
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen-doped porous carbon sphere supported Pt nanoparticles for methanol and ethanol electro-oxidation in alkaline media
Cuiping Zhai*a,
Shenshen Liab,
Jianhong Wanga and
Yong Liu *b
*b
aHenan Engineering Laboratory of Flame-Retardant and Functional Materials, College of Chemistry and Chemical Engineering, Henan University, Kaifeng, 475004, PR China. E-mail: zhaicuiping@henu.edu.cn
bHenan Key Laboratory of Polyoxometalate Chemistry, Henan University, Kaifeng, 475004, PR China. E-mail: liuyong79@126.com
First published on 25th October 2018
Abstract
A simple method for the preparation of porous nitrogen-doped carbon spheres (MNCS) with honeycomb-like structures through the self-assembly of colloidal silica by using polyaniline as the nitrogen source and a carbon precursor is reported. The BET surface area calculated from N2 adsorption/desorption measurement of the MNCS is up to 882 m2 g−1. The synthesized MNCS were employed as a support for Pt nanoparticles for the oxidation of methanol and ethanol in alkaline media. Compared to Pt supported on commercial Vulcan XC-72R carbon (Pt/C), Pt/MNCS exhibit higher electro-catalytic performance and greater stability, indicating that MNCS have potential application prospects as electro-catalyst supports for alcohol oxidation in alkaline media. This enhancement in performance is due to the honeycomb-like porous structure of the MNCS and the doping with nitrogen.
1. Introduction
Direct alcohol fuel cells (DAFCs) have attracted more and more attention owing to their several benefits like high energy conversion efficiency, high volumetric energy density, easy storage and transport, and low operating temperature.1 Among various kinds of DAFCs, direct methanol fuel cells (DMFCs) and direct ethanol fuel cells (DEFCs) are considered to be the most promising for mobile and portable electronic devices.2–4 In particular, alkaline DAFCs equipped with anion-exchange membranes stimulate greater interest because of the faster reaction kinetics of alcohol oxidation, lower alcohol permeability, and simple water management by comparison with those in acidic DAFCs.5,6 Platinum and platinum-based materials (e.g. Pt–Ru, Pt–Pd, and Pt–Au) have been used as electro-catalysts for alcohol oxidation in alkaline DAFCs because of their high catalytic activities and stabilities.7–11 However, the commercialization of alkaline DAFCs is limited by the high cost and scarcity of platinum. Thus, reducing the use of platinum and increasing the performance of electro-catalysts for alcohol oxidation are still among the greatest challenges in this field. In order to solve these problems, many studies have been carried out with non-Pt catalysts or supported Pt catalysts using excellent support materials.12,13The supports for electro-catalysts for DAFCs can significantly affect the electro-catalytic activity and utilization of noble metals in the catalysts. An ideal electro-catalyst support in DAFCs should possess a high surface area and outstanding conductivity to promote the transport of electrons and reactants. To date, a number of carbon-based supporting materials, such as carbon black, graphene, carbon nanotubes, and mesoporous carbon, have been reported in the relevant literature.14–17 Among the reported carbon-based materials, porous carbon materials as electro-catalyst supports have received great attention because of their large effective surface area and excellent electrical conductivity. These advantages led to their wide application as supports for electro-catalysts in alcohol oxidation and the oxygen reduction reaction.18–20
Recently, nitrogen-doped carbon materials have been used as electro-catalyst supports for alcohol oxidation because of the ability of the nitrogen doping on the carbon support to promote the electro-catalytic activities and stabilities of Pt-based catalysts.21–23 Many nitrogen-doped carbon materials have been prepared by the carbonization and activation of nitrogen-containing precursors, such as polyaniline, polypyrrole, and dopamine.24,25 For example, Liu et al.26 and Mai et al.27 have prepared nitrogen-doped carbon spheres employing dopamine as a carbon and nitrogen precursor and a high-molecular-weight block copolymer as a soft template. Huang et al.28 and Wang et al.29 reported the preparation of mesoporous carbon materials with a combination of polymerization of aniline and co-assembly of colloidal silica nanoparticles.
In this work, using polyaniline as carbon and nitrogen sources, we successfully synthesized porous nitrogen-doped carbon spheres (MNCS) with honeycomb-like structures by the method of self-assembly of colloidal silica. The synthesized MNCS were employed as supports for Pt nanoparticles for methanol and ethanol oxidation in alkaline media. Pt supported on MNCS (Pt/MNCS) electro-catalysts exhibited better electrocatalytic activity compared with Pt supported on commercial Vulcan XC-72R (Pt/C) electro-catalysts.
