
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton conductive ionic liquid crystalline poly(ethyleneimine) polymers functionalized with oxadiazole†
Alberto Concellón‡
 a,
Silvia Hernández-Ainsabc,
Joaquín Barberáa,
Pilar Romeroa,
José Luis Serranob and
Mercedes Marcos*a
a,
Silvia Hernández-Ainsabc,
Joaquín Barberáa,
Pilar Romeroa,
José Luis Serranob and
Mercedes Marcos*a
aDpto. Química Orgánica, Facultad de Ciencias-Instituto de Ciencia de Materiales de Aragón, Universidad de Zaragoza-CSIC, 50009, Zaragoza, Spain. E-mail: mmarcos@unizar.es
bDpto. Química Orgánica, Instituto de Nanociencia de Aragón, Universidad de Zaragoza, 50009, Zaragoza, Spain
cARAID Foundation, Government of Aragon, 50018, Zaragoza, Spain
First published on 8th November 2018
Abstract
Two novel series of ionic liquid crystal polymers that display proton conductive properties are presented here. These materials are based on linear (l-PEI) or branched (b-PEI) poly(ethyleneimine) functionalized with unsymmetrical oxadiazole carboxylic acids derived from 1,3,4-oxadiazole (1,3,4-OXAm) or 1,2,4-oxadiazole (1,2,4-OXAm). The subscript “m” indicates the length of the spacer between the rigid moiety and the carboxyl group, namely m = 4 and 10. The occurrence of proton transfer from the carboxylic acid to the amine groups was confirmed by FTIR and NMR measurements. The liquid crystalline properties were investigated by differential scanning calorimetry (DSC), polarizing optical microscopy (POM), and X-ray diffraction (XRD). All ionic complexes displayed enantiotropic smectic A mesophases and in the case of the l-PEI derivatives a nematic phase was also observed at high temperatures. All investigated derivatives presented good proton conductivity values as determined by electrochemical impedance spectroscopy (EIS). Therefore, these ionic LC hyperbranched polymers represent an effective approach for the preparation of proton-transporting polymeric materials with potential applications in electrochemical devices.
Introduction
Ionic liquid crystals (ILCs) are fascinating materials that combine the ion-conductive properties of ionic liquids with the anisotropic properties provided by the liquid crystal (LC) state.1,2 In fact, the self-organization of LC materials into various mesophases (nematic, smectic or columnar) provides well-organized channels for the transport of electrons, holes or ions.3–8 Thus, this strategy seems promising for the development of a wide range of efficient ion-conductive materials.9–11ILCs derived from dendritic polymers have attracted special interest because the large number of functional peripheral groups enables the attachment of different moieties that modify the macromolecule properties. In these ionic dendritic polymers, the LC order arises from the nanosegregation between charged groups and non-ionic regions of the molecule.12 This strategy has been widely employed in amine-terminated dendrimers and hyperbranched polymers, such as poly(propyleneimine) (PPI), poly(amidoamine) (PAMAM), or poly(ethyleneimine) (PEI) complexed with different carboxylic acids by a proton transfer reaction between the carboxylic acids and the basic amine groups.13–18
In this regard, our research group has a large record on studying the LC properties of ionic dendrimers. We reported several examples of ionic LC dendrimers based on different generations of PAMAM and PPI that exhibited different properties depending on the characteristics of the attached carboxylic acids. Specifically, the functionalization with aliphatic carboxylic acids19–22 or with an azobenzene containing carboxylic acid produced high and stable photoinduced birefringence.23 Other derivatives bearing 1,3,4-oxadiazole or 1,2,4-oxadiazole rings24 exhibited good luminescent properties, whereas the introduction of carbazole rings25 resulted into good luminescent and electrochemical properties. The well-defined segregation in layers is responsible for the smectic LC behavior exhibited by most of these dendrimers.
We recently developed a new versatile approach for the preparation of proton-conductive materials by using ionic LC dendrimers synthesized from PAMAM dendrimers surrounded by carboxylic acid dendrons bi-functionalized with a promesogenic unit (cholesteryl hemisuccinate) and coumarin moieties.26 All the materials showed good proton conductive properties as the LC arrangement resulted in ionic nanosegregated areas (formed by the ion pairs) that favoured proton conduction.
Aiming to expand the library of ionic LC dendritic materials showing proton conductive properties, we present here two novel series of ionic LC polymers. These are derived from linear (l-PEI) or branched (b-PEI) poly(ethyleneimine) containing 1,3,4-oxadiazole (1,3,4-OXAm) or 1,2,4-oxadiazole (1,2,4-OXAm) carboxylic acids (the subscript “m” indicates the length of the spacer between the rigid moiety and the carboxyl group, namely m = 4 or 10), with unsymmetrical substitutions (Scheme 1). In this work b-PEI was chosen because hyperbranched polymers are likely to have more practical applications than dendrimers as they are prepared very easily (by a one-pot synthesis), while retaining some of the structural features and properties of dendrimers (yielded through tedious and costly multi-step reactions).27 Herein, we report the synthesis, characterization and mesomorphic properties of these ionic LC polymers (linear and branched) with the objective of assessing their possible use as new proton conductive materials for electrochemical devices.
