crystallography study
Single crystal X-ray diffraction analyses of MOF-N7, α-Pb(N3)2 and Pb-FBA were performed on a Bruker SMART APEX II CCD diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at room temperature. The empirical absorption corrections based on psi-scan were carried out. The crystal diffraction data were integrated with the Bruker SAINT program. Sadabs program was used for absorption correction. The crystal structures were solved via direct methods and all non-hydrogen atoms were refined anisotropically using the full-matrix least-squares technique on F2 using the SHELXL-2014.34 All hydrogen atoms on the carbon atoms were calculated in their idealized positions and refined using the riding model. The largest diffraction peak and hole are attributed to the ghost peaks of the heavy lead atom. The ISOR restraint was applied to the disordered N1, C5 and C6 atoms in MOF-N7 (PLAT342_ALERT_3_B checkCIF alert for MOF-N7 due to the disordered C5 and C6 atoms). At the same time, the SIMU and DELU restraints were also applied to the N1, N2, N3 and N4 atoms in MOF-N7. All these restraints can make the ADP values of the disordered atoms in MOF-N7 more reasonable. Furthermore, the guest water molecules in MOF-N7 and Pb-FBA and hydronium ions in MOF-N7 in the corresponding frameworks are highly distorted and impossible to refine via conventional discrete atom models (PLAT601_ALERT_2_A checkCIF alert for MOF-N7, and PLAT601_ALERT_2_B checkCIF alert for Pb-FBA). The final formulas for MOF-N7 and Pb-FBA were obtained from the PLATON results in combination with the experimental results obtained from elemental analysis (EA) and thermogravimetric analysis (TGA). The pertinent crystal parameters, data collection and refinement as well as the selected bond lengths and angles with their estimated deviations for MOF-N7, α-Pb(N3)2 and Pb-FBA are summarized in the Tables 1 and S1–S3,† respectively.
Table 1 Crystallographic data and structure refinement summary for MOF-N7, α-Pb(N3)2 and Pb-FBA
| Complexes |
MOF-N7 |
α-Pb(N3)2 |
Pb-FBA |
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = |∑w(|Fo|2 − |Fc|2)|/∑|w(Fo)2|1/2, w = 1/[σ2(Fo2) +(aP)2 + bP]. P = (Fo2 + 2Fc2)/3. |
| Formula |
C34H51N7O27Pb8 |
N18Pb3 |
C14H12F2O7Pb2 |
| Formula weight |
2647.41 |
873.75 |
744.64 |
| Crystal system |
Monoclinic |
Orthorhombic |
Tetragonal |
| Space group |
C2/m |
Pnma |
I41/a |
| a (Å) |
8.1312(6) |
6.6300(2) |
24.6059(4) |
| b (Å) |
29.931(3) |
16.2630(6) |
24.6059(4) |
| c (Å) |
11.5601(9) |
11.3428(4) |
10.7731(2) |
| β (°) |
102.974(5) |
90 |
90 |
| Volume (Å3) |
2741.7(4) |
1223.02(7) |
6522.6(2) |
| Z |
2 |
4 |
16 |
| T (K) |
293(2) |
296(2) |
296(2) |
| Dcalcd (mg m−3) |
3.096 |
4.745 |
2.886 |
| F(000) |
2250 |
1488 |
5024 |
| θ range (°) |
3.268 to 25.050 |
2.189 to 25.042 |
1.655 to 25.049 |
| Reflections collected |
6743 |
3906 |
34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 531 531 |
| Independent reflections |
2468[Rint = 0.0476] |
1111[Rint = 0.0426] |
2888[Rint = 0.0384] |
| Data/restraints/parameters |
2468/41/170 |
1111/0/104 |
2888/0/208 |
| Goodness of fit on F2 |
1.003 |
1.000 |
1.001 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0419a |
R1 = 0.0276a |
R1 = 0.0183a |
| wR2 = 0.1255b |
wR2 = 0.0711b |
wR2 = 0.0462b |
| R indices (all data) |
R1 = 0.0541a |
R1 = 0.0319a |
R1 = 0.0268a |
| wR2 = 0.1310b |
wR2 = 0.0731b |
wR2 = 0.0489b |
| Largest diff. peak and hole (e Å−3) |
2.526 and −1.818 |
2.084 and −1.259 |
0.548 and −0.875 |
| CCDC reference number |
1487965 |
1487966 |
1487967 |
Computational details
All computations reported in the present work were performed using the Gaussian09 D.01 program package.35 Density-functional theory (DFT) calculations were carried out using the popular B3LYP method.36,37 The Stuttgart–Dresden (SDD)38 effective core potential (ECP) was employed for Pb atoms, while the 6-31+G(d,p) basis set was used for C, H, O and N atoms. To verify the activation of the azide ion by the Pb2+ ion, we considered two different activated models of Pb2+ with N3− and 4-FBN. The DFT calculations were performed on the above Pb⋯N3 and Pb⋯4-FBN associations (Fig. S1†). The additional calculations were also performed on monomeric Pb2+, N3− and 4-FBN. For all the species studied here, the geometries were optimized and the final compounds correspond to minima since no imaginary frequencies were found. The SMD39 solvation model was selected for H2O effects. The interaction energies of both associations were determined from the energy difference between the association and monomers, considering the zero point vibrational energy, ZPE. The basis-set superposition error, BSSE was computed using the counterpoise method of Boys and Bernardi.40 Additionally, to further aid in the interpretation of the reaction mechanism of 4-FBN and NaN3 with Pb2+ ion as the Lewis acid catalyst, the molecular electrostatic potential as well as the electrostatic potential basins analysis by means of the Multiwfn program41 were obtained based on the optimized stable Pb⋯N3 and 4-FBN, respectively. Additionally, to clarify the nature of N73− anion and MOF-N7, DFT calculations were performed on the N73− anion (N73−-C2h) and a minor calculation model was selected, [Pb16(O)4(ox)4(N7)(CH3COO)14(CH3COOH)2]− (MOF-N7′) from the crystal structure of MOF-N7 (Fig. S2†). The optimized stable structure of the N73− anion was also obtained. The stable N73− anion (N73−-C2v) possessed C2v symmetry. For MOF-N7′, only the hydrogen positions were optimized. Based on the Multiwfn procedure, the characteristics of the bond critical points (BCPs) of all the N–N and Pb–N bonds in MOF-N7′ were also analysed with the atoms in molecules (AIM) methodology.42 Furthermore, the dissociation energies obtained for N73−-C2h and N73−-C2v were established via ΔEdis = E(N73−) − 7/2E(N2) at the B3LYP/6-31+G(d,p) level of theory.