2. Experimental
2.1. Chemicals
Aniline, ammonium persulfate (APS), and chloroplatinic acid (H2PtCl6·6H2O) were obtained from Aladdin Chemistry Co., Ltd. (Shanghai, China). Silica colloid (Ludox AS-30, 12 nm SiO2 30 wt% suspension in H2O) was purchased from Sigma-Aldrich. Nafion solution (5 wt%) was obtained from DuPont. All chemicals were directly used without any further treatment. Deionized water was used in all the experiments.2.2. Preparation of MNCS and Pt/MNCS electro-catalyst
The MNCS support was prepared through polymerization of aniline using colloidal silica as a template. In the first step, 1 g of aniline monomer, 15 g of colloidal silica, and 20 mL of 1 M HCl solution were mixed. After this mixture had been stirred for 1 h, a solution of 2.26 g of APS in 5 mL of 1 M HCl, was added dropwise under vigorous stirring. The solution was stirred for 8 h in an ice bath. After completion of the reaction, the products were collected by centrifugation, washed three times with deionized water, and dried at 60 °C for 24 h. The obtained products were then carbonized in a tube furnace under flowing N2 gas at 900 °C for 2 h with a ramp of 4 °C min−1. Subsequently, the silica template was removed by etching treatment in 10 wt% HF solution. Finally, the mixtures were centrifuged, and the solid products were collected and washed with deionized water until their supernatants became neutral. After drying, MNCS with honeycomb-like structures were obtained.Pt/MNCS were synthesized using the ethylene glycol (EG) reduction method. First, 0.04 g of MNCS were dispersed into 60 mL of EG under ultrasonic treatment for 30 min to form a uniform slurry. Then, 1.32 mL of H2PtCl6 (10 mg total Pt) solution in EG was added drop by drop into the MNCS slurry under vigorous stirring. After agitating for 1 h, the pH value of the mixture was adjusted to 10 by dropwise addition of 0.5 M NaOH solution in EG. The mixture was heated at 130 °C for 3 h in an oil bath. After completion of the reaction, the products were filtered, and washed three times with deionized water. The obtained Pt/MNCS electro-catalysts were dried for 12 h at 50 °C. For comparison, the commercial Vulcan XC-72R carbon supported Pt catalysts (Pt/C) were also prepared by the same procedure.
2.3. Material characterization
Surface morphologies of the electro-catalysts were obtained by scanning electron microcopy (SEM, JSM-7610F JEOL, Japan) and transmission electron microscopy (TEM, HT7700 HITACHI, Japan). The Brunauer–Emmett–Teller (BET) surface area of the MNCS was examined through a nitrogen adsorption experiment at 77 K by using a Quadrasorb SI (Quantachrome, USA). Powder X-ray diffraction (XRD) analysis was recorded on a Bruker D8 Advance (Bruker AXS, Germany) with a Cu Kα radiation source. X-ray photoelectron spectroscopy (XPS) measurement was carried out with an Escalab 250Xi (Thermo Scientific, USA) to determine the surface properties. Raman spectra were obtained by using an RM-1000 spectrometer (Renishaw Instruments, England) using an Ar ion laser as the excitation source.2.4. Electrochemical measurements
The electrochemical performances of the catalysts were studied using an electrochemical workstation (CHI660D, CH Instruments, China) with a three-electrode cell at room temperature. The standard three-electrode cell was fabricated by modifying a glassy carbon electrode with electro-catalysts for use as the working electrode along with using a Pt wire as the counter electrode, and Ag/AgCl as the reference electrode. The working electrode was prepared as follows: 3.0 mg of Pt/MNCS electro-catalysts were dispersed in 3.0 mL of a mixed solution of isopropanol, deionized water, and Nafion solution (5 wt%) (volume ratio 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:3) under ultrasonic treatment for 30 min to form a uniform catalyst slurry. 35 μL of catalyst slurry was pipetted onto a clean glassy carbon electrode of 3 mm in diameter. The prepared electrode was dried at room temperature. The mass loading of Pt was 0.1 mg cm−2. Following a similar method to the above, the working electrode was also prepared using Pt/C as electro-catalysts. Cyclic voltammograms (CVs) for methanol and ethanol oxidation were recorded within a potential range from −1.0 to 0.4 V (vs. Ag/AgCl) at a scan rate of 50 mV s−1. Chronoamperometry curves for methanol and ethanol oxidation were obtained at −0.20 V. Before all the experiments, the electrolyte solution was saturated with N2 gas for 30 min to expel oxygen out of the solution.
:7:3) under ultrasonic treatment for 30 min to form a uniform catalyst slurry. 35 μL of catalyst slurry was pipetted onto a clean glassy carbon electrode of 3 mm in diameter. The prepared electrode was dried at room temperature. The mass loading of Pt was 0.1 mg cm−2. Following a similar method to the above, the working electrode was also prepared using Pt/C as electro-catalysts. Cyclic voltammograms (CVs) for methanol and ethanol oxidation were recorded within a potential range from −1.0 to 0.4 V (vs. Ag/AgCl) at a scan rate of 50 mV s−1. Chronoamperometry curves for methanol and ethanol oxidation were obtained at −0.20 V. Before all the experiments, the electrolyte solution was saturated with N2 gas for 30 min to expel oxygen out of the solution.
