
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural heterogeneity in polymeric nitric oxide donor nanoblended coatings for controlled release behaviors†
Hyejoong Jeonga,
Kyungtae Parka,
Jae-Chan Yoob and
Jinkee Hong *a
*a
aDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul 03722, Republic of Korea. E-mail: jinkee.hong@yonsei.ac.kr
bBiotechnology Research Center, JCBIO Co., LTD, Avison Biomedical Research Center (ABMAC), Yonsei University, Seoul 03722, Republic of Korea
First published on 19th November 2018
Abstract
Nitric oxide (NO) gas delivery has attracted extensive interest due to its endogenous therapeutic functions and potential biomedical applications for the treatment of various diseases. The important thing about NO delivery is the emission control due to the fast diffusion rate of gas molecules. To develop NO delivery platforms using macromolecules and to comprehend the chemical NO donor generation and release mechanisms, we studied branched polyethyleneimine/alginate (BPEI/ALG) nanoblended coatings fabricated by giving structural heterogeneity to the structure through a self-assembly process for the controlled release of gas molecules. NO release could be remarkably expected via the well-organized coating structures and explained by quantification of the NO donors. Taking advantage of these polymeric coatings, this process could be applied to the treatment of various diseases based on the biocompatibility of materials and the fine control of NO release rate and its amount.
Introduction
Over the past tens of years, nitric oxide delivery carriers have developed into various biomedical application forms, such as nanoparticles,1–4 thin film coatings,5,6 hydrogels,7,8 metal–organic frameworks (MOFs).9,10 Nitric oxide (NO) is an indispensable cell signaling gas molecule, which is generated by nitric oxide synthases (NOSs) by consuming L-arginine.11,12 It is involved everywhere in the human body, in particular in blood vessels, neurons, ocular system, wound healing, and so on.13 To control various biomedical functions, NO is delivered by carriers with an exogenous class of chemical NO donors. One of the most extensively used NO donors is N-diazeniumdiolate, which spontaneously releases NO triggered by protons.14 Since NO acts on physiological systems depending on its concentration, it is necessary to develop carriers to control the NO release behaviors and concentration.15,16 In an event of particles, the NO-releasing efficiency is higher but a rapid release behavior is inevitable due to their higher surface area. However, a nanoblended coating based on macromolecular scaffolds could release NO in a controlled manner because of their lower surface area and controlling structures on substrates.To fabricate nanoblended coatings for the controlled release of NO, a layer-by-layer (LbL) assembly technique can be used. Basically, two different polyelectrolytes are assembled onto substrates by electrostatic interactions and by repeating the process, nanometer to micrometer nanoblended coatings are formed.17 Nowadays, materials are expanded to not only polyelectrolytes but also every molecule that has a molecular interaction, such as graphene oxide,18 drugs,7,19 nanoparticles,20 proteins,21,22 and DNAs.23 This is the only way to fabricate nanoblended coatings using biomaterials in aqueous solution. Also, by controlling the ionization of polyelectrolytes via adjusting the pH of solutions for weak polyelectrolytes, both the surface and internal structures of coatings can be precisely changed at the nano and micro level. Furthermore, since coatings could be applied for surface modification without a restriction of substrates, including the size, formation, and materials, it has the potential to be useful in a broad range of biomedical applications. Thus, making full use of a LbL assembly technique, biocompatible coatings can be formed into highly organized structures as a substrate for the controlled release of NO.
In this report, we fabricated nanoblended coatings which could perform the controlled release of NO. Initially, we assumed that the nanoblended coatings would exhibit different NO releasing behaviors depending on the structures. Therefore, we experimentally investigated the relationship between the nanoblended coating structures and NO gas release because we were confident in the hypothesis. In previous studies, mostly materials were formed to be NO-releasing substances at the beginning and then manufactured into coatings or particles specific for certain applications. However, this way has some limitations. For example, N-diazeniumdiolate formed on the materials cannot be manufactured in aqueous solution due to its instability. Thus, modification should be processed under organic solvents, not a biocompatible way. Also in organic solvents, there will be many losses of NO during the process. Additionally, the N-diazeniumdiolate of materials has a rapid release behavior (half-life of a few minutes) because there is a lack of opportunity to be stabilized. In this study, we prepared nanoblended coatings with a biocompatible method of LbL and then induced a controlled release of NO with different amounts and were able to retard NO release rate (half-life of a few hours). For NO-releasing coatings, branched polyethylene imine (BPEI) was chosen as a macromolecular scaffold of NO donors due to its amine-rich property for donor generation, while alginate (ALG) was used as a counter polymer due to its biocompatibility. BPEI and ALG were electrostatically combined in the assembled nanoblended coatings in two different pH conditions for controlling the structures upon different molecular interaction forces.24,25 The generation of NO donors depending on heterogeneity of the nanoblended coatings was demonstrated using UV-vis absorption and FT-IR spectroscopy, including the quantification of NO donors inside the coating structures. NO release behaviors through the coatings were further investigated via a real-time chemiluminescent detecting system. The relation between the coating structures and NO-release characteristics is discussed herein based on our chemical investigations.
Materials and methods
Materials
Branched polyethylene imine (BPEI, MW 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), alginic acid, anhydrous ethanol, and methanol were purchased from Aldrich. Based on the information of FMC Corporation, we supposed the molecular weight of alginic acid to be 75000 based on the viscosity of 15–25 cps in 1% of H2O (Pronova UP VLVM, viscosity: <20 mPa, Ms < 75 kDa). A non-oxidized silicon wafer was obtained from Namkang Hitech Co., Ltd (Korea). The electrode of a quartz crystal microbalance (QCM) was obtained from Stanford Research Systems (SRS). Sodium methoxide was purchased from Acros.
