
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ investigation of the kinetics and microstructure during photopolymerization by positron annihilation technique and NIR-photorheology†
Helena Švajdlenková *a,
Ondrej Šaušab,
Gernot Peercd and
Christian Gorschec
*a,
Ondrej Šaušab,
Gernot Peercd and
Christian Gorschec
aPolymer Institute of SAS, Dúbravská cesta 9, 845 41 Bratislava, Slovakia. E-mail: helena.svajdlenkova@savba.sk
bInstitute of Physics of SAS, Dúbravská cesta 9, 845 11 Bratislava, Slovakia
cInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163 MC, 1060 Vienna, Austria
dChristian-Doppler-Laboratory for Photopolymers in Digital and Restorative Dentistry, Getreidemarkt 9, 1060 Vienna, Austria
First published on 2nd November 2018
Abstract
The microstructural free volume evolution during a photopolymerization process was studied on a commercial photopolymer (SPOT LV) in situ by positron annihilation lifetime spectroscopy (PALS) and concomitant NIR-photorheology. Analysis of the positron lifetime spectra revealed a high sensitivity of the PALS technique to the different phases of photopolymerization associated with different reaction rates as well as to the evolution of microstructural free-volume shrinkage, which was described at the molecular level by the Kohlrausch–Williams–Watts equation. The in situ PALS study of microstructural changes in photopolymerization was related to the vitrification (gel point) accompanied by shrinkage stress registered via NIR-photorheology. The simultaneous NIR measurements yield information on the monomer conversion of SPOT LV, which can be correlated to the occurrence of the gel point and the evolution of the microstructural free volume. This combined study allows us to see deeper into the crosslinking process and its influence on the resulting material characteristics.
1. Introduction
Photopolymerization is an efficient technique for the rapid production of thermosets with low energy consumption and unprecedented physical and mechanical properties. One of its advantages is the possibility to modify the material properties for a wide variety of applications, such as protective and decorative coatings,1 dental medicine2 and 3D-printing.3 The process of photopolymerization is a very complex phenomenon, which has been studied very intensively using different techniques. The most extensively studied factors are the compound composition, the curing conditions (e.g. the time, wavelength spectrum and intensity of radiation, reaction temperature), and macroscopic physical and mechanical properties of the final materials. It is possible to investigate the curing process, accompanied by volume reduction, via photo-DSC,4 and photodilatometry.5,6 Another suitable method for studying photopolymerization is real-time infrared spectroscopy (RT-IR) due to its continuous and exact determination of the rate of reaction and the conversion of monomers.7,8 An important and versatile tool for the in situ characterization of photopolymerization reactions is the simultaneous combination of techniques, for instance, real-time near-/mid-infrared (NIR/MIR)-photorheology.9 The coupled photorheometry with NIR spectroscopy simultaneously provides molecular information, such as double bond conversion (DBC) and shrinkage stress data.9,10 In addition, NIR-ultrasonic reflectometry,11 dynamic shrinkage measurement with NIR spectroscopy12 or the dielectric spectroscopy (DS) also appears to be a suitable technique for the monitoring of photopolymerization.13,14 Each of these techniques contribute to our knowledge of both, the crosslinking process and the resulting properties of materials.One of the most important factors studied is shrinkage stress, a long-term problem influencing the practical use of photopolymers, which also contributes to the increased brittleness of materials.15 The negative impact of shrinkage and shrinkage stress on the material properties has been mainly studied in dental resins.16,17 It is known that the crosslinking process for (meth)acrylates proceeds very fast and inhomogeneously, yielding incomplete conversion of monomers. A very promising direction is regulated photopolymerization using chain transfer agents such as thiols18 or addition–fragmentation chain transfer agents (AFCTs).19 Employing such chemistry leads to a more homogeneous crosslinking reaction obtaining materials with tailored network architecture, thus reduced shrinkage stress and high toughness.20,21
As outlined above, many studies focus on the macroscopic properties of photopolymers but a deeper view into the microstructure of cured samples is limited. In recent decades, the PALS technique has also begun to be used in the study of free volume microstructures in photopolymers.22 This technique measures the lifetime of positrons injected into the investigated material. Part of the positrons can create a bound state with an electron, called a positronium (Ps), whose triplet state, ortho-positronium (o-Ps) as a local electron density probe, is trapped in the local free volume (e.g. cavities, pores). The o-Ps is annihilated by the electrons from the wall of the cavity and this mode of annihilation is called a pick-off annihilation, which is dominant for the material with short lifetimes (shorter than a few ns). The three gamma annihilation as a competitive process is in this case negligible. The “o-Ps lifetime” used in the text means the lifetime of o-Ps annihilated via pick-off process. The average lifetime of o-Ps, τo-Ps, depends on the dimension of the cavity. Using a suitable model, we can determine the size of a cavity. The technique is relatively simple and non-destructive. It is a useful tool for exploring the microstructural free volume in the range of 0.2–50 nm and the dynamics of processes in the free volume, which brings new insights into the material properties of polymers.
