
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced oxygen reduction activity of Pt shells on PdCu truncated octahedra with different compositions†
Xingqiao Wu‡
,
Qingfeng Xu‡,
Yucong Yan,
Jingbo Huang,
Xiao Li,
Yi Jiang ,
Hui Zhang* and
Deren Yang
,
Hui Zhang* and
Deren Yang
State Key Laboratory of Silicon Materials and School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: msezhanghui@zju.edu.cn
First published on 11th October 2018
Abstract
Pd@Pt core–shell nanocrystals with ultrathin Pt layers have received great attention as active and low Pt loading catalysts for oxygen reduction reaction (ORR). However, the reduction of Pd loading without compromising the catalytic performance is also highly desired since Pd is an expensive and scarce noble-metal. Here we report the epitaxial growth of ultrathin Pt shells on PdxCu truncated octahedra by a seed-mediated approach. The Pd/Cu atomic ratio (x) of the truncated octahedral seeds was tuned from 2, 1 to 0.5 by varying the feeding molar ratio of Pd to Cu precursors. When used as catalysts for ORR, these three PdxCu@Pt core–shell truncated octahedra exhibited substantially enhanced catalytic activities compared to commercial Pt/C. Specifically, Pd2Cu@Pt catalysts achieved the highest area-specific activity (0.46 mA cm−2) and mass activity (0.59 mA μgPt−1) at 0.9 V, which were 2.7 and 4.5 times higher than those of the commercial Pt/C. In addition, these PdxCu@Pt core–shell catalysts showed a similar durability with the commercial Pt/C after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles due to the dissolution of active Cu and Pd in the cores.
000 cycles due to the dissolution of active Cu and Pd in the cores.
Introduction
Over the past few decades, proton-exchange membrane fuel cells (PEMFCs) have been deemed to be one of the most promising clean and sustainable energy technologies due to their high-energy conversion efficiency, low pollutant emission, and high-energy density.1,2 The difficulty in widespread commercialization of PEMFCs is the lack of high-efficiency catalysts, especially those used for oxygen reduction reaction (ORR) at the cathodes since the sluggish ORR is the rate-determining step in use.3–5 In PEMFCs, Pt is proved to be the most efficient monometallic catalyst for ORR.6 However, the extremely scarce abundance and high cost associated with Pt limit its extensive use as industrial catalysts.7,8 A solution which can reduce the cost as much as possible while ensuring ORR activity and stability is a goal pursued by scientific researchers.Recently, tremendous efforts have been devoted to the development of bimetallic nanocrystals by combining Pt with another more abundant and/or much less expensive metal in an effort to address the above-mentioned issues.9,10 This design not only reduces the loading of Pt, but also improves the activity and stability of the catalyst by both ligand and geometry effects arising from the interaction between different components.11–13 One of the most promising methods is to synthesize Pt–M (M = Ni, Co, Cu, Fe, and so on) alloyed nanocrystals with certain facets, showing extremely excellent ORR activity.14–17 For instance, Stamenkovic and co-workers reported that the ORR activity of Pt3Ni(111) was 10 and 90 times higher than that of the Pt(111) and commercial Pt/C, respectively.18 Nevertheless, the nonprecious metal (e.g., Ni) in the alloy is unstable in the extremely harsh environments, resulting in poor durability of the catalysts.19 Another effective approach is to generate core–shell nanocrystals by depositing Pt shells on cores made of other metals. For example, our group have reported the synthesis of the Au@Pt core–shell star-shaped decahedra by epitaxial growth of Pt ultrathin shells on five-fold twinned Au cores, showing remarkably enhanced ORR properties relative to commercial Pt/C.20 However, Au is not an optimal core material for designing Pt-based core–shell catalysts according to the previously reported volcano plot.21 It is clear that Pd is probably the best core material to form Pt-based core–shell catalysts owing to the suitable electron coupling and synergetic effect between Pd and Pt.22 As such, many research groups have demonstrated the synthesis of the Pd@Pt nanocrystals with different shapes by manipulating the reaction kinetics and thermodynamics.23–25 To this end, we have demonstrated a thermodynamically-controlled approach to the synthesis of icosahedral, octahedral, and cubic Pd@Pt core–shell nanocrystals by simply using Pd icosahedra, octahedra, and cubes as the seeds, respectively.26 Compared to the Pd@Pt core–shell octahedra and cubes, such core–shell icosahedra exhibited remarkably enhanced catalytic properties for ORR due to the facet and strain effects. Although the incorporation of Pd into Pt can boost the ORR properties of the catalysts in combination with the improved utilization efficiency of Pt, the reduction of Pd loading is highly desired since Pd is also belong to noble-metals. As such, alloying Pd with cheap transition metal can meet such criteria required for the desirous core material. In addition, the component of Pd-based alloy cores provides additional handles for tuning the catalytic properties of Pt-based core–shell catalysts for ORR. For instance, Pd–M (M = Ni, Co, Cu, Fe and so on) was used as cores to build Pt-based core–shell nanocrystals, which significantly reduce cost of the catalysts while ensuring their catalytic performance.27–30 It is found that most of these Pd–M@Pt nanocrystals were synthesized by electrochemical dealloying, underpotential deposition (UPD), or galvanic replacement methods.31–33 However, these methods remain tremendous challenge in the synthesis of such core–shell nanocrystals with well-defined shapes, which have a great influence on the catalytic properties.
