
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nitroalkane-based approach to one-pot three-component synthesis of isocryptolepine and its analogs with potent anti-cancer activities†
Nicolai A. Aksenov *a,
Alexander V. Aksenova,
Alexander Kornienkob,
Annelise De Carvalhocd,
Véronique Mathieucd,
Dmitrii A. Aksenova,
Sergei N. Ovcharova,
Georgii D. Griaznove and
Michael Rubin*ae
*a,
Alexander V. Aksenova,
Alexander Kornienkob,
Annelise De Carvalhocd,
Véronique Mathieucd,
Dmitrii A. Aksenova,
Sergei N. Ovcharova,
Georgii D. Griaznove and
Michael Rubin*ae
aDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355009, Russian Federation. E-mail: naksenov@ncfu.ru
bDepartment of Chemistry and Biochemistry, Texas State University, San Marcos, Texas 78666, USA
cDepartment of Pharmacotherapy and Pharmaceutics, Faculté de Pharmacie, Université Libre de Bruxelles, Brussels, Belgium
dULB Cancer Research Center, Université Libre de Bruxelles, Brussels, Belgium
eDepartment of Chemistry, University of Kansas, 1251 Wescoe Hall Dr., Lawrence, KS 66045-7582, USA. E-mail: mrubin@ku.edu; Tel: +1-785-864-5071
First published on 1st November 2018
Abstract
A second generation polyphosphoric acid-mediated one-pot three-component synthesis of indoloquinoline scaffold is developed. This improved version of the process involves electrophilically activated nitroalkanes for the installation of strategic C–C and C–N bonds and ring C assembly. This modification allows the elimination of unnecessary solvent change operations and all steps are carried out in a true, uninterrupted one-pot manner. A further improvement involves the possibility to install an ortho-amino group in situ. A synthetic application of this method is showcased by the concise synthesis of an isocryptolepine alkaloid and its synthetic analogs with potent anticancer activities.
Introduction
Isocryptolepine (cryptosanguinolentine, 1), one of the four types of indoloquinoline alkaloids isolated from West African plant Cryptolepis sanguinolenta,1 has been investigated as a promising antimalarial and anticancer agent (Fig. 1),2 and has been the target of numerous synthetic efforts.3 All these synthetic routes required multistep linear sequences with the isolation and purification of intermediates. Recently, we disclosed a highly efficient metal-free three-component synthesis of isocryptolepine and its bioactive analogs involving a sequential assembly of the indoloquinoline core in one-pot with a single isolation operation.4 This synthetic approach was inspired by the 2009 Kundu total synthesis, featuring Fischer indolization followed by an oxidative Pictet–Spengler type reaction to assemble ring C.5 Our featured protocol (Generation 1) involved the formation of hydrazones 4 from hydrazines 2 and ortho-aminoacetophenones 3 (step A), followed by polyphosphoric acid (PPA)-assisted Fischer indolization (step B) to furnish indole 5 (Scheme 1).4 Subsequent acid-mediated acylation of ortho-aniline substituent at C-2 (step C) with an appropriate carbonyl compound equivalent (carboxylic acid, nitrile or triazine) afforded