Results and discussion
Synthesis
It should be noted that the first N73− anion stabilized in the MOF-N7 was isolated via the in situ reaction of 4-fluorobenzonitrile (4-FBN) and sodium azide using Pb2+ ion as the Lewis acid catalyst under hydrothermal condition. Therefore, a few comparative experiments were considered to show the predominant influence factors to generate the N73− anion. The results demonstrate that the in situ reaction can be affected by several factors such as the metal ions and the auxiliary organic carboxylate ligands. Under the similar reaction conditions used for the preparation of MOF-N7, when the Pb2+ ion in the in situ reaction was replaced by other metal ions, e.g. Zn2+, Cd2+, Co2+, Ni2+, Cu2+, Ag+, Mn2+ and so on, as the Lewis acid catalyst, the N73− anion could not be isolated, so the Pb2+ ion plays a key role in the in situ generated N73− anion. Additionally, the auxiliary H2CHDA also plays a crucial role in this reaction, as the N73− anion was not obtained when the H2CHDA was substituted by other organic carboxylate ligands or removed from the reaction mixture. However, when the 4-FBN in the reaction was replaced by other nitriles, e.g. benzonitrile, 4-cyanopyridine, 4-aminobenzonitrile, 4-methylbenzonitrile, 4-methoxybenzonitrile, 4-(imidazol-1-ylmethyl)benzonitrile, 1-(4-cyanophenyl)imidazole, 4-chlorobenzonitrile and 1,4-benzenedicarbonitrile, the target MOF-N7 can also be obtained under the same reaction conditions.
Description of crystal structure
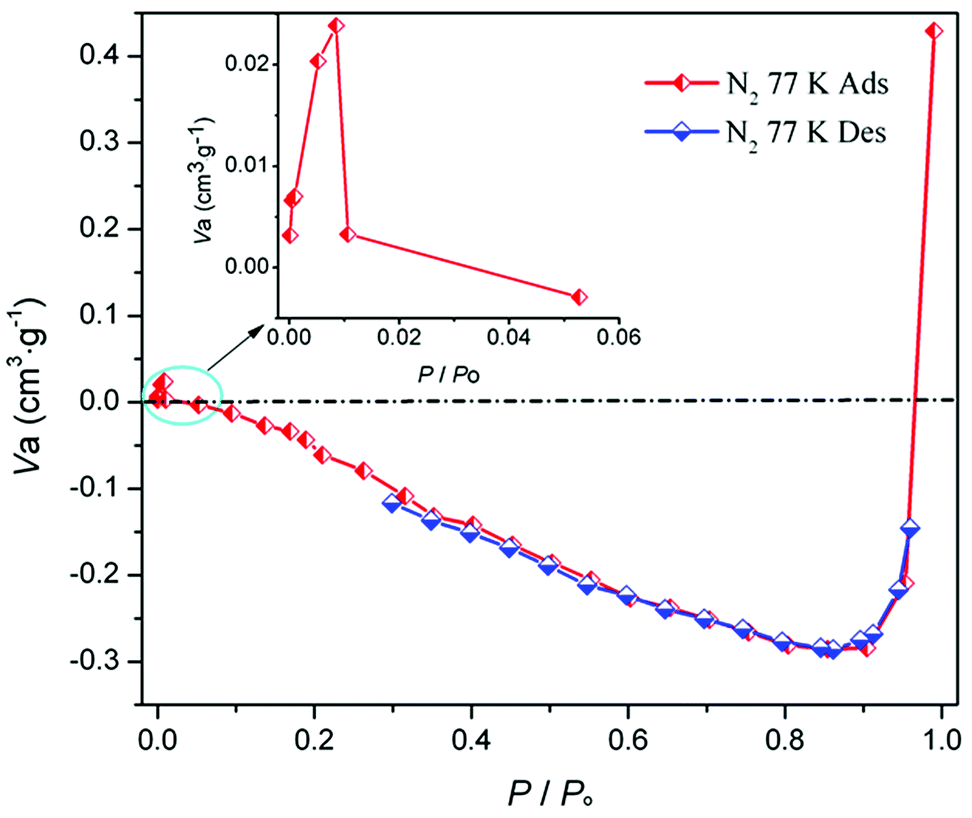
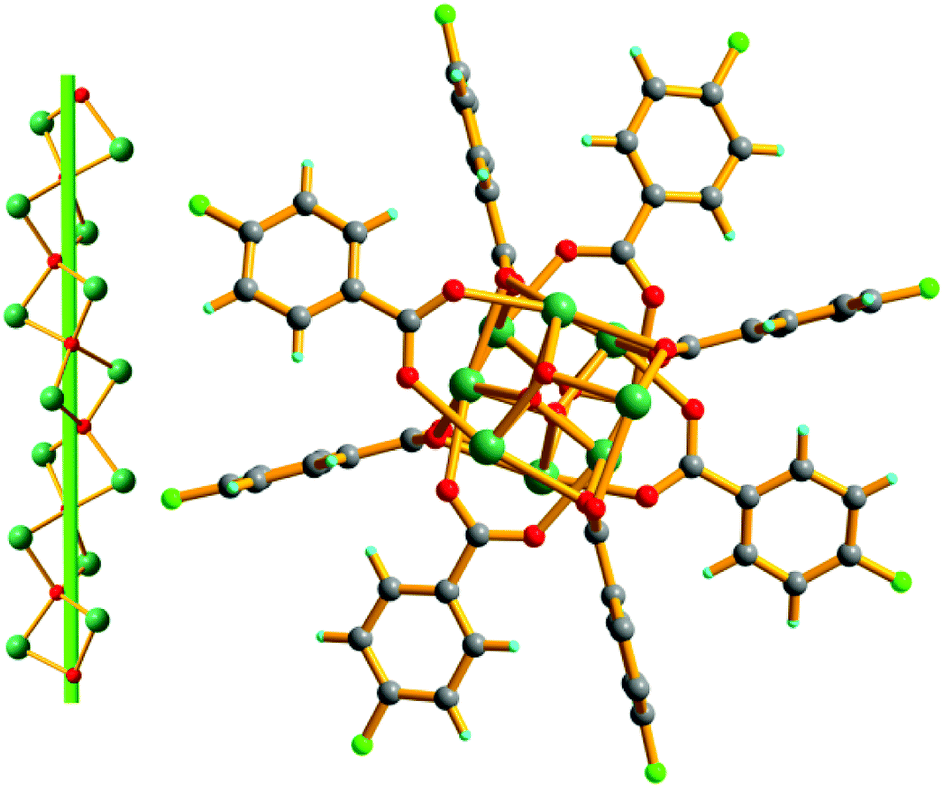
Crystal structure of MOF-N7. Crystals of MOF-N7 were achieved via the hydrothermal reaction of Pb(NO3)2 with H2CHDA, 4-FBN and NaN3 in water at 160 °C for 96 h (see the ESI†). The phase purity of MOF-N7 was examined using powder X-ray diffraction (Fig. S3†). MOF-N7 crystallizes in the monoclinic space group C2/m and presents a 3D pillared-layer metal–organic framework containing the in situ formed N73− anion. The asymmetric unit in MOF-N7 (Fig. S4a†) was comprised of three independent Pb2+ ions (Pb1 and Pb2 located on the crystallographic mirror plane and two-fold axis, respectively), one CHDA2− anion, one half O2− anion located on the two-fold axis and one quarter in situ generated ox2− anion and the N73− anion all located on the crystallographic mirror plane and two-fold axis. The Pb1 and Pb3 ions are seven-coordinated whereas the Pb2 ion was eight-coordinated. The