Our first pilot PALS study on both pure and regulated (using chain transfer agents) dimethacrylate-based networks combined a dynamic mechanical thermal analysis (DMTA), calculation of bulk density and PALS experiments. It was found that the reduced shrinkage stress in the modified networks led to closer packing, higher bulk density and smaller voids with a narrow void size distribution.23 Next, their microstructure (of CTA-based formulations) in terms of cooperative length, relaxation, thermal macro and micro expansion and free volume characteristics below and above Tg were elucidated.24
It is essential to investigate the crosslinking process more in detail, i.e. factors which influence network formation, as well as the final properties of materials. An in situ investigation of photopolymerization offers a closer look at occurring events as they take place and this can play a crucial role in the understanding of final material properties.
Previously, physical changes in slow processes, such as solidification/crystallization in polymers and in low molecular materials were very well characterized by annihilation parameters, mostly in the o-Ps lifetime parameter.25 In addition, changes in the free volume during the recovery process in shape memory polymers,26 in mechanically deformed elastomers27 and the positronium formation on trapped excess electrons and radical formation at the irradiation of polymers28–30 are well-studied issues. In the case of time-dependent studies, annihilation parameters show changes in the physical and chemical aging/degradation of polymers,31 in the post curing of the epoxy resin system,32 as well as in the discontinuous investigation of cured processes ex situ.33–35 For the first time, in our work, the in situ time evolution of microstructural shrinkage during photopolymerization was shown by PALS technique and related to parallel NIR-photorheology measurements giving output on macrostructural changes. Also, the kinetics of photopolymerization were described via the free-volume concept.
2. Experimental
2.1. Materials
The commercially available resin SPOT LV, produced by Sonnaya Ulitka S.L., division Spot-A Materials was used. The mixture consists of three types of acrylates: a cycloaliphatic monofunctional acrylate (10–20 wt%), an aliphatic diacrylate (10–30 wt%), and an aliphatic triacrylate (60–80 wt%), some of which were ethoxylated. The molecular weight of the components was 190–430 g mol−1 and the photoinitiator was a type of phosphine oxide (1.5 wt%). More information about this resin is available on the website.362.2. PALS experiment
In both containers, holes for the LEDs were made. The geometry and the intensity of the irradiation of the samples were identical for both containers. The sandwich assembly of samples is shown in the Fig. S2 of ESI.†
The positron lifetime measurements were performed by the fast–fast type of coincidence spectrometer with a time resolution of about 350 ps (full width at half of the maximum of the time instrumental curve). The time-resolving function and the correction for the annihilation in the positron source was performed by the defect-free pure Al sample. The lifetime spectra were evaluated using an LT program37 and decomposed into three discrete lifetime components or two discrete and one largest continuous components by the complex mode of data evaluation. This means that the ratio of relative intensities I1 and I3 was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and the lifetime τ1 was set at 125 ps. More information about the lifetime distribution is given in the ESI.† The first two short components were connected with the annihilation of para-positronium (p-Ps, singlet state of Ps) and the direct electron-positron annihilation in the bulk sample. The longest-lived component from the annihilation of o-Ps corresponds to the average void size in a network structure, determined as follows:
:3 and the lifetime τ1 was set at 125 ps. More information about the lifetime distribution is given in the ESI.† The first two short components were connected with the annihilation of para-positronium (p-Ps, singlet state of Ps) and the direct electron-positron annihilation in the bulk sample. The longest-lived component from the annihilation of o-Ps corresponds to the average void size in a network structure, determined as follows:
The Tao–Eldrup's equation38,39 for the estimation of the microstructural free volume in the photopolymer was applied. This commonly used equation assumes that a positronium is trapped in the spherical free-volume holes of the polymers. The o-Ps lifetime τo-Ps is related to the radius rh of a free-volume hole in eqn (1):
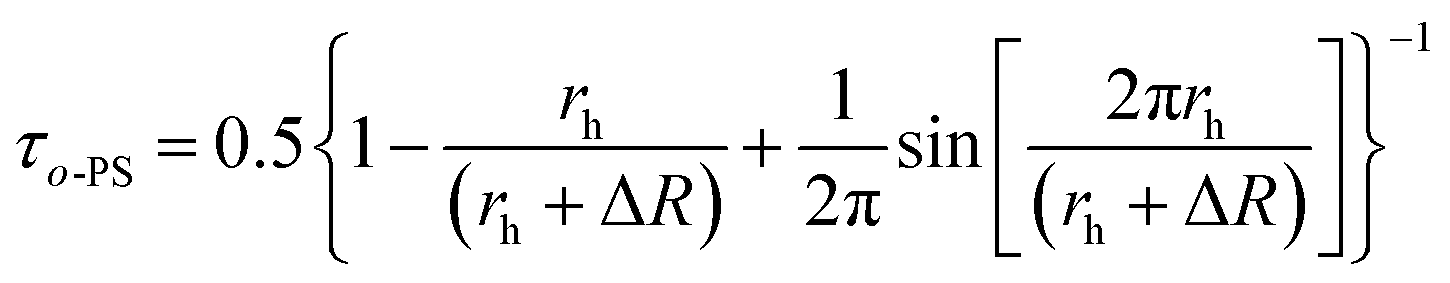 | (1) |
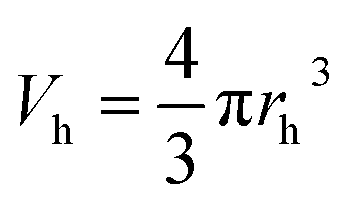 | (2) |
We have to note that this approach of the free volume evaluation is a great simplification of the real state in specific materials with the complicated shape of voids and their dynamics. However, such approximation for the description of the photopolymerization dynamics is appropriate for this case.