3. Results and discussion
3.1. Characterization of MNCS and Pt/MNCS
The morphology of the MNCS was examined by SEM and TEM. As shown in Fig. 1A and B, the MNCS are obtained as spherical particles with a honeycomb-like structure after heat treatment and HF solution etching of SiO2 nanoparticles. The substantial size of the homogeneous pores could be clearly observed on the surface of the MNCS. The nitrogen adsorption–desorption isotherms for MNCS are shown in Fig. 1C. The isotherms of MNCS displayed type IV isotherm characteristics with hysteresis loops, indicating the existence of mesopores. The pore size distribution is shown in the inset of Fig. 1C. Moreover, the BET surface area of the MNCS was 882 m2 g−1, which is far higher than that of the commercial Vulcan XC-72R carbon (254 m2 g−1). The high surface area of MNCS can provide a lot of active sites for the loading of Pt nanoparticles to increase the catalytic activity for alcohol oxidation in alkaline media. The Raman spectrum was used to further confirm the formation of the carbon structure. As shown in Fig. 1D, the MNCS have two distinctive D and G band scattering peaks at 1334 and 1585 cm−1, respectively. The presence of the G-band indicates the graphitic nature of the MNCS, which is beneficial to an improvement in electrical conductivity.30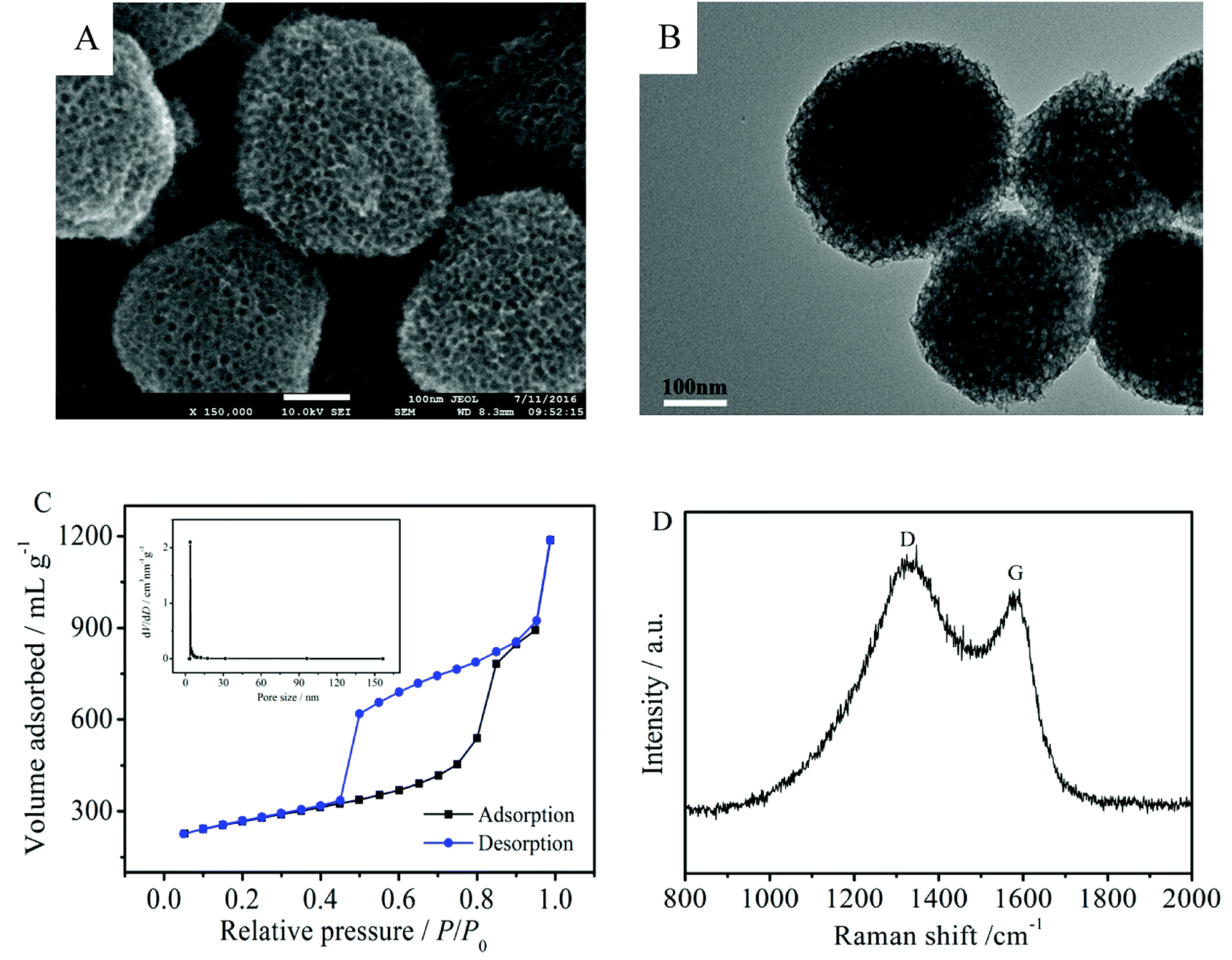 | ||
| Fig. 1 SEM (A) and TEM (B) images of the MNCS. (C) Nitrogen adsorption–desorption isotherms of the MNCS; insert: pore size distribution. (D) Raman spectrum of the MNCS. | ||
The morphology and nanostructural characteristics of Pt/MNCS were examined by TEM. As shown in Fig. 2A, it can clearly be observed that Pt nanoparticles are uniformly deposited on the MNCS support without forming aggregates. The crystalline structures of Pt/MNCS were characterized by XRD. As shown in Fig. 2B, obvious diffraction peaks at 2θ values of 40.2°, 46.8°, 68.0°, 81.8°, and 86.4° are observed, which are indexed to Pt(111), (200), (220), (311), and (222), respectively. These results indicate that Pt formed a face-centered cubic (fcc) crystal structure. A weak and broad peak of the electro-catalyst is observed at 24°, which is the C(002) plane.
 | ||
| Fig. 2 (A) TEM images of Pt/MNCS. (B) XRD patterns of Pt/MNCS. | ||
XPS tests were carried out to probe the surface composition and chemical states of the Pt/MNCS. As can be seen in Fig. 3A, four remarkable peaks were observed at 73.4, 284.9, 400.9, and 531.5 eV, which are assigned to Pt 4f, C 1s, N 1s, and O 1s signals, respectively,31,32 indicating the existence of Pt, C, N, and O elements. In the high-resolution spectrum, O 1s could be separated into three peaks: O–C at 531.1 eV, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 532.4 eV, and OC–O at 533.5 eV, as shown in Fig. 3B. The N 1s spectrum (Fig. 3C) of the Pt/MNCS was deconvoluted into three peaks at 398.5, 401.0 and 404.6 eV, which were assigned to pyridinic N, graphitic N, and pyridine N-oxide.33 As can be seen in Fig. 3D, the Pt 4f spectrum of the Pt/MNCS can be distinguished as two pairs of peaks, which can be attributed to Pt0 and Pt2+, respectively. The ratio of Pt content to that of support is 19.68 wt%, which is consistent with the theoretical value of 20 wt%.
C at 532.4 eV, and OC–O at 533.5 eV, as shown in Fig. 3B. The N 1s spectrum (Fig. 3C) of the Pt/MNCS was deconvoluted into three peaks at 398.5, 401.0 and 404.6 eV, which were assigned to pyridinic N, graphitic N, and pyridine N-oxide.33 As can be seen in Fig. 3D, the Pt 4f spectrum of the Pt/MNCS can be distinguished as two pairs of peaks, which can be attributed to Pt0 and Pt2+, respectively. The ratio of Pt content to that of support is 19.68 wt%, which is consistent with the theoretical value of 20 wt%.
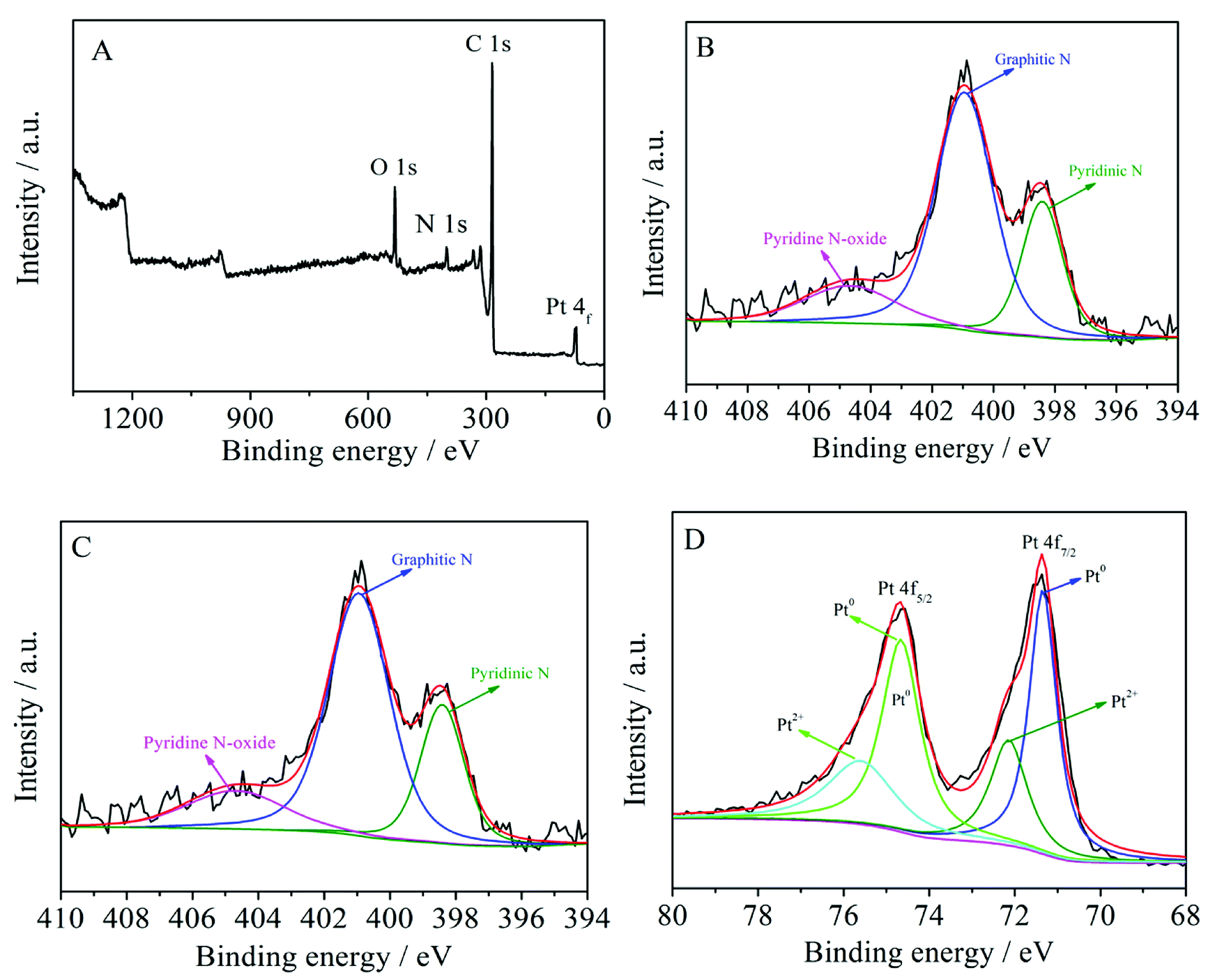 | ||
| Fig. 3 (A) XPS survey spectrum of Pt/MNCS; the high-resolution XPS spectra of O 1s (B), N 1s (C), and Pt 4f (D). | ||
3.2. Electrochemical activity
Cyclic voltammetry (CV) measurements of Pt/MNCS and Pt/C were carried out in 1.0 M NaOH solution from −1.0 V to 0.4 V (vs. Ag/AgCl) at a scan rate of 50 mV s−1, as can be seen in Fig. 4. The electrochemically active surface area (ECSA) can be calculated by measuring the charge with hydrogen desorption curves (QH) according to the following equation:34where [Pt] represents the loading of Pt (mg cm−2) on the electrode and QH (mC cm−2) represents the charge exchanged during the desorption of hydrogen on Pt. The value 0.21 (mC cm−2) is the assumed monolayer charge from a surface density of 1.3 × 1015 atoms cm−2. The ECSA of the synthetized Pt/MNCS electro-catalysts was 96 m2 g−1, which is higher than that of the Pt/C electro-catalysts (38 m2 g−1). Compared to Pt/C electro-catalysts, the larger ECSA of Pt/MNCS may be due to the higher surface area and more porous structure of the MNCS support, which improves the distribution of Pt nanoparticles on the MNCS.
