
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethane adsorption and methanol desorption of copper modified boron silicate†
Xueting Wang ab,
Alexander Shishkinab,
Felix Hemmingssonab,
Magnus Skoglundhab,
Francisco Javier Martinez-Casadoc,
Lorenz Bocka,
Alexander Idströma,
Lars Nordstiernaa,
Hanna Härelindab and
Per-Anders Carlsson*ab
ab,
Alexander Shishkinab,
Felix Hemmingssonab,
Magnus Skoglundhab,
Francisco Javier Martinez-Casadoc,
Lorenz Bocka,
Alexander Idströma,
Lars Nordstiernaa,
Hanna Härelindab and
Per-Anders Carlsson*ab
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden. E-mail: per-anders.carlsson@chalmers.se
bCompetence Centre for Catalysis, Chalmers University of Technology, 412 96 Gothenburg, Sweden
cMAX IV Laboratory, Lund University, 221 00 Lund, Sweden
First published on 26th October 2018
Abstract
Boron silicate (BS) with a chabazite framework structure was synthesised using a direct route and rigorously characterized before it was ion-exchanged with copper to form Cu-BS. Employing in situ infrared spectroscopy, we show that Cu-BS is capable of oxidising methane to methoxy species and methanol interacts with the boron sites without deprotonation.
1 Introduction
At oil and gas production sites, methane rich gas is wasted through gas flaring because infrastructure for liquefaction and/or transportation of gas is lacking.1 A potential alternative method to gas flaring, which would yield a useful product while still mitigating methane emissions, is that of direct conversion of methane to methanol (DCMM). To produce methanol through DCMM, a quasi-catalytic reaction sequence has been most commonly used. Here, the catalyst is first oxidised (activation), then exposed to methane (reaction) and finally exposed to, e.g., water or ethanol (extraction) whereby methanol is achieved. Copper containing zeolites, e.g. Cu-ZSM-5,2,3 Cu-SSZ-13,4,5 and Cu-MOR,6,7 have been studied and proposed as possible candidates for DCMM. Recently, Cu-zeolites possessing 8-membered rings, such as Cu-SSZ-13, were shown to exhibit higher methanol production compared to framework structures with other ring sizes.5,8 A main limitation, however, is the strong interaction between the methoxy reaction intermediate and the zeolite framework structure, which requires protonic extraction to obtain methanol.9 Interestingly, zeolites are not the only type of support for copper species active for DCMM. Methanol formation has also been observed for Cu/silica10 and on copper containing metal organic frameworks,11 which suggests a certain flexibility in the choice of support materials. These findings inspired us to investigate the interactions of methane and methanol with copper exchanged boron silicate (BS) with a chabazite framework structure (CHA), a zeotype that is less acidic compared to the corresponding Al containing zeolite, e.g. SSZ-13,12–14 with the aim of decreasing the strength whereby methoxy interacts with framework sites.In the present study, boron silicate was synthesised using a direct route and its physicochemical properties were characterized with inductively coupled plasma atomic emission spectroscopy (ICP-AES), high-resolution X-ray diffraction (HE-XRD), nuclear magnetic resonance (NMR) spectroscopy, nitrogen sorption, ammonia adsorption and temperature programmed desorption (NH3–TPD) using combined differential scanning calorimetry (DSC) and mass spectrometry (MS), and scanning electron microscopy (SEM). Copper was then ion-exchanged into the BS and the interaction of methanol with the resulting Cu-BS, as well as BS and H-SSZ-13, was studied in situ using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). Moreover, the formation of methoxy species upon exposure to methane was studied over pre-oxidised Cu-BS.
2 Experimental
2.1 Sample preparation
For the synthesis of the BS sample, 2 g NaOH (1 N), 2.78 g N,N,N-trimethyl-1-adamantanammonium hydroxide (0.72 M, AlCheTech) and 3.22 g Milli-Q water (18 MΩ cm) were added to a Teflon lined stainless steel 0.16 L autoclave (Parr) and mixed for 15 min. Subsequently, 0.22 g B2O3 (Sigma-Aldrich) was added and the solution was mixed for another 15 min. This was followed by addition of 0.6 g fumed SiO2 (Sigma-Aldrich) and mixing for another 15 min. The resultant solution was kept in the autoclave at 140 °C for 7 days under static conditions. The resulting product was washed several times with Milli-Q water, vacuum filtered and dried in air at room temperature for 12 h. The dried sample was finally calcined in air at 550 °C for 3 h using an initial heating rate of 2 °C min−1 starting from room temperature. An H-SSZ-13 reference sample (Si/Al = 10) was synthesised according to the method described elsewhere.15,16In order to functionalise the BS sample for DCMM, a Cu-BS sample was prepared using aqueous ion-exchange by mixing aqueous solutions of Cu(NO3)2 (0.1 M, 100 mL g−1 sample) with the BS sample at room temperature for 24 hours. The slurry was then filtered and the solid fraction was washed with Milli-Q water and dried at 120 °C in air overnight. Characterisation of the Cu-BS sample with in situ DRIFTS during CO/NO adsorption and XRD confirms the existence of well dispersed Cu species in the Cu-BS sample (shown in ESI†). A Cu-SSZ-13 sample was prepared using the same ion-exchange method from the reference H-SSZ-13 for comparison of the acidity.