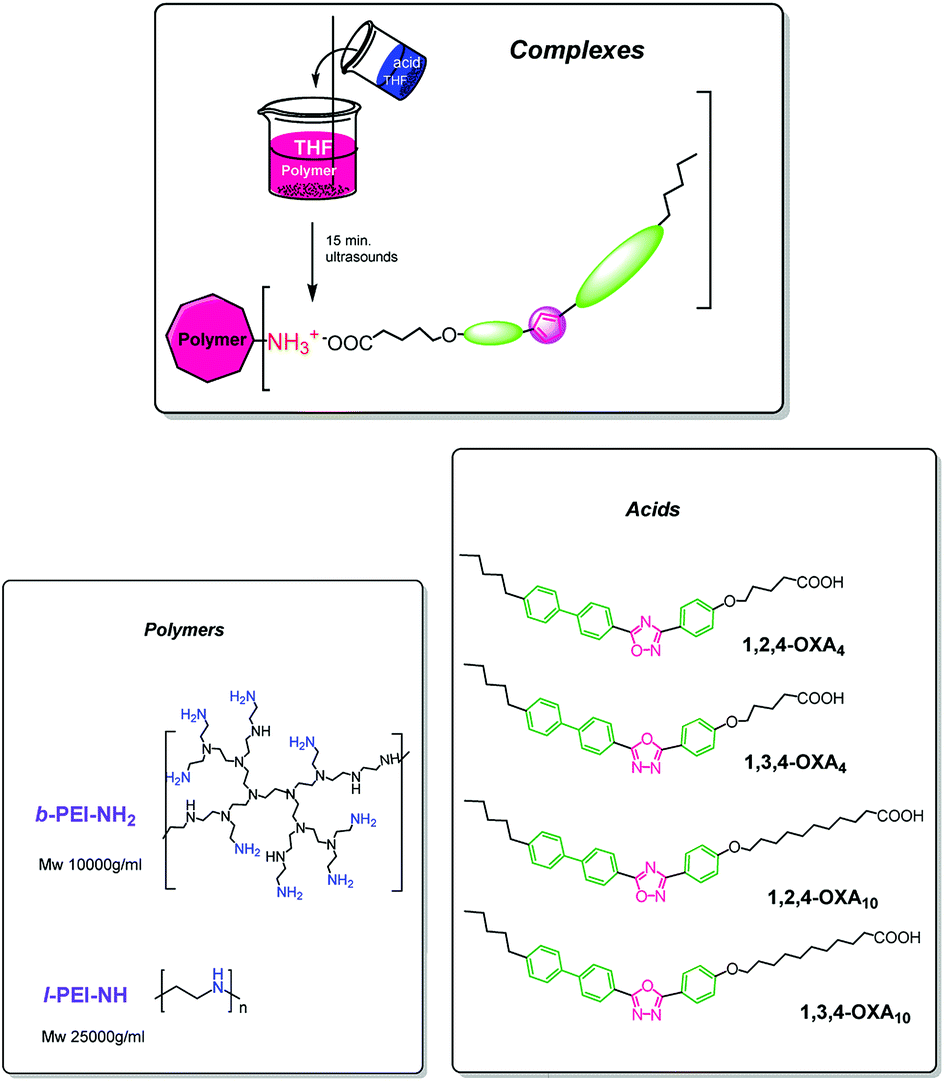 | ||
| Scheme 1 Schematic representation of the synthetic route to give the ionic complexes and their chemical structure. | ||
Experimental section
Synthesis and nomenclature of the oxadiazole acids
Two oxadiazole isomers (1,3,4- and 1,2,4-) were used to prepare the acids. They are denoted as 1,3,4-OXAm and 1,2,4-OXAm. The subscript “m” indicates the length of the spacer between the rigid moiety and the carboxyl group, namely m = 4 and 10. Scheme 1 shows the structure of the different acids. The preparation of the acids was reported previously.24Synthesis and nomenclature of the ionic polymers
Ionic polymers were synthesized as schematically represented in Scheme 1, following a previously described method.19 n equiv. of the corresponding oxadiazole acid were dissolved in anhydrous tetrahydrofuran (THF) and were added to a solution of 1 equiv. of the corresponding PEI polymer in anhydrous THF. The mixture was sonicated for 15 minutes, and then it was slowly evaporated at room temperature and dried under vacuum for ca. 12 h at 40 °C until the weight remains constant. These compounds are named b-PEI-(1,3,4-OXA4)n, b-PEI-(1,2,4-OXA4)n, b-PEI-(1,3,4-OXA10)n, b-PEI-(1,2,4-OXA10)n, l-PEI-(1,3,4-OXA4)n and l-PEI-(1,2,4-OXA4)n.Results and discussion
Characterization of the ionic polymers
Selected examples of NMR spectra of the ionic complexes derived from 1,2,4-OXA4 are shown in Fig. 1 and 2. The broad signal at 9.20–9.60 ppm corresponding to the carboxylic acid proton (HZ) of the oxadiazole derivative disappeared in the 1H NMR spectra of the complexes. In addition, the 13C signal of the carboxylic group was slightly shifted upfield in the spectra of the ionic complexes. A shifting of −0.06 ppm was also observed for the methylene protons (HA) in alpha position to the carboxylic group of the oxadiazole derivatives. In the same way, the ionic complexes formation was also corroborated by the displacement of the methylene CA carbon signal from 33.6 to 34.6–35.1 ppm.
