
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupported binary CuOx–Pt catalysts with high activity and thermal stability for the combustion of NH3 as a carbon-free energy source†
Saaya Kiritoshia,
Takeshi Iwasa b,
Kento Arakia,
Yusuke Kawabataa,
Tetsuya Taketsugub,
Satoshi Hinokuma*ac and
Masato Machidaa
b,
Kento Arakia,
Yusuke Kawabataa,
Tetsuya Taketsugub,
Satoshi Hinokuma*ac and
Masato Machidaa
aField of Environmental Chemistry and Materials, Division of Materials Science and Chemistry, Faculty of Advanced Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan. E-mail: hinokuma@kumamoto-u.ac.jp; Tel: +81-96-342-3653
bDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan
cInternational Research Organization for Advanced Science and Technology, Kumamoto University, Japan
First published on 12th December 2018
Abstract
Recently, NH3 has been thought to be a renewable and carbon-free energy source. The use of NH3 fuel, however, is hindered by its high ignition temperature and N2O/NO production. To overcome these issues, in this study, the combustion of NH3 over copper oxide (CuOx) and platinum (Pt) catalysts supported on aluminium silicates (3Al2O3·2SiO2), aluminium oxides (Al2O3), and silicon oxides (SiO2) were compared. To achieve high catalytic activity for the combustion of NH3 and high selectivity for N2 (or low selectively for N2O/NO), conditions for the preparation of impregnated binary catalysts were optimised. With respect to the binary catalysts, sequentially impregnated CuOx/Pt/Al2O3 exhibited relatively higher activity, N2 selectivity, and thermal stability. From XRD and XAFS analyses, CuOx and Pt in CuOx/Pt/Al2O3 were present as CuAl2O4 and metallic Pt, respectively. Given that the combustion activity was closely associated with the Pt nanoparticle size, which was estimated from the Scherrer equation and the pulsed CO technique, highly dispersed Pt nanoparticles were crucial for the low-temperature light-off of NH3. For single and binary catalysts, although NH (imide) deformation modes as a key species for N2O production were detected by in situ FTIR spectral analysis, the band intensity of CuOx/Pt/Al2O3 was less than those of CuOx/Al2O3 and Pt/Al2O3. Therefore, CuOx/Pt/Al2O3 exhibits high selectivity for N2 in NH3 combustion.
Introduction
Recent studies have reportedly considered NH3 to serve not only as a H2 carrier material but also as a renewable, carbon-free energy source because of its high energy density (3160 W h L−1) and negligible thermal NOx emission.1 Currently, NH3 is industrially produced at a high pressure using N2 (produced from air) and H2 (produced from natural gas) by employing the Haber–Bosch process, which consumes a large amount of energy. However, the energy and costs associated with H2 production and natural gas is 70–90% of that required for NH3 production.2 Therefore, the low energy and costs that are associated with H2 production along with the fact that it is a renewable energy source should be emphasised to ensure that the society utilises H2 along with NH3. As a proof-of-concept for using NH3 as a fuel, an NH3-fuelled micro-gas turbine has been employed to demonstrate the potential of NH3-fired power plants at the Fukushima Renewable Energy Institute in Japan, as well as an NH3-fuelled industrial furnace.3 Compared to fossil fuels, however, NH3 exhibits disadvantages of a high ignition temperature, low combustion rate, and N2O/NOx production. Therefore, to overcome these issues, it is imperative to develop a novel NH3 combustion system. First, a catalytic NH3 combustion system, which decreases the ignition temperature and N2O/NOx emissions, was developed. Previously, our group has successfully developed a catalytic NH3 combustion system and novel catalysts exhibiting high activity, N2 selectivity, and thermal stability.4 For example, copper oxides (CuOx) supported on Al2O3-based composite oxide materials (such as 10Al2O3·2B2O3 and 3Al2O3·2SiO2) exhibit high N2 selectivity and thermal stability, whereas binary CuOx and silver (Ag) supported on Al2O3 exhibit high activity and N2 selectivity.4 However, the former catalyst exhibits a low activity (ignition temperature at ∼300 °C), while the latter catalyst exhibits low thermal stability (deactivation temperature at ∼900 °C likely because of the low melting point of Ag).4 Therefore, it is imperative to develop catalysts to exhibit high activities and thermal stabilities for NH3 combustion.On the other hand, binary systems comprising supported CuOx–Ag,5 CuOx–Pt,6 CuOx–Au,7 CuOx–CeO28 and –Li2O38 catalysts exhibiting a high performance for the selective catalytic oxidation of NH3 (NH3-SCO) have been previously reported. In particular, CuOx–Pt systems are expected to exhibit high thermal stability because of the high melting point of Pt (∼1770 °C). Recently, Sun et al. have synthesised impregnated CuOx–Pt/ZSM-5 catalysts and reported that CuOx on Pt species can lead to higher NH3-SCO performance compared to single catalysts.6 On the other hand, Jabłońska has synthesised Al2O3-, TiO2-, and ZrO2-supported CuOx–Pt, –Pd and –Rh catalysts6 and found that the supported CuOx–Pt catalyst exhibits high activity and N2 selectivity for NH3-SCO. However, in these studies, NH3 is considered to cause air pollution; therefore, the thermal stability is investigated at temperatures of less than 900 °C.
In this study, supported binary CuOx–Pt catalysts were synthesised, and their catalytic properties for the combustion of NH3 (as an energy source) at high reaction temperatures were examined. To achieve high catalytic activity for the combustion of NH3, N2 (low N2O/NO) selectivity and thermal stability, preparation conditions for impregnated binary catalysts were optimised. Moreover, the relationship between the local structure and catalytic properties for the combustion of NH3 over supported CuOx–Pt catalysts was discussed.
Experimental
Catalyst preparation
3Al2O3·2SiO2 (3A2S) of the catalyst support material was prepared by using an alkoxide method. According to previous studies,4,9 Si(OC2H5)4 (TEOS, Wako Pure Chemicals) was first dissolved in C2H5OH (Wako Pure Chemicals) at a concentration of 1 M at room temperature (RT). Second, after the addition of H2O and HCl (Wako Pure Chemicals), the solution was stirred at 70 °C for 5 h, and a Si solution was prepared. Third, the alcohol was dehydrated. The H2O and HCl concentrations were H2O/TEOS = 2 (mol mol−1) and HCl/TEOS = 0.1 (mol mol−1), respectively. Next, Al[OCH(CH3)2]3 (AIP, Sigma-Aldrich) was subjected to reflux conditions and dissolved in (CH3)2CHCH2OH (Nacalai Tesque) to prepare an Al solution. The precursor solution was prepared by mixing the Al and Si solutions in a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 at RT. H2O was added, and the solution was stirred for 1 h to prepare the precursor solution. The precursor sols were dried at 110 °C for 48 h, affording xerogel precursor powders. These powders were pulverised and calcined at 600 °C for 3 h and finally at 1200 °C for 5 h in air. In addition, α-Al2O3 (Wako Pure Chemicals), γ-Al2O3 (JRC-ALO-8, Catalysis Society of Japan), SiO2 (JRC-SIO-10, Catalysis Society of Japan), and CuO (Wako Pure Chemicals) were used as support materials.