2.3. NIR-photorheology
The NIR-photorheology measurements were performed on an Anton Paar MCR 302 WESP with a P-PTD 200/GL Peltier glass plate and a disposable PP25 measuring system, which is hyphenated with a Bruker Vertex 80 FTIR spectrometer, equipped with NIR optics. A detailed description on the experimental set-up and the underlying method can be found in literature.9 For each measurement 150 μL of sample was applied between the glass and steel parallel plates at 20 °C with a gap of 200 μm. The formulations were oscillated with a strain of 1% and a frequency of 1 Hz. As UV light source an Exfo OmniCure LX 400 spot curing system was used and the light was projected via a dual-waveguide onto the glass plate with the sample (LED with 400 nm, 0.05 mW cm−2 on the surface of the sample). During the experimental run, the storage modulus, loss modulus and normal force were measured via rheology. From NIR data collected during the measurements the double bond conversion (DBC) of the acrylate double bonds can be obtained in situ. The data were collected for 65 s in the dark and then during interrupted irradiation of the sample consisting of intervals with 6 s light impulses and 30 s periods in the dark. The data from the end of the 30 s intervals were plotted into a graph.3. Results and discussion
3.1. PALS experiment in dark
In the first step, the influence of the radioactive source on the crosslinking process was tested. The sample was measured in the dark for 7 d at a temperature of 298 K.These long-term measurements did not show a significant change in the o-Ps lifetime, τo-Ps, over time (Fig. 1). This confirms that the radiation energy from the positron source does not initiate the polymerization, thus an influence during the irradiation periods of the sample by LED diodes can be excluded.
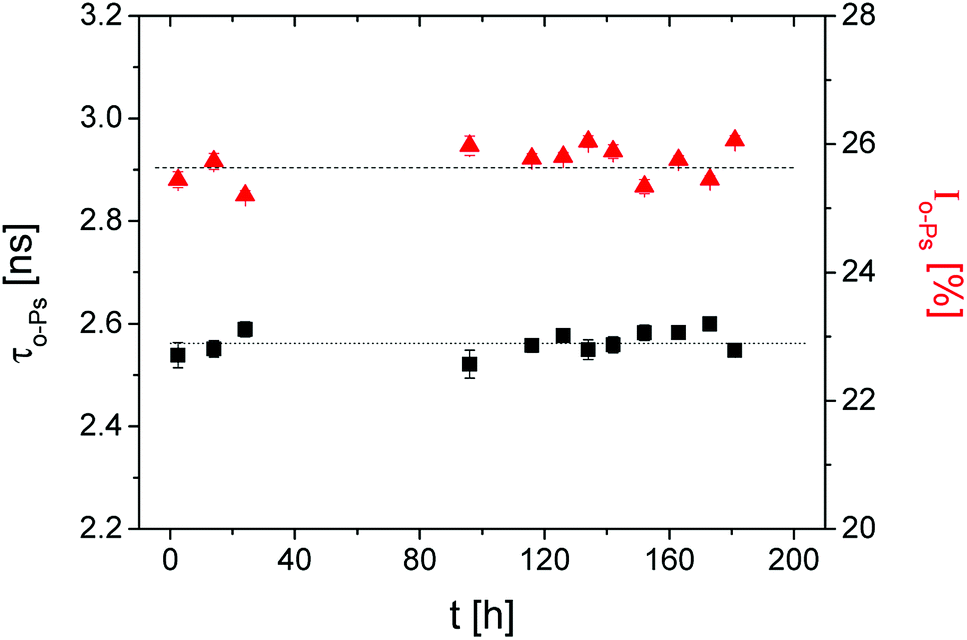 | ||
Fig. 1 The ortho-positronium lifetime τo-Ps (■) and the relative intensity Io-Ps ( ) of the sample in the dark over 7 d. ) of the sample in the dark over 7 d. | ||
3.2. PALS measurements of the irradiated sample
It is known that the network formation during photopolymerization proceeds very fast, commonly within a few seconds (<5 s). However, a minimum one-hour-long PALS measurement for each data point was required to achieve a reasonable error in the evaluation of the lifetime spectrum. Therefore, the PALS experiment had to be performed with discontinuous irradiation of the sample by the LEDs. In addition, the low intensity of the light source slowed the photopolymerization. The flux density value of 0.27 mW cm−2 specified in the light source characterization above for the PALS sample container, measured at air, seems to be high. In fact, the average light intensity in the real thick bulk sample due to the different geometry of the measurement, sample size as well as the location of light guide specified in PALS experiment was substantially reduced compared to the thin sample in the rheological measurements. The optimization of the light source and chamber for PALS experiments is one of the objectives for further research.Both factors, low intensity and discontinuous irradiation of the sample, allow to detect fine changes within the forming microstructure during the crosslinking polymerization process.
After each defined short irradiation period (6 s), the measurement of lifetimes took place in the dark for several hours, to achieve a sufficient number of annihilation events. Eight shorter (6 s) and three longer irradiation periods (60 s) were used then the continuous irradiation was followed. The accumulated (total) irradiation time is shown on the x axis.
In the first dark experiment, the total count number (tcn) for PALS spectra was about 4 M (million) where the sum of several spectra was used. Tcn for PALS spectra was varied in the sample irradiation experiment. But tcn in the discontinuous irradiation was minimal at the level of 0.5 M, except for two spectra during the change to the continuous irradiation. At the end of the photopolymerization reaction, without obvious changes in response, a large number of spectra were summed.
In Fig. 2, we can see the irradiation time evolution of the average microstructural free volume of a network structure via two parameters of an o-Ps lifetime component, i.e. τo-PS, and Vh, calculated by eqn (1) and (2), respectively. Detection of microstructural shrinkage, which directly affects the strength of a material, was observed in situ by positron annihilation technique. The shrinkage and shrinkage stress observed during the crosslinking is due to the shortening of chemical bonds.
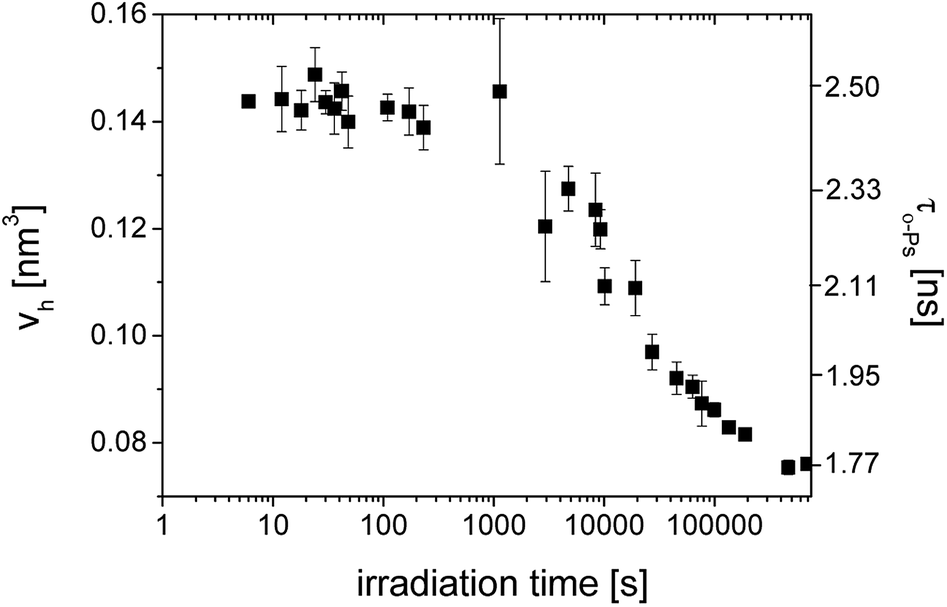 | ||
| Fig. 2 The o-Ps lifetime, τo-Ps (■) and the calculated microstructural free volume Vh (left scale) (■) versus irradiation time during photopolymerization of SPOT LV. | ||
Larger errors at two points above 1000 s were caused by a lower total count of impulses in spectra as a result of the switching to the continuous mode of irradiation and the shorter measured time period. In this case, the sum of spectra was not appropriate.
From the literature, it is known that the evolution of a macroscopic shrinkage can be described by the Kohlrausch–Williams–Watts equation (KWW):41
| ΔV/V0 = 1 − exp(t/τ)β | (3) |
372 s and the stretch exponent β = 0.635.
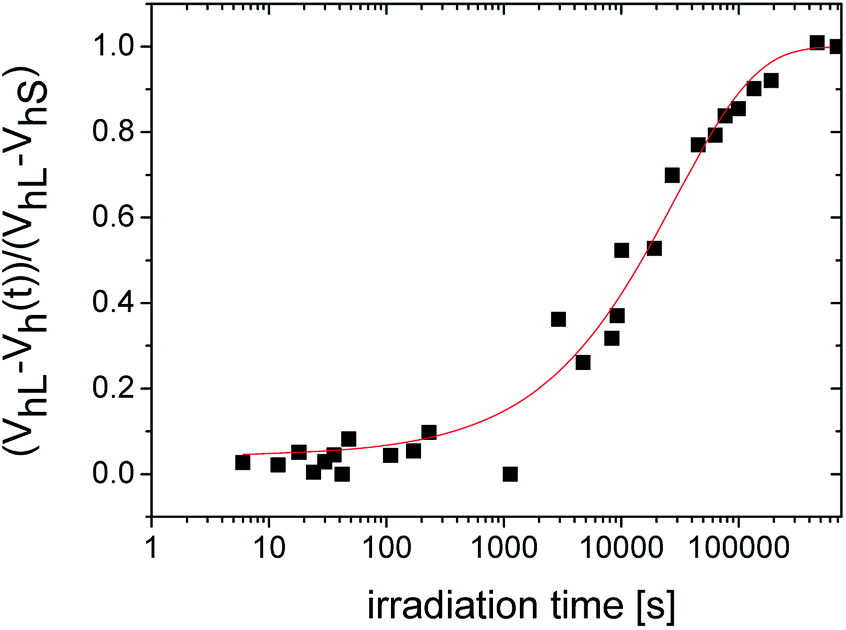 | ||
| Fig. 3 The relative change of the microstructural free volume described by the KWW equation with two characteristic parameters, i.e. the overall time-constant τ = 29372 s and the stretch exponent β = 0.635. | ||
It has to be noted that the Tao–Eldrup equation (eqn (2)) is commonly used for solid and viscous materials, e.g. glasses. However, in the case of a liquid state, such as the uncured mixture of monomers, the bubble effect occurs at the formation of a Ps.22,42,43 The zero point motion of Ps creates bubble, a spherical cavity around himself whereupon the free volume “seen” by the positrons is greater than the real intermolecular space in liquids measured by other methods such as the sound velocity.44 Therefore, the free volume determined by PALS measurements in the early stages of network formation deviates from the actual value. After the initiation of the network formation and the densification of the structure, it is expected that the Vh values are correct in the framework of the selected model for the free-volume calculation. However, this fact does not diminish the usefulness of the PALS technique for the investigation of photopolymerization reactions.