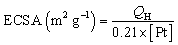
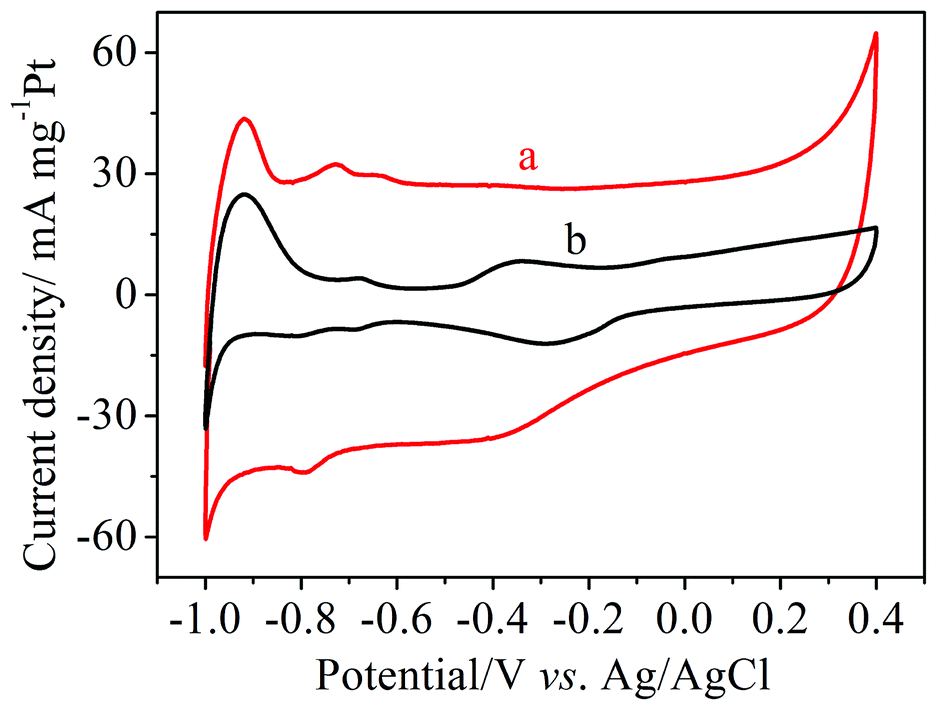 | ||
| Fig. 4 CV curves of different electro-catalyst modified electrodes measured in 1.0 M NaOH at a scan rate of 50 mV s−1: (a) Pt/MNCS, (b) Pt/C. | ||
The catalytic activity of the electro-catalysts was studied by cyclic voltammetry in a 1.0 M NaOH solution containing 1.0 M methanol or ethanol at a scan rate of 50 mV s−1. As can be seen in Fig. 5, the typical characteristics of alcohol oxidation on the Pt/MNCS and Pt/C catalysts were observed from the CV curves. The peaks in the forward scan correspond to the oxidation of alcohol on the catalyst, while those in the backward scan are due to the oxidation of intermediate products produced from the forward scan.35 It can clearly be observed that Pt/MNCS exhibits a higher electro-catalytic activity than Pt/C for the oxidation of both methanol and ethanol. The electrochemical performance parameters of different catalysts were compared and are summarized in Table 1. For methanol oxidation (Fig. 5A), the onset potential on Pt/MNCS (−0.56 V) shifts to a more negative value compared to that on Pt/C (−0.50 V) while the forward peak current density on Pt/MNCS (799 mA mg−1 Pt) is about twice that of Pt/C (387 mA mg−1 Pt). As shown in Fig. 5B, the onset potential of ethanol oxidation is found to be −0.61 V on Pt/MNCS, which is 40 mV more negative than that on Pt/C. From Table 1, compared with some similar electro-catalysts which have been reported in the literature, Pt/MNCS also have good electro-catalytic activity. These results suggest that the enhancement in alcohol oxidation on Pt/MNCS may be due to the honeycomb-like porous structure and nitrogen-doping of the support. MNCS have a higher surface area and more porous structure compared with the commercial support Vulcan XC-72R, which can improve the distribution of Pt nanoparticles on the MNCS and promote catalytic activity. In addition, nitrogen-doping may cause some changes in Pt nucleation, growth behavior, electronic structure, and support/catalyst binding, which mean that more uniform, highly dispersed, and higher activity Pt nanoparticles can be generated.39 These can also improve the alcohol oxidation in alkaline media.
