
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimulation of nitrogen transformation in pressurized oxy-fuel combustion of pulverized coal
Xiaorui Liang,
Qinhui Wang *,
Zhongyang Luo,
Heng Zhang,
Kaikun Li,
Yi Feng,
Abdul Rahim Shaikh and
Jianmeng Cen
*,
Zhongyang Luo,
Heng Zhang,
Kaikun Li,
Yi Feng,
Abdul Rahim Shaikh and
Jianmeng Cen
State Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310027, Zhejiang, China. E-mail: qhwang@zju.edu.cn; Fax: +86 571 87952802
First published on 18th October 2018
Abstract
Chemical kinetic modeling was applied to simulate N transformation in the pressurized oxy-fuel combustion process of pulverized coal. Modeling accuracy was validated by experimental data at different operation pressures. The key reaction paths from fuel-N to different N products were revealed by analyzing the rate of production. NO formation was synergistically affected by six elementary reactions, in which NCO and other intermediate species were involved. The reactions among N, NH, NH2, and NO were the key paths of N2 formation. After pressurizing the combustion system, NO and N2 contents decreased and increased, respectively. High operation pressure inhibited the diffusion of NO from the internal to the external part of char. This condition prolonged the residence time of NO inside the char, triggered a typical heterogeneous reaction between gaseous NO and unburned char, and reduced the conversion from fuel-N to NO. Moreover, modeling was performed to predict NOx emission in pressurized oxy-fuel combustion as a function of various operating parameters, including temperature and excess air and recycling ratios. This study may provide guidance for reducing NOx emissions and improving combustion efficiency in oxy-fuel combustion, and it can serve as a reference for industrial applications that involve pulverized coal combustion.
1 Introduction
Greenhouse gases emitted from coal-fired power plants, industrial boilers, and other energy utilization equipment are preferentially enrolled in the blacklist for emission reduction due to the pressure posed by increasing environmental awareness.1–3 Researchers worldwide are developing new combustion techniques for energy conservation and emission reduction. Oxy-fuel combustion is one of the most promising approaches due to its low CO2, NOx, and SOx emissions and high combustion efficiency of pulverized coal.4–6In oxy-fuel combustion, a blend of nearly pure oxygen is used as an oxidant instead of air. This usage results in a flue gas that consists mainly of CO2 and H2O and allows the occurrence of a simple gas purification process before CO2 capture. Oxy-fuel combustion always involves flue gas recirculation (FGR) in practice. The formation mechanism of air pollutants is remarkably different from that of air-firing combustion due to the influence of a low flame temperature and CO2-rich atmosphere. The transformation of N from fuel-N to NOx has elicited widespread attention.7–9 Several studies have reported that NOx emissions in oxy-fuel combustion with the FGR process are inferior to those under air-firing conditions because the combustion process has lower temperatures and higher char concentrations than air-fired combustion.10–13
The formation mechanism of NOx during oxy-fuel combustion has been investigated mainly under atmospheric pressure conditions, and only a few studies have reported N transformation under pressurized oxy-fuel combustion. Lasek et al.14 set up a pressurized oxy-fuel combustion process and observed reduced NO emission in air-fired and oxy-fuel pressurized combustion. The authors concluded that the positive influence of pressure on NO reduction is the creation of preferential conditions for the NO–char reaction. Lei et al.15 investigated the release behavior of NOx during pressurized oxy-fuel combustion of Datong bituminous coal and discovered that NO2 emission declines at 3 MPa due to the accelerated decomposition rate of NO2 at high combustion temperatures. However, the characterizations applied in these studies limited the demonstrations, and the elementary reactions involving the combustion process were unclear. Therefore, the N transformation mechanism during pressurized oxy-fuel combustion still requires clarification. Moreover, modeling studies on NOx emissions in oxy-fuel combustion focused on gaseous fuels.12,16 Modeling of pulverized coal relied on computational fluid dynamics codes with simplified schemes of nitrogen transformation and exhibited limited accuracy when compared with experimental results.17
In the present study, a chemical engineering model was applied to simulate NOx formation and consumption during pressurized oxy-fuel combustion. This work aims to predict NOx emission as a function of various operating parameters, including operation pressure, temperature, and excess air and recycling ratios. Rate of production (ROP) and sensitivity analyses were performed to determine the key reaction paths from fuel-N to different N products. The analysis results were compared with and validated against experimental data. This study may provide guidance for reducing pollutant emissions and improving oxy-fuel combustion efficiency, and it can serve as a reference for industrial applications that involve pulverized coal combustion.
2 Experimental and modeling
2.1 Modeling
Simulations of the oxy-fuel combustion of pulverized coal were performed using the plug flow reactor model of CHEMKIN-PRO. Mixing, pyrolysis, combustion, and gas–solid reactions were represented by expressions that can be handled in CHEMKIN.The physical model was based on conservation equations for mass, energy, and species in a specified system. The influence of thermal diffusion was considered in the simulations, and the multicomponent method was used to calculate the transport parameters. The initial conditions, including initial temperature, operation pressure, flow rate, and reactant fractions, were inputted for computation. The coal sample used in this study was a type of bituminous coal, and the pyrolysis process was conducted in oxy-fuel (O2/CO2) atmosphere at a high heating rate. According to previous research,18–20 pyrolysis of bituminous coal at a high heating rate produces HCN, especially in oxy-fuel atmosphere. Therefore, HCN was considered as the main pyrolysis product of fuel-N in this study.