000), alginic acid, anhydrous ethanol, and methanol were purchased from Aldrich. Based on the information of FMC Corporation, we supposed the molecular weight of alginic acid to be 75000 based on the viscosity of 15–25 cps in 1% of H2O (Pronova UP VLVM, viscosity: <20 mPa, Ms < 75 kDa). A non-oxidized silicon wafer was obtained from Namkang Hitech Co., Ltd (Korea). The electrode of a quartz crystal microbalance (QCM) was obtained from Stanford Research Systems (SRS). Sodium methoxide was purchased from Acros.
Coating process
First, branched polyethylene imine and alginic acid were dissolved in distilled (DI) water at 1 mg mL−1. The pH of the BPEI solution was adjusted to 9 using 1 M of HCl. Alginic acid solutions were prepared at pH 4 and 8 by carefully adding 0.1 M of HCl or NaOH. DI water was prepared at pH 4, 8, and 9 in an equal way for washing. To clean the substrates, we sonicated 1 × 4 cm of silicon wafers under ethanol. Quartz glass and QCM electrodes were cleaned in a piranha solution (H2SO4:H2O2, 3:1 vol/vol%) for 5 min. Following the conventional dipping method for preparing nanoblended coatings, we soaked the substrates in the BPEI, the positively charged solution for 10 min, and sequentially dipped the substrates into pH 9 DI water, which had an equal pH with the polymer solution, three times for 2, 1, 1 min, respectively. Subsequently, the substrates soaked into alginic acid solution and went through equal washing steps. Through this process, we prepared two compositions of nanoblended coatings: (BPEI9/ALG8)n and (BPEI9/ALG4)n (n = number of bilayers). The number flanked to the name of the polymers indicates the pH condition, in other words, BPEI9 means pH 9 of BPEI solution, “n” indicates the repeat of the above process. In the case of pH 4 alginic acid, we added one more washing solution, which had an equal pH with the next polymer solution at the end of the washing steps. Due to the absence of a drying step, the washing solution with different pH conditions could change the pH of the polymer solution and induce an irregular assembly of polymers on the surface. At the end of the process, each coating was dried by compressed air.
Characterization of coatings
For analysis of the thickness, FT-IR, AFM, and SEM, the nanoblended coatings were characterized on the silicon wafer due to its perfect surface uniformity. The thickness of coatings was measured using a profilometer (Dektak 150, Veeco) and functional groups were detected by a Fourier transform infrared (FT-IR) spectrometer (FT/IR 4700, Jasco). The top-view micrographs were obtained from field emission scanning electron microscopy (SEM) (SIGMA, Carl Zeiss) and atomic force microscopy (AFM) (NX10, Park Systems). UV-vis absorbance of coatings was obtained using a UV-vis spectrophotometer (Evolution 300, Thermo Fisher Scientific) on the quartz glass substrates. Each layer of nanoblended coatings was quantitatively analyzed by a QCM (QCM 200, 5 MHz, SRS).Synthesis of NO-releasing coatings
The nanoblended coatings were converted to having nitric oxide-releasing properties via high pressure reaction. The prepared nanoblended coatings were dipped into the anhydrous ethanol and methanol mixture (4:1 vol/vol%) with 0.232 mmol of sodium methoxide (the equal molar number of secondary amine in 10 mg of BPEI) in the vial. Subsequently, the prepared vials were placed in the high-pressure reactor (custom-made, Hanwoul Engineering Co., Ltd.). To remove reactive gas molecules such as oxygen, the chamber was rapidly flushed with 10 atm of argon gas three times and then slowly three times for 10 min each. After the washing steps, the chamber was filled with 10 atm of nitric oxide gas and kept for 3 days at RT. Before taking the coatings from the chamber, unreacted nitric oxide gas was purged by rapid washing steps as mentioned before. The obtained coatings should be washed with anhydrous ethanol to eliminate the residual sodium methoxide. Then the coatings were rapidly dried under vacuum condition and stored in a vacuum-packed bag at −20 °C.