:2 at RT. H2O was added, and the solution was stirred for 1 h to prepare the precursor solution. The precursor sols were dried at 110 °C for 48 h, affording xerogel precursor powders. These powders were pulverised and calcined at 600 °C for 3 h and finally at 1200 °C for 5 h in air. In addition, α-Al2O3 (Wako Pure Chemicals), γ-Al2O3 (JRC-ALO-8, Catalysis Society of Japan), SiO2 (JRC-SIO-10, Catalysis Society of Japan), and CuO (Wako Pure Chemicals) were used as support materials.
Single and binary supported CuOx (CuO loading of 6 wt%) and/or Pt (Pt loading of 2 wt%) catalysts were prepared by the co-impregnation (CuOx–Pt) of an aqueous solution of Cu(NO3)2 (Wako Pure Chemicals) and [Pt(NH3)2(NO3)2] (Tanaka Kikinzoku Kogyo), followed by drying and calcination at 600 °C for 3 h in air. In addition, the sequentially impregnated binary catalysts, i.e. Pt and subsequent CuOx (CuOx/Pt) and/or CuOx and subsequent Pt (Pt/CuOx), were synthesised in a similar fashion. As CuOx/Pt/Al2O3 exhibited a high catalytic activity for the combustion of NH3, physically mixed catalysts (CuO + Pt/Al2O3, CuAl2O4 + Pt/Al2O3, and CuOx/Al2O3 + Pt/Al2O3) with the same composition ratios were also prepared. To evaluate their thermal stabilities and catalytic properties, the as-prepared catalysts were subjected to thermal aging at 900 °C for 100 h in air and/or at 1000 °C for 5 h in air.
Characterisation
Powder X-ray diffraction (XRD) patterns were recorded using monochromatic Cu Kα radiation (30 kV, 20 mA, Multiflex, Rigaku). The average size of the Pt particles was calculated by the XRD line broadening method by using the Scherrer equation.10 The chemical composition was determined by X-ray fluorescence (XRF; EDXL-300, Rigaku) measurements. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) mapping images were recorded on a JEM-ARM200CF (JEOL) system. Brunauer–Emmett–Teller (BET) surface area (SBET) values were calculated from the N2 adsorption isotherms, which were recorded at −196 °C (Belsorp, Bel Japan, Inc.). Pt dispersion and particle size were determined by the pulsed CO technique (BELCAT-B, BEL Japan, Inc.) at 50 °C after O2 oxidation and subsequent H2 reduction at 400 °C according to previous studies.11 Pt dispersion and particle size were calculated by assuming hemispherical particles with the stoichiometric adsorption of CO on Pt at a CO:Pt ratio of 1:1. Cu K-edge and Pt L3-edge X-ray absorption fine structure (XAFS) spectral analyses were carried out at BL9A of PF and BL01B1 of SPring-8. XAFS spectra were recorded in the transmission mode at RT using an ionisation chamber filled with N2 for the incident beam, another chamber filled with 75% N2/Ar for the Cu K-edge and 50% N2/Ar for Pt L3-edge for the transmitted beam, and a Si(111) double-crystal monochromator. Reference samples (e.g. Cu2O, CuO, CuAl2O4, and PtO2) were mixed with boron nitride (BN) powder to achieve an appropriate absorbance at the edge energy, whereas the catalysts were used without mixing with BN. XAFS data were processed using the IFEFFIT software package (Athena and Artemis).
Catalytic NH3 combustion tests
The catalytic combustion of NH3 was performed in a flow reactor at atmospheric pressure. Catalysts (10–20 mesh, <0.3 mm thickness, 50 mg) were fixed in a quartz tube (OD: 6 mm, ID: 4 mm) with quartz wool at both ends of the catalyst bed. The temperature dependence of the catalytic activity was evaluated by heating the catalyst bed from RT to 900 °C at a constant rate of 10 °C min−1 while a gas mixture containing 1.0% NH3, 1.5% O2 and He balance at 100 cm3 min−1 (W/F = 5.0 × 10−4 g min cm−3) was supplied. The O2 excess ratio for NH3 combustion was expressed as λ = (pO2/pNH3)exp./(pO2/pNH3)stoichiom., where p denotes pressure. The NH3/N2O/NO, N2 and NO2 gas concentrations were analysed using a nondispersive infrared gas analyser (VA-3011, Horiba), chromatography (GC-8A, Shimadzu) and chemiluminescence (NOA-7000, Shimadzu) measurements. The ESI† provides the calculation formulae used to determine the concentration ratios.In situ Fourier transform infrared (FTIR) spectra were recorded on a Nicolet 6700 spectrometer using a diffuse-reflectance reaction cell with a BaF2 window connected to a gas supply and a heating system to enable measurements at atmospheric pressure. First, the catalysts were preheated in situ in flowing He at 400 °C for 30 min prior to each experiment. After pre-treatment, the temperature of the catalyst was decreased to 200 °C, followed by the subsequent purging of the cell with He and then filling with 0.3% NH3/He mixed gas. Finally, FTIR spectra were recorded while the catalysts were maintained under a stream of NH3/He.