μ4-O2− anions link the adjacent Pb2+ ions (Pb1 and Pb3) to give a rare cationic cluster [Pb6O2]8+ located on the crystallographic inverse center. Furthermore, these cationic clusters are linked via the μ6-ox2− ligands to present a definite 1D cationic chain (Fig. S4b†). The adjacent Pb2 ions are also bridged via μ7-CHDA2− carboxylates to form a distinct 1D anionic chain (Fig. S4c†). The adjacent cationic and anionic chains are not isolated but linked together by the carboxylate groups of the CHDA2− ligands into a 2D cationic layer parallel to the ab plane (Fig. S4d†). These layers are further linked via the linear μ6-N73− anion via the Pb1–N1 and Pb3–N1 bonds to generate a 3D pillared-layer open framework (Fig. 1 and S4e†). The disordered H2O molecules and H3O+ ions are trapped in the channels and further reinforce the open framework via weak intermolecular interactions. This can be also supported by the N2 sorption isotherm measured at 77 K (Fig. 2). After removal of the disordered H2O molecules and H3O+ ions, the MOF-N7 framework can be only stable up to approximately 0.009 P0 and then started to abruptly decompose, accompanied by the release of N2, which was due to the decomposition of the N73− anion. The above result presents that the disordered groups in the channels of MOF-N7 play a crucial role in the stabilizing N73− anion via weak intermolecular interactions as well as the coordination interaction between the Pb2+ ion and N73− anion.
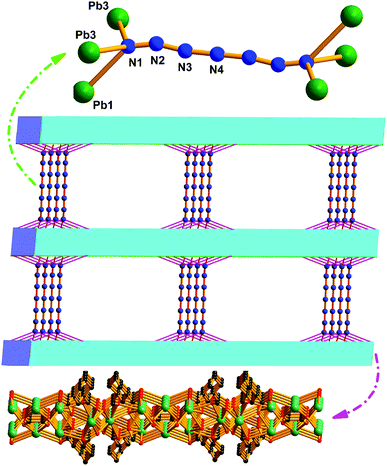 |
| | Fig. 1 The 3D open framework constructed from the 2D cationic layers and N73− anionic pillars of MOF-N7. | |
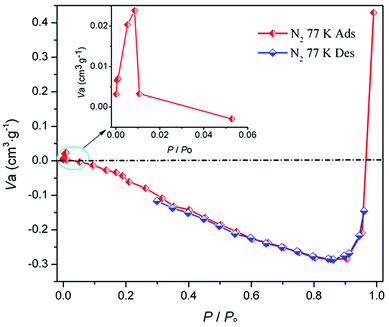 |
| | Fig. 2 The N2 sorption isotherm of MOF-N7 at 77 K. | |
Moreover, this is the first N73− anion stabilized in a MOF via coordination to the Pb2+ ions located on the 2D metal–organic cationic layers, which possesses C2h symmetry. The N1–N2, N2–N3 and N3–N4 bond lengths are 1.198(31), 1.261(39) and 1.191(30) Å respectively. The bond angles of ∠N–N–N are at the range of 165.2(3)–180.0(2)° (Table S1†). The N73− anion structure can be also further confirmed by the IR spectra data (Fig. S5†). The single signal occurring at 2061 cm−1 in the FT-IR spectrum of MOF-N7, which was not observed in the IR spectra of both NaN3 or 4-FBN, was attributed to the anti-symmetric stretching vibration of the N73− anion and such an assignment was in good agreement with the C2h symmetry open-chain N73− anion in MOF-N7.