Here we report a facile approach to the synthesis of three kinds of PdxCu@Pt truncated octahedra by a seed-mediated growth, in which the atomic ratio (x) of Pd/Cu is tuned from 2, 1 to 0.5. Moreover, we also investigated the effect of compositional changes in the cores on the electrocatalytic properties of Pt-based core–shell catalysts with the same morphology. We found that Pd2Cu@Pt truncated octahedra exhibited remarkably enhanced catalytic properties for ORR compared to PdCu@Pt and PdCu2@Pt nanocrystals as well as commercial Pt/C.
Experimental section
Materials and chemicals
Palladium(II) acetylacetonate (Pd(acac)2, 99%), copper(II) acetylacetonate (Cu(acac)2), poly(vinylpyrrolidone) (PVP, MW = 29000), chloroplatinic(IV) acid (H2PtCl6, 99.9%) were all purchased from Sigma Aldrich. Commercial Pt/C (20 wt%) were purchased from Alfa Aesar. Oleylamine (OAm, 80–90%) was purchased from Aladdin. Benzyl alcohol (BA), dimethylformamide (DMF), ethanol, acetone, chloroform and toluene were purchased from Sinopharm Chemical Reagent. All the chemicals and materials were used as received. All aqueous solutions were prepared using ultrapure water with a resistivity of 18.2 MΩ cm.
Synthesis of PdxCu truncated octahedra
Taking Pd2Cu as an example, 20.1 mg of Pd(acac)2, 8.6 mg of Cu(acac)2, and 50 mg of PVP were dissolved in a mixed solution of 5 mL of DMF and 5 mL of benzyl alcohol and stirred for 30 min at room temperature. The homogeneous solution was then transferred into a 25 mL glass flask. The sealed flask was heated at 140 °C under magnet stirring for 2 h and then cooled to room temperature. The product was collected by centrifugation, washed with ethanol and acetone for three times, and then re-dispersed in 8 mL of ethanol for further use. For the synthesis PdCu truncated octahedra, the amount of Pd(acac)2 and Cu(acac)2 was adjusted to 15 and 13 mg, respectively. When synthesizing PdCu2 truncated octahedra, 10 mg of Pd(acac)2 and 17.3 mg of Cu(acac)2 were used, together with the use of 2.5 mL of DMF and 7.5 mL of benzyl alcohol.Phase transfer of PdxCu truncated octahedra
In a standard procedure, 9 mL of ethanol solution containing the PdxCu truncated octahedra was mixed with 6 mL of OAm in a 20 mL glass vial. The phase transfer was conducted by magnetic stirring of the mixture at 80 °C for 12 h. The product was collected by centrifugation, washed with ethanol, and then re-dispersed in OAm serving as the seeds for the epitaxial growth of ultrathin Pt shells.Epitaxial growth of ultrathin Pt shells on PdxCu truncated octahedra
In a standard preparation, 5 mL of OAm solution containing PdxCu seeds was added into a 25 mL single-necked flask and preheated to 180 °C in an oil bath for 10 min. After that, 3 mL of OAm solution containing a certain amount of H2PtCl6 was added into the flask by a pipette, where the mass ratio of Pt is 25% of PdCu seed. The reaction was proceeded at 180 °C for 3 h. Finally, the final product was collected by centrifugation, washed with ethanol and cyclohexane for several times.Preparation of carbon-supported catalysts
In a standard preparation, carbon black (Vulcan XC-72) was dispersed in chloroform and sonicated for 30 min. The as-prepared PdxCu@Pt (x = 2, 1, 0.5) nanocrystals (20 wt%) were added to this dispersion. This mixture was further sonicated for 10 min and stirred for 12 h. After that, the resultant was precipitated out by centrifugation and re-dispersed in n-butylamine at a concentration of 0.5 mg-catalyst per mL. The mixture was kept under magnetic stirring for 3 days, and then centrifuged and washed four times with methanol. Finally, the PdxCu@Pt/C catalysts were obtained by the freeze drying in DI water.Morphological, structural, and compositional characterizations