The thermodynamic and kinetic parameters modified by Hashemi et al.17 was applied in this work. The detailed elementary reactions used were proposed by the Lawrence Livermore National Laboratory, and these reactions have been widely tested and verified by several studies on pulverized coal combustion.21–24
2.2 Materials
The Shenhua coal utilized in this study was purchased from the largest coal producer (Shenhua Group Corporation Ltd.) in China. The proximate and ultimate analyses that were conducted are presented in Table 1. Coal samples were pulverized into 60–100 meshes by using a coal mill before the combustion experiments. Then, the coal powders were dried at 105 °C for 12 h to remove moisture.| Proximate analysis (%) | Qb,ad (kJ g−1) | Ultimate analysis (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mad | Aad | Vad | FCad | Cad | Had | Nad | Sad | Oad | |
| 1.66 | 10.18 | 30.83 | 57.33 | 283.27 | 73.57 | 4.23 | 0.9 | 0.53 | 8.93 |
The major pyrolysis products of pulverized coal consisted of conventional light volatiles (H2O, CO2, CH4, CO, etc.) and char. HCN was the main pyrolysis product of fuel-N. In this modeling, we assumed that char is largely converted into soot and C2H2 according to the C/H ratio, as reported in similar studies.25,26 The molar volatile composition of the present coal was estimated in accordance with Mackrory correlations,27 which are illustrated in Table 2.
| Species | H2O | CO2 | CH4 | CO | Csoot | C2H2 | HCN |
| Molar fraction | 17.2 | 6.7 | 6 | 8.1 | 36.8 | 23.8 | 1.4 |
2.3 Experimental
The experimental apparatus and procedure are illustrated in Fig. 1. The pressurized oxy-fuel combustion process was implemented in a fixed-bed reactor. The reactor tube was made of nickel-base alloy steel to ensure device stability under high operation pressures and temperatures. The lengths of the furnace and tube were 660 and 1000 mm, respectively. The inner and external diameters of the tube were 50 and 85 mm, respectively. Furthermore, the furnace consisted of three heating zones to stabilize the temperature of the combustion zone. The combustion zone was in the middle of the furnace, and the two ends of the furnace worked as heat preservation areas. The temperature of the furnace bed was monitored automatically by thermocouples. The monitoring point was set inside the steel tube where the coal samples were placed to ensure accurate temperature control. | ||
| Fig. 1 Schematic of the experimental apparatus. | ||
Initially, CO2 and O2 with 99.99% purity were used as the carrier gas and oxidant in oxy-fuel combustion, respectively. The O2/CO2 volume ratio was set to 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :79 in the carrier gas, and the total feed flow rate was maintained at 1000 mlN min−1 with a mass flow controller. The operation pressure of the system was monitored by two pressure sensors located at the inlet and outlet of the steel tube. During each test, the reactor was heated to the corresponding temperature and fed with O2/CO2 for 30 min. Thereafter, 1.0 g of coal sample was uniformly spread in a quartz boat (length of 200 mm and width of 35 mm) and rapidly pushed into the center of the steel tube. The combustion experiments were conducted at a temperature of 900 °C for 10 min. A Fourier transform infrared (FTIR) gas analyzer, Gasmet DX4000 (Finland), was utilized to measure NO and CO2 concentrations. The FTIR instrument was calibrated by selecting the appropriate positions of characteristic peaks before the experiments to ensure highly accurate species concentrations. During combustion, the gaseous species were collected into a gas bag for subsequent analysis. After combustion, the gas bag was transferred and connected to the FTIR instrument. The pipeline that connected the gas bag and FTIR instrument was maintained at 180 °C to prevent the gaseous species from condensing. The measurement frequency of the FTIR instrument was set to 3 s. The species concentration of each sampling point was measured by the FTIR instrument three times and averaged to ensure high accuracy. Additionally, a gas chromatograph (Agilent 7890A, USA) equipped with a thermal conductivity detector (TCD) was utilized to measure the N2 concentration. Each experiment was conducted three times and averaged to ensure that the relative standard deviations were below 3%.
:79 in the carrier gas, and the total feed flow rate was maintained at 1000 mlN min−1 with a mass flow controller. The operation pressure of the system was monitored by two pressure sensors located at the inlet and outlet of the steel tube. During each test, the reactor was heated to the corresponding temperature and fed with O2/CO2 for 30 min. Thereafter, 1.0 g of coal sample was uniformly spread in a quartz boat (length of 200 mm and width of 35 mm) and rapidly pushed into the center of the steel tube. The combustion experiments were conducted at a temperature of 900 °C for 10 min. A Fourier transform infrared (FTIR) gas analyzer, Gasmet DX4000 (Finland), was utilized to measure NO and CO2 concentrations. The FTIR instrument was calibrated by selecting the appropriate positions of characteristic peaks before the experiments to ensure highly accurate species concentrations. During combustion, the gaseous species were collected into a gas bag for subsequent analysis. After combustion, the gas bag was transferred and connected to the FTIR instrument. The pipeline that connected the gas bag and FTIR instrument was maintained at 180 °C to prevent the gaseous species from condensing. The measurement frequency of the FTIR instrument was set to 3 s. The species concentration of each sampling point was measured by the FTIR instrument three times and averaged to ensure high accuracy. Additionally, a gas chromatograph (Agilent 7890A, USA) equipped with a thermal conductivity detector (TCD) was utilized to measure the N2 concentration. Each experiment was conducted three times and averaged to ensure that the relative standard deviations were below 3%.
