
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel nanoporous covalent organic frameworks for the selective extraction of endogenous peptides†
Xiaofei Zhang‡
 ,
Guangyan Qing‡,
Long Yu,
Hongjian Kang,
Cheng Chen,
Xiuling Li* and
Xinmiao Liang*
,
Guangyan Qing‡,
Long Yu,
Hongjian Kang,
Cheng Chen,
Xiuling Li* and
Xinmiao Liang*
Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, 116023, China. E-mail: lixiulign@dicp.ac.cn; liangxm@dicp.ac.cn
First published on 7th November 2018
Abstract
Endogenous peptides are important biomarkers, but their low abundance and abundant interference in biosamples impede their analysis. In this study, a novel nanoporous covalent organic framework (COF) was prepared and successfully applied for selective extraction of endogenous peptides from human serum. This novel COF exhibited strong retention and high adsorption capacity toward peptides, as well as efficient exclusion of large proteins, ascribed to its strong hydrophobicity, uniform pore size (∼2.5 nm) and large surface area (826.5 m2 g−1). These features facilitated the extraction of endogenous peptides from complex biosamples, resulting in 27 identified peptides from tryptic digests of bovine serum albumin (BSA) mixed with 1000 mass folds of BSA protein. Moreover, the adsorption rate of the peptides was 3.6-fold faster than that of proteins on this novel COF. After application the novel COF to 5 μL human serum, 416 unique peptides were unambiguously identified. These results demonstrated the excellent properties of the novel COF in extraction of endogenous peptides. We envisage that COFs with adjustable organic building units and unique physicochemical properties will qualify their potential applications in peptidomics research.
1. Introduction
Endogenous, native peptides such as neuropeptides and peptide hormones play pivotal roles in bioprocesses including signal transmission,1 inflammatory response2 and endocrine processes.3 Some endogenous peptides are important biomarkers, e.g. amyloid peptides correlated with Alzheimer's diseases,4 natriuretic peptides in heart diseases5 and proinsulin in diabetes.6 Thus it is significant to identify and analyze of endogenous peptides. However, it is challenging to analyze endogenous peptides owing to their trace amount, the co-existence of abundant proteins and the high complexity of biological matrices. Selective extraction of endogenous peptides is essential prior to the routine mass spectrometry (MS) analysis. In the past decade, a variety of extraction methods have been developed, such as acetonitrile (ACN) precipitation,7,8 ultrafiltration,9 and reversed phase adsorbents-based solid-phase extraction (SPE).10,11 ACN precipitation and ultrafiltration are widely used to remove high molecular weight (MW) proteins and enrich endogenous peptides. However, both methods suffer from low peptide yields.8,11 Reversed phase adsorbents-based SPE mainly relies on hydrophobic interaction, which has been increasingly used.12,13 Based on this, various nanomaterials were developed to the enrichment of endogenous peptides. These nanomaterials include silica nanospheres,14 carbon nanotubes,15 silica-carbon composite nanofibers,16 zeolite nanocrystals,17 polymeric functionalized nanoparticles,18 magnetic nanoparticles19 and so on. But nonspecific proteins were often adsorbed on the outer surface of these nanomaterials inevitably. For eliminating the co-enrichment of proteins, porous materials attracted more and more attentions. Inorganic porous materials, which were represented by mesoporous silica20 and mesoporous carbon,12 have demonstrated commendable performance in this field. Recently, metallic organic framework materials (MOFs)21 and covalent organic framework materials (COFs)22 were employed in peptidomics analysis and showed great potentials.Covalent organic frameworks (COFs) are an emerging class of ordered porous crystalline materials formed by strong covalent linkages between H, B, C, N, O, and Si.23,24 The unique features of COFs including large surface areas, tunable pore sizes and facilely tailored functionalities offer significant opportunities for many applications including gas separation,25,26 drug delivery,27 energy storage,28 catalysis29 and particularly, bioseparation.22 However, the limited stability of the employed COFs hindered the further application of COFs in this field.
Incorporating methoxy groups into the topological structures of COFs would help fight interlayered electrostatic repulsion and greatly enhance the stability.30 In this work, we prepared a novel covalent organic porous material named TPB–DMTP-COF (TPB, triphenylbenzene; DMTP, dimethoxyterephthaldehyde). This COF contained methoxy groups through a condensation of 2,5-dimethoxyterephthalaldehyde (DMTA) and 1,3,5-tri-(4-aminophenyl) benzene (TAPB). We applied this COF into selective extraction of endogenous peptides from human serum. Its strong hydrophobic forces toward peptides, efficient exclusion of large proteins, high adsorption capacity and specific selectivity of peptides were also studied.