species 6, that underwent intramolecular Vilsmeier reaction (step D) to provide the indoloquinoline core of the alkaloid (Scheme 1).4 Remarkably, this transformation is carried out in a one-pot fashion requiring a single isolation operation at the end of the sequence, leading to an efficient synthesis of an anticancer drug candidate library for SAR studies. However, this synthetic protocol depended on the availability of ortho-aminoacetophenones 3 and carbonyl compound equivalents (acylating agents used in step C: carboxylic acids, nitriles or 1,3,5-triazines). We also discovered an alternative protocol involving unusual cyclization with nitroalkenes accompanied by extrusion of HCN (Scheme 2).6 Still, these previously developed methods did not provide any synthetically meaningful results in reactions of acylating agents bearing functional group as substituent R5. In certain cases, these issues rendered synthesis of some types of compounds for our SAR studies impossible or cost prohibitive. Herein, we disclose a further modified synthetic protocol (Generation 2) addressing these problems, and results of our second series of screening for antiproliferative activities that allowed to identify highly potent lead structures.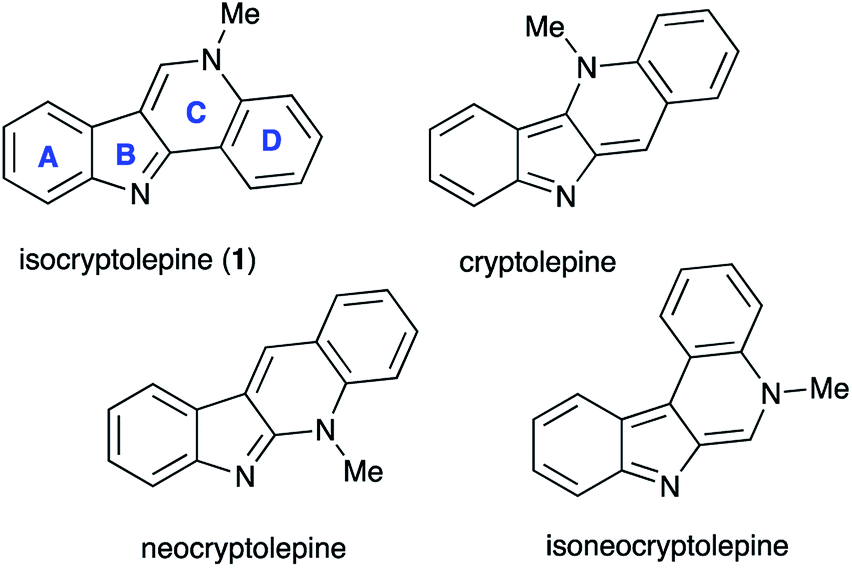 | ||
| Fig. 1 Natural indoloquinoline alkaloids isolated from Cryptolepis sanguinolenta. | ||
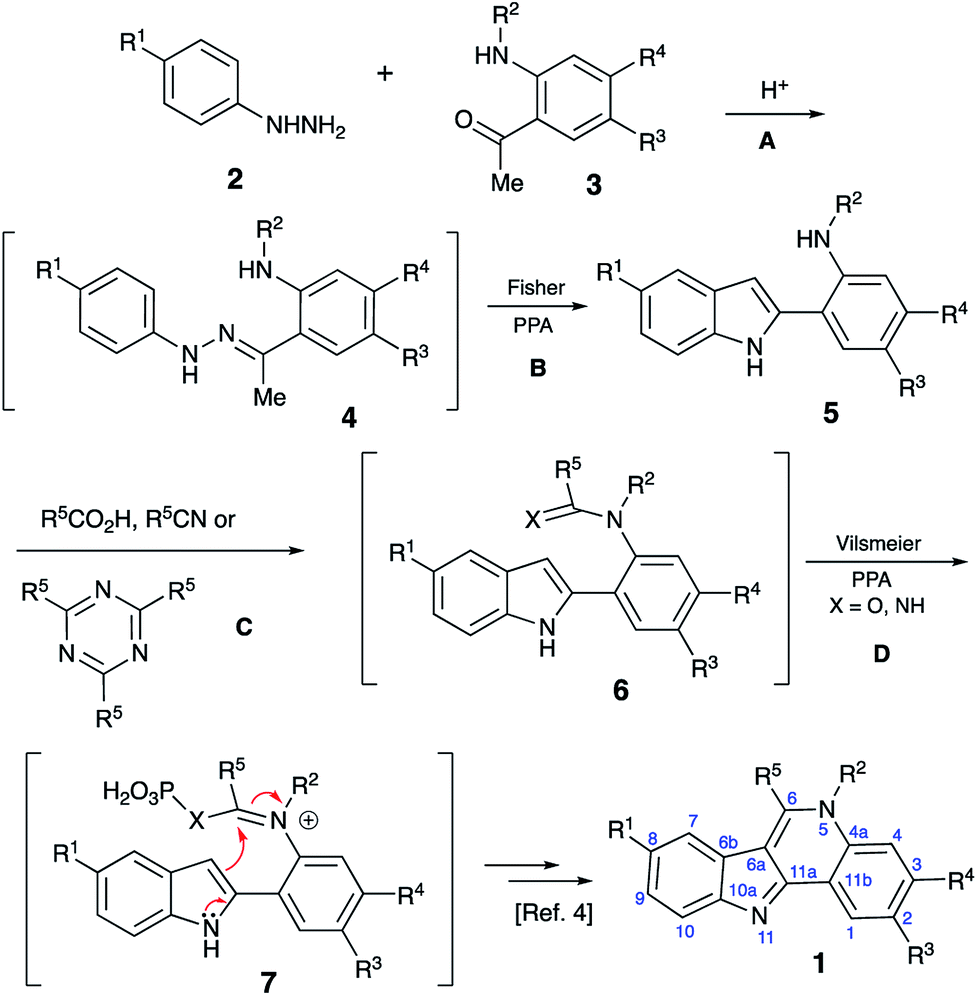 | ||
| Scheme 1 | ||
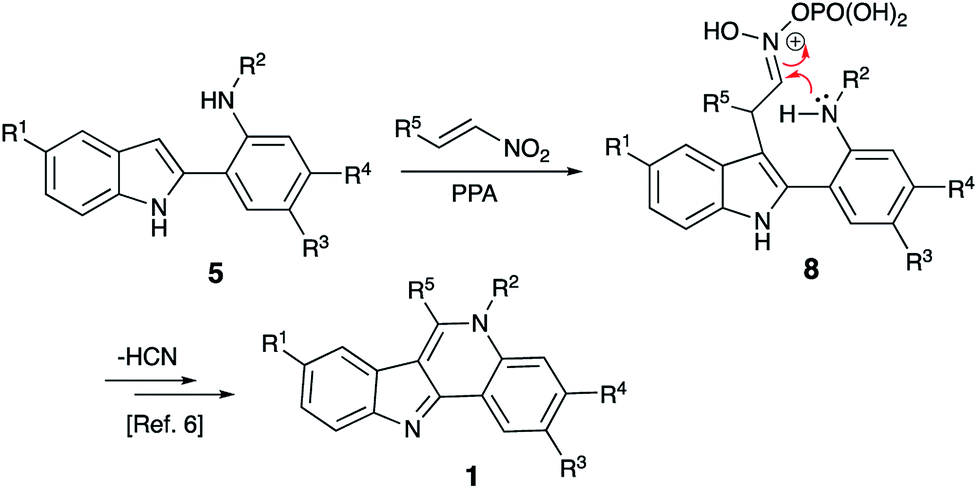 | ||
| Scheme 2 | ||
Results and discussion
It should be pointed out that cyclization of intermediate 8 shown in Scheme 2 involves an unusual electrophilic component formed via phosphorylation of a nitroalkane in nitronate form. Mechanistically, this process is related to the classical Nef reaction,7 employing nucleophilic amine species instead of water. We have discovered a number of innovative one-pot transformations involving nitrogen8 or carbon9 nucleophile-based versions of the Nef reaction and wondered if nitroalkane species 9, electrophilically activated by PPA, could serve as an alternative acylating agent to trigger cyclization of the in situ generated intermediate 5 into indoloquinoline species 1. Indeed, a nucleophilic attack of indole 5 across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the phosphorylated nitronate 10 would afford (azanediylbis(oxy))bis(phosphonic) species 11, which after 1,2-elimination of ortho-phosphoric acid would produce O-phosphorylated oxime 12 (Scheme 3). Intramolecular nucleophilic attack of aniline moiety would produce cyclic aminal 13, which after 1,2-elimination of hydroxylamine O-phosphonate would aromatize into product 1 (Scheme 3). Alternatively, nitronate 10 can be attacked by the nucleophilic aniline moiety in 5 to form an O-phosphorylated N′-hydroxyacetimidamide species that can be viewed as a direct analog of intermediate 6 (with X = NOPO(OH)2). This intermediate can undergo a subsequent Vilsmeier-type intramolecular attack involving the nucleophilic C-3 center of indole moiety to produce indoloquinoline 1 in a way similar to the one described in Scheme 1.