Computational details
Spin-restricted density functional theory (DFT) computations were carried out for NH3/Ptx (x = 13 and/or 20) at the M06 level using the def-SV(P) basis sets, with the 60-electron relativistic effective core potential for Pt, as implemented in TURBOMOLE under the resolution of the identity approximation.12 The model clusters of Pt13 with Ih symmetry and Pt20 with Td symmetry having four (111) faces were locally optimised until imaginary frequencies were removed. One atop geometry for NH3–Pt13 and three initial geometries for NH3–Pt20 were considered, i.e. atop the (111) face; bridge-to-edge site; and atop the vertex site, respectively. These geometries were optimised until no imaginary frequencies were observed. The adsorption energy of NH3 on Ptx was estimated as Eads = E(NH3) + E(Ptx) − E(NH3–Ptx), where E(A) denotes the electronic energy of species A.Results & discussion
Local structures of supported CuOx and Pt catalysts
Fig. 1 shows the XRD patterns of sequentially impregnated binary (CuOx/Pt) catalysts, because CuOx/Pt/Al2O3 exhibited higher activity, N2 (lower N2O/NO) selectivity, and thermal stability for the combustion of NH3 compared to all binary catalysts before and after thermal ageing at 900 °C (Table 1). ESI† shows the XRD patterns of the other catalysts. The XRD patterns of as-prepared CuOx/Pt/3A2S and CuOx/Pt/SiO2 before and after aging revealed peaks corresponding to CuO, whereas peaks corresponding to CuAl2O4 were observed for CuOx/Pt/3A2S and CuOx/Pt/Al2O3 (900 °C), possibly related to the solid-state reaction between γ-Al2O3 and CuOx. Diffraction peaks for Cu species were not observed for as-prepared CuOx/Pt/Al2O3, possibly related to the high dispersion of CuOx. The Al2O3 phase for the binary catalysts was transformed from γ to α, and the diffraction peaks for Pt became sharper after thermal aging at temperatures of higher than 900 °C. Therefore, the growth of particles and sintering of Al2O3 and Pt are induced by thermal aging. Indeed, the Pt particle sizes calculated by the Scherrer equation increased with the aging temperature (Table 1). On the other hand, as peaks corresponding to CuO were observed for the binary catalysts prepared using α-Al2O3 (CuOx/Pt/α-Al2O3) before and after aging, it was possibly difficult to form CuAl2O4 from the solid-state reaction between α-Al2O3 and CuOx (ESI†).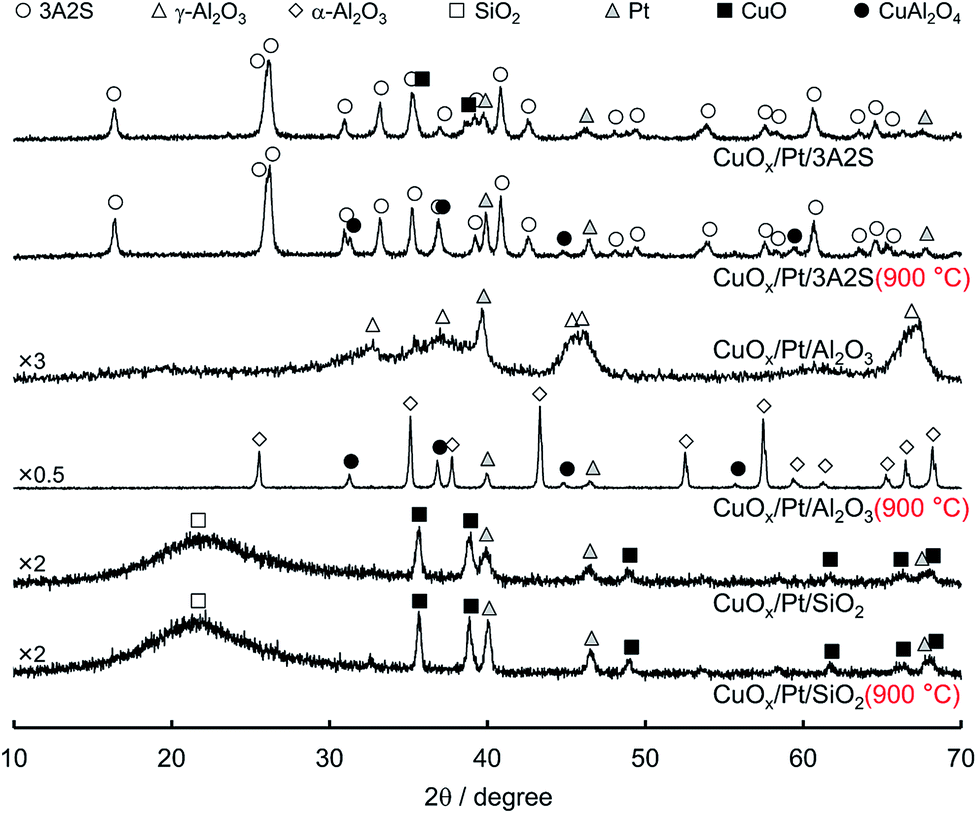 | ||
| Fig. 1 XRD patterns of sequentially impregnated catalysts before and after thermal aging 900 °C for 100 h in air. | ||
| Catalyst | Phase | T10 a/°C | T90 a/°C | Selectivity at T90 a/% | SBET /m2 g−1 | Pt particle size/nm | Desorbed gas d/μmol m−2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N2 | N2O | NO | b XRD | c Pulsed CO | NH3 | NO | |||||
| a Temperature at which NH3 conversion reached 10% and 90%.b Calculated from XRD line broadening method.c Calculated from pulsed CO chemisorption.d Estimated by NH3- and NO-TPD ranging from 50 °C to 500 °C.e The amount of CO chemisorption was not detected (n. d.). | |||||||||||
| CuOx/Pt/3A2S | CuO/Pt/3A2S | 210 | 356 | 90 | 8 | 2 | 32 | 18 | |||
| CuOx/Pt/3A2S (900 °C) | CuAl2O4/Pt/3A2S | 203 | 344 | 89 | 10 | 1 | 27 | 50 | |||
| CuOx/Al2O3 | CuAl2O4/γ-Al2O3 | 303 | 476 | 92 | 6 | 2 | 149 | 1.3 | 0.050 | ||
| CuOx/Al2O3 (900 °C) | CuAl2O4/α, γ-Al2O3 | 295 | 450 | 91 | 8 | 1 | 102 | 0.4 | 0.063 | ||
| Pt/Al2O3 | Pt/γ-Al2O3 | 188 | 289 | 81 | 19 | <1 | 156 | 11 | 11 | 0.6 | 0.051 |
| Pt/Al2O3 (900 °C) | Pt/γ, θ-Al2O3 | 213 | 266 | 87 | 12 | <1 | 99 | 31 | e n. d. | 2.0 | 0.140 |
| CuOx/Pt/Al2O3 | CuAl2O4/Pt/γ-Al2O3 | 189 | 278 | 86 | 13 | <1 | 139 | 15 | 5 | 1.6 | 0.064 |
| CuOx/Pt/Al2O3 (900 °C) | CuAl2O4/Pt/α-Al2O3 | 220 | 339 | 86 | 13 | 1 | 14 | 33 | 176 | 1.5 | 0.444 |
| CuOx/Pt/SiO2 | CuO/Pt/SiO2 | 234 | 316 | 93 | 7 | <1 | 182 | 13 | e n. d. | ||
| CuOx/Pt/SiO2 (900 °C) | CuO/Pt/SiO2 | 301 | 456 | 96 | <1 | 3 | 81 | 24 | e n. d. | ||
Fig. 2 shows the normalised Cu K-edge X-ray absorption near-edge structure (XANES) spectra and k-space extended X-ray absorption fine structure (EXAFS) oscillations of the catalysts and the three reference compounds (Cu2O, CuO and CuAl2O4, respectively). Cu K-edge XANES spectra and k-space EXAFS oscillations of CuOx/Pt/3A2S (900 °C) and CuOx/Pt/Al2O3 before and after thermal aging at 900 °C were similar to those of CuAl2O4, whereas those of CuOx/Pt/3A2S and CuOx/Pt/SiO2 before and after thermal aging at 900 °C were similar to those of CuO. In the XANES spectra of CuOx/Pt/3A2S and CuOx/Pt/SiO2 before and after aging, the pre-edge (at ∼8985 eV, corresponding to Cu2+ 1s → 4p + ligand and Cu2+ 1s → Cu2+ charge-transfer excitation)13 for CuO was observed. XAFS analyses were consistent with the XRD patterns of the catalysts. The Pt L3-edge XAFS profiles of the catalysts and two references (i.e. Pt foil and PtO2, respectively) are also depicted in Fig. 3. For as-prepared CuOx/Pt/Al2O3, the XANES profile was slightly similar to that observed for PtO2. According to the calculated thermodynamic Pt–PtOx phase equilibrium (ESI†), PtO2 and metallic Pt exhibited thermodynamic stability at RT and 600 °C, respectively. Therefore, Pt nanoparticles (especially, their surfaces) in CuOx/Pt/Al2O3 are thought to be oxidised during the cooling process of calcination. By contrast, the XANES profiles of the other catalysts were similar to that of Pt foil (metallic Pt) as against the calculated phase equilibrium. This disagreement is supposedly caused by the calcination and/or thermal aging-induced sintering of metallic Pt nanoparticles in these catalysts; hence, the bulk metallic Pt particles are stable at RT.