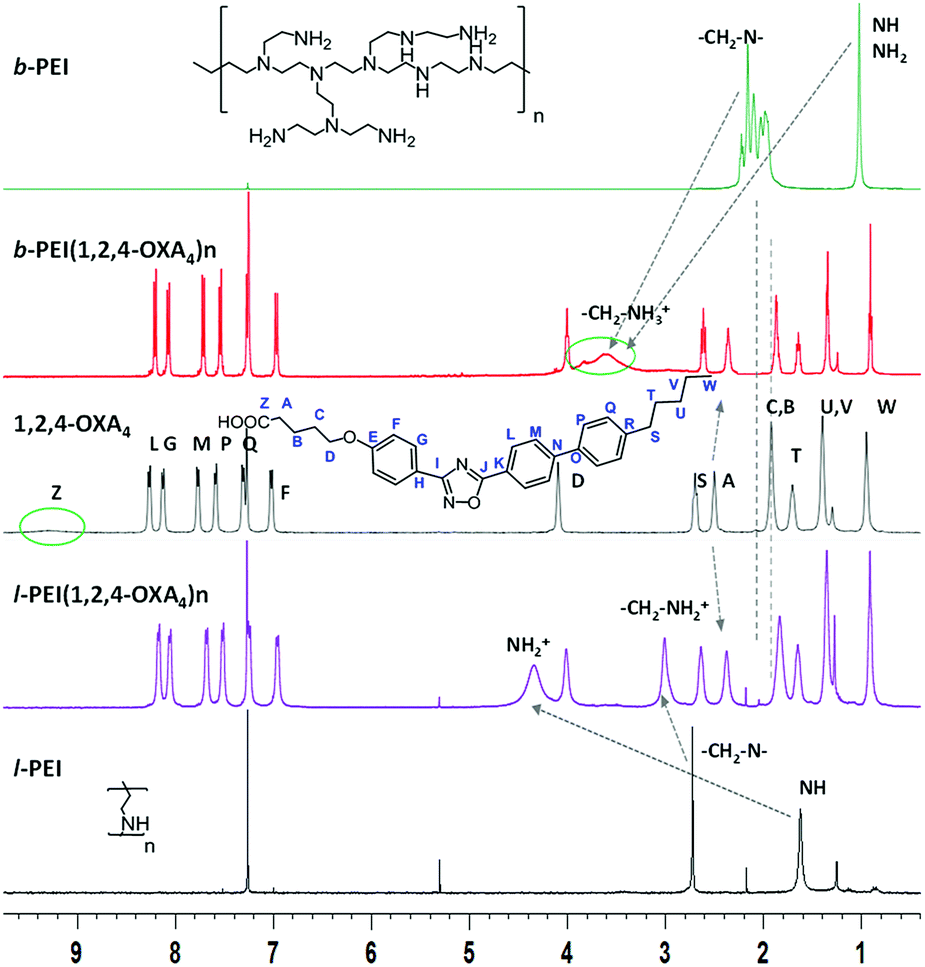 | ||
| Fig. 1 1H NMR spectra in CDCl3 of ionic complexes b-PEI(1,2,4-OXA4)n and l-PEI(1,2,4-OXA4)n as well as of the (1,2,4-OXA4) acid and the hyperbranched (b-PEI) and linear (l-PEI) poly(ethyleneimine) polymers. | ||
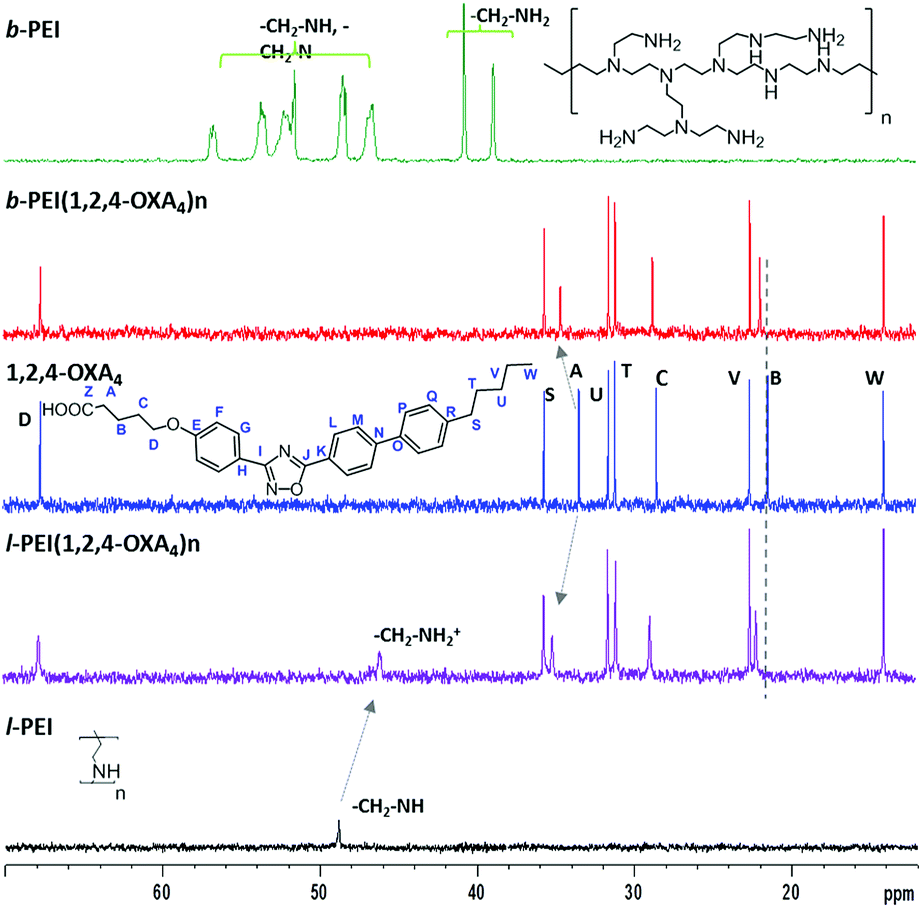 | ||
| Fig. 2 13C NMR spectra in CDCl3 of complexes b-PEI(1,2,4-OXA4)n and l-PEI(1,2,4-OXA4)n as well as of the (1,2,4-OXA4) acid and the hyperbranched (b-PEI) and linear (l-PEI) poly(ethyleneimine) polymers. | ||
Moreover, protons of the terminal amine groups and methylene protons of the dendritic part underwent a considerable deshielding in the 1H NMR spectra. The 13C signals of these methylene groups also shifted from 49.1 to 46.1 ppm for l-PEI complex. In the case of b-PEI(1,2,4-OXA4)n complex, the shifts of these methylene groups could not be determined due to its low solubility in CDCl3, but it was confirmed by HSQC experiments in DMSO-d6 (see Fig. S1†).