N bond of the phosphorylated nitronate 10 would afford (azanediylbis(oxy))bis(phosphonic) species 11, which after 1,2-elimination of ortho-phosphoric acid would produce O-phosphorylated oxime 12 (Scheme 3). Intramolecular nucleophilic attack of aniline moiety would produce cyclic aminal 13, which after 1,2-elimination of hydroxylamine O-phosphonate would aromatize into product 1 (Scheme 3). Alternatively, nitronate 10 can be attacked by the nucleophilic aniline moiety in 5 to form an O-phosphorylated N′-hydroxyacetimidamide species that can be viewed as a direct analog of intermediate 6 (with X = NOPO(OH)2). This intermediate can undergo a subsequent Vilsmeier-type intramolecular attack involving the nucleophilic C-3 center of indole moiety to produce indoloquinoline 1 in a way similar to the one described in Scheme 1.
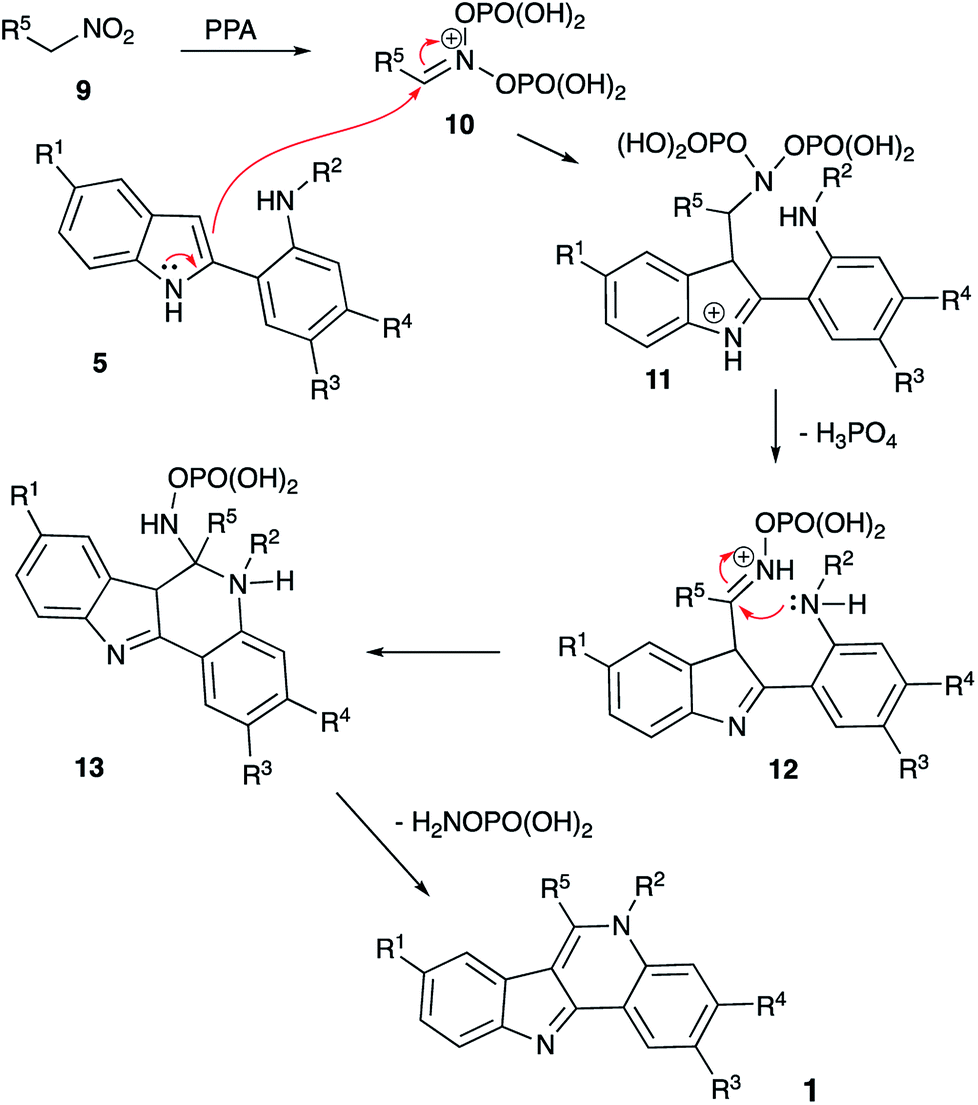 | ||
| Scheme 3 | ||
To test this idea, we subjected 2-phenyl-1H-indole (5aa) to the reaction with excess nitromethane (9a) (ca. 5 equiv.) in PPA (80% P2O5). Gratifyingly, the reaction proceeded smoothly at 100 °C forming the desired indoloquinoline 1aaa as the sole product, which was isolated in 92% yield (Scheme 4). Large excess of nitromethane was used in this case to offset its high volatility at high temperature, and probably its amount could be reduced upon scale up or in the case when the process is carried out in a closed pressurized reactor. For this study, however, to simplify the set up and reduce the reagent cost, we preferred to use an open vessel and excess of nitromethane. Reactions with nitroethane (9b, ca. 3 equiv.) and 1-nitrohexane (9c, 1.2 equiv.) also proceeded uneventfully at 110 °C affording the corresponding tetracyclic products 1aab and 1aac in 91 and 88% yields, respectively (Scheme 4). Expectedly, lower volatility of nitroalkane 9c allowed a nearly stoichiometric amount of this reagent to be used.