2.2 Ex situ characterisation
The morphology of the BS sample was studied using a Zeiss Ultra 55 FEG scanning electron microscope and the SEM images of the sample were collected with an acceleration voltage of 20 kV at a working distance of 10.2 mm.The composition of the BS sample was determined using inductively coupled plasma optical emission spectroscopy (ICP-OES) of acid digested samples using a Perkin Elmer Optima 7300 DV instrument. The acid digestion was carried out for 20 min in a mixture of HCl, HNO3 and HF at 200 °C using a microwave digestion unit.
The coordination of boron in the BS sample was analysed using NMR of 11B. The NMR experiments were performed with a Varian Inova-600 spectrometer operating at 14.7 T and equipped with a 3.2 mm solid-state magic angle spinning (MAS) probe. The measurements were conducted at 25 °C with a spinning rate of 15 kHz. All spectra were recorded using a simple detection-pulse sequence. Acquisition parameters for the 11B-spectra included a 0.46875 μs 11B π/16 pulse, 20 ms acquisition time, 2 s recycle delay to allow for full thermal equilibrium, and 2048 acquisitions for each spectrum. The short pulse lengths were chosen in order to reduce the experimental time. The spectra were processed using the MestreNova 8.1 software suite. For all spectra, zero-filling with 8192 points, phase correction and a first order polynomial baseline correction were used in the processing.
The HR-XRD analysis was performed at beamline I711 at Max II (MAX IV Laboratory, Lund, Sweden) to determine the crystalline phases of the BS sample. The sample was measured in transmission mode using a 0.3 mm spinning capillary and a Newport diffractometer equipped with a Pilatus 100 K area detector at a distance of 765 mm from the sample. The detector was scanned continuously from 0 to 120° for approximately 6–10 min, recording 62.5 images per angle (step size 0.016°) for each measurement. The true 2θ position of each pixel was recalculated, giving an average number of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pixels contributing to each 2θ value. Integration, applying no corrections for the tilt of the detector, provided FWHM values of 0.03–0.08°, from 0 to 120°. For the Rietveld refinement, the FullProf program17 was used, employing the model of chabazite (Si/Al = 12) reported by Fickel et al.18 The unit cell of chabazite is hexagonal (space group R
000 pixels contributing to each 2θ value. Integration, applying no corrections for the tilt of the detector, provided FWHM values of 0.03–0.08°, from 0 to 120°. For the Rietveld refinement, the FullProf program17 was used, employing the model of chabazite (Si/Al = 12) reported by Fickel et al.18 The unit cell of chabazite is hexagonal (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Disordered water molecules were modelled in the pores of the structure of the chabazite with low occupancy (0.25 in total).
m). Disordered water molecules were modelled in the pores of the structure of the chabazite with low occupancy (0.25 in total).
The specific surface area and pore size distribution of the samples were determined by nitrogen sorption at −196 °C using a Micromeritics ASAP 2010 instrument. Prior to analysis, the sample was degassed under vacuum at 250 °C for 12 h. Respective surface areas were then determined according to the standard Brunauer–Emmett–Teller (BET) method using P/P0 = 0.06–0.20.
The acidic properties of the BS sample were characterised by adsorption of NH3 followed by TPD using a DSC-MS setup. Briefly, the experimental setup consists of a gas mixing system, which includes several mass flow controllers, a calorimeter (Setaram Sensys DSC) and a mass spectrometer (Hiden HPR-20 QIC). The calorimeter consists of two quartz tubes. In one of them, a certain amount of the sample (BS: 100.0 mg, Cu-BS: 30.7 mg, H-SSZ-13: 31.3 mg and Cu-SSZ-13: 30.1 mg) was placed on a sintered quartz bed, while the other tube was used as the reference. Before the measurements, the sample was pretreated in 8% O2 at 500 °C for 10 min. The temperature was then decreased to 150 °C. After 20 min, the sample was exposed to 1000 ppm NH3 for 60 min, followed by exposure to Ar for 40 min. Thereafter, the temperature was linearly increased at a rate of 10 °C min−1 to 500 °C. The total flow through the sample was 100 mL min−1 for the BS sample and 20 mL min−1 for the Cu-BS, H-SSZ-13 and Cu-SSZ-13 sample. Argon was used as a balance. The gas composition after the calorimeter was continuously measured using mass spectrometry. The average heat of adsorption was calculated as ΔH = −QmaxVm/(c![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) ) × 103, where ΔH is the heat of adsorption (kJ mol−1), Qmax is the maximum value of the heat flow (mW), Vm is the gas molar volume at 298.15 K and 1 atm using the ideal gas law (24.5 L mol−1), c is the ammonia volume concentration (ppm) and is the volumetric flow rate (mL s−1).
) × 103, where ΔH is the heat of adsorption (kJ mol−1), Qmax is the maximum value of the heat flow (mW), Vm is the gas molar volume at 298.15 K and 1 atm using the ideal gas law (24.5 L mol−1), c is the ammonia volume concentration (ppm) and is the volumetric flow rate (mL s−1).
