
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient one-pot conversion of carboxylic acids into benzimidazoles via an HBTU-promoted methodology†
Leonard Barasa and
Sabesan Yoganathan *
*
Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John's University, Queens, NY 11439, USA. E-mail: yoganats@stjohns.edu; Tel: +1-718-990-5215
First published on 19th October 2018
Abstract
Benzimidazole is a privileged, and routinely used pharmacophore in the drug discovery process. Herein, we report a mild, acid-free and one-pot synthesis of indole, alkyl and alpha-amino benzimidazoles through a novel HBTU-promoted methodology. An extensive library of indole-carboxylic acids, alkyl carboxylic acids and N-protected alpha-amino acids has been converted into the corresponding benzimidazoles in 80–99% yield. Since alpha-aminobenzimidazoles are highly useful synthons as chiral ligands for chemical catalysis, as well as for drug discovery endeavors, our reported method provides direct access to this scaffold in a simple, one-pot operation from commercially available carboxylic acids.
Introduction
Heterocyclic structures have been extensively utilized during the process of drug development.1–3 The presence of heterocycles modulates physicochemical properties and the pKa profile of therapeutic leads. Additionally, nitrogen substitution enables a useful functional handle for further derivatization. In this vein, the benzimidazole core (1, Fig. 1) has become a highly sought after and privileged pharmacophore in drug discovery.4–8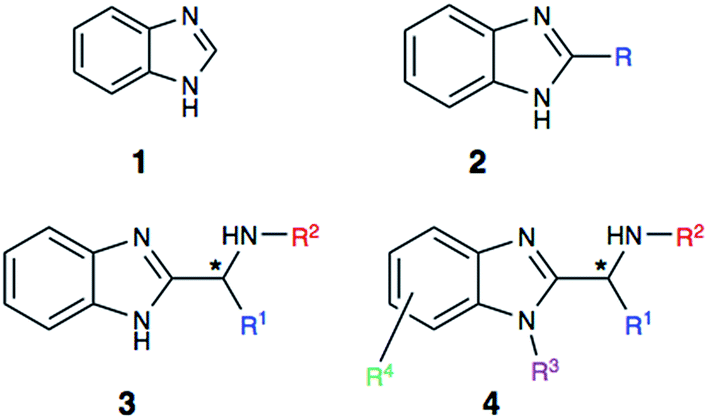 | ||
| Fig. 1 Structures of benzimidazole core (1), and benzimidazole derivatives (2–4). | ||
Benzimidazole's structural similarity to purine makes it a key structural motif in drug design.6 Hence, this important pharmacophore is commonly encountered in drugs used for the treatment of cancer, infectious diseases, hypertension and other illnesses (Fig. 2). Benzimidazole is also actively used in drug leads that exhibit a broad range of pharmacological activities, including anticancer, antibacterial, and anti-inflammatory activities.4–8
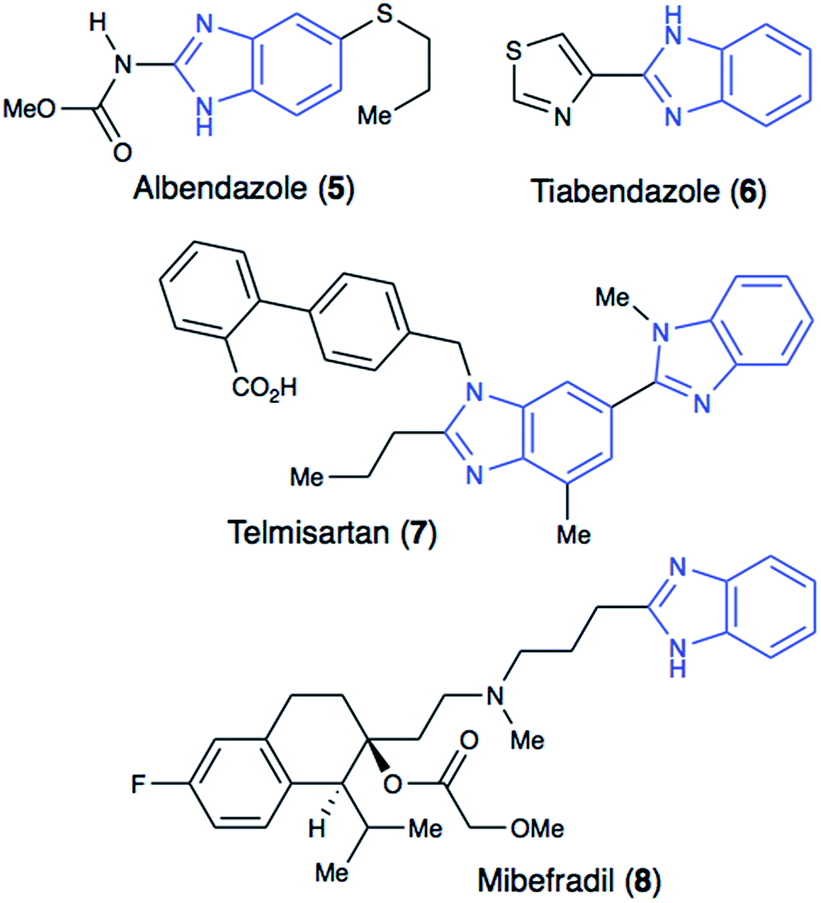 | ||
| Fig. 2 Structures of benzimidazole-containing clinically used drugs. | ||
Due to the high prevalence of benzimidazoles within medicinal organic molecules, there has been a considerable interest in developing efficient approaches for their synthesis.9–14 One of the common synthetic approaches employed to access benzimidazoles involves condensation–dehydration sequence of o-aryldiamine with an aryl-aldehyde substrate under mild conditions. A second approach involves reaction of o-aryldiamine with carboxylic acid derivatives under forcing conditions, in the presence of a mineral acid or acetic acid, and under refluxing temperatures. Other recent methods for the preparation of benzimidazole derivatives include several examples of transition metal-catalysed C–N coupling of N-(2-haloary)amidines with 1,2-phenylenediamines, and the intramolecular oxidative C–N couplings of arylamidines with N-substituted 1,2-phenylenediamines via the TEMPO-air promoted oxidative coupling.12–14
A most commonly used (Phillip's method)9d method involves the condensation of o-aryldiamine with carboxylic acids or its derivatives, including heating the reagents together in the presence of aqueous hydrochloric acid (Scheme 1). Due to the harsh nature of reaction condition, substrate scope for this method is very limited; as sensitive functional groups are less likely to survive such harsh conditions. Additionally, limited availability of substrates for other available methods often impedes their application during medicinal chemistry efforts.