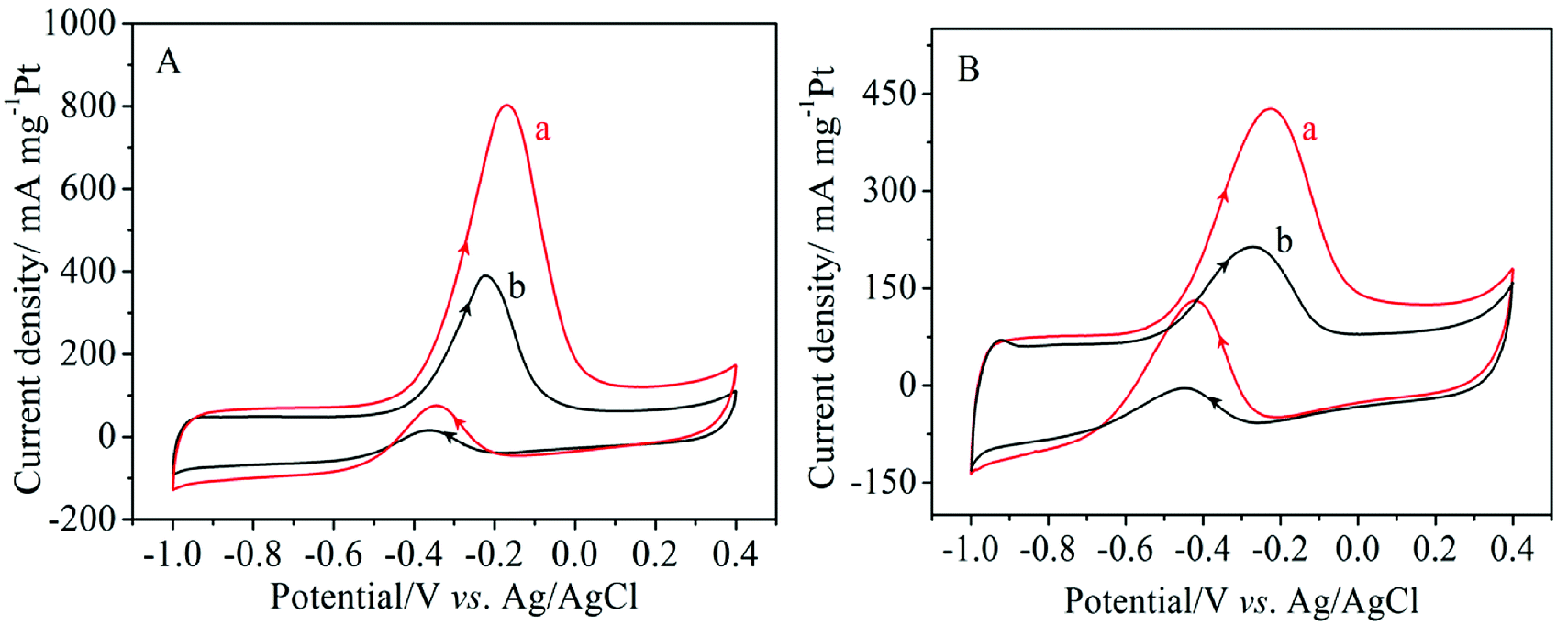 | ||
| Fig. 5 Cyclic voltammograms of different catalysts in 1.0 M NaOH + 1.0 M CH3OH (A) and 1.0 M NaOH + 1.0 M C2H5OH (B) at a scanning rate of 50 mV s−1: (a) Pt/MNCS, (b) Pt/C. | ||
| Catalyst | Methanol oxidation | Ethanol oxidation | Ref. | ||
|---|---|---|---|---|---|
| Onset potentials (V) | Current density (mA mg−1 Pt) | Onset potentials (V) | Current density (mA mg−1 Pt) | ||
| Pt/MNCS | −0.56 | 799 | −0.61 | 426 | This work |
| Pt/C | −0.50 | 387 | −0.57 | 213 | This work |
| Pt/CMS | −0.51 | 610 | −0.51 | 120 | 8 |
| Pt/C + PpPDA/TiO2 | −0.42 | 191 | −052 | 389 | 37 |
| Pt/TiO2/G | — | 272.4 | — | 207.7 | 38 |
The electro-catalytic stability of Pt/MNCS and Pt/C catalysts for methanol and ethanol oxidation were also investigated using chronoamperometric curves at a constant potential of −0.20 V for 1800 s. As shown in Fig. 6A, the current densities of the two catalysts decayed rapidly in the initial stage because the electrode was poisoned by reaction intermediates during the methanol oxidation.36 Then the current density gradually decayed, and a pseudo-steady state was obtained. The Pt/MNCS exhibits a higher initial current and a much slower current decay than the Pt/C catalyst. At the end of the 1800 s, the oxidation current density on the Pt/MNCS catalyst is higher than that on the Pt/C catalyst. For ethanol oxidation, the current evolution on the different catalysts shows a similar trend to the oxidation of methanol. As can be seen in Fig. 6B, the current density decay rate on Pt/C is higher than that on Pt/MNCS. From these results, we can see that the Pt/MNCS catalysts have good stability for both methanol and ethanol oxidation in alkaline media.
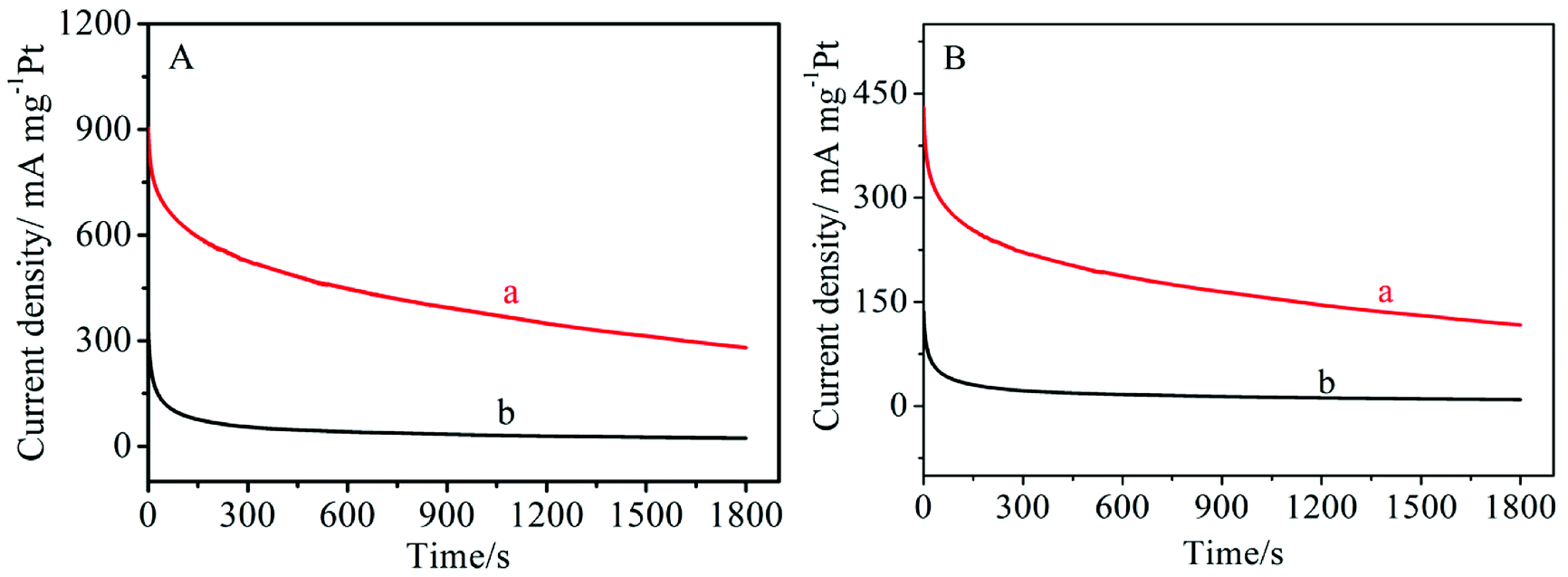 | ||
| Fig. 6 Chronoamperometric curves for alcohol oxidation on different modified electrodes in 1.0 M NaOH with 1.0 M methanol (A) and 1.0 M NaOH with 1.0 M ethanol at −0.20 V (vs. Ag/AgCl): (a) Pt/MNCS, (b) Pt/C. | ||
4. Conclusions
In this work, honeycomb-like porous nitrogen-doped carbon spheres (MNCS) were successfully prepared through the self-assembly of colloidal silica, using polyaniline as a carbon and nitrogen precursor. The obtained MNCS exhibit an extremely high surface area of 882 m2 g−1, as confirmed through N2 adsorption/desorption measurement. The high surface area and porous structure are beneficial for the uniform dispersion of Pt nanoparticles. Pt nanoparticles supported on MNCS (Pt/MNCS) were used as electro-catalysts for methanol and ethanol oxidation in alkaline media. The Pt/MNCS catalysts show higher electro-catalytic activity and more stability than Pt/C catalysts. This enhancement in performance may be due to the porous structure, high surface area, and nitrogen doping of the MNCS.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are thankful to the Natural Science Foundation of Henan Province (182300410244), and Foundation for University Youth Key Teachers from Henan Province (No. 2017GGJS018).References
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J. M. Leger, J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- J. N. Tiwari, R. N. Tiwari, G. Singh and K. S. Kim, Nano Energy, 2013, 2, 553–578 CrossRef CAS.
- S. Basri, S. K. Kamarudin, W. R. W. Daud and Z. Yaakub, Int. J. Hydrogen Energy, 2010, 35, 7957–7970 CrossRef CAS.
- M. Z. F. Kamarudin, S. K. Kamarudin, M. S. Masdar and W. R. W. Daud, Int. J. Hydrogen Energy, 2013, 38, 9438–9453 CrossRef CAS.
- S. S. Gupta and J. Datta, J. Power Sources, 2005, 145, 124–132 CrossRef.
- Y. Xiong, Q. L. Liu, A. M. Zhu, S. M. Huang and Q. H. Zeng, J. Power Sources, 2009, 186, 328–333 CrossRef CAS.
- Y. Liu, H. Yang, X. Y. Li and L. Q. Mao, Mater. Lett., 2013, 106, 287–289 CrossRef CAS.
- C. W. Xu, L. Q. Cheng, P. K. Shen and Y. L. Liu, Electrochem. Commun., 2007, 9, 997–1001 CrossRef CAS.
- F. F. Ren, C. Y. Zhai, M. S. Zhu, C. Q. Wang, H. W. Wang, D. Bin, J. Guo, P. Yang and Y. K. Du, Electrochim. Acta, 2015, 153, 175–183 CrossRef CAS.