Analysis of NO release
The release profiles of nitric oxide from the nanoblended coatings were obtained in the phosphate-buffered saline (PBS, 0.01 M, pH 7.4) at 37 °C as model physiological conditions, by a nitric oxide analyzer based on the principle of chemiluminescence (Sievers NOA 280i, GE Analytical Instruments). The analyzer was calibrated every time prior to measuring the samples with a zero filter (0 ppm NO) and the standard nitric oxide gas (45 ppm balanced with N2, AIRKOREA Inc.). Nitric oxide released from the nanoblended coatings in the sealed flask was delivered to the analyzer along with the argon gas flow (70 mL min−1) passed through the reaction cell. Measurements were carried out until the NO release was below 5 ppb.Results and discussion
Structural optimization of the nanoblended coatings by pH
As aforementioned in the Introduction, a nitric oxide-release coating can be applied to biomedical applications as a treatment for various diseases (Fig. 1). NO-release coatings on the stent can play the role of a vasodilator. NO signals the surrounding smooth muscle to relax and results in increasing blood flow and inhibiting restenosis.26 Contact lenses releasing NO can prevent the onset of glaucoma. The primary risk factor of glaucoma is elevated intraocular pressure (IOP) and reducing it is the only treatment to mitigate further worsening.27 NO can decrease the IOP by reducing the outflow resistance of the eye and an eye drop involving NO has been commercialized. Also, NO promotes wound healing by taking part in the phases, including inflammation, proliferation, and remodeling.28 It can be used as an efficient and biocompatible therapeutic for wound healing. For this application, NO-release coatings are required, and since the treatment effect depends on the NO concentration, the NO-release behavior should be precisely controlled.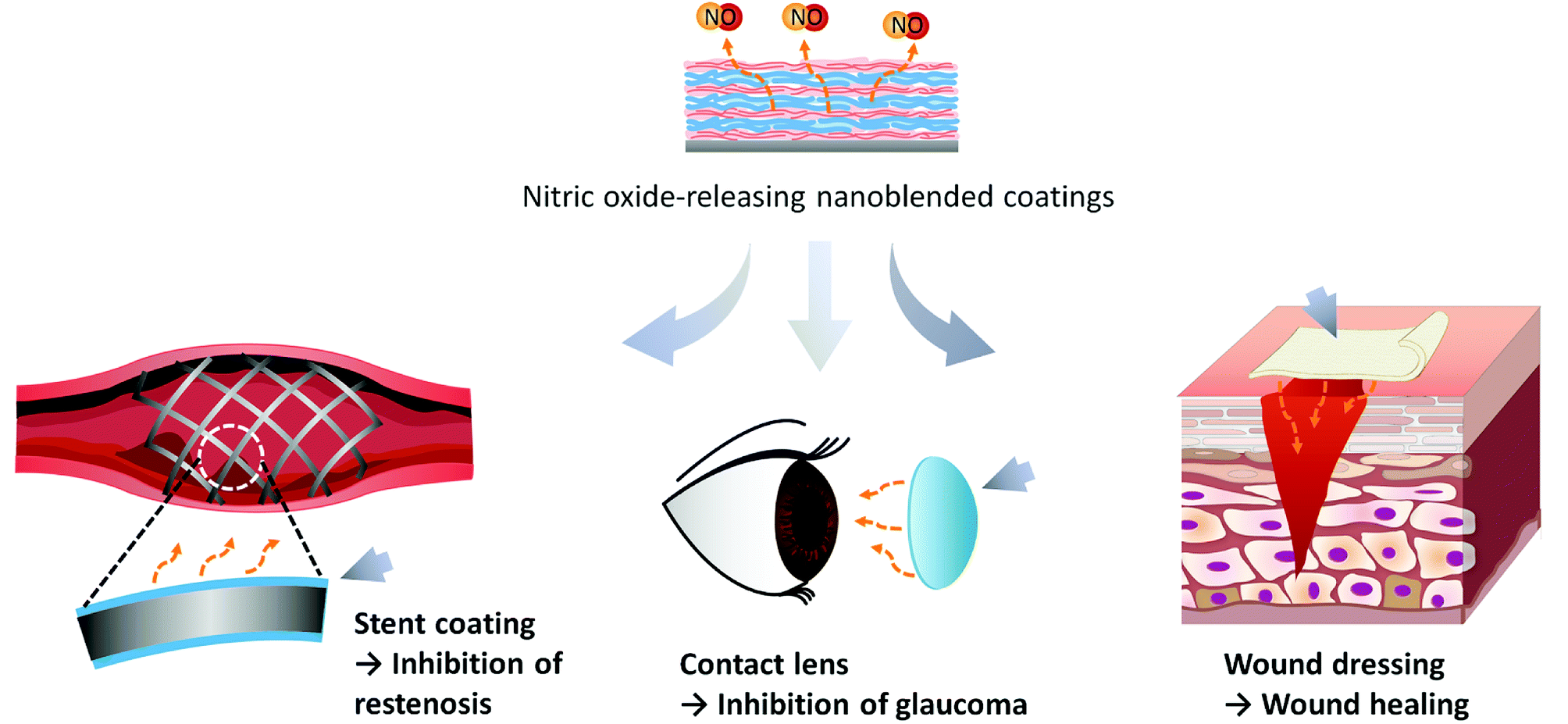 | ||
| Fig. 1 An illustration of the biomedical uses of nitric oxide-releasing coatings by nanoblended coatings. | ||
To develop polymeric coatings for the controlled release of NO, we fabricated two different nanoblended coating structures. BPEI, which is a biocompatible polymer, was chosen as a reservoir of nitric oxide donors and the structures of coatings were constructed by LbL assembly technique. On the basis of the BPEI and ALG, which are weak polyelectrolytes, two different coatings were constructed depending on the degree of ionization. In the ESI (Table S1†), we summarize the detailed information on the polymers; nanoblended coatings were assembled between amines of BPEI and carboxylic acid of ALG based on mostly electrostatic interactions and additional hydrogen bonding. The molecular weight of BPEI is 25000 and in the case of ALG, we estimated that the molecular weight of ALG was three times higher than BPEI based on the viscosity, which has a relationship with the molecular weight. We compared two coating structures: Film1-(BPEI9/ALG8)n and Film2-(BPEI9/ALG4)n films. (BPEI9/ALG4) indicates that the coating was prepared under the conditions of a BPEI solution of pH 9 (ref. 29) and an alginate solution of pH 4.25,30 The degree of ionization of BPEI was fixed at pH 9, near the pKa value and only the pH of alginate was changed to pH 4 and 8. We expected that the condition of Film2 would form more heterogeneous coatings with a higher thickness based on the stronger affinity between BPEI and alginate.