2.3 In situ DRIFTS
The in situ DRIFTS measurements were carried out using a VERTEX70 spectrometer (Bruker) equipped with a liquid nitrogen cooled mercury cadmium telluride detector with the band width 600–12000 cm−1, a Praying Mantis™ diffuse reflectance accessory and a high-temperature stainless steel reaction chamber (Harrick Scientific Products Inc.). All spectra were measured between 900 and 4000 cm−1 with a spectral resolution of 1 cm−1. Approximately 85 μl of sample was loaded into the reaction chamber.
The interaction of methane with the three samples, i.e., H-SSZ-13, BS and Cu-BS, was studied using methane adsorption experiments using the in situ DRIFTS set-up. The samples were pre-treated in an oxidising (500 ppm N2O) or a reducing (1% H2 following calcination in 10% O2) atmosphere at 550 °C for 1 h. The samples were then exposed to 2% CH4 in Ar at 250 °C and the IR spectra were recorded at various exposure times in Ar. The reference spectrum was taken at 250 °C in 500 ppm N2O for the oxidised samples and in pure Ar for the reduced samples.
The interaction of methanol with the H-SSZ-13, BS and Cu-BS samples was studied in situ during methanol–TPD using the same DRIFTS set-up. For the methanol–TPD experiment, after pre-treatment of the samples with 10% O2 at 450 °C for one hour, the samples were cooled to 30 °C and a reference spectrum was recorded. A few droplets of methanol (99.8%, Sigma-Aldrich) were then added to the sample followed by a TPD in 100 mL min−1 flow of pure Ar with stepwise temperature increases from 30 to 450 °C. Each spectrum was taken 10 minutes after reaching the targeted temperature.
3 Results and discussion
3.1 Characterisation of the BS sample
Fig. 1–4 present ex situ characterisation results of the BS sample. The SEM images (Fig. 1) show the presence of 9.1 μm large crystals with a typical cubic-shaped morphology, with a rhombohedral, almost cube shaped, morphology, typical for the CHA framework structure. The ICP-AES analysis of the BS sample gives an elemental composition of 0.3% Na, 1.2% B and 30% Si in weight percentage. Thus the Si/B molar ratio is 9.6, which is in accordance with the corresponding ratio reported by Guth et al.19 | ||
| Fig. 1 SEM images of the BS sample. The red box in the left image (a) indicates the zoomed in area as shown in the right image (b). | ||
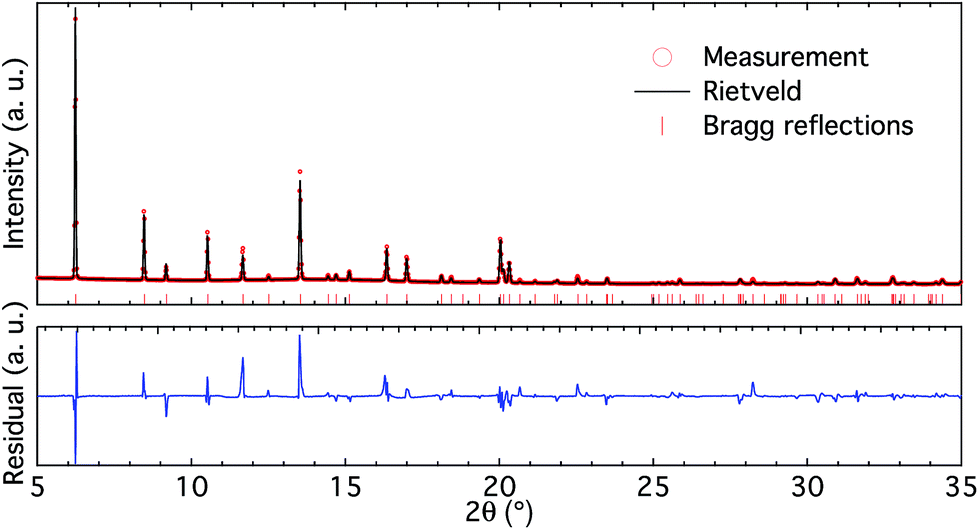 | ||
| Fig. 2 High-resolution X-ray diffractogram (upper panel) and Rietveld refinement (lower panel) of the BS sample. | ||
 | ||
| Fig. 3 11B magic angle spinning NMR spectrum of the BS sample. | ||