3 Results and discussion
3.1 Model validation
Modeling accuracy was validated by comparing the molar fraction of the major combustion products at different operation pressures in the numerical simulation and experimental results. The experiments and modeling were conducted at a temperature of 900 °C. As shown in Fig. 2, the NO and N2 contents in the experimental data and modeling predictions decreased and increased, respectively, as the operation pressure increased. However, the modeling results of NO content were slightly underestimated compared with those of the experimental data. This result can be ascribed to the temperature variation during the combustion process. Although the temperatures in the modeling and experiment part were set to be similar (900 °C), the actual temperature in the experimental part might have slightly exceeded 900 °C due to the exothermic reaction of combustion. Therefore, the oxidation of fuel-N into NO accelerated, resulting in high values of NO content in the experimental results. Additionally, the concentration of CO2 emitted from the flue gas was nearly constant in the entire pressure range, implying that the majority of volatiles were completely combusted in pressurized oxy-fuel combustion. As demonstrated in Fig. 2, the simulation results are in good agreement with the experimental data. Therefore, the reaction mechanism and calculation method applied in this work are reasonable and convincing.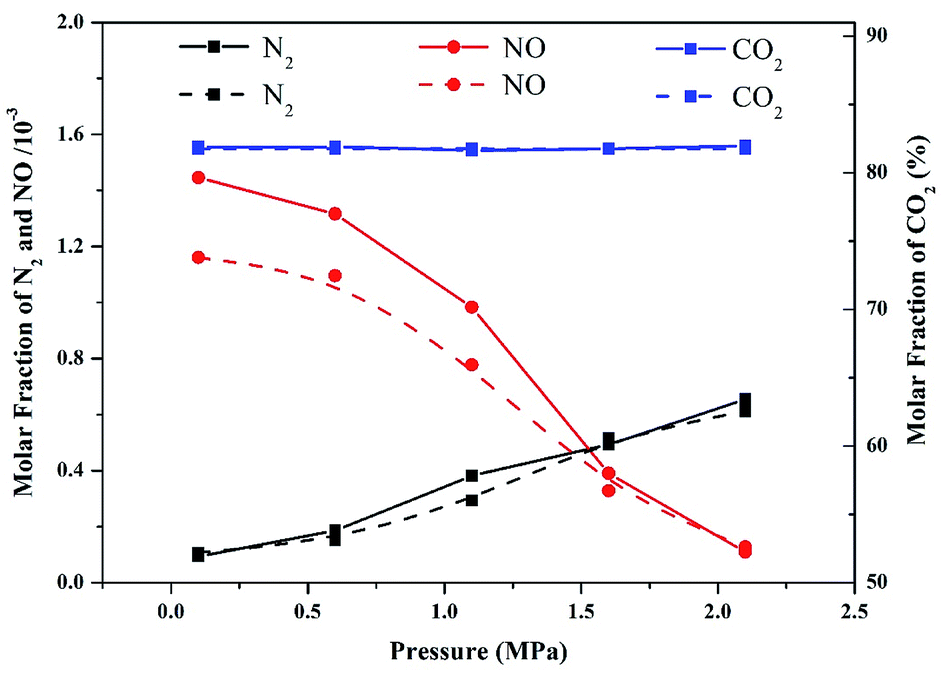 | ||
| Fig. 2 Comparison of experimental data (solid lines) and modeling results (dashed lines) of major combustion products as a function of operation pressure in pressurized oxy-fuel combustion. | ||
3.2 Nitrogen products and free radicals
Fuel-N is the only source of final nitrogen products in oxy-fuel combustion. In the current modeling of pulverized coal, HCN was added as fuel-N. The modeling results showed that NO, NO2, N2O, and N2 were the primary N products. The molar fractions of the four products varied along the homothermal length of the reactor, as illustrated in Fig. 3. The coal sample was rapidly pushed into the center of the steel tube after the reactor was preheated, as indicated in Section 2.3. Thus, the start point of the distance in Fig. 3 is the center of the steel tube. | ||
| Fig. 3 Molar fraction of main nitrogen products as a function of reactor distance in (a) atmospheric oxy-fuel combustion and (b) pressurized (1.1 MPa) oxy-fuel combustion. The modeling is simulated at a temperature of 900 °C and excess air ratio of 1.0. | ||
As displayed in Fig. 3a, the molar fraction of NO increased sharply to 1.16 × 10−3 at the initial stage of oxy-fuel combustion then remained almost unchanged until the end of the combustion. The molar fraction of N2 increased to 1.08 × 10−4, indicating that N2 was the second most abundant product. Two other conventional N compounds, namely, NO2 and N2O, were observed among the products. The molar fractions of NO2 and N2O were 3.43 × 10−6 and 1.18 × 10−5, respectively. After the initial stage of oxy-fuel combustion, the molar fractions of the four N products remained constant, indicating that the combustion system was stable.
However, the yields of the four N products differed remarkably after introducing operation pressure (1.1 MPa) into this combustion system. As indicated in Fig. 3b, the molar fraction of NO decreased from 1.16 × 10−3 to 7.78 × 10−4 during atmospheric and pressurized oxy-fuel combustion. Meanwhile, the molar fraction of N2 increased gradually and stabilized at 2.93 × 10−4. Notably, the yield of NO declined by 30%, whereas that of N2 increased by nearly three times compared with atmospheric pressure. The NO2 and N2O contents were 1.78 × 10−5 and 1.33 × 10−5, respectively, suggesting that the production of NO2 was promoted and slightly exceeded that of N2O.
The molar fraction changes of H, O, and OH radicals in the combustion process are also shown in Fig. 4. The maximum molar fractions of the three radicals (Fig. 4) are much higher than those of the N products in atmospheric and pressurized oxy-fuel combustion (Fig. 3). This result implies that H, O, and OH radicals are vital to the ignition process at the initial stage of combustion. Studies28,29 have proven that in the ignition process of fuel combustion, the chain branch reaction R1: H + O2 ⇔ O + OH (Table 3) plays the leading role in promoting combustion. Additionally, all three radicals' molar fractions in the initial stage of pressurized oxy-fuel combustion were superior to those in atmospheric oxy-fuel combustion. This result indicates that the reaction rate is much faster in pressurized oxy-fuel combustion than in atmospheric oxy-fuel combustion. This result can be explained by classic chemical reaction collision theory.30 The quantity of activated molecules in the reaction system increases as the pressure increases, thereby increasing the number of effective collisions and improving the rate of chain branch reaction R1. Furthermore, R1 consumes one H radical and generates two radicals (O and OH) simultaneously. Thus, the increase in pressure results in an exponential increase in free radical concentration and promotes the establishment of a radical pool. This radical pool then triggers numerous elementary reactions, sustains the entire combustion process, involves N conversion, and consumes fuel to the end.