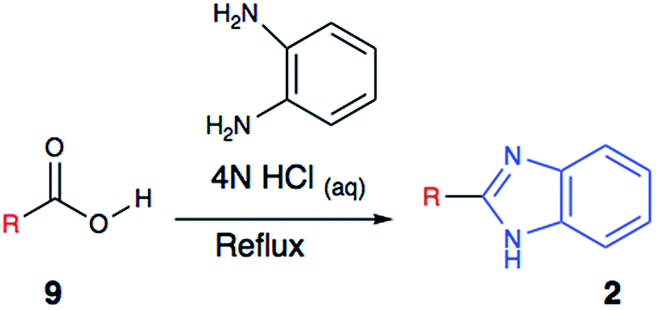 | ||
| Scheme 1 A most commonly used synthetic method of benzimidazoles (Phillip's Method). | ||
In recent years, alpha-aminobenzimidazoles (3 and 4) have emerged as potent drug leads for infections, cancer and autoimmune diseases,15–17 as well as metal-binding motifs.5 Due to the limited synthetic utility of existing methods, we envisioned developing a mild, functional group tolerant method for accessing a diverse class of benzimidazole synthons (Scheme 2). Such a methodology would greatly benefit our research group and others who are interested in developing benzimidazole-containing peptides as drug leads,16 and chiral benzimidazoles as ligands. The reported study explores a simple, yet reliable approach to access a structurally diverse library of benzimidazoles that includes amino acid derived alpha-aminobenzimidazoles, alkyl benzimidazoles and indole benzimidazoles.
 | ||
| Scheme 2 Proposed one-pot, two-step synthesis of Boc-protected amino acid derived benzimidazole (12). | ||
Result and discussion
We began our exploration by synthesizing the ‘amide’ intermediate via a standard O-(benzotriazole-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) assisted coupling approach. Once we have the amide intermediate, we isolated it and proceeded to screen mild conditions to perform a dehydrative cyclization of the amide into the benzimidazoles. Our initial attempts to perform the dehydrative cyclization under a mild basic or acidic condition (Table 1, entry 2–4) failed to yield the desired product. We then realized that carbodiimides are known for their ability to promote oxidation or dehydration.20,21
|
||
|---|---|---|
| Entry | Reagent | Yield |
| 1 | No additives | No reaction |
| 2 | DIPEA | No reaction |
| 3 | DBU | No reaction |
| 4 | HCl | No reaction |
| 5 | DIC | No reaction |
| 6 | EDCI | No reaction |
| 7 | HBTU | 94% |
| 8 | HCTU | 5% |
| 9 | PyBOP | 94% |
| 10 | DIPEA/HCl | No reaction |
| 11 | Tetramethylurea | No reaction |
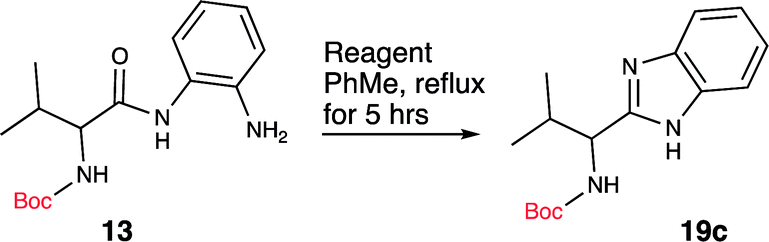
Based on this knowledge, we selected several commonly used carbodiimide-based coupling agents, including N,N′-diisopropylcarbodiimide (DIC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) and HBTU to study the dehydrative cyclization (Table 1, entry 5–9). For this investigation, Boc-valine derived amide (13) was used as a model substrate. We discovered that under an optimized condition, HBTU (1 equiv.) yielded the best conversion of the amide 13 into the corresponding benzimidazole 19c (Table 1, entry 7). Additionally, we found that catalytic amount of HBTU (0.3 equiv.) is ineffective in providing the desired product in high yield. Although DIC, EDCI and HCTU are useful carbodiimide agents for amide formation, these agents did not afford the desired product. This is perhaps due to the reduced reactivity of these coupling agents. Finally, we also found that (benzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyBOP), a more reactive coupling agent compared to DIC and EDCI, indeed exhibited desirable reactivity towards the dehydrative cyclization. Phosphonium-based coupling agents are useful activating reagents for amide formation and cyclization of thioureas.22
Since the amide precursor is synthesized via an HBTU activated process, we attempted a one-pot strategy to form the amide and a subsequent benzimidazole formation by using 2 equivalents of HBTU. This approach worked remarkable well as a one-pot process. Initial formation of aryl-amide occurred with high efficiency within 4 hours at room temperature, and then a one-pot HBTU promoted cyclization under refluxing temperature yielded the desired product in less than 3 hours. The two-step, one-pot synthesis worked extremely well in PhMe, yielding 19c in 96% isolated yield (Scheme 3). To further confirm the role of HBTU in this process, we performed a series of control reactions to rule out the role of other components present in the reaction mixture from the amidation step. We observed that none of the components in the reaction mixture, which are part of the amidation reaction or byproducts formed during the amide formation promoted the conversion of amide into the benzimidazole (Table 1, entries 10 and 11). These observations led us to conclude that HBTU is an effective promoter of benzimidazole synthesis, and it is presumably behaving like an activating agent.