Crystal structure of α-Pb(N3)2. α-Pb(N3)2 crystallizes in the orthorhombic space group Pnma, an asymmetric unit contains two independent Pb2+ atoms Pb1 and Pb2 (Pb2 located on the crystallographic mirror plane), four kinds of azide anions N3− (two of them are located on the crystallographic mirror plane) (Fig. S6†). Each Pb2+ ion is coordinated to eight N atoms from eight independent azide anions. Through the bridges comprised of the azide anions, the Pb2+ ions are linked together to form a 3D framework (Fig. 3).
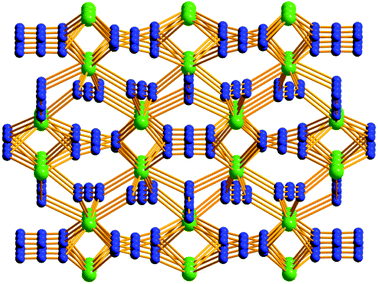 |
| | Fig. 3 A view of the 3D framework of α-Pb(N3)2. | |
Crystal structure of Pb-FBA. Pb-FBA crystallizes in the tetragonal space group I41/a. In the asymmetric unit of Pb-FBA, there are two independent Pb2+ ions, one O2− ion and two FBA− ligands (Fig. S7†). Each Pb2+ ion coordinates to five O atoms from two O2− ions and three FBA− ligands, respectively. The μ4-O2− anions link the adjacent Pb2+ ions to generate a [Pb2O]2+ cationic chain. The μ2-FBA− ligands encompass the cationic chain and lead to the formation of a neutral 41-helical chain via coordination to the Pb2+ ions running along the c-axis with a pitch of 10.773 Å (Fig. 4).
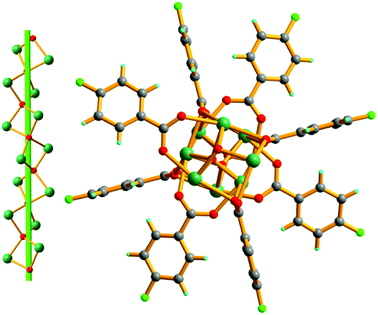 |
| | Fig. 4 A view of the 1D neutral 41-helical chain (right) constructed via the inorganic [Pb2O]2+ chain (left) and FBA− ligands along the c-axis of Pb-FBA. | |
Mechanism of formation of the N73− anion
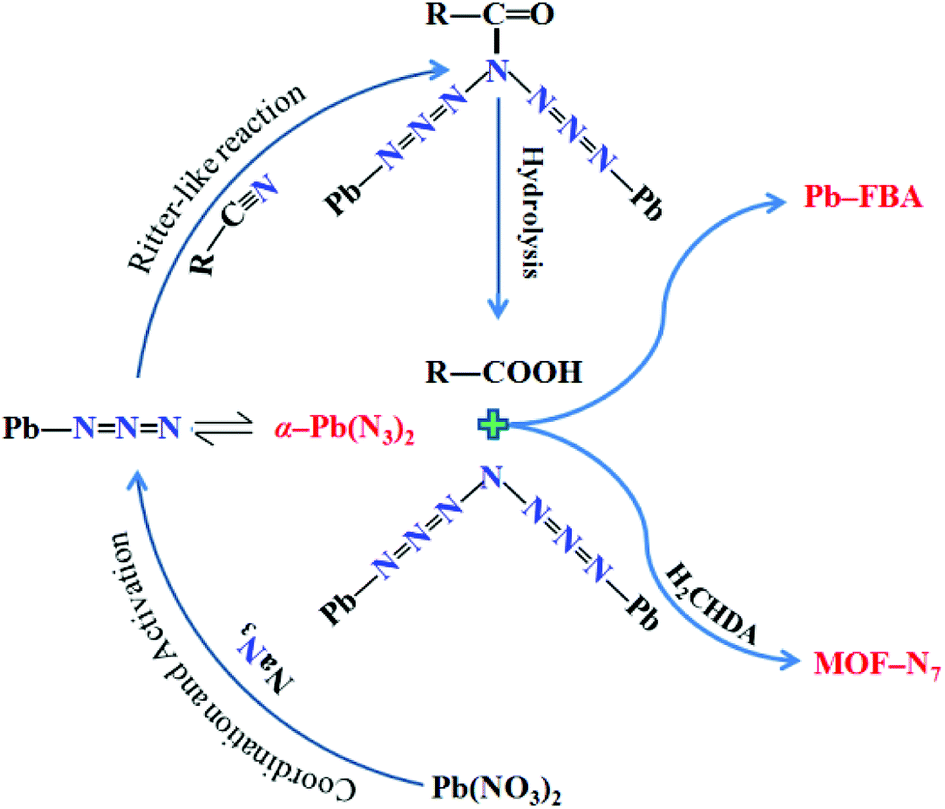
As is well known, under hydrothermal conditions, the reaction of nitrile and azide using transition metal ions as a Lewis acid catalyst can lead to tetrazole ligands via an in situ [2 + 3] cycloaddition reaction mechanism,28–33 in distinct contradiction with the in situ formed N73− anion in MOF-N7. The reaction mechanism of N73− anion formation is proposed as follows (Scheme 1). The Lewis acid Pb2+ ion activates the azide via coordination to form an intermediate, which undergoes a reaction process, which is similar to the Ritter reaction43,44 (a Ritter-like reaction) to generate an amide. Under the hydrothermal conditions, the amide was further hydrolyzed to give an amine and carboxylic acid, which were captured via coordination to the Pb2+ ions to form MOF-N7 and Pb-FBA, respectively. This is the first example of metal ions being used as a Lewis acid catalyst in the reaction between nitrile and azide for the in situ preparation of polynitrogens.