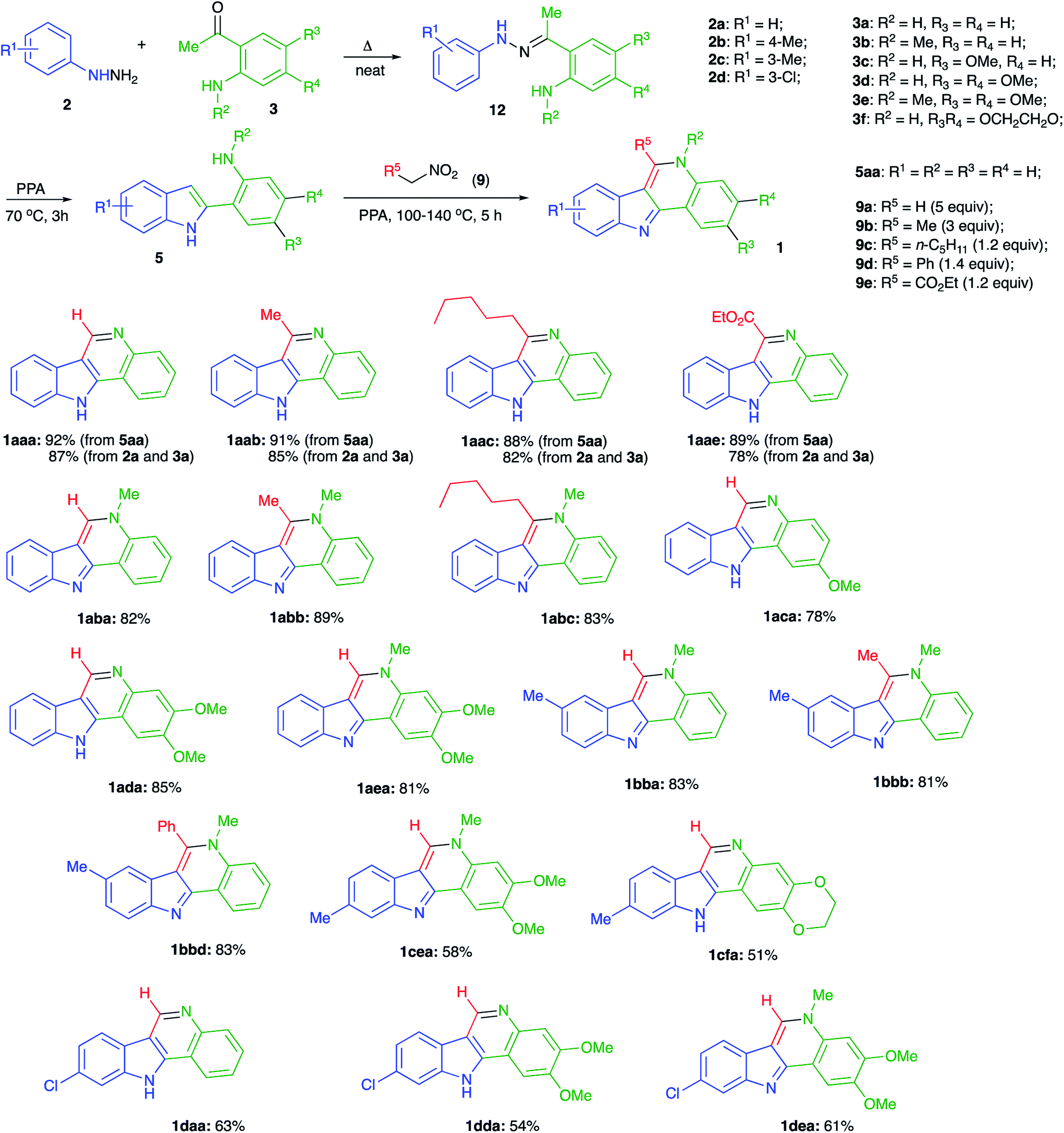 | ||
| Scheme 4 | ||
Also, we tested if the featured method is compatible with the ester functionality. To this end, reaction of 5aa in PPA was carried at 130 °C in the presence of nitroacetic ester (9e, 1.2 equiv.). We were pleased to find that the corresponding functionalized indoloquinoline 1aae was obtained smoothly in high yield (Scheme 4).
With this reaction protocol in hand, we proceeded with the development of a one-pot method allowing for an in situ assembly of indole moiety 5 via Fischer indolization of readily available arylhydrazines 2 and ortho-aminoacetophenones 3. To this end, phenylhydrazine 2a was first heated with an equimolar amount of acetophenone 3a for a short period of time without solvent to assemble hydrazine 12. The addition of PPA and heating at 70 °C triggers the Fischer indolization, which proceeded within 3 h to afford indole 5aa. Nitroalkanes could be then added directly to this mixtures and the temperature of the mixture should be raised (100 °C for nitromethane (9a), 110 °C for longer linear nitroalkanes (9b, c), 130 °C for ethyl nitroacetate (9e), 140 °C for α-nitrotoluene (9d)). The optimal excess amounts of the nitroalkanes are shown in Scheme 4. We were pleased to find that via this one-pot protocol indoloquinoline products were obtained in high yields, which were only slightly lower than those obtained from indole precursor 5a (Scheme 4).
Next, we evaluated the possibility of employing N-methyl protected aminoacetophenone 3b as one of the key reactants. If successful, the application of this starting material would provide access to derivatives of 5H-indolo[3,2-c]quinoline, including the natural alkaloid isocryptolepine. To our delight, a three component one-pot reaction between hydrazine 2a, acetophenone 3b, and nitromethane (9a) afforded isocryptolepine (1aba) in high yield (Scheme 4). The replacement of nitromethane with nitroethane (9b) or 1-nitrohexane (9c) allowed for the efficient preparation of homologs 1abb and 1acc, respectively (Scheme 4). To further elaborate on the newly developed method we decided to synthesize a small library of isocryptolepine analogs by varying hydrazine and acetophenone reagents, capitalizing mostly on the use of nitromethane to take advantage of its low cost. Also, taking into account the results of our preliminary SAR studies, we concentrated on the preparation of structures bearing alkoxy substituents in ring D, as they demonstrated the highest antiproliferative potency. Reactions involving p-tolylhydrazine 2b proceeded smoothly affording the corresponding indoloquinolines 1bba, 1bbb, and 1bbd in good yields. It is important to mention that straight after aqueous work up of the reaction mixtures all of these products were sufficiently pure for all practical purposes.