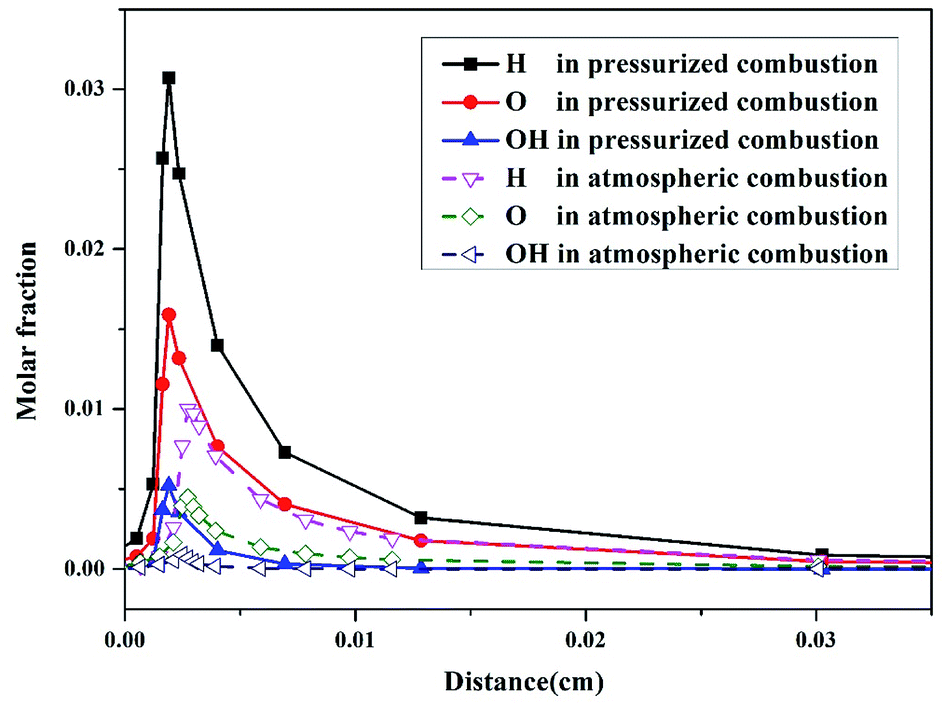 | ||
| Fig. 4 Molar fraction of H, O, and OH radicals as a function of reactor distance in atmospheric oxy-fuel combustion (dashed lines) and pressurized (1.1 MPa) oxy-fuel combustion (solid lines). The modeling is simulated at a temperature of 900 °C and excess air ratio of 1.0. | ||
| No.a | Chemical reaction equation | No.a | Chemical reaction equation |
|---|---|---|---|
| a The sequence numbers (no.) of elementary reactions are derived from the reaction mechanism database.17 | |||
| R1 | H + O2 ⇔ O + OH | R442 | NH + NO ⇔ NNH + O |
| R351 | HNO + H ⇔ NO + H2 | R514 | HCN + M ⇔ HNC + M |
| R361 | NO + HO2 ⇔ NO2 + OH | R516 | HCN + O ⇔ NCO + H |
| R391 | N2O + H ⇔ N2 + OH | R521 | HCN + OH ⇔ HNCO + H |
| R417 | NH2 + NO ⇔ N2 + H2O | R526 | HNC + O ⇔ NH + CO |
| R418 | NH2 + NO ⇔ NNH + OH | R527 | HNC + OH ⇔ HNCO + H |
| R422 | NH + H ⇔ N + H2 | R537 | CN + NO2 ⇔ N2 + CO2 |
| R423 | NH + O ⇔ NO + H | R544 | HNCO + H ⇔ NH2 + CO |
| R430 | NH + NO ⇔ N2O + H | R546 | HNCO + O ⇔ NCO + OH |
| R432 | NH + NO ⇔ N2 + OH | R547 | HNCO + O ⇔ NH + CO2 |
| R434 | NH + NO2 ⇔ N2O + OH | R548 | HNCO + O ⇔ HNO + CO |
| R435 | N + OH ⇔ NO + H | R571 | NCO + O ⇔ NO + CO |
| R436 | N + O2 ⇔ NO + O | R697 | C + NO ⇔ CN + O |
| R437 | N + NO ⇔ N2 + O | R698 | C + NO ⇔ CO + N |
| R438 | NNH ⇔ N2 + H | ||
3.3 ROP analysis of NO
ROP analysis of the modeling calculations was performed to study the transformation of N and obtain an improved understanding of N transformation in pressurized oxy-fuel combustion. The major elementary reactions involving N transformation are presented in Table 3. Positive and negative ROP values indicate the rates of formation and consumption of NO, respectively.In atmospheric oxy-fuel combustion, the six major elementary reactions involving NO production are R423, R435, R436, R571, R697, and R698, as shown in Fig. 5a. The first four elementary reactions (R423, R435, R436, and R571) were crucial in promoting NO formation. The last two elementary reactions (R697 and R698) mainly worked on NO consumption. R571 participated in the entire reaction process and displayed the highest reaction rate (with a maximum reaction rate of 1.48 × 10−3 mol cm−3 s−1) of NO production, implying that NCO might be the prior intermediate product with regard to NO formation. R423, R435, and R436 also exhibited remarkable acceleration in NO formation, verifying that NH and N acted as the two other chief intermediate products. On the contrary, a fraction of generated NO was consumed mainly through R697 and R698. This result can be ascribed to the reduction atmosphere at the initial stage of combustion. The generated NO diffused and transferred to the vicinity of unburned char then triggered a typical heterogeneous reaction between gaseous NO and unburned char. After adsorption on the surface of char, the NO molecules combined with the active species on the carbon surface and were reduced into low-valence species thereafter.31,32 However, the maximum reaction rates of R697 and R698 were relatively lower than those of R423, R435, R436, and R571. This result implies that considerable quantities of NO existed in the flue gas, which is in accordance with the results in Fig. 3a.