2. Material and methods
2.1 Material and reagents
Tetrahydrofuran (THF), acetic acid, o-dichlorobenzene (o-DCB), n-BuOH, 1,3,5-tri-(4-aminophenyl)benzene (TAPB), 2,5-dimethoxyterephthalaldehyde (DMTA), bovine serum albumin (BSA), cytochrome c (Cyt c), trypsin, ammonium formate, ammonium acetate, ammonium bicarbonate (NH4HCO3), urea, dithiothreitol (DTT) and iodoacetamide (IAA) were ordered from Sigma-Aldrich (St. Louis, MO). The standard peptide (with sequence of SLHTLFGELCK) was synthesized by Jietai biotechnology Co. Ltd. (Shanghai, China). HPLC grade acetonitrile (ACN) was from Merck Life Science Pvt. Ltd. (Worli, Mumbai, India). Formic acid (FA) was obtained from Acros Organics (Geel, Belgium). The mesoporous MCM-41 was purchased from Nankai catalyst factory (Tianjin, China). GELoader tips were from Eppendorf AG (Hamburg, Germany). Deionized water was purified by Milli-Q pure water system (Millipore, Bedford, MA, USA).2.2 Instruments and characterization
Fourier transformation-infrared spectroscopy (FT-IR) was performed with a Bruker Vertex 80V FT-IR spectrometer (Bruker Optics Inc., Billerica, MA. USA). Mass spectra were acquired with a nano electrospray ionization-quadrupole time-of-flight mass spectrometer (ESI Q-TOF MS) (Waters, and Milford, MA, USA) coupled with a Nano Acquity UPLC (Waters, Milford, MA, USA) or with a LTQ Orbitrap Velos coupled with an Accela 600 HPLC system (Thermo, San Jose, California). Zeta potential was measured on a Malvern ZETASIZER 2000/3000 Instrument (Malvern Instruments Ltd., Malvern Worcestershire, UK). High-resolution transmission electron microscopy (HR-TEM) images were obtained on a JEOL model JEM-2100 microscopy (JEOL, Akishima, Tokyo, Japan). BET adsorption isotherms were recorded on a Micrometrics Quadrasorb SI nitrogen adsorption–desorption apparatus (Quantachrome Instruments, Boynton Beach, FL, USA). The peptides/protein adsorption of TPB–DMTP-COF was measured on a Multiskan GO 1510 microplate reader (Thermo Fisher Scientific Inc., Vantaa, Finland).2.3 Experimental details
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30. Digestion was stopped with FA at a final concentration of 0.5%.
:30. Digestion was stopped with FA at a final concentration of 0.5%.The control material MCM-41 was subjected to the same experiment process.
The endogenous peptide fractions from human serum were identified with LTQ-Orbitrap Velos coupled with Accela 600 HPLC system (Thermo, San Jose, California). The full scan mass data were obtained from m/z 400 to 2000 (R = 60000 at m/z 400).
3. Results and discussion
3.1 Characterization
The TPB–DMTP-COF was synthesized through the condensation of DMTA and TAPB.30 N2 sorption–desorption isotherm of TPB–DMTP-COF revealed a type-IV curve (Fig. S1, ESI†), indicating the presence of mesostructure. The Brunauer–Emmett–Teller (BET) surface area was 826.5 m2 g−1 and the pore size peaked at 2.5 nm (Fig. 1A), which was further confirmed with a high resolution transmission electron microscopy (HRTEM) (Fig. 1B). HRTEM also demonstrated the existence of the highly ordered hexagon mesopore and the uniform pore size. The Fourier transform infrared (FTIR) spectra implied the presence of essential functional groups in TPB–DMTP-COF (Fig. 1C). The peaks located at 3000.79 cm−1 and 3031.67 cm−1 were attributed to the benzene ring C–H stretching vibrations. The peaks located at 2960.41 cm−1, 2870.28 cm−1 and 2838.52 cm−1 corresponded to methyl C–H stretching vibrations. The peaks located at 1599.91 cm−1 and 1507.98 cm−1 were attributed to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C skeletal vibrations. The peak located at 1248.90 cm−1 was assigned to the specific absorption of C–O–C ether bond stretching vibration. The peak located at 2820.27 cm−1 was ascribed to the C–H stretching vibrations of –OCH3 which connected to benzene ring. Meanwhile, the peak appeared at 829.41 cm−1 originated from the characteristic absorption of the para-substitution on benzene ring. In the PXRD pattern of TPB–DMTP-COF (Fig. 1D), six measurable diffraction peaks could be observed. They were respectively at 2.76°, 4.82°, 5.58°, 7.39°, 9.67° and 25.18°, which were assigned to the (100), (110), (200), (210), (220) and (001) facets, respectively.30 It indicated the good crystallinity of TPB–DMTP-COF. The pH-dependent zeta potential of TPB–DMTP-COF (Fig. 1A inset) monotonously decreases as pH increases and becomes zero at pH 5.9. The water contact angles for TPB–DMTP-COF was 51.1° (Fig. S2B†), which was larger than that of MCM-41 (30.8°, Fig. S2A†). It suggested that TPB–DMTP-COF is more hydrophobic than MCM-41. The above results indicated that TPB–DMTP-COF with uniform pore size, large surface area and tunable surface charge was successfully synthesized.