The employment of meta-substituted arylhydrazines 2c and 2d proved to be more challenging as the Fischer indolization reaction in this case suffered from the lack of regioselectivity. When carried out at elevated temperatures (above 90 °C) it afforded nearly equimolar mixtures of 4- and 6-substituted indole intermediates, which were further converted into a mixture of 9- and 7-substituted indoloquinolines. The problem was solved by carrying out the indolization at lower temperatures (below 70 °C), which led to 6-substituted indoles predominantly (the typical product ratio was about 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for both hydrazines 2c and 2d). The subsequent reaction with nitroalkanes resulted in the formation of 9-methyl- (1cea, 1cfa) or 9-chloro- (1daa, 1dda, 1dea) substituted indoloquinolines along with minor amounts of 7-substituted isomers. This impurity was removed by preparative column chromatography, which lowered the yields of purified materials (Scheme 4).
:1 for both hydrazines 2c and 2d). The subsequent reaction with nitroalkanes resulted in the formation of 9-methyl- (1cea, 1cfa) or 9-chloro- (1daa, 1dda, 1dea) substituted indoloquinolines along with minor amounts of 7-substituted isomers. This impurity was removed by preparative column chromatography, which lowered the yields of purified materials (Scheme 4).
In continuation of our studies of isocryptolepine and its analogs as potential anticancer agents, we evaluated the synthesized compounds against a panel of six cancer cell lines that included the human A549 non-small cell lung cancer (NSCLC), MCF-7 breast carcinoma, U373 glioblastoma, HS683 oligodendroglioma, SKMEL-28 melanoma as well as murine B16F10 melanoma using cryptolepine as control (Table 1). The A549, U373 and SKMEL-28 cell lines represent in vitro cell models of the corresponding aggressive cancer types, i.e. NSCLCs, glioma and melanoma because they have been shown to display various degrees of resistance to pro-apoptotic insults.10 It was found that methylated compounds, including isocryptolepine (1aba) itself, in general had higher activities than their demethylated counterparts. A number of such compounds were equally or more potent than the parent alkaloid itself. These included 1abb, 1aea, 1bbb, 1cea, 1dea. It also appeared that alkoxy groups in the quinoline part of the molecule, such as in compounds 1aea, 1cea, 1cfa, 1dea were beneficial for activity. Of note however, demethylated analog 1cfa, which contains bridged alkoxy groups in the quinoline part of the molecule showed nanomolar antiproliferative activity, more than two orders of magnitude more potent than that of isocryptolepine (1aba). Synthesis of a methylated analogue of 1cfa, which is expected to have even higher potency is underway.
| Compound | Carcinoma | Glioma | Melanoma | Mean + SEM | |||
|---|---|---|---|---|---|---|---|
| A549 | MCF-7 | U373n | HS638 | SKMEL-28 | B16F10 | ||
| a Mean concentration required to reduce the cell viability by 50% after a 72 h treatment relative to a control as determined by MTT assay. Each experiment was performed once in sextuplicate.b These data were previously reported in ref. 4. | |||||||
| 1aaab | 8 | 27 | 16 | 4 | 24 | 7 | 14 ± 4 |
| 1aab | 3 | 4 | 6 | 3 | 6 | 3 | 4.0 ± 0.5 |
| 1aac | 17 | 27 | 24 | 11 | 22 | 4 | 17 ± 4 |
| 1aae | 4 | 5 | 5 | 4 | 4 | 3 | 4.0 ± 0.3 |
| 1abab | 0.6 | 1.0 | 1.3 | 0.4 | 0.9 | 0.7 | 0.8 ± 0.1 |
| 1abb | 0.5 | 1.3 | 3.3 | 0.5 | 1.0 | 0.7 | 1.2 ± 0.4 |
| 1aeab | 0.4 | 0.5 | 0.6 | 0.3 | 0.4 | 0.3 | 0.4 ± 0.05 |
| 1bba | 3 | 4 | 4 | 1 | 4 | 2 | 3.0 ± 0.4 |
| 1bbb | 0.4 | 0.6 | 1.2 | 0.4 | 0.7 | 0.3 | 0.6 ± 0.1 |
| 1bbd | 4 | 6 | 4 | 4 | 4 | 3 | 4.1 ± 0.4 |
| 1cea | 0.4 | 0.7 | 1.6 | 0.4 | 0.5 | 0.4 | 0.7 ± 0.2 |
| 1cfa | 0.008 | 0.05 | 0.27 | 0.01 | 0.007 | 0.008 | 0.06 ± 0.04 |
| 1daa | 5 | 6 | 2 | 4 | 6 | 3 | 4.0 ± 0.7 |
| 1dda | 2 | 3 | 3 | 1 | 2 | 0.6 | 2.0 ± 0.4 |
| 1dea | 0.04 | 0.18 | 0.24 | 0.06 | 0.65 | 0.21 | 0.2 ± 0.1 |
Conclusion
In conclusion, we designed and developed an improved one-pot assembly of 5H- and 11H-indolo[3,2-c]quinoline skeleton. The method involved an in situ formation of hydrazones, followed by a PPA-assisted Fischer indolization reaction and intramolecular cycloaddition of electrophilically activated nitroalkanes. Using this protocol, we synthesized the natural alkaloid isocryptolepine and a small library of its analogs, which were evaluated for anti-proliferative activity against six cancer cell lines. Several analogs demonstrated activity higher than that of the natural alkaloid, with one compound (1cfa) having significant activity at nanomolar concentrations.Experimental part
General information
1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3 or DMSO-d6, using TMS as internal standard. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart smp30 apparatus. All reactions were performed in oven-dried 5 mL round-bottomed flasks open to the atmosphere, employing overhead stirring. The reaction progress and purity of isolated compounds were controlled by TLC on Silufol UV-254 plates, with EtOAc as eluent. All reagents and solvents were purchased from commercial vendors and used as received. Indoloquinoline products 1aaa,11 1aab,12 1aac,4 1aba,10 1abb,11 1abc,4 1aca,13 1ada,3g 1aea,3g 1bba,4 1bbb,12 1bbd6 are known compounds and their physical and spectral properties matched those previously reported in literature.Preparation of indoloquinolines via three-component one-pot protocol (general procedure A)
Reaction vessel was charged with arylhydrazine 2 (1.0 mmol) and o-aminoacetophenone 3 (1.0 mmol), and the mixture was heated at 120 °C for 10 min. Then, the mixture was cooled down to ca. 70 °C, and polyphosphoric acid (80% PPA, 2.0 g) was added. The mixture was stirred at the same temperature for 3 h, then the nitroalkane of choice was added in the listed excess (5.0 mmol (305 mg, 368 μL) of nitromethane (9a), 3.0 mmol (225 mg, 214 μL) of nitroethane (9b), 1.2 mmol (157 mg, 167 μL) of 1-nitrohexane (9c), 1.4 mmol (192 mg, 160 μL) of α-nitrotoluene (9d), or 1.2 mmol (160 mg, 133 μL) of ethyl α-nitroacetate (9e)). The reaction mixture was stirred at the appropriate temperature (100 °C for reactions with 9a, 110 °C for 9b, c, 130 °C for 9e, and 140 °C for 9d) for 5 h, then poured into water (50 mL) and neutralized with 25% aqueous ammonia. The formed precipitate was filtered out and dried on air. Usually such material was sufficiently pure for all practical application, but for analytical purposes it could be additionally purified by flash column chromatography on silica gel, eluting with mixture acetone/benzene (1:3) doped with aqueous ammonia (1 drop of per 10 mL of eluent).
Preparation of indoloquinolines from indole (general procedure B)
The reaction vessel was charged with 2-phenyl-1H-indole (5aa) (193 mg, 1.0 mmol) and polyphosphoric acid (80% PPA, 1.5 g). The mixture was stirred at 70 °C, and nitroalkane was added according to the specifications described in general procedure A. Temperature regimen, post-reaction work up, isolation and purification of the products was performed in the same manner as described in general procedure A.Ethyl 11H-indolo[3,2-c]quinoline-6-carboxylate (1aae)
Yield 78% (general procedure A), 89% (general procedure B). Light-yellow crystalline solid, mp 91–92 °C (acetone–hexane), Rf 0.46 (Hex/EtOAc/EtOH 7:2:1). 1H NMR (400 MHz, CDCl3) δ 11.39 (s, 1H), 8.63 (d, J = 7.8 Hz, 1H), 8.37 (d, J = 8.1 Hz, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.50–7.42 (m, J = 15.6, 7.8 Hz, 2H), 7.36 (t, J = 7.6 Hz, 1H), 4.61 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 166.6, 144.0, 143.4, 142.6, 139.6, 129.4, 129.3, 127.6, 126.8, 124.2, 121.8, 121.6, 121.5, 117.4, 113.5, 111.8, 62.7, 14.4; IR (KBr, film, cm−1): 2930, 2855, 2368, 1730, 1565, 1501, 1460, 1355, 1310, 1238, 1193, 1081, 1017, 744; HRMS (ES TOF) calcd for C18H15N2O2 (M + H)+ 291.1128, found 291.1127 (0.4 ppm).
2,3-Dimethoxy-5,9-dimethyl-5H-indolo[3,2-c]quinoline (1cea)
Yield 58% (via general procedure A). Yellow crystalline solid, Rf 0.20 (acetone/benzene/ammonia 8:8:1), mp 252–253 °C (CH2Cl2–benzene–hexane), Lit mp 252–253 °C.3g 1H NMR (400 MHz, DMSO) δ 9.14 (s, 1H), 7.95–7.88 (m, 2H), 7.48 (s, 1H), 7.25 (s, 1H), 7.05 (d, J = 7.8 Hz, 1H), 4.19 (s, 3H), 3.98 (s, 3H), 3.96 (s, 3H); 13C NMR (101 MHz, DMSO) δ 151.5, 149.9, 148.2, 136.5, 135.4, 131.1 (2C), 121.9, 121.4, 119.4, 116.5, 114.9, 113.6, 103.0, 99.2, 56.1, 55.8, 42.8, 21.9; HRMS (ES TOF) calcd for C19H19N2O2 (M + H)+ 307.1441, found 307.1443 (0.8 ppm).
10-Methyl-2,3-dihydro-12H-[1,4]dioxino[2,3-g]indolo[3,2-c]quinoline (1cfa)
Yield 51% (via general procedure A). Colorless crystalline solid, Rf 0.60 (acetone/benzene/ammonia 8:8:1). Decomposed without melting at 380 °C (DMF–dioxane). Lit decomp. 380 °C.3g 1H NMR (400 MHz, DMSO) δ 12.29 (s, 1H), 9.33 (s, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.90 (s, 1H), 7.50 (c, 1H), 7.47 (c, 1H), 7.12 (dd, J = 8.0, 0.7 Hz, 1H), 4.39–4.41 (m, 4H), 2.51 (s, 3H); 13C NMR (101 MHz, DMSO) δ 145.2, 143.4, 142.8, 141.3, 139.3, 139.0, 134.8, 122.0, 119.9, 119.6, 114.4, 113.2, 112.3, 111.6, 106.9, 64.4, 64.2, 21.7; HRMS (ES TOF) calcd for C18H15N2O2 (M + H)+ 291.1128, found 291.1126 (0.7 ppm).