Furthermore, the 1H–1H NOESY experiment showed cross-peaks between the methylene groups in alpha (CA) or beta (CB) position to the carboxylate and the PEI-amine protons, confirming the formation of the ion pairs (see Fig. S2†).
13C CPMAS experiments were carried out in the case of insoluble complexes. In these spectra, the branched PEI (b-PEI) can be observed forming part of the ionic complex. The signal of the carboxylic acid is spread out in the frequency domain and the packing of oxadiazole core differs with regard to the packing of the initial acids (Fig. 3).
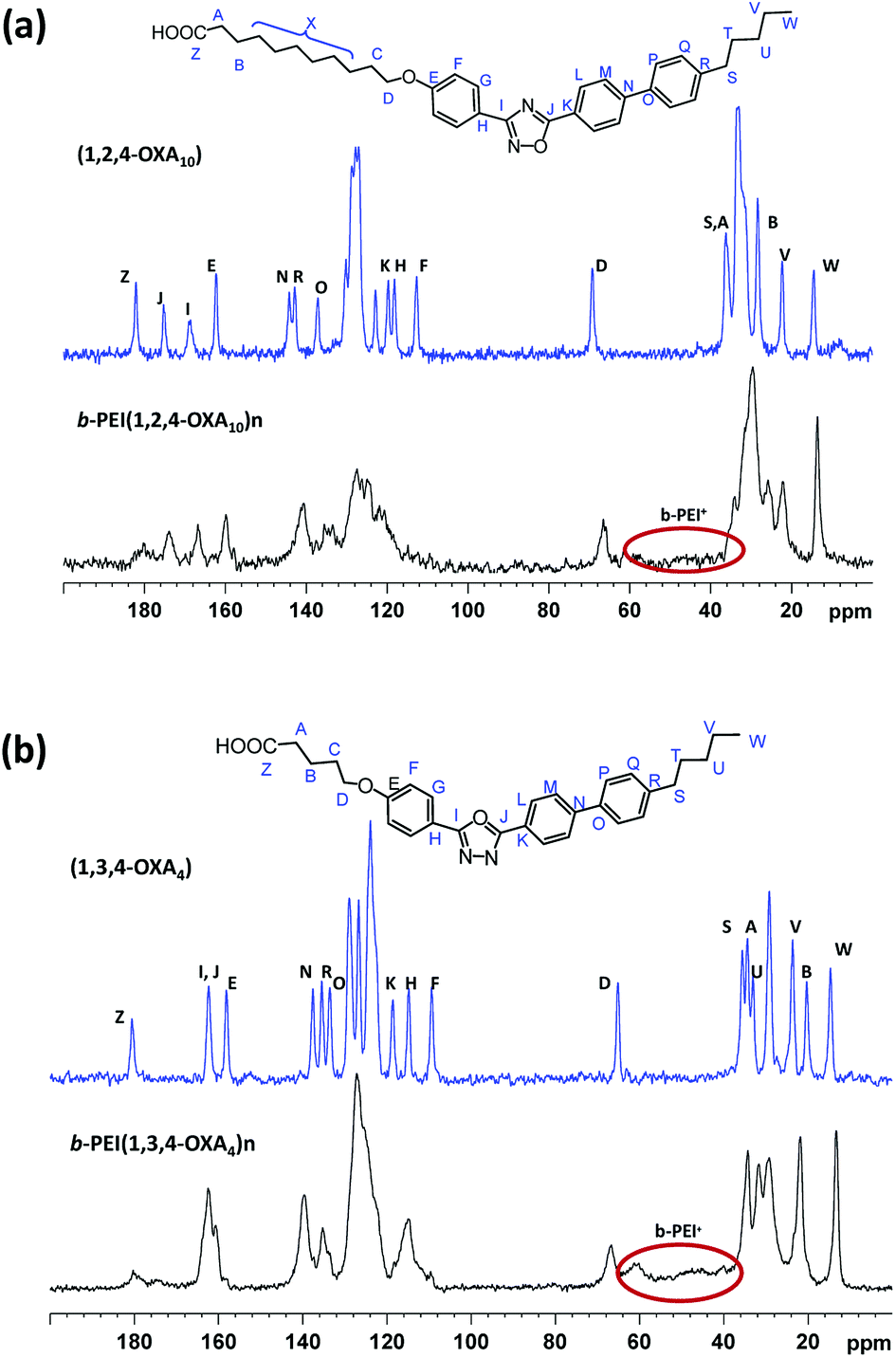 | ||
| Fig. 3 13C CPMAS spectra of b-PEI complexes (bottom) and their corresponding acids (top): (a) b-PEI(1,2,4-OXA10)n and (b) b-PEI(1,3,4-OXA4)n. | ||
Thermal stability of the polymers
The thermal stability of the ionic dendrimers was studied by thermogravimetric analysis (TGA). All the samples showed good thermal stability and in all cases the 5% weight loss temperature (T5%) was detected at temperatures above the isotropization point (Table S4, Fig. S9a†).Liquid crystal properties
The mesomorphic behavior of the compounds was analyzed by POM and DSC. Three cycles were carried out in DSC experiments and data were taken from the second cycle. In some cases, the isotropization temperatures were taken from POM observations because no transition peaks were detected in the DSC curves (Table 1). The nature of the mesophase was also confirmed by X-ray diffraction (XRD).| Compound | Transitions (°C), ΔHa [J g−1] | Texpc (°C) | Mesophased | dexpe (Å) | df (Å) |
|---|---|---|---|---|---|
| a Data from the second scan and taken at the maximum of the peak. C = crystal, g = mesomorphic glass, N = nematic mesophase, I = isotropic liquid.b Data taken from POM.c Temperature of the XRD experiment.d Mesophase exhibited by the compounds at the given temperature.e Measured spacings.f d = layer spacing (Å) of the smectic phase. | |||||
| 1,2,4-OXA4 | C 153 [26.7] SmA 190 [0.5] N 217 [1.0] I | — | — | — | — |
| b-PEI-(1,2,4-OXA4)n | g 102 SmA 204–230b I | 150 | SmA | 49/24.5 | 49 |
| l-PEI-(1,2,4-OXA4)n | C 127 [17.6] SmA 210 N 224 [1.2] I | 150 | SmA | 65/32.5 | 65 |
| 1,2,4-OXA10 | C 142 [35.3] SmA 181 [0.7] N 193 [0.8] I | — | — | — | — |
| b-PEI-(1,2,4-OXA10)n | g 39 SmA 218–230b I | — | — | — | — |
| 1,3,4-OXA4 | C 186 [54.0] I; I 166b N 162 [50.4] C | — | — | — | — |
| b-PEI-(1,3,4-OXA4)n | C 135 SmA 180–195b I | 140 | SmA | 55 | 55 |
| l-PEI-(1,3,4-OXA4)n | g 60 SmA 160 N 194b I | — | — | — | — |
| 1,3,4-OXA10 | C 175 [47.9] I | — | — | — | — |
| b-PEI-(1,3,4-OXA10)n | g 120 SmA 170 b I | — | — | — | — |
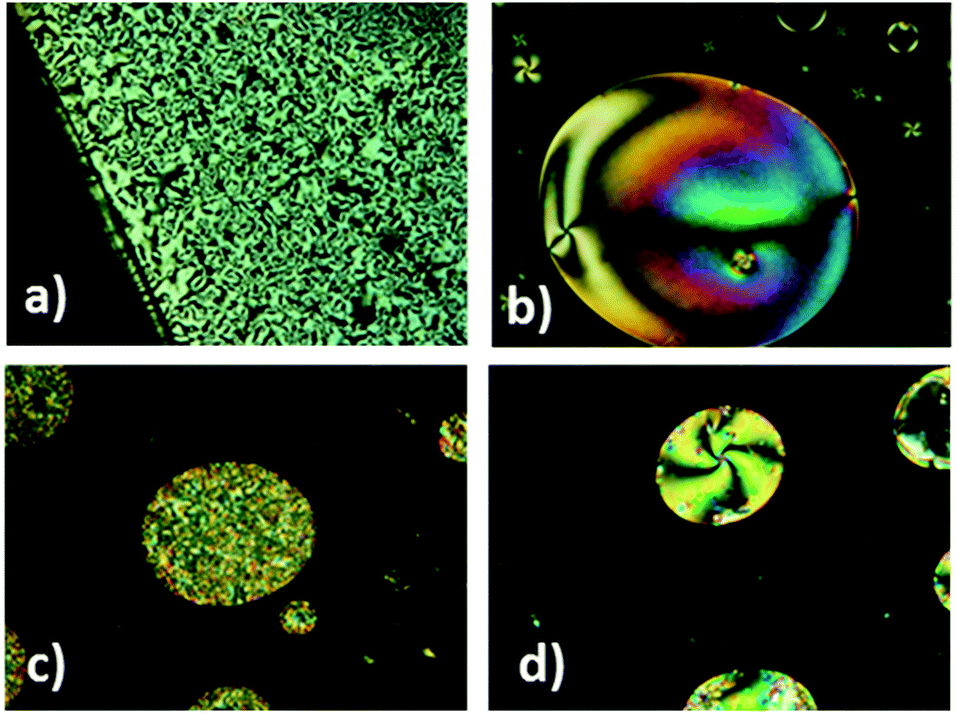 | ||