C skeletal vibrations. The peak located at 1248.90 cm−1 was assigned to the specific absorption of C–O–C ether bond stretching vibration. The peak located at 2820.27 cm−1 was ascribed to the C–H stretching vibrations of –OCH3 which connected to benzene ring. Meanwhile, the peak appeared at 829.41 cm−1 originated from the characteristic absorption of the para-substitution on benzene ring. In the PXRD pattern of TPB–DMTP-COF (Fig. 1D), six measurable diffraction peaks could be observed. They were respectively at 2.76°, 4.82°, 5.58°, 7.39°, 9.67° and 25.18°, which were assigned to the (100), (110), (200), (210), (220) and (001) facets, respectively.30 It indicated the good crystallinity of TPB–DMTP-COF. The pH-dependent zeta potential of TPB–DMTP-COF (Fig. 1A inset) monotonously decreases as pH increases and becomes zero at pH 5.9. The water contact angles for TPB–DMTP-COF was 51.1° (Fig. S2B†), which was larger than that of MCM-41 (30.8°, Fig. S2A†). It suggested that TPB–DMTP-COF is more hydrophobic than MCM-41. The above results indicated that TPB–DMTP-COF with uniform pore size, large surface area and tunable surface charge was successfully synthesized.
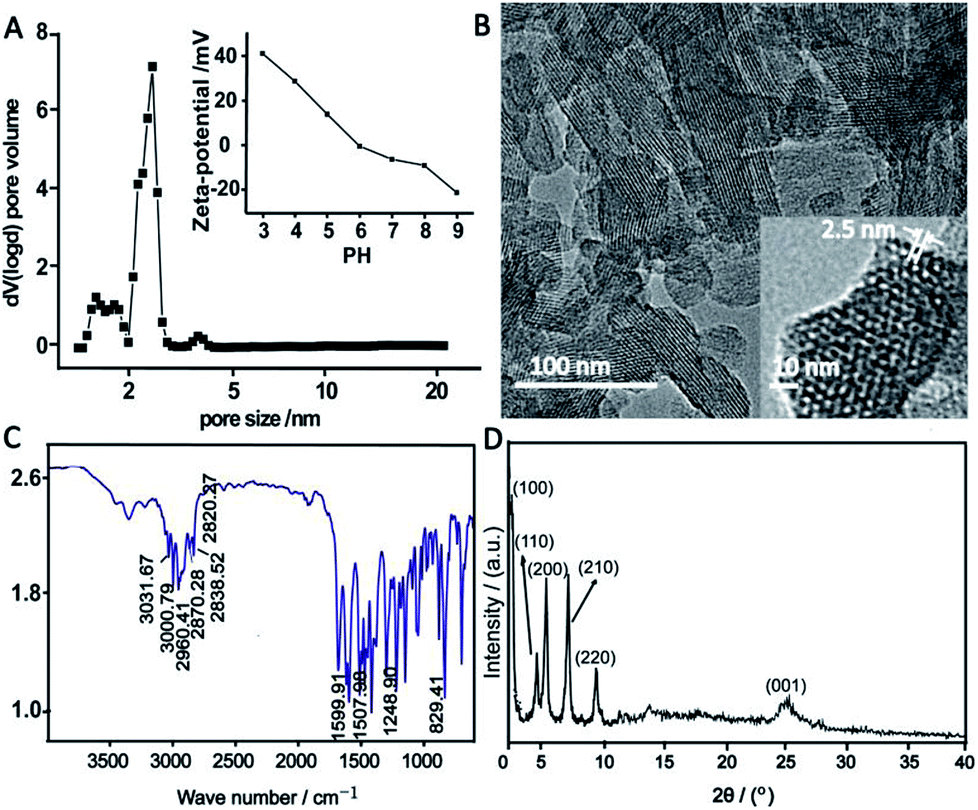 | ||
| Fig. 1 The pore size distribution (A), pH-dependent zeta potential change (A inset), TEM images (B), FTIR spectrum (C) and PXRD pattern (D) of TPB–DMTP-COF. | ||
3.2 The effect of ACN content to the retention of peptides on TPB–DMTP-COF
TPB–DMTP-COF has abundant benzene rings and methoxy groups in its structure (ESI Scheme S1†), which confer the high hydrophobicity. We investigated whether tryptic digests of BSA could be captured by TPB–DMTP-COF under different ACN content (0–50% ACN, pH = 5.9) conditions (Fig. 2A). To facilitate further discussion, 6 representative peptides covered different molecular weights and GRAVY from BSA trypsin digests were chosen (detailed information, e.g. molecular weight, pI and GRAVY see Table S1†). All of the target peptides could be strongly retained on TPB–DMTP-COF under aqueous solution and the bound peptides could be gradually