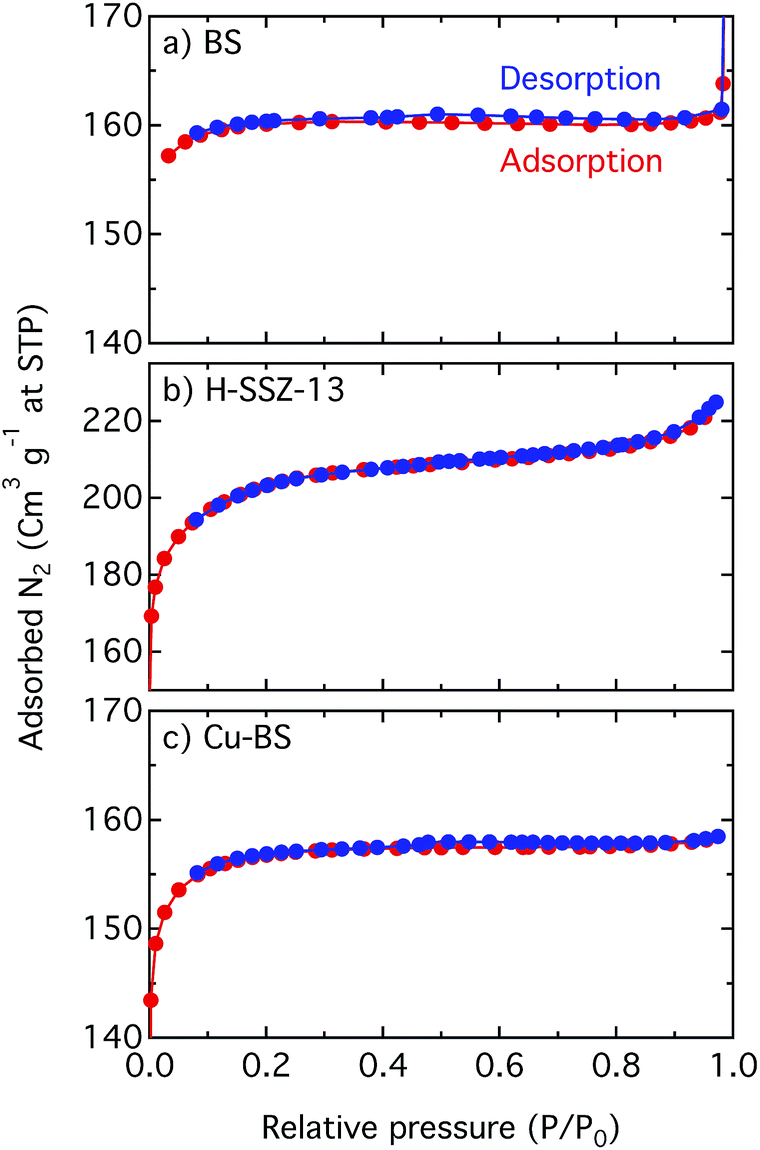 | ||
| Fig. 4 Linear plots of the N2 adsorption–desorption isotherms for the BS (a), H-SSZ-13 (b) and Cu-BS (c) samples. | ||
The high-resolution X-ray diffractogram of the BS sample (Fig. 2 upper panel) shows clear reflections characteristic of the CHA framework structure and is free from additional peaks. Rietveld refinement of the diffractogram (Fig. 2 lower panel) reveals that the unit cell parameters for the BS sample are a = 13.4517 and b = 14.6562 Å. These cell parameters are smaller than those for pure silica chabazite (i.e. a = 13.68 and b = 14.77 Å (ref. 20)), which is in accordance with the different ionic radii for tetrahedrally coordinated boron (0.11 Å) and silicon (0.26 Å).
The 11B MAS NMR spectrum of the BS sample (Fig. 3) mainly reveals two narrow and sharp peaks at −3.1 and −3.9, respectively. These peaks are assigned to tetrahedral BO4 sites within a crystalline framework based on their shift and low quadrupolar coupling interaction.21,22 The other peaks in the solid-state spectrum are spinning sidebands located at a distance of ± multiples of the MAS frequency of 15 kHz from the central peaks.
Fig. 4 shows linear plots of the adsorption–desorption isotherms for the BS, H-SSZ-13 and Cu-BS samples. The adsorption isotherms show the typical shape for microporous materials where at low relative pressure, when the micropores are filled, a steep increase of the isotherm can be seen. However, when the surface is completely covered with the adsorbate, the isotherm reaches a plateau. The BET surface area and the micropore volume of the BS sample are 533 m2 g−1 and 0.24 mL g−1, respectively. These values are lower in comparison with the corresponding values for the H-SSZ-13 sample (657 m2 g−1 and 0.26 mL g−1). This is in line with the study by Liang et al. showing decreasing BET values with increased boron content for chabazite zeolites.23 For the Cu-BS sample the BET surface area and micropore volume are 499 m2 g−1 and 0.23 mL g−1, respectively. This decrease is reasonable as Cu is introduced into the cages, limiting the amount of nitrogen that can be taken up. The results, however, indicate no detrimental change of the microporous structure upon copper ion-exchange. We mention that the surface area of microporous materials, especially those with small pores such as materials with the CHA framework structure, derived from BET analysis of N2 sorption should not be considered absolute but rather used for comparisons (see Shishkin et al.15 and references therein for details).