| Fig. 4 POM textures of (a) 1,2,4-OXA4 (taken at 215 °C in the 1st cooling), (b) 1,2,4-OXA10 (taken at 191 °C, in the 1st cooling), (c) 1,3,4-OXA4 (taken at 160 °C in the 4th cooling), and (d) 1,3,4-OXA4 (nematic droplets taken at 160 °C in the 1st cooling). | ||
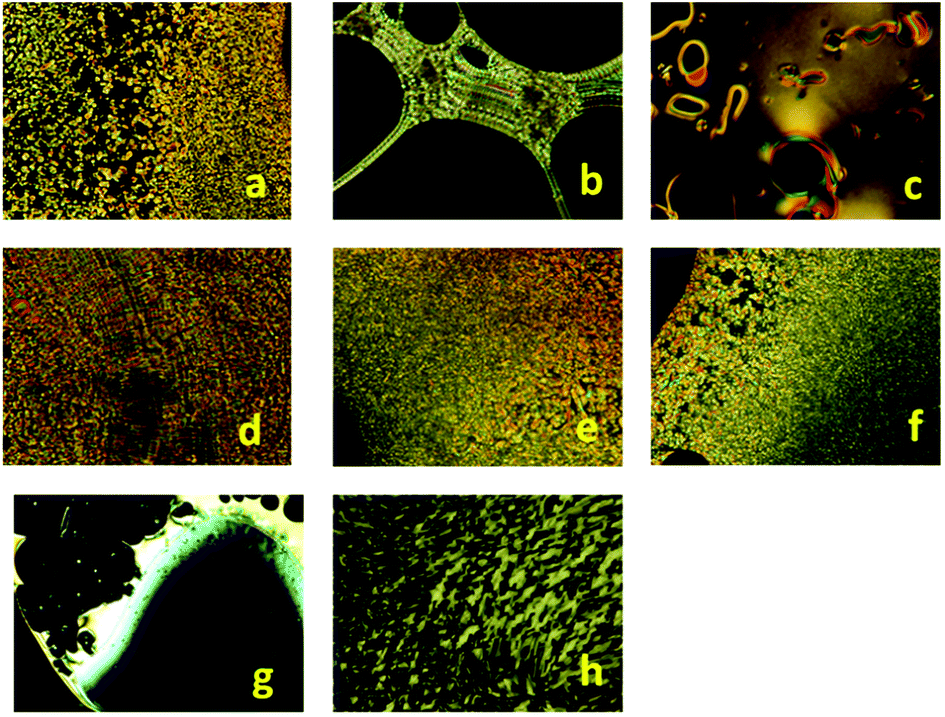 | ||
| Fig. 5 POM textures of: (a) b-PEI-(1,2,4-OXA4)n (taken at 179 °C in the 2nd cooling), (b) l-PEI-(1,2,4-OXA4)n (taken at 152 °C in the 1st heating), (c) l-PEI-(1,2,4-OXA4)n (taken at 222 °C, in the 1st cooling), (d) b-PEI-(1,2,4-OXA10)n (taken at 229 °C in the 1st heating), (e) b-PEI-(1,3,4-OXA4)n (taken at 170 °C in the 2nd cooling), (f) b-PEI-(1,3,4-OXA10)n (taken at 155 °C in the 2nd cooling), (g) l-PEI-(1,3,4-OXA4)n (taken at 163 °C, in the 1st cooling), (h) l-PEI-(1,3,4-OXA4)n (taken at 105 °C in the 2nd heating). | ||
The type of oxadiazole isomer also influences the liquid crystal properties due to the different exocyclic bond angles and the different polarity of the 1,3,4-oxadiazole or 1,2,4-oxadiazole central heterocyclic rings. For instance, LC transition temperatures are strongly affected by the oxadiazole isomers. The compounds derived from 1,2,4-oxadiazole exhibited lower melting points but higher isotropization temperatures compared to those displayed by the 1,3,4-oxadiazole homologues. Consequently, 1,2,4-oxadiazole produced broader mesomorphic temperature ranges (see Table 1).
The layer spacing values (d) shown in Table 1 depend on the compound. The layer thickness value displayed by the branched b-PEI-(1,2,4-OXA4)n derivative is contracted compared to homologous lineal derivative (l-PEI-(1,2,4-OXA4)n). On the other hand for compound b-PEI-(1,3,4-OXA4)n, containing the other regioisomeric oxadiazole ring, the layer spacing is intermediate between those of b-PEI-(1,2,4-OXA4)n and l-PEI-(1,2,4-OXA4)n. The experimentally-measured layer thickness for the mesophase of these compounds is consistent with a molecular arrangement in which the poly(ethyleneimine) part of the molecule is located in the inner part of each layer and the mesogenic units statistically extend upwards and downwards to both layer boundaries. This structural model has been previously described by us for similar ionic and non-ionic mesogenic dendrimers.24