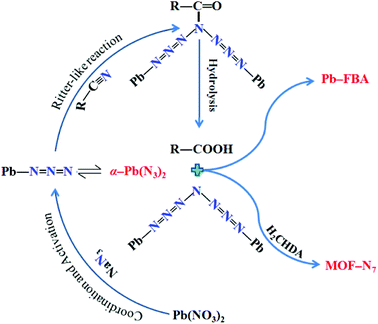 |
| | Scheme 1 The proposed mechanism of the formation of N73− anion. | |
To verify the activation of azide by the Pb2+ ion, we considered two different activated models of Pb2+ with N3− and 4-FBN. The optimized stable structures were successfully obtained based on quantum chemical calculations. The optimized structures and interaction energies are summarized in Fig. S1 and Table S4,† respectively. The corrected interaction energies observed for Pb⋯N3 and Pb⋯4-FBN were found to be −316.48 and −106.18 kcal mol−1, respectively, indicating that Pb⋯N3 was more favored in the reaction mixture of Pb2+, N3− and 4-FBN. Furthermore, the calculated analysis was supported by the following experiments. Interestingly, a Pb2+ complex containing azide ions, α-Pb(N3)2 was isolated. The preparation of α-Pb(N3)2 was carried out using the procedure for preparing MOF-N7 with the addition of 4-FBN not included. Moreover, we also attempted to prepare the possible product containing Pb2+ and 4-FBN. Unfortunately, we failed to obtain the Pb2+ complex involving the 4-FBN ligand. It was noted that only a few complexes of Pb2+ and nitrile is available in the literature, which is probably due to the existence of only a weak coordination interaction between Pb2+ ion and the CN group. The above results suggest the possibility that the activation of azide by the Pb2+ ion occurred during the hydrothermal reaction process.
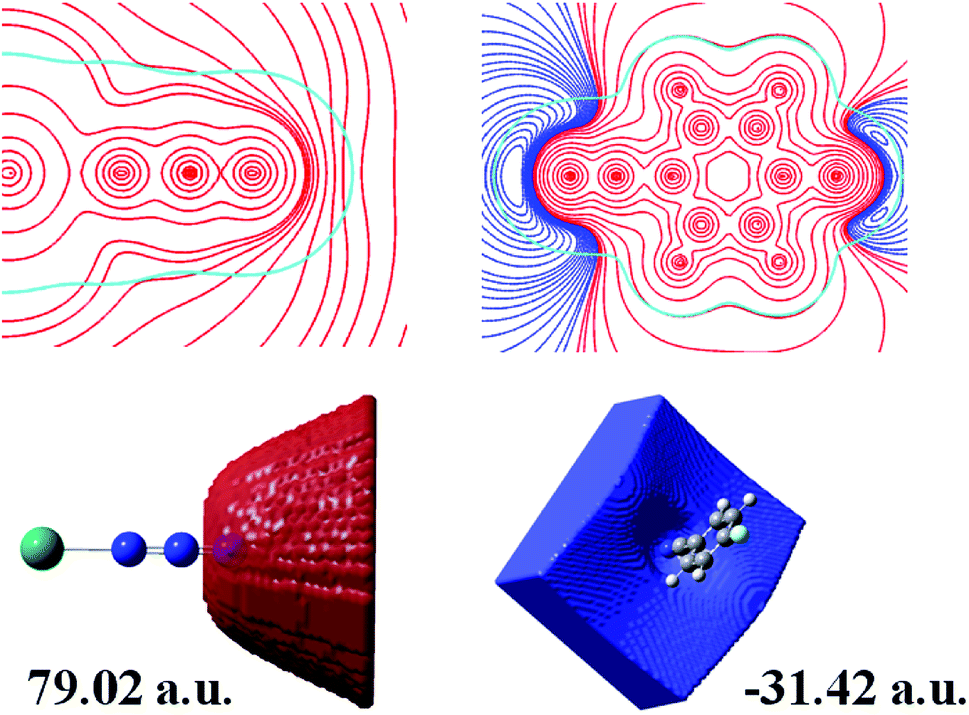
To further aid the understanding of the reaction mechanism of 4-FBN and NaN3 with Pb2+ ion as a Lewis acid catalyst, electrostatic potential and electrostatic potential basins analyses were made for Pb⋯N3 and 4-FBN, which were based on theoretical calculations. As shown in Fig. 5, a positive potential occurs around the uncoordinated terminal N atom in Pb⋯N3 and a negative potential encompasses the N atom of the CN group in 4-FBN. The electrostatic potential values in the corresponding basins were 79.02 and −31.42 a.u., respectively. This suggests the possibility that the N atom of the CN group is an electron donor that interacts with the terminal N atom of Pb⋯N3. The computational results also disclose that the activated Pb⋯N3 can attack the N atom of the CN group via the terminal N atom to form an amide by means of a Ritter-like reaction process (Scheme 1). Unfortunately, we have not directly isolated the amide product due to the amide produced being susceptible to hydrolysis under the hydrothermal reaction conditions to yield the amine and carboxylic acid. We only captured the amine and carboxylic acid yielded via coordination in MOF-N7 and Pb-FBA, respectively. Pb-FBA was obtained via the reaction of Pb(NO3)2 with 4-FBN and NaN3 under the same reaction conditions used for the preparation of MOF-N7. In addition, under the above reaction conditions, the prepared α-Pb(N3)2 can also directly react with 4-FBN to form Pb-FBA. In the crystal of Pb-FBA, the in situ formed FBA− ligand is the hydrolysis product of the amide intermediate produced. Based on the above three crystalline interrelated intermediates, MOF-N7, α-Pb(N3)2 and Pb-FBA, we further confirmed that the azide ion, rather than the nitrile was activated by the Pb2+ ion and this mechanism is a distinct contradiction with the previous results in which the nitrile reacts with azide in the presence of transition metal ions.28–33
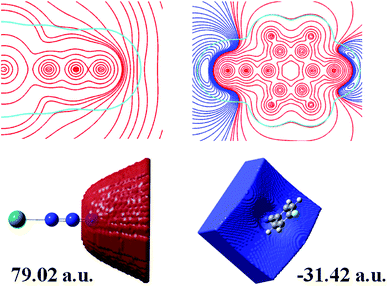 |