 | ||
| Fig. 2 Cu K-edge (upper) normalised XANES spectra and (lower) EXAFS oscillations of sequentially impregnated catalysts before and after thermal aging and three references (i.e. Cu2O, CuO, and CuAl2O4, respectively). | ||
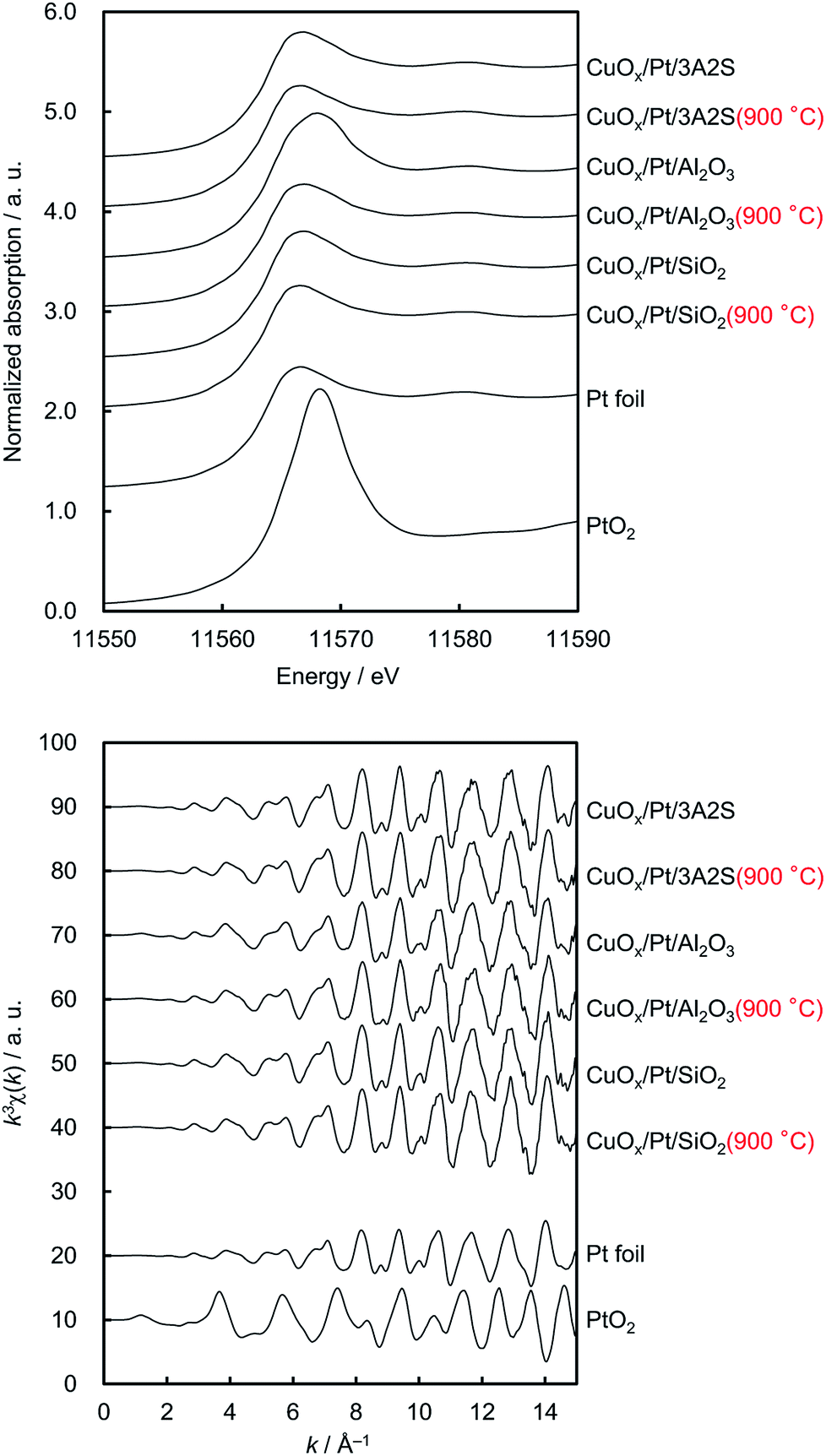 | ||
| Fig. 3 Pt L3-edge (upper) normalised XANES spectra and (lower) EXAFS oscillations of sequentially impregnated catalysts before and after thermal aging and two references (i.e. Pt foil and PtO2, respectively). | ||
Next, HAADF-STEM and EDS mapping images of sequentially impregnated CuOx/Pt/Al2O3 (900 °C) were recorded (Fig. 4). From the HAADF-STEM image, dispersed nanoparticles with a bright contrast supported on Al2O3 particles (blue for Al) were observed, and the EDS mapping images revealed the presence of Pt (red). On the other hand, for the CuOx species of CuOx/Pt/Al2O3 (900 °C), the EDS mapping images revealed the presence of CuOx aggregates (green). However, because the overlap of the fluorescence lines of Pt-L and Cu-K, as denoted by the solid arrows in the overlaid image, a physical proximity between Pt and CuOx nanoparticles in CuOx/Pt/Al2O3 was supposed after aging at 900 °C. On the other hand, Pt particles with sizes larger than 200 nm were clearly observed for CuOx/Pt/SiO2 (900 °C) (ESI†).