The heat-flow, and the effluent NH3 concentration during adsorption and temperature programmed desorption of NH3 for the BS, Cu-BS, H-SSZ-13 and Cu-SSZ-13 samples are shown in the ESI Fig. S3.† The corresponding maximum value of heat signal and heat of adsorption are listed in Table 1. The heat of adsorption for the BS sample (−41.2 kJ mol−1) compared with that for the H-SSZ-13 sample (−110.2 kJ mol−1) is much lower in absolute value, indicating a considerably lower acidity of the BS sample. Introducing Cu to the BS sample, however, results in an increase in the absolute value of ΔH (−88.2 kJ mol−1), suggesting a higher acidity of the Cu sites in the Cu-BS sample. Such an acidity increase is also obvious for the Cu-SSZ-13 sample (−132.3 kJ mol−1) compared with its parent H-SSZ-13. During the TPD of the BS sample (Fig. S3c†), most NH3 desorbs before 300 °C with a maximum at 230 °C, presenting weak interactions between ammonia and the boron silicate framework structure. For the Cu-BS sample (Fig. S3f†), however, besides the ammonia desorption with a maximum at 230 °C, additional ammonia desorbs at temperatures higher than 300 °C, suggesting a stronger interaction between ammonia and the Cu sites in the Cu-BS sample. Therefore, the DSC measurements together with the TPD results demonstrate that the Cu sites in the Cu-BS sample possess stronger acidity than the boron silicate framework sites. Moreover, the BS sample is considerably less acidic than its aluminium counterpart, the H-SSZ-13 sample. Both conclusions suggest that Cu-containing boron silicate is a promising material for direct methane conversion to methanol as the important reaction intermediate, methoxy groups, may stay on the Cu sites and not be consumed by the weak acidic boron silicate framework structure.
| BS | Cu-BS | H-SSZ-13 | Cu-SSZ-13 | |
|---|---|---|---|---|
| Qmax (mW) | 2.8 | 1.2 | 1.5 | 1.8 |
| ΔH (kJ mol−1) | −41.2 | −88.2 | −110.2 | −132.3 |
In summary, boron silicate with the chabazite framework structure has been synthesised. Even though no thermal stability test was done in this work, previous studies have reported that boron silicate can withstand at least up to 500 °C13,24 which is much higher than the methane oxidation temperature (typically below 250 °C). Moreover, despite the possible deboronation of boron silicate via hydration treatments, reoccupation of boron into the framework position can be achieved upon dehydration.24 Such hydrothermal property of boron silicate makes it possible as a potential catalyst for steam-assisted extraction of methanol from methane oxidation.
3.2 Methane oxidation over the Cu-BS sample
Fig. 5 shows IR spectra for the pre-oxidised (a) and pre-reduced (b) Cu-BS samples under exposure to methane at 250 °C during 7 h. For the pre-oxidised sample two bands at 2924 and 2855 cm−1 appear and increase in intensity upon exposure to methane, whereas no obvious features can be seen for the pre-reduced sample. These two bands were not observed in the spectra of methanol adsorbed on the Cu-BS, BS or H-SSZ-13 samples (Fig. 6). Therefore, they are not associated with framework methoxy groups. Similar features, however, have previously been assigned to C–H stretching vibrations originating from methoxy or methyl adsorbed on Cu particles and single crystals (2918–2927 cm−1 for νas(CH3) and 2821–2890 cm−1 for νs(CH3)).25–28 Moreover, bands in similar regions (2920–2935 cm−1 and 2820–2830 cm−1) have been assigned to methoxy formed from methane on Fe29–31 or Co32 sites in zeolite systems. Therefore, we presumably assign the bands at 2924 and 2855 cm−1 to asymmetric and symmetric C–H stretching vibrations respectively for methoxy species adsorbed on well dispersed copper sites. The accumulation of methoxy groups on copper sites indicates that the copper species in the BS sample are capable of catalysing the oxidation of methane. Unlike Cu-zeolites, where methoxy groups are observed on the framework structure and zeolite defects,6,10 no methoxy groups on boron or silicon sites can be observed for the Cu-BS sample during methane oxidation. This can be explained by the weaker acidity of the boron sites compared to the aluminium sites. For the reference samples shown in ESI Fig. S5–S10,† i.e., the BS and H-SSZ-13 samples, no methoxy species develop on the sample surface during methane exposure.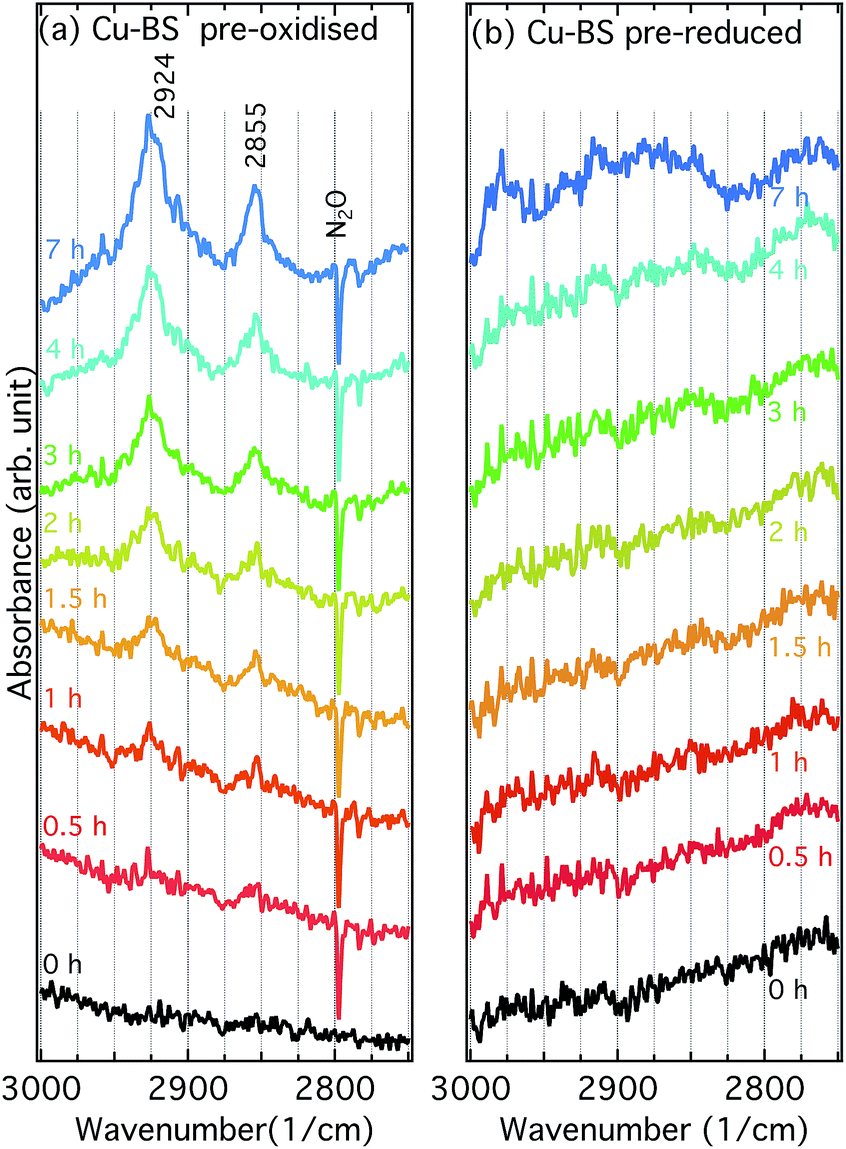 | ||
| Fig. 5 Infrared spectra for the pre-oxidised (a) and pre-reduced (b) Cu-BS samples under exposure to methane at 250 °C during 0 to 7 h. The spectrum at 0 h for pre-oxidised Cu-BS was taken in the presence of 500 ppm of N2O whereas the remaining spectra were recorded in Ar. | ||
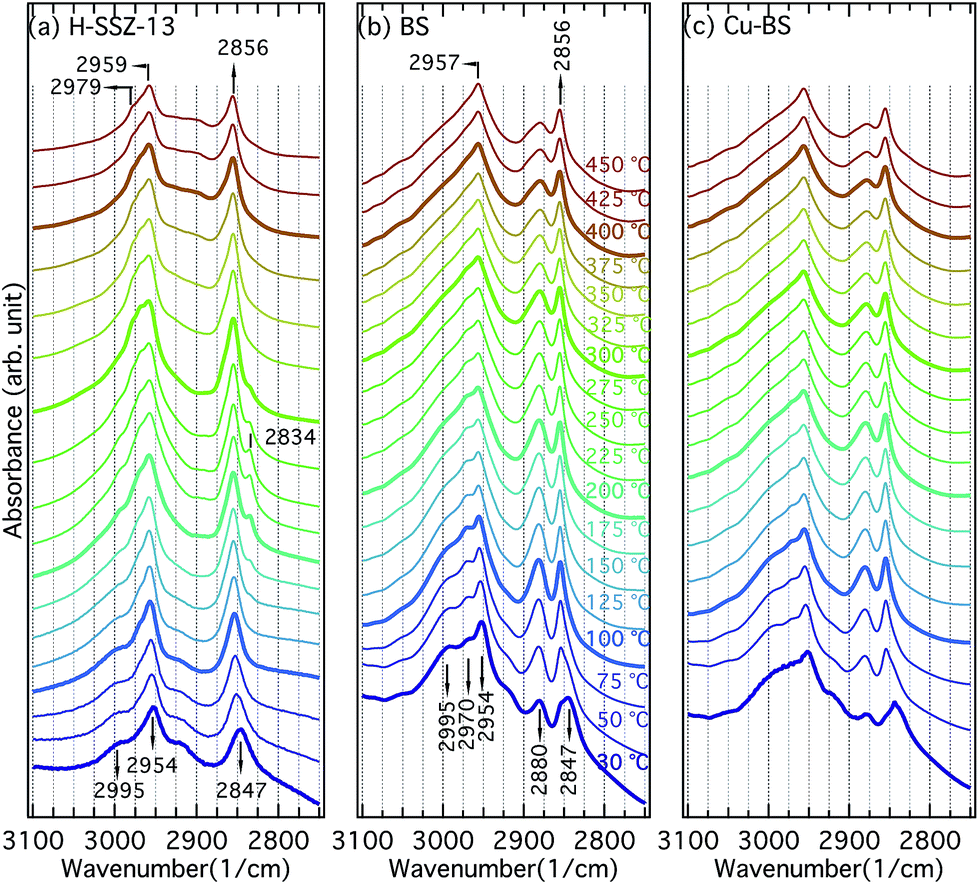 | ||
| Fig. 6 Infrared spectra collected for the H-SSZ-13 (a), BS (b) and Cu-BS (c) samples during methanol desorption from 30 to 450 °C. | ||
3.3 Methanol–TPD
Fig. 6 presents the IR spectra for the H-SSZ-13 (a), BS (b) and Cu-BS (c) samples recorded during the methanol–TPD experiments. The discussion will be focused on the C–H stretching region (3100–2750 cm−1) of the spectra. Upon methanol adsorption, two bands centred at 2954 (with obvious shoulders) and 2847 cm−1 appear for all three samples. These two bands are associated with asymmetric and symmetric C–H stretching vibrations of hydrogen-bonded methanol, respectively.9,12,13 For the BS and Cu-BS samples, additional absorption bands at 2995, 2970 and 2880 cm−1 are evident, which are presumably due to methanol interacting with boron.12,13 With temperature increases, additional bands at 2979 and 2834 cm−1 become visible at 175 °C for the H-SSZ-13 sample. We assign these two bands to methoxy groups and methanol bound to Brønsted acid sites.9 With further temperature increases, methanol converts to methoxy groups, resulting in decreasing intensity of the bands at 2834 cm−1 (methanol on Brønsted acid sites). At 450 °C, only methoxy groups remain on Brønsted acid sites (2979 cm−1) and extra framework Si (2959 and 2856 cm−1)9,32,33 in the H-SSZ-13 sample. For the BS and the Cu-BS samples, the absorption bands at 2954 and 2847 cm−1 (hydrogen-bonded methanol)9,34,35 blue-shift to 2957 and 2856 cm−1 (methoxy on silicon)9,32–34 during heating, indicating conversion of methanol to methoxy on the silicon sites. Moreover, the intensity of the bands at 2995, 2970 and 2880 cm−1 (methanol/methoxy on the boron sites) first increases (30 to 100 °C) and then decreases (above 100 °C) with increasing temperature. Though the spectra for the BS and Cu-BS samples resemble each other, the absorption bands characteristic for the boron sites (2995, 