
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient conversion of CO2 to cyclic carbonates with a binary catalyst system in a microreactor: intensification of “electrophile–nucleophile” synergistic effect†
Ming-Ran Li,
Ming-Chao Zhang,
Tian-Jun Yue,
Xiao-Bing Lu and
Wei-Min Ren *
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Road, Dalian, 116024, China. E-mail: wmren@dlut.edu.cn
First published on 23rd November 2018
Abstract
An intensification of the “electrophile–nucleophile” synergistic effect was achieved in a microreactor for the coupling reaction of CO2 and epoxides mediated by the binary Al complex/ternary ammonium salt catalyst system. The microreactor technology is proven to be a powerful tool for the preparation of cyclic carbonates with an improved reaction rate and a wide substrate scope.
Introduction
The chemical fixation of carbon dioxide (CO2) into economically competitive products has attracted much attention, as CO2 is one of the most important greenhouse gases, as well as being an abundant, inexpensive, nontoxic, and renewable C1 resource.1–6 One of the most studied reactions for the use of CO2 is the coupling reaction of CO2 with epoxides to afford cyclic carbonates,7–10 which are widely used as electrolytes in lithium-ion secondary batteries11,12 and polar aprotic solvents,13,14 as well as intermediates in organic synthesis.15–19 Successful industrial processes for the preparation of cyclic carbonates from the CO2/epoxides coupling reaction have been realized for more than 50 years, however, efficient transformations usually required high catalyst loadings, elevated CO2 pressures, and long reaction time.In the past decades, various catalysts have been developed for the coupling reaction of CO2 with epoxides, such as metal oxides,20,21 alkali metal salts,22,23 organic bases,24,25 ionic liquids,26–28 metal complexes,29–31 and so on. Prominent among these are the homogeneous catalyst systems based on well-defined metal complexes.32–37 These catalyst systems provide a high activity and product selectivity for the coupling reaction of CO2 with various epoxides even under mild reaction conditions. For example, Ema et al. developed a bifunctional magnesium porphyrin catalyst showing a very high turnover number (TON up to 103![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) for this coupling reaction.38 Kleij and coworkers also reported an aluminium complex exhibiting an unprecedented high activity (TOF up to 36000 h−1) in the formation of cyclic carbonates.39 It is worth noting that the presence of a nucleophilic co-catalyst, such as organic base or quaternary ammonium salt, is beneficial to obtain cyclic carbonates efficiently for most metal complexes.40 A widely accepted mechanism concerning epoxide ring-opening and CO2 activation was shown in Scheme 1 when a binary metal complex/quaternary ammonium salt system was employed. According to this mechanism, the activation of epoxide coordinated to the electrophilic metal center and further ring-opened by the attack of nucleophilic agent is the rate-determining step during the coupling reaction. In this context, an intensification of the “electrophile–nucleophile” synergistic effect might help to further improve the catalytic activity of the binary systems.
000) for this coupling reaction.38 Kleij and coworkers also reported an aluminium complex exhibiting an unprecedented high activity (TOF up to 36000 h−1) in the formation of cyclic carbonates.39 It is worth noting that the presence of a nucleophilic co-catalyst, such as organic base or quaternary ammonium salt, is beneficial to obtain cyclic carbonates efficiently for most metal complexes.40 A widely accepted mechanism concerning epoxide ring-opening and CO2 activation was shown in Scheme 1 when a binary metal complex/quaternary ammonium salt system was employed. According to this mechanism, the activation of epoxide coordinated to the electrophilic metal center and further ring-opened by the attack of nucleophilic agent is the rate-determining step during the coupling reaction. In this context, an intensification of the “electrophile–nucleophile” synergistic effect might help to further improve the catalytic activity of the binary systems.
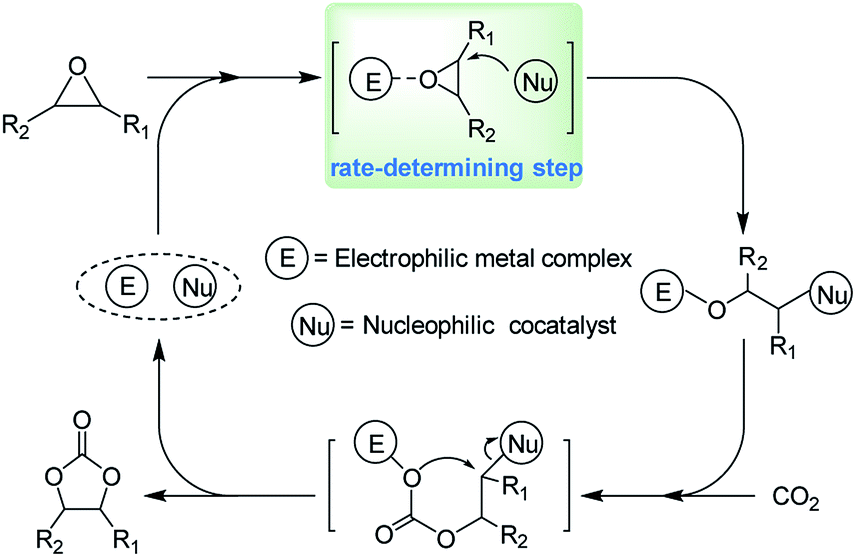 | ||
| Scheme 1 Catalytic cycle for CO2/epoxide coupling reaction mediated by the binary “electrophile–nucleophile” system. | ||
Microreactor technology is regarded as a promising process intensification strategy for chemical synthesis due to its unique characteristics, such as high effective surface-to-volume ratio, enhanced heat- and/or mass-transfer rates, and excellent process safety.41–45 In 2013, Chen and co-workers studied the coupling reaction of CO2 with propylene oxide (PO) in a microreactor using a hydroxyl-functionalized quaternary ammonium salt as catalyst.46 The reaction rate is significantly improved compared to the conventional stirred reactor with the use of the same reaction conditions. Herein, we wise to further explore the application of microreactor technology in the coupling reaction of CO2 with epoxides. The aluminium salen complex in conjunction with a quaternary ammonium salt was chosen as a binary catalyst system for this transformation since it is robust, easily prepared and recyclable. The main purpose of this study is to verify the intensification of the “electrophile–nucleophile” synergistic effect for epoxides ring-opening, decrease the reaction time from hours to seconds, and expand the substrate scope.
Experimental
Materials
PO and tetrabutylammonium bromide (TBAB) purchased from Sinopharm Chemical Reagent Co. Ltd. Carbon dioxide (99.99%) was used as received.Experimental procedure
The microreactor is consisted of two stainless-steel plates with microstructures that combine to form a complex sloped structure channel (Fig. 1). The geometric size of the mixing channel is 300 μm × 300 μm × 1 cm (length). The fluid continuously rises and falls as it flows through the channel, achieving multiple separation and recombination to complete the ample mixing process.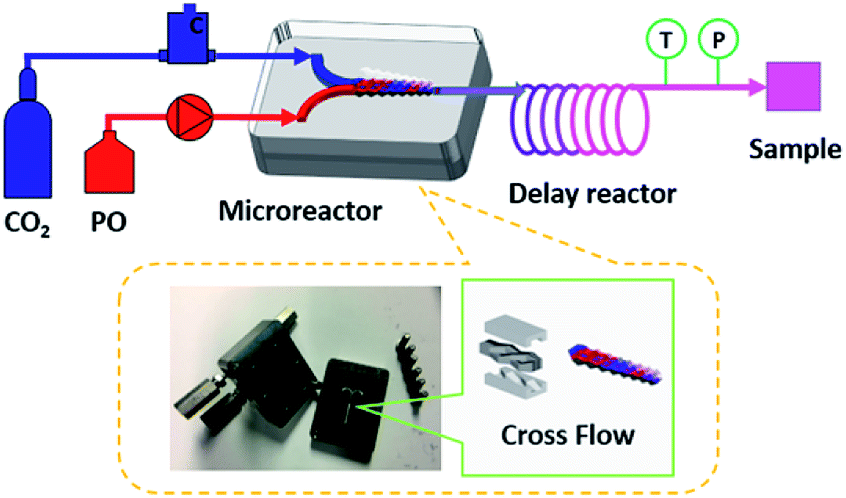 | ||
| Fig. 1 The experimental setup and illustration of the microreactor inner structure. | ||
The aluminum catalyst and TBAB were dissolved in epoxides and pumped by a flow pump into the reaction system. On the other hand, the CO2 high-pressure gas cylinder was connected to the pipeline via a gas flow meter and the flow rate was controlled by the gas quality controller. The two-phase materials were mixed in microreactor and fully reacted via a delay reactor (1/8 inch 316L stainless steel tube, wall thickness 0.5 mm, 22.83 mL). Both the microreactor and the delay reactor located in the oil bath were regarded as the residence time unit for regulating the reaction time. The temperature and pressure controlled by the back pressure valve of the reaction system were detected by sensors and transmitted to the data acquisition system, making the data fluctuation observation more intuitive. Then, the gas–liquid reaction mixture was separated in the gas–liquid separator. The liquid product sample was collected and weighted for the calculation of the yield. Additionally, a small amount of the crude product was dissolved in methanol, and analyzed by Agilent 7890B gas chromatography (GC with flame ionization detector, HP-5 column, 30 m × 0.32 mm × 0.25 μm) for determining the cyclic product selectivity. Each set of experiments was repeated three times to ensure the accuracy of the data.
Results and discussion
We initially chose PO as a model substrate to test the catalytic activity of the binary system in the microreactor. Neither the aluminum complex (0.5% mol) nor TBAB (1% mol) alone can mediate the PO/CO2 coupling reaction with satisfactory conversion (0 and 36%, respectively) at 150 °C and 2.0 MPa CO2 pressure (Table 1, entries 1 and 2). When the aluminum complex (0.5% mol) in combination with TBAB (1% mol) was employed as a binary catalyst system for the coupling reaction, the conversion of PO increased dramatically (entry 3). With a further increase in the loading of TBAB, quantitative conversion of PO was achieved with a residence time of 48.8 s (entry 5). In this case, the turnover frequency (TOF) value was up to 14700 h−1, which is significantly higher than the value (5160 h−1) in a conventional stirred reactor (entry 6).47 In contrast, a yield of 90% was observed under the same conditions when TBAB (5% mol) was used exclusively (entry 7). These results suggest that the “electrophile–nucleophile” synergistic effect of the binary catalyst system can be intensified in the microreactor.
|
||||
|---|---|---|---|---|
| Entry | [Al]/[PO] | [TBAB]/[PO] | Yieldb (%) | TOFc (h−1) |
| a Typical reaction conditions: 150 °C, 2.0 MPa, [CO2]/[PO] = 2:1, (CO2 flow rate of 385 mL min−1 under standard conditions and PO flow rate of 0.6 mL min−1) residence time 48.8 s for entries 1–5. The propylene carbonate selectivity is >99% based on GC.b Yield of propylene carbonate.c Turnover frequency (TOF) = moles of cyclic carbonate per mole of Al-catalyst per hour.d The coupling reaction was performed in a conventional stirred reactor for 49 s. |
||||
| 1 | 0.5% | 0 | <1 | — |
| 2 | 0 | 1% | 36 | — |
| 3 | 0.5% | 1% | 62 | 9150 |
| 4 | 0.5% | 3% | 85 | 12540 |
| 5 | 0.5% | 5% | >99 | 14700 |
| 6d | 0.5% | 5% | 35 | 5160 |
| 7 | 0 | 5% | 90 | — |
In order to further verify the intensification, the effects of the residence time on the conversion of PO was studied. The residence time can be defined as the total volume of the reaction line divided by the gas–liquid mixing flow rate in the microreactor. In this text, changing the flow rate of CO2 and PO in the same proportion can adjust the residence time of the reaction while maintaining other conditions unchanged. For example, the residence time of 48.8 s corresponded to CO2 flow rate of 385 mL min−1 and PO flow rate of 0.6 mL min−1, while the residence time of 6.1 s can be got by increasing the CO2 flow rate to 3080 mL min−1 and PO flow rate to 4.8 mL min−1. Thus, a series of plots of conversion versus residence time was obtained with a reaction condition of 150 °C and 2.0 MPa CO2 pressure (Fig. 2). The results show that the yield of propylene carbonate reaches >98% when the residence time is 30 s. More importantly, the reaction rate did not show an obvious decrease even the yield of propylene carbonate up to 95%. By contrast, the reaction rate was gradually decreased after the yield of cyclic carbonate reaches 60% in a conventional stirred reactor, and it may take about half of the total reaction time to convert the remaining ∼20% PO.48
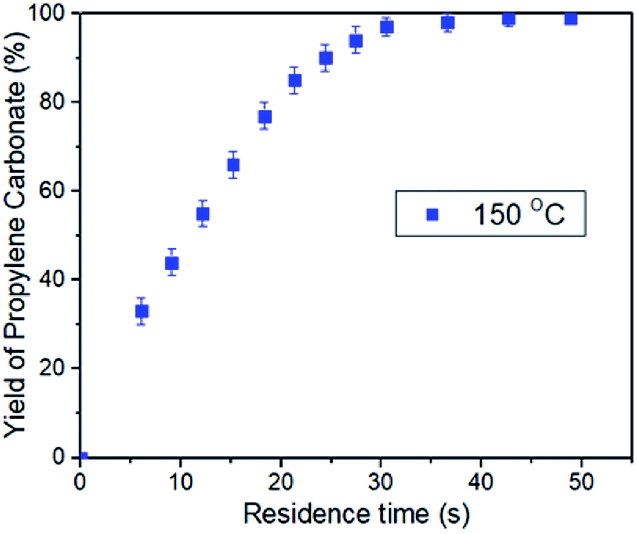 | ||
| Fig. 2 Plots of PC conversion vs. time, with the use of the binary catalyst system at 150 °C and 2.0 MPa CO2 pressure. [Al]/[TBAB]/[PO] = 0.5%/5%/1, [PO]/[CO2] = 1/2. | ||
Another advantage of the microreactor, compared with the conventional stirred reactor, is that changing the ratio of CO2 to PO does not affect the reaction pressure.49 Therefore, we examined the effects of the molar ratio of CO2 to PO on the reaction rate under the same CO2 pressure. When the coupling reaction was performed at 140 °C and 2.0 MPa CO2 pressure with the residence time of 48.8 s or 24.4 s, an increase in the molar ratio of CO2 to PO from 2/1 to 4/1 does not lead to an observable change in the yield of propylene carbonate (Fig. 3). These results imply the variation of the CO2/PO molar ratio has little effect on the “electrophile–nucleophile” synergistic effect for epoxides ring-opening. The very slight fluctuation in the yield of propylene carbonate may be due to the small change of PO concentration in the liquid phase with the different CO2/PO molar ratios. Unfortunately, we did not obtained the data with a CO2/PO molar ratio of 1/1, because the consumption of CO2 may lead to a loss of accuracy in the calculation of the residence time.
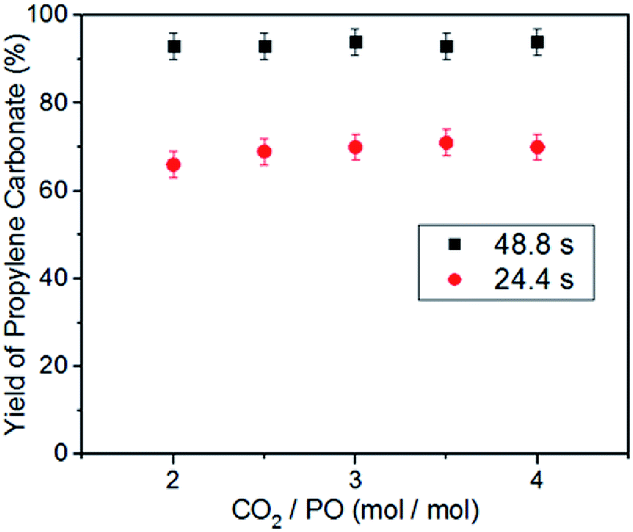 | ||
| Fig. 3 Effect of the molar ratio of CO2/PO on the yield of propylene carbonate, with the use of the binary catalyst system at 140 °C and 2.0 MPa CO2 pressure. [Al]/[TBAB]/[PO] = 0.5%/5%/1. | ||
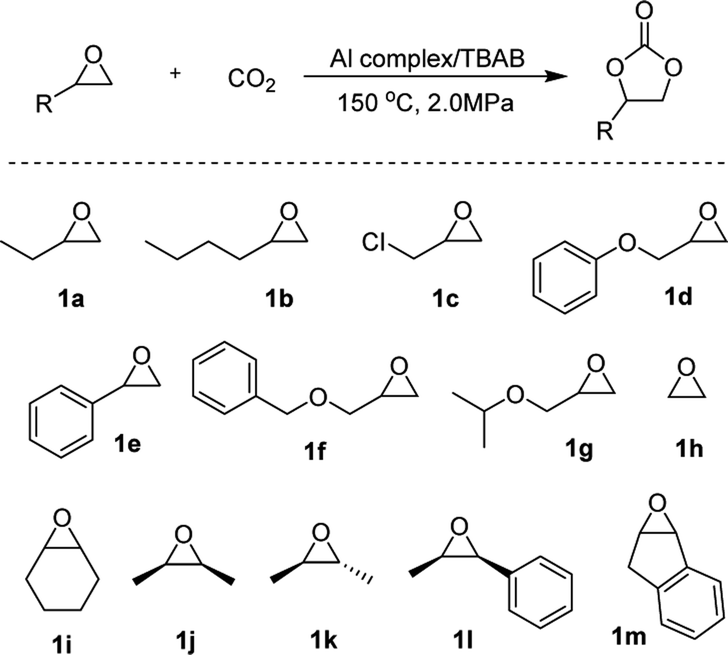
To explore the wider applicability of the microreactor, a series of monosubstituted terminal epoxides and ethylene oxide were tested. All the terminal cyclic carbonates were obtained with excellent yields (>90%) and high selectivity (99%) under 150 °C and 2.0 MPa CO2 pressure within the residence time of less than 100 s (Table 2, entries 1–7). When ethylene oxide was employed as a substrate, the quantitative formation of ethylene carbonate with a high reaction rate of TOF up to 29000 h−1 was achieved under a residence time of 24.4 s (entry 8). For further expanding the substrate scope, some disubstituted or internal epoxides were chosen as reaction partners. All of the substrates studied were conveniently converted into the corresponding carbonates with high selectivity (>99%) and good to excellent yields, indicating the high versatility of the microreactor technology.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Residence time (s) | Yieldb (%) | TOFc (h−1) |
| a Typical reaction conditions: 150 °C, 2.0 MPa, [Al]/[TBAB]/[PO] = 0.5%/5%/1, [CO2]/[PO] = 2:1. The cyclic carbonate selectivity is >99% based on GC or NMR spectroscopy.b Yield of cyclic carbonate.c Turnover frequency (TOF) = moles of cyclic carbonate per mole of Al-catalyst per hour.d Cis/trans = 86:14.e Cis/trans = 12:88.f Cis/trans = 38:62. The cis/trans ratios were determined by 1H NMR spectroscopy. |
||||
| 1 | 1a | 48.8 | >99 | 14700 |
| 2 | 1b | 97.6 | >99 | 7300 |
| 3 | 1c | 48.8 | >99 | 14700 |
| 4 | 1d | 97.6 | 97 | 7100 |
| 5 | 1e | 97.6 | 96 | 6860 |
| 6 | 1f | 97.6 | 96 | 7000 |
| 7 | 1g | 97.6 | 98 | 7200 |
| 8 | 1h | 24.4 | >99 | 29000 |
| 9 | 1i | 97.6 | 97 | 7100 |
| 10 | 1j | 97.6 | 91d | 6700 |
| 11 | 1k | 97.6 | 92e | 6800 |
| 12 | 1l | 97.6 | 87f | 6400 |
| 13 | 1m | 97.6 | 81 | 6000 |
Conclusions
In summary, the conversion of CO2 to various cyclic carbonates proceeds effectively in a microreactor using a binary Al complex/quaternary ammonium salt catalyst system. All the tested epoxides can be transformed to the corresponding cyclic carbonates with >95% yield under the residence time of less than 100 s due to the intensification of “electrophile–nucleophile” synergistic effect for epoxides ring-opening. It is clear that the microreactor technology presented here might be ideally suited for the industrial preparation of cyclic carbonates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gratitude is expressed to the Fundamental Research Funds for the Central Universities (DUT18ZD105).Notes and references
- A. W. Kleij, M. North and A. Urakawa, ChemSusChem, 2017, 10, 1036–1038 CrossRef CAS PubMed.
- A. Farran, C. Cai, M. Sandoval, Y. Xu, J. Liu, M. J. Hernaiz and R. J. Linhardt, Chem. Rev., 2015, 115, 6811–6853 CrossRef CAS PubMed.
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- J. A. Martens, A. Bogaerts, N. De Kimpe, P. A. Jacobs, G. B. Marin, K. Rabaey, M. Saeys and S. Verhelst, ChemSusChem, 2017, 10, 1039–1055 CrossRef CAS PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- J. Liang, Y. Q. Xie, X. S. Wang, Q. Wang, T. T. Liu, Y. B. Huang and R. Cao, Chem. Commun., 2018, 54, 342–345 RSC.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2017, 8, 419–450 CrossRef.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- Y. Wang, J. Zhao, J. Qu, F. Wei, W. Song, Y. G. Guo and B. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 26008–26012 CrossRef CAS PubMed.
- J. Mindemark, L. Imholt, J. Montero and D. Brandell, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2128–2135 CrossRef CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- B. Schaeffner, F. Schaeffner, S. P. Verevkin and A. Boerner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- W. Guo, J. E. Gomez, A. Cristofol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS PubMed.
- A. Kaithal, M. Holscher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449–13453 CrossRef CAS PubMed.
- B. R. James, J. A. Boissonnault, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, RSC Adv., 2018, 8, 2132–2137 RSC.
- B. Yu and L. N. He, ChemSusChem, 2015, 8, 52–62 CrossRef CAS PubMed.
- J. Zhang, Q. Cai, J. Zhao and S. Zang, RSC Adv., 2018, 8, 4478–4482 RSC.
- M. N. Timofeeva, A. E. Kapustin, V. N. Panchenko, E. O. Butenko, V. V. Krupskaya, A. Gil and M. A. Vicente, J. Mol. Catal. A: Chem., 2016, 423, 22–30 CrossRef CAS.
- H. Chang, Q. Li, X. Cui, H. Wang, Z. Bu, C. Qiao and T. Lin, J. CO2 Util., 2018, 24, 174–179 CrossRef CAS.
- Z. Yang, J. Sun, X. Liu, Q. Su, Y. Liu, Q. Li and S. Zhang, Tetrahedron Lett., 2014, 55, 3239–3243 CrossRef CAS.
- L. Ji, Z. Luo, Y. Zhang, R. Wang, Y. Ji, F. Xia and G. Gao, Mol. Catal., 2018, 446, 124–130 CrossRef CAS.
- M. Ahmed and A. Sakthivel, J. CO2 Util., 2017, 22, 392–399 CrossRef CAS.
- C. Yang, M. Liu, J. Zhang, X. Wang, Y. Jiang and J. Sun, Mol. Catal., 2018, 450, 39–45 CrossRef CAS.
- Y. Ma, C. Chen, T. Wang, J. Zhang, J. Wu, X. Liu, T. Ren, L. Wang and J. Zhang, Appl. Catal., A, 2017, 547, 265–273 CrossRef CAS.
- D.-H. Lan, N. Fan, Y. Wang, X. Gao, P. Zhang, L. Chen, C.-T. Au and S.-F. Yin, Chin. J. Catal., 2016, 37, 826–845 CrossRef CAS.
- J. Peng, H.-J. Yang, Y. Geng, Z. Wei, L. Wang and C.-Y. Guo, J. CO2 Util., 2017, 17, 243–255 CrossRef CAS.
- H. Buttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- X. B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- C. Miceli, J. Rintjema, E. Martin, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, ACS Catal., 2017, 7, 2367–2373 CrossRef CAS.
- J. A. Castro-Osma, K. J. Lamb and M. North, ACS Catal., 2016, 6, 5012–5025 CrossRef CAS.
- C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS PubMed.
- D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS.
- L. Wang, C. Xu, Q. Han, X. Tang, P. Zhou, R. Zhang, G. Gao, B. Xu, W. Qin and W. Liu, Chem. Commun., 2018, 54, 2212–2215 RSC.
- R. Ma, L.-N. He and Y.-B. Zhou, Green Chem., 2016, 18, 226–231 RSC.
- T. Ema, Y. Miyazaki, S. Koyama, Y. Yano and T. Sakai, Chem. Commun., 2012, 48, 4489–4491 RSC.
- C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed.
- However, the bimetallic Aluminum (salen) complexes was proven to be efficient for the synthesis of cyclic carbonates from the coupling reaction of epoxides and CO2 in the absence of a halide cocatalyst, see: J. A. Castro-Osma, M. North, W. K. Offermans, W. Leitner and T. E. Muller, ChemSusChem, 2016, 9, 791–794 CrossRef CAS PubMed .Similarly, the Aluminum complex based on an amino triphenolate ligand could alone mediate the coupling reaction of epoxy alcohols with CO2, see: J. Rintjema, R. Epping, G. Fiorani, E. Martin, E. C. Escudero-Adan and A. W. Kleij, Angew. Chem., Int. Ed., 2016, 55, 3972–3976 CrossRef PubMed.
- P. L. Suryawanshi, S. P. Gumfekar, B. A. Bhanvase, S. H. Sonawane and M. S. Pimplapure, Chem. Eng. Sci., 2018, 189, 431–448 CrossRef CAS.
- M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2018, 118, 4541–4572 CrossRef CAS PubMed.
- Y. Song, M. Shang, G. Li, Z.-H. Luo and Y. Su, AIChE J., 2018, 64, 1828–1840 CrossRef CAS.
- H. Yao, Y. Wang and G. Luo, Ind. Eng. Chem. Res., 2017, 56, 4993–4999 CrossRef CAS.
- S. Zhao, Z. Dong, C. Yao, Z. Wen, G. Chen and Q. Yuan, AIChE J., 2018, 64, 1412–1423 CrossRef CAS.
- Y. Zhao, C. Yao, G. Chen and Q. Yuan, Green Chem., 2013, 15, 446–452 RSC.
- The reaction was carried out in a 75 mL conventional stirred reactor, with the reaction conditions: 150 °C, 2.0 MPa CO2 pressure, PO 15 mL, [Al]/[TBAB]/[PO] = 0.5%/5%/1 and performed for 49 s..
- W. M. Ren, Y. Liu and X. B. Lu, J. Org. Chem., 2014, 79, 9771–9777 CrossRef CAS PubMed.
- Reaction pressure was regulated by a back pressure valve, and the flow rate of the mixture mainly depended on the CO2. Therefore, the same pressure, same residence time with different molar ratio of PO and CO2 can be realized, which is more controllable than the conventional stirred reactor..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07236a |
| This journal is © The Royal Society of Chemistry 2018 |