The variations in the layer thickness as a function of each particular compound are probably due to the high conformational freedom of the poly(ethyleneimine) polymer. As a consequence, the thickness of the central slab of the layer occupied by the poly(ethyleneimine) moiety can change to a great extent and this produces an important variation in the total layer thickness. Moreover, there can be some interpenetration between neighbouring layers through interdigitation of the mesogenic units. This phenomenon seems to be more favored in the case of the b-PEI derivatives, as can be deduced from their smaller layer thickness compared to l-PEI-(1,2,4-OXA4)n, probably as a consequence of the different conformational behavior of each type of poly(ethyleneimine) unit. In addition to this, the nature of the oxadiazole exerts an influence on the layer spacing with b-PEI-(1,3,4-OXA4)n showing larger value than b-PEI-(1,2,4-OXA4)n. This phenomenon is probably related to a larger degree of interdigitation for the mesogenic unit based on 1,2,4-oxadiazole.
It is interesting to note that, while the second order layer reflection is absent for b-PEI-(1,3,4-OXA4)n, this reflection is clearly visible for b-PEI-(1,2,4-OXA4)n and l-PEI-(1,2,4-OXA4)n. Moreover its intensity is higher than usual, and for compound b-PEI-(1,2,4-OXA4)n it is even stronger than the first order reflection. This unusual feature found for the two polymers containing the mesogenic unit derived form 1,2,4-oxadiazole must arise from the presence of a period d/2 in the electron-density wave in the direction perpendicular to the layers. Similar behavior has been previously reported for some side-chain liquid-crystalline polymers and it is explained by the confinement of the polymeric backbones in a thin sublayer, so that the polymeric backbones produce an electron-density maximum comparable to that of the mesogenic cores.28,29
Proton conductive properties
The proton conductivity was measured using electrochemical impedance spectroscopy (EIS) in samples consisting of films sandwiched between ITO-coated electrodes. The typical EIS response (Nyquist plots) consisted of a suppressed semicircle in the high-frequency region and an incline straight line in the low-frequency range. Since diffusible ions apart from protons did not exist in the compounds, the observed EIS responses were ascribed to proton conduction, which was calculated from the EIS responses and the cell constant (see Experimental Section in the ESI†).The proton conductivities of some selected complexes were measured to investigate the effect of the dendrimer core, oxadiazole regioisomer type and length of the spacer on this property. Namely, the proton conductivities of b-PEI-(1,2,4-OXA4)n, b-PEI-(1,2,4-OXA10)n, b-PEI-(1,3,4-OXA10)n and l-PEI-(1,3,4-OXA4) were measured as a function of the temperature (Fig. 6). For all the ionic compounds, the conductivities increased as the temperature increased from 30 °C to 225 °C (from 303 K to 498 K). A plateau was observed once the isotropic state was reached.