released from TPB–DMTP-COF with increased ACN content (Fig. 2B), indicating the hydrophobic interaction between TPB–DMTP-COF and peptides. In the Fig. S3,† with the GRAVY value of peptides increased from −0.557 to 0.429, the hydrophobicities of peptides enhanced. When the GRAVY value were low (−0.557 to 0.175), the corresponding peptides mainly distributed in the fractions with a low ACN content (10% ACN). With the GRAVY value raised, the distribution of corresponding peptides moved toward the fractions with higher ACN content. When the GRAVY value was 0.429 (Pep 4), the distribution of Pep 4 mainly concentrated in the fractions with ≥30% ACN. It revealed that, with the GRAVY value of peptides increased, the hydrophobic interaction between peptides and TPB–DMTP-COF enhanced.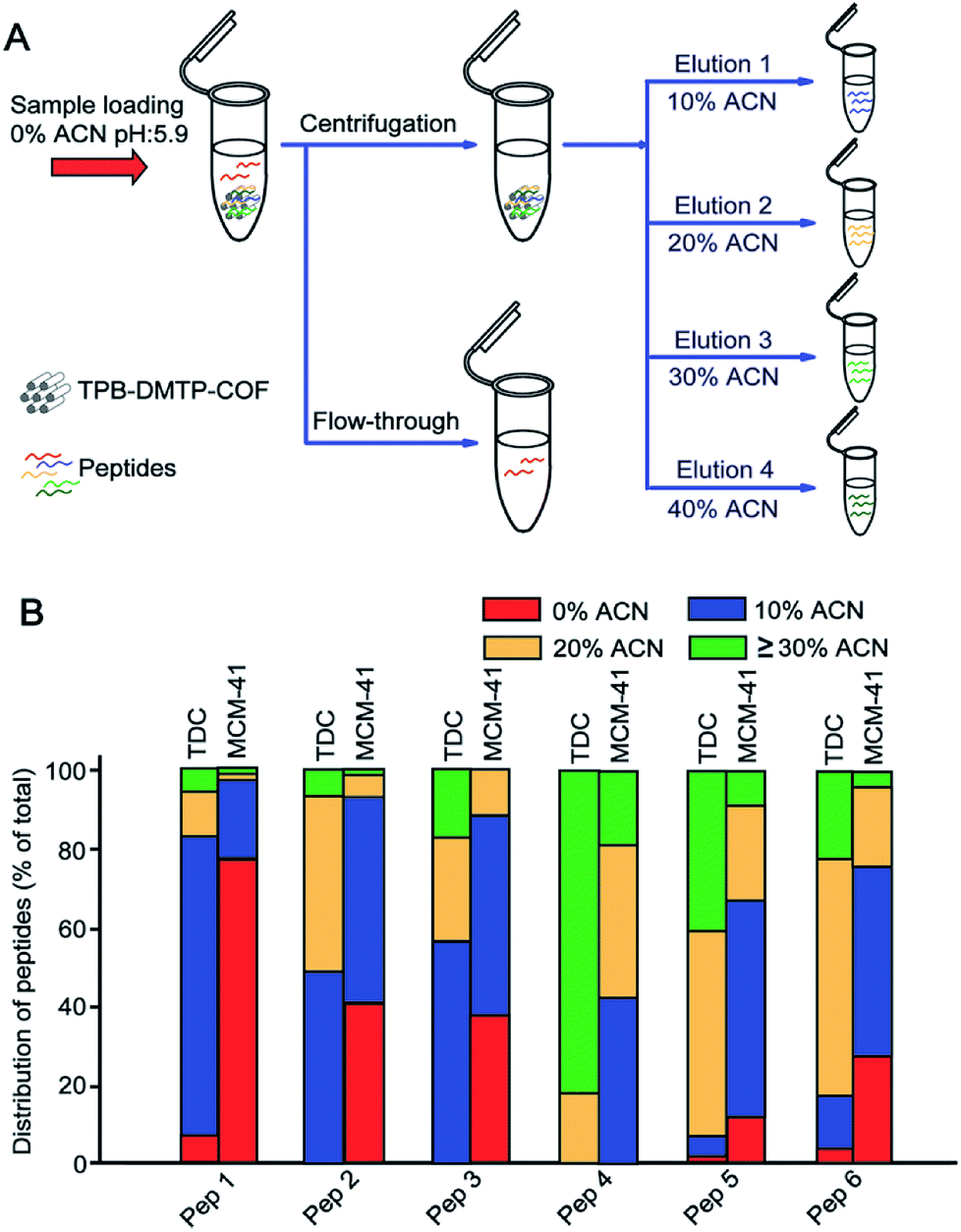 | ||
| Fig. 2 Workflow of the peptides extraction with TPB–DMTP-COF (A) and the distribution of 6 peptides from tryptic digests of BSA in fractions eluted with 0%, 10%, 20%, ≥30% ACN from TPB–DMTP-COF and MCM-41 (B). The distribution of peptides is defined as the percentage of the signal intensity of target peptide in one fraction to its summarized signals intensity. | ||
We further compared the differences on hydrophobic interaction toward peptides between TPB–DMTP-COF and MCM-41. After treatment with TPB–DMTP-COF, only three target peptides with low percentage could be characterized in the flow-through, in sharp contrast to five target peptides with relatively high percentage in that treated with MCM-41 (Fig. 2B). Taking Pep 1 (GRAVY is −0.557) as an example, the percentage of total amount in the flow-through was 7.1 with TPB–DMTP-COF versus 77.3 with MCM-41. Regarding the bound peptides on the adsorbents, overwhelming majority of corresponding peptides distribute in the higher ACN content (10–40%) eluent from TPB–DMTP-COF than from MCM-41 (Fig. S4†). This result indicated that TPB–DMTP-COF has stronger hydrophobic interaction toward peptides than MCM-41, implying the potential high coverage and recovery of peptides with TPB–DMTP-COF based method.
3.3 Exclusion of proteins with TPB–DMTP-COF
The finely uniform and proper pore structure of TPB–DMTP-COF benefit to the exclusion of large proteins (size-exclusion). To test this, two proteins with different sizes were treated with TPB–DMTP-COF in dispersive solid phase extraction mode. These two proteins were bovine serum albumin (BSA) and cytochrome c (Cyt c).32 After treatment with TPB–DMTP-COF, the signals of BSA and Cyt c were detected in flow-through (Fig. 3A and C, upper panel), while no signals of proteins could be found in the eluate (Fig. 3A and C, lower panel). In comparison, the same experiments were also performed with MCM-41 (pore diameter: 3.8 nm). After extraction with MCM-41, the signals of BSA were also detected in flow-through, but the signals of Cyt c could be only observed in the eluate (Fig. 3B and D). The size of BSA (5.0 × 7.0 × 7.0 nm) is larger than the pore size of TPB–DMTP-COF and MCM-41, leading to the exclusion of BSA; whereas the size of Cyt c (2.6 × 3.2 × 3.3 nm) is larger than the pore size of TPB–DMTP-COF but smaller than that of MCM-41, therefore Cyt c enters the pore of MCM-41 but is excluded by TPB–DMTP-COF. The above results indicated that TPB–DMTP-COF possess excellent size-exclusion effect toward proteins. Besides exclusion of majority of BSA in aqueous solution, 8.5% of BSA was found in 50% ACN/1% FA eluent (Fig. S5†). This result can be explained that the high hydrophobicity of TPB–DMTP-COF particles inevitably leads to the adsorption of BSA on their external surface (less than 10% of the total surface area, ESI†). However, the notable difference in the elution conditions of peptides (≤40% ACN) and BSA (50% ACN) facilitate the separation of peptides and proteins with TPB–DMTP-COF. The N2 sorption result (Fig. S6†) revealed that the pore volume of PB–DMTP-COF decreased by about 27.8% after the adsorption of the BSA tryptic peptides, but those were almost unchanged after the adsorption of BSA.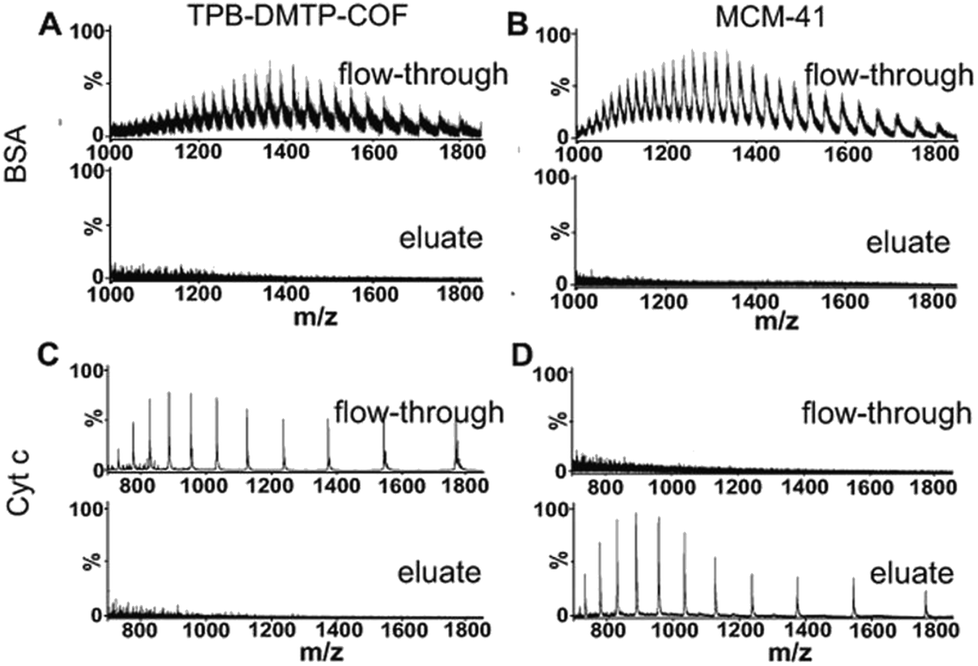 | ||
| Fig. 3 Mass spectra of standard proteins BSA (A and B) and Cyt c (C and D) after treatment with TPB–DMTP-COF (A and C) and MCM-41 (B and D). TPB–DMTP-COF efficiently excludes BSA and Cyt c with its small pore size, while small protein Cyt c enters the pores of MCM-41. | ||
3.4 Measurement of the adsorption capacity of peptides and protein on TPB–DMTP-COF
The large surface areas of PB–DMTP-COF indicate its potential high adsorption capacity. We further examined the adsorption capacity of BSA tryptic peptides on TPB–DMTP-COF. The equilibrium adsorption curves indicate that the adsorption capacity of TPB–DMTP-COF toward BSA tryptic peptides is 0.9 × 10−4 mol g−1, which is much higher than that toward BSA protein, 1.0 × 10−6 mol g−1 (Fig. 4). Moreover, the adsorption equilibrium of BSA tryptic peptides on TPB–DMTP-COF could be reached within 20 min, rather than 80 min for BSA protein. The adsorption rate constant of BSA tryptic peptides on TPB–DMTP-COF was calculated from pseudo-second-order rate equations (R2 > 0.99) (Fig. S7†) and k2 is 2.44 × 10−3 g (mg min)−1, which was 3.6-fold faster than that of BSA protein. This result further confirmed the adsorption rate of peptides was much faster than that of proteins on TPB–DMTP-COF. The high adsorption capacity and high adsorption rate provided by TPB–DMTP-COF further benefit the discrimination of peptides and protein.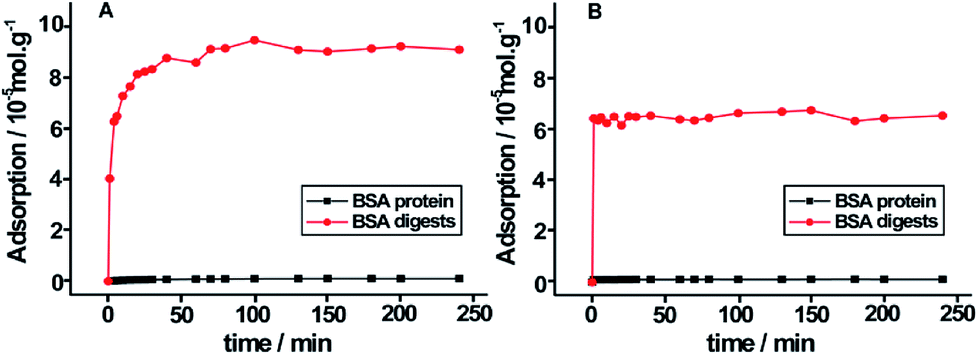 | ||
| Fig. 4 Equilibrium adsorption curves of tryptic digests of BSA and BSA protein on TPB–DMTP-COF (A) and MCM-41 (B). | ||
3.5 The effect of solution pH to the retention of peptides on TPB–DMTP-COF