 | ||
| Fig. 5 Reaction rate of NO as a function of reactor distance in (a) atmospheric oxy-fuel combustion and (b) pressurized (1.1 MPa) oxy-fuel combustion. The modeling is simulated at a temperature of 900 °C and excess air ratio of 1.0. | ||
After pressurizing the combustion process, the six major elementary reactions that involved NO production and consumption were unchanged, as illustrated in Fig. 5b. Notably, the reaction rates of these six elementary reactions were improved greatly. With regard to the main NO production reaction (R571), the maximum reaction rate increased to 3.82 × 10−2 mol cm−3 s−1. The maximum reaction rates of the main NO consumption reactions (R697 and R698) were also significantly enhanced. Furthermore, the maximum reaction rates of R697 and R698 increased to the same magnitude order as R571, which is slightly different from the situation in atmospheric combustion. Thus, we infer that the NO content in the flue gas decreased distinctly, whereas the low-valence N species (N2) increased, as verified by the results in Fig. 3b.
The rapid increase in reaction rate under the pressurized condition can be explained by chemical reaction rate (1), Arrhenius (2), and Clapeyron (3) equations.
| r = k[HCN]M[O2]N, | (1) |
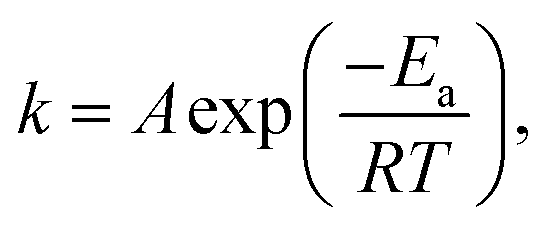 | (2) |
| PV = nRT, | (3) |
We assumed that the gaseous components in this reactor are ideal. In this modeling, the reaction rate constant (k) was unchanged because the temperature (T) was constant according to eqn (2). As the pressure (P) increased, the molar amount (n) and/or concentration [x] of the reactants increased because the gas volume (V) was constant according to eqn (3). Thereafter, the reaction rate (r) augments exponentially with the increase of [x] according to eqn (1). This conclusion has been proven by many studies and is in accordance with our modeling results.33–35
In summary, after pressurizing the combustion system, the promotion of NO consumption rate exceeded that of NO production rate. The decrease in emitted NO content in pressurized oxy-fuel combustion synergistically resulted from the six major elementary reactions, namely, R423, R435, R436, R571, R697, and R698.
3.4 Key reaction paths
The key reaction paths involving N transformation in oxy-fuel combustion were described in accordance with the ROP and sensitivity analyses in this modeling, as demonstrated in Fig. 6. The reaction paths were complicated processes that involved multiple elementary reactions toward multiple potential directions. Nevertheless, fuel-N (HCN) generally reacted initially with H, O, and OH radicals to generate NCO, HNCO, and HNC. These three species are pivotal products to start HCN conversion, not only to NOx but also to N2.36–39 | ||
| Fig. 6 Reaction paths of nitrogen transformation in pressurized oxy-fuel combustion. | ||
During the entire oxidation process from HCN to NO, the O radical acted as the main oxidant. Initially, HCN was converted to NCO and HNCO through R516 and R521, followed by the oxidation of HNCO to NCO via R546. Plenty of NCO was formed and accumulated by these three reaction paths. According to Glarborg,36 the subsequent reaction of NCO largely determines the fate of N atoms in the NO formation process, which is in good agreement with our modeling results. As the major precursor of NO, the NCO generated from R516, R521, and R546 was further oxidized by the O radical through R571. However, a small fraction of the generated HNCO from R521 was converted to NH, N, and HNO through R547, R422, and R548, respectively. The generated NH, N, and HNO were further oxidized to NO through R423, R435, and R351. In this manner, the migration and conversion of N atoms from HCN to NO was completed. Thereafter, the conversion from NO to NO2 and N2O was triggered through R361, R430, and R434. According to these reaction paths, the free radical pool played a crucial role throughout the entire oxidation process involving NOx formation.
Fig. 6 also illustrates the conversion from HCN to N2. Initially, HCN was converted to HNCO and HNC through R521 and R514. The generated HNC was partially converted into HNCO through R527. Then, conversion from HNCO and HNC to N, NH, and NH2 was realized through R547, R544, R526, and R422. Notably, the subsequent reactions between N/NH/NH2 produced from the abovementioned reactions and the NO generated in the aforementioned paths were the key step of N2 formation in the process.37,38 As shown in Fig. 6, N, NH, and NH2 reacted with NO through R437, R432, and R417. Another intermediate product, NNH, which participates in the N transformation of most oxy-fuel combustion,39 was also evident. NNH was produced through R442 and R418. As an unstable species, NNH dissociated automatically into N2 through R442 and R418. However, a small fraction of NO2 and N2O generated in the aforementioned paths was reduced into N2 through R537 and R391. Herein, the migration and conversion of N atoms from HCN to N2 were demonstrated.