To explain the coating process of Film1 at a molecular level, BPEI(1) was ionized 21.5% at pH 9 (ref. 31) and formed a coiled structure. BPEI could be adsorbed on the negatively-charged substrate by electrostatic interaction. Sequentially, in the alginate solution at pH 8 (Film1 shown in Fig. 2a), adsorbed BPEI was ionized 31.9% and had electrostatic affinity to the fully-charged alginate(2) with a linear structure due to the intermolecular repulsion among ionized carboxylic groups. Since the linear alginate polymer was bound to the positively-charged BPEI on the substrate, we expected that the coating was certainly uniform. Next, in the BPEI solution, adsorbed alginate was still fully-charged at pH 9 and 21.5% charged BPEI(3) was exhibited preferentially on the alginate surface. To maintain the charge balance, coiled BPEI polyelectrolyte was adsorbed and induced a slightly exponential growth. Film1 described in Fig. 2a presented the uniform structure based on the normal adsorption of polyelectrolytes depending on their ionization.
 | ||
| Fig. 2 (a) An illustration of two different nanoblended coating cross-sectional structures controlled by the pH condition of the polymers: Film1-(BPEI9/ALG8)n and Film2-(BPEI9/ALG4)n, the numbers flanked by the name of the polymers indicate the pH value of each solution. The Arabic numbers and Roman numerals next to the polymeric layers indicate the adsorption step of each layer during the LbL assembly. Each step is explained in the main text. Analysis of coating growth: (b) thickness growth curves of Film1 and Film2, the measurements were carried out at 2.5 bilayers interval by a profilometer (n = 2.5, 4.5, 6.5, 8.5, 10.5), each coating was analyzed quantitatively using a QCM; (c) frequency shifting tendency of whole coatings measured at 1 layer interval, (d) mass of each layer. (BPEI: branched polyethylene imine, ALG: alginate.) | ||
In the case of Film2, first BPEI (i) displayed an equal adsorption property on the substrate at pH 9. But, in the pH 4 alginate solution, the surface amine groups were highly protonated and half-ionized alginate (ii) presented large adsorption on the surface due to the charge compensation. Consequently, in the BPEI solution at pH 9, 21.5% of ionized BPEI (iii) was also dominantly deposited on the fully ionized carboxylic surface until reaching charge balance. In this way, Film2 described in Fig. 2a would present a highly exponential growth pattern and rough surface structures due to the large adsorption behaviors of both BPEI and alginate in the continuous charge compensation process.
Characterization of nanoblended coatings
To characterize Film1 and Film2, the growth patterns of two kinds of nanoblended coatings prepared by the aforementioned process above were analyzed for their thickness and mass increase by using a profilometer and QCM. In Fig. 2b, we measured the thickness of the coatings at 2.5 bilayer intervals because these coatings were supposed to generate nitric oxide donors on the amine groups of BPEI. Thus, the outermost surface should be BPEI to generate donors with high efficiency and coatings would have thickness differences (different amounts of BPEI) at 2.5 bilayer intervals. Using the properties of the coatings, we intended to control the nitric oxide release behaviors. In the results, the thickness of Film2 was over 5 times higher than Film1 due to the consecutive large adsorption of polymers for the charge compensation. Furthermore, we confirmed the details of the coating formation through frequency shifts of every layer in Fig. 2c. The results presented an equal pattern with thickness growth. To compare the mass change of each layer in Fig. 2d, the alginate layer displayed a higher mass increase compared to the BPEI layer in both coatings, because the molecular weight of alginate might be higher than BPEI. Based on the thickness and mass of each coating, we compared the estimated polymer packing density. The densities of Film1 and Film2 were calculated and are listed in Table S2.† The density of Film2 presented 1.5 to 2 times higher density compared to Film1.The surface morphologies of Film1 were observed using both SEM and AFM. As shown in Fig. 3f–j, we cannot perceive any surface structure in the SEM microscopy because of its surface uniformity. The AFM microscopy in Fig. 3a–e presented an accumulation of polymer coatings by the polymer assembling process. Also, roughness values were obtained from each image; Rq values were 2.9 nm at 2.5 bi, 8.1 nm at 4.5 bi, 2.5 nm at 6.5 bi, 2.6 nm at 8.5 bi, and even 0 nm at 10.5 bi, respectively. From this result, we demonstrated that all the surfaces had uniformity. While, Film2 was observed using only a SEM due to the micron-scale massive structures. Notably, in Fig. 3n–o, we readily found circular microstructures in a few micron diameter on the top of the nanoblended coatings over 8.5 bilayers. The micron structures were grown as a function of the number of bilayers. As we already explained in Fig. 2a, we demonstrated that in Film2, low ionized polyelectrolytes were largely deposited onto the surface and induced high roughness with a micron-scale.