Here it is also necessary to note that the common used value of the empirical parameter ΔR = 0.166 nm can vary for different types of matter. For instance some measurements showed45,46 that smaller value of ΔR (about 0.15 nm) should be more suitable for polymers. In this work we used the simple approach and the value ΔR = 0.166 nm. It would be interesting in the future to clarify the question of an appropriate value of ΔR for this type of material.
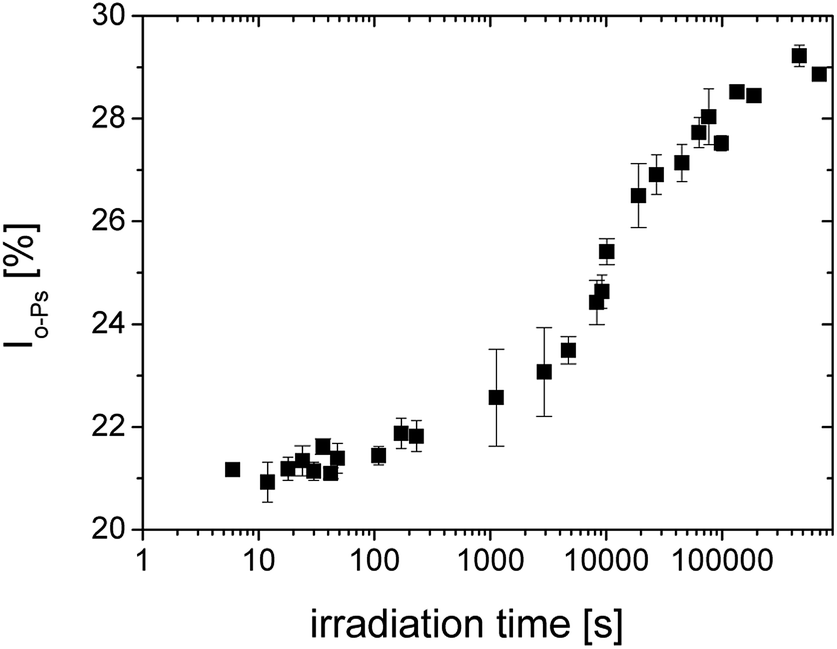 | ||
| Fig. 4 The relative o-Ps intensity during photopolymerization of SPOT LV. | ||
After the initial state of the system with slow increase of intensity, the acceleration of the curing process can be seen. The change occurs at the time of about 5000 s. A first slowdown of the crosslinking reaction was observed above 20000 s and then the reaction was gradually terminated.
By the comparison to Fig. 2, the changes of the both parameters τo-Ps and Io-Ps occur at comparable times.
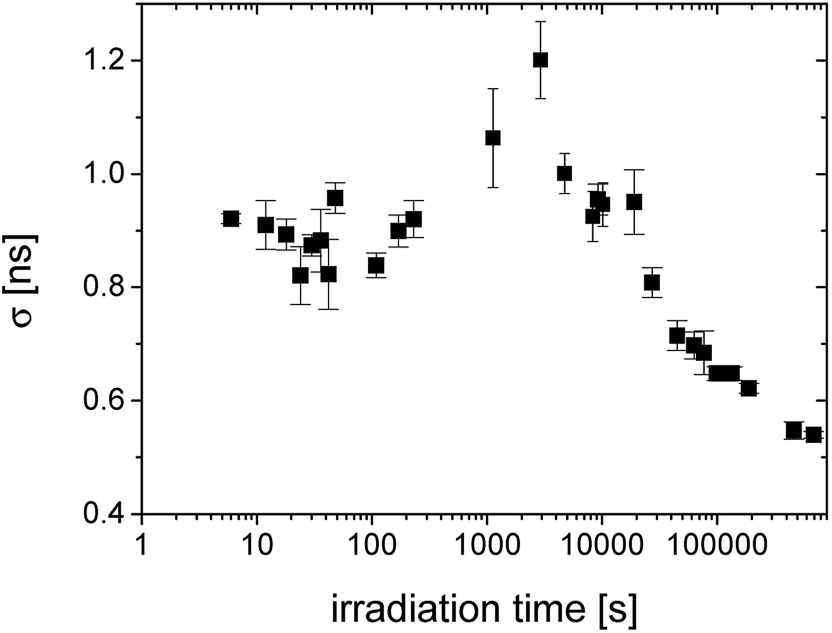 | ||
| Fig. 5 The irradiation time evolution of the standard deviation of lifetime distributions (σ) given by the width of distribution. | ||
The width of distribution characterized by σ is a measure of homogeneity of an internal structure of the material. Fig. 5 shows the regions with unchanged distribution width, the broad peak around 1000–5000 s and then the gradual narrowing of distribution reflecting homogenization of the structure with the increasing crosslinking of the material.