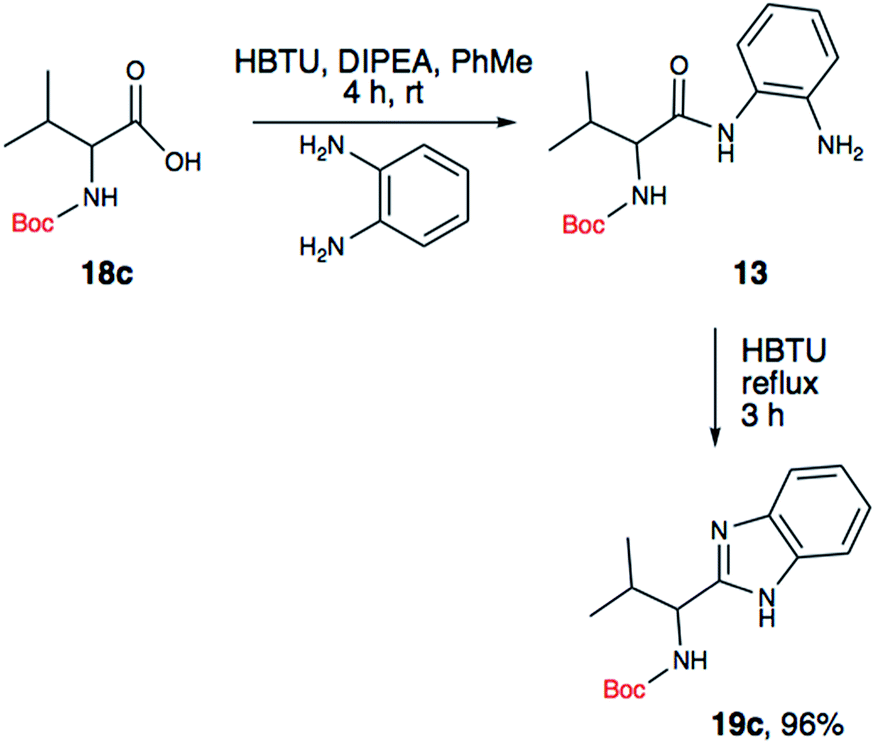 | ||
| Scheme 3 One pot synthesis of Boc-valine derived benzimidazole. | ||
To investigate the versatility of solvents, we performed the benzimidazole synthesis in 1,4-dioxane, DMF or PhMe. The coupling was performed in one of these solvents, and subsequently the crude reaction mixture was subjected to cyclization under refluxing temperature in the same solvent. Based on the high yield obtained, all three solvents were suitable for this operation, providing great flexibility with the choice of solvents. Since solubility of substrates (i.e. peptides and indoles) in toluene tends to be a limitation for synthesis, we have demonstrated that either DMF or 1,4-dioxane can be employed to overcome this limitation.
In comparison to reported methods for benzimidazole synthesis, our approach highlights several important improvements and advantages. First, the benzimidazole synthesis is a one pot process and high yielding, where current methods require isolation of the aryl-amide prior to dehydrative cyclization. Second, there is no need to perform the cyclization in the presence of an acid as a co-solvent, which greatly broadens the substrate scope and the functional group tolerability, including various protecting groups found in amino acids and peptides. Third, the reaction works extremely well in three different solvents enabling synthetic flexibility for substrates with limited solubility.
We then investigated the substrate scope using a small library of commercially available carboxylic acids (Scheme 4). Boc-Asp-OMe (14b) was successfully converted to the beta-benzimidazole derivative (15b) in 92% yield, providing a unique non-protein amino acid that is useful for medicinal chemistry. Additionally, four different aliphatic carboxylic acids (14c–14f) were converted into the corresponding benzimidazoles in high yield. As peptide substrates are of prime interest to others and us in the field, a Cbz-protected dipeptide (14g) was successfully transformed into the C-terminal benzimidazole derivative (15g) in good yield as well. This demonstrates the utility of reported method for the synthesis of peptide-based benzimidazoles for drug discovery.
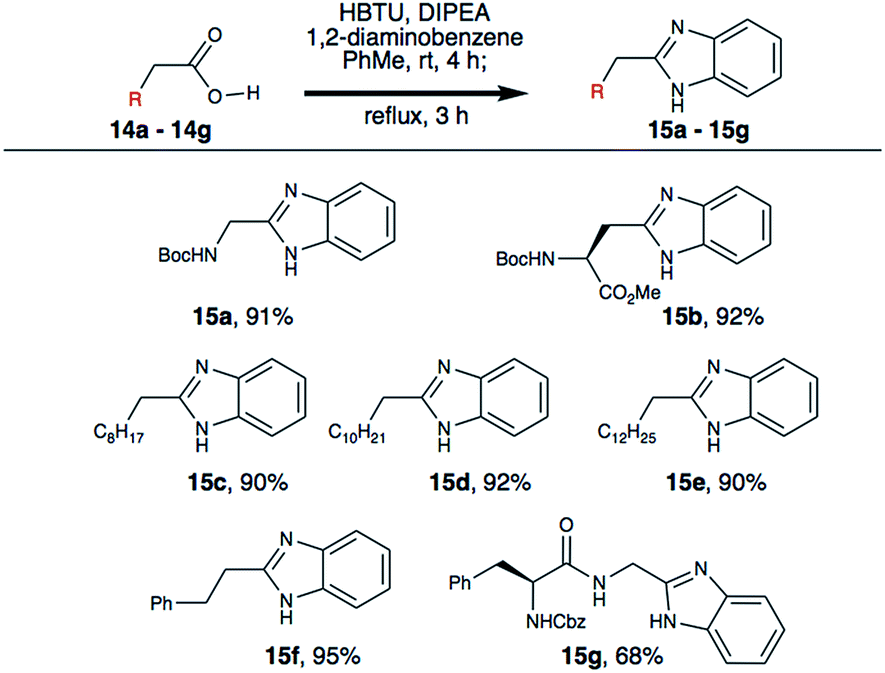 | ||
| Scheme 4 Synthesis of aliphatic benzimidazoles. | ||
During our investigation, we realized that indole-2-carboxylic acid is a privileged substrate and the corresponding benzimidzole is widely used in drug discovery efforts.18,19 We have successfully synthesized various indole-2-benzimidazoles (17a–17f), where the aryl ring of the benzimidazole is substituted with different functional groups (Scheme 5). We envisioned that having halogen substitution on the aryl-ring provides a useful chemical handle for further structure diversification via Pd-catalysed cross-coupling reactions. We also noticed that electron rich 1,2-diaminobenzene derivatives yielded better yield than those that are electron deficient. This may be due to change in nucleophilicity of the diamine.