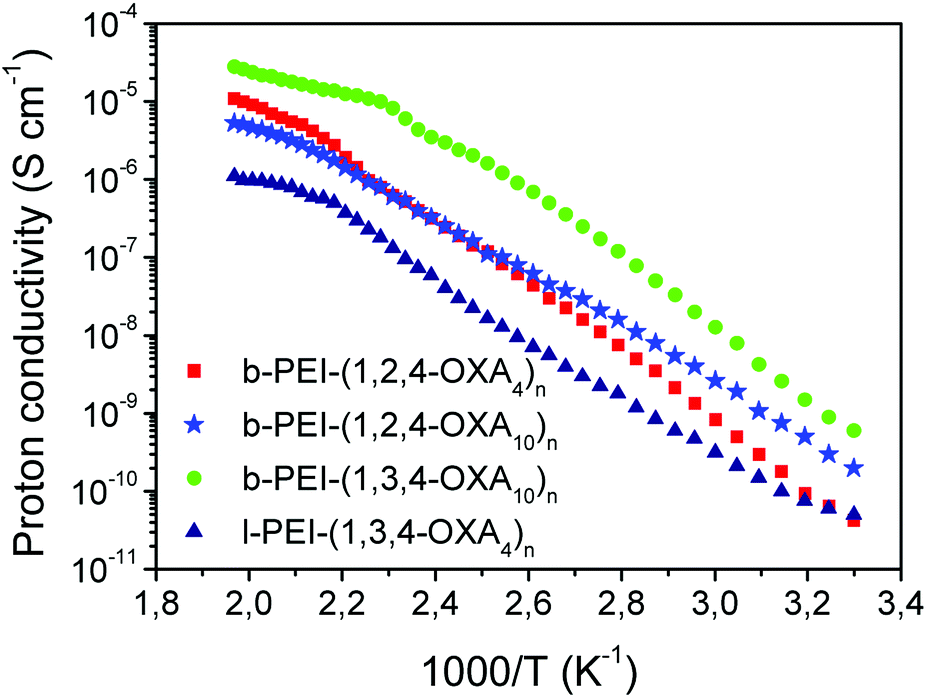 | ||
| Fig. 6 Proton conductivities as a function of the temperature. | ||
At low temperatures, conductivities of b-PEI-(1,2,4-OXA4)n (see red data in Fig. 6) increased respect to those measured in b-PEI-(1,2,4-OXA10)n (see light blue data in Fig. 6), indicating that the longer alkyl spacer reduces conductivity. For instance, conductivity values of b-PEI-(1,2,4-OXA10)n and b-PEI-(1,2,4-OXA4)n at 50 °C (323 K) were 3 × 10−10 and 1 × 10−9 S cm−1, respectively. This enhanced conductivity in b-PEI-(1,2,4-OXA10)n may be related to the more efficient molecular packing produced by this derivative in the smectic mesophase. It has been previously described that oxadiazole moieties with longer spacers favor a more efficient interaction between neighboring oxadiazoles, while shorter spacers preclude such interactions.31 Thus, the mean effective hopping distances in b-PEI-(1,2,4-OXA4)n are bigger than those of the analogous derivatives with longer alkyl spacers, and therefore a decrease in conductivity is expected.
Oxadiazole type also exerts a significant influence in the conductivity values. Indeed, b-PEI-(1,3,4-OXA10)n ionic polymer (green data in Fig. 6), exhibited larger conductivity values at any temperature compared to b-PEI-(1,2,4-OXA10)n (light blue data in Fig. 6). For instance, the proton conductivity value for b-PEI-(1,3,4-OXA10)n at 100 °C (4 × 10−7 S cm−1) was 20 times higher than that of b-PEI-(1,2,4-OXA10)n (2 × 10−8 S cm−1). This result indicates that the 1,3,4-oxadiazole isomer allows for more effective intermolecular interactions, thereby favoring a more ordered supramolecular arrangement that may enhance the proton transport by reducing the hopping distances.
On the other hand, the dendritic polymer core greatly influences the ionic conductivity. Namely, l-PEI-(1,3,4-OXA4)n side-chain polymer exhibited the lowest proton conductivity at any temperature. The hyperbranched PEI (b-PEI) core enables a more congested packing than its lineal analogue (l-PEI). Therefore, in the derivatives b-PEI-(1,2,4-OXA4)n, b-PEI-(1,2,4-OXA10)n, b-PEI-(1,3,4-OXA10)n, the mobility of the ionic pairs is restricted compared to the l-PEI-(1,3,4-OXA4)n side-chain LC polymer, which favors their supramolecular alignment and hence contributes to enhance proton conductivities.
Because protons have to travel between electrodes, in liquid crystalline materials the measured proton conductivity depends on the macroscopic alignment of the phase with respect to the electrodes. Therefore, several alignment procedures (shearing and thermal treatments) were carried out in an effort to uniform planar alignment of the smectic layers. However, these attempts were not successful, and all measurements were performed on polydomain samples. Thus, the measured values can be considered as a lower estimation of the proton conductivity. Nonetheless, despite the fact that the conductivity values obtained for the hyperbranched ionic polymers are 3–4 orders of magnitude lower than those of obtained for non-mesogenic polymers (e.g. Nafion, poly(ethylene oxide), or poly(sulfonic) polymers),30,31 they are still high compared to other liquid crystalline materials,10,11,32 providing an attractive strategy to prepare proton conductors.