 | ||
| Fig. 3 Morphologies of nanoblended coatings observed from top by SEM and AFM. Film1; (a and f) n = 2.5, (b and g) n = 4.5, (c and h) n = 6.5, (d and i) n = 8.5, (e and j) n = 10.5, Film2; (k) n = 2.5, (l) n = 4.5, (m) n = 6.5, (n) n = 8.5, (o) n = 10.5. Every scale bar indicates 1 μm. (n, number of bilayers of nanoblended coatings.) | ||
NO donor generation on the nanoblended coatings
The prepared nanoblended coatings were used as two-dimensional platforms for NO delivery. We generated N-diazeniumdiolate groups in the amine sites of Film1 and 2 by the high pressure reaction of nitric oxide gas (Fig. 4b). As shown in Fig. 4a, it is a well-known method for the generation N-diazeniumdiolate, which converts amine under high pressure of NO gas by adding a strong basic catalyst, which readily reacts via deprotonation of the amines.32,33 The detailed chemical structures during the synthesis of N-diazeniumdiolate is provided in the Fig. S1.† | ||
| Fig. 4 Analysis of NO-releasing Film1 and Film2. (a) A simple chemistry shown mechanism of N-diazeniumdiolate formation at amine site of BPEI included in coatings via high pressure NO gas. (b) A scheme presenting the difference in the formation of N-diazeniumdiolate in Film1 and 2. Analysis of N-diazeniumdiolate in NO-releasing Film1 and 2: (c and f) FT-IR spectra, (d and g) UV-vis absorbance spectra, gray dotted lines indicate the peaks of functional groups in N-diazeniumdiolate. (e and h) UV-vis absorbance at 252 nm summarized as square points and the difference from the coatings' own absorbance at 252 nm indicated by circle points. | ||
As shown in Fig. 4b, we assumed that Film1 possessed more N-diazeniumdiolate not only on the surface but also in the inner structures because the structure of this coating was well-organized by a linear form of highly ionized polyelectrolytes. In terms of the physical views, nitric oxide gas can be readily penetrated and reacted with amine groups located inside the coatings. We successfully generated N-diazeniumdiolate groups in the amine sites of Film1 and 2 by the above method. In Fig. 4c–e, we detected N-diazeniumdiolate functional groups in Film1 by comparing the coatings before and after high pressure reaction. In Fig. 4c, we determined N-diazeniumdiolate group by FT-IR spectra by comparing the graph with that from before the reaction (Fig. S2†). The peak of O–N–N–O asymmetric stretch at 1360 cm−1,34,35 N–O stretches at 1220 cm−1,36 and the in-plane N2 symmetric stretch of the N2O2− group at 945 cm−1 (ref. 34) were detected, respectively. Additionally, N-diazeniumdiolate groups were readily detected by UV-vis absorbance at 252 nm in Fig. 4d.37,38 The difference of UV absorbance between before and after N-diazeniumdiolate generation is obviously displayed in Fig. 4e (UV-vis spectra before reaction provided in Fig. S3†). The difference of UV absorbance presented the quantity of N-diazeniumdiolate produced in each coating and it was proportionally increased as a function of the coating thickness. From this result, we clearly see that our first hypothesis was correct.
As an opposite coating design, we produced N-diazeniumdiolate to Film2 and analyzed it through an equal experimental process. As shown in Fig. 4b, it was assumed that Film2 had a smaller amount of N-diazeniumdiolate even though it was thicker than Film1. It was also based on the structure of coating. As mentioned in Fig. 2, Film1 was prepared by the strong electrostatic interaction between two polymers and the polymer structures were coiled due to a lack of intermolecular repulsion forces. Thus, although the final coatings had a higher thickness and roughness, the coating structure was not appropriate to generate N-diazeniumdiolate due to insufficient physical spaces (densely packed coatings). Therefore, we detected a small peak generation in the FT-IR spectra corresponding to the N-diazeniumdiolate at the equal part of Film1, shown in Fig. 4f (FT-IR spectra before reaction provided in Fig. S4†). UV-vis absorbance was proportional to the thickness in the whole range shown in Fig. 4g, depending on the amount of materials. To summarize, the absorbance of N-diazeniumdiolate seemed to increase according to the number of bilayers shown as square points in Fig. 4h. However, the difference of UV-vis absorbance between before and after reaction did not present a tendency as a function of the thickness or number of bilayers (UV-vis absorbance before reaction provided in Fig. S5†). Also, the difference of UV absorbance was much smaller than for Film1. We determined that the results well reflected our assumption and Film2 was not a good platform to materialize the controlled-release of NO gas.
Modulation of NO release profiles
Based on the previous results of the quantitative analysis of N-diazeniumdiolate in the coatings, we confirmed it by analyzing NO release from each coating structure in the model physiological environment (PBS at pH 7.4, 37 °C). NO gas was detected in real time by a commercialized NO analyzer based on the principle of chemiluminescence, as shown in Fig. 5a and c.39 The total NO release profile was calculated by sum of real-time release amount in Fig. 5b and d. Film1 displayed almost a proportionally increasing tendency to the number of bilayers. While, Film2 had ambiguous NO release behaviors. To compare the NO release results to yields of NO donors, we collected UV-vis absorbance of N-diazeniumdiolate of both Film1 and Film2, as shown in Fig. 5e. Also, the total NO-release amounts from each coating are summarized in Fig. 5f. According to both graphs, the total NO amount of each coating corresponded to the UV-vis absorbance. From this, we determined that the UV-vis absorbance can reflect the quantification of NO donors. Moreover, as previously expected, the Film1 structure could be used to increase the coating thickness and to accurately control the NO release amounts. | ||
| Fig. 5 A comparison of NO-releasing efficiency of (BPEI/ALG)n nanoblended coatings obtained from NO analyzer in model physiological conditions (0.01 M PBS at pH 7.4 and 37 °C). NO-release profiles of Film1 (n = 2.5, 4.5, 6.5, 8.5, 10.5); (a) NO flux detected in real-time and (b) accumulated NO release behaviors (t[NO] = total released NO concentration), Film2 (n = 2.5, 4.5, 6.5, 8.5, 10.5); (c) NO flux detected in real-time and (d) accumulated NO release behaviors, (e) comparison of differences in UV-vis absorbance between before and after N-diazeniumdiolate formation and (f) summary of total NO release amount. | ||
The detail information of NO-release behaviors is shown in Table 1. The total release amount of NO from Film1 was in a range of 0.04 to 0.3 μmol cm−2 and their half-life time was 3.6 to 5.4 h. In the case of Film2, the total NO release was calculated to be 0.04 to 0.09 μmol cm−2 and the half-life was 3.2 to 5.9 h. Both coatings presented a prolonged half-life time but only Film1 had a sufficient range of controlled NO release. In common, N-diazeniumdiolate rapidly releases NO gas with a few minutes of half-life because it is a quite thermodynamically unstable molecule, triggered by protons. However, in our nanoblended coatings, the half-life was obviously prolonged to a few hours regardless of the coating structures. It is considered that the main reason for the prolonged half-life is stabilization of N-diazeniumdiolate by hydrogen bonding with near amine groups.37 In addition, the retarded release rate of NO due to the low surface area is an advantage of the nanoblended coating structure.