| | Fig. 5 A 2D representation of the molecular electrostatic potential (MEP) of Pb⋯N3 and 4-FBN in the molecular plane (top) and electrostatic potential basins of the attractor and repulsor around the terminal N atom in Pb⋯N3 and the N atom of the CN group in 4-FBN, respectively (bottom) (red, positive; blue, negative; cyan, 0.001 a.u. electron density isopotential line). | |
Chemical and thermal stability of the N73− anion
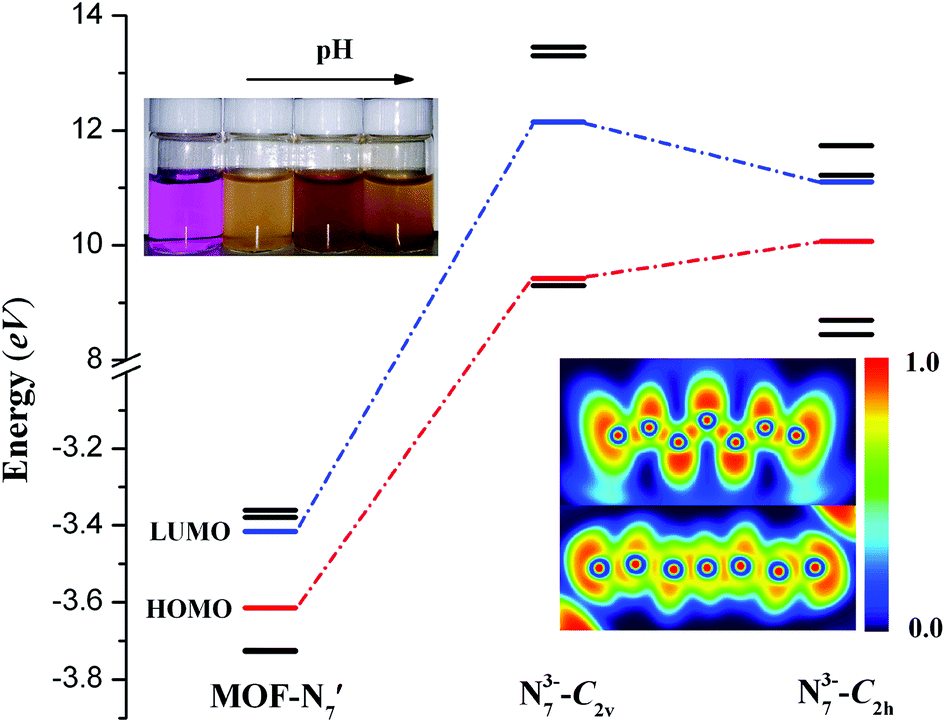
Furthermore, deeper insight into the nature of the N73− anion and theoretical calculations were performed for MOF-N7. In the DFT calculations, MOF-N7′ was taken from the crystal structure of MOF-N7, where the symmetry of N73− anion was C2h and a minor calculation model was selected. The optimized stable structure of the N73− anion was also obtained, which shows that the vibrationally stable N73− anion possesses C2v symmetry (N73−-C2v) and this calculated result was comparable to the theoretical analysis of N7− anion reported by others.45,46 Fig. 6 exhibits the energies of the frontier molecular orbitals of three models, respectively. The energies of the HOMO of N73− anion (N73−-C2h) in MOF-N7 and N73−-C2v are all larger, while that of MOF-N7′ is the smallest, indicating that both the formers can easily donate electrons. As expected, the free N73− anion was unstable in the chemical process with electron transfer. For the latter, MOF-N7′ cannot readily donate electrons and possesses higher chemical stability, showing that N73− anion can be captured via coordination. However, it can be easily oxidized by KMnO4 solution (Fig. 6). Especially, it can react with basic KMnO4 solution, meaning that MOF-N7 may be a good reducing agent in chemical reactions, which is also further evidence for the formation of N73− anion stabilized in MOF-N7.
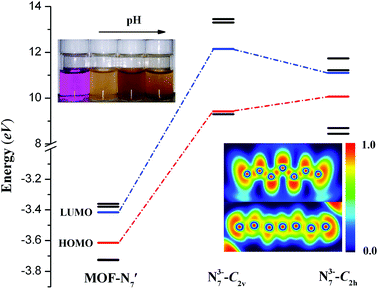 |
| | Fig. 6 The energy diagram for N73−-C2h, N73−-C2v and MOF-N7′ (inset: the ELF contours illustrated on a plane of N73−-C2v (top), the N73− anion fragment in MOF-N7 (bottom) and the colorimetric changes in the KMnO4 solution before and after the addition of MOF-N7). | |
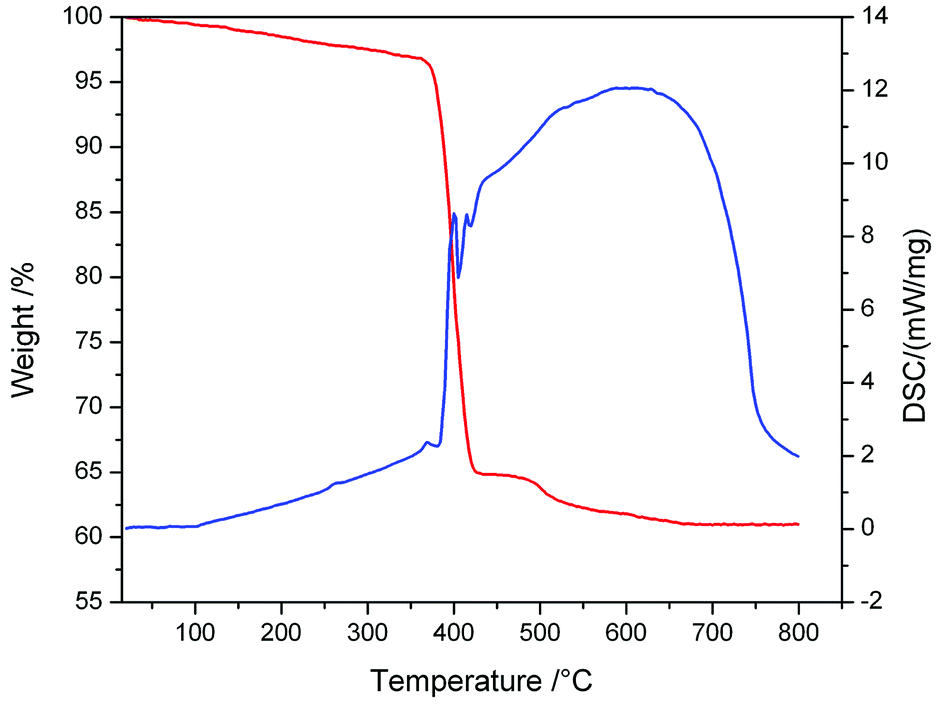
Besides, the bonding patterns of N73− anion and MOF-N7′ were explored via an intuitive map of the electron localization function (ELF). As shown in Fig. 6, for N73-C2v, the regions containing the maximum ELF values on the molecular plane can be identified as the lone pairs and six σ bonds. The π electrons located above and below the molecular plane are highly delocalized, which can also be directly observed via the EFL-π isosurface map (value = 0.70) contributed to from all the occupied π orbitals (Fig. S8†). When compared with N73−-C2v, the bonding features of the N73− anion fragment in MOF-N7′ have distinct diversification. The regions with maximum ELF values for the lone pairs and six σ bonds are smaller, except for the lone pairs of the two terminal N atoms, indicating that the lone pairs and σ electrons as well as the π electrons were more highly delocalized and distributed around the whole N73− anion chain (Fig. S8†). Clearly, the corresponding N–N bond distances in N73−-C2v (1.259, 1.278 and 1.407 Å) are also different from those of the N73− anion fragment (1.198(31), 1.261(39) and 1.191(30) Å) in MOF-N7 (Fig. S2†). The experimental N–N bond lengths of the N73− anion fragment, which are all within the range expected for predominantly covalent N–N bonds and indicate that the N73− anion