In summary, H, O, and OH radicals were vital to sustaining and accelerating the entire reaction process. NCO acted as the major precursor to form NO. NO could be further converted into NO2 and N2O. Furthermore, the reactions among N, NH, NH2, and NO were the key steps of N2 formation. NO2 and N2O could also be converted into N2 through particular paths. Generally, the migration and conversion of N atoms in oxy-fuel combustion are complicated with regard to the formation and consumption of N species.
3.5 Effect of operation pressure
Fig. 7 demonstrates the trend of N products as a function of operation pressure in pressurized oxy-fuel combustion. The NO molar fraction displayed a significant reduction when the operation pressure increased from 0.1 MPa to 2.1 MPa. On the contrary, the molar fraction of N2 showed an opposite trend as the pressure increased. In this modeling, oxy-fuel combustion was conducted under O2/CO2 atmosphere with a temperature of less than 1000 °C. Thus, almost all of the NO can be considered to have been produced from fuel-N. | ||
| Fig. 7 Effect of operation pressure on the main nitrogen products simulated at a temperature of 900 °C and excess air ratio of 1.0. | ||
The increasing operation pressure exerted two effects on NO emission. First, the concentration [x] of reactants increased with the increase in total pressure, leading to the acceleration of the oxidation reaction rate from fuel-N to NO, as discussed in Section 3.3. Therefore, the molar fraction of NO in the flue gas increased accordingly. Second, the partial pressure and concentration of CO2 also increased with the increase in total pressure.40–42 The high CO2 partial pressure resulted in an elevated concentration of CO through the following reactions.
| CO2 + C → 2CO | (4) |
| 2CO2 → 2CO + O2 | (5) |
Thereafter, the generated CO promoted the reduction of NO through the following reaction.
| 2NO + 2CO → N2 + 2CO2 | (6) |
The high pressure inhibited the diffusion of NO from the internal to the external part of char, thereby prolonging the residence time of NO inside the char. As discussed in Section 3.3, the high NO concentration triggered a typical heterogeneous reduction with unburned char through the following reaction.
| 2NO + 2C → N2 + 2CO | (7) |
According to these demonstrations, the increase in total pressure was beneficial to NO reduction and N2 formation. The molar fraction of NO2 also increased slightly with the increase in pressure. This condition can be ascribed to the promotion of the following reaction.
| 2NO + O2 → 2NO2 | (8) |
However, this reaction was limited because the pressure and residence time were insufficient to realize complete oxidation to NO2.43
In summary, operation pressure exerted a joint effect on NO formation and reduction. The molar fraction of NO decreased with the increase in total pressure, whereas that of N2 increased.
3.6 Effect of temperature
Temperature is an important factor that influences the conversion of combustion products, especially N oxides. Improving the temperature of a combustion system exerts complex effects on N evolution.44 The oxidation of N into NO is accelerated, and the reduction of N from high valence to low valence is facilitated. Thus, the NO molar fraction in flue gas is an integrated result of multiple reactions.As shown in Fig. 8, the final molar fraction of NO emitted from the reactor increased with the increase in temperature, whereas N2 and NO2 displayed the opposite trend. Generally, not all of N atoms can be deeply oxidized to high-valence N (NO and NO2) at a low-temperature range due to the relatively low reaction rate and reactivity. Moreover, reductive N species, such as HCN, NH3, and HNCO, that are not timely oxidized certainly react with NO to generate a considerable amount of N2 through a similar path of the SNCR process.45,46 Therefore, the oxidation products consist of medium-valence N (N2) and high-valence N (NO and NO2) at a low-temperature range.
 | ||
| Fig. 8 Effect of temperature on the main nitrogen products simulated at an operation pressure of 1.1 MPa and excess air ratio of 1.0. | ||
After the temperature range moved to a high value, the reaction rate and reactivity of the oxidant were activated, and many nitrogen species were oxidized to a high valence. Meanwhile, a large amount of NO was retained because the reductive N species were rapidly consumed. Therefore, the molar fraction of NO increased with increasing temperature, whereas a reverse trend was obtained for N2 conversion, which is consistent with the results of previous studies.47–49 In a similar combustion,45 the amount of fuel-N converted to NO2, N2O, and NH3 is relatively small, with only NO being formed in large amounts, which is in accordance with this study. In summary, the formation of NO is thermodynamically favored as the temperature increases.
3.7 Effect of excess air ratio
Excess air ratio is another crucial factor that affects pulverized coal combustion and N conversion. The conversion ratio from fuel-N to NO (denoted as CRNO) was calculated with the following equation to evaluate the conversion degree of fuel-N.
 | (9) |
 | ||
| Fig. 9 Effect of excess air ratio on the main nitrogen products simulated at an operation pressure of 1.1 MPa and temperature of 900 °C. | ||
As shown in the figure, CRNO increased gradually with the increase in the excess air ratio. When the excess air ratio exceeded 1.0, the concentration of inlet oxygen was higher than the required molar ratio for complete combustion of N and C species. In this case, the reductive species, such as HCN, NH3, and HNCO, were rapidly consumed and adequately oxidized into high-valence N (NO and NO2). Meanwhile, the incomplete combustion carbon species, such as CO and unburned char, were reduced due to the high oxygen content, thus decreasing the chance of CO/NO and char/NO reduction. In short, the reduction reactions in the combustion system were greatly suppressed, leading to the high CRNO in the excess air ratio range over 1.0.
However, the N conversion ratio to NO decreased substantially as the excess air ratio declined. A 17% decrease in CRNO was observed as the excess air ratio decreased from 1.0 to 0.8, but the N2 molar fraction increased by 7%. The formation of NO was inhibited under the low excess air ratio condition, and the generation of N2 was promoted. In other words, appropriately reducing the excess air ratio is conducive to lowering the NO emission.
3.8 Effect of recycling ratio
In practice, oxy-fuel combustion always involves the FGR process. As discussed in Section 3.1, the flue gas of oxy-fuel combustion mainly consists of CO2 (approximately 80%, as shown in Fig. 2) and H2O (nearly 20%, not shown here). Therefore, a part of the flue gas is recycled into the intake system to replace proportional inlet CO2 after simple drying and filtration. Accordingly, the cost of CO2 capture for oxy-fuel combustion is reduced, and the net power generation efficiency of the system is improved. A series of modeling with different recycling ratios ranging from 0 to 0.8 was conducted to obtain insights into the actual N evolution process during pressurized oxy-fuel combustion.Fig. 10 presents the effect of recycling ratio on the main N products. The NO molar fraction in the flue gas generally increased as the recycling ratio increased. In the experiments with FGR, a high concentration of NO was recycled along with CO2 into the inlet mixture, thereby improving the NO molar fraction in the final flue gas. Thus, the higher the recycling ratio is, the more concentrated the emitted NO is.
 | ||
| Fig. 10 Effect of recycling ratio on the main nitrogen products simulated at an operation pressure of 1.1 MPa, temperature of 900 °C, and excess air ratio of 1.0. | ||
Additionally, the conversion ratio from fuel-N to NO (CRNO) decreased with the increase in recycling ratio. This result can be ascribed to the reaction between reductive N species (HCN, NCO, and HNCO) and recycled NO. Large amounts of reductive N species were rapidly consumed by recycled NO as the recycling ratio increased. These N species are vital precursors to form NO, as discussed in Section 3.4. Thus, the oxidation paths to form NO were inhibited, leading to a continuous reduction of CRNO as the recycling ratio increased. Moreover, the reactions between reductive N species and recycled NO produced abundant N2, resulting in an evident increase in N2 molar fraction in the flue gas, as illustrated in Fig. 10. These results are in accordance with those reported by Yang in a similar investigation on the coal reburning process.50 The authors concluded that an increase in NO concentration in inlet gas might lead to a decrease in HCN and an increase in N2 in the exhausted gas. In this study, fuel-derived HCN accounted for 10% of the NO reduction in the flue gas at the initial stage of combustion. However, the detailed mechanism still needs to be explored.
Notably, the conversion ratio from fuel-N to NO (CRNO) displayed an opposite trend of final NO concentration. However, no contradiction was noted. A part of the flue gas was separated to be recycled and serve as inlet CO2 after involving FGR, and the rest was emitted to the vent. The FGR process decreased the flow rate of the final emitted flue gas. With regard to combustion with FGR, although the NO concentration in the flue gas was high, the final NO emission load was lower than the non-recycle runs.
4 Conclusions
In this study, chemical kinetic modeling was applied to simulate N transformation in the pressurized oxy-fuel combustion process of pulverized coal. Modeling accuracy was validated by experimental data at different operation pressures, and an ROP analysis was performed to determine the key reaction paths from fuel-N to different nitrogen products. NO emission was synergistically affected by six elementary reactions, in which NCO and other intermediate species were involved. The reactions among N, NH, NH2, and NO were the key paths of N2 formation. NO and N2 contents decreased and increased after pressurizing the combustion system, respectively. Accordingly, the high operation pressure inhibited the diffusion of NO from the internal to the external part of char. This condition prolonged the residence time of NO inside the char, triggered a typical heterogeneous reaction between gaseous NO and unburned char, and reduced the conversion from fuel-N to NO. Moreover, modeling was performed to predict NOx emission in pressurized oxy-fuel combustion as a function of various operating parameters, including temperature and excess air and recycling ratios. This study may provide guidance for reducing pollutant emissions and improving combustion efficiency in oxy-fuel combustion, and it can serve as a reference for industrial applications involving pulverized coal combustion.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 51661125012).References
- M. B. Toftegaard, J. Brix, P. A. Jensen, P. Glarborg and A. D. Jensen, Prog. Energy Combust. Sci., 2010, 36, 581–625 CrossRef CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- Y. Lu, Y. Wang, Y. Zhao, Z. Wei, Y. Li, W. Hao and Y. Zhang, RSC Adv., 2017, 7, 51036–51045 RSC.
- Q. Shen, Y. Zheng, C. Luo, N. Ding, C.-G. Zheng and M. Thern, RSC Adv., 2015, 5, 39785–39790 RSC.
- M. C. Stern and T. A. Hatton, RSC Adv., 2014, 4, 5906–5914 RSC.
- J. Sun, Z. Yi, X. Zhao, Y. Zhou and C. Gao, RSC Adv., 2017, 7, 14678–14687 RSC.
- Z. Sun, S. Su, J. Xu, K. Xu, S. Hu, Y. Wang, L. Jiang, N. Si, Y. Zhou and S. S. A. Syed-Hassan, Energy Fuels, 2017, 31, 8392–8399 CrossRef CAS.
- L. Álvarez, J. Riaza, M. V. Gil, C. Pevida, J. J. Pis and F. Rubiera, Greenhouse Gases: Sci. Technol., 2011, 1, 180–190 CrossRef.
- M. C. Stewart, R. T. Symonds, V. Manovic, A. Macchi and E. J. Anthony, Fuel, 2012, 92, 107–115 CrossRef CAS.
- H. I. Mathekga, B. Oboirien, A. Engelbrecht, B. North and K. Premlal, Energy Fuels, 2016, 30, 6756–6763 CrossRef CAS.
- L. Duan, C. Zhao, W. Zhou, C. Qu and X. Chen, Fuel Process. Technol., 2011, 92, 379–384 CrossRef CAS.
- K. Andersson, F. Normann, F. Johnsson and B. Leckner, Ind. Eng. Chem. Res., 2008, 47, 1835–1845 CrossRef CAS.