 | ||
| Fig. 4 HAADF-STEM image and EDS mapping analysis of CuOx/Pt/Al2O3 (900 °C). Red, green, and blue points denote the Pt-L, Cu-K, and Al-K fluorescence lines, respectively. | ||
Combustion properties of supported CuOx and Pt catalysts
Fig. 5 shows the comparison of the temperature dependence on the selectivities of products obtained over supported CuOx/Pt, CuOx/Al2O3 and Pt/Al2O3 before and after thermal aging at 900 °C under O2 excess conditions (λ = 2). ESI† shows those of the other catalysts. With respect to the supported CuOx/Pt catalysts before and after aging, CuOx/Pt/Al2O3 exhibited a comparatively higher catalytic NH3 combustion activity and N2 (lower N2O/NO) selectivity. For CuOx/Pt/Al2O3 and Pt/Al2O3 before and after aging, the light-off curves for NH3 were obtained at ∼200 °C, whereas CuOx/Al2O3 exhibited lower catalytic activity (T10: ∼300 °C). On the other hand, before and after aging, CuOx/Pt/Al2O3 and CuOx/Al2O3 exhibited lower NO selectivity compared to Pt/Al2O3 although NO selectivities for all catalysts were observed after the NH3 conversion reached ∼90%. In contrast, the selectivity of N2O over the catalysts before and after aging increased in the order of CuOx/Al2O3 < CuOx/Pt/Al2O3 < Pt/Al2O3. Finally, CuOx/Pt/Al2O3 exhibited higher N2 selectivity. Therefore, binary CuOx/Pt/Al2O3 renders synergistic effects of Pt (enhancing the NH3–O2 reaction) and CuOx (increasing N2 selectivity). To investigate the low NO selectivity of CuOx/Pt/Al2O3 and CuOx/Al2O3, an NH3–NO–O2 reaction test (0.8% NH3, 0.2% NO, 1.4% O2, He balance) was also performed (shown in ESI†). For the NH3–NO reaction, CuOx/Pt/Al2O3 also exhibited higher activity compared to CuOx/Al2O3 and Pt/Al2O3. NH3 was supposedly consumed not only by O2 but also by NO, which was formed as an intermediate during the light-off of NH3. However, the selectivity of N2O for the NH3–NO–O2 reaction over CuOx/Pt/Al2O3 was higher than that for the combustion of NH3 (Fig. 5), indicating that the N2O is produced from the NH3–NO reaction.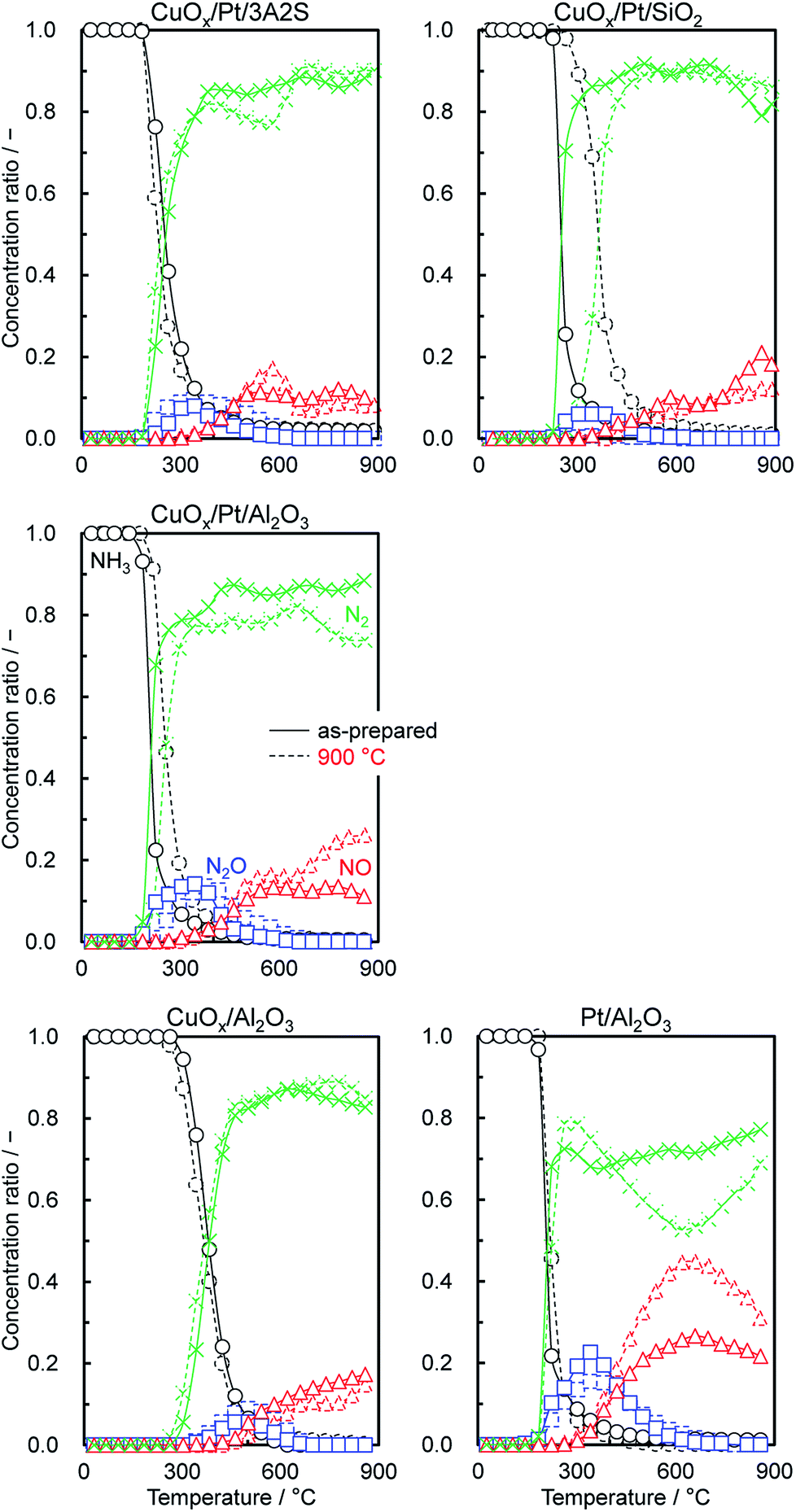 | ||
| Fig. 5 Product selectivities for the catalytic combustion of NH3 (NH3–O2) over catalysts before and after thermal aging in air at 900 °C for 100 h. Reaction conditions: 1.0% NH3, 1.5% O2, λ = 2, He balance, W/F = 5.0 × 10−4 g min cm−3. | ||
For N2O production, on the other hand, according to the kinetic model for NH3 oxidation, the elementary reaction of NH (imide) + NO → N2O + H exhibited high sensitivity for N2O production.14 Moreover, for catalytic NH3 oxidation and NH3–NO reactions, NH was also regarded as a key species for N2O production.15 Therefore, to verify the adsorption of NH on the catalysts, in situ FTIR spectra of NH3 adsorbed on the catalysts before and after aging were recorded at 200 °C (Fig. 6); this temperature is the approximate initiation temperature for NH3 combustion (Fig. 5). As has been reported previously,15 FTIR bands were observed at 1250 and 1625 cm−1, corresponding to the deformation modes of NH3 adsorbed on Lewis acid sites, for all catalysts. Moreover, for Pt/Al2O3 and CuOx/Pt/Al2O3, a set of bands were slightly observed at 1395 and 1695 cm−1, corresponding to the asymmetric and symmetric bending vibrations of NH3 species on the Brønsted acid sites. Although the single band at 1458 cm−1 corresponding to the NH (considered as a species for N2O production) deformation modes was observed for all catalysts, the band intensity for CuOx/Pt/Al2O3 was less than that for Pt/Al2O3. As CuOx/Pt/Al2O3 prevented the production of NO from the combustion of NH3 and the dissociative adsorption of NH3, the catalyst finally achieved high N2 selectivity.