can be stabilized in the MOF-N7. Additionally, the positive ∇2ρ values, negative H(r) values and |V(r)|/G(r) > 1 demonstrate that the Pb–N bonds have dominant covalent character whereas classical covalent character is observed for all the N–N bonds with negative ∇2ρ and H(r) values and |V(r)|/G(r) > 2 (Table S5†). Thus, the MOF-N7 and the N73− anion have high thermal stability. The aforementioned results show that the formation of a metal complex can stabilize N73− anion, which coincides with those ideas of other predicted metal complexes, such as M4+(η6-N64−) (M = Ti, Zr, Hf, Th),47 Sc3+(η7-N73−)48 and (η5-N5−)M4+(η7-N73−) (M = Ti, Zr, Hf, Th).49 Therefore, the calculated dissociation energies of N73− into N2, for N73−-C2h and N73−-C2v are 645.08 and 440.13 kcal mol−1, respectively, indicating that the N73− anion lies much higher energetically than 7/2 N2. All these findings coincide with the results obtained during TGA/DSC. The TGA shows that the framework of MOF-N7 was stable up to 369 °C and the N73− anion starts to decompose up until 430 °C, accompanied by an exothermic enthalpy change of ca. 8.61 kJ g−1 (Fig. 7), which was more than two times greater than heats of detonation of TNT (3.75 kJ g−1).19,50 The high energy content of N73− anion shows that MOF-N7 may be useful as an explosive just like other nitrogen-rich MOFs.
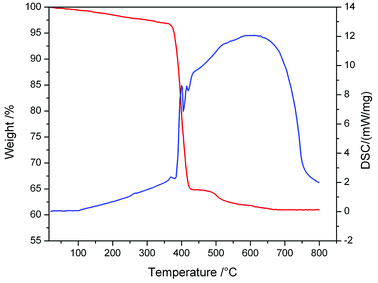 |
| | Fig. 7 The TGA/DSC plots obtained for MOF-N7 at a heating rate of 10 °C min−1 under a flow of N2. | |
Conclusions
In summary, we have successfully prepared and captured a new N73− anion for the first time, which has been studied using FT-IR, single-crystal XRD and theoretical calculations. The N73− anion in MOF-N7 possesses excellent thermal stability and the high energy N73− anion was stabilized by coordination interactions with Pb2+ ions. Our study is the first to reveal that transition metal ions and main group Pb2+ ions show different activation mechanisms in the in situ reaction of metal ions with azide and nitrile, and this distinction was due to the different interactions between the metal ion and azide/nitrile. The weak interaction between transition metal ions and azide results in the metal ion activating the nitrile, and thus, giving the tetrazole product. Whereas the strong interactions between Pb2+ ion and azide gives rise to the azide ion being activated via a coordination interaction with the Pb2+ ion, accordingly, producing polynitrogen anions. The successful synthesis and coordination capture of N73− anion as well as the reaction mechanism study will not only be an important milestone towards one ultimate goal of preparing new polynitrogens, but also provides promising insights into the mechanism of metal catalysis in the in situ formation of polynitrogens. In addition, this work brings fresh design ideas to polynitrogen molecules and nitrogen-rich MOFs as energetic materials of the future.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We thank the Natural Science Foundation of the Education Commission of Anhui Province of China (No. KJ2017A347) and the Program for Innovative Research Team in Anqing Normal University for financial support.
Notes and references
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- R. B. Getman, Y. S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef CAS PubMed.
- W. J. Shi, L. Y. Du, H. Y. Yang, K. Zhang, L. Hou and Y. Y. Wang, Inorg. Chem., 2017, 56, 10090–10098 CrossRef CAS PubMed.