The surface charge of TPB–DMTP-COF depends on the pH-dependent zeta potential of TPB–DMTP-COF and solution pH value, which will affect the interactions between peptides and materials.To investigate this, we compared the retentions of Pep 1–6 under three solution pH (4.0, 5.9 and 8.4) conditions by considering the distributions of them in eluents with 10–50% ACN (Fig. S8†). According to the pH-dependent zeta potential of TPB–DMTP-COF, When solution pH value was 4.0, both peptides and material had positive surface charges, the electrostatic repulsion weakened the interaction between peptides and the material. This resulted in a portion of peptides ran off in the flow-through (0% ACN) on the loading step. Therefore, the distributions of Pep 1–6 in eluents with 10–50% ACN were not higher than 90%, especially Pep 1 and Pep 6, the two peptides with high pI (8.75 and 9.79). When solution pH value was 5.9, the distributions of Pep 2–4 in eluents with 10–50% ACN were all one hundred percent. Those of Pep 1, 5, and 6 were also nearly one hundred percent. This phenomenon can be explained that the surface charge of TPB–DMTP-COF is theoretically neutral at solution pH 5.9, the adsorption of peptides on TPB–DMTP-COF is independent of electrostatic interaction, but mainly driven by hydrophobic interaction. For the case of solution pH 8.4, the material possessed negative surface charges. The distributions of Pep 2–5 (pI < 8.4, with negative charge) in eluents with 10–50% ACN were reduced because the competence between electrostatic repulsion and hydrophobic interaction. However, the positive charged Pep 1 and Pep 6 with pI > 8.4 showed high distributions (89.1% and 99.5%) in eluents with 10–50% ACN resulting from the synergy between electrostatic attraction interaction and hydrophobic interaction.
3.6 Application to extract of peptides from model samples and endogenous peptides from human serum
Based on the above results, the extraction method was optimized. The selectivity of TPB–DMTP-COF for peptides was assessed via the mixture of BSA protein and tryptic digests of BSA at different mass ratios. As shown in Fig. 5A, with a mass ratio of 1:20 (BSA tryptic digests:BSA protein), the protein signals dominated the mass spectrum and the peptide signals were severely suppressed and barely detectable before extraction. After extraction with TPB–DMTP-COF, interfering protein signals were completely removed and the signal intensities of peptides were notably enhanced. More than 30 of BSA peptides, including those of very low abundance, were identified (Fig. 5B). Even though the weight ratio of peptides/protein was reduced to 1:1000 (Fig. 5C), up to 27 BSA peptides were still detected. The extraction recovery of TPB–DMTP-COF toward a standard peptide (sequence: SLHTLFGELCK) was 84.0% ± 2.6% (n = 3). The detection limit of the standard peptide could reach the level of 5 fmol μL−1. The further application of TPB–DMTP-COF to 5 μL human serum resulted in a total of 416 characterized unique peptides (Table S2†). The isoelectric point (pI) and hydropathy (GRAVY) value distribution for all of the identified endogenous peptides are shown in Fig. 6. GRAVY values distribution revealed that more than 50% of the identified peptides were hydrophilic, with a GRAVY value in the range of −2 to 0. It indicated that TPB–DMTP-COF could capture not only hydrophobic peptides but also those hydrophilic peptides effectively. This result was consistent with that in the previous report.16 The molecular weight distribution of the identified peptides mainly falls in the ranges 1000 to 1500 Da and 1500 to 2000 Da (Fig. S9†). These results suggest the TPB–DMTP-COF is an efficient adsorbent for selective extraction of endogenous peptides from biosamples.