Transmission electron microscopy (TEM) images of the obtained samples were taken using a HITACHI HT-7700 microscope operated at 100 kV. High-resolution transmission electron microscopy (HRTEM) was performed using a FEI Tecnai F30 G2 microscope operated at 300 kV. High-angle annular dark-field scanning TEM (HAADF-STEM) and Energy Dispersive X-ray (EDX) mapping analyses were taken on a FEI Titan ChemiSTEM equipped with a probe-corrector and a Super-X EDX detector system. This microscope was operated at 200 kV with a probe current of 50 pA and a convergent angle of 21.4 mrad for illumination. The X-ray diffraction (XRD) patterns were recorded on a Miniflex600 X-ray diffractometer in a scan range of 10–80° at a scan rate of 10° min−1. The percentages of Pd, Pt, and Cu in the samples were determined using inductively coupled plasma atomic emission spectrometry (ICP-AES, IRIS Intrepid II XSP, TJA Co., USA). TGA analysis was performed with a thermogravimetric analyzer (SDT Q600). The temperature was scanned from room temperature to 800 °C with a scan rate of 10 °C min−1.Electrochemical measurement
The electrochemical performances of PdxCu@Pt catalysts including commercial Pt/C were measured by a three-electrode cell using a CHI760E electrochemical analyser with a glassy-carbon rotating disk electrode (RDE, area: ∼0.196 cm2), a Pt mesh (1 × 1 cm2), and a Ag/AgCl electrode as the working, counter, and reference electrode, respectively. The as-received data were finally converted to reversible hydrogen electrode (RHE) as the reference. To make catalyst ink, 2.5 mg of PdxCu@Pt/C catalysts was dispersed in 2.5 mL of a mixed solvent and sonicated for 10 min. The solvent contained a mixture of de-ionized water, isopropanol, and 5% Nafion 117 solution at the volumetric ratio of 8:2:0.05. 30 μL of the catalyst ink was added onto the RDE and dried under the air flow for 30 min to make the working electrode. The loading amount of PdxCu@Pt core–shell catalysts on the RDE was determined to be ∼20 μgmetal cm−2. The CV measurement was carried in Ar-saturated 0.1 M HClO4 solution at room temperature with a scan rate of 50 mV s−1. The electrochemical active surface area (ECSA) was determined from the CV curves, calculating the amount of charges by integrating hydrogen desorption region after double layer correction. ORR linear scan voltammetry (LSV) curves were measured at a rotating rate of 1600 rpm in a 0.1 M HClO4 solution, which was purged with oxygen for 30 min prior to, and during testing. The scan rate for ORR measurements was set at 10 mV s−1. Data were used without iR-drop correction. The accelerated stability test (ADT) was carried out between 0.6 and 1.0 V at a scan rate of 100 mV s−1 for 10000 cycles in O2-saturated 0.1 M HClO4 solution.
Results and discussion
Fig. 1a, c and e show TEM images of PdxCu nanocrystals with different compositions prepared by simultaneous reduction of Pd(acac)2 and Cu(acac)2 in a mixture containing DMF and benzyl alcohol, respectively. One can see that most of the nanocrystals have uniform size with a truncated octahedral shape. ICP-AES data (Table S1†) indicate that the Pd/Cu atomic ratios of these truncated octahedra are measured to be 2 (i.e., Pd2Cu), 1 (i.e., PdCu), and 0.5 (i.e., PdCu2), respectively. Clearly, the Pd/Cu atomic ratios of the former two samples (Pd2Cu and PdCu) are close to the feeding molar ratios of the Pd to Cu precursors, while the third sample (PdCu2) needs the additional amount of the Cu precursor due to its much slower reduction rate than the Pd precursor. We randomly selected 100 particles to calculate the average sizes of these Pd–Cu nanocrystals, as shown in Fig. S1.† It can be seen that the average sizes of Pd2Cu, PdCu and PdCu2 nanocrystals are 17.92, 19.98 and 15.73 nm, respectively. The crystal structure of these three nanocrystals was characterized by XRD analysis, as shown in Fig. S2.† From the XRD patterns, there are three main diffraction peaks located between those of pure Pd and Cu, implying the formation of alloy structure with a face-centered cubic (fcc) structure. In addition, there is an obvious diffraction peak corresponding to pure Pd(111) in Pd2Cu nanocrystals, indicating the formation of the segregated Pd phase. | ||
| Fig. 1 TEM images of (a) Pd2Cu, (c) PdCu and (e) PdCu2 truncated octahedra and (b) Pd2Cu@Pt, (d) PdCu@Pt and (f) PdCu2@Pt truncated octahedra. | ||
The as-prepared PdxCu truncated octahedra were transferred in OAm, and then used as seeds for synthesizing PdxCu@Pt core–shell nanocrystals by the epitaxial growth. From the TEM images in Fig. 1b, d, and f, the final products remain the truncated octahedral shapes of the seeds after the epitaxial growth of Pt shells. Similarly, the average sizes of Pd2Cu@Pt, PdCu@Pt and PdCu2@Pt are measured to be 18.62, 20.65, 16.30 nm (Fig. S3†). Fig. 2 shows the HAADF-STEM image, HRTEM image, EDX mapping image and line-scan spectrum of Pd2Cu@Pt core–shell nanocrystals using the Pd2Cu truncated octahedra as seeds. From the HAADF-STEM image in Fig. 2a, one can see a clear bright Pt shell was deposited on the surface of the Pd2Cu cores, implying a Pd2Cu@Pt core–shell structure. The HRTEM image (Fig. 2b) reveals the well-resolved, continuous fringes in the same orientation from the core to shell, indicating the Pt shell was grown epitaxially on the Pd2Cu seed. The fringes with a lattice spacing of 2.2 Å can be indexed to the {111} planes of Pd–Cu alloy with a fcc structure. The distribution of Pt, Pd and Cu in the truncated octahedra was determined using EDX analysis. EDX mapping images (Fig. 2c) clearly show a color difference between the core and shell, confirming the Pd2Cu@Pt core–shell nanocrystals. In addition, Pd color pattern is larger than Cu color pattern with an overlap in the interior, suggesting that the cores consist of the PdCu alloy and Pd interlayer. The formation of Pd2Cu@Pt core–shell nanocrystals with a Pd interlayer is also confirmed by the EDX line-scan spectrum (Fig. 2d), in which the Cu trace has one broad peak in the center, the Pt trace has two narrow peaks at the two ends, while the Pd trace has one broad peak enclosing that of Cu trace and located between Pt peaks. Obviously, the formation of Pd interlayer can be attributed to its segregation from the alloy in the presence of the excess Pd precursor during the synthesis. Moreover, the XRD pattern of Pd2Cu@Pt (Fig. S4†) also demonstrates the existence of Pd interlayer after epitaxial growth of Pt. When using PdCu and PdCu2 truncated octahedra as the seeds, the Pt-based core–shell nanocrystals were also generated by a seed-mediated growth. The same electron microscopic characterizations have been employed for PdCu@Pt (Fig. 3) and PdCu2@Pt (Fig. S5†) nanocrystals, which display the epitaxial growth of Pt shells on the seeds. In addition, the complete overlap between Pd and Cu color patterns (Fig. 3c and Fig. S5c†) in combination with the peaks associated with Pd and Cu traces (Fig. 3d and S5d†) indicates that there is no Pd interlayer existing in these two samples. The amount of Pt in all these samples reaches ∼20 wt%, which were determined by ICP-AES (Table S1†). According to our previous report,26 the thickness of Pt shells was easily calculated from geometry analysis and ICP-AES data. From Table S2,† the thickness of Pt shells in these three samples was all 1–2 atomic layer.
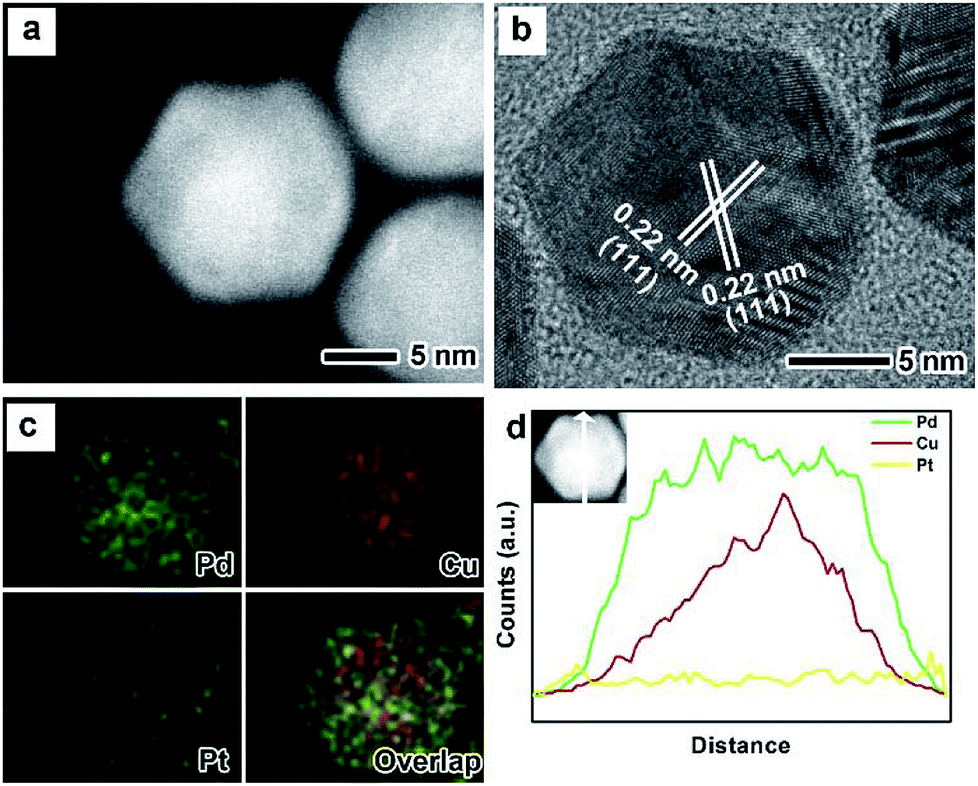 | ||
| Fig. 2 Morphological, structural, and compositional characterizations of truncated octahedral Pd2Cu@Pt nanocrystals: (a) HAADF-STEM image, (b) HRTEM image, (c) EDX mapping image, and (d) line-scanning profiles. | ||
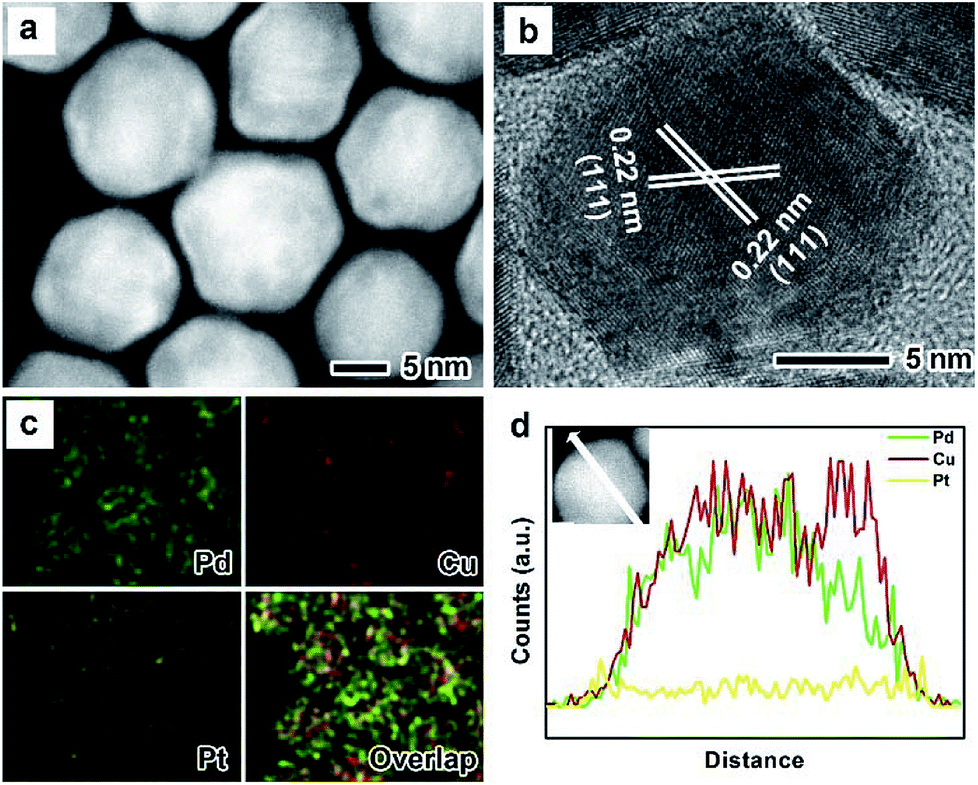 | ||
| Fig. 3 (a) HAADF-STEM image, (b) HRTEM image, (c) EDX mapping image and (d) line-scanning profiles of PdCu@Pt truncated octahedra. | ||
The surface composition and valance states of PdxCu@Pt nanocrystals were analyzed using the XPS technique (Fig. S6†). The Pt4f, Pd3d, Cu2p XPS spectra were calibrated with respect to the binding energy (BE) of the C 1s peak of graphite at 284.5 eV. Owing to the ORR properties of the catalysts arising from the Pt and Pd components, we only discuss the evolution of Pd3d5/2 and Pt4f7/2 with the amount of Cu for simplicity. After the peak splitting in Fig. S6a,† the red, pink, blue spectra correspond to the Pd3d peaks associated with Pd0, PdO and Pd2+, respectively. Taking Pd2Cu@Pt nanocrystals as an example, the three peaks at 335.3, 336.0, and 336.9 eV can be attributed to Pd0, PdO and Pd2+, respectively. As a result, the peak of Pd0 (335.3 eV) shifts positively compare to the standard one of Pd metal (334.9 eV, black dotted line in Fig. S6a†). Meanwhile, with the increase in the amount of Cu, the peak associated with Pd0 shifts to the higher binding energy. Fig. S6b† shows the XPS spectra associated with Pt4f of these three samples. A trend similar to Pd can also be observed in the XPS spectra of Pt. As the amount of Cu increases, the peaks of Pt0 (71.1, 71.15, and 71.39 eV) shift more positively relative to the standard one of Pt metal (70.9 eV, black dotted line in Fig. S6b†), indicating the electron coupling between them (i.e., ligand effect). The reason for such shifts is the larger work function of Pt and Pd than Cu (5.65, 5.12, and 4.65 eV for Pt, Pd and Cu, respectively).34–36 Moreover, the lattice constant of Cu and Pd is smaller than that of Pt, leading to the lattice contraction of Pt shell (i.e., geometry effect). It is clear that the suitable electron coupling and compressive strain can contribute a lowered d-band center of Pt, thereby reducing the binding strength of the adsorbed intermediates (e.g., OH·) on the catalysts and improving their ORR activities.37 However, the calculation performed by Chorkendorff and co-workers indicated that the catalysts with little compressive strain have an optimal value of ORR activities.38 As such, it is expected that the PdxCu@Pt core–shell nanocrystals with various compositions may show different catalytic properties towards ORR.
The as-synthesized PdxCu@Pt nanocrystals were loaded on carbon support (Vulcan XC-72), and then evaluated as the catalysts for ORR with commercial Pt/C as a reference. The cyclic voltammetry (CV) curves of these four carbon-supported catalysts were recorded at room temperature in Ar-purged 0.1 M HClO4 solutions at a sweep rate of 50 mV s−1 between 0.05 and 1.0 V versus reversible hydrogen electrode (RHE), as shown in Fig. S7.† The electrochemical active surface areas (ECSAs) of these catalysts were calculated by measuring the charge collected in the hydrogen adsorption region assuming an adsorbed hydrogen monolayer on metal surface, which is 210 μC cm−2. As observed from Table S3,† the ECSAs of Pd2Cu@Pt, PdCu@Pt, and PdCu2@Pt core–shell catalysts are about 129.0, 106.2 and 70.3 m2 gPt−1, respectively. The first two samples have much higher ECSAs than the commercial Pt/C (78.2 m2 gPt−1), implying an increase in the utilization efficiency of Pt owing to the high dispersion of Pt atoms. Fig. 4a shows the positive-going ORR polarization curves of the PdxCu@Pt catalysts including the commercial Pt/C. One can see that the onset potentials of these PdxCu@Pt catalysts have positive shifts relative to the Pt/C. The kinetic currents of the ORR polarization curves were calculated by the Koutecky–Levich equation and normalized against the ECSAs and Pt mass to get the area-specific (is, Fig. 4b) and mass activities (im, Fig. 4c). In the potential region of 0.88–0.94 V, these three core–shell catalysts exhibit substantially enhanced is and im compared to the Pt/C. Fig. 4d shows a comparison of specific and mass activities of these four catalysts at 0.9 V. Clearly, the Pd2Cu@Pt catalysts display the highest area-specific activity (0.46 mA cm−2) and mass activity (0.59 mA μgPt−1) at 0.9 V, which are 2.7 and 4.5 times higher than those of the Pt/C (0.17 mA cm−2 and 0.13 mA μgPt−1), respectively. In addition, the PdxCu@Pt nanocrystals gradually decrease with the increase in the amount of Cu component. As well-known, the too strong or weak strains of catalysts are both harmful to the ORR activity, showing a volcano relationship between them.39 The incorporation of Pd can induce the compressive strain with a suitable strength in the Pt shell, resulting in the excellent ORR activity. As such, introducing Cu component with a smaller atom size leads to additional compressive strain in the Pt shell, and thus the decrease in ORR activity. The Pd interlayer between the Pt shell and PdCu core can buffer the additional compressive strain arising from the introduction of Cu, leading to the best ORR activity of the Pd2Cu@Pt catalysts.40
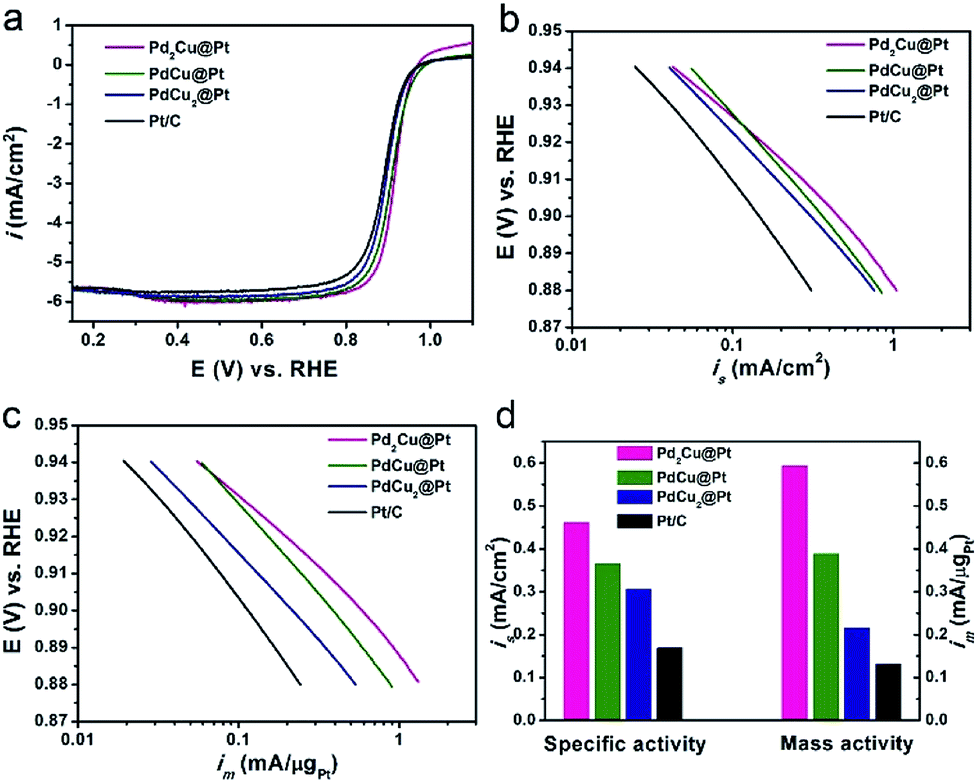 | ||
| Fig. 4 Electrochemical characterizations of catalysts towards ORR. (a) ORR polarization curves, (b) area-specific (is), and (c) mass (im) activities in the potential range of 0.88–0.94 V for Pd2Cu@Pt, PdCu@Pt, PdCu2@Pt, and Pt/C catalysts measured in O2 saturated 0.1 M of HClO4 solution with a scan rate of 10 mV s−1 at room temperature. (d) Area-specific and mass activities of these four catalysts at 0.9 V. | ||
In addition to the activity, long-term stability is another critically important parameter for evaluating an ORR catalyst. We conducted an accelerated durability test (ADT) for 10000 cycles in O2-saturated 0.1 M HClO4 solution to investigate the ORR stability of our three PdxCu@Pt core–shell catalysts with the commercial Pt/C as a reference. The polarization curves in Fig. 5a–d show that the Pd2Cu@Pt catalysts have smaller loss in half-wave potential relative to the PdCu@Pt and PdCu2@Pt after ADT, which are similar to the commercial Pt/C. The detailed data for their ORR stability are summarized in Fig. 5e and f. After 10000 cycles, Pd2Cu@Pt catalysts remain 71.3% of the ORR activities, which are comparable to the commercial Pt/C, but little better than the PdCu@Pt and PdCu2@Pt catalysts. The reason for the decrease in ORR activity of the Pd2Cu@Pt catalysts during the reaction was revealed by morphological characterization. We compared the TEM images of the PdCu2@Pt catalysts before and after 10000 cycles, as shown in Fig. S8.† As observed, most of the Pd2Cu cores were removed in the acid media due to the highly active metal component (e.g., Cu and Pd), leading to the degradation of durability of the PdxCu@Pt catalysts. The ordered PdCu intermetallic nanocrystals with a higher chemical stability are expected to become a promising substitute relative to their alloyed counterpart serving as cores for Pt-based core–shell catalysts for ORR.41
 | ||
| Fig. 5 Polarization curves of (a) Pd2Cu@Pt, (b) PdCu@Pt, (c) PdCu2@Pt, and (d) Pt/C catalysts before and after ADT for 10000 cycles. A comparison of the loss in (e) area-specific and (f) mass activities at 0.9 V versus RHE for these four catalysts after ADT. The ADT was carried out between 0.6 and 1.0 V at a scan rate of 100 mV s−1 for 10000 cycles in O2 saturated 0.1 M HClO4 solution. | ||
Conclusions
In summary, we have developed a simple seed-mediated approach to synthesize PdxCu@Pt core–shell truncated octahedra consisting of ultrathin Pt shells. The atomic ratios of Pd/Cu in the cores were tuned by using the feeding molar ratio of Pd to Cu precursors, which have a great influence on the ORR properties of the Pt-based core–shell catalysts. The Pd2Cu@Pt truncated octahedra with the segregation of the Pd interlayers exhibited the best activities towards ORR in an acid media due to the suitable ligand and geometry effects. The Pd2Cu@Pt truncated octahedra showed the similar stability relative to the commercial Pt/C due to the dissolution of the active metals in the cores. This work not only provides a facile approach to the synthesis of ternary core–shell nanocrystals with ultrathin Pt shells, but also offers great opportunities to design economical and efficient Pt-based ORR catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Electron microscopy was carried out in the Center for Electron Microscopy of Zhejiang University. This work was supported by the National Science Foundation of China (51522103, 61721005 and 51871200) and the National Program for Support of Top-notch Young Professionals.Notes and references
- M. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- H. Mistry, A. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- M. Shao, Q. Chang, J. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- H. Gasteiger, S. Kocha, B. Sompalli and T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- H. Gasteiger and N. Marković, Science, 2009, 324, 48–49 CrossRef CAS PubMed.
- J. Nørskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- P. Ferreira, G. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. Gasteiger, J. Electrochem. Soc., 2005, 152, 65–78 CrossRef.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- K. Gilroy, A. Ruditskiy, H. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- X. Liu, D. Wang and Y. Li, Nano Today, 2012, 7, 448–466 CrossRef CAS.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- H. Zhang, M. Jin and Y. Xia, Chem. Soc. Rev., 2012, 41, 8035–8049 RSC.
- J. Kitchin, J. Nørskov, M. Barteau and J. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed.
- V. Stamenkovic, B. Fowler, B. Mun, G. Wang, P. Ross, C. Lucas and N. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543–18547 CrossRef CAS PubMed.
- C. Li, T. Liu, T. He, B. Ni, Q. Yuan and X. Wang, Nanoscale, 2018, 10, 4670–4674 RSC.
- S. Choi, R. Choi, S. Han and J. Park, Chem. Commun., 2010, 46, 4950–4952 RSC.
- V. Stamenkovic, B. Mun, M. Arenz, K. Mayrhofer, C. Lucas, G. Wang, P. Ross and N. Marković, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasché and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 3273–3291 CrossRef CAS.
- T. Bian, H. Zhang, Y. Jiang, C. Jin, J. Wu, H. Yang and D. Yang, Nano Lett., 2015, 15, 7808–7815 CrossRef CAS PubMed.
- J. Greeley, I. Stephens, A. Bondarenko, T. Johansson, H. Hansen, T. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. C. Camargo, E. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 40, 1302–1305 CrossRef PubMed.
- S. Xie, S. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- J. Park, L. Zhang, S. Choi, L. Roling, N. Lu, J. A. Herron, S. Xie, J. Wang, M. Kim, M. Mavrikakis and Y. Xia, ACS Nano, 2015, 9, 2635–2647 CrossRef CAS PubMed.
- X. Wang, S. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- Y. Xiong, H. Shan, Z. Zhou, Y. Yan, W. Chen, Y. Yang, Y. Liu, H. Tian, J. Wu, H. Zhang and D. Yang, Small, 2017, 13, 1603423 CrossRef PubMed.
- D. Wang, H. Xin, Y. Yu, H. Wang, E. Rus, D. A. Muller and H. D. Abruña, J. Am. Chem. Soc., 2010, 132, 17664–17666 CrossRef CAS PubMed.
- W. Xiao, M. Cordeiro, G. Gao, A. Zheng, J. Wang, W. Lei, M. Gong, R. Lin, E. Stavitski, H. Xin and D. Wang, Nano Energy, 2018, 50, 70–78 CrossRef CAS.
- M. Shao, B. Smith, S. Guerrero, L. Protsailo, D. Su, K. Kaneko, J. Odell, M. Humbert, K. Sasaki, J. Marzullo and R. Darling, Phys. Chem. Chem. Phys., 2013, 15, 15078 RSC.
- T. Cochell and A. Manthiram, Langmuir, 2012, 28, 1579–1587 CrossRef CAS PubMed.
- M. Shao, K. Shoemaker, A. Peles, K. Kaneko and L. Protsailo, J. Am. Chem. Soc., 2010, 132, 9253–9255 CrossRef CAS PubMed.
- D. Wang, H. Xin, H. Wang, Y. Yu, E. Rus, D. Muller, F. DiSalvo and H. Abruña, Chem. Mater., 2012, 24, 2274–2281 CrossRef CAS.
- K. Sasaki, H. Naohara, Y. Choi, Y. Cai, W. Chen, P. Liu and R. Adzic, Nat. Commun., 2012, 3, 1115 CrossRef PubMed.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489 CrossRef CAS PubMed.
- M. Weinert and R. Watson, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 17168–17180 CrossRef CAS.
- D. Chen, P. Sun, H. Liu and J. Yang, J. Mater. Chem. A, 2017, 5, 4421–4429 RSC.
- J. Zhang, M. Vukmirovic, Y. Xu, M. Mavrikakis and R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132–2135 CrossRef CAS PubMed.
- I. Stephens, A. Bondarenko, U. Gronbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744 RSC.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley and K. More, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- Q. Xu, W. Chen, Y. Yan, Z. Wu, Y. Jiang, J. Li, T. Bian, H. Zhang, J. Wu and D. Yang, Sci. Bull., 2018, 63, 494–501 CrossRef CAS.
- Y. Yan, J. Du, K. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07415a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |