
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel information diffusion method based on network consistency for identifying disease related microRNAs
Min Chen†
 ab,
Yan Peng†c,
Ang Li*a,
Zejun Liab,
Yingwei Denga,
Wenhua Liua,
Bo Liao*b and
Chengqiu Daia
ab,
Yan Peng†c,
Ang Li*a,
Zejun Liab,
Yingwei Denga,
Wenhua Liua,
Bo Liao*b and
Chengqiu Daia
aCollege of Computer Science and Technology, Hunan Institute of Technology, 421002 Hengyang, China. E-mail: liang@hnit.edu.cn
bCollege of Information Science and Engineering, Hunan University, Changsha 410082, China. E-mail: dragonbw@163.com
cCollege of International Communication, Hunan Institute of Technology, 421002 Hengyang, China
First published on 30th October 2018
Abstract
The abnormal expression of miRNAs is directly related to the development of human diseases. Predicting the potential candidate miRNAs associated with diseases can contribute to the detection, diagnosis, treatment and prevention of human complex diseases. The effective inference of the calculation method of the relationship between miRNAs and diseases is an effective supplement to biological experiments. It is of great help in the prevention, treatment and prognosis of complex diseases. This paper proposes a novel information diffusion method based on network consistency (IDNC) for identifying disease related microRNAs. The model first synthesizes the miRNA family information and the miRNA function similarity to reconstruct the miRNA network, and reconstruct the disease network by using the known disease and miRNA-related information and the semantic score between diseases. Then the global similarity of the two networks is obtained by using the Laplacian score of graphs. The global similarity score is a measure of the similarity between diseases and miRNAs. The disease–miRNA relation network was reconstructed by integrating the global similarity relation. The network consistency diffusion seed is then obtained by combining the global similarity network with the reconstructed disease–miRNA association network. Thereafter, the stable diffusion spectrum is generated as the prediction score by using the restarted random walk algorithm. The AUC value obtained by performing the LOOCV in the gold benchmark dataset is 0.8814. The AUC value obtained by performing the LOOCV in the predictive dataset is 0.9512. Compared with other frontier methods, our method has higher accuracy, which is further illustrated by case studies of breast neoplasms and colon neoplasms to prove that IDNC is valuable.
Introduction
RNA is the intermediate between DNA and encoded protein. It has a variety of important functions and is ubiquitous in organisms. The RNA that is not involved in the process of encoding protein is called non-coding RNA. About 98% of the human genome sequences are non-coding regions.1 miRNA means the single-strand endogenous non-coding RNA with a length of about 20–25 nucleotides and is evolutionarily conserved. miRNAs are widely distributed in eukaryotes. In human genes, the number of miRNAs can account for 1–4%2–5 of the total. The discovery of miRNA has been initially considered and has not received much attention. However, in recent years, there has been increasing evidence that shows the correlation between miRNAs and many life processes, such as cell growth,6,7 tissue differentiation,8 cell proliferation,9 embryonic development,10 apoptosis,11 metabolism12,13 and so on.Recently, miRNAs have been found to be closely related to human tumors, especially the changes in the expression of miRNAs are involved in the occurrence, progression and metastasis of various types of human cancer.14 For example, hsa-mir-10b is unregulated in breast neoplasms compared with benign breast lesions;15 hsa-mir-126 and hsa-mir-145 can inhibit the growth of colonic tumor cells;16,17 hsa-mir-21 has higher expression level in colon cancer cells;18 Gao et al.19 found that the expression of hsa-mir-155 in serum of lung cancer patients was much higher than that of normal samples by PCR test; Johnson20 found that the down regulation of the let-7 family led to the development of lung cancer.
The identification of disease-related miRNAs is of great importance to human health. Identifying the interaction between miRNAs and diseases have become a key issue. Many researchers have worked hard to identify the interaction. The association between miRNAs and diseases can be accurately mined through sophisticated biological experiments but it is subject to high cost, long experimental period, and high requirements on equipment.21–24 In recent years, with the discovery of a large number of miRNAs, researchers have developed a variety of databases to store related information about miRNAs. With these data as the background, more and more bioinformatics calculation methods have been developed to predict their relationship.25–30 This kind of calculation method is the best supplement to biological experiments. The advantages include reducing the blindness of biologists' biological experiments, reducing the cost of biological experiments, and saving the human and material resources of biological experiments. At present, this method can be roughly divided into machine learning method and biological network method.31–33 The methods of predicting disease-associated miRNA are elaborated below from these two aspects.
In 2010, Jiang et al.34 extracted positive sample data from negative sample data by using support vector machine. The method extracted features from miRNA target data and phenotypic similarity data, which achieved good prediction results. In 2011, Xu et al.35 took prostate cancer as an example and proposed an MTDN calculation method to predict prostate cancer related miRNA by using miRNA target topology imbalance network. In 2016, Zeng et al.36 adopted two multipath methods to predict the association between miRNA and disease. All of these methods require the negative sample information of known disease-related miRNA, while negative miRNA–disease association is hard to obtain.
In 2014, Chen et al. proposed a regularized least squares semi supervised algorithm (RLSMDA)to predict potential miRNA–disease association.37 This method is built on semi supervised learning framework to predict potential disease–miRNA association and does not require related negative miRNA–disease information. In 2017, Chen et al.38 proposed LRSSLMDA model to predict miRNA–disease association with Laplacian Regularized Sparse Subspace Learning. In 2017, Peng et al.39 proposed a new information fusion strategy RLSSLP based on the regularization framework and the idea of Kronecker's regularized least squares based on multi-core learning. In 2017, Chen et al.40 established a MKRMDA model that can automatically optimize the combination of disease and miRNA multi-source data and efficiently use multi-core data to predict the potential association of miRNA–diseases. In 2017, Luo et al.41 used Kronecker regularized least squares to predict miRNA related diseases based on heterogeneous omics data.
Matrix factorization is designed to predict the association between disease and miRNA.42 In 2016, Lan et al.43 proposed a computational model kbmf-md to predict the association between miRNA and disease based on the improved microRNA and disease similarities. In 2016, Lan et al.44 used nuclear Bayesian matrix factorization to forecast potential miRNA–disease association. In 2018, Xiao et al.45 integrated the semantic information of diseases with the functional information of the miRNA to obtain the isomeric data, and then used the isomeric data to regularize the non-negative matrix factorization of the graph to predict the potential association between miRNA and the disease, which is called GRNMF. In 2018, Zhong et al.46 constructed a double layer network to express the complex relationship between miRNA, disease and miRNA–disease, and then sorted out the non-negative matrix factorization method to predict the potential disease miRNA. In 2018, Chen et al.47 developed a computational model of matrix decomposition and heterogeneous graph inference for miRNA–disease association prediction.
In addition, neural network and other algorithms are designed to predict the association between disease and miRNA. In 2017, Chen et al.48 proposed model EGBMMDA based on the relationship between Extreme Gradient Boosting Machine to predict association between miRNA and disease. In 2017, Chen et al.49 developed a miRNA–disease association prediction model DRMDA based on depth representation. After data extraction and depth representation, the unsupervised hierarchical layer-by-layer greedy pre-training and Support Vector Machine were used to predict the miRNA–disease association. In 2017, Fu et al.50 proposed a deep integration model, DeepMDA, which used an automatic encoder to extract advanced features from similar information, and then used a three layer neural network to predict the association between miRNAs and diseases. In 2015, Chen et al.51 used a Restricted Boltzmann Machine (RBM) to predict different types of miRNA–disease associations by applying RBMMMDA method. In 2017, Luo et al.52 developed a predictive method CPTL based on transduction learning.
However, previous studies are not adequate and have some disadvantages, such as the lack of miRNAs similarity data and the facts that known relationship between miRNAs and diseases is scarce and that there are few negative samples. In 2016, Zeng et al.53 proposed a method to predict miRNA–disease association by matrix completion algorithm based on miRNA–miRNA network and disease–disease network. In 2017, Li et al.54 propose MCMDA method to predict miRNA–disease association by using matrix completion algorithm. In 2017, Peng et al.55 used the improved low rank matrix recovery (ILRMR) algorithm to predict the correlation between miRNAs and diseases. In this method, it is possible to predict diseases which are not associated with any known miRNA. In 2018, Chen et al.56 presents a novel model of inductive matrix completion for miRNA–disease association prediction. Zhao et al.57 used symmetric nonnegative matrix factorization to reveal the relation of miRNA–disease pairs.
Bioinformatics researchers also utilized recommendation system.58 In 2014, Li et al.59 developed a computational system toxicology framework which used the recommendation system to predict the new association of environmental factors, miRNA and diseases by integrating the structural similarity of environmental factors and the phenotype similarity of diseases. In 2017, Gu et al.60 applies collaborative filtering recommendation algorithms to the miRNA–disease association prediction. In 2017, Peng et al.61 combined rating-based recommendation algorithm with negative-aware algorithm to predict miRNA–disease association. In 2017, Chen et al.62 proposed a new computational model HAMDA for miRNA–disease association by using hybrid graph-based recommendation algorithm. HAMDA not only considered the network structure and information dissemination, but also discussed the problem of node assignment. A satisfactory prediction result was achieved.
In 2015, inspired by social network analysis, Zou et al.63 proposed to using the methods based on social network analysis for the prediction of miRNA–disease association. They used two social network analysis methods, KATZ and CATAPULT, to analyze heterogeneous networks. CATAPULT is a deformation of supervised learning algorithm and can overcome the shortcoming that there are only positive samples and unmarked samples in miRNA–disease association. In 2018, Chen et al.64 proposed a computational model of Ensemble Learning and Link Prediction for miRNA–disease association prediction.
Based on the hypothesis, that functionally related miRNA tends to associate with phenotypically similar diseases, many calculation methods have been proposed to predict the potential association between miRNA and disease.25–27
In 2009, Jiang et al.65 first proposed a hypergeometric distribution model to predict miRNA–disease correlation. In 2010, Jiang et al.66 proposed a new method based on genomic data integration, integrating a variety of data resources with naive Bayes model and establishing a functional prediction model among genes. In 2011, Li et al. put forward a method of genes' functional consistency to predict carcinogenic miRNA.67 In 2013, Shi et al. further proposed a computational model that exploits the functional association between miRNAs and diseases.68 They integrated the disease–target association, the known disease–gene association, the protein inter-association to create a complex network. Then they made use of the random walk algorithm on the network and achieved a good prediction effect. In 2014, Xu et al.69 proposed a disease-associated miRNA prediction method which integrated the phenotypically similar miRNAs with mRNAs expression profiles. However, these methods depend on the prediction of miRNA–target association, and the false positive of the target gene is high. So they cannot obtain high predictive performance.
In 2011, Rossi et al.70 proposed a method called OMiR to predict the association of diseases in miRNA and OMIM. They calculated the degree of overlap between miRNA loci and disease loci in OMIM as the correlation between miRNA and disease. Xuan et al. proposed a prediction method based on weighted k most similar neighbors, which is called HDMP.71 However, HDMP cannot be applied to the prediction of isolated diseases. In 2017, Chen et al.72 designed a novel KNN-based disease-related sorting algorithm (RKNNMDA). In 2015, Le et al.73 used PageRank and k-step Markov algorithm, a classic algorithm for web page ranking in link analysis to predict the association between disease and miRNAs.
In 2012, Chen et al.74 proposed a RWRMDA computing model based on the similarity of global networks to predict the miRNA–disease association. They utilized the restarted random walk method to predict the pathogenetic miRNA. The results demonstrated that the global similarity network can improve the prediction accuracy more than the local similarity network. However, this method cannot predict new diseases without any known association. In 2013 and 2016, Shi68,75 integrated data such as protein–protein and gene ontology data to build heterogeneous networks where the random walk algorithm can also be employed to predict. In 2015, Xuan et al.76 designed a computing model named MIDP based on random walk algorithm. In 2015, Liao et al.77 designed a global similarity prediction model based on information diffusion, which is known as NDBM. In 2017, Luo et al.78 implemented the unbalanced bi-random walk algorithm (BRWH) on heterogeneous networks to search two-part graph sub-graphs to discover potential miRNA–disease associations. In 2017, Mugunga et al.79 combined the path-based features and the random walk algorithm to predict the association between miRNA and disease.
In 2013, Chen et al. proposed Net-CBI method to predict the relationship between miRNA and disease by using the consistency of disease network.80 In 2016, Gu et al.81 designed a network consistency method to predict miRNA–disease association (NCPMDA). In 2017, Li et al.82 proposed an integrated network similarity method (NSIM).
In 2015, Nalluri et al.83 designed two scientific methods from the perspective of graph theory: one is to choose the maximum weighted matching inference model of the dominant disease by solving an equation; the other is based on the model of motivation analysis. In 2016, Chen et al.84 constructed a heterogeneous graph method to predict miRNA–disease association method, which is called HGIMDA. In 2017, You et al.85 proposed A novel and effective path-based miRNA–disease association prediction method, PBMDA, which uses a unique depth-first search algorithm to search in the isomeric graph. In 2016, Sun et al.86 proposed a method to predict the association between them by using network topological similarity of miRNA–disease correlation network, which is called NTSMDA. In 2018, Chen et al.87 proposed a novel computational model of triple layer heterogeneous network based inference for miRNA–disease association prediction. Chen et al.88 proposed a method of graph regression to predict the miRNA–disease association.
In 2016, Chen et al.89 developed the model of within and between score to predict potential miRNAs associated with various complex diseases. In 2017, Chen et al.90 used the graphlet interaction of miRNAs (diseases) to represent the complex relationship between any two miRNAs (diseases), and established a GIMDA model for predicting the potential miRNA–diseases association by calculating the number of interactions of different types. In 2017, Chen et al.91 introduced the concepts of “super miRNA” and “super disease” to strengthen the similarity measurement of disease and miRNA. In 2018, Li et al.92 present a label propagation model with linear neighborhood similarity to predict unobserved miRNA–disease associations.
To sum up, due to the complexity of biological systems and the limitations of existing research methods, some problems and challenges exist in the field of disease–miRNA association prediction: firstly, the prediction accuracy is not high; secondly, many algorithms isolate disease and new miRNA prediction without known association; thirdly, the method of similarity construction is not reasonable in most of the current models; the fourth is the problem of model defects. At present, many machine-learning methods either need negative samples or have difficulties in model training. Some methods based on biological networks use local information instead of global information, which results in poor prediction accuracy. Many methods have data dependence problem. The generalization ability of some methods is not strong. Some methods have good prediction ability for a data set but not satisfactory for other data sets. It is urgent to develop simple, effective and universal models for disease-related miRNA prediction.
In view of the shortcomings of the algorithm described above, we designed an information diffusion disease association prediction method based on network consistency to reveal the potential relationship between miRNA and disease. On the basis of building disease and miRNA global similarity network, this method reconstructs two disease–miRNA association networks. By using the consistency of the network to capture the comprehensive information of the vector, the information diffusion method is used to forecast the correlation. The experimental results show that the proposed method has some advantages: no need for negative samples; the ability to predict isolated disease and new miRNA, the simple design of the algorithm and so on. In the comparison of methods, our method is superior to other methods on different data sets, and case studies show better prediction ability of the algorithm.
Materials and methods
Data preparation
We first downloaded 270 miRNA–disease pairs from the literature,27 removed 19 miRNAs that could not be found in the literature,27 and kept 99 miRNAs and 51 diseases including 242 disease–miRNA pairs, which we refer to as the gold standard dataset.To verify that our method has better universality, we downloaded another disease–miRNA association data set from the literature,27 which contains 1616 experimentally verified human miRNA–disease associations. After merging different miRNA records and unifying the names of miRNA and disease, the data set eventually retained 1395 disease–miRNA associations, including 271 miRNA and 137 diseases. We refer to the data set as predictive dataset.
miRNA–miRNA functional similarity score is downloaded from the literature.27 The data set is successfully applied to multiple methods.80,93–95 We use matrix SM to represent the adjacency matrix of miRNA, and SM(i, j) is the score of functional similarity score between miRNA i and miRNA j.
Disease similarity data are downloaded from the literature.96 We use matrix SD to represent the adjacency matrix of disease, SD(i, j) representing the similarity score between di and disease dj. The family information of miRNA is obtained from miRBase database.97 The family information of miRNA is represented by matrix SMfam. If two miRNAs are in the same family, the corresponding set SMfam(i, j) is 1, otherwise it will set 0.
Algorithm flow
The basic work flow of disease-related miRNA prediction method based on network consistency has four steps (Fig. 1). Namely: | ||
| Fig. 1 The flow chart of information diffusion method based on network consistency is divided into four steps: (1) building a global similarity network. (2) Reconstruction of disease–miRNA association network. (3) Information diffusion based on network consistency. (4) Information fusion. | ||
Step 1: similarity network construction
We integrate the known information of disease–miRNA association and the similarity of the disease semantic to obtain the similarity network of the disease. Then we use the Laplacian score of graphs to find the similarity of the disease to express the similarity between the diseases. We use the miRNA family information and the miRNA function similarity data to construct the miRNA similarity network. Laplacian score of graphs is used to find the global similarity of miRNA to represent the similarity between miRNA.Based on the hypothesis that the phenotype resemblance of disease tendency is associated with function related miRNA,27 we believe that the more common miRNA of two diseases are, the more similar the two diseases are. When the two diseases share the same amount of miRNA, if the miRNA of these two diseases is less, the two diseases are more similar. When there is no common miRNA between disease di and disease dj, the score of known association network structure is set to 0 directly. The formula is as follows:
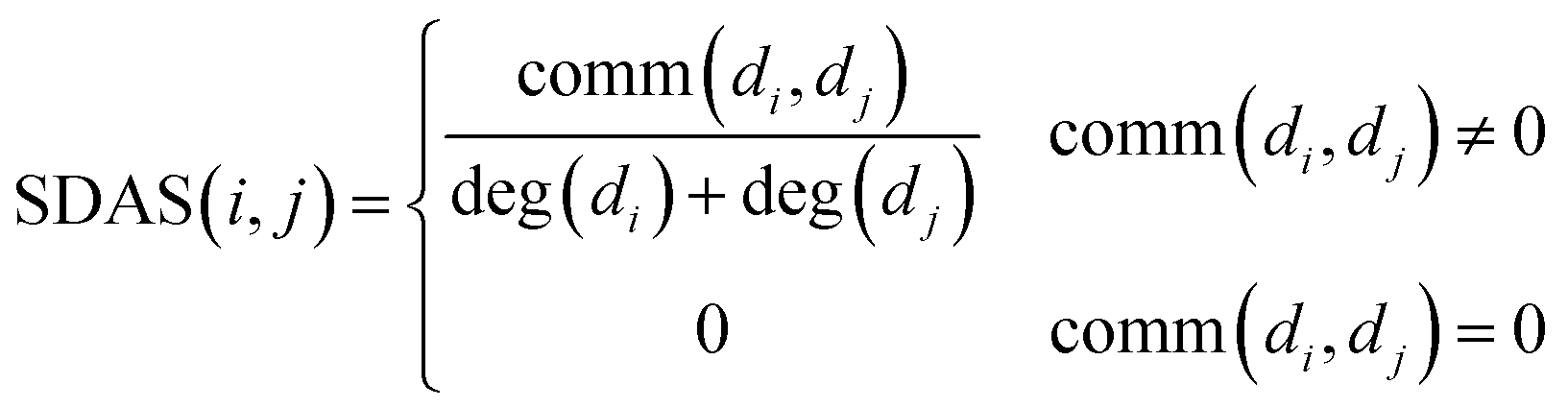 | (1) |
Then we integrate the semantic correlation information of the disease and the score of the known association network structure to get the weighted score.
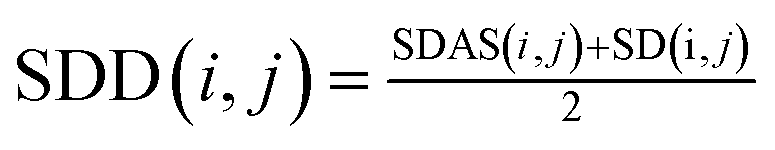 | (2) |
Then we seek global similarity. Binary vector d = {d1, d2, …, dn} is used to represent the initial vector of disease di. The corresponding di values were set to 1 and the rest were 0. The global similarity between diseases is obtained by Laplacian score of graphs ![[d with combining tilde]](https://www.rsc.org/images/entities/i_char_0064_0303.gif) . It can be solved by the optimization problem of formula (3).98
. It can be solved by the optimization problem of formula (3).98
 | (3) |
In formula (3), the previous one is a smooth penalty item, 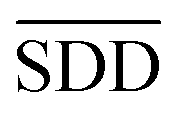 is the normalized matrix of the matrix SDD, which guarantees similar score for similar diseases. The second penalty items obtained the consistency between the disease and other diseases. α is a balance factor with a range of α ∈ (0,1). This factor is used to balance two penalty items' weight in formula (3). The approximate solution of formula (3) is as follows98:
is the normalized matrix of the matrix SDD, which guarantees similar score for similar diseases. The second penalty items obtained the consistency between the disease and other diseases. α is a balance factor with a range of α ∈ (0,1). This factor is used to balance two penalty items' weight in formula (3). The approximate solution of formula (3) is as follows98:
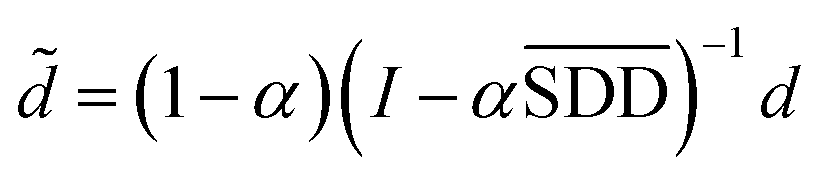 | (4) |
With the above method, we can get the global similarity score of all diseases in the disease network, which is represented by matrix SDLAPLACIAN.
Bandyopadhyay et al.26 found that the more the shared mRNA target targets were in the same family miRNA, the more similar their functions were. In order to make full use of family information of miRNA, we give higher weight to miRNA belonging to the same family when constructing miRNA network.
We use the following formula to calculate the similarity score of miRNA:
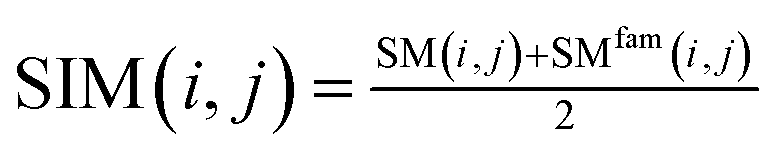 | (5) |
Among them, SIM(i, j) represents the similarity score between miRNA node mi and miRNA node mj after merging two kinds of information. SIM(i, j) is a functional similarity score between miRNA mi and miRNA mj calculated by Wang et al.27 SMfam is the miRNA family information matrix. When miRNA mi and miRNA mj are in the same family, SMfam(i, j) equals 1, which gives a higher score between two miRNA.
Then the global similarity weight matrix of miRNA is obtained by finding Laplacian score of graphs:
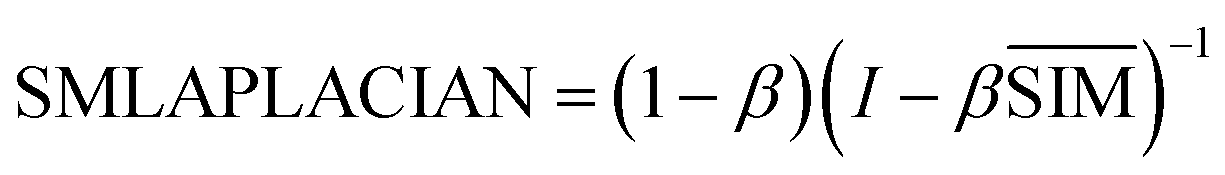 | (6) |
SMLAPLACIAN represents miRNA global similarity network score matrix. I is a nm dimensional unit matrix, and nm is the total number of miRNA. 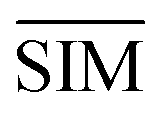 is the normalization matrix of miRNA similarity score SIM. β is a balance factor and β ∈ (0, 1).
is the normalization matrix of miRNA similarity score SIM. β is a balance factor and β ∈ (0, 1).
Step2: the reconstruction of disease–miRNA association network
From the previous analysis, we know that the known experimentally validated disease–miRNA association network is a Boolean bipartite network, which cannot fully characterize the tightness of the disease–miRNA association. We restructured the disease–miRNA association network by using the global similarity of the disease and the global similarity of the miRNA. Respectively, they are accounted as the disease–miRNA correlation network ASm based on the global similarity information of miRNA and the disease–miRNA correlation network ASd based on the global similarity information of the disease.
 | (7) |
Among them, ASm(j, i) is the weight of disease node dj to miRNA node mi in disease–miRNA bipartite network after reconstruction. AS(i, j) is the weight of miRNA nodes mi and disease nodes dj in the bipartite network before reconstruction. (In the experimentally verified disease–miRNA Boolean bipartite network, if the miRNA node mi is known to be associated with the disease node dj, the value is 1. Otherwise the value is 0.) AS′ is the transposed matrix of AS. SMLAPLACIAN(mi, mk) is the weight between the miRNA node mi and the miRNA node mk in the miRNA global similarity network. nm is the total number of miRNA, and λ is a balance parameter.
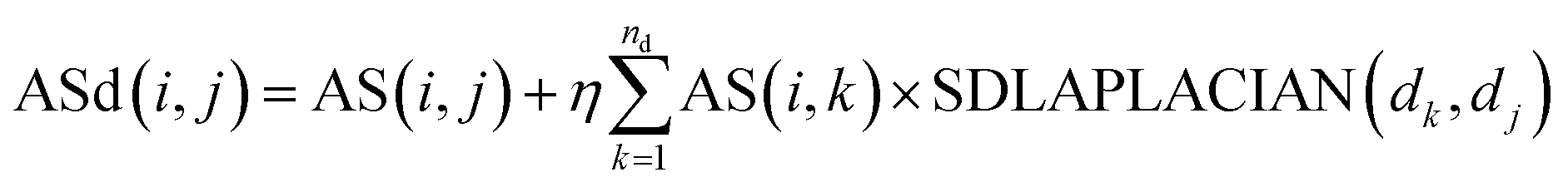 | (8) |
Among them, ASd(i, j) is the weight of miRNA node mi and disease node dj in the reconstructed miRNA–disease bipartite network. AS(i, j) is used to reconstruct the weight of miRNA nodes mi and disease nodes dj in the miRNA–disease bipartite network before reconstruction. SDLAPLACIAN(dk, dj) is the weight of disease nodes dk and disease nodes dj in the global similarity network of diseases. nd is the total number of diseases. η is a balance parameter.
Step3: information diffusion based on network consistency
Based on the hypothesis that functionally similar miRNA is usually associated with phenotypically similar diseases, we designed an information diffusion method based on network consistency to reveal the potential association between miRNA and disease. We use network consistency to describe the relationship between two vectors in the same order and the same object. By using the similarity in the change rule of these two vectors, we can get comprehensive information of two heterogeneous networks. The projection of vectors can be used to express the degree of association between two vectors.| SEEDM = SDLAPLACIAN × ASm | (9) |
Next, in order to accurately describe the degree of association between miRNA nodes and disease nodes, we used random walk algorithm to walk in the global similarity network of disease, and captured the stable distribution of information called stable spread spectrum. Then the data of stable diffusion spectrum are utilized to represent the correlation between miRNA nodes and disease nodes. After the matrix normalization, each column is the seed sequence of associations between the miRNA node mi and all the disease nodes. The stable diffusion spectrum is obtained by SDLAPLACIAN random diffusion of these seed sequences in the adjacency matrix of the disease consistency network.
 | (10) |
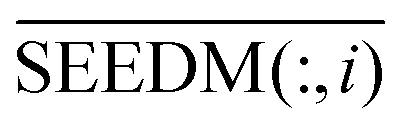 is the information of column I after the normalization of SEEDM matrix. The column vector is the seed sequence of the associations between miRNA node mi and all disease nodes.
is the information of column I after the normalization of SEEDM matrix. The column vector is the seed sequence of the associations between miRNA node mi and all disease nodes.  is the normalized matrix of the adjacency matrix SDLAPLACIAN of the disease consistency network. γ is the restart probability. MTDt(i) vector represents the information distribution after t iterations. After several iterations, the probability space can reach the steady state MTD∞(i) (|MTDt+1(i) − MTDt(i)| < 10−6) and stop the iteration. When the state is stable, the value of the vector is the correlation score between miRNA node mi and each disease. The correlation scores of all miRNA nodes and disease nodes are expressed by matrix MTD.
is the normalized matrix of the adjacency matrix SDLAPLACIAN of the disease consistency network. γ is the restart probability. MTDt(i) vector represents the information distribution after t iterations. After several iterations, the probability space can reach the steady state MTD∞(i) (|MTDt+1(i) − MTDt(i)| < 10−6) and stop the iteration. When the state is stable, the value of the vector is the correlation score between miRNA node mi and each disease. The correlation scores of all miRNA nodes and disease nodes are expressed by matrix MTD.
| SEEDD = SMLAPLACIAN × ASd | (11) |
The seed matrix of the disease node dj is obtained through the above formula. After normalization of the matrix, each column is used as the seed sequence of the disease node dj and all miRNA. These seed sequences are SMLAPLACIAN randomly spread in the adjacency matrix of the miRNA consistency network in order to obtain stable diffusion spectra:
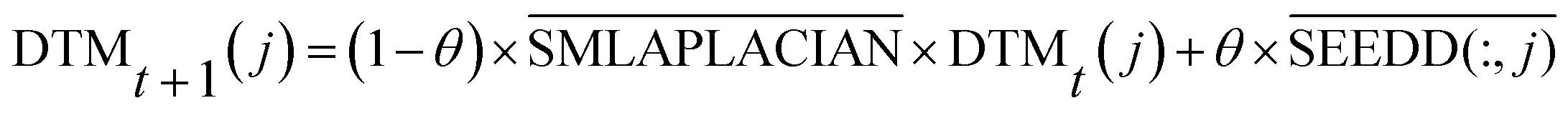 | (12) |
 is the normalized matrix of the adjacency matrix SMLAPLACIAN of the miRNA consistency network. θ is the restart probability. DTMt(j) vector represents information distribution after t iterations. After several iterations, the probability space can reach a stable state DTM∞(j) (|DTMt+1(j) − DTMt(j)| < 10−6), and then the iteration can be stopped. Each value of the vector represents the correlation score of disease j and each miRNA. The correlation score of all diseases and each miRNA is expressed by matrix DTM.
is the normalized matrix of the adjacency matrix SMLAPLACIAN of the miRNA consistency network. θ is the restart probability. DTMt(j) vector represents information distribution after t iterations. After several iterations, the probability space can reach a stable state DTM∞(j) (|DTMt+1(j) − DTMt(j)| < 10−6), and then the iteration can be stopped. Each value of the vector represents the correlation score of disease j and each miRNA. The correlation score of all diseases and each miRNA is expressed by matrix DTM.
Step4: information fusion
Finally, we integrated the two prediction scores obtained in the third step to form the final prediction score.| MD(i, j) = w × MTD′(i, j) + (1 − w) × DTM(i, j) | (13) |
Results
Parameter selection
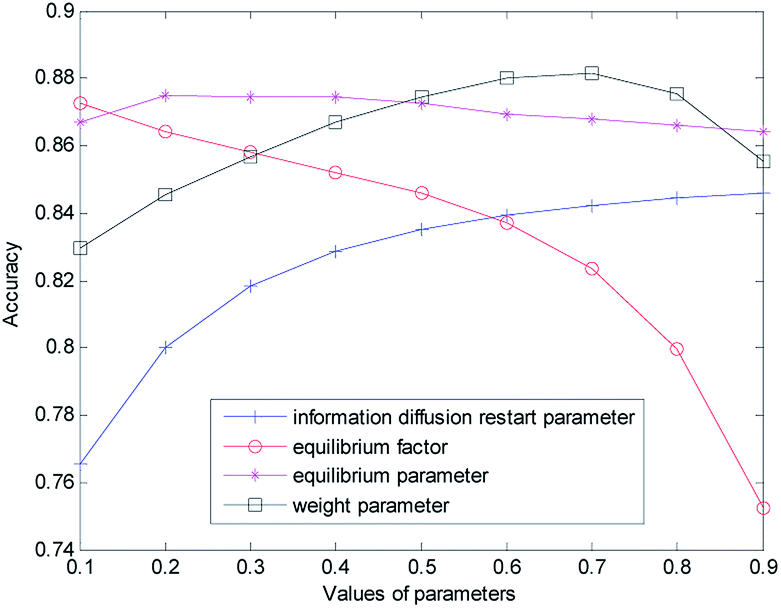
The proposed method has four kinds of parameters: the information diffusion restart parameters γ and θ; the equilibrium factor α constructing the disease global similarity network, the equilibrium factor β constructing the miRNA global similarity network; equilibrium parameter λ based on global similarity network information for reconstructing the disease–miRNA association network ASm of miRNA, equilibrium parameter η based on disease global similarity network information for reconstructing the miRNA–disease association network ASd; the weight parameter w of information diffusion disease-related miRNA prediction score based on network consistency.The selection and influence of these four kinds of parameters are discussed respectively. In the process of information diffusion, γ and θ indicate the probability of repetitive random walks that represent random callbacks to the source node. The greater γ and θ are, the greater the probability of returning the node for each step is. For the sake of simplicity, we set γ and θ to the same size. To verify the impact of γ and θ on the performance of the prediction algorithm, the other parameters are fixed (α = β = λ = η = w = 0.5) while the values of γ and θ are changed (0.1 for step length, from 0.1 to 0.9) to do cross-validation on the gold benchmark dataset and to calculate the AUC value. The experimental results are shown in Fig. 2. In the experiment, we found that when γ and θ increased from 0.1 to 0.9, the AUC value increased gradually from 0.7656 to 0.8460. The best prediction performance was obtained when the maximum value was obtained at 0.9.
 | ||
| Fig. 2 Influence of parameter variation on model prediction accuracy. | ||
Then we set the balance factor α of the disease global similarity network and the balance factor β of the miRNA consistency network as the same. To verify the impact of such parameters on the predictive performance of the algorithm, other parameters are fixed on the basis of the previously obtained parameters (γ = θ = 0.9, λ = η = w = 0.5), and then the α and β values are changed (with 0.1 for step length, from 0.1 to 0.9). As you can see from Fig. 2 with the increase of α and β, the AUC value gradually decreases. When α = β = 0.1, the AUC value is the largest and the prediction performance is the best.
In order to measure the degree of disease–miRNA association more accurately, we used the global similarity of the disease and the global similarity of miRNA to reconstruct the disease–miRNA association network respectively. The balance parameters λ and η determine the contribution rate of other diseases and other miRNA to the disease–miRNA association network. To verify the impact of the two parameters on the predictive performance of the algorithm, other parameters are fixed on the basis of the previously obtained parameters (γ = θ = 0.9, α = β = 0.1, w = 0.5), and then the λ and η values are changed (from 0 to 0.9) for cross-validation. In the experiment, it was found that the AUC value was 0.8670 when the set value is 0.1 (0.8748 when the set value is 0.2; 0.8745 when the set value is 0.3; 0.8743 when the set value is 0.4). At this time, the AUC value was not very different. When the set value changes from 0.4, AUC decreased slowly. With the increase of λ and η, the AUC value became smaller and decreased to 0.8618 when the set value is 0.9.
In order to obtain the best prediction performance, we got the final correlation prediction score of the miRNA–disease association by weighting the miRNA–disease association prediction algorithm score (based on miRNA network consistency information diffusion) and the disease–miRNA association prediction algorithm score (based on disease network consistency information diffusion). The score weight parameter of miRNA–disease correlation prediction based on miRNA network consistency information diffusion is set as w (0 ≤ w ≤ 1), then 1 − w is the weight of disease–miRNA association prediction score based on disease network consistency information diffusion. When the w is larger, the weight of the miRNA–disease correlation prediction score based on miRNA network consistency information diffusion is greater, which means that the prediction results take more consideration of the miRNA–disease correlation prediction score based on miRNA network consistency information diffusion. When the w is smaller, the prediction results take more consideration of the disease–miRNA association prediction score based on disease network consistency information diffusion. Based on the previous discussion, we fixed the values of other parameters (γ = θ = 0.9, α = β = 0,1, λ = η = 0.3), and then changed the value of w (from 0 to 0.9). When w increases from 0.1 to 0.7, the AUC value increases gradually. When the w increases from 0.7 to 0.9, the AUC value gradually decreases. When w is 0.7, the prediction effect is the best, and AUC achieves the maximum value of 0.8814. When λ and η are set as 0.2 and 0.4, the experiment result is similar, that is, when w is 0.7, the prediction effect is the best.
Finally, we determine that the parameters are: γ = θ = 0.9, α = β = 0,1, λ = η = 0.3, w = 0.7.
Performance evaluation
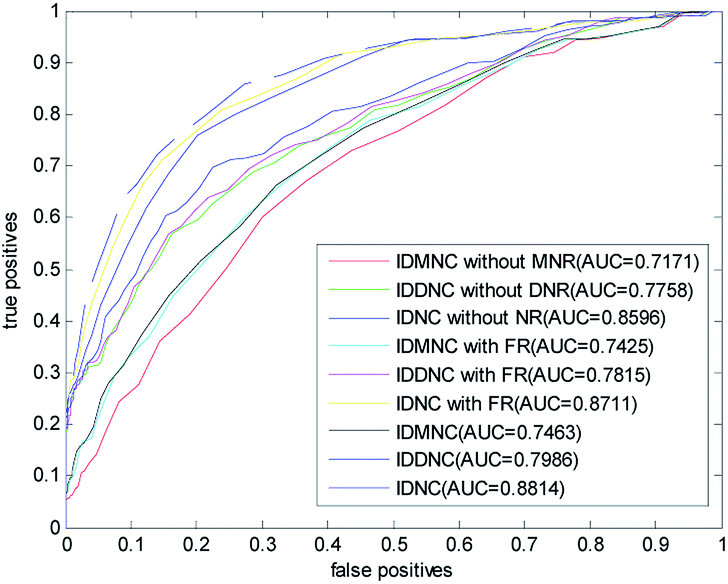
In this paper, a disease-related miRNA prediction model based on network consistency information diffusion is proposed, which is the integration of the miRNA–disease correlation prediction score (based on miRNA network consistency information diffusion) and the disease–miRNA correlation prediction score (based on disease network consistency information diffusion). In the construction of the similarity network, we restructured the disease and miRNA in different ways, and used the global similarity score as the similarity score to measure the relationship between the nodes. When we tested the model, we considered the following nine cases falling into three categories: (1) information diffusion method based on miRNA network consistency without considering the miRNA network reconstruction (IDMNC without MNR); (2) information diffusion method based on disease network consistency without considering the disease network reconstruction (IDDNC without DNR); (3) information diffusion method based on network consistency without considering the network reconstruction (IDNC without NR); (4) information diffusion method based on miRNA network consistency by considering the miRNA network reconstruction with family information (IDMNC with FR); (5) information diffusion method based on disease network consistency by considering the miRNA network reconstruction with family information (IDDNC with FR); (6) information diffusion method based on network consistency by considering the miRNA network reconstruction with family information (IDNC with FR); (7) information diffusion method based on miRNA network consistency by considering the network reconstruction (IDMNC); (8) information diffusion method based on disease network consistency by considering the network reconstruction (IDDNC); (9) information diffusion method based on network consistency by considering the network reconstruction (IDNC). Based on the above conditions, parameters are selected on the gold standard dataset: γ = θ = 0.9, α = β = 0,1, λ = η = 0.3, w = 0.7. The calculated ROC curve and the AUC value are shown in Fig. 3.From Fig. 3, Information diffusion based on miRNA network consistency method, information diffusion based on disease network consistency method and information diffusion method based on network consistency method are gradually improved in the prediction accuracy.
 | ||
| Fig. 3 The ROC curve and AUC value for LOOCV in three classes of nine cases. | ||
The prediction accuracies of non network reconstruction, reconstruction of miRNA network with family information, reconstruction of both disease and miRNA network are gradually improved. When using all the information, the AUC value is 0.8814. When the method is information diffusion based on miRNA network consistency without network reconstruction, AUC value is only 0.7171. This fully demonstrated the effectiveness of our method of restructuring network and the feasibility of integrating the two scoring methods with the weighted method.
Comparison with other methods
We compared the algorithm proposed in this paper with three classical methods RLSMDA,37 NetCBI,99 GSTRW. In the LOOCV assessment, each known miRNA–disease association is considered as a test sample, while other known associations are considered as training samples. The miRNA–disease association without known evidence is considered to be a candidate sample. In the gold datum data set, the AUC value of NetCBI is 0.8001; the AUC value of RLSMDA is 0.8059; the AUC value of GSTRW is 0.8479; and the AUC value of the algorithm proposed in this paper is 0.8841, which is far superior to the other three methods. The ROC curves and AUC values of the four methods on the gold datum data set are shown in Fig. 4.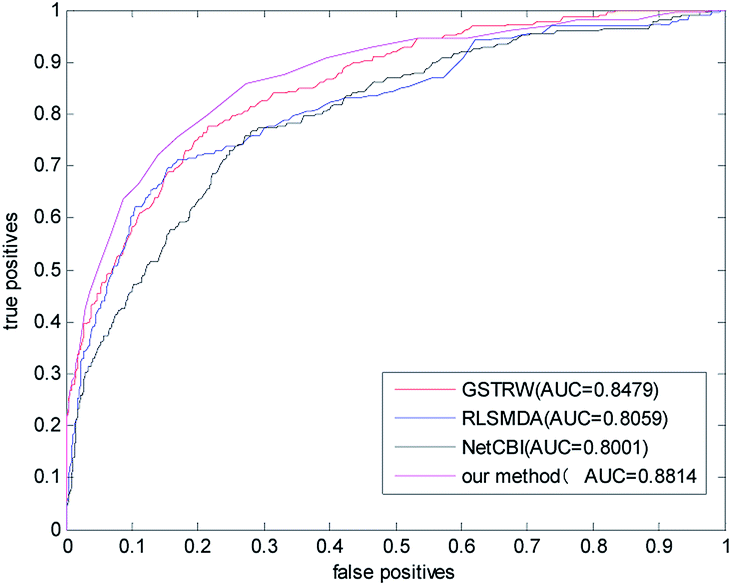 | ||
| Fig. 4 The ROC curve and AUC value of our method compared with other methods on the gold benchmark dataset. | ||
In order to avoid data dependence, we further verified the four methods on the forecast data set, and the AUC values of the four methods in the forecast dataset have been greatly improved. As shown in Fig. 5, the AUC value of NetCBI is 0.9053; the AUC value of RLSMDA is 0.9232; the AUC value of GSTRW is 0.9434; and the AUC value of the algorithm proposed in this paper is 0.9512. This is mainly due to the increase in the number of available disease–miRNA associations, and the higher accuracy of the constructed similarity network, which makes the prediction accuracy increase. Both in the gold datum data set, or in the forecast data set, the methods presented in this paper have shown strong predictive ability, especially in the case of less number of disease–miRNA associations. Because the method proposed in this paper takes advantage of global similarity and network consistency, the algorithm proposed in this paper has more advantages.
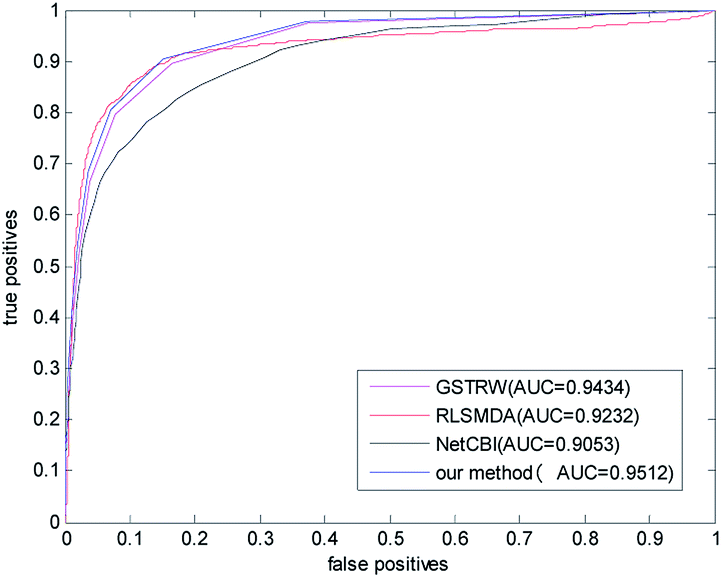 | ||
| Fig. 5 The ROC curve and AUC value of our method compared with other methods in the predictive dataset. | ||
The prediction of new miRNA and isolated disease
The new miRNA refers to the unknown miRNA associated with the disease. With the discovery of a large number of unknown miRNA, the new miRNA prediction becomes more important in the prediction of disease–miRNA association. This paper also used the LOOCV to predict the new miRNA. One by one, we removed the association information of verified miRNA with all other diseases and simulated them as new miRNA. In the gold benchmark dataset, the AUC value of our method is 0.8087. Its ROC curve and the AUC value are shown in Fig. 6, which is higher than the AUC value predicted by RLSMDA and NetCBI for the common disease. This shows that our method has a better prediction ability for the new miRNA.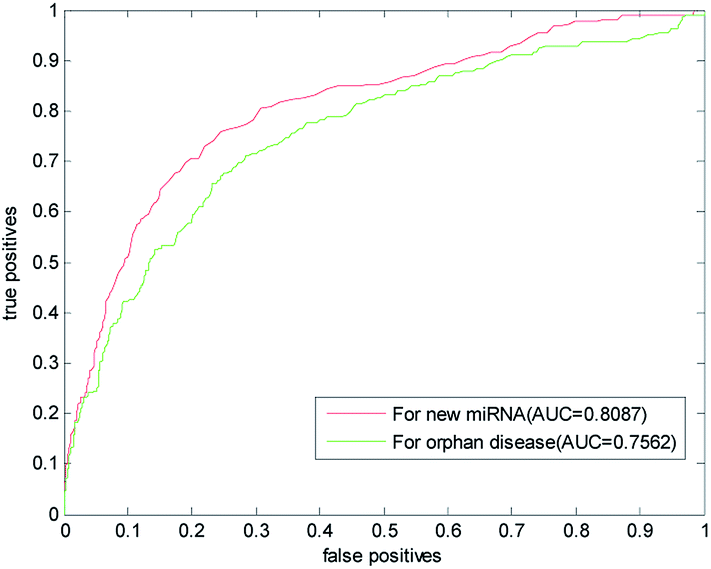 | ||
| Fig. 6 Results of our prediction method of new miRNA and isolated diseases in gold datum dataset. | ||
Isolated diseases refer to diseases whose associations with miRNA are unknown. Prediction of isolated diseases is also a difficult problem to be solved in the prediction of disease–miRNA associations. Similarly, in order to test the predictive performance of this article on isolated diseases, we removed the associations between disease and miRNA. The ROC curve and AUC value obtained with LOOCV are listed in Fig. 6,It can be seen from the figure that the AUC predicted by this algorithm for isolated diseases is 0.7562. This shows that our method has certain predictive ability for isolated diseases, but the accuracy of prediction needs to be further improved.
Case studies
In 2017, 135![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 430 new cases of colon neoplasms were reported in the United States. Among them, 50260 cases of colon neoplasms led to death.100 Colon neoplasms is associated with many miRNA, such as miR-126, which inhibits the growth of colon tumor cells;16 miR-21 has a high expression level in colon neoplasms cells.18 Using the calculation method to predict the association between colon neoplasms and miRNA can help us to diagnose the cancer patients in the early stage. This is of great importance to increase the survival rate of colon neoplasms patients. Therefore, it is urgent to develop a scientific method to forecast the miRNA which is related to colon neoplasms disease. In the forecast dataset, 37 miRNA related to colon tumors were experimentally verified. We used the method proposed in this article to experiment with colon tumors by using these 37 known associations and considering both disease similarity and miRNA similarity. Among the first 50 unknown disease–miRNA associations got in the experiment, 45 miRNA got supporting evidence in the updated HMDD, miR2Disease, and dbDEMC data sets (shown in Table 1). Only 5 miRNA have not been verified, which are hsa-mir-199a, hsa-mir-92b, hsa-mir-200a, hsa-mir-373 and hsa-mir-216b. However, in previous literatures, we have found supporting evidence, for example: Nonaka et al.101 found that miR-199a could be used as a biomarker for colorectal cancer; Mussnich et al.102 found that miR-199a and miR-375 affect the sensitivity of colon neoplasms cells to cetuximab by targeting PHLPP1. Niu et al.103 stated that hsa-miR-92b can be used as a reference gene for circulating microRNA in colorectal cancer. Pichler et al.104 found that miR-200a regulates the prognosis of patients with rectal cancer by regulating the expression of epithelial mesenchymal metastasis related genes. Tanaka et al.105 found that the apparent silencing of microRNA-373 played an important regulatory role in the proliferation of colon neoplasms cells. Previous studies also suggested that these miRNA are closely related to colon neoplasms, such as hsa-mir-199a and hsa-mir-200a, which are predicted to be associated with colon tumors in PBMDA,85 MCMDA,54 EGBMMDA.48 The two miRNA, hsa-mir-92b and hsa-mir-200a, were predicted to be associated with colon neoplasms in the case analysis of RLSMDA. These documents are published after the latest update date of the three databases, which fully demonstrates the strong predictive power of our method.
430 new cases of colon neoplasms were reported in the United States. Among them, 50260 cases of colon neoplasms led to death.100 Colon neoplasms is associated with many miRNA, such as miR-126, which inhibits the growth of colon tumor cells;16 miR-21 has a high expression level in colon neoplasms cells.18 Using the calculation method to predict the association between colon neoplasms and miRNA can help us to diagnose the cancer patients in the early stage. This is of great importance to increase the survival rate of colon neoplasms patients. Therefore, it is urgent to develop a scientific method to forecast the miRNA which is related to colon neoplasms disease. In the forecast dataset, 37 miRNA related to colon tumors were experimentally verified. We used the method proposed in this article to experiment with colon tumors by using these 37 known associations and considering both disease similarity and miRNA similarity. Among the first 50 unknown disease–miRNA associations got in the experiment, 45 miRNA got supporting evidence in the updated HMDD, miR2Disease, and dbDEMC data sets (shown in Table 1). Only 5 miRNA have not been verified, which are hsa-mir-199a, hsa-mir-92b, hsa-mir-200a, hsa-mir-373 and hsa-mir-216b. However, in previous literatures, we have found supporting evidence, for example: Nonaka et al.101 found that miR-199a could be used as a biomarker for colorectal cancer; Mussnich et al.102 found that miR-199a and miR-375 affect the sensitivity of colon neoplasms cells to cetuximab by targeting PHLPP1. Niu et al.103 stated that hsa-miR-92b can be used as a reference gene for circulating microRNA in colorectal cancer. Pichler et al.104 found that miR-200a regulates the prognosis of patients with rectal cancer by regulating the expression of epithelial mesenchymal metastasis related genes. Tanaka et al.105 found that the apparent silencing of microRNA-373 played an important regulatory role in the proliferation of colon neoplasms cells. Previous studies also suggested that these miRNA are closely related to colon neoplasms, such as hsa-mir-199a and hsa-mir-200a, which are predicted to be associated with colon tumors in PBMDA,85 MCMDA,54 EGBMMDA.48 The two miRNA, hsa-mir-92b and hsa-mir-200a, were predicted to be associated with colon neoplasms in the case analysis of RLSMDA. These documents are published after the latest update date of the three databases, which fully demonstrates the strong predictive power of our method.
| Rank | miRNA name | Evidences | Rank | miRNA name | Evidences |
|---|---|---|---|---|---|
| 1 | hsa-mir-196a | dbDEMC, miR2Disease | 26 | hsa-mir-421 | dbDEMC |
| 2 | hsa-mir-199a | Unconfirmed | 27 | hsa-mir-15b | dbDEMC, miR2Disease |
| 3 | hsa-mir-448 | dbDEMC | 28 | hsa-mir-30d | dbDEMC |
| 4 | hsa-mir-25 | dbDEMC | 29 | hsa-mir-29a | HMDD, dbDEMC, miR2Disease |
| 5 | hsa-mir-122 | dbDEMC | 30 | hsa-mir-451 | dbDEMC, miR2Disease |
| 6 | hsa-mir-181b | dbDEMC, miR2Disease | 31 | hsa-mir-203 | dbDEMC |
| 7 | hsa-mir-18b | dbDEMC | 32 | hsa-mir-212 | dbDEMC |
| 8 | hsa-mir-224 | dbDEMC | 33 | hsa-mir-30b | dbDEMC |
| 9 | hsa-mir-15a | HMDD, dbDEMC | 34 | hsa-mir-106b | HMDD, miR2Disease, dbDEMC |
| 10 | hsa-mir-92b | Unconfirmed | 35 | hsa-mir-214 | dbDEMC |
| 11 | hsa-mir-372 | dbDEMC, miR2Disease | 36 | hsa-mir-98 | dbDEMC |
| 12 | hsa-mir-34c | dbDEMC | 37 | hsa-mir-220 | dbDEMC |
| 13 | hsa-mir-200a | Unconfirmed | 38 | hsa-mir-137 | HMDD, dbDEMC, miR2Disease |
| 14 | hsa-mir-190 | dbDEMC | 39 | hsa-mir-33a | dbDEMC |
| 15 | hsa-mir-217 | dbDEMC | 40 | hsa-mir-216b | Unconfirmed |
| 16 | hsa-mir-222 | dbDEMC | 41 | hsa-mir-33b | dbDEMC |
| 17 | hsa-mir-205 | HMDD, dbDEMC | 42 | hsa-mir-216a | dbDEMC |
| 18 | hsa-mir-93 | dbDEMC | 43 | hsa-mir-199b | dbDEMC |
| 19 | hsa-mir-20b | dbDEMC | 44 | hsa-mir-429 | dbDEMC |
| 20 | hsa-mir-135b | HMDD, miR2Disease, dbDEMC | 45 | hsa-mir-376c | dbDEMC |
| 21 | hsa-mir-34b | dbDEMC | 46 | hsa-mir-16 | HMDD, dbDEMC |
| 22 | hsa-mir-29c | dbDEMC | 47 | hsa-mir-146b | dbDEMC |
| 23 | hsa-mir-373 | Unconfirmed | 48 | hsa-mir-302b | HMDD, dbDEMC |
| 24 | hsa-mir-125b | dbDEMC | 49 | hsa-mir-125a | dbDEMC, miR2Disease |
| 25 | hsa-mir-9 | dbDEMC | 50 | hsa-mir-95 | dbDEMC |
Breast neoplasms is one of the most important causes of cancer death in women every year. So many scientists have been studying the pathology of breast neoplasms. The study of the relationship between microRNA and breast neoplasms can help us understand the development of the disease at a molecular point of view. Of the first 50 unknown associations for breast neoplasms, only 3 were not verified: hsa-mir-518b, hsa-mir-612 and hsa-mir-657, which are shown in Table 2. hsa-miR-21 is significantly associated with many diseases, which can be proved by breast neoplasms related evidences in HMDD, miR2Disease and dbDEMC. Persson et al.106 stated that hsa-miR-4656 is associated with breast neoplasms. hsa-miR-21, hsa-miR-612 and hsa-miR-4656 share many common target genes.107 This indicates that these miRNA may have similar biological processes. So we highly believe that hsa-miR-612 is associated with breast neoplasms. In addition, we found that the three miRNA appeared in the breast neoplasms related miRNA collection in SDMMDA.91 Among them, hsa-mir-518b is located in the fifth position while hsa-mir-612 and hsa-mir-657 are located in the 22nd and 23rd positions respectively.
| Rank | miRNA name | Evidences | Rank | miRNA name | Evidences |
|---|---|---|---|---|---|
| 1 | hsa-mir-518b | Unconfirmed | 26 | hsa-mir-658 | dbDEMC |
| 2 | hsa-mir-518c | dbDEMC | 27 | hsa-mir-575 | dbDEMC |
| 3 | hsa-mir-612 | Unconfirmed | 28 | hsa-mir-423 | HMDD, dbDEMC |
| 4 | hsa-mir-600 | dbDEMC | 29 | hsa-mir-500 | dbDEMC |
| 5 | hsa-mir-629 | HMDD, dbDEMC | 30 | hsa-mir-346 | HMDD, dbDEMC |
| 6 | hsa-mir-622 | dbDEMC | 31 | hsa-mir-99a | dbDEMC |
| 7 | hsa-mir-638 | HMDD, dbDEMC | 32 | hsa-mir-130b | dbDEMC |
| 8 | hsa-mir-486 | HMDD, dbDEMC | 33 | hsa-mir-208b | dbDEMC |
| 9 | hsa-mir-596 | dbDEMC | 34 | hsa-mir-134 | dbDEMC |
| 10 | hsa-mir-557 | dbDEMC | 35 | hsa-mir-433 | dbDEMC |
| 11 | hsa-mir-642 | dbDEMC | 36 | hsa-mir-484 | dbDEMC |
| 12 | hsa-mir-769 | dbDEMC | 37 | hsa-mir-663 | dbDEMC |
| 13 | hsa-mir-602 | dbDEMC | 38 | hsa-mir-365 | HMDD, dbDEMC |
| 14 | hsa-mir-611 | dbDEMC | 39 | hsa-let-7e | HMDD, dbDEMC |
| 15 | hsa-mir-185 | dbDEMC | 40 | hsa-mir-494 | dbDEMC |
| 16 | hsa-mir-583 | dbDEMC | 41 | hsa-let-7i | HMDD, miR2Disease, dbDEMC |
| 17 | hsa-mir-615 | dbDEMC | 42 | hsa-let-7b | HMDD, dbDEMC |
| 18 | hsa-mir-654 | dbDEMC | 43 | hsa-mir-198 | dbDEMC |
| 19 | hsa-mir-662 | dbDEMC | 44 | hsa-mir-373 | HMDD, miR2Disease, dbDEMC |
| 20 | hsa-mir-601 | dbDEMC | 45 | hsa-mir-203 | HMDD, miR2Disease, dbDEMC |
| 21 | hsa-mir-324 | HMDD, dbDEMC | 46 | hsa-mir-223 | HMDD, dbDEMC |
| 22 | hsa-mir-608 | HMDD | 47 | hsa-let-7g | HMDD, dbDEMC |
| 23 | hsa-mir-637 | dbDEMC | 48 | hsa-mir-101 | HMDD, dbDEMC |
| 24 | hsa-mir-657 | Unconfirmed | 49 | hsa-mir-92b | dbDEMC |
| 25 | hsa-mir-197 | HMDD, dbDEMC | 50 | hsa-let-7c | HMDD, dbDEMC |
The prediction of isolated disease and new miRNA
In order to verify our algorithm's ability to predict isolated diseases, we removed the known associations of miRNAs with the proven diseases, which ensures that we only use the similarity information of the confirmed disease and other diseases and the miRNAs information associated with other diseases. We used colon neoplasms and breast neoplasms as case studies. The results are shown in Tables 3 and 4 respectively.| Rank | miRNA name | Evidences | Rank | miRNA name | Evidences |
|---|---|---|---|---|---|
| 1 | hsa-mir-21 | HMDD, miR2Disease, dbDEMC | 26 | hsa-mir-19b | HMDD, miR2Disease, dbDEMC |
| 2 | hsa-mir-15a | HMDD, dbDEMC | 27 | hsa-mir-92a | HMDD, dbDEMC |
| 3 | hsa-mir-451 | dbDEMC, miR2Disease | 28 | hsa-let-7a | HMDD, miR2Disease, dbDEMC |
| 4 | hsa-mir-373 | Unconfirmed | 29 | hsa-mir-10a | dbDEMC, miR2Disease |
| 5 | hsa-mir-16 | HMDD, dbDEMC | 30 | hsa-mir-205 | HMDD, dbDEMC |
| 6 | hsa-mir-155 | HMDD, miR2Disease, dbDEMC | 31 | hsa-mir-211 | Unconfirmed |
| 7 | hsa-mir-29c | dbDEMC | 32 | hsa-mir-200b | HMDD, dbDEMC |
| 8 | hsa-mir-34a | HMDD, miR2Disease, dbDEMC | 33 | hsa-mir-196a | dbDEMC, miR2Disease |
| 9 | hsa-mir-19a | HMDD, miR2Disease, dbDEMC | 34 | hsa-mir-181a | dbDEMC, miR2Disease |
| 10 | hsa-mir-17 | HMDD, dbDEMC | 35 | hsa-mir-141 | HMDD, miR2Disease, dbDEMC |
| 11 | hsa-mir-221 | HMDD, miR2Disease, dbDEMC | 36 | hsa-let-7e | HMDD, dbDEMC |
| 12 | hsa-mir-125b | dbDEMC | 37 | hsa-mir-145 | HMDD, miR2Disease, dbDEMC |
| 13 | hsa-mir-302b | HMDD, dbDEMC | 38 | hsa-mir-223 | HMDD, miR2Disease, dbDEMC |
| 14 | hsa-mir-372 | dbDEMC, miR2Disease | 39 | hsa-let-7d | HMDD, dbDEMC |
| 15 | hsa-mir-143 | HMDD, miR2Disease, dbDEMC | 40 | hsa-let-7b | HMDD, miR2Disease, dbDEMC |
| 16 | hsa-mir-20a | HMDD, miR2Disease, dbDEMC | 41 | hsa-mir-9 | dbDEMC |
| 17 | hsa-mir-184 | dbDEMC | 42 | hsa-let-7c | HMDD, dbDEMC |
| 18 | hsa-mir-181b | dbDEMC, miR2Disease | 43 | hsa-let-7i | HMDD, dbDEMC |
| 19 | hsa-mir-29a | HMDD, dbDEMC, miR2Disease | 44 | hsa-let-7f | HMDD, dbDEMC |
| 20 | hsa-mir-122 | dbDEMC | 45 | hsa-let-7g | HMDD, miR2Disease, dbDEMC |
| 21 | hsa-mir-18a | HMDD, miR2Disease, dbDEMC | 46 | hsa-mir-15b | dbDEMC, miR2Disease |
| 22 | hsa-mir-146a | HMDD, dbDEMC | 47 | hsa-mir-92b | Unconfirmed |
| 23 | hsa-mir-222 | dbDEMC | 48 | hsa-mir-30a | HMDD, dbDEMC |
| 24 | hsa-mir-212 | dbDEMC | 49 | hsa-mir-126 | HMDD, dbDEMC |
| 25 | hsa-mir-137 | HMDD, dbDEMC, miR2Disease | 50 | hsa-mir-19b | HMDD, miR2Disease, dbDEMC |
| Rank | miRNA name | Evidences | Rank | miRNA name | Evidences |
|---|---|---|---|---|---|
| 1 | hsa-mir-21 | HMDD, miR2Disease, dbDEMC | 26 | hsa-mir-10a | HMDD, miR2Disease, dbDEMC |
| 2 | hsa-mir-146a | HMDD, miR2Disease, dbDEMC | 27 | hsa-mir-211 | dbDEMC |
| 3 | hsa-mir-125b | HMDD, miR2Disease, dbDEMC | 28 | hsa-mir-137 | HMDD, dbDEMC |
| 4 | hsa-mir-373 | HMDD, miR2Disease, dbDEMC | 29 | hsa-mir-141 | HMDD, miR2Disease, dbDEMC |
| 5 | hsa-mir-155 | HMDD, miR2Disease, dbDEMC | 30 | hsa-mir-223 | HMDD, dbDEMC |
| 6 | hsa-mir-16 | HMDD, dbDEMC | 31 | hsa-let-7e | HMDD, dbDEMC |
| 7 | hsa-mir-451 | HMDD, miR2Disease | 32 | hsa-mir-200b | HMDD, miR2Disease, dbDEMC |
| 8 | hsa-mir-29c | HMDD, dbDEMC | 33 | hsa-mir-146b | HMDD, miR2Disease |
| 9 | hsa-mir-34a | HMDD, dbDEMC | 34 | hsa-let-7b | HMDD, dbDEMC |
| 10 | hsa-mir-19a | HMDD, dbDEMC | 35 | hsa-mir-181a | HMDD, miR2Disease, dbDEMC |
| 11 | hsa-mir-17 | HMDD, dbDEMC | 36 | hsa-let-7d | HMDD, miR2isease, dbDEMC |
| 12 | hsa-mir-184 | Unconfirmed | 37 | hsa-let-7c | HMDD, dbDEMC |
| 13 | hsa-mir-221 | HMDD, miR2Disease | 38 | hsa-let-7i | HMDD, miR2Disease, dbDEMC |
| 14 | hsa-mir-15a | HMDD, dbDEMC | 39 | hsa-mir-9 | HMDD, dbDEMC |
| 15 | hsa-mir-302b | HMDD, miR2Disease | 40 | hsa-let-7f | HMDD, miR2Disease, dbDEMC |
| 16 | hsa-mir-20a | HMDD, dbDEMC | 41 | hsa-let-7g | HMDD, dbDEMC |
| 17 | hsa-mir-29a | HMDD, dbDEMC | 42 | hsa-mir-143 | HMDD, miR2Disease, dbDEMC |
| 18 | hsa-mir-372 | HMDD, dbDEMC | 43 | hsa-mir-145 | HMDD, miR2Disease, dbDEMC |
| 19 | hsa-mir-18a | HMDD, dbDEMC | 44 | hsa-mir-92b | dbDEMC |
| 20 | hsa-mir-222 | HMDD, dbDEMC | 45 | hsa-mir-30a | HMDD, dbDEMC |
| 21 | hsa-mir-181b | HMDD, miR2Disease, dbDEMC | 46 | hsa-mir-150 | HMDD, dbDEMC |
| 22 | hsa-mir-19b | HMDD, dbDEMC | 47 | hsa-mir-15b | dbDEMC |
| 23 | hsa-mir-92a | HMDD, dbDEMC | 48 | hsa-mir-127 | HMDD, miR2Disease, dbDEMC |
| 24 | hsa-let-7a | HMDD, miR2Disease, dbDEMC | 49 | hsa-mir-203 | HMDD, miR2Disease, dbDEMC |
| 25 | hsa-mir-205 | HMDD, miR2Disease, dbDEMC | 50 | hsa-mir-126 | HMDD, miR2Disease, dbDEMC |
For colon neoplasms, 37 known associations of miRNAs with colon neoplasms were removed. Among the first 50 miRNAs predicted, 47 miRNAs were identified in the three databases while three miRNAs, hsa-mir-373, hsa-mir-211 and hsa-mir-92b, failed to find support in the three databases, which is shown in Table 3. However, Cai et al.108 found that hsa-miR-211 promoted the growth of colorectal cancer cells through targeting CHD5. The other two miRNAs were predicted in previous cases about colon tumor. As mentioned above, a number of references to the association of these miRNAs and colonic tumors are also introduced. Therefore, we think our algorithm performs well for the prediction of isolated diseases.
For breast neoplasms, we deleted 78 known associations of breast neoplasms with miRNAs. We used this method to predict a potential association between miRNAs and breast neoplasms. In the first 50 miRNAs projections, 49 were found in the HMDD, miR2Disease, and dbDEMC databases, and only hsa-mir-184 had not been found in the three databases. However, when Yang et al.109 studied the classification of breast tumor subtypes by immunohistochemical markers, it was found that there were differences in expression of hsa-miR-365, hsa-miR-1238 and hsa-miR-184.
Next, we studied the new miRNA association prediction. hsa-mir-21 plays a crucial role in carcinogenesis and can be used as a biomarker for detecting various cancers. In this section, we removed all the associations of hsa-mir-21 with diseases in the forecast data set. Among the first 50 projected diseases related to hsa-mir-21, 40 diseases are verified in the above three databases while 10 kinds of diseases that are not verified, which is shown in Table 5. But previous literature show that these diseases are associated with hsa-mir-21. For example, Han et al.110 discovered that hsa-mir-21 can slow down the apoptosis of cortical neurons by promoting PTEN-Akt signaling pathway in vitro after traumatic brain injury. Montalban et al.111 found that hsa-mir-21 could regulate the growth factor signal and regulate the degeneration of neurons in PC12 cells. Smigielska et al.112 found that hsa-mir-21 plays a role in supporting the survival of T cells in CD4+T cells. Zhang et al.113 found that hsa-mir-21 is associated with the development of liver fibrosis. Ding et al.114 found that hsa-miR-21 could be used as a new biomarker for diagnosing HBV related acute liver failure through real-time quantitative PCR technology. Liao et al.115 found that 80% of the patients with hepatocellular carcinoma have the background of chronic hepatitis B or type C hepatitis and cirrhosis, and hsa-miR-21 can be used for subdivision of hepatocellular carcinoma and chronic hepatitis. Yao et al.116 found that compared with patients with obstructive spermatozoa, miRNA in spermatocytes, such as hsa-miR-21, was decreased in patients with non obstructive spermatozoa. Gutsaeva et al.117 found that hsa-mir-21 is closely related to new vascularization in ischemic retina. Andrade et al.118 found differential expression of 11 kinds of miRNA (such as hsa-miR-424 and hsa-miR-21) in the muscles of the patients with amyotrophic lateral sclerosis (rapidly progressive neurodegenerative disease) by microarray. miR-21 plays a crucial role in carcinogenesis,119 which can be used as a diagnostic and prognostic marker for digestive cancers for Asians. These documents were published after the last update date of these three databases, which fully demonstrates the effectiveness of our method.
| Rank | miRNA name | Evidences | Rank | miRNA name | Evidences |
|---|---|---|---|---|---|
| 1 | Heart failure | HDMM | 26 | Lymphoma, B-cell | HMDD, miR2Disease |
| 2 | Breast neoplasms | HMDD, miR2Disease, dbDEMC | 27 | Colorectal eoplasms | HMDD, miR2Disease, dbDEMC |
| 3 | Lung neoplasms | HMDD, miR2Disease, dbDEMC | 28 | Hodgkin disease | HMDD, miR2Disease |
| 4 | Ovarian neoplasms | HDMM | 29 | Carcinoma, renal cell | HMDD, miR2Disease, dbDEMC |
| 5 | Neoplasms | HDMM | 30 | Hepatitis, chronic | Unconfirmed |
| 6 | Melanoma | HMDD, dbDEMC | 31 | Lymphoma | HDMM |
| 7 | Adrenocortical carcinoma | dbDEMC | 32 | Azoospermia | Unconfirmed |
| 8 | Muscular disorders, atrophic | HDMM | 33 | Hepatitis C | Unconfirmed |
| 9 | Stomach neoplasms | HDMM | 34 | Lymphoma, primary effusion | dbDEMC |
| 10 | Pancreatic neoplasms | HMDD, dbDEMC | 35 | Sarcoma, kaposi | dbDEMC |
| 11 | Lupus vulgaris | HDMM | 36 | Cardiomyopathy, hypertrophic | HMDD, miR2Disease |
| 12 | Colonic neoplasms | HMDD, dbDEMC | 37 | Pituitary neoplasms | Unconfirmed |
| 13 | Autistic disorder | HDMM | 38 | Uterine cervical neoplasms | HMDD, dbDEMC |
| 14 | Prostatic neoplasms | HDMM | 39 | Waldenstrom macroglobulinemia | Unconfirmed |
| 15 | Head and neck neoplasms | HDMM | 40 | Polycythemia vera | HDMM |
| 16 | Carcinoma, hepatocellular | HMDD, miR2Disease, dbDEMC | 41 | Digestive system neoplasms | Unconfirmed |
| 17 | Salivary gland neoplasms | HDMM | 42 | Urinary bladder neoplasms | HDMM |
| 18 | Adenocarcinoma | HDMM | 43 | Leukemia, B-cell | dbDEMC |
| 19 | Schizophrenia | Unconfirmed | 44 | Leukemia, promyelocytic, acute | dbDEMC |
| 20 | Endometriosis | HDMM | 45 | Precursor B-cell lymphoblastic leukemia-lymphoma | miR2Disease |
| 21 | Leukemia, lymphocytic, chronic, B-cell | HMDD, miR2Disease, dbDEMC | 46 | Retinal neovascularization | Unconfirmed |
| 22 | Medulloblastoma | HDMM | 47 | ACTH-secreting pituitary adenoma | HDMM |
| 23 | Leukemia, myeloid, acute | miR2Disease, dbDEMC | 48 | Neurodegenerative diseases | Unconfirmed |
| 24 | Leukemia | HDMM | 49 | Multiple myeloma | HMDD, dbDEMC |
| 25 | Thyroid neoplasms | HMDD, dbDEMC | 50 | Hepatitis B | Unconfirmed |
Discussion and conclusions
miRNA has been found associated with the development of many complex diseases. miRNA imbalance can be regarded as a biomarker for complex disease diagnosis. Although biological experiments can be used to predict disease–related miRNA, it takes much time and lots of efforts to use biological experiments. The calculation method for predicting potential associations between miRNAs and diseases is an effective complement to biological experiments. A reasonable similarity relationship of diseases and miRNAs can improve the prediction accuracy of the calculation method. In order to build a reasonable similarity relationship, we first reconstructed the miRNA network by combining the miRNA family information and the miRNA function similarity, and reconstructed the disease network by using the semantic scores between the known disease and the association information of the miRNA and the disease. Then the global similarity of the two networks is obtained by Laplasse operator. The similarity between diseases and miRNA is measured by global similarity score. Thereafter, the disease–miRNA association network ASm based on the global similarity information of miRNA was constructed by using the global similarity of the miRNA nodes and the known diseases–miRNA relationship. The disease–miRNA correlation network ASd based on disease global similarity information was constructed by using the global similarity of disease nodes and the known disease–miRNA relationship. Then the consistency information between vectors is obtained by projection of vectors. By using this information to diffuse the disease and miRNA global network respectively, a stable diffusion spectrum was obtained as a corresponding prediction score. Finally, the weighted average of two prediction scores was used as the final score of disease–miRNA association miRNA prediction. This method does not need negative samples and can predict isolated disease and new miRNA. The design of the algorithm is simple. The AUC value of the LOOCV experiment in the gold datum dataset is up to 0.8814, and the AUC value in the forecast data set is up to 0.9512, which is superior to the methods of others. In the case study, we also chose breast tumor and colon tumor for experimental research. Among the top 50 and the corresponding disease related miRNAs predictions, the accuracy rate in the updated HDMM, miR2Disease and dbDEMC databases were 94% and 90% respectively. In the prediction of isolated disease cases, 98% and 94% of the top 50 were confirmed by the three databases mentioned above. Finally, we simulated hsa-mir-21 as a new miRNA for prediction. Of the top 50 diseases predicted, 40 were verified by the database. The rests have found supporting evidence in the latest literature, showing predictive capability of our method.The algorithm presented in this paper shows strong predictive capability, mainly due to the following reasons. Firstly, we added family information to reconstruct the miRNA similarity network, and integrate the known miRNA related disease information and the disease phenotype similarity information to reconstruct the disease network; secondly, we used the Laplasse operator to obtain the global similarity of both miRNA network and disease network; thirdly, we reconstructed the disease–miRNA correlation network by adding the global similarity information of the network; the fourth is the use of network consistency to get data association between miRNA and disease. Although the disease-related miRNA prediction model based on IDNC has achieved a satisfactory prediction performance, there are still some defects. Firstly, there are too many parameters. It takes a lot of time to find the best parameter for different data sets; secondly, the construction of disease and miRNA similarity network needs more data to be integrated for accuracy; thirdly, the accuracy of prediction for isolated diseases and new miRNA needs to be improved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research of this paper has been sponsored by National Nature Science Foundation of China (Grant No. 61772192, 61672214, 61672223), Nature Science Foundation of Hunan Province, China (Grant No. 2018JJ2085), Science-Technology of Hunan Province, China(Grant No. 2015GK3029), Science-Technology of Hengyang City, China (Grant No. 2016KJ17, 2012KS19), Major cultivation projects of Hunan Institute of Technology (Grant No. 2017HGPY001).References
- J. S. Mattick and I. V. Makunin, Hum. Mol. Genet., 2006, 15, R17–R29 CrossRef CAS PubMed.
- G. Meister and T. Tuschi, Nature, 2004, 431, 343 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- V. Ambros, Cell, 2001, 107, 823–826 CrossRef CAS PubMed.
- V. Ambros, Nature, 2004, 431, 350 CrossRef CAS PubMed.
- L. Zhu, J. Zhao, J. Wang, C. Hu, J. Peng, R. Luo, C. Zhou, J. Liu, J. Lin and Y. Jin, PLoS Pathog., 2016, 12, e1005423 CrossRef PubMed.
- T. R. Fernando, N. I. Rodriguez-Malave and D. S. Rao, J. Hematol. Oncol., 2012, 5, 7 CrossRef CAS PubMed.
- E. A. Miska, Curr. Opin. Genet. Dev., 2005, 15, 563–568 CrossRef CAS PubMed.
- A. M. Cheng, M. W. Byrom, J. Shelton and L. P. Ford, Nucleic Acids Res., 2005, 33, 1290–1297 CrossRef CAS PubMed.
- V. Ambros, Cell, 2003, 113, 673–676 CrossRef CAS PubMed.
- P. Xu, M. Guo and B. A. Hay, Trends Genet., 2004, 20, 617–624 CrossRef CAS PubMed.
- M. Alshalalfa and R. Alhajj, BMC Bioinf., 2013, 14, S1 CrossRef PubMed.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- S. Volinia, M. Galasso, S. Costinean, L. Tagliavini, G. Gamberoni, A. Drusco, J. Marchesini, N. Mascellani, M. E. Sana and R. A. Jarour, Genome Res., 2010, 20, 589–599 CrossRef CAS PubMed.
- L. Yong, Z. Jing, P. Y. Zhang, Z. Yu, S. Y. Sun, S. Y. Yu and Q. S. Xi, Med. Sci. Monit., 2012, 18, BR299 Search PubMed.
- C. Guo, F. J. Sah, L. Beard, J. K. V. Willson, S. D. Markowitz and K. Guda, Genes, Chromosomes Cancer, 2008, 47, 939–946 CrossRef CAS PubMed.
- B. Shi, L. Sepp-Lorenzino, M. Prisco, P. Linsley and R. Baserga, J. Biol. Chem., 2007, 282, 32582–32590 CrossRef CAS PubMed.
- A. J. Schetter, S. Y. Leung, J. J. Sohn, K. A. Zanetti, E. D. Bowman, N. Yanaihara, S. T. Yuen, T. L. Chan, D. L. Kwong and G. K. Au, JAMA, J. Am. Med. Assoc., 2008, 299, 425–436 CAS.
- F. Gao, J. Chang, H. Wang and G. Zhang, Oncol. Rep., 2014, 31, 351–357 CrossRef CAS PubMed.
- S. M. Johnson, H. Grosshans, J. Shingara, M. Byrom, R. Jarvis, A. Cheng, E. Labourier, K. L. Reinert, D. Brown and F. J. Slack, Cell, 2005, 120, 635–647 CrossRef CAS PubMed.
- C. C. Pritchard, H. H. Cheng and M. Tewari, Nat. Rev. Genet., 2012, 13, 358 CrossRef CAS PubMed.
- H. Dong, J. Lei, L. Ding, Y. Wen, H. Ju and X. Zhang, Chem. Rev., 2013, 113, 6207 CrossRef CAS PubMed.
- X. Li, Bioinformatics, 2017, 33, 2829–2836 CrossRef PubMed.
- X. Li, Curr. Bioinf., 2018, 13, 367–372 CrossRef CAS.
- M. Lu, Q. Zhang, M. Deng, J. Miao, Y. Guo, W. Gao and Q. Cui, PLoS One, 2008, 3, e3420 CrossRef PubMed.
- S. Bandyopadhyay, R. Mitra, U. Maulik and M. Q. Zhang, Silence, 2010, 1, 6 CrossRef PubMed.
- D. Wang, J. Wang, M. Lu, F. Song and Q. Cui, Bioinformatics, 2010, 26, 1644–1650 CrossRef CAS PubMed.
- X. Chen, L. Y. Wang and L. Huang, J. Cell. Mol. Med., 2018, 22, 2884–2895 CrossRef CAS PubMed.
- L.-H. Peng, C.-N. Sun, N.-N. Guan, J.-Q. Li and X. Chen, Mol. Genet. Genomics, 2018, 1–13 Search PubMed.
- G. Li, J. Luo, Q. Xiao, C. Liang, P. Ding and B. Cao, IEEE Access, 2017, 5, 24032–24039 Search PubMed.
- X. Zeng, X. Zhang and Q. Zou, Briefings Bioinf., 2016, 17, 193–203 CrossRef CAS PubMed.
- Q. Zou, J. Li, L. Song, X. Zeng and G. Wang, Briefings Funct. Genomics, 2016, 15, 55–64 CAS.
- X. Chen, D. Xie, Q. Zhao and Z.-H. You, Briefings Bioinf., 2017, 10, 1–25 CAS.
- Q. Jiang, G. Wang, T. Zhang and Y. Wang, Predicting human microRNA-disease associations based on support vector machine, in 2010 IEEE International Conference On Bioinformatics and Biomedicine (BIBM), 2010, pp. 467–472 Search PubMed.
- J. Xu, C.-X. Li, J.-Y. Lv, Y.-S. Li, Y. Xiao, T.-T. Shao, X. Huo, X. Li, Y. Zou and Q.-L. Han, Mol. Cancer Ther., 2011, 10, 1857–1866 CrossRef CAS PubMed.
- X. Zeng, Z. Xuan, Y. Liao and L. Pan, Biochim. Biophys. Acta, 2016, 1860, 2735–2739 CrossRef CAS PubMed.
- X. Chen and G.-Y. Yan, Sci. Rep., 2014, 4, 5501 CrossRef CAS PubMed.
- X. Chen and L. Huang, PLoS Comput. Biol., 2017, 13, e1005912 CrossRef PubMed.
- L. Peng, M. Peng, B. Liao, Q. Xiao, W. Liu, G. Huang and K. Li, RSC Adv., 2017, 7, 44447–44455 RSC.
- X. Chen, Y. W. Niu, G. H. Wang and G. Y. Yan, J. Transl. Med., 2017, 15, 251 CrossRef PubMed.
- J. Luo, Q. Xiao, C. Liang and P. Ding, IEEE Access, 2017, 5, 2503–2513 Search PubMed.
- G. Li, J. Luo, Q. Xiao, C. Liang and P. Ding, RSC Adv., 2018, 8, 4377–4385 RSC.
- W. Lan, J. Wang, M. Li, J. Liu, F. X. Wu and Y. Pan, IEEE/ACM Trans. Comput. Biol. Bioinf., 2016, 1 CAS.
- W. Lan, J. Wang, M. Li, J. Liu and Y. Pan, Predicting microRNA-disease associations by integrating multiple biological information, in IEEE International Conference on Bioinformatics and Biomedicine, 2015, pp. 183–188 Search PubMed.
- Q. Xiao, J. Luo, C. Liang, J. Cai and P. Ding, Bioinformatics, 2018, 34, 239–248 CrossRef PubMed.
- Y. Zhong, P. Xuan, X. Wang, T. Zhang, J. Li, Y. Liu and W. Zhang, Bioinformatics, 2018, 34, 267–277 CrossRef PubMed.
- X. Chen, J. Yin, J. Qu and L. Huang, PLoS Comput. Biol., 2018, 14, e1006418 CrossRef PubMed.
- X. Chen, L. Huang, D. Xie and Q. Zhao, Cell Death Dis., 2018, 9, 3 CrossRef PubMed.
- X. Chen, Y. Gong, D. H. Zhang, Z. H. You and Z. W. Li, J. Cell. Mol. Med., 2018, 22, 472–485 CrossRef CAS PubMed.
- L. Fu and Q. Peng, Sci. Rep., 2017, 7, 14482 CrossRef PubMed.
- X. Chen, C. C. Yan, X. Zhang, Z. Li, L. Deng, Y. Zhang and Q. Dai, Sci. Rep., 2015, 5, 13877 CrossRef PubMed.
- J. Luo, P. Ding, L. Cheng, B. Cao and X. Chen, IEEE/ACM Trans. Comput. Biol. Bioinf., 2017, 14, 7 Search PubMed.
- X. Zeng, N. Ding, A. Rodríguez-Patón, Z. Lin and Y. Ju, Curr. Proteomics, 2016, 13, 151–157 CrossRef CAS.
- J. Q. Li, Z. H. Rong, X. Chen, G. Y. Yan and Z. H. You, Oncotarget, 2017, 8, 21187–21199 Search PubMed.
- L. Peng, M. Peng, B. Liao, G. Huang, W. Liang and K. Li, Sci. Rep., 2017, 7, 6007 CrossRef PubMed.
- X. Chen, L. Wang, J. Qu, N.-N. Guan and J.-Q. Li, Bioinformatics, 2018, 2, 503 Search PubMed.
- Y. Zhao, X. Chen and J. Yin, Front. Genet., 2018, 9, 324 CrossRef PubMed.
- X. Chen, D. Xie, L. Wang, Q. Zhao, Z.-H. You and H. Liu, Bioinformatics, 2018, 1, 9 Search PubMed.
- J. Li, Z. Wu, F. Cheng, W. Li, G. Liu and Y. Tang, Sci. Rep., 2014, 4, 5576 CrossRef CAS PubMed.
- C. Gu, B. Liao, X. Li, L. Cai, H. Chen, K. Li and J. Yang, RSC Adv., 2017, 7, 44961–44971 RSC.
- L. Peng, Y. Chen, N. Ma and X. Chen, Mol. BioSyst., 2017, 2650–2659 RSC.
- X. Chen, Y. W. Niu, G. H. Wang and G. Y. Yan, J. Biomed. Inf., 2017, 76, 50–58 CrossRef PubMed.
- Q. Zou, J. Li, Q. Hong, Z. Lin, Y. Wu, H. Shi and Y. Ju, BioMed Res. Int., 2015, 2015, 810514 Search PubMed.
- X. Chen, Z. Zhou and Y. Zh ao, RNA Biology, 2018, 1–50 CrossRef PubMed.
- Q. Jiang, Y. Hao, G. Wang, L. Juan, T. Zhang, M. Teng, Y. Liu and Y. Wang, BMC Syst. Biol., 2010, 4(suppl. 1), S2 CrossRef PubMed.
- Q. Jiang, G. Wang and Y. Wang, An approach for prioritizing disease-related microRNAs based on genomic data integration, in International Conference on Biomedical Engineering and Informatics, 2010, pp. 2270–2274 Search PubMed.
- X. Li, Q. Wang, Y. Zheng, S. Lv, S. Ning, J. Sun, T. Huang, Q. Zheng, H. Ren and J. Xu, Nucleic Acids Res., 2011, 39, e153 CrossRef CAS PubMed.
- H. Shi, J. Xu, G. Zhang, L. Xu, C. Li, L. Wang, Z. Zhao, W. Jiang, Z. Guo and X. Li, BMC Syst. Biol., 2013, 7, 101 CrossRef PubMed.
- C. Xu, Y. Ping, X. Li, H. Zhao, L. Wang, H. Fan, Y. Xiao and X. Li, Mol. BioSyst., 2014, 10, 2800–2809 RSC.
- S. Rossi, A. Tsirigos, A. Amoroso, N. Mascellani, I. Rigoutsos, G. A. Calin and S. Volinia, Genomics, 2011, 97, 71–76 CrossRef CAS PubMed.
- P. Xuan, K. Han, M. Guo, Y. Guo, J. Li, J. Ding, Y. Liu, Q. Dai, J. Li and Z. Teng, PLoS One, 2013, 8, e70204 CrossRef CAS PubMed.
- X. Chen, Q. F. Wu and G. Y. Yan, RNA Biology, 2017, 1 Search PubMed.
- D. H. Le, Comput. Biol. Chem., 2015, 58, 139–148 CrossRef CAS PubMed.
- X. Chen, M.-X. Liu and G.-Y. Yan, Mol. BioSyst., 2012, 8, 2792–2798 RSC.
- H. Shi, G. Zhang, M. Zhou, C. Liang, H. Yang, J. Wang, J. Sun and Z. Wang, PLoS One, 2016, 11, e0148521 CrossRef PubMed.
- P. Xuan, K. Han, Y. Guo, J. Li, X. Li, Y. Zhong, Z. Zhang and J. Ding, Bioinformatics, 2015, 31, 1805–1815 CrossRef CAS PubMed.
- B. Liao, S. Ding, H. Chen, Z. Li and L. Cai, J. Bioinf. Comput. Biol., 2015, 13, 1550014 CrossRef CAS PubMed.
- J. Luo and Q. Xiao, J. Biomed. Inf., 2017, 66, 194–203 CrossRef PubMed.
- I. Mugunga, Y. Ju, X. Liu and X. Huang, Oncotarget, 2017, 8, 58526–58535 CrossRef PubMed.
- H. Chen and Z. Zhang, BMC Med. Genomics, 2013, 6, 12 CrossRef CAS PubMed.
- C. Gu, L. Bo, X. Li and K. Li, Sci. Rep., 2016, 6, 36054 CrossRef CAS PubMed.
- X. Li, Y. Lin and C. Gu, RSC Adv., 2017, 7, 32216–32224 RSC.
- J. J. Nalluri, B. K. Kamapantula, D. Barh, N. Jain, A. Bhattacharya, S. S. de Almeida, R. T. Juca Ramos, A. Silva, V. Azevedo and P. Ghosh, BMC Genomics, 2015, 16, S12 CrossRef PubMed.
- X. Chen, C. C. Yan, Z. Xu, Z. H. You, H. Yuan and G. Y. Yan, Oncotarget, 2016, 7, 65257–65269 Search PubMed.
- Z. H. You, Z. A. Huang, Z. Zhu, G. Y. Yan, Z. W. Li, Z. Wen and X. Chen, PLoS Comput. Biol., 2017, 13, e1005455 CrossRef PubMed.
- D. Sun, A. Li, H. Feng and M. Wang, Mol. BioSyst., 2016, 12, 2224 RSC.
- X. Chen and J. Qu, Front. Genet., 2018, 9, 234 CrossRef PubMed.
- X. Chen, J.-R. Yang, N.-N. Guan and J.-Q. Li, Frontiers in Physiology, 2018, 9, 92 CrossRef PubMed.
- X. Chen, C. C. Yan, X. Zhang, Z. H. You, L. Deng, Y. Liu, Y. Zhang and Q. Dai, Sci. Rep., 2016, 6, 21106 CrossRef CAS PubMed.
- X. Chen, N. Guan, J. Li and G. Yan, J. Cell. Mol. Med., 2017, 1548–1561 Search PubMed.
- X. Chen, Z. C. Jiang, D. Xie, D. S. Huang, Q. Zhao, G. Y. Yan and Z. H. You, Mol. BioSyst., 2017, 13, 1202–1212 RSC.
- G. Li, J. Luo, Q. Xiao, C. Liang and P. Ding, J. Biomed. Inf., 2018, 82, 169–177 CrossRef PubMed.
- X. Chen, M. X. Liu and G. Y. Yan, Mol. BioSyst., 2012, 8, 2792–2798 RSC.
- C. Gu, B. Liao, X. Li and K. Li, Sci. Rep., 2016, 6, 36054 CrossRef CAS PubMed.
- M. Chen, X. Lu, B. Liao, Z. Li, L. Cai and C. Gu, PLoS One, 2016, 11, e0166509 CrossRef PubMed.
- M. A. Van Driel, J. Bruggeman, G. Vriend, H. G. Brunner and J. A. Leunissen, Eur. J. Hum. Genet., 2006, 14, 535 CAS.
- A. Kozomara and S. Griffithsjones, Nucleic Acids Res., 2011, 39, D152–D157 CrossRef CAS PubMed.
- D. Zhou, O. Bousquet, T. N. Lal, J. Weston, and B. Schölkopf, Learning with local and global consistency, in Advances in neural information processing systems, 2004, pp. 321–328 Search PubMed.
- H. Chen and Z. Zhang, BMC Med. Genomics, 2013, 6, 12 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller, S. A. Fedewa, D. J. Ahnen, R. G. Meester, A. Barzi and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 177 CrossRef PubMed.
- R. Nonaka, J. Nishimura, Y. Kagawa, H. Osawa, J. Hasegawa, K. Murata, S. Okamura, H. Ota, M. Uemura and T. Hata, Oncol. Rep., 2014, 32, 2354–2358 CrossRef CAS PubMed.
- P. Mussnich, R. Ros, R. Bianco, A. Fusco and D. D'Angelo, Expert Opin. Ther. Targets, 2015, 19, 1017–1026 CrossRef CAS PubMed.
- Y. Niu, Y. Wu, J. Huang, Q. Li, K. Kang, J. Qu, F. Li and D. Gou, Sci. Rep., 2016, 6, 35611 CrossRef CAS PubMed.
- M. Pichler, A. L. Ress, E. Winter, V. Stiegelbauer, M. Karbiener, D. Schwarzenbacher, M. Scheideler, C. Ivan, S. W. Jahn and T. Kiesslich, Br. J. Cancer, 2014, 110, 1614–1621 CrossRef CAS PubMed.
- T. Tanaka, M. Arai, S. Wu, T. Kanda, H. Miyauchi, F. Imazeki, H. Matsubara and O. Yokosuka, Oncol. Rep., 2011, 26, 1329 CAS.
- H. Persson, A. Kvist, N. Rego, J. Staaf, J. Vallon-Christersson, L. Luts, N. Loman, G. Jonsson, H. Naya and M. Hoglund, Cancer Res., 2011, 71, 78–86 CrossRef CAS PubMed.
- J. Shou, S. Gu and W. Gu, Exp. Ther. Med., 2015, 9, 167–171 CrossRef CAS PubMed.
- C. Cai, H. Ashktorab, X. Pang, Y. Zhao, W. Sha, Y. Liu and X. Gu, PLoS One, 2012, 7, e29750 CrossRef CAS PubMed.
- L. Yang, X. Q. Tang, Z. Bai and X. Dai, Sci. Rep., 2016, 6, 35773 CrossRef PubMed.
- Z. Han, F. Chen, X. Ge, J. Tan, P. Lei and J. Zhang, Brain Res., 2014, 1582, 12 CrossRef CAS PubMed.
- E. Montalban, N. Mattugini, R. Ciarapica, C. Provenzano, M. Savino, F. Scagnoli, G. Prosperini, C. Carissimi, V. Fulci and C. Matrone, NeuroMol. Med., 2014, 16, 415–430 CrossRef CAS PubMed.
- K. Smigielska-Czepiel, d. B. A. Van, P. Jellema, I. Slezak-Prochazka, H. Maat, d. B. H. Van, V. D. L. Rj, J. Kluiver, E. Brouwer and A. M. Boots, PLoS One, 2013, 8, e76217 CrossRef CAS PubMed.
- Z. Zhang, Y. Zha, W. Hu, Z. Huang, Z. Gao, Y. Zang, J. Chen, L. Dong and J. Zhang, J. Biol. Chem., 2013, 288, 37082 CrossRef CAS PubMed.
- W. Ding, J. Xin, L. Jiang, Q. Zhou, T. Wu, D. Shi, B. Lin, L. Li and J. Li, Sci. Rep., 2015, 5, 13098 CrossRef CAS PubMed.
- Q. Liao, P. Han, Y. Huang, Z. Wu, Q. Chen, S. Li, J. Ye and X. Wu, PLoS One, 2015, 10, e0130677 CrossRef PubMed.
- C. Yao, Q. Yuan, M. Niu, H. Fu, F. Zhou, W. Zhang, H. Wang, L. Wen, L. Wu and Z. Li, Mol. Ther.--Nucleic Acids, 2017, 9, 182–194 CrossRef CAS PubMed.
- D. R. Gutsaeva, M. Thounaojam, S. Rajpurohit, F. L. Powell, P. M. Martin, S. Goei, M. Duncan and M. Bartoli, Oncotarget, 2017, 8, 103568–103580 CrossRef PubMed.
- H. Andrade, M. Albuquerque, T. Peluzzo, D. Dogni, A. Nucci, I. Lopes-Cendes, M. Franca Jr and S. Avansini, Neurology, 2015, 84 Search PubMed.
- C. Yin, X. Zhou, Y. Dang, Y. Jin and G. Zhang, Medicine, 2015, 94, e2123 CrossRef CAS PubMed.
Footnote |
| † The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first authors. |
| This journal is © The Royal Society of Chemistry 2018 |