2970 and 2880 cm−1) are clearly less intense for the copper containing sample, indicating that some boron sites constitute ion-exchange sites for Cu ions. The methanol–TPD experiments show strong interactions between methanol/methoxy and the framework/defect sites in all samples. For the H-SSZ-13 sample, the conversion of methanol to methoxy on the Brønsted acid sites is fairly distinct, while no clear evidence of methoxy formation on the boron sites can be isolated from the absorption bands observed for the BS or the Cu-BS samples. This can be explained by the weaker acidity, therefore the weaker ability of the boron sites to deprotonate methanol compared to the aluminium sites. This further elaborates that boron containing zeotypes have the potential to realise DCMM without the extraction step. It is noticeable, however, that methoxy is strongly adsorbed on defect silicon sites in the CHA structure, which, for example, is not observed on the MFI structure.9 This may be due to the shape of the CHA cage.4 Conclusions
Boron silicate with a chabazite framework structure was synthesised using a direct route and characterised using HR-XRD, ICP-AES, SEM, NMR and DSC-MS during NH3 adsorption and TPD. The latter revealed the weaker acidity of the BS sample compared to the aluminium containing counterpart. Cu-BS was then prepared by copper ion-exchange of the BS sample and characterised in situ using DRIFTS during methane oxidation and methanol–TPD. The methane oxidation experiment shows that Cu-BS is capable of oxidising methane to methoxy species, which is likely a necessary step towards methanol formation. These methoxy species are presumably adsorbed on the copper sites as no interaction between methoxy species and framework/defect sites could be discerned. The methanol-TPD study suggests that methanol interacts with the boron sites without deprotonation, which is important for the production of methanol. Hence, copper containing boron zeotypes are potential candidates for the direct catalytic conversion of methane to methanol, as protonic extraction may be avoided and DME formation is suppressed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank MAX IV Laboratory (Lund, Sweden) for providing the beamtime. This work is supported by the Swedish Research Council through the Röntgen-Ångström Cluster [grant number 349-2013-567 and 2017-06709], the Knut and Alice Wallenberg Foundation [grant number 2015.0058] and the Competence Centre for Catalysis, which is financially supported by Chalmers University of Technology, the Swedish Energy Agency and the member companies: AB Volvo, ECAPS AB, Johnson Matthey AB, Preem AB, Scania CV AB, Umicore Denmark ApS and Volvo Car Corporation AB.Notes and references
- Global Gas Flaring Reduction Partnership (GGFR), 2011.
- P. Vanelderen, R. G. Hadt, P. J. Smeets, E. I. Solomon, R. A. Schoonheydt and B. F. Sels, J. Catal., 2011, 284, 157–164 CrossRef CAS PubMed.
- G. Li, P. Vassilev, M. Sanchez-Sanchez, J. A. Lercher, E. J. M. Hensen and E. A. Pidko, J. Catal., 2016, 338, 305–312 CrossRef CAS.
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed.
- M. B. Park, S. H. Ahn, A. Mansouri, M. Ranocchiari and J. A. van Bokhoven, ChemCatChem, 2017, 9, 3705–3713 CrossRef CAS.
- V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed.
- S. Grundner, M. A. C. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. M. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- B. Ipek, M. J. Wulfers, H. Kim, F. Goltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS.
- X. Wang, A. A. Arvidsson, M. O. Cichocka, X. Zou, N. M. Martin, J. Nilsson, S. Carlson, J. Gustafson, M. Skoglundh, A. Hellman and P.-A. Carlsson, J. Phys. Chem. C, 2017, 121, 27389–27398 CrossRef CAS.
- X. Wang, N. M. Martin, J. Nilsson, S. Carlson, J. Gustafson, M. Skoglundh and P.-A. Carlsson, Catalysts Search PubMed , under revision.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuno, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- L. Regli, C. Lamberti, C. Busco, A. Zecchina, C. Prestipino, K. P. Lillerud, S. I. Zones and S. Bordiga, Proc. Int. Zeolite Conf., 15th, 2007, 585–593 CAS.
- L. Regli, S. Bordiga, C. Lamberti, K. P. Lillerud, S. I. Zones and A. Zecchina, J. Phys. Chem. C, 2007, 111, 2992–2999 CrossRef CAS.
- Q. Zhu, J. N. Kondo, T. Yokoi, T. Setoyama, M. Yamaguchi, T. Takewaki, K. Domen and T. Tatsumi, Phys. Chem. Chem. Phys., 2011, 13, 14598–14605 RSC.
- A. Shishkin, H. Kannisto, P.-A. Carlsson, H. Härelind and M. Skoglundh, Catal. Sci. Technol., 2014, 4, 3917–3926 RSC.
- D. W. Fickel, J. M. Fedeyko and R. F. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- D. Fickel and R. Lobo, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- J.-L. Guth and H. Kessler, Synthesis of Aluminosilicate Zeolites and Related Silica-Based Materials, Springer, Berlin Heidelberg, 1999, vol. 151, pp. 1–52 Search PubMed.
- E. L. First, C. E. Gounaris, J. Wei and C. A. Floudas, Phys. Chem. Chem. Phys., 2011, 13, 17339–17358 RSC.
- M. Stöker, Review on Recent NMR Results, Elsevier, Amsterdam, 1994, pp. 429–498 Search PubMed.
- M. R. Hansen, T. Vosegaard, H. J. Jakobsen and J. Skibsted, J. Phys. Chem. A, 2004, 108, 586–594 CrossRef CAS.
- J. Liang, J. Su, Y. Wang, Z. Lin, W. Mu, H. Zheng, R. Zou, F. Liao and J. Lin, Microporous Mesoporous Mater., 2014, 194, 97–105 CrossRef CAS.
- S.-J. Hwang, C.-Y. Chen and S. I. Zones, J. Phys. Chem. B, 2004, 108, 18535–18546 CrossRef CAS.
- M. D. Driessen and V. H. Grassian, J. Phys. Chem., 1995, 99, 16519–16522 CrossRef CAS.
- M. A. Chesters and E. M. McCash, J. Electron Spectrosc. Relat. Phenom., 1987, 44, 99–108 CrossRef CAS.
- R. Burch, S. Chalker and J. Pritchard, J. Chem. Soc., Faraday Trans., 1991, 87, 1791–1794 RSC.
- M. P. Andersson, P. Uvdal and A. D. MacKerell, J. Phys. Chem. B, 2002, 106, 5200–5211 CrossRef CAS.
- B. R. Wood, J. A. Reimer, A. T. Bell, M. T. Janicke and K. C. Ott, J. Catal., 2004, 225, 300–306 CrossRef CAS.
- T. Nobukawa, M. Yoshida, S. Kameoka, S.-i. Ito, K. Tomishige and K. Kunimori, J. Phys. Chem. B, 2004, 108, 4071–4079 CrossRef CAS.
- E. V. Starokon, M. V. Parfenov, S. S. Arzumanov, L. V. Pirutko, A. G. Stepanov and G. I. Panov, J. Catal., 2013, 300, 47–54 CrossRef CAS.
- M. C. Kung, S. S. Y. Lin and H. H. Kung, Top. Catal., 2012, 55, 108–115 CrossRef CAS.
- B. R. Wood, J. A. Reimer, A. T. Bell, M. T. Janicke and K. C. Ott, J. Catal., 2004, 225, 300–306 CrossRef CAS.
- T. Nobukawa, M. Yoshida, S. Kameoka, S.-i. Ito, K. Tomishige and K. Kunimori, J. Phys. Chem. B, 2004, 108, 4071–4079 CrossRef CAS.
- S. M. Campbell, X.-Z. Jiang and R.F. Howe, Microporous Mesoporous Mater., 1999, 29, 91–108 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD for the H-SSZ-13 and Cu-BS samples; HR-XRD for the BS sample with full angle range; DSC-MS measurements for the BS, Cu-BS, H-SSZ-13 and Cu-SSZ-13 samples; CO/NO adsorption on the Cu-BS sample using DRIFTS; full infrared spectra for the methane exposure experiments; full infrared spectra for the methanol desorption experiments. See DOI: 10.1039/c8ra08038k |
| This journal is © The Royal Society of Chemistry 2018 |