9-Chloro-11H-indolo[3,2-c]quinoline (1daa)
Yield 63% (via general procedure A). Colorless crystalline solid, Rf 0.60 (acetone/benzene/ammonia 8:8:1), mp 325 °C with sublimation (dioxane), Lit mp 325 °C.3g 1H NMR (400 MHz, DMSO) δ 12.89 (br. s, 1H), 9.61 (s, 1H), 8.52 (dd, J = 8.0, 1.2 Hz, 1H), 8.35 (d, J = 8.4 Hz, 1H), 8.15 (dd, J = 8.3, 0.6 Hz, 1H), 7.70–7.79 (m, 3H), 7.38 (dd, J = 8.4, 1.9 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 145.6, 144.9, 140.4, 139.4, 130.0, 129.7, 128.4, 126.0, 122.2, 121.6, 121.0, 120.8, 117.0, 113.9, 111.6; HRMS (ES TOF) calcd for C15H10ClN2 (M + H)+ 253.0527, found 253.0528 (0.6 ppm).
9-Chloro-2,3-dimethoxy-5-methyl-5H-indolo[3,2-c]quinoline (1dea)
Yield 61% (via general procedure A). Yellow crystalline solid, Rf 0.39 (acetone/benzene/ammonia 8:8:1), mp 270–271 °C (dioxane–EtOH), Lit mp 270–271 °C.3g 1H NMR (400 MHz, DMSO) δ 9.17 (s, 1H), 8.04–7.98 (m, 2H), 7.70 (d, J = 1.8 Hz, 1H), 7.31 (s, 1H), 7.16 (dd, J = 8.2, 1.9 Hz, 1H), 4.20 (s, 3H), 4.00 (s, 2H), 3.99 (s, 3H); 13C NMR (101 MHz, DMSO) δ 155.5, 153.7, 151.2, 148.0, 136.8, 131.0, 129.5, 124.3, 120.6, 118.7, 117.3, 115.1, 115.0, 103.3, 99.5, 56.0, 55.9, 42.5; HRMS (ES TOF) calcd for C18H16ClN2O2 (M + H)+ 327.0895, found 327.0886 (2.8 ppm).
9-Chloro-2,3-dimethoxy-11H-indolo[3,2-c]quinoline (1dda)
Yield 54% (via general procedure A). Off-white crystalline solid, mp 309–310 °C (acetone/hexane), Lit mp 309–310 °C.3g Rf 0.55 (acetone/benzene/ammonia 8:8:1). 1H NMR (400 MHz, DMSO) δ 12.53 (s, 1H), 9.39 (s, 1H), 8.26 (d, J = 8.4 Hz, 1H), 7.91 (s, 1H), 7.71 (d, J = 1.6 Hz, 1H), 7.53 (s, 1H), 7.32 (dd, J = 8.4, 1.5 Hz, 1H), 3.99 (s, 3H), 3.95 (s, 3H); 13C NMR (101 MHz, DMSO) δ 150.9, 148.8, 142.2, 142.2, 140.3, 139.3, 129.6, 121.3, 121.0, 120.5, 113.1, 111.3, 111.0, 109.2, 101.2, 55.8, 55.6; HRMS (ES TOF) calcd for C17H14ClN2O2 (M + H)+ 313.0738, found 313.0732 (1.9 ppm).
Cancer cell growth inhibition
Cell lines used to evaluate the growth inhibitory effects of the compounds were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), the European Collection of Cell Culture (ECACC, Salisbury, UK) and the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). The human cell lines breast carcinoma MCF-7 (DSMZ ACC115), oligodendroglioma Hs683 (ATCC HTB138), non-small cell lung cancer A549 (DSMZ ACC107), glioblastoma U373 cells (ECACC 08061901), melanoma SKMEL-28 (ATCC HTB72) and the murine melanoma B16F10 (ATCC CRL-6475) cells were cultured in RPMI supplemented with 10% FBS, 4 mM glutamine, 100 μg mL−1 gentamicin, and 200 units per mL to 200 μg mL−1 penicillin–streptomycin. The cell lines were cultured in flasks, maintained and grown at 37 °C, 95% humidity and 5% CO2. The evaluation of the antiproliferative effects of the compounds on these cell lines was performed through the colorimetric assay MTT.14 Firstly, cells were trypsinized and seeded in 96 well plates. After 24 h, cells were treated with the compounds at different concentrations, ranging from 10 nM to 100 μM or left untreated for 72 h. The compounds' dilutions in culture medium were prepared from a stock solution in DMSO (10 mM). Estimation of cell viability was performed by means of the MTT – (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (Sigma, Bornem, Belgium) mitochondrial reduction into formazan by living cells. The optical density of the untreated control after 72 h was normalized as 100% of viable cells allowing determination of the concentration that reduced their global growth by 50% after 72 h of treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Russian Science Foundation (grant #17-73-10301).Notes and references
- (a) P. T. Parvatkar and P. S. Parameswaran, Curr. Org. Synth., 2016, 13, 58–72 CrossRef CAS; (b) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Curr. Org. Chem., 2011, 15, 1036–1057 CrossRef CAS; (c) K. H. Lee, J. Nat. Prod., 2010, 73, 500–516 CrossRef CAS PubMed; (d) P. T. Parvatkar and M. S. Majik, RSC Adv., 2014, 4, 22481–22486 RSC.
- (a) P. Aroonkit, C. Thongsornkleeb, J. Tummatorn, S. Kra-jangsri, M. Mungthin and S. Ruchirawat, Eur. J. Med. Chem., 2015, 56–62 CrossRef CAS PubMed; (b) N. Wang, K. J. Wicht, K. Imai, M. Wang, T. Anh Ngoc, R. Kiguchi, M. Kaiser, T. J. Egan and T. Inokuchi, Bioorg. Med. Chem., 2014, 22, 2629–2642 CrossRef CAS PubMed; (c) S. Van Miert, S. Hostyn, B. U. W. Maes, K. Cimanga, R. Brun, M. Kaiser, P. Matyus, R. A. Domisse, G. Lemière, A. Vlietinck and L. Pieters, J. Nat. Prod., 2005, 68, 674–677 CrossRef CAS PubMed; (d) L. R. Whittell, K. T. Batty, R. P. M. Wong, E. M. Bolitho, S. A. Fox, T. M. E. Davis and P. E. Murray, Bioorg. Med. Chem., 2011, 19, 7519–7525 CrossRef CAS PubMed.