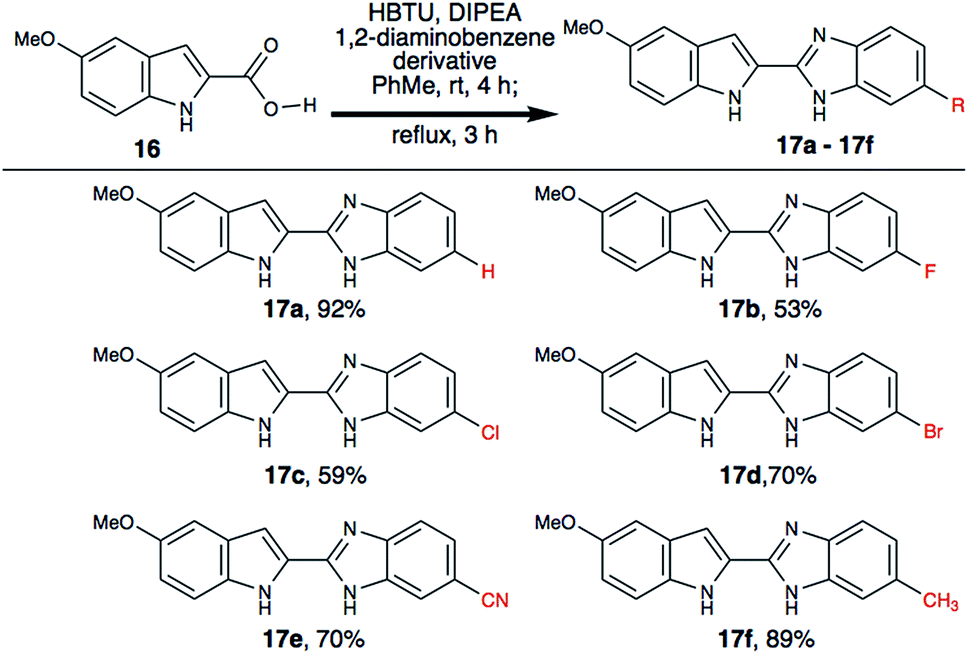 | ||
| Scheme 5 Synthesis of indole-2-carboxylic acid derived benzimidazoles. | ||
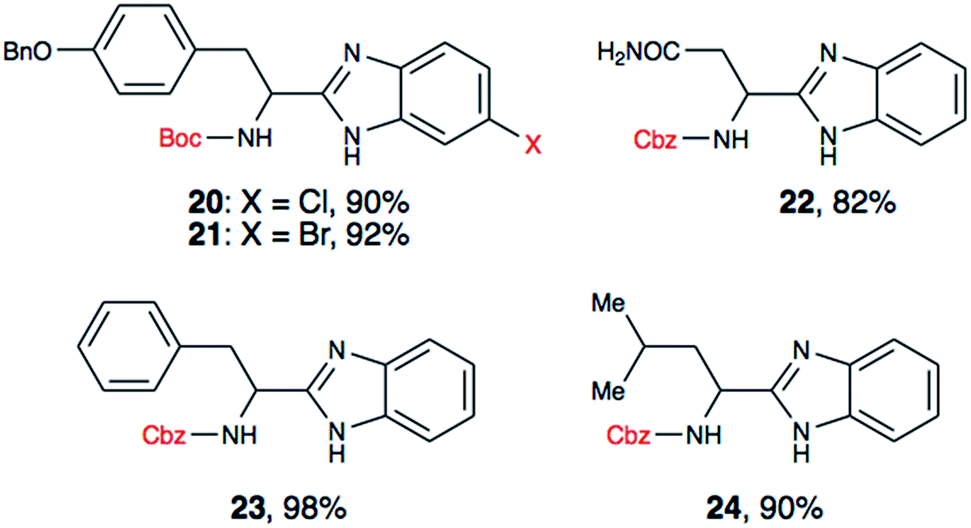
To further validate our method, we proceeded to synthesize an extensive library of alpha-amino acid derived benzimidazoles. There are two reasons for this endeavor: (i) we wanted to access a structurally diverse collection of alpha-amino benzimidazoles from commercially available amino acids with suitable protecting groups; and (ii) we envisioned accessing alpha-amino acids precursors for the synthesis of peptide-based benzimidazoles. This library includes thirteen Boc-protected amino acids (18a–18n, Table 2) and three Cbz-protected amino acids (22–24, Fig. 3). We reacted all Boc-amino acids with 1,2-diaminobenzene under optimized, one-pot reaction condition, and isolated the desired benzimidazoles (19a–19n) in excellent yield. We also learned that many side chains protecting groups, including benzyl ether, benzyl thioether, and benzyl esters are stable to the reaction condition. Additionally, the side chain of Boc-Asn-OH (18l) required no protecting group to generate the Boc-Asn derived benzimidazole 19l. It is also interesting to note that compound 19d is structurally similar to veliparib, a poly(ADP-ribose)polymerase (PARP) inhibitor that is in clinical trials.23
|
|||
|---|---|---|---|
| Entry | Amino acid | Product | Yield |
| 1 | Boc-Leu-OH (18a) | 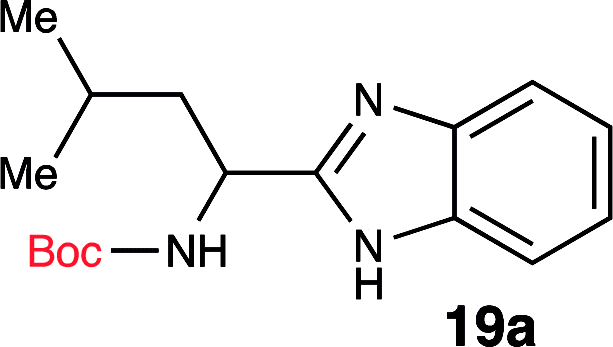 |
96% |
| 2 | Boc-Ileu-OH (18b) | 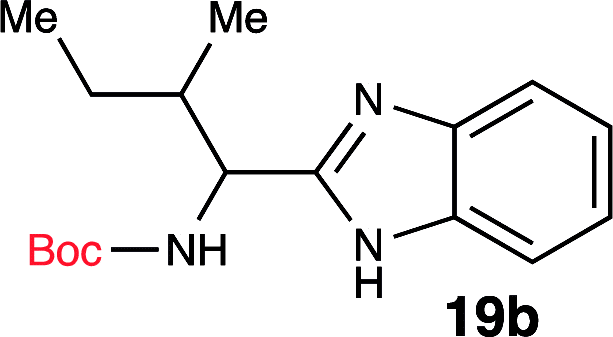 |
95% |
| 3 | Boc-Val-OH (18c) | 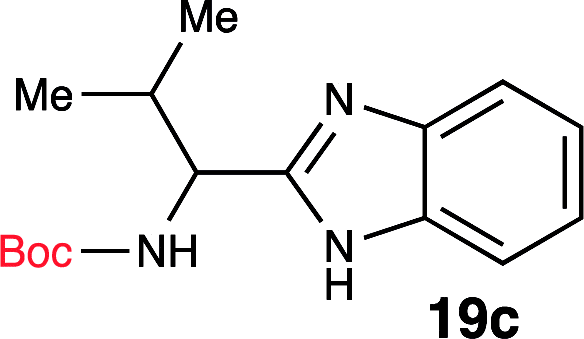 |
96% |
| 4 | Boc-Pro-OH (18d) | 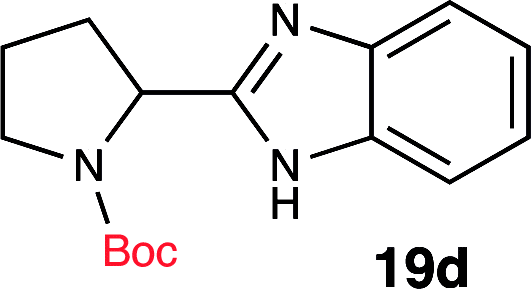 |
99% |
| 5 | Boc-Tyr(OBn)-OH (18e) | 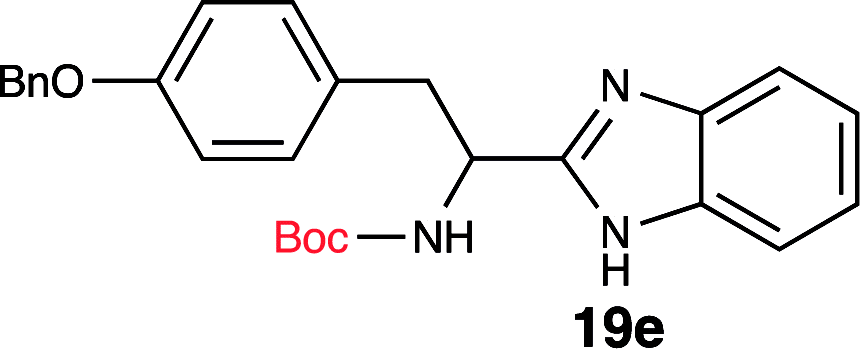 |
84% |
| 6 | Boc-Ser(OBn)-OH (18f) | 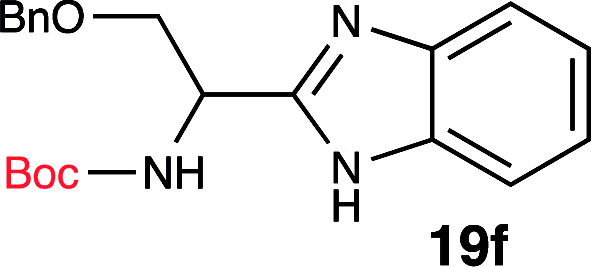 |
84% |
| 7 | Boc-Cys(SBn)-OH (18g) | 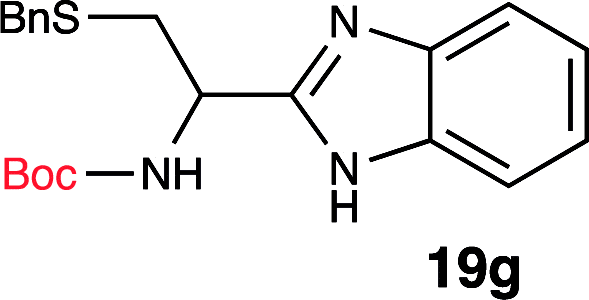 |
99% |
| 8 | Boc-Thr(OBn)-OH (18h) | 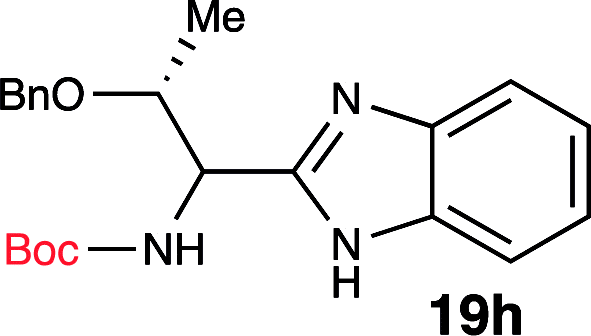 |
94% |
| 9 | Boc-Lys(Boc)-OH (18i) | 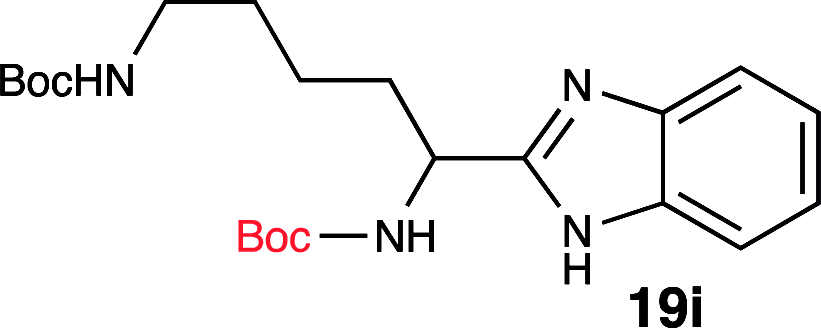 |
99% |
| 10 | Boc-Asp(OBn)-OH (18j) |  |
90% |
| 11 | Boc-Glu(OBn)-OH (18k) | 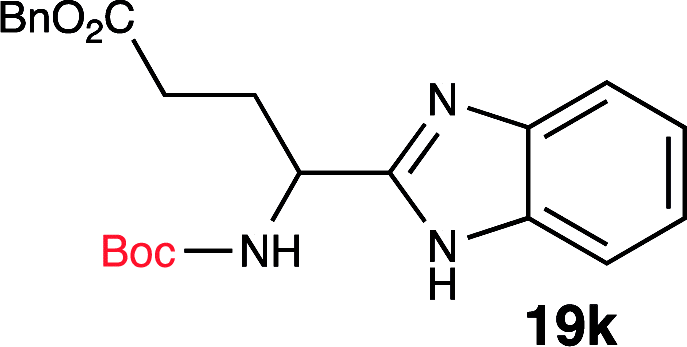 |
80% |
| 12 | Boc-Asn-OH (18l) | 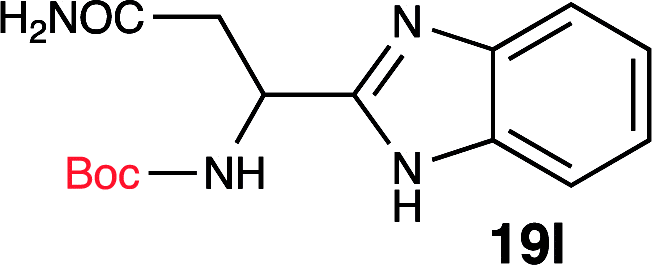 |
96% |
| 13 | Boc-Gln-OH (18m) | 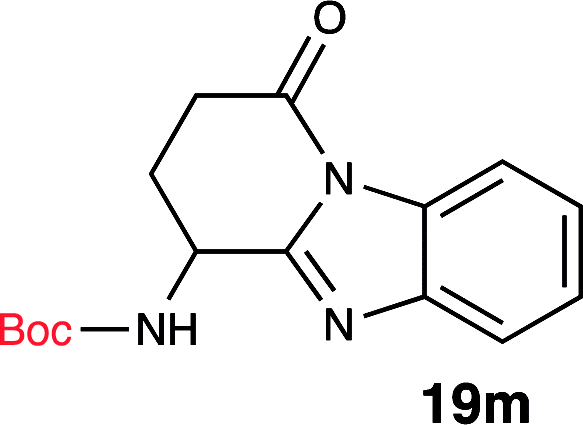 |
96% |
| 14 | Boc-Phe-OH (18n) | 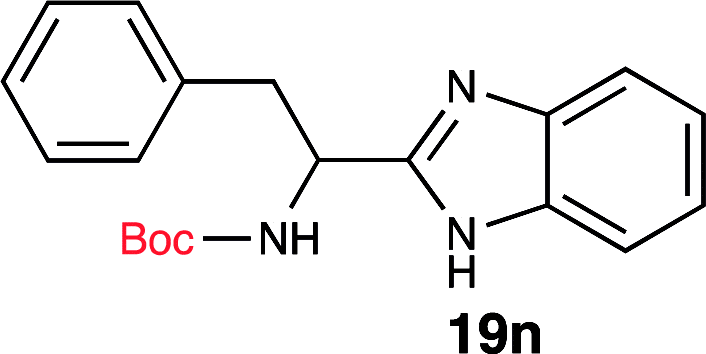 |
99% |

 | ||
| Fig. 3 Structures of halogenated and N-Cbz protected amino acid based benzimidazoles. | ||
One interesting observation was made when Boc-Gln-OH (18m) was reacted to form the corresponding benzimidazole. Absence of protecting group on the side chain yielded an interesting tricyclic structure (19m). Based on literature precedent,24 we propose that the side chain amide underwent a transamidation reaction with the benzimidazole nitrogen, generating the unique tricyclic product 19m. Since compound 19m has an amine handle, and a conformationally distinct tricyclic structure, it could be a useful synthon for medicinal chemistry efforts.