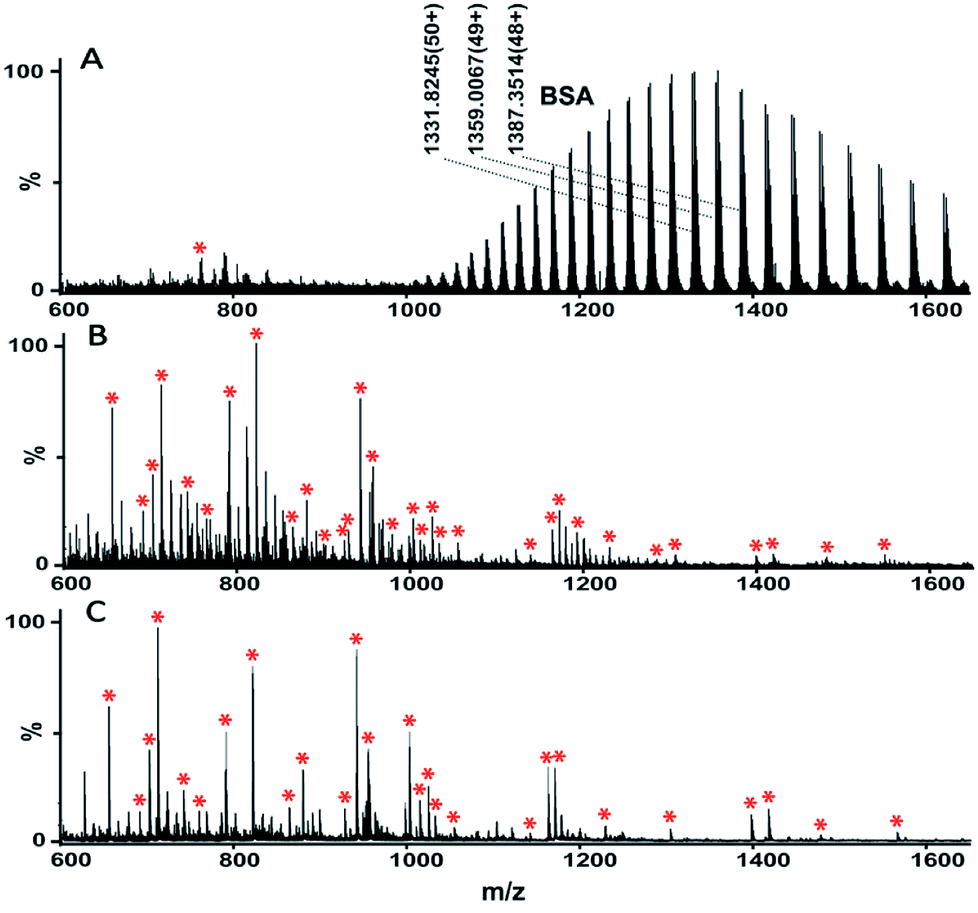 | ||
| Fig. 5 Mass spectra of the mixture of BSA and tryptic digests of BSA before (A) and after (B and C) treatment with TPB–DMTP-COF. The mixture contains BSA and tryptic digests of BSA at weight ratios of 20:1 (A and B) and 1000:1 (C). Tryptic digests of BSA were marked with asterisks in the mass spectra. | ||
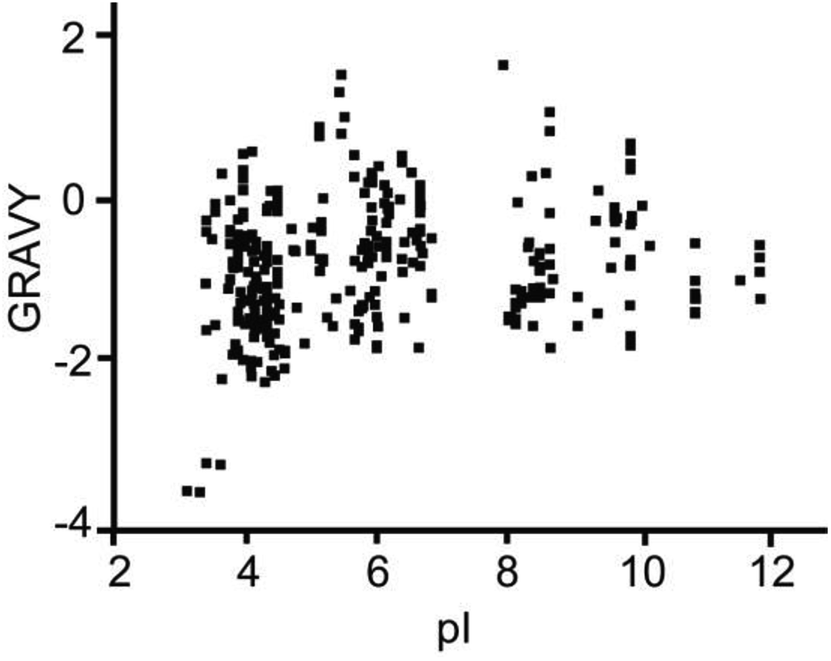 | ||
| Fig. 6 The pI and GRAVY value distribution for all of the identified endogenous peptides from human serum. | ||
4. Conclusions
In summary, nanoporous covalent organic frameworks material TPB–DMTP-COF was prepared. Due to the unique characteristics including strong hydrophobicity, uniform porous structure, large surface area and tunability of surface charge, the material exhibited excellent ability in trapping endogenous peptides. This material could selectively extract endogenous human serum peptides. In addition, the adjustable organic building units and abundant customizable functional groups qualified the potential applications of COFs in peptidomics research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (51533007, 21775148 and 21475129) for funding support.References
- M. W. Schwartz, S. C. Woods, D. Porte, R. J. Seeley and D. G. Baskin, Nature, 2000, 404, 661–671 CrossRef CAS PubMed.