Fig. 6 displays lifetime distributions for different stages of curing process given by the selected spectra with high total counts. The representative curve for the first 6 s of the irradiation time (squares, ■) considers the liquid state where the bubble effect is still present.
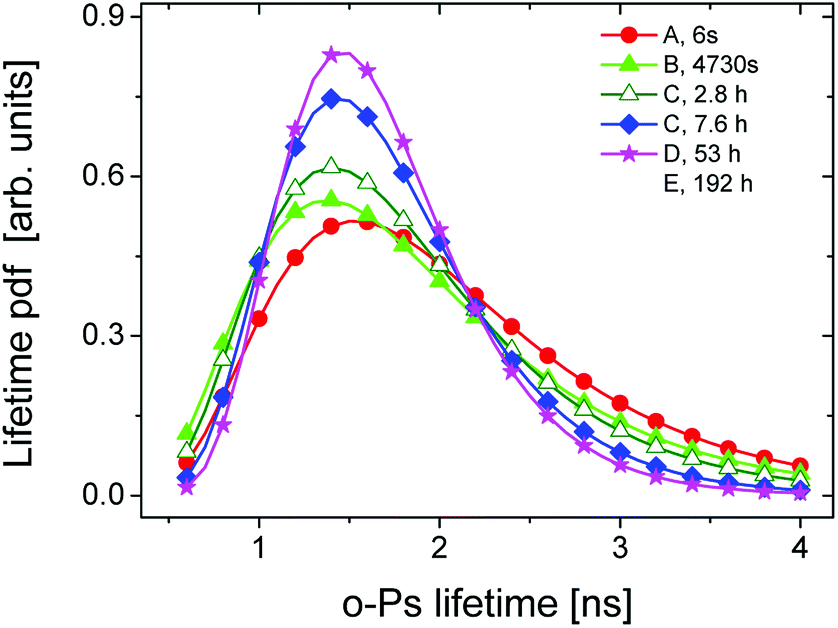 | ||
| Fig. 6 The o-Ps lifetime distribution for different stages of network formation. | ||
In this state the large width of distribution can be ascribed to the inhomogeneous system consisting of the mixture of monomers and created oligomers.
The lifetime distribution near at 5000 s of the irradiation time (circles, ●) starts to narrow and the mean lifetime shifts towards lower values. This trend was also confirmed in the length scale (Fig. S3 in ESI†).
The narrowing of the o-Ps lifetime distribution as well as the hole radius distribution can reflect the reduction of intermolecular space (free volume) due to crosslinking and the shift of lifetime/radii to lower values is attributed to the microstructural shrinkage and the reduction of the liquid phase during solidification.
In the Table 1, the mean lifetime τ and the standard deviation σ of lifetime distributions are listed for the representative spectra of various degrees of curing.
| t/h | τ/ns | σ/ns |
|---|---|---|
| 0.002 | 2.48(0.01) | 0.92(0.01) |
| 1.3 | 2.32(0.04) | 1.00(0.04) |
| 2.8 | 2.13(0.04) | 0.95(0.04) |
| 7.6 | 2.00(0.04) | 0.81(0.03) |
| 53 | 1.83(0.01) | 0.62(0.01) |
| 192 | 1.77(0.01) | 0.54(0.01) |
The distributions are able to reveal characteristic features of the network formation, such as appearance of crosslinked domains, homogeneity of network formation during photopolymerization that can bring more light to the understanding of the crosslinking mechanism.
From these results one can conclude that PALS is a highly sensitive technique to the different stages of reaction via the free volume concept. The sufficient total number of impulses in the lifetime spectrum can bring more detailed information about microstructural shrinkage and the homogeneity of the crosslinked structure.
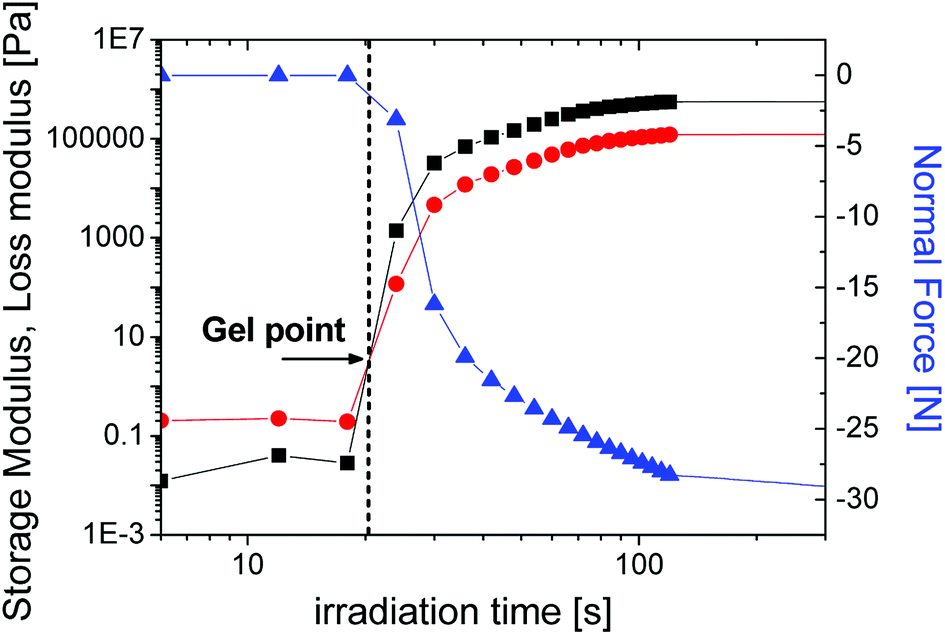 | ||
| Fig. 7 The storage (■) and loss modulus (●) as well as normal force (▲) as a function of the irradiation time for SPOT LV sample. | ||
We note that the experimental conditions are not identical in PALS versus NIR-photorheology, due to limitations in sample thickness and for different light intensity on the surface of the sample from which follows a different irradiation time on the x-axis. These two independent sets of experiments were performed at slightly varying conditions.