During the synthesis of various alpha-aminobenzimidazoles (Table 2), we investigated the extent of epimerization of the alpha-chiral centre. Current synthetic methods limit the access to amino acid derived benzimidazoles in high enantiopurity and high yield. Although our approach is superior in terms of mild reaction condition and high yield, we were unable to obtain these benzimidazoles in high enantiopurity. Our initial analysis showed that the amino acid derived products are obtained as a racemic mixture or with poor ee. We believe that the basic components (i.e. DIPEA, 1,2-diaminobenzene) used during coupling condition are perhaps epimerizing the relatively acidic alpha-proton of the benzimidazoles. Alternatively, the epimerization may be occurring during the dehydrative cyclization process where the pKa of the alpha-proton is significantly lowered. Our initial efforts to overcome this limitation by replacing the base used to a milder base (i.e. N-methylmorpholine or pyridine) failed to improve the enantiopurity of benzimidazoles. We are continuing to investigate various conditions to overcome the problem of epimerization.
We have also utilized the reported method for the synthesis of two halogenated analogues of Boc-tyrosine derived benzimidazoles (20 and 21), and three N-Cbz protected amino acid derived benzimidazoles (22–24). Using these substrates, we demonstrated that both Boc and Cbz carbamates are tolerated. Additionally, having a halogen handle on the aryl ring of benzimidazole provides a new venue to diversify the benzimidazole core for medicinal chemistry purposes.