| Kind of coatings | Number of bilayers (n) | t[NO]a (μmol cm−2) | t1/2b (hr) | [NO]mc (ppb cm−2) | tmd (min) | tde (hr) |
|---|---|---|---|---|---|---|
| a Total moles of NO generated per square of coatings.b Half-life time of NO release.c Maximum NO concentration of real time release profile.d The time of maximum concentration NO.e Duration of NO release. | ||||||
| Film1: (BPEI9/ALG8)n | 2.5 | 0.04 ± 0.01 | 3.9 ± 0.4 | 145.3 ± 36.6 | 9.3 ± 3.1 | 11.4 ± 2.2 |
| 4.5 | 0.12 ± 0.00 | 5.4 ± 0.5 | 227.6 ± 0.6 | 23.8 ± 20.4 | 22.6 ± 1.5 | |
| 6.5 | 0.11 ± 0.04 | 4.3 ± 1.0 | 341.3 ± 78.9 | 14.5 ± 11.1 | 19.0 ± 0.7 | |
| 8.5 | 0.26 ± 0.01 | 4.8 ± 0.5 | 1044.9 ± 111.7 | 15.6 ± 2.0 | 27.6 ± 6.0 | |
| 10.5 | 0.30 ± 0.02 | 3.6 ± 0.4 | 1236.8 ± 41.6 | 13.1 ± 1.8 | 30.1 ± 4.6 | |
| Film2: (BPEI9/ALG4)n | 2.5 | 0.04 ± 0.01 | 4.5 ± 0.3 | 111.8 ± 26.7 | 7.0 ± 2.7 | 12.6 ± 0.2 |
| 4.5 | 0.09 ± 0.01 | 5.9 ± 0.5 | 216.9 ± 10.7 | 10.5 ± 5.0 | 19.5 ± 2.1 | |
| 6.5 | 0.07 ± 0.04 | 3.2 ± 0.1 | 273.4 ± 33.9 | 11.2 ± 3.8 | 14.1 ± 1.4 | |
| 8.5 | 0.07 ± 0.00 | 5.2 ± 1.3 | 159.1 ± 63.2 | 11.1 ± 7.1 | 18.5 ± 3.0 | |
| 10.5 | 0.08 ± 0.01 | 5.8 ± 0.6 | 130.1 ± 2.8 | 18.2 ± 0.3 | 19.1 ± 2.1 | |
Finally, we developed two-dimensional platforms for controlled-NO delivery. Our outcome has valuable advantages over previous reports; first, we enhanced the NO release amount by generating NO donors inside of the coating structures to overcome the disadvantage of coatings with a low release amount of NO due to a small surface area. Also, there was no loss of NO during the process because we did not use the materials that have N-diazeniumdiolate. Second, the smaller surface area and stabilizing effect of NO donors of coatings indicated a sustained release of NO accompanied by a prolonged half-life time. Exquisite coating structures fabricated by well-organized polymers have great potential to be applied to biomedical uses for a sufficient amount of time with controlled NO release manners aided by their structures.
For the clinical use of NO-releasing nanoblended coatings, the fact regarding nitrosamine formation should be noted. In the case of using N-diazeniumdiolate as a NO donor, carcinogenic N-nitrosamine formation can restrict NO-releasing materials for clinical use.40 According to the hypothesis of the mechanism of diazeniumdiolate formation (Fig. S1†), first NO forms the nitrosamine radical anion at the amine and then the nitrosamine subsequently forms N-diazeniumdiolate by reacting with another NO.41–43 In this process, polyamines present undesired nitrosamine via a competitive formation of nitrosamines and diazeniumdiolates in anaerobic reactions of NO, because intramolecular hydrogen bonding can stabilize the intermediate nitrosamine and impede the sequential addition of NO to form diazeniumdiolate for polyamines, but not monoamines. Also, released NO is easily oxidized to nitrite species and nitrite ions react with amine compounds to induce nitrosamines.44 To investigate the N-nitrosamine formation of NO-releasing nanoblended coatings, first a sample of Film1, which was (BPEI9/ALG8)10.5 film, on a quartz glass was kept in PBS solution at 37 °C to release overall NO and was monitored by NOA at the same time. Although NO was released until the limit of detection (under 10 ppb) for 43 h, 13.7% of N-diazeniumdiolate remained according to the UV-vis absorbance at 252 nm (provided as Fig. S6†). The peak of N-nitrosamine could be detected at UV-vis absorbance values between 330–350 nm associated with the n → π* transition of N-nitroso compounds.45 In Fig. S6,† the peak of N-nitrosamine was detected at around 350 nm of UV-vis absorbance. For the NO analysis released from the film, the produced NO was carried by inert gas immediately to the NOA. Therefore, nitrite, an oxidized molecule of NO, was not formed and nitrosamine also was not produced by the reaction between nitrite and amine at the polymers. For this reason, N-nitrosamine detected in the UV-vis spectrum was not produced during the release of NO and probably was already present on the film. This result indicated that the potential toxicity concerns associated with the nitrosamine of (BPEI9/ALG8) nanoblended coatings should be investigated for clinical use. Fortunately, since N-diazeniumdiolate is generated at covalently immobilized amines with a polymeric matrix, the (BPEI/ALG) nanoblended coatings can provide the localized release of NO without concern for N-nitrosamine byproduct leaching from the materials. Therefore, (BPEI/ALG) nanoblended coatings possibly would not be carcinogenic in this research. For clarity, N-nitrosamines leaching from the coatings (degraded or detached BPEI) will be investigated in future studies.