- P. Geppetti, R. Nassini, S. Materazzi and S. Benemei, BJU Int., 2008, 101, 2–6 CrossRef CAS PubMed.
- B. Yusta, D. Matthews, G. B. Flock, J. R. Ussher, B. Lavoie, G. M. Mawe and D. J. Drucker, Mol. Metab., 2017, 6, 503–511 CrossRef CAS PubMed.
- O. Lazarov, J. Y. Robinson, P. Tang, I. S. Hairston, Z. Korade-Mirnics, V. M. Y. Lee, L. B. Hersh, R. M. Sapolsky, K. Mirnics and S. S. Sisodia, Cell, 2005, 120, 701–713 CrossRef CAS PubMed.
- M. Dorkhan, A. Frid and L. Groop, Diabetes Res. Clin. Pract., 2008, 82, 340–345 CrossRef CAS PubMed.
- J. Weng, Y. Li, W. Xu, L. Shi, Q. Zhang, D. Zhu, Y. Hu, Z. Zhou, X. Yan, H. Tian, X. Ran, Z. Luo, J. Xian, L. Yan, F. Li, L. Zeng, Y. Chen, L. Yang, S. Yan, J. Liu, M. Li, Z. Fu and H. Cheng, Lancet, 2008, 371, 1753–1760 CrossRef CAS.
- H. R. Sobhi, B. Vatansever, A. Wortmann, E. Grouzmann and B. Rochat, J. Chromatogr. A, 2011, 1218, 8536–8543 CrossRef CAS PubMed.
- Y. Kawashima, T. Fukutomi, T. Tomonaga, H. Takahashi, F. Nomura, T. Maeda and Y. Kodera, J. Proteome Res., 2010, 9, 1694–1705 CrossRef CAS PubMed.
- F. Liu, C. Zhao, L. Liu, H. Ding, R. Huo and Z. Shi, J. Proteomics, 2016, 139, 38–44 CrossRef CAS PubMed.
- J. Villanueva, J. Philip, C. A. Chaparro, Y. B. Li, R. Toledo-Crow, L. DeNoyer, M. Fleisher, R. J. Robbins and P. Tempst, J. Proteome Res., 2005, 4, 1060–1072 CrossRef CAS PubMed.
- L. P. Aristoteli, M. P. Molloy and M. S. Baker, J. Proteome Res., 2007, 6, 571–581 CrossRef CAS PubMed.
- Y. Du, D. Wu, Q. Wu and Y. Guan, Anal. Bioanal. Chem., 2015, 407, 1595–1605 CrossRef CAS PubMed.
- A. Secher, C. D. Kelstrup, K. W. Conde-Frieboes, C. Pyke, K. Raun, B. S. Wulff and J. V. Olsen, Nat. Commun., 2016, 7, 11436 CrossRef CAS PubMed.
- H. Chen, X. Xu, N. Yao, C. Deng, P. Yang and X. Zhang, Proteomics, 2008, 8, 2778–2784 CrossRef CAS PubMed.
- S. Ren and Y. Guo, J. Am. Soc. Mass Spectrom., 2006, 17, 1023–1027 CrossRef CAS PubMed.
- G. Zhu, X. Chen, X. He, H. Wang, Z. Zhang and Y. Feng, Chem.–Eur. J., 2015, 21, 4450–4456 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, W. Shan, B. Wu, H. Fan, X. Yu, Y. Tang and P. Yang, Angew. Chem., Int. Ed., 2005, 44, 615–617 CrossRef CAS PubMed.
- H. Chen, C. Deng and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 607–611 CrossRef CAS PubMed.
- M. Zhao, C. Deng and X. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 13104–13112 CrossRef CAS PubMed.
- R. Tian, H. Zhang, M. Ye, X. Jiang, L. Hu, X. Li, X. Bao and H. Zou, Angew. Chem., Int. Ed., 2007, 46, 962–965 CrossRef CAS PubMed.
- Z. Gu, Y. Chen, J. Jiang and X. Yan, Chem. Commun., 2011, 47, 4787–4789 RSC.
- C. H. Gao, G. Lin, Z. X. Lei, Q. Zheng, J. S. Lin and Z. A. Lin, J. Mater. Chem. B, 2017, 5, 7496–7503 RSC.
- S. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310–17313 CrossRef CAS PubMed.
- Z. Yin, S. Xu, T. Zhan, Q. Qi, Z. Wu and X. Zhao, Chem. Commun., 2017, 53, 7266–7269 RSC.
- R. Gomes and A. Bhaumik, RSC Adv., 2016, 6, 28047–28054 RSC.
- P. Pachfule, S. Kandmabeth, A. Mallick and R. Banerjee, Chem. Commun., 2015, 51, 11717–11720 RSC.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- X. L. Li, Y. T. Xiong, G. Y. Qing, G. Jiang, X. Q. Li, T. L. Sun and X. M. Liang, ACS Appl. Mater. Interfaces, 2016, 8, 13294–13302 CrossRef CAS PubMed.
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07500j |
| ‡ The first author |
| This journal is © The Royal Society of Chemistry 2018 |