In the case of distributions from the PALS, the gel point is accompanied by the broadening of the lifetime distribution. The nearest representative point from this time region with appropriate number of impulses is characterized with σ = 2.32 ns and the τo-Ps = 2 ns appearing around 4730 s of irradiation time, in Fig. 6. Moreover, the KWW fit begins to increase sharply in this time region (Fig. 3).
The mutual comparison of in situ studies by PALS and NIR-photorheology at the phenomenological level clarifies stages of the crosslinking process and the gel point, which helps to see deeper into the underlying mechanisms.
Further optimization of experimental conditions will allow such interconnection techniques to achieve other original results important for characterization of sample properties at macro and microstructural levels. The PALS technique thus appears to be a unique tool for investigating changes in free volumes at the microstructural level even over time under defined external conditions.
4. Conclusions
In this paper, we present an in situ PALS study of photopolymerization on the photoresin SPOT LV. We reveal that the time evolution of the o-Ps lifetime, which is proportional to the microstructural free volumes, exhibits three basic regions of the curing process with different reaction kinetics for the tested material. The nonlinear time dependence of the relative changes in the microstructural free volumes is well described by the KWW equation and is characterized by the overall time constant and the stretched exponent, which reflects shrinkage stress. Moreover, the time dependence of relative intensity Io-PS shows the evolution from the liquid to crosslinked state of photopolymer. The lifetime or hole radius distributions determined from the PALS spectra reflect the reduction of intermolecular space during the curing process.Here the gel point was ascribed to the broadening of distribution which is indicated by the intersection point of storage and loss modulus determined from photorheology. The advantage of PALS technique is in the possibility to investigate microstructural changes, i.e. microstructural shrinkage during photopolymerization through the free volume parameters (τo-Ps, Io-Ps) and distributions can reveal detailed features of different stages of the crosslinking process (e.g. gel point).
The PALS technique shows a new promising approach for a deeper understanding of network formation from the point of view of free volume and dynamic factors. These factors can lead to a reduction in the microstructural shrinkage stress, with a significant impact on the material development and the tunability of material properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Agency VEGA Slovakia for funding project no. 2/0157/17 and the Christian Doppler Research Association and the company Ivoclar Vivadent AG for funding the framework of a Christian Doppler Laboratory for “Photopolymers in Digital and Restorative Dentistry”. The financial support by the Austria Federal Ministry for Digital and Economic Affairs and the National Foundation for Research, Technology and Development is gratefully acknowledged. We also thank the following: the manufacturer Sonnaya Ulitka S.L., DIVISION Spot-A Materials, Barcelona, Spain, for providing information about the sample; the firm Edico Sk, Bratislava, Slovakia, for technical consultations and for giving samples; Ing. Vojtech Nádaždy, CSc., at the Institute of Physics, Slovak Academy of Science, Bratislava, Slovakia, for the flux density measurements of the light sources.References
- Y. Abe, DIC Tech. Rev., 2005, 11, 1–20 CAS.
- C. Dworak, T. Koch, F. Varga and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2916–2924 CrossRef CAS.
- J. Torgersen, X.-H. Qin, Z. Li, A. Ovsianikov, R. Liska and J. Stampfl, Adv. Funct. Mater., 2013, 23, 4542–4554 CrossRef CAS.
- K. S. Anseth, C. N. Bowman and N. A. Peppas, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 139–147 CrossRef CAS.
- F. Catalina, C. Peinado, R. Sastre, J. L. Mateo and N. S. Allen, J. Photochem. Photobiol., A, 1989, 47, 365–377 CrossRef CAS.
- V. Y. Voytekunas, F. L. Ng and M. J. M. Abadie, Eur. Polym. J., 2008, 44, 3640–3649 CrossRef CAS.
- C. Decker and K. Moussa, Makromol. Chem., 1988, 189, 2381–2394 CrossRef CAS.
- C. Decker and K. Moussa, J. Coat. Technol., 1990, 36(786), 55–61 Search PubMed.
- C. Gorsche, R. Harikrishna, S. Baudis, P. Knaack, B. Husar, J. Laeuger, H. Hoffmann and R. Liska, Anal. Chem., 2017, 89, 4958–4968 CrossRef CAS PubMed.
- A. Botella, J. Dupuy, A. A. Roche, H. Sautereau and V. Verney, Macromol. Rapid Commun., 2004, 25, 1155–1158 CrossRef CAS.
- I. Alig, D. Lellinger, S. Agarwal and H. Oehler, React. Funct. Polym., 2013, 73, 316–322 CrossRef CAS.
- Y. Lin and J. W. Stansbury, RadTech e|5, Technical Proceedings, 2004 Search PubMed.