Furthermore, Film1, which presented NO release duration from 11 to 30 h, can be applied to wound dressings because typically the dressing should be changed every 48 h (every 12 h for infected wounds). NO is well-known as an efficient antibacterial agent and also wound healing treatment based on the function of enhancing the collagen deposition and anti-inflammatory effect.46 Depending on the thickness, controllable NO release of Film1 can be used to find the appropriate film system that has both antibacterial effects and cytocompatibility. In addition, by taking full advantage of the LbL assembly method, the film system can be applied to a variety of substrates such as contact lenses, surgical sutures, bandages as well as skin wound dressings.
For clinical use, now all diazeniumdiolate materials should be stored below −20 °C due to its thermal instability, but we will be able to store them at a 4 °C relatively higher temperature by using a thorough vacuum packing with NO gas saturation. For the future follow-up studies, we will solve the limitation of short NO release duration and thermal instability of N-diazeniumdiolate. In addition, N-diazeniumdiolate can be replaced with more stable materials with longer half-release times or stimuli-responsive NO-releasing materials, such as β-Gal-NONOate.47 Also, NO release from the films can be blocked by coating with hydrophobic materials to retard the rapid hydration of films48 with graphene oxide to block the release of NO by sieving effects.49
Conclusions
To give a summary, we fabricated nitric oxide gas releasing nanoblended coatings based on the BPEI and alginate polymers and controlled release depending on the coating design. By adjusting the pH of alginate solution, we concisely controlled the coating structures based on the intramolecular interaction between two polymers. Although Film1-(BPEI9/ALG8) coatings were thinner than Film2-(BPEI9/ALG4) coatings, they generated more NO donors inside of the coating structures and presented a well-controlled NO releasing tendency depending on the thickness. In conclusion, we suggest nanoblended coatings that have a lower polymer packing density and well-organized structures can possess N-diazeniumdiolate inside of the structures with a controlled manner. This research is significant in that the developed nanoblended coatings would be a promising platform for NO delivery from surfaces for the purposes of the treatment of various diseases in clinics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017R1E1A1A01074343) and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C-1653).Notes and references
- J. F. Quinn, M. R. Whittaker and T. P. Davis, J. Controlled Release, 2015, 205, 190–205 CrossRef CAS PubMed.
- X. Zhang, G. Tian, W. Yin, L. Wang, X. Zheng, L. Yan, J. Li, H. Su, C. Chen and Z. Gu, Adv. Funct. Mater., 2015, 25, 3049–3056 CrossRef CAS.
- R. Chandrawati, J. Y. H. Chang, E. Reina-Torres, C. Jumeaux, J. M. Sherwood, W. D. Stamer, A. N. Zelikin, D. R. Overby and M. M. Stevens, Adv. Mater., 2017, 29, 1604932 CrossRef PubMed.
- J. S. Rink, W. Sun, S. Misener, J.-J. Wang, Z. J. Zhang, M. R. Kibbe, V. P. Dravid, S. Venkatraman and C. S. Thaxton, ACS Appl. Mater. Interfaces, 2018, 10, 6904–6916 CrossRef CAS PubMed.
- A. K. Winther, B. Fejerskov, M. Ter Meer, N. B. Jensen, R. Dillion, J. E. Schaffer, R. Chandrawati, M. M. Stevens, L. J. Schultze Kool and U. Simonsen, ACS Appl. Mater. Interfaces, 2018, 10, 10741–10751 CrossRef CAS PubMed.
- A. Lutzke, J. B. Tapia, M. J. Neufeld and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 2104–2113 CrossRef CAS PubMed.
- J. Park, S. Pramanick, D. Park, J. Yeo, J. Lee, H. Lee and W. J. Kim, Adv. Mater., 2017, 29, 1702859 CrossRef PubMed.
- T. T. H. Thi, Y. Lee, P. Le Thi and K. D. Park, Acta Biomater., 2018, 67, 66–78 CrossRef PubMed.
- M. J. Neufeld, A. Lutzke, J. B. Tapia and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 5139–5148 CrossRef CAS PubMed.
- M. J. Neufeld, A. Lutzke, W. M. Jones and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 35628–35641 CrossRef CAS PubMed.
- L. J. Ignarro, G. M. Buga, K. S. Wood, R. E. Byrns and G. Chaudhuri, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9265–9269 CrossRef CAS.
- E. Culotta and D. E. Koshland Jr, Science, 1992, 258, 1862–1865 CrossRef CAS PubMed.
- M. Rosselli, P. J. Keller and R. K. Dubey, Hum. Reprod. Update, 1998, 4, 3–24 CrossRef CAS PubMed.
- L. K. Keefer, ACS Chem. Biol., 2011, 6, 1147–1155 CrossRef CAS PubMed.
- K.-D. Kröncke, K. Fehsel and V. Kolb-Bachofen, Nitric Oxide, 1997, 1, 107–120 CrossRef PubMed.