- (a) B. Boganyi and J. Kaman, Tetrahedron, 2013, 69, 9512–9519 CrossRef CAS; (b) T. H. M. Jonckers, B. U. W. Maes, G. L. F. Lemière, G. Rombouts, L. Pieters, A. Haemers and R. A. Dommisse, Synlett, 2003, 615–618 CAS; (c) G. Timári, T. Soós and G. Hajós, Synlett, 1997, 1067–1068 CrossRef; (d) P. S. Volvoikar and S. G. Tilve, Org. Lett., 2016, 18, 892–895 CrossRef CAS PubMed; (e) K. Hayashi, T. Choshi, K. Chikaraishi, A. Oda, R. Yoshinaga, N. Hatae, M. Ishikura and S. Hibino, Tetrahedron, 2012, 68, 4274–4279 CrossRef CAS; (f) X. Chen, P. Sun, J. Xu, X. Wu, L. Kong, H. Yao and A. Lin, Tetrahedron Lett., 2014, 55, 7114–7117 CrossRef CAS; (g) M. G. Uchuskin, A. S. Pilipenko, O. V. Serdyuk, I. V. Trushkov and A. V. Butin, Org. Biomol. Chem., 2012, 10, 7262–7265 RSC; (h) J. Tummatorn, C. Thongsornkleeb and S. Ruchirawat, Tetrahedron, 2012, 68, 4732–4739 CrossRef CAS; (i) R. N. Kumar, T. Suresh and P. S. Mohan, Tetrahedron Lett., 2002, 43, 3327–3328 CrossRef CAS.
- A. V. Aksenov, D. A. Aksenov, N. A. Orazova, N. A. Aksenov, G. D. Griaznov, A. De Carvalho, R. Kiss, V. Mathieu, A. Kornienko and M. Rubin, J. Org. Chem., 2017, 82, 3011–3018 CrossRef CAS PubMed.
- P. K. Agarwal, D. Sawant, S. Sharma and B. Kundu, Eur. J. Org. Chem., 2009, 292–303 CrossRef CAS.
- A. V. Aksenov, D. A. Aksenov, G. D. Griaznov, N. A. Aksenov, L. G. Voskressensky and M. Rubin, Org. Biomol. Chem., 2018, 16, 4325–4332 RSC.
- For reviews, see: (a) R. Ballini and M. Petrini, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS; (b) R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS.
- (a) N. A. Aksenov, A. V. Aksenov, O. N. Nadein, D. A. Aksenov, A. N. Smirnov and M. Rubin, RSC Adv., 2015, 5, 71620–71626 RSC; (b) A. V. Aksenov, A. N. Smirnov, N. A. Aksenov, A. S. Bijieva, I. V. Aksenova and M. Rubin, Org. Biomol. Chem., 2015, 13, 4289–4295 RSC; (c) A. V. Aksenov, N. A. Aksenov, D. S. Ovcharov, D. A. Aksenov, G. Griaznov, L. G. Voskressensky and M. Rubin, RSC Adv., 2016, 6, 82425–82431 RSC; (d) A. V. Aksenov, N. A. Aksenov, S. V. Scherbakov, A. N. Smirnov, I. V. Aksenova, V. I. Goncharov and M. A. Rubin, Russ. J. Org. Chem., 2017, 53, 1081–1084 CrossRef CAS; (e) A. I. Konovalov, I. S. Antipin, V. A. Burilov, T. I. Madzhidov, A. R. Kurbangalieva, A. V. Nemtarev, S. E. Solovieva, I. I. Stoikov, V. A. Mamedov and L. Y. Zakharova, et al., Russ. J. Org. Chem., 2018, 54, 157–371 CrossRef CAS.
- (a) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synth. Commun., 2012, 42, 541–547 CrossRef CAS; (b) A. V. Aksenov, N. A. Aksenov, O. N. Nadein and I. V. Aksenova, Synlett, 2010, 2628–2630 CrossRef CAS.
- (a) V. Mathieu, M. Remmelink, N. D'Haene, S. Penant, J. F. Gaussin, R. Van Ginckel, F. Darro, R. Kiss and I. Salmon, Cancer, 2004, 101, 1908–1918 CrossRef PubMed; (b) F. Branle, F. Lefranc, I. Camby, J. Jeuken, A. Geurts-Moespot, S. Sprenger, F. Sweep, R. Kiss and I. Salmon, Cancer, 2002, 95, 641–655 CrossRef CAS PubMed; (c) F. Lefranc, G. Nuzzo, N. A. Hamdy, I. Fakhr, L. M. Y. Banuls, G. V. Van Goietsenoven, G. Villani, V. Mathieu, R. Van Soest, R. Kiss and M. L. Ciavatta, J. Nat. Prod., 2013, 76, 1541–1547 CrossRef CAS PubMed; (d) V. Mathieu, C. Pirker, E. Martin de Lasalle, M. Vernier, T. Mijatovic, N. De Neve, J. F. Gaussin, M. Dehoux, F. Lefranc, W. Berger and R. Kiss, J. Cell. Mol. Med., 2009, 13, 3960–3972 CrossRef PubMed; (e) H. Brenner, Lancet, 2002, 360, 1131–1135 CrossRef PubMed.
- P. K. Agarwal, D. Sawant, S. Sharma and B. Kundu, Eur. J. Org. Chem., 2009, 292–303 CrossRef CAS.
- T. Dhanabal, R. Sangeetha and P. S. Mohan, Tetrahedron, 2006, 62, 6258–6263 CrossRef CAS.
- E. S. Ibrahim, A. M. Montgomerie, A. H. Sneddon, G. R. Proctor and B. Green, Eur. J. Med. Chem., 1988, 23, 183–188 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data. See DOI: 10.1039/c8ra08155g |
| This journal is © The Royal Society of Chemistry 2018 |