In addition to being an effective coupling agent, we believe that HBTU (26) is playing a very important role in the cyclization process. Herein, we propose a plausible mechanistic pathway, which may explain the HBTU-promoted formation of the benzimidazole (Scheme 6). The intermediate aryl-amide (25) is relatively stable, and for it to undergo dehydration, it needs to be activated. We propose that HBTU (26) helps in the activation of amide, where the oxygen atom of the amide reacts with the carbodiimide motif first. Following the attack of the amide oxygen, a molecule of 1-hydroxybenzotriazole (HOBt, 29) is lost from HBTU. In the subsequent step, the second aryl-amine motif (28) reacts to kick-out a molecule of tetramethylurea (30) and forms the desired benzimidazole (2). Based on LC-MS and 1H NMR analyses, we confirmed the formation of the key byproducts, HOBt (29) and tetramethylurea (30) during the conversation of amide substrate into benzimidazole. This finding provides experimental support for this mechanistic proposal.
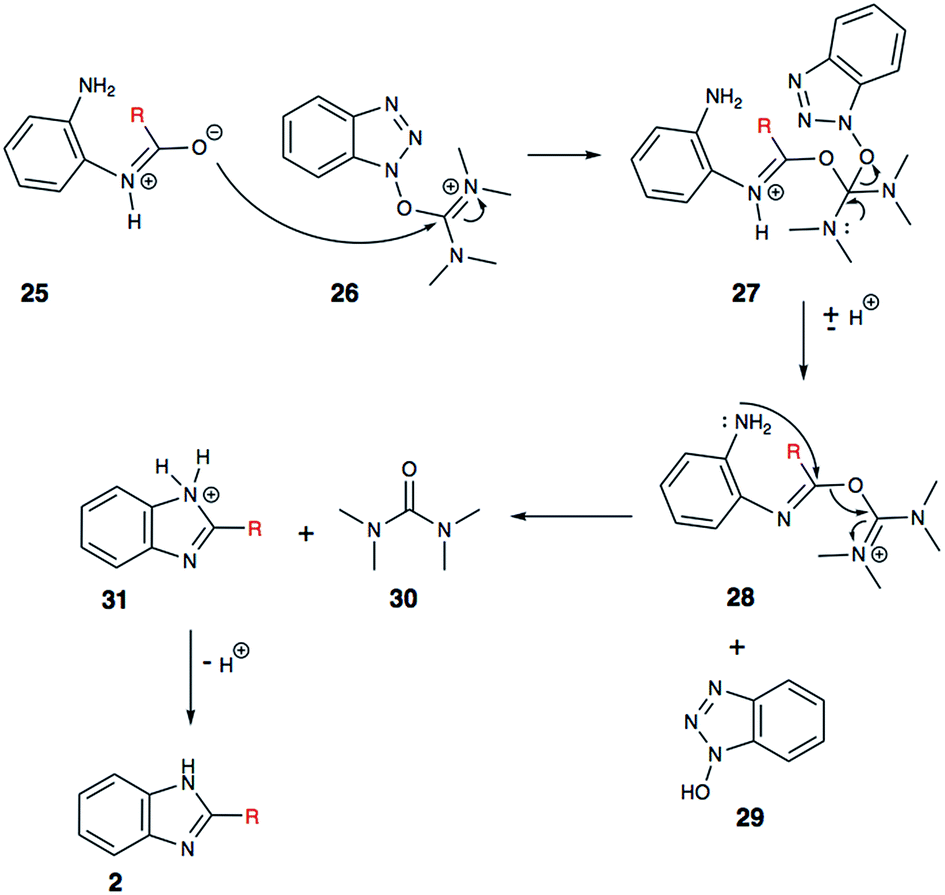 | ||
| Scheme 6 Proposed mechanism of HBTU promoted cyclization. | ||
In a recent report, amino acid derived thiazoles have been shown to be potent modulators of P-glycoprotein, which contributes to drug resistance in cancer cells.25 Since the amino acid benzimidazoles we have generated, including 20, 21, 23 and 24 are isosteres of reported thiazole derivatives, we plan to evaluate these compounds for potential P-glycoprotein binding affinity, and reversal of anticancer drug resistance. The proposed synthetic approach provides convenient entry to prepare and evaluate potentially bioactive P-glycoprotein modulators.
Experimental section
General procedure for the conversion of carboxylic acids into benzimidazoles
To a solution of commercially available carboxylic acid (1.0 equiv.) in 30 mL of toluene or DMF was added N-ethyldiisopropylamine (1.9 equiv.) and the solution was stirred for 10 minutes at room temperature. To the stirring solution, HBTU (2 equiv.) was added and the reaction mixture was stirred for another 10 min. To the reaction mixture, O-phenylenediamine (1 equiv.) was added and stirred for 4 hours. Thereafter, the reaction mixture was heated under reflux for 3 hours. The reaction was cooled to room temperature, after which the solvent was removed in vacuo in the case of toluene, but for DMF, the reaction mixture was diluted with water and the desired product was extracted using ethyl acetate (EtOAc). The organic layer was dried over anhydrous sodium sulphate, filtered and concentrated in vacuo. The crude product was purified using column chromatography using hexanes/EtOAc in an increasing polarity up to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture. The fractions containing the desired product were concentrated and recrystallized in hexanes/EtOAc (1:1) to yield the desired product. All compounds are fully characterized using 1H NMR, 13C NMR, LC-MS and IR (see ESI† for detail).
:1 mixture. The fractions containing the desired product were concentrated and recrystallized in hexanes/EtOAc (1:1) to yield the desired product. All compounds are fully characterized using 1H NMR, 13C NMR, LC-MS and IR (see ESI† for detail).
Conclusions
We have reported a convenient and mild methodology to readily convert commercially available carboxylic acids, including indole-carboxylic acids, aliphatic-carboxylic acids and alpha-amino acids into corresponding benzimidazoles. Due to increased interest of such synthons in drug discovery, our approach provides access to structurally diverse benzimidazoles in a one-pot operation. The methodology is high yielding, acid-free and tolerates various common functional groups. As our research group and several other groups are highly interested in structurally unique benzimidazoles, our methodology is a considerable addition to the field of organic chemistry and medicinal chemistry.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the College of Pharmacy and Health Sciences, the Department of Pharmaceutical Sciences and the Office of Grants and Sponsored Research at St. John's University for their financial support for research.Notes and references
- L. Ting-Tang, X. Dan, Z. Cheng-He and C. Gui-Xin, J. Org. Chem., 2016, 81, 8806–8815 CrossRef PubMed
.