- A. W. Carpenter and M. H. Schoenfisc, Chem. Soc. Rev., 2012, 41, 3742–3752 RSC.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. Heo, M. Choi, J. Chang, D. Ji, S. W. Kang and J. Hong, Sci. Rep., 2017, 7, 456 CrossRef PubMed.
- U. Han, Y. Seo and J. Hong, Sci. Rep., 2016, 1, 24158 CrossRef PubMed.
- S. Hwangbo, J. Heo, X. Lin, M. Choi and J. Hong, Sci. Rep., 2016, 6, 19178 CrossRef CAS PubMed.
- M. Choi, H. H. Park, D. Choi, U. Han, T. H. Park, H. Lee, J. Park and J. Hong, Adv. Healthcare Mater., 2017, 6, 1700216 CrossRef PubMed.
- U. Han, H. H. Park, Y. J. Kim, T. H. Park, J. H. Park and J. Hong, ACS Appl. Mater. Interfaces, 2017, 9, 25087–25097 CrossRef CAS PubMed.
- H. Jeong, S. Ranallo, M. Rossetti, J. Heo, J. Shin, K. Park, F. Ricci and J. Hong, Small, 2016, 12, 5572–5578 CrossRef CAS PubMed.
- H. Jeong, J. Hwang, H. Lee, P. T. Hammond, J. Choi and J. Hong, Sci. Rep., 2017, 7, 9481 CrossRef PubMed.
- J. M. Silva, S. G. Caridade, R. R. Costa, N. l. M. Alves, T. Groth, C. Picart, R. L. Reis and J. F. Mano, Langmuir, 2015, 31, 11318–11328 CrossRef CAS PubMed.
- M. A. Elnaggar, S. H. Seo, S. Gobaa, K. S. Lim, I. H. Bae, M. H. Jeong, D. K. Han and Y. K. Joung, Small, 2016, 12, 6012–6023 CrossRef CAS PubMed.
- R. Chandrawati, J. Y. H. Chang, E. Reina-Torres, C. Jumeaux, J. M. Sherwood, W. D. Stamer, A. N. Zelikin, D. R. Overby and M. M. Stevens, Adv. Mater., 2017, 29, 1604932 CrossRef PubMed.
- Y. Kang, J. Kim, Y. M. Lee, S. Im, H. Park and W. J. Kim, J. Controlled Release, 2015, 220, 624–630 CrossRef CAS PubMed.
- S. Choosakoonkriang, B. A. Lobo, G. S. Koe, J. G. Koe and R. Middaugh, J. Pharm. Sci., 2003, 92, 1710–1722 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- S. L. Clark and P. T. Hammond, Langmuir, 2000, 16, 10206–10214 CrossRef CAS.
- R. S. Drago and B. R. Karstetter, J. Am. Chem. Soc., 1961, 83, 1819–1822 CrossRef.
- R. S. Drago, Adv. Chem., 1962, 36, 143–149 CAS.
- Z. Zhou, G. M. Annich, Y. Wu and M. E. Meyerhoff, Biomacromolecules, 2006, 7, 2565–2574 CrossRef CAS PubMed.
- D. Park, J. Kim, Y. M. Lee, J. Park and W. J. Kim, Adv. Healthcare Mater., 2016, 5, 2019–2024 CrossRef CAS PubMed.
- R. S. Drago and F. E. Paulik, J. Am. Chem. Soc., 1960, 82, 96–98 CrossRef CAS.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, J. Org. Chem., 1993, 58, 1472–1476 CrossRef CAS.
- E. M. Hetrick, J. H. Shin, N. A. Stasko, C. B. Johnson, D. A. Wespe, E. Holmuhamedov and M. H. Schoenfisch, ACS Nano, 2008, 2, 235–246 CrossRef CAS PubMed.
- J. N. Bates, NeuroProtocols, 1992, 1, 141–149 CrossRef CAS.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- D. S. Bohle and K. N. Smith, Inorg. Chem., 2008, 47, 3925–3927 CrossRef CAS PubMed.
- H. Zhang, G. M. Annich, J. Miskulin, K. Stankiewicz, K. Osterholzer, S. I. Merz, R. H. Bartlett and M. E. Meyerhoff, J. Am. Chem. Soc., 2003, 125, 5015–5024 CrossRef CAS PubMed.
- R. S. Drago, R. O. Ragsdale and D. P. Eyman, J. Am. Chem. Soc., 1961, 83, 4337–4339 CrossRef CAS.
- J. E. Saavedra and L. K. Keefer, J. Chem. Educ., 2002, 79, 1427–1434 CrossRef CAS.
- P. N. Coneski and M. H. Schoenfisch, Org. Lett., 2009, 11, 5462–5465 CrossRef CAS PubMed.
- M. Rizk, M. B. Witte and A. Barbul, World J. Surg., 2004, 28, 301–306 CrossRef PubMed.
- A. K. Winther, B. Fejerskov, M. ter Meer, N. B. S. Jensen, R. Dillion, J. E. Schaffer, R. Chandrawati, M. M. Stevens, L. J. Schultze Kool, U. Simonsen and A. N. Zelikin, ACS Appl. Mater. Interfaces, 2018, 10, 10741–10751 CrossRef CAS PubMed.
- R. J. Soto, E. P. Merricks, D. A. Bellinger, T. C. Nichols and M. H. Schoenfisch, Biomaterials, 2018, 157, 76–85 CrossRef CAS PubMed.
- J. Heo, M. Choi, J. Chang, D. Ji, S. W. Kang and J. Hong, Sci. Rep., 2017, 7, 456–463 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07707j |
| This journal is © The Royal Society of Chemistry 2018 |