- G. Zhang, C. Huang, R. Qin, Z. Shao, D. An, W. Zhang and Y. Wang, J. Mater. Chem. A, 2015, 3, 5204–5211 RSC.
- Y. Liu, S. S. Li, Y. M. Zhang, W. H. Liu, J. H. Wang and C. P. Zhai, J. Solid State Electrochem., 2018, 22, 817–824 CrossRef CAS.
- P. K. Shen, Z. Yan, H. Meng, M. Wu, G. Cui, R. Wang, L. Wang, K. Si and H. Fu, RSC Adv., 2011, 1, 191–198 RSC.
- Y. H. Qin, Y. Zhang, R. L. Ren, T. L. Wang, W. G. Wang and C. W. Wang, Electrochim. Acta, 2015, 154, 77–82 CrossRef CAS.
- Y. Li, L. Q. Zhang, A. R. Liu, Y. J. Zhang and S. Q. Liu, J. Electroanal. Chem., 2014, 730, 65–68 CrossRef CAS.
- M. Liu, C. Peng, W. Yang, J. Guo, Y. Zheng, P. Chen, T. Huang and J. Xu, Electrochim. Acta, 2015, 178, 838–846 CrossRef CAS.
- P. Song, L. Zhu, X. Bo, A. Wang, G. Wang and L. Guo, Electrochim. Acta, 2014, 127, 307–314 CrossRef CAS.
- Y. H. Qin, Z. Y. Xiong, J. Y. Ma, Y. Li, Z. K. Wu, W. L. Feng, T. L. Wang, W. G. Wang and C. W. Wang, Int. J. Hydrogen Energy, 2017, 42, 1103–1112 CrossRef CAS.
- H. W. Liang, X. Zhuang, S. Bruller, X. Feng and K. Mullen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- Y. Liu, Y. M. Zhang, C. P. Zhai, X. Y. Li and L. Q. Mao, Mater. Lett., 2016, 166, 16–18 CrossRef CAS.
- A. Navaee, A. Salimi, S. Soltanian and P. Servati, J. Power Sources, 2015, 277, 268–276 CrossRef CAS.
- S. J. Jiang, L. Zhu, Y. W. Ma, X. Z. Wang, J. G. Liu, J. M. Zhu, Y. N. Fan, Z. G. Zou and Z. Hu, J. Power Sources, 2010, 195, 7578–7582 CrossRef CAS.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- L. M. Zhang, Z. B. Wang, J. J. Zhang, X. L. Sui, L. Zhao and D. M. Gu, Carbon, 2015, 9, 1050–1058 CrossRef.
- M. Sevilla, L. Yu, T. Fellinger, A. Fuertes and M. M. Titirici, RSC Adv., 2013, 3, 9904–9910 RSC.
- Y. M. Zhang, Y. Liu, W. H. Liu, X. Y. Li and L. Q. Mao, Appl. Surf. Sci., 2017, 407, 64–71 CrossRef CAS.
- J. Tang, J. Liu, C. Li, Y. Li, M. O. Tade, S. Dai and Y. Yamauchi, Angew. Chem., Int. Ed., 2015, 54, 588–593 CAS.
- H. Tian, Z. Lin, F. Xu, J. Zheng, X. Zhuang, Y. Mai and X. Feng, Small, 2016, 12, 3155–3163 CrossRef CAS PubMed.
- X. X. Huang, L. J. Zhou, D. Voiry, M. Chhowalla, X. X. Zou and T. Asefa, ACS Appl. Mater. Interfaces, 2016, 8, 18891–18903 CrossRef CAS PubMed.
- G. Wang, Y. Sun, D. Li, H. W. Liang, R. Dong, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2015, 54, 15191–15196 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- X. Zhang, J. Zhu, C. S. Tiwary, Z. Ma, H. Huang, J. Zhang, Z. Lu, W. Huang and Y. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 10858–10865 CrossRef CAS PubMed.
- K. G. Nishanth, P. Sridhar and S. Pitchumani, Int. J. Hydrogen Energy, 2013, 38, 612–619 CrossRef CAS.
- C. Liu, J. Wang, J. S. Li, R. Luo, J. Y. Shen, X. Y. Sun, W. Q. Han and L. J. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 18609–18617 CrossRef CAS PubMed.
- S. Wu, J. Liu, Z. Tian, Y. Cai, Y. Ye, Q. Yuan and C. Liang, ACS Appl. Mater. Interfaces, 2015, 7, 22935–22940 CrossRef CAS PubMed.
- H. L. Liao, Z. P. Qiu, Q. J. Wan, Z. J. Wang, Y. Liu and N. J. Yang, ACS Appl. Mater. Interfaces, 2014, 7, 18055–18062 CrossRef PubMed.
- X. Chen, Z. Cai, X. Chen and M. Oyama, J. Mater. Chem. A, 2014, 2, 315–320 RSC.
- H. Rostami, A. A. Rostami and A. Omrani, Electrochim. Acta, 2016, 191, 536–547 CrossRef CAS.
- H. Hua, C. G. Hu, Z. H. Zhao, H. Liu, X. Xie and Y. Xi, Electrochim. Acta, 2013, 105, 130–136 CrossRef CAS.
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Energy Environ. Sci., 2010, 3, 1437–1446 RSC.
| This journal is © The Royal Society of Chemistry 2018 |