- A. Trapti, K. Sonu and K. V. Akhilesh, Org. Biomol. Chem., 2016, 14, 7639–7653 RSC
- B. Marcus, R. B. Ian, V. L. Steven and N. Nikzad, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef PubMed
-
(a) Y. Geeta and G. Swastika, Eur. J. Med. Chem., 2015, 97, 419–443 CrossRef PubMed
- S. Di and M. Shutao, ChemMedChem, 2016, 11, 646–659 CrossRef PubMed
- B. H. Scott, P. K. Min, L. Clare, X. Yusheng, T. Jim, D. J. Bennett, A. Cooke, J. Cai, E. Carswell, J. Robinson, J. MacLean, L. Brown, S. Belshaw, T. R. Clarkson, K. Liu, G. B. Liang, M. Struthers, D. Cully, T. Wisniewsi, N. Ren, C. Bopp, A. Sok, T. Q. Cai, S. Stribling, L. Y. Pai, X. Ma, J. Mertzger, A. Verras, D. McMasters, Q. Chen, E. Tung, W. Tang, G. Salituro, N. Buist, J. Kuethe, N. Rivera, J. Clemas, G. Zhou, D. Szeto, G. Forrest, R. Hajdu, M. Rosenbach, J. Gibson, C. A. Maxwell, M. Lassman, T. McLaughlin and A. Ali, ACS Med. Chem. Lett., 2015, 6, 573–578 CrossRef PubMed
- K. Tatani, M. Hiratochi, Y. Kuramochi, S. Watanabe, Y. Yamauchi, F. Itoh, M. Isaji and S. Shuto, J. Med. Chem., 2016, 59, 3719–3731 CrossRef CAS PubMed
-
(a) A. M. Hamad, Y. A. Abdullahi and W. Shudong, Bioorg. Med. Chem. Lett., 2012, 22, 1317–1321 CrossRef PubMed
-
(a) A. V. Ivachtchenko, P. M. Yamanushkin, O. D. Mit'kin, E. V. Ezhova, O. M. Korzinov, E. A. Bulanova, V. V. Bichko and A. A. Ivashchenko, Pharm. Chem. J., 2015, 49, 10–19 CrossRef
- C. Wenge, B. K. Robert, S. H. Zohreh, A. Feryan and F. Jolicia, Synlett, 2012, 23, 247–250 Search PubMed
- S. Xiang, L. Xiao-Hui, Y. Lin-Miao, H. Yu, C. Yan-Yan, Z. Xue-Jing and Y. Ming, Org. Biomol. Chem., 2015, 13, 7381–7383 RSC
- Y. Venkateswarlu, S. R. Kumar and P. Leelavathi, Org. Med. Chem. Lett., 2013, 3, 7 CrossRef PubMed
- B. Gulluzar, E. Christophe and E. M. Istvan, Org. Lett., 2012, 14, 3502–3505 CrossRef PubMed
- D. Xiaoqiong, W. Yuji, J. Yongwen and M. Dawei, J. Org. Chem., 2009, 74, 7974–7977 CrossRef PubMed
- S. Youngsook, S. Julia, C. Mario, D. Jason, H. Xiao, H. Kirk, H. Yi-Ling, C. K. Ron, M. John, R. M. Lawrence, C. M. Julio, M. Daniela, S. M. Tisha, M. Deanna, T. Thuy, V. Christine, W. Simon, W. Sharon, A. W. Douglas, W. John, Y. Gang, Z. Leeanne, Z. Xuxia and D. C. Timothy, J. Med. Chem., 2016, 59, 431–447 CrossRef PubMed
- J. A. Henderson, D. Bilimoria, M. Bubenik, C. Cadilhac, K. M. Cottrell, F. Denis, E. Dietrich, N. Ewing, G. Falardeau, S. Giroux, L. L'Heureux, B. Liu, N. Mani, M. Morris, O. Z. Pereira, C. Poisson, T. J. Reddy, S. Selliah, R. S. Shawgo, L. Vaillancourt, J. Wang, J. Xu, N. Chauret, F. Berlioz-Seux, L. C. Chan, S. K. Das, A. Grillot, Y. L. Bennani and P. J. Maxwell, Bioorg. Med. Chem. Lett., 2015, 25, 948–951 CrossRef CAS PubMed
-
(a) A. Muth, V. Subramanian, E. Beaumont, M. Nagar, P. Kerry, P. McEwan, H. Srinath, K. Clancy, S. Parelkar and P. R. Thompson, J. Med. Chem., 2017, 60, 3198–3211 CrossRef CAS PubMed
- D. V. Jannifer, C. Hui, C. Wenying, A. D. Curt, A. G. Cheryl, A. J. Jill, R. S. Chandra, K. K. Annette, S. L. Kiev, P. Barbara, W. Jianmei, J. D. Pragnya, J. Wen, N. Steven, L. Ping, J. W. Sandy, J. D. Paul, L. T. Robin, W. L. Timothy, K. Lars, I. C. Nicholas and P. E. James, J. Med. Chem., 2005, 48, 8289–8298 CrossRef PubMed
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed
- K. E. Pfitzner and J. G. Moffatt, J. Am. Chem. Soc., 1963, 85(19), 3027–3028 Search PubMed
- L. Stevens, G. H. Singhal and A. B. Ash, J. Org. Chem., 1967, 32(9), 2895 CrossRef
- Z.-K. Wan, E. F. Ousman, N. Papaioannou and E. Saiah, Tetrahedron Lett., 2011, 52, 4149–4152 CrossRef CAS
- L. M. Wagner, OncoTargets Ther., 2015, 8, 1931–1939 CrossRef CAS PubMed
- M. Kazuyuki and J. Ohtani, Agric. Biol. Chem., 1978, 42, 483–484 Search PubMed
- S. Singh, N. R. Prasad, E. E. Chufan, B. A. Patel, Y. J. Wang, Z. S. Chen, S. V. Ambudkar and T. T. Talele, J. Med. Chem., 2014, 57, 4058–4072 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra available for all compounds reported. See DOI: 10.1039/c8ra07773h |
| This journal is © The Royal Society of Chemistry 2018 |