
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of sulfur-containing hybrid materials based on sodium silicate
O. A. Dudarko *a and
S. Baranybcd
*a and
S. Baranybcd
aChuiko Institute of Surface Chemistry NAS of Ukraine, 17, General Naumov str., Kyiv 03164, Ukraine. E-mail: odudarko80@gmail.com
bInstitute of Chemistry, University of Miskolc, Miskolc Egyetemvaros, H-3515, Hungary
cThe Transcarpathian II. Ferenc Rakoczi Hungarian Institute, 6, Kossuth sq., Beregovo, 90200, Ukraine
dMTA-ME "Materials Science" Research Group, 3515 Miskolc-Egyetemvaros, Hungary
First published on 7th November 2018
Abstract
An original method for the one-stage synthesis of sulfur-containing silica of SBA-15 type is developed and described. Instead of tetraethylorthosilicate (TEOS) typically used as a source of silica, the inexpensive sodium metasilicate has been applied in our method. The mesoporous silica material was first functionalized with thiol groups then oxidized by concentrated nitric acid to produce sulfonic groups. The samples obtained possess developed specific surface area (Ssp = 320–675 m2 g−1) and porous structure with an effective pore diameter of 3.5–5.7 nm. The orderliness of the structure and presence of surface sulfur-containing acidic groups of various natures in the synthesized materials were determined using XRD, TEM, N2 adsorption, conductivity and potentiometric titration methods. Based on the results of the measurements of the zeta potential vs. pH and electrolyte concentration, conclusions about the electro-surface properties and aggregation stability of sample dispersion have been drawn. The obtained samples are environmentally-friendly and could be used in green chemistry.
Introduction
Mesoporous sulfoxide catalysts are alternative materials to sulphuric acid for the acceleration of numerous liquid-phase organic transformations. Enhancement of the electron donor properties of the sulfonic acid groups was accomplished by using phenyl-linked groups.1,2 In addition, higher polarity of the catalyst surface can be achieved by using additional functional groups or by selecting the substituent near the sulfur-containing group of organosilicates.3 Such modifications can significantly increase the activity of the catalyst, promoting the adsorption of nonpolar reagents on the acidic sites.In recent years, mesoporous materials functionalized with sulfur-derivatives have increasingly been used as effective catalysts for acidic catalytic reactions.1–5 Incorporation of the sulfonic acid into the structure of mesoporous silica is of special interest. Such materials can be proposed as solid state acidic catalysts, adsorbents for sorption and separation of biomolecules,6 as cation-exchangers in the form of membranes,7 and fillers for nanocomposites intended for application in fuel cells.5
For the synthesis of such materials, the method of direct sulfonation of alkyl or aryl groups fixed on the substrate2,8,9 has been proposed. In most cases, surface sulfonic groups were obtained by non-selective oxidation of thiol groups on the silica surface.2,3,10 The hydrothermal stability of such materials is very important for practical application, because the most acidic-catalyzed reactions, such as etherification, hydrolysis and condensation, are carried out with the participation of water. It was previously reported that mesoporous materials modified by sulfonic acid are unstable to hydrothermal treatment, and their structure tends to break down at prolonged contact with water.11
Synthesis of SBA-15 with thiol groups based on TEOS (tetraethyl orthosilicate) by post-synthetic oxidation of the surface groups with hydrogen peroxide2,12–14 or concentrated nitric acid,5,15 or single stage oxidation of SH-groups during the template synthesis12,14,16,17 are quite widespread. Also for this purpose different structure-forming agents, such as 1,2-bis(trimethoxysilyl)ethane (BTME),5 1,4-bis (triethoxysilyl)benzene [(BTEB)]18 and BTME mixture with TMOS (tetramethylorthosilicate)5 and others, have been applied. Enhancement of the catalytic properties was accomplished by introducing aryl radicals14,19 or fluorine derivatives14,20 into the silica matrix.
An original method14,21 was proposed to synthesize disulfide intermediate material with closely spaced sulfonic groups on the surface. In this case, a cooperative effect was observed, which enhanced the catalytic properties of the material in comparison with the alkyl- or aryl-sulfur-containing analogues.
Mesoporous organo-inorganic hybrid materials functionalized by sulfonic acids were also synthesized by the method of radical polymerization with atomic transfer (ATRP). The SBA-15 material was first modified with 4-(chloromethyl)phenyl-trimethoxysilane. Surface initiation polymerization of sodium p-styrene sulfonate inside the functionalized walls of SBA-15 pores resulted in producing mesoporous materials functionalized with p-styrene sulfonic acid (PSS-SBA-15).22
Positive experience in the synthesis of SBA-15 type materials based on metasilicate without a functional layer was described in papers,23–29 and those with different types of functional groups in papers.30–35
Based on the analysis of the literature data, the main conclusions about sulfonic acid catalysts can be summarized as follows: (a) the acidic and catalytic properties of materials synthesized in different ways are similar; (b) there is a relationship between the specific activity of the attached sulfonic acid groups and the concentration of these groups on the substrate surface. It is also possible to form clusters of interacting acid groups, with increasing concentration on the substrate. From the practical point of view, this means that the rising concentration of acid groups on the substrate increases the duration of catalytic activity.36
In this work a new method of synthesis of SBA-15 on the basis of sodium metasilicate with thiol groups is proposed. It is based on the surface treatment with concentrated nitric acid and transformation of thiol groups into sulfonic ones. These materials are intended to apply as selective and capacitive materials in organic catalysis.
The novelty of our work is to maximally simplify and reduce the process of synthesis of catalysts with sulfur-containing groups, to achieve maximum conversion of thiol groups into sulfonic ones, to characterize the porous structure and surface properties of the materials produced while maintaining their efficiency and selectivity.
Experimental
Materials
Chemicals used for the synthesis of functionalized mesoporous silicates include: sodium metasilicate, Na2SiO3·5H2O (SM, ATK Ukraine, Ukraine); mercapto-propyltrimethoxysilane (CH3O)3Si(CH2)3SH (MPTMS), (95%, Aldrich, USA), poly(ethyleneoxide)-poly(propyleneoxide)-poly(ethyleneoxide) triblock copolymer, pluronic EO20PO70EO20 (P123, 99%, BASF, USA), concentrated nitric acid, ethanol abs. and acidified (1.5 cm3 HClconc. per 200 mL ethanol) for the template extraction, as well as solutions of HCl, KCl and NaOH (Reanal, Hungary), AgNO3 (99,5% puriss. p.a., Sigma-Aldrich, USA).Methods
Transmission electron microscopy (TEM) was performed using a JEM-1230 (JEOL, Japan) instrument. For analysis, the powdery samples were dispersed in ethanol using a moderate ultrasound treatment (22 kHz 30 min) at concentration of ∼5 wt% of solids. The diffractograms of the synthesized materials in the region of 0.5–5 2θ° were registered by DRON-4-07 diffractometer with CuKα radiation. Scanning Electron Microscopy (SEM) images of the samples were obtained by a JEOL JSM-6060LA (Jeol, Tokyo, Japan) device in the secondary electron mode with accelerating voltage of 30 kV. The samples were mounted on the surface of the subject table, with a pre-applied adhesive coating. To prevent the accumulation of the surface charge and to obtain a contrast image, a thin coating of gold layer was applied on the surface of the samples by cathode sputtering method in vacuum. Isotherms of low-temperature nitrogen adsorption were obtained by volumetric method using the device “Kelvin-1042” (Costech Microanalytical). In this case, the samples were pre-degassed in a stream of helium at 120 °C for 2 hours. The volume of the adsorbed gas was determined at the time of quasi-equilibrium in the gas flow controlled by thermal conductivity detector (the accuracy of the measurement was ±3%). The specific surface area of the samples, Ssp, was determined by the BET method.37 The pore diameters were calculated using the desorption branch of the isotherm by the modified DFT method,38 and, in some cases, by the Hurwich formula (d = 4 Vs/Ssp).39 Potentiometric titration was employed to determine the concentration of active acidic centers of the synthesized materials. The measurements were carried out both in 10−3 mol L−1 NaNO3 solution and in pure water. Based on conductometric and potentiometric titration data we have estimated the surface coverage of samples by sulfonic groups (eqn (1)).| af.gr. = (νNaOHVsol/VNaOH)/m, | (1) |
The concentration of immobilized groups of strongly acidic nature was determined by conductometric method in the alkaline concentration range of (1 × 10−5–1 × 10−2 M).
The pKa value of surface functional groups was determined by eqn 2
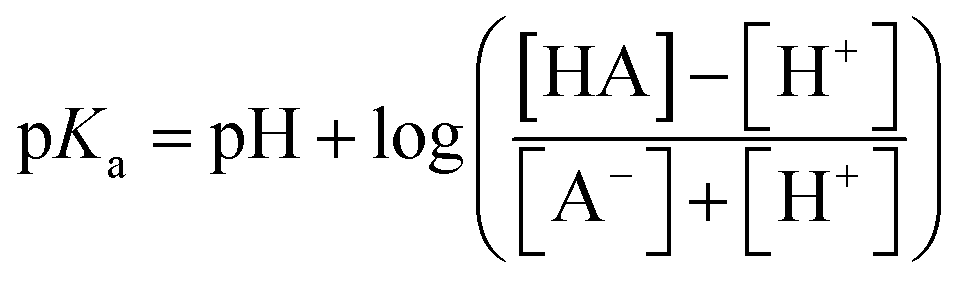 | (2) |
Electrophoretic mobility measurements were carried out with ZetaSizer Nano ZS Analyser (Malvern Instruments, UK) at room temperature and external electric field gradients of 6–15 V cm−1. All measurements were performed in diluted suspensions of adsorbents in aqueous solutions, at different ionic strength values and in broad pH range. Solutions of NaOH and HCl were employed to adjust the pH values of the suspensions. The electrophoretic mobility values were converted into electrokinetic potentials by the classical Smoluchowski formula using the software package supplied with the instrument. The resulting ζ-potentials were obtained by averaging the data of three to five individual measurements, the error of which can be estimated at 2–3%.
Preparation
DSH1 (SM/MPTMS = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (mol)). Firstly, a template solution (1.6 g P123 plus 26 cm3 water and 14.7 cm3 concentrated HCl) was prepared (solution no. 1). Separately, 3.4 g of sodium metasilicate was dissolved in 10 cm3 of water (solution no. 2). Then 0.28 cm3 of MPTMS dissolved in 2 cm3 of 0.0024 M HCl was added to the transparent template solution. Slowly growing clouding of the solution was observed. In 5 min, the solution no. 2 in the form of a thin jet was poured into the solution no. 1 with MPTMS. At once the mixture became transparent, later opalescence appeared which in