- Y. Tan, E. Croiset, M. A. Douglas and K. V. Thambimuthu, Fuel, 2006, 85, 507–512 CrossRef CAS.
- J. A. Lasek, M. Janusz, J. Zuwała, K. Głód and A. Iluk, Energy, 2013, 62, 105–112 CrossRef CAS.
- M. Lei, X. Huang, C. Wang, W. Yan and S. Wang, J. Therm. Anal. Calorim., 2016, 126, 1067–1075 CrossRef CAS.
- T. Mendiara and P. Glarborg, Energy Fuels, 2009, 23, 3565–3572 CrossRef CAS.
- H. Hashemi, S. Hansen, M. B. Toftegaard, K. H. Pedersen, A. D. Jensen, K. Dam-Johansen and P. Glarborg, Energy Fuels, 2011, 25, 4280–4289 CrossRef CAS.
- P. Glarborg, A. Jensen and J. E. Johnsson, Prog. Energy Combust. Sci., 2003, 29, 89–113 CrossRef CAS.
- J. Leppälahti, Fuel, 1995, 74, 1363–1368 CrossRef.
- Y. Duan, L. Duan, E. J. Anthony and C. Zhao, Fuel, 2017, 189, 98–106 CrossRef CAS.
- M. Østberg, P. Glarborg, A. Jensen, J. E. Johnsson, L. S. Pedersen and K. Dam-Johansen, Symposium (International) on Combustion, 1998, 27, 3027–3035 CrossRef.
- M. U. Alzueta, R. Bilbao, A. Millera, P. Glarborg, M. Østberg and K. Dam-Johansen, Energy Fuels, 1998, 12, 329–338 CrossRef CAS.
- P. Glarborg, P. G. Kristensen, K. Dam-Johansen, M. Alzueta, A. Millera and R. Bilbao, Energy Fuels, 2000, 14, 828–838 CrossRef CAS.
- E. Vilas, U. Skifter, A. D. Jensen, C. López, J. Maier and P. Glarborg, Energy Fuels, 2004, 18, 1442–1450 CrossRef CAS.
- S. Niksa and S. Cho, Energy Fuels, 1996, 10, 463–473 CrossRef CAS.
- J. C. Chen and S. Niksa, Energy Fuels, 1992, 6, 254–264 CrossRef CAS.
- A. J. Mackrory, Ph.D. thesis, Brigham Young University, 2008.
- J. Huang and W. Bushe, Combust. Flame, 2006, 144, 74–88 CrossRef CAS.
- P. Zhang, J. Ran, C. Qin, X. Du, J. Niu and L. Yang, Energy Fuels, 2018, 32, 3900–3907 CrossRef CAS.
- M. S. Child, Molecular collision theory, Courier Corporation, 1996 Search PubMed.
- W. Shangguan, Y. Teraoka and S. Kagawa, Appl. Catal., B, 1997, 12, 237–247 CrossRef CAS.
- M. Higashi, S. Uchida, N. Suzuki and K. Fujii, IEEE Trans. Plasma Sci., 1992, 20, 1–12 CrossRef CAS.
- T. F. Wall, G.-s. Liu, H.-w. Wu, D. G. Roberts, K. E. Benfell, S. Gupta, J. A. Lucas and D. J. Harris, Prog. Energy Combust. Sci., 2002, 28, 405–433 CrossRef CAS.
- D. Roberts and D. Harris, Energy Fuels, 2000, 14, 483–489 CrossRef CAS.
- R.-L. Du, K. Wu, D.-A. Xu, C.-Y. Chao, L. Zhang and X.-D. Du, Fuel Process. Technol., 2016, 148, 295–301 CrossRef CAS.
- P. Glarborg and P. Marshall, Energy Fuels, 2017, 31, 2156–2163 CrossRef CAS.
- Y. He, X. Zheng, J. Luo, H. Zheng, C. Zou, G. Luo and C. Zheng, Energy Fuels, 2017, 31, 10093–10100 CrossRef CAS.
- H. Xiao, Z. Wang, A. Valera-Medina and P. J. Bowen, J. Therm. Sci., 2018, 27, 270–276 CrossRef CAS.
- S. J. Klippenstein, L. B. Harding, P. Glarborg and J. A. Miller, Combust. Flame, 2011, 158, 774–789 CrossRef CAS.
- J. A. Lasek, K. Głód, M. Janusz, K. Kazalski and J. Zuwała, Energy Fuels, 2012, 26, 6492–6500 CrossRef CAS.
- K. Okazaki and T. Ando, Energy, 1997, 22, 207–215 CrossRef CAS.
- Y. Hu, N. Kobayashi and M. Hasatani, Energy Convers. Manage., 2003, 44, 2331–2340 CrossRef CAS.
- F. Normann, K. Andersson, B. Leckner and F. Johnsson, Prog. Energy Combust. Sci., 2009, 35, 385–397 CrossRef CAS.
- Y. Hu, N. Kobayashi and M. Hasatani, Fuel, 2001, 80, 1851–1855 CrossRef CAS.
- F. Winter, C. Wartha and H. Hofbauer, Bioresour. Technol., 1999, 70, 39–49 CrossRef CAS.
- L. Duan, Y. Duan, C. Zhao and E. J. Anthony, Fuel, 2015, 150, 8–13 CrossRef CAS.
- F. Kapteijn, J. Rodriguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- A. Hayhurst and A. Lawrence, Combust. Flame, 1996, 105, 341–357 CrossRef CAS.
- C. J. Tullin, S. Goel, A. Morihara, A. F. Sarofim and J. M. Beer, Energy Fuels, 1993, 7, 796–802 CrossRef CAS.
- Y. B. Yang, E. Hampartsoumian and B. M. Gibbs, Symposium (International) on Combustion, 1998, 27, 3009–3017 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |