
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution processed organic light-emitting diodes using a triazatruxene crosslinkable hole transporting material†
Azin Babaeia,
Kasparas Rakstysb,
Simon Guelenc,
Vahid Fallah Hamidabadiae,
Maria-Grazia La-Placaa,
Laura Martínez-Sartia,
Michele Sessolo a,
Huckaba Aron Joelb,
Olivier P. M. Gaudind,
Vincent Schanenc,
Mohammad Khaja Nazeeruddinb and
Henk J. Bolink*a
a,
Huckaba Aron Joelb,
Olivier P. M. Gaudind,
Vincent Schanenc,
Mohammad Khaja Nazeeruddinb and
Henk J. Bolink*a
aInstituto de Ciencia Molecular, Universidad de Valencia, C/J. Beltrán 2, 46980, Paterna, Spain. E-mail: henk.bolink@uv.es
bGroup for Molecular Engineering of Functional Materials, Ecole Polytechnique Federale de Lausanne, Valais Wallis, CH-1951 Sion, Switzerland
cCentre de Recherche Solvay, 85 rue des Freres Perret, 69190 Saint-Fons, France
dSolvay, rue de Ransbeek 310, B-1120 Brussels, Belgium
eDepartment of Physics, Faculty of Basic Sciences, University of Mazandaran, Babolsar 47416-95447, Iran
First published on 19th October 2018
Abstract
A cross-linkable triazatruxene that leads to insoluble films upon thermal annealing at temperatures compatible with flexible substrates is presented. The films were used as the hole transporting and electron blocking layer in partially solution processed phosphorescent organic light-emitting diodes, reaching power conversion efficiencies of 24 lm W−1, an almost 50% improvement compared to the same OLEDs without the cross-linkable hole transporting layer.
Organic light-emitting diodes (OLEDs) have become the new standard in the portable display industry, thanks to their high color quality and contrast, high brightness and wide viewing angle, and most importantly their low power consumption. The latter property is of particular importance in lighting applications, as lighting accounts for about 20% of global energy consumption. While white OLEDs with very high efficiency have been demonstrated and are under development,1,2 their cost is still not competitive with other current lighting technologies. Efficient OLEDs are multilayer thin-film devices prepared by vacuum thermal deposition, implying a high initial capital investment and limited throughput. For this reason, solution processing methods for multilayer OLEDs are still being investigated, as they might allow the preparation of efficient and inexpensive light sources.3–7 The processing of multilayer thin-film structures from solution requires avoiding the dissolution or intermixing of subsequent layers. This can be achieved either by using orthogonal solvents,8,9 or by cross-linking the organic semiconductor based thin films in order to minimize their solubility.10,11 The first route, using orthogonal solvents for subsequent layers, limits the materials choice as most of organic semiconductors present very similar solubility properties. On the other hand, one of the advantages of organics is the possibility of introducing desired functionalities through chemical synthesis without undermining the semiconducting properties. For this reason the photo- or thermal cross-linking is the widest adopted route towards the fabrication of organic multilayer devices. Most reports focus on the development of cross-linkable hole transport materials (HTMs).12–19 Besides the more common arylamine-based HTMs, triazatruxenes are recently being intensively studied for their interesting solid-state assembly and charge transport properties.20 Due to the C3 symmetry present in the molecule, discotic donor–acceptor (D–A) materials can easily be targeted. These charge transfer materials benefit from the three electron rich indolyl phenyl rings and the three acceptor moieties. Some of these discotic structures have been shown to preferentially stack in a “face-on” orientation in neat films.21 The electron rich triazatruxenes have destabilized ground state energy values relative to polyaromatic hydrocarbon with similar structures, which renders them excellent candidates for hole transport materials. Selective modification of either the indolyl nitrogens or the phenyl rings can yield densely functionalized molecules with good hole mobilities.22,23 In this work, we describe the synthesis and application to multilayer OLEDs of a new cross-linkable triazatruxene derivative. To facilitate solution processing a triazatruxene HTM containing alkyl chains was designed. This molecule was functionalized with the versatile styrene moiety that enables the crosslinking of the HTM without the need for photo-initiators. This is beneficial for semiconducting properties as photo-initiators are not easily removed after reaction in the thin films. However, since styrene groups have a low-energy triplet state capable of quenching triplet excitons in the adjacent light-emitting layer in OLEDs,24 we chose a weakly accepting phenyl group as the acceptor to the indolyl phenyl groups to ensure a wide molecular bandgap and a high energy triplet state. We show proof-of-principle multilayer OLEDs using the cross-linked HTM in combination with a solution-processed, small molecular weight phosphorescent emissive layer. The results highlight the potential of this class of HTM, which can further find applications in OLEDs, organic or perovskite solar cells and organic photodiodes.
Experimental
We synthesized the cross-linkable HTM KR386 in four steps starting from the commercially available 2-oxindole (Scheme 1). POCl3 mediated triple condensation of 2-oxindole yielded the triazatruxene core 1, which was alkylated with 1-iodohexane to yield triazatruxene 2 and then brominated using N-bromosuccinimide (NBS). Finally, palladium-catalyzed Suzuki coupling of the triply-brominated 3 with 4-vinylphenylboronic acid yielded KR386.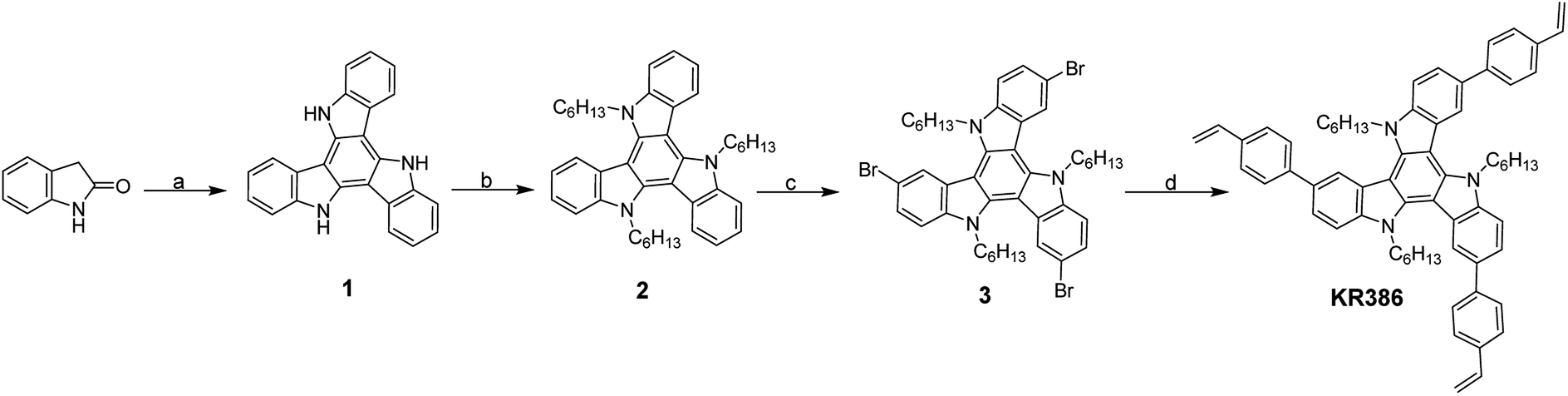 | ||
| Scheme 1 (a) POCl3, 100 °C; (b) 1-iodohexane, NaH, DMF, refluxed 2 h; (c) NBS in DMF, CHCl3, 0–25 °C; (d) 4-vinylphenylboronic acid, Pd(PPh3)4, 2 M aq. K2CO3, THF, 80 °C. | ||
All reagents were used as received. Unless otherwise noted, all reactions were performed under the dry N2 atmosphere, all reactions requiring air-free techniques were performed in oven-dried glassware or in glassware that was heated with a heat gun under vacuum for 5–10 minutes. Thin-layer chromatography (TLC) was conducted with pre-coated TLC-sheets ALUGRAM® SIL G/UV254 and visualized with UV. Flash column chromatography was performed using Sili-cycle P60, 40–63 μm (230–400 mesh) silica particles. 1H and 13C NMR spectra were recorded on a Bruker AVIII-HD (400 MHz) and are reported in ppm using solvents as an internal standards (CDCl3 at 7.26 ppm and 77.16 ppm, acetone-d6 at 2.05 ppm and 29.84 ppm for 1H and 13C NMR spectra respectively). Data reported as: s = singlet, d = doublet, t = triplet, m = multiplet, p = pentet, dd = doublet of doublets, ddd = doublet of doublet of doublets, br = broad signal, coupling constant(s) in Hz; integration.
Synthesis of 10,15-dihydro-5H-diindolo[3,2-a:3′,2′-c]carbazole (1)
A mixture of 2-indolinone (10 g, 75 mmol) and POCl3 (50 mL) was heated at 100 °C and allowed to stir for 8 h. Then, the reaction mixture was poured into ice and carefully neutralized with NaOH. After neutralization, the precipitate was filtered to give the crude product as a brown solid. The crude solution in MeOH was absorbed on silica-gel, dried, loaded and eluted through a thick silica-gel pad using DCM as the eluent. After concentration of the eluent at reduced pressure and recrystallization from acetone, a pure pale yellow solid was obtained (5.5 g, 63%). 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 3H), 8.66 (d, J = 7.4 Hz, 3H), 7.71 (d, J = 7.9 Hz, 3H), 7.41–7.28 (m, 6H). 13C NMR (100 MHz, acetone-d6) δ 141.0, 136.4, 124.8, 124.5, 121.5, 121.4, 112.9, 103.3. C24H15N3[M+] exact mass = 345.1266, MS (ESI-TOF) = 345.1034.Synthesis of 5,10,15-trihexyl-10,15-dihydro-5H-diindolo[3,2-a:3′,2′-c]carbazole (2)
To a solution of 500 mg (1) (1.45 mmol, 1 eq.) in DMF (10 mL), 0.1 g NaH (5.1 mmol, 3.5 eq.) was added at room temperature and allowed to stir. After 30 m, 1.23 g 1-iodohexane (5.8 mmol, 4 eq.) was added via syringe and the mixture was then heated at reflux for 2 h. The cooled mixture was poured into water and extracted with DCM. The organic phase was dried over MgSO4. The crude product was subjected to silica gel column chromatography and was eluted using 20% DCM in hexane. The eluent was concentrated to yield a pale yellow solid (650 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 8.0 Hz, 3H), 7.64 (d, J = 8.0 Hz, 3H), 7.45 (t, J = 7.4 Hz, 3H), 7.34 (t, J = 7.6 Hz, 3H), 4.92 (m, 6H), 1.99 (p, J = 7.9 Hz, 6H), 1.38–1.16 (m, 18H), 0.81 (t, J = 7.1 Hz, 9H). 13C NMR (100 MHz, CDCl3) δ 137.25, 127.47, 120.96, 138.29, 119.80, 118.79, 110.30, 107.14, 44.76, 31.15, 29.17, 25.70, 21.56, 13.80. C42H51N3[M+] exact mass = 597.4083, MS (ESI-QTOF) = 597.4080.Synthesis of 3,8,13-tribromo-5,10,15-trihexyl-10,15-dihydro-5H-diindolo[3,2-a:3′,2′-c]carbazole (3)
To a solution of 350 mg (2) (0.58 mmol, 1 eq.) in 30 mL CHCl3, 320 mg NBS (1.8 mmol, 3.1 eq.) in 5 mL DMF was added dropwise via syringe at 0 °C. After addition, the reaction mixture was allowed to stir for 1 h at room temperature. The mixture was diluted with DCM, washed with water, and the organic phase was dried over MgSO4. The crude product was subjected to silica gel column chromatography and eluted with 10% DCM in hexane. The eluent was concentrated to yield a pale-yellow solid (400 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.6 Hz, 3H), 7.71 (s, 3H), 7.42 (d, J = 8.6 Hz, 3H), 4.81–4.70 (m, 6H), 1.96–1.76 (m, 6H), 1.12–1.28 (m, 18H), 0.80 (t, J = 7.0 Hz, 9H). 13C NMR (100 Hz, CDCl3) δ 125.9, 124.9, 124.2, 124.0, 123.1, 122.7, 113.4, 112.2, 47.4, 31.8, 30.4, 26.5, 22.8, 14.2. C42H48Br3N3[M+] exact mass = 831.1398, MS (ESI-QTOF) = 831.1389.Synthesis of 5,10,15-trihexyl-3,8,13-tris(4-vinylphenyl)-10,15-dihydro-5H-diindolo[3,2-a:3′,2′-c]carbazole (KR386)
To a degassed mixture of 1 g, (3) (1.2 mmol, 1 eq.), 0.9 g 4-vinylphenylboronic acid (6 mmol, 5 eq.) in THF (25 mL) and 2 M aqueous K2CO3 (5 mL), 300 mg Pd(PPh3)4 (20%) was added under N2, the resulting solution was heated to 80 °C and allowed to stir overnight. After allowing the reaction to cool to room temperature, the mixture was poured into water and extracted with DCM. The organic layer was concentrated and the crude product was subjected to silica gel column chromatography and eluted with 40% DCM in hexane. The eluent was concentrated to yield a yellow solid (650 mg, 59%). 1H NMR (400 MHz, tetrahydrofuran-d8) δ 8.40 (d, J = 8.4 Hz, 3H), 8.01 (s, 3H), 7.85 (d, J = 8.2 Hz, 6H), 7.68 (d, J = 8.4 Hz, 3H), 7.61 (d, J = 8.3 Hz, 6H), 6.85 (dd, J = 17.6, 10.9 Hz, 3H), 5.88 (d, J = 17.7 Hz, 3H), 5.28 (d, J = 11.4 Hz, 3H), 5.17–4.99 (m, 6H), 2.00 (p, J = 8.5, 7.8 Hz, 6H), 1.46–1.12 (m, 18H), 0.81 (t, J = 7.2 Hz, 9H). C66H69N3[M+] exact mass = 903.5491, MS (MALDI-TOF) = 903.614. 13C NMR (101 MHz, tetrahydrofuran-d8) δ 141.85, 141.32, 139.26, 136.69, 136.22, 135.41, 127.04, 126.53, 122.77, 121.74, 118.78, 112.49, 108.56, 103.36, 46.55, 31.37, 29.55, 26.16, 24.87, 24.67, 22.36, 13.27.OLEDs preparation and characterization
Pre-patterned indium tin oxide (ITO) coated glass substrates were purchased from Naranjo Substrates. Poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS, CLEVIOS™ P VP CH 8000) was purchased from Heraeus Holding GmbH. 1,3-Bis[2-(4-tert-butylphenyl)-1,3,4-oxadiazo-5-yl]benzene (OXD-7), 4,4′,4′′-tris(carbazol-9-yl)triphenylamine (TcTa), tris[2-(p-tolyl)pyridine]iridium(III) (Ir(mppy)3) and 1,3-bis[3,5-di(pyridin-3-yl)phenyl]benzene (BmPyPhB) were purchased from Luminescence Technology Corp.ITO-coated glass slides were first cleaned using chemical and UV-ozone methods. The hole-injection layer (HIL), hole-transport layer (HTL), and emissive layer (EML) were spin-coated from their solutions. A 40 nm thick PEDOT:PSS film (HIL) was spin coated (1500 rpm, 60 s) and annealed (180 °C, 15 min) in air. Then the substrates were transferred in a nitrogen-filled glove box where the rest of the organic film deposition was carried out. KR386 was spin coated (7000 rpm, 60 s) from a toluene solution (10 mg mL−1) on top of the PEDOT:PSS layer. To crosslink the KR386 the film was annealed for 30 minutes at temperature ranging from 130 to 180 °C. The final film thickness was 20 nm. The emissive layer consisted of a mixture of TcTa, OXD-7, and Ir(mppy)3 at 5 wt%, and was deposited by spin-coating (1500 rpm, 60 s) from chlorobenzene solutions (10 mg mL−1) to a thickness of 30 nm. The electron transport layer (ETL, BmPyPhB) was deposited by thermal vacuum deposition (40 nm) and the devices were finished by deposition of the top cathode, composed by barium (5 nm) and silver (70 nm). Thermal vacuum deposition was carried out in an Edwards Auto 500 chamber integrated into a glovebox.
The devices were characterized in an inert atmosphere without encapsulation. The current-versus-voltage (J–V) and luminance-versus-voltage (L–V) curves were obtained by using a Keithley 2400 source meter and a photodiode coupled to a Keithley 6485 picoammeter using a Minolta LS100 to calibrate the photocurrent. The absorption and electroluminescent spectra were measured using an Avantes AvaSpec-2048 Fiber Optic Spectrometer. The highest occupied molecular orbital of the KR386 was determined with an Ambient Pressure Photoemission Spectroscopy Systems (APS03) from KP Technology Ltd.
Results and discussion
Thin films of KR386 were spin-coated onto PEDOT:PSS from toluene solutions, obtaining a thickness of 20 nm. The films were annealed at different temperature in order to evaluate the cross-linking kinetics. The absorption spectra of the as-deposited and annealed KR386 films are reported in Fig. 1a. As-deposited films show an absorption onset at about 450 nm, which corresponds to an optical bandgap of about 2.7 eV. Annealing at 130–140 °C for 30 minutes results in a narrower absorption feature, slightly blue-shifted by approximately 15 nm for higher annealing temperature (150–180 °C). All films were subsequently rinsed twice with toluene and the excess solvent was removed by spin-coating. In order to evaluate the insolubility of KR386, the relative absorption of the films at each temperature was estimated by normalizing its absorbance to the corresponding spectra recorded before the toluene rinsing (Fig. 1b). The resistance to the solvent rinsing and hence the extent of cross-linking was found to increase substantially from 130 °C to 150 °C, and remained almost constant for higher temperatures. The film insolubility for each temperature was expressed as the relation between the maximum optical absorbance measured after and before rinsing the KR386 films with toluene. The trend of this film insolubility versus the annealing temperature (Fig. 1c) shows that below 140 °C more than half of the material is still removed, while the KR386 thin films are fully insoluble as a results of extensive crosslinking by annealing at 180 °C for 15 minutes. The degree of crosslinking is not easily determined. The infrared spectrum of the as-deposited film was measured, however, no clearly identifiable changes could be observed before and after annealing the film (Fig. S1†). This is likely due to the overlap of absorption of similar groups with those that are reacting. However, for the OLED performance the film integrity is the main importance as this allows for the construction of neat multilayer devices. Hence the film morphology was investigated by atomic force microscopy, before and after crosslinking (Fig. S2†). As-deposited KR386 films showed a rather flat surface with small aggregates, resulting in a root-mean-square (RMS) roughness of approximately 1 nm. After cross-linking the surface at 180 °C for 15 minutes, the surface morphology was found to be much more homogeneous and pinhole-free, with only a slightly larger roughness (2 nm). This demonstrates that the film uniformity stays intact after thermal annealing. The cross-linked films were characterized by measuring the ionization potential of the KR386 in air by UV photoelectron spectroscopy in the 4.5–6.0 eV range (Fig. 2a).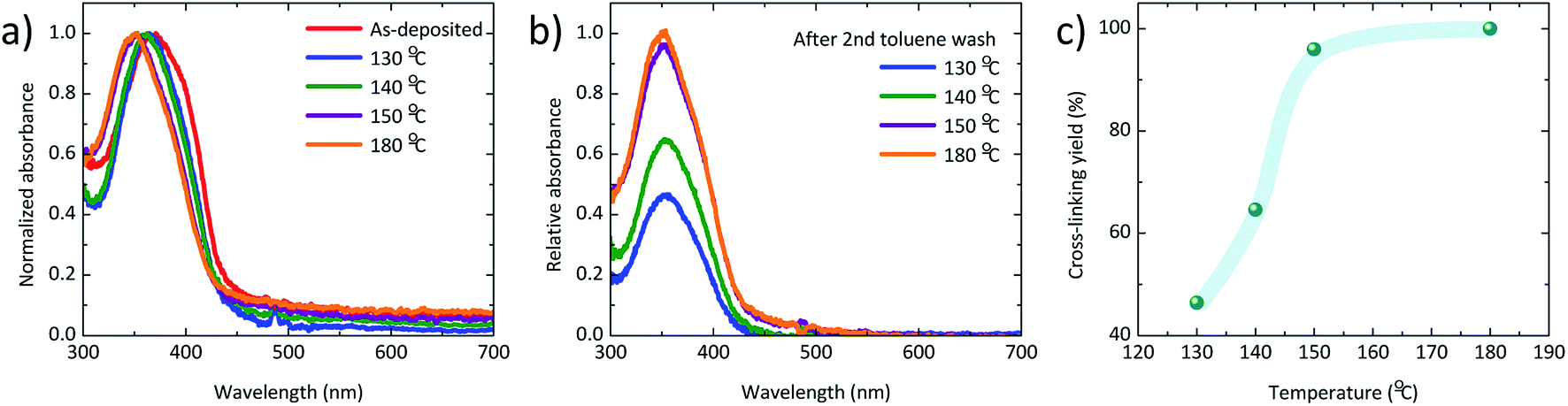 | ||
| Fig. 1 (a) Normalized absorption spectra for KR386 films annealed at different temperature. (b) Relative absorption spectra normalized to the initial maximum value for films annealed at different temperatures and washed twice with toluene. (c) Cross-linking yield of the same sample series. | ||
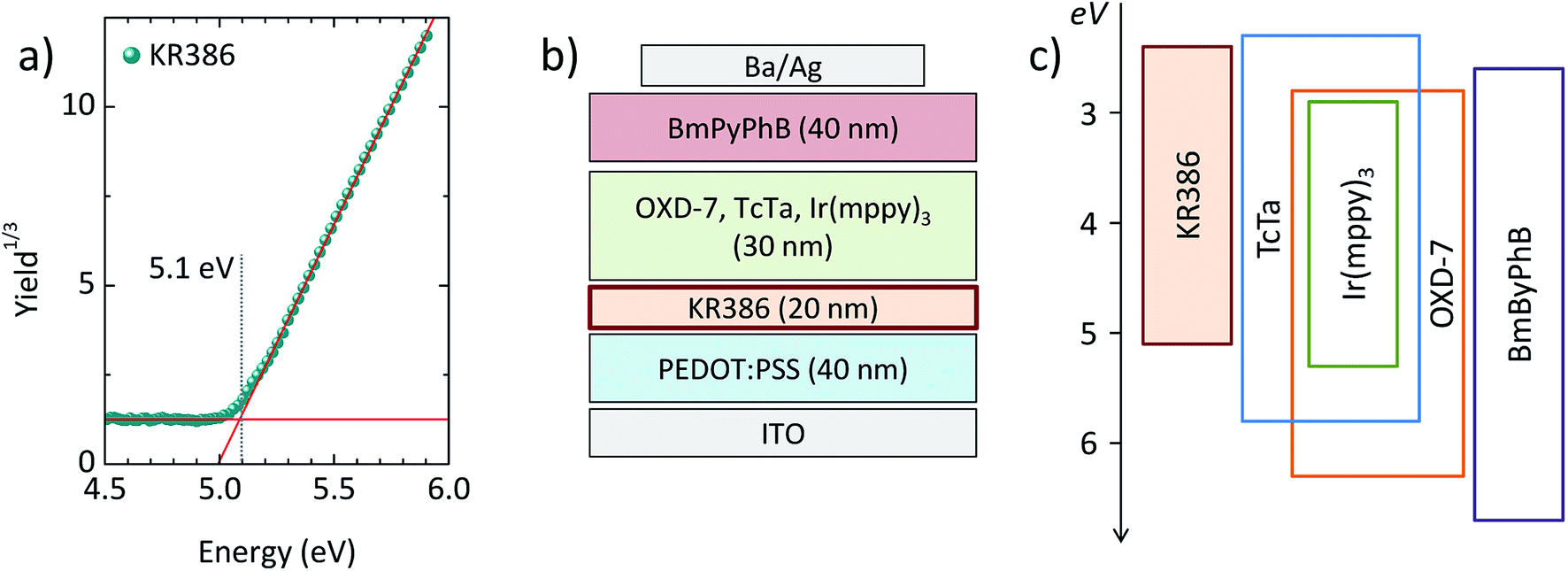 | ||
| Fig. 2 (a) Air photoemission spectrum for a KR386 thin film. (b) Scheme of the OLEDs configuration with (c) the corresponding flat band energy diagrams for the organic materials employed. The energy values are referred to the vacuum level. | ||
With this value and taking into account its bandgap energy (2.7 eV as from Fig. 1a), it is possible to estimate its energy levels. The highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO) of KR386 have a value of −5.1 eV and −2.4 eV from the vacuum level, respectively. KR386 was used to prepare multilayer OLEDs (Fig. 2b), using PEDOT:PSS as the HIL between the ITO anode and the HTL, and by using a small molecular weight solution processed phosphorescent emissive layer (EML). The latter is composed of TcTa, OXD-7 and Ir(mppy)3 as the emitter. In order to avoid exciton recombination at the cathode interface, a thin layer (40 nm) of the wide bandgap electron transport material BmPyPhB was deposited prior to the metallic cathode (Ba/Ag). A flat band energy diagram of the materials used in the OLEDs is depicted in Fig. 2c.
Fig. 3a shows the typical current density–voltage (J–V–L) characteristics of the above described OLEDs both with and without KR386. All devices show a very low current leakage at low bias, indicating a high morphological quality of the diode. Current injection starts at 2–2.5 V for the two type of devices. OLEDs without HTL show, however, a lower current density at higher applied voltage, due to the energy mismatch between the PEDOT:PSS work function (about −5 eV) and the HOMO of the emitter (−5.4 eV). In the presence of the HTL, the current injection is enhanced, even though a faster current saturation above 6 V was observed for the OLEDs using KR386. The luminance–voltage curves (L–V, Fig. 3b) exhibit a similar trend as for the current density, with steeper rise of the electroluminescence when KR386 is used. Nevertheless, the net result is an enhanced power conversion efficiency (PCE, Fig. 3c) when using the KR386 HTL, with a maximum of 23.7 lm W−1 at about 3 V as compared to 15.8 lm W−1 for the OLEDs without the HTL. Some triazatruxenes have been used as light emitting layers and to ensure that KR386 does not interfere with the OLED stack also a single layer OLED with only KR386 has been prepared and analyzed. A very low electroluminescence signal was observed as shown in Fig. S3† confirming that this does not interfere with the multi-layer OLED presented here.
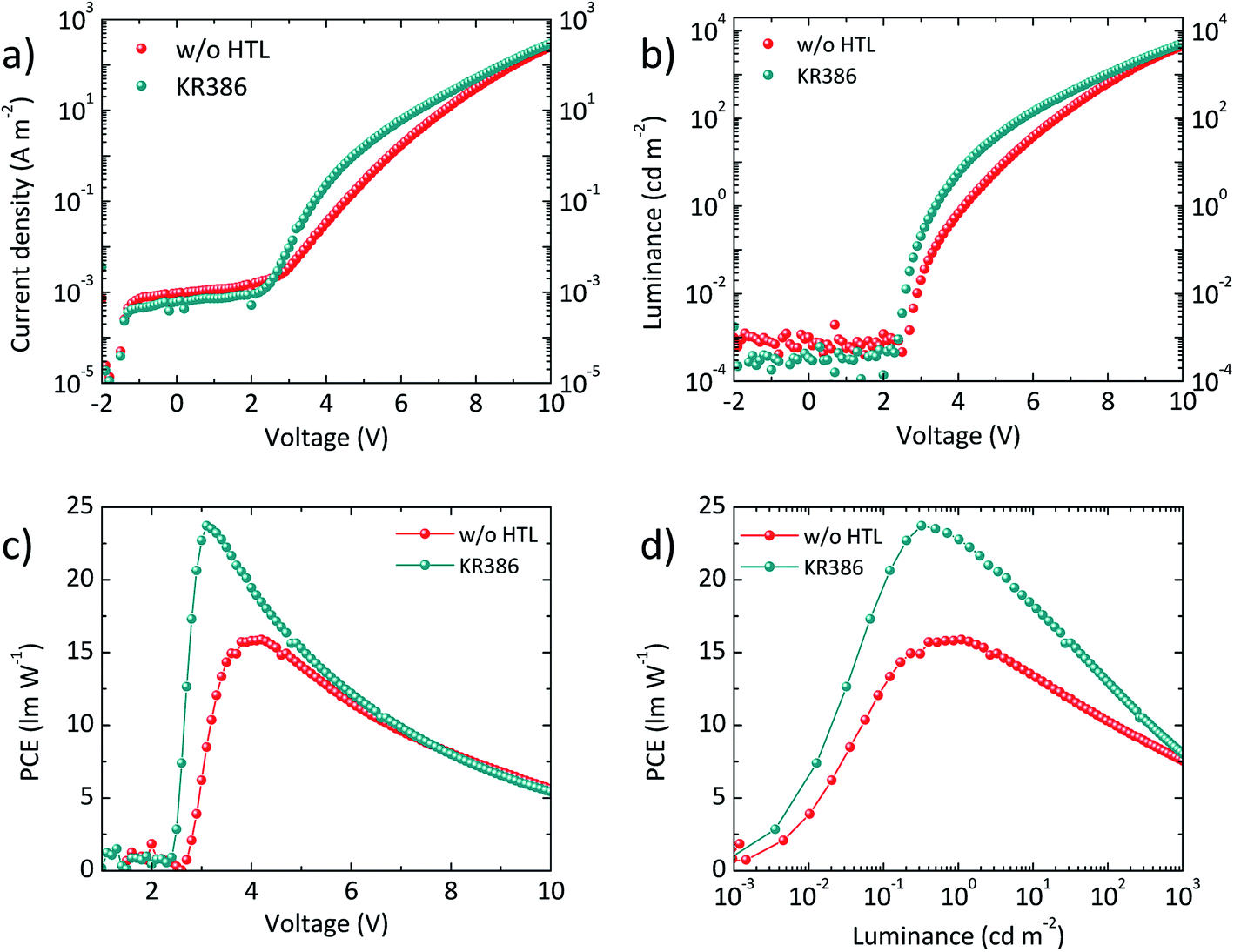 | ||
| Fig. 3 (a) J–V and (b) L–V characteristics for a series of OLEDs with and without the KR386 HTL. (c) PCE versus applied bias or (d) versus the measured luminance for the same device series. | ||
In summary, we presented a new cross-linkable triazatruxene derivative, KR386, which can be used as an hole transport material in solution processed multilayer optoelectronic devices. By using phenylstyrene groups, we ensure quantitative cross-linking in absence of any initiator. Also, these groups avoid possible triplet quenching encountered when using bare styrene moieties. Thanks to its favorable energy level alignment with common phosphorescent emitters, KR386 leads to OLEDs with high brightness and efficiency.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge financial support from the European Union H2020 project SOLEDLIGHT (grant 643791), the Spanish Ministry of Economy and Competitiveness (MINECO) via the Unidad de Excelencia María de Maeztu MDM-2015-0538, MAT2017-88821-R, PCIN-2015-255 and MAT2017-88905-P, and the Generalitat Valenciana (Prometeo/2016/135). H. B. acknowledges the support of ERA NET PCIN-2017-014. M. S. thanks the MINECO for his post-doctoral RyC contracts.References
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, J. Mater. Chem. C, 2013, 1, 1699–1707 RSC.
- C. Zhong, C. Duan, F. Huang, H. Wu and Y. Cao, Chem. Mater., 2011, 23, 326–340 CrossRef CAS.
- N. Aizawa, Y.-J. Pu, M. Watanabe, T. Chiba, K. Ideta, N. Toyota, M. Igarashi, Y. Suzuri, H. Sasabe and J. Kido, Nat. Commun., 2014, 5, 5756 CrossRef PubMed.
- S. Ho, S. Liu, Y. Chen and F. So, J. Photonics Energy, 2015, 5, 057611 CrossRef.
- T. Chiba, Y.-J. Pu and J. Kido, J. Mater. Chem. C, 2015, 3, 11567–11576 RSC.
- S. Wang, X. Wang, B. Yao, B. Zhang, J. Ding, Z. Xie and L. Wang, Sci. Rep., 2015, 5, 12487 CrossRef CAS PubMed.
- X. Gong, S. Wang, D. Moses, G. C. Bazan and A. J. Heeger, Adv. Mater., 2005, 17, 2053–2058 CrossRef CAS.
- H. Wu, F. Huang, Y. Mo, W. Yang, D. Wang, J. Peng and Y. Cao, Adv. Mater., 2004, 16, 1826–1830 CrossRef CAS.
- C. A. Zuniga, S. Barlow and S. R. Marder, Chem. Mater., 2011, 23, 658–681 CrossRef CAS.
- C. D. Müller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829 CrossRef PubMed.
- E. Bacher, M. Bayerl, P. Rudati, N. Reckefuss, C. D. Müller, K. Meerholz and O. Nuyken, Macromolecules, 2005, 38, 1640–1647 CrossRef CAS.
- O. Solomeshch, V. Medvedev, P. R. Mackie, D. Cupertino, A. Razin and N. Tessler, Adv. Funct. Mater., 2006, 16, 2095–2102 CrossRef CAS.
- S. Jungermann, N. Riegel, D. Müller, K. Meerholz and O. Nuyken, Macromolecules, 2006, 39, 8911–8919 CrossRef CAS.
- P. Zacharias, M. C. Gather, M. Rojahn, O. Nuyken and K. Meerholz, Angew. Chem., Int. Ed., 2007, 46, 4388–4392 CrossRef CAS PubMed.
- O. Solomeshch, Y.-J. Yu, V. Medvedev, A. Razin, B. Blumer-Ganon, Y. Eichen, J.-I. Jin and N. Tessler, Synth. Met., 2007, 157, 841–845 CrossRef CAS.
- F. Huang, Y.-J. Cheng, Y. Zhang, M. S. Liu and A. K. Y. Jen, J. Mater. Chem., 2008, 18, 4495–4509 RSC.
- Y.-J. Cheng, M. S. Liu, Y. Zhang, Y. Niu, F. Huang, J.-W. Ka, H.-L. Yip, Y. Tian and A. K. Y. Jen, Chem. Mater., 2008, 20, 413–422 CrossRef CAS.
- A. Köhnen, N. Riegel, J. H. W. M. Kremer, H. Lademann, D. C. Müller and K. Meerholz, Adv. Mater., 2009, 21, 879–884 CrossRef.
- X.-C. Li, C.-Y. Wang, W.-Y. Lai and W. Huang, J. Mater. Chem. C, 2016, 4, 10574–10587 RSC.
- K. Rakstys, S. Paek, P. Gao, P. Gratia, T. Marszalek, G. Grancini, K. T. Cho, K. Genevicius, V. Jankauskas, W. Pisula and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 7811–7815 RSC.
- T. Bura, N. Leclerc, S. Fall, P. Lévêque, T. Heiser and R. Ziessel, Org. Lett., 2011, 13, 6030–6033 CrossRef CAS PubMed.
- G. Hu, S. P. Kitney, S. M. Kelly, W. Harrison, B. Lambert and M. O'Neill, RSC Adv., 2018, 8, 8580–8585 RSC.
- D. J. Unett and R. A. Caldwell, Res. Chem. Intermed., 1995, 21, 665–709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07184e |
| This journal is © The Royal Society of Chemistry 2018 |