- D. Schuele, R. Renner and D. Coleman, Mol. Cryst. Liq. Cryst., 1997, 299, 343–352 CrossRef CAS.
- K. Zahouily, C. Decker, E. Kaisersberger and M. Gruener, Eur. Coat. J., 2003, 11, 14–18 Search PubMed.
- H. Lu, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2004, 20, 979–986 CrossRef CAS PubMed.
- R. R. Braga, R. Y. Ballester and J. L. Ferrance, Dent. Mater., 2005, 21, 962–970 CrossRef CAS PubMed.
- L. F. J. Schneider, L. M. Cavalcante and N. Silikas, J. Dent. Biomech., 2010, 1, 1–14 Search PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49(9), 1540–1573 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49(5), 1079–1131 CrossRef CAS.
- K. Seidler, M. Griesser, M. Kury, H. Reghunathan, P. Dorfinger, T. Koch, A. Svirkova, M. Marchetti-Deschmann, J. Stampfl, N. Moszner, C. Gorsche and R. Liska, Angew. Chem., Int. Ed., 2018, 57, 9165–9169 CrossRef CAS PubMed.
- C. Gorsche, T. Koch, N. Moszner and R. Liska, Polym. Chem., 2015, 6, 2038–2047 RSC.
- T. Goworek, Annales Universitatis Mariae Curie-Sklodowska, 2014, 69, 1–110 Search PubMed.
- H. Švajdlenková, O. Šauša, J. Steindl, T. Koch and C. Gorsche, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2476–2484 CrossRef.
- H. Švajdlenková, O. Šauša, I. Maťko, T. Koch and C. Gorsche, Macromol. Chem. Phys., 2018, 219, 1800119 CrossRef.
- V. Majerník, J. Krištiak, O. Šauša and M. Iskrová-Miklošovičová, Mater. Sci. Forum, 2013, 733, 84–87 Search PubMed.
- N. García-Huete, E. Axpe, J. M. Cuevas, D. Merida, J. M. Laza, J. A. García, J. L. Vilas, F. Plazaola and L. M. Leon, Polymer, 2017, 109, 66–70 CrossRef.
- K. Krzemien, J. Kansy and J. E. Frackowiak, Acta Phys. Pol., A, 2005, 107, 837–841 CrossRef CAS.
- T. Hirade, F. H. J. Maurer and M. Eldrup, Radiat. Phys. Chem., 2000, 58, 465–471 CrossRef CAS.
- T. Goworek, B. Zgardzinska, M. Pietrow and J. Wawryszczuk, Mater. Sci. Forum, 2013, 733, 67–74 CAS.
- M. Pietrow and J. Wawryszczuk, Mater. Sci. Forum, 2013, 733, 75–79 CAS.
- A. J. Hill, AIP Conf. Proc., 1999, 497, 699–705 CrossRef CAS.
- J. Kanzow, V. Zaporojtchenko, H. Nabika, M. Mizuhata, S. Deki and F. Faupel, Mater. Sci. Forum, 2004, 445–446, 313–315 CAS.
- M. Hyla, J. Filipecki, J. Swiatek, R. I. Mervinskii, L. A. Gudzowska and V. Savitsky, J. Non-Cryst. Solids, 2001, 290, 20–24 CrossRef CAS.
- J. Filipecki, K. Chamerski, O. Boyko and K. Kotynia, Polim. Med., 2014, 44(1), 21–28 CAS.
- O. Shpotyuk, S. Adamiak, E. Bezvushko, J. Cebulski, M. Iskiv, O. Shpotyuk and V. Balitska, Nanoscale Res. Lett., 2017, 12, 75 CrossRef PubMed.
- http://spotamaterials.com/wp/wp-content/uploads/2015/07/Spot-LV_MSDS_tmp.pdf.
- J. Kansy, Nucl. Instrum. Methods Phys. Res., Sect. A, 1996, 374, 235–244 CrossRef CAS.
- M. Eldrup, D. Lightbody and J. N. Sherwood, Chem. Phys., 1981, 63, 51–58 CrossRef CAS.
- S. J. Tao, J. Chem. Phys., 1972, 56, 5499–5510 CrossRef CAS.
- H. Nakanishi, S. Wang and Y. Jean, Microscopic surface tension studies by positron annihilation, in Int. Symp. Positron Annihil. Stud. Fluids, ed. S.C. Sharma, World Scientific Publishing, Singapore, 1988, p. 292 Search PubMed.
- G. Wiliams and D. C. Watts, Trans. Faraday Soc., 1970, 66, 80–85 RSC.
- R. A. Ferrell, Phys. Rev., 1957, 108, 167–175 CrossRef CAS.
- B. Zgardzińska, Mater. Sci. Forum, 2013, 733, 29–32 Search PubMed.
- J. F. Kincaid and H. Eyring, J. Chem. Phys., 1938, 6, 620–629 CrossRef CAS.
- R. Zaleski, J. Goworek and M. Maciejewska, Phys. Status Solidi C, 2009, 6, 2445–2447 CrossRef CAS.
- C. He, B. Xiong, W. Mao, Y. Kobayashi, T. Oka, N. Oshima and R. Suzuki, Chem. Phys. Lett., 2013, 590, 97–100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07578f |
| This journal is © The Royal Society of Chemistry 2018 |