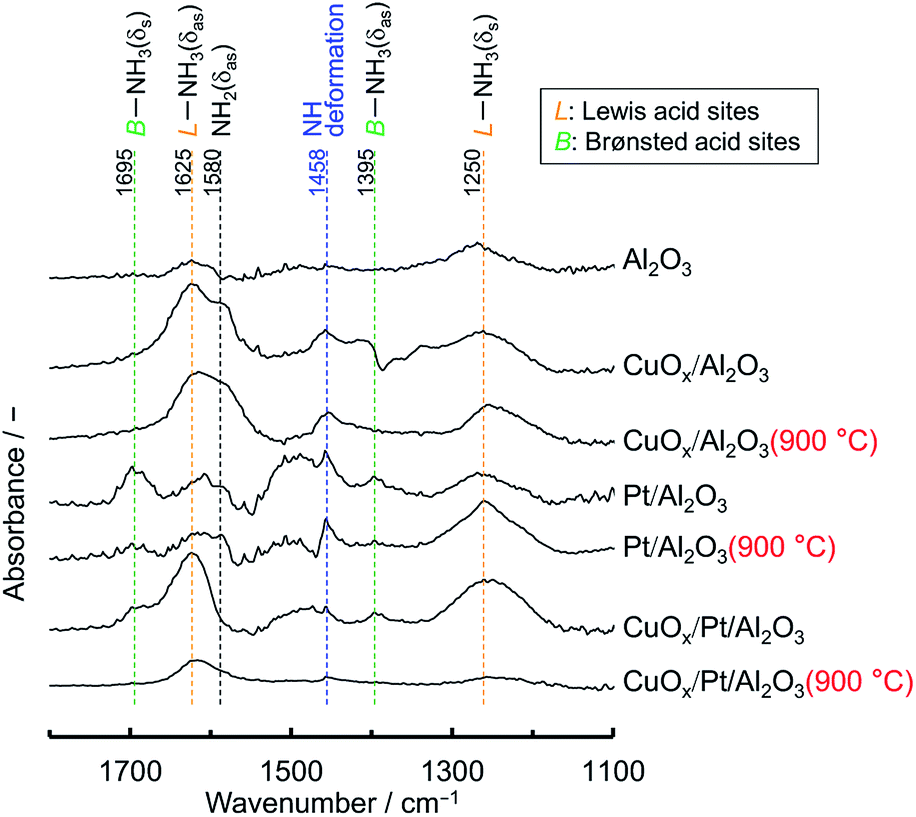 | ||
| Fig. 6 In situ FTIR spectra of NH3 adsorbed on supports and catalysts before and after thermal aging in air at 900 °C for 100 h. The spectra were recorded at 200 °C in gas feeds of 0.3% NH3 with He balance. | ||
Catalytic properties of supported CuOx and/or Pt
Table 1 summarises the catalytic properties of supported CuOx/Pt, CuOx/Al2O3 and Pt/Al2O3 before and after thermal aging at 900 °C, and ESI† shows those of the other catalysts. Catalytic activity was expressed in terms of the light-off temperature at which 10% conversion of NH3 was achieved (T10), and the product selectivities were evaluated at the reaction temperature at which 90% NH3 conversion was achieved (T90). ESI† shows the temperature dependence of the product selectivities for the combustion of NH3 over the catalysts. For single catalysts supported on each material (i.e. 3A2S, Al2O3 and SiO2), supported CuOx exhibited low activity and N2O/NO selectivities, whereas supported Pt exhibited high activity and N2O/NO selectivity (ESI†). On the other hand, binary supported catalysts exhibited the same high activity as the single supported Pt, albeit a slightly lower N2O/NO selectivity. As the combustion activity was closely associated with the Pt particle size, which was calculated by the Scherrer equation (plot shown in the ESI†), highly dispersed Pt nanoparticles supposedly played a key role in the low-temperature light-off of NH3. In this study, for pulsed CO chemisorption, it is expected that CO is adsorbed not only on Pt but also on CuOx in case of binary system catalysts.16 Therefore, the correlation between the NH3 combustion activity (T10) and the Pt particle size can be estimated using the XRD line broadening method by employing the Scherrer equation.To elucidate the correlation between the activity and size of Pt nanoparticles, the adsorption energy (Eads) between NH3 and Pt13 and/or Pt20 clusters (Pt–N) were estimated by DFT computations (Fig. 7). Compared with the Pt–N Eads of the vertex for the Pt20 (larger) cluster, that of the vertex for the Pt13 (smaller) cluster was higher, indicating that NH3 preferentially adsorbs on highly dispersed Pt nanoparticles as well as coordinatively unsaturated (cus) Pt atoms. Indeed, for the Pt20 cluster, the Pt–N Eads of three initial geometries (see inset in Fig. 7) increased in the order of atop < edge < vertex. Moreover, the bond distance of N–H in NH3 adsorbed on Pt13 was longer than that on Pt20, suggesting that cus Pt exhibits high activity for the dissociation of the N–H bond in adsorbed NH3. Compared with the activity of binary CuOx–Pt catalysts prepared by co-impregnation, those of the sequentially impregnated CuOx/Pt catalysts were higher probably because Pt nanoparticles can be highly dispersed onto each support material without inhibition from the presence of CuOx.
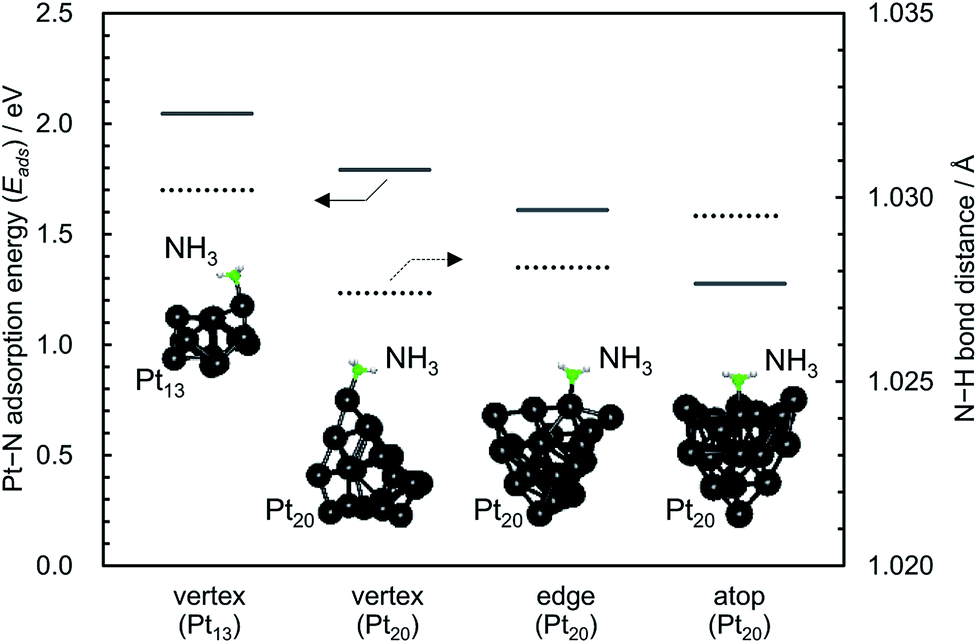 | ||
| Fig. 7 Adsorption energy (Eads) between NH3 and Pt13 and/or Pt20 clusters (Pt–N) and bond distances of N–H in NH3 adsorbed on Pt13 and/or Pt20 obtained by the DFT computations. | ||
Among the CuOx/Pt catalysts supported on various support materials, CuOx/Pt/Al2O3 exhibited a relatively higher activity, N2 selectivity, and thermal stability (Table 1). However, a significant decrease in SBET was observed for CuOx/Pt/Al2O3 (900 °C) (139 → 14 m2 g−1) in comparison with that observed for CuOx/Al2O3 (900 °C) and Pt/Al2O3 (900 °C). For this reason, first, it is considered that the significant decrease in specific surface area can be attributed to the phase transition of (γ → α) Al2O3 as well as thermal aging at 900 °C. However, in case of only γ-Al2O3, the phase transformation to α-Al2O3 was observed at approximately 1200 °C.17 Next, however, according to a previous report related to the effects of divalent cation additives on the phase transition of (γ → α) Al2O3, the additive Cu2+ species that coexist with γ-Al2O3 accelerates its phase transition to α-Al2O3 at a low temperature of 1200 °C, which can be attributed to the fact that CuAl2O4 is formed at a temperature of lower than 1200 °C.18 Indeed, in this study, α-Al2O3 was observed in CuOx/Al2O3 after thermal aging at 900 °C (Table 1). Finally, it is probable that the additive and co-existing Pt also accelerates the formation of CuAl2O4 as well as the phase transition to α-Al2O3. Although no concrete data are available, a similar significant decrease of the specific surface area could be observed in both Cu–Al–O and Cu–Pt–Al–O system catalysts after thermal aging occurred at 1000 °C.19
Notably, the high performance for CuOx/Pt/Al2O3 was maintained after thermal aging at 1000 °C despite the phase transition (γ → α) and decrease in the surface area. On the other hand, CuOx/Pt supported on α-Al2O3 exhibited slightly higher N2O/NO selectivities compared to CuOx/Pt/Al2O3 (using γ-Al2O3), the results of which can be explained from the difference in the CuOx phase of CuO in CuOx/Pt/α-Al2O3 and CuAl2O4 in CuOx/Pt/γ-Al2O3. Previously, our group has reported that supported CuAl2O4 exhibits a higher NH3–NO–O2 reaction activity to N2 compared to supported CuO because supported CuAl2O4 exhibits a higher fraction of the Cu2+ active species for the reaction.4 Therefore, the trend for N2O/NO selectivities over CuOx/Pt/α-Al2O3 and CuOx/Pt/Al2O3 can be explained in a similar manner. Moreover, physically mixed catalysts (CuO + Pt/Al2O3, CuAl2O4 + Pt/Al2O3 and CuOx/Al2O3 + Pt/Al2O3) exhibited lower N2 selectivity at high reaction temperatures (ca. > 600 °C) compared to binary catalysts (ESI†), indicating that highly dispersed Pt and proximate CuOx particles are required to achieve high N2 selectivity for catalytic NH3 combustion. On the other hand, Pt/CuO exhibited high N2 selectivity, albeit low catalytic activity (ESI†), probably because of low Pt dispersion. To examine the acid and base properties of these catalysts, the amount of desorbed gas per surface area of the catalyst was estimated using NH3- and NO-temperature-programmed desorption (TPD) over a range of 50–500 °C (ESI†). Table 1 summarises this data. For all catalysts after aging, the amount of desorbed NO increased noticeably, likely because of decreased surface area. However, before and after aging, CuOx/Pt/Al2O3 exhibited relatively higher NH3 and NO adsorption properties compared to CuO/Al2O3 and Pt/Al2O3. These results indicated that CuOx/Pt/Al2O3 exhibits higher acidity and basicity compared to single supported catalysts; therefore, the reactivity of NH3–NO to N2 is enhanced on the CuOx/Pt/Al2O3 surface.
Finally, the effects of CuO, Cu2O and metallic Cu on the catalytic NH3 combustion properties are discussed. ESI† denotes the comparison of the temperature dependence on the selectivities of the products that were obtained when compared to those of the commercially available CuO, Cu2O and metallic Cu. The metallic Cu was prepared from CuO that was reduced at 400 °C for 30 min in 5% H2/He because CuO can be reduced to metallic Cu at an approximate temperature of 300 °C, as demonstrated from the H2-TPR measurement.4 For observing the NH3 combustion activity, the light-off curve for CuO was obtained at approximately 300 °C; further, Cu2O and metallic Cu exhibited lower activity when compared to that exhibited by CuO. Therefore, it can be considered that CuO (Cu2+ oxidation state) acts as an active site during the reaction. However, while considering the selectivities of the products, Cu2O and metallic Cu exhibited low N2O/NO selectivities because their activities (NH3 conversions) were observed to be low. In the O2-excess condition for NH3 combustion (λ = 2.0), it can be considered that the combustion over CuOx catalysts proceeds because of redox cycling between CuO and Cu2O.4 Further, the XRD patterns of CuO and Cu2O before and after the reaction (λ = 2.0) also indicate redox cycling because the patterns of Cu2O after the reaction revealed diffraction peaks corresponding to those of CuO (ESI†). In addition, based on our previous studies related to operando XAFS, similar redox behaviours could be observed in the supported CuO catalysts (CuOx/SiO2).4 Therefore, it can be assumed that the effect of metallic Cu on combustion is negligible.
Conclusions
In this study, binary CuOx and Pt catalysts supported on 3A2S, Al2O3 and SiO2 suppressed the production of N2O/NOx via the catalytic combustion of NH3 as a carbon-free energy source. Among the binary supported CuOx and Pt catalysts, sequentially impregnated CuOx/Pt catalysts tended to exhibit higher activity because Pt nanoparticles were highly dispersed onto each support material without inhibition due to the presence of CuOx. Compared to sequentially impregnated CuOx/Pt/3A2S and CuOx/Pt/SiO2, CuOx/Pt/Al2O3 exhibited high NH3 combustion activity, N2 selectivity, and thermal stability. Before and after aging, CuOx/Pt/Al2O3 was characterised as proximate CuAl2O4 and dispersed metallic Pt particles. As the combustion activity was closely associated with the Pt particle size, highly dispersed Pt nanoparticles were thought to play a key role in the low-temperature light-off of NH3. For selectivity, the proximate CuAl2O4 in CuOx/Pt/Al2O3 enhanced the NH3–NO to N2 reaction, and the catalyst prevented the dissociative adsorption of NH3 to NH, which was regarded as an intermediate species obtained during the N2O production. Therefore, CuOx/Pt/Al2O3 exhibits high N2 selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JST, PRESTO (JPMJPR1344) and JSPS (18K14326). This work was partly supported by JST-Mirai Program and the Advanced Characterization Platform of the Nanotechnology Platform Japan sponsored by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The XAFS experiments were performed at BL9A with the approval of PF, KEK (Proposal No. 2016G527), and at the BL01B1 of SPring-8 with the approval of JASRI (Proposal No. 2017A1050).Notes and references
- P. Glarborg, A. Jensen and J. Johnsson, Prog. Energy Combust. Sci., 2003, 29, 89–113 CrossRef CAS.
- (a) J. Andersson and J. Lundgren, Appl. Energy, 2014, 130, 484–490 CrossRef CAS; (b) Y. Bicer, I. Dincer, C. Zamfirescu, G. Vezina and F. Raso, J. Cleaner Prod., 2016, 135, 1379–1395 CrossRef CAS; (c) S. Giddey, S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- (a) A. Hayakawa, T. Goto, R. Mimoto, T. Kudo and H. Kobayashi, Mech. Eng. J., 2015, 2, 14–00402 CAS; (b) R. Murai, R. Omori, R. Kano, Y. Tada, H. Higashino, N. Nakatsuka, J. Hayashi, F. Akamatsu, K. Iino and Y. Yamamoto, Energy Procedia, 2017, 120, 325–332 CrossRef CAS.
- (a) S. Hinokuma, et al., Chem. Lett., 2016, 45, 179–181 CrossRef CAS; (b) S. Hinokuma, et al., J. Phys. Chem. C, 2016, 120, 24734–24742 CrossRef CAS; (c) S. Hinokuma, et al., J. Phys. Chem. C, 2017, 121, 4188–4196 CrossRef CAS; (d) S. Hinokuma, et al., J. Ceram. Soc. Jpn., 2017, 125, 770–772 CrossRef CAS; (e) S. Hinokuma, et al., Catal. Commun., 2018, 105, 48–51 CrossRef CAS; (f) S. Hinokuma, et al., Catal. Today, 2018, 303, 2–7 CrossRef CAS; (g) S. Hinokuma, et al., J. Catal., 2018, 361, 267–277 CrossRef CAS.
- (a) L. Gang, B. Anderson, J. van Grondelle, R. van Santen, W. van Gennip, J. Niemantsverdriet, P. Kooyman, A. Knoester and H. Brongersma, J. Catal., 2002, 206, 60–70 CrossRef; (b) M. Yang, C. Wu, C. Zhang and H. He, Catal. Today, 2004, 90, 263–267 CrossRef CAS; (c) J. Lee, Y. Lim, B. Park, A. Adelodun and Y. Jo, Bull. Korean Chem. Soc., 2015, 36, 162–167 CrossRef CAS.
- (a) R. Burch and B. Southward, J. Catal., 2000, 195, 217–226 CrossRef CAS; (b) R. Burch and B. Southward, Chem. Commun., 2000, 1115–1116 RSC; (c) H. Kusar, A. Ersson, M. Vosecky and S. Jaras, Appl. Catal., B, 2005, 58, 25–32 CrossRef CAS; (d) G. Olofsson, A. Hinz and A. Andersson, Chem. Eng. Sci., 2004, 59, 4113–4123 CrossRef CAS; (e) G. Olofsson, L. Wallenberg and A. Andersson, J. Catal., 2005, 230, 1–13 CrossRef CAS; (f) M. Sun, S. Wang, Y. Li, Q. Wang, H. Xu and Y. Chen, J. Taiwan Inst. Chem. Eng., 2017, 78, 401–408 CrossRef CAS; (g) M. Jabłońska, Catal. Commun., 2015, 70, 66–71 CrossRef.
- S. Lin, A. Gluhoi and B. Nieuwenhuys, Catal. Today, 2004, 90, 3–14 CrossRef CAS.
- M. Lippits, A. Gluhoi and B. Nieuwenhuys, Catal. Today, 2008, 137, 446–452 CrossRef CAS.
- H. Suzuki, M. Shimizu, H. Kamiya, T. Ota and M. Takahashi, Adv. Powder Technol., 1997, 8, 311–323 CrossRef.
- B. D. Cullity, Elements of X-RAY DIFFRACTION, Addison-Wesley Publishing Company, Inc., Philippines, 1978 Search PubMed.
- (a) M. Hatanaka, N. Takahashi, T. Tanabe, Y. Nagai, A. Suda and H. Shinjoh, J. Catal., 2009, 266, 182–190 CrossRef CAS; (b) J. Anderson, R. Daley, S. Christou and A. Efstathiou, Appl. Catal., B, 2006, 64, 189–200 CrossRef CAS; (c) Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef; (c) D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS; (d) R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS; (e) TURBOMOLE, A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com; (f) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- (a) F. Giordanino, E. Borfecchia, K. Lomachenko, A. Lazzarini, G. Agostini, E. Gallo, A. Soldatov, P. Beato, S. Bordiga and C. Lamberti, J. Phys. Chem. Lett., 2014, 5, 1552–1559 Search PubMed; (b) U. Deka, A. Juhin, E. Eilertsen, H. Emerich, M. Green, S. Korhonen, B. Weckhuysen and A. Beale, J. Phys. Chem. C, 2012, 116, 4809–4818 CrossRef CAS.
- Z. Tian, Y. Li, L. Zhang, P. Glarborg and F. Qi, Combust. Flame, 2009, 156, 1413–1426 CrossRef CAS.
- (a) S. Yang, Y. Liao, S. Xiong, F. Qi, H. Dang, X. Xiao and J. Li, J. Phys. Chem. C, 2014, 118, 21500–21508 CrossRef CAS; (b) Y. Liu, T. Gu, X. Weng, Y. Wang, Z. Wu and H. Wang, J. Phys. Chem. C, 2012, 116, 16582–16592 CrossRef CAS; (c) L. Darvell, K. Heiskanen, J. Jones, A. Ross, P. Simell and A. Williams, Catal. Today, 2003, 81, 681–692 CrossRef CAS; (d) G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS; (e) J. Amores, V. Escribano, G. Ramis and G. Busca, Appl. Catal., B, 1997, 13, 45–58 CrossRef; (f) G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. Willey, J. Catal., 1995, 157, 523–535 CrossRef CAS; (g) B. Duffy, H. Curryhyde, N. Cant and P. Nelson, J. Catal., 1994, 149, 11–22 CrossRef CAS.
- (a) V. Dasireddy, B. Likozar and J. Valand, Appl. Catal., B, 2018, 237, 1044–1058 CrossRef CAS; (b) S. Lambert, C. Cellier, P. Grange, J. Pirard and B. Heinrichs, J. Catal., 2004, 221, 335–346 CrossRef CAS.
- G. Busca, Catal. Today, 2014, 226, 2–13 CrossRef CAS.
- K. Okada, A. Hattori, T. Taniguchi, A. Nukui and R. Das, J. Am. Ceram. Soc., 2000, 83, 928–932 CrossRef CAS.
- M. Ferrandon, B. Ferrand, E. Bjornbom, F. Klingstedt, A. Neyestanaki, H. Karhu and I. Vayrynen, J. Catal., 2001, 202, 354–366 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation formulae, calculated thermodynamic Pt–PtOx phase equilibrium, XRD patterns, HAADF-STEM/EDX mapping images, product selectivities for the NH3–O2 and NH3–NO–O2 reactions, relationship between NH3 combustion activity and Pt particle size, NH3- and NO-TPD profiles. See DOI: 10.1039/c8ra07969b |
| This journal is © The Royal Society of Chemistry 2018 |