Conclusions
We have prepared new ionic LC dendrimers by complexation between a hyperbranched or linear PEI polymer and oxadiazole-containing acids. The non-covalent architectures were obtained by the formation of the ionic salts between the carboxylic acid group and the terminal amine groups of the PEI polymers. All the compounds exhibited smectic A mesogenic behavior and linear polymers also presented a nematic mesophase. Electrochemical impedance spectroscopy measurements revealed that the LC phases obtained in these ionic hyperbranched dendrimers clearly favored proton conduction due to the presence of ionic continuous nanosegregated areas.Our results suggest that these ionic LC dendritic polymers provide a simple and versatile strategy for the development of proton-conductive materials with potential applications in electrochemical devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the MINECO-FEDER funds (project CTQ2015-70174), Gobierno de Aragón-FSE (Research Group E47_17R). The authors acknowledge the use of the SAI (UZ) and CEQMA (UZ-CSIC) Services.References
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- M. Mansueto and S. Laschat, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2014, ch. 8, vol. 6, pp. 231–280 Search PubMed.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- V. Iguarbe, A. Concellón, R. Termine, A. Golemme, J. Barberá and J. L. Serrano, ACS Macro Lett., 2018, 7, 1138–1143 CrossRef CAS.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- A. Concellón, M. Marcos, P. Romero, J. L. Serrano, R. Termine and A. Golemme, Angew. Chem., Int. Ed., 2017, 56, 1259–1263 CrossRef PubMed.
- M. O'Neill and S. M. Kelly, Adv. Mater., 2011, 23, 566–584 CrossRef PubMed.
- E. K. Fleischmann and R. Zentel, Angew. Chem., Int. Ed., 2013, 52, 8810–8827 CrossRef CAS PubMed.
- M. Yoshio and T. Kato, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2014, ch. 23, vol. 8, pp. 727–749 Search PubMed.
- L. Vanti, S. Mohd Alauddin, D. Zaton, N. F. K. Aripin, M. Giacinti-Baschetti, C. T. Imrie, A. Ribes-Greus and A. Martinez-Felipe, Eur. Polym. J., 2018, 109, 124–132 CrossRef CAS.
- T. Liang, H. P. C. van Kuringen, D. J. Mulder, S. Tan, Y. Wu, Z. Borneman, K. Nijmeijer and A. P. H. J. Schenning, ACS Appl. Mater. Interfaces, 2017, 9, 35218–35225 CrossRef CAS PubMed.
- S. Hernández-Ainsa, M. Marcos and J. L. Serrano, in Handbook of Liquid Crystals, ed. J. W. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Wiley-VCH Verlag GmbH & Co. KGaA, 2nd edn, 2014, ch. 7, vol. 7, pp. 259–300 Search PubMed.
- P.-J. Yang, C.-W. Wu, D. Sahu and H.-C. Lin, Macromolecules, 2008, 41, 9692–9703 CrossRef CAS.
- N. Canilho, E. Kasëmi, A. D. Schlüter and R. Mezzenga, Macromolecules, 2007, 40, 2822–2830 CrossRef CAS.
- Y. Chen, Z. Shen, L. Gehringer, H. Frey and S.-E. Stiriba, Macromol. Rapid Commun., 2006, 27, 69–75 CrossRef.
- A. G. Cook, U. Baumeister and C. Tschierske, J. Mater. Chem., 2005, 15, 1708–1721 RSC.
- D. Tsiourvas, T. Felekis, Z. Sideratou and C. M. Paleos, Liq. Cryst., 2004, 31, 739–744 CrossRef CAS.
- A. F. Thünemann and J. Beyermann, Macromolecules, 2000, 33, 6878–6885 CrossRef.
- R. Martín-Rapún, M. Marcos, A. Omenat, J. Barberá, P. Romero and J. L. Serrano, J. Am. Chem. Soc., 2005, 127, 7397–7403 CrossRef PubMed.
- S. Hernández-Ainsa, J. Barberá, M. Marcos and J. L. Serrano, Chem. Mater., 2010, 22, 4762–4768 CrossRef.
- S. Hernández-Ainsa, M. Marcos, J. Barberá and J. L. Serrano, Angew. Chem., Int. Ed., 2010, 49, 1990–1994 CrossRef PubMed.
- M. Marcos, R. Martín-Rapún, A. Omenat and J. L. Serrano, Chem. Soc. Rev., 2007, 36, 1889–1901 RSC.
- M. Marcos, R. Alcalá, J. Barberá, P. Romero, C. Sánchez and J. L. Serrano, Chem. Mater., 2008, 20, 5209–5217 CrossRef CAS.
- S. Hernández-Ainsa, J. Barberá, M. Marcos and J. L. Serrano, Macromolecules, 2012, 45, 1006–1015 CrossRef.
- S. Castelar, P. Romero, J. L. Serrano, J. Barberá and M. Marcos, RSC Adv., 2015, 5, 65932–65941 RSC.
- A. Concellón, T. Liang, A. P. H. J. Schenning, J. L. Serrano, P. Romero and M. Marcos, J. Mater. Chem. C, 2018, 6, 1000–1007 RSC.
- A. M. Caminade, D. Yan and D. K. Smith, Chem. Soc. Rev., 2015, 44, 3870–3873 RSC.
- P. Davidson, A. M. Levelut, M. F. Achard and F. Hardouin, Liq. Cryst., 1989, 4, 561–571 CrossRef CAS.
- J. Barberá, L. Giorgini, F. Paris, E. Salatelli, R. M. Tejedor and L. Angiolini, Chem.–Eur. J., 2008, 14, 11209–11221 CrossRef PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS PubMed.
- D.-J. Mulder, T. Liang, Y. Xu, J. ter Schiphorst, L. M. W. Scheres, B. M. Oosterlaken, Z. Borneman, K. Nijmeijer and A. P. H. J. Schenning, J. Mater. Chem. C, 2018, 6, 5018–5024 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08253g |
| ‡ Present address: Department of Chemistry, Massachusetts Institute of Technology, MA 02139, Cambridge, USA. |
| This journal is © The Royal Society of Chemistry 2018 |