- M. H. Teplensky, M. Fantham, P. Li, T. C. Wang, J. P. Mehta, L. J. Young, P. Z. Moghadam, J. T. Hupp, O. K. Farha, C. F. Kaminski and D. Fairen-Jimenez, J. Am. Chem. Soc., 2017, 139, 7522–7532 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- Z. Zheng, X. Jiang and J. Zhao, Chem. Phys. Lett., 2015, 628, 76–80 CrossRef CAS.
- L. H. Blair, A. Colakel, R. M. Vrcelj, I. Sinclair and S. J. Coles, Chem. Commun., 2015, 51, 12185–12188 RSC.
- X. Qu, S. Zhang, Q. Yang, Z. Su, Q. Wei, G. Xie and S. Chen, New J. Chem., 2015, 39, 7849–7857 RSC.
- S. Zhang, X. Liu, Q. Yang, Z. Su, W. Gao, Q. Wei, G. Xie, S. Chen and S. Gao, Chem.–Eur. J., 2014, 20, 7906–7910 CrossRef CAS PubMed.
- O. S. Bushuyev, G. R. Peterson, P. Brown, A. Maiti, R. H. Gee, B. L. Weeks and L. J. Hope-Weeks, Chem.–Eur. J., 2013, 19, 1706–1711 CrossRef CAS PubMed.
- Z. Liu, T. Zhang, J. Zhang and S. J. Wang, J. Hazard. Mater., 2008, 154, 832–838 CrossRef CAS PubMed.
- S. Li, Y. Wang, C. Qi, X. Zhao, J. Zhang, S. Zhang and S. Pang, Angew. Chem., Int. Ed., 2013, 52, 14031–14035 CrossRef CAS PubMed.
- W. Gao, X. Liu, Z. Su, S. Zhang, Q. Yang, Q. Wei, S. Chen, G. Xie, X. Yang and S. Gao, J. Mater. Chem., 2014, 2, 11958–11965 RSC.
- S. Chen, S. Shu and S. Gao, Inorg. Chim. Acta, 2009, 362, 3043–3408 CrossRef CAS.
- K. L. Zhang, Z. C. Pan, Y. Chang, W. L. Liu and S. W. Ng, Mater. Lett., 2009, 63, 2136–2138 CrossRef CAS.
- J. D. Lin, S. H. Wang, L. Z. Cai, F. K. Zheng, G. C. Guo and J.-S. Huang, CrystEngComm, 2013, 15, 903–910 RSC.
- Y. Feng, X. Liu, L. Duan, Q. Yang, Q. Wei, G. Xie, S. Chen, X. Yang and S. Gao, Dalton Trans., 2015, 44, 2333–2339 RSC.
- J. Zhang, H. Su, Y. Dong, P. Zhang, Y. Du, S. Li and S. Pang, Inorg. Chem., 2017, 56, 10281–10289 CrossRef CAS PubMed.
- L. Batzdorf, F. Fischer, M. Wilke, K. J. Wenzel and F. Emmerling, Angew. Chem., Int. Ed., 2015, 54, 1799–1802 CrossRef CAS PubMed.
- R. Uematsu, E. Yamamoto, S. Maeda, H. Ito and T. Taketsugu, J. Am. Chem. Soc., 2015, 137, 4090–4099 CrossRef CAS PubMed.
- P. Verma, K. D. Vogiatzis, N. Planas, J. Borycz, D. J. Xiao, J. R. Long, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2015, 137, 5770–5781 CrossRef CAS PubMed.
- D. C. Zhong, Y. Q. Wen, J. H. Deng, X. Z. Luo, Y. N. Gong and T. B. Lu, Angew. Chem., Int. Ed., 2015, 54, 11795–11799 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2110–2113 CrossRef CAS PubMed.
- Z. P. Demko and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2113–2116 CrossRef CAS PubMed.
- R. G. Xiong, X. Xue, H. Zhao, X. Z. You, B. F. Abrahams and Z. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800–3803 CrossRef CAS PubMed.
- H. Zhao, Z. R. Qu, H. Y. Ye and R. G. Xiong, Chem. Soc. Rev., 2008, 37, 84–100 RSC.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983–9987 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-2014/7, Program for the refinement of crystal structures, Acta Crystallogr., 2015, 71, 9–18 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1998, 38, 3098–3100 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- D. Andrae, U. Häuβerman, M. Dolg, H. Stoll and H. Preuβ, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. F. W. Bader Atoms in molecules, a quantum theory, Oxford University Press, Oxford, 1990 Search PubMed.
- J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045–4048 CrossRef CAS PubMed.
- J. J. Ritter and J. Kalish, J. Am. Chem. Soc., 1948, 70, 4048–4050 CrossRef CAS PubMed.
- H. H. Michels, J. A. Montgomery, K. O. Christe and D. A. Dixon, J. Phys. Chem., 1995, 99, 187–194 CrossRef CAS.
- Y. D. Liu, J. F. Zhao and Q. S. Li, Theor. Chem. Acc., 2002, 107, 140–146 Search PubMed.
- M. Straka, Chem. Phys. Lett., 2002, 358, 531–536 CrossRef CAS.
- L. Gagliardi and P. Pyykkö, J. Am. Chem. Soc., 2001, 123, 9700–9701 CrossRef CAS PubMed.
- L. Gagliardi and P. Pyykkö, J. Phys. Chem. A, 2002, 106, 4690–4694 CrossRef CAS.
- O. S. Bushuyev, P. Brown, A. Maiti, R. H. Gee, G. R. Peterson, B. L. Weeks and L. J. Hope-Weeks, J. Am. Chem. Soc., 2012, 134, 1422–1425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1487965–1487967. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08486f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Chao Zhanga,
Da Yana,
Zhi Xionga,
Heng Xu*a and
Xiao-Ming Ren*b
*a,
Chao Zhanga,
Da Yana,
Zhi Xionga,
Heng Xu*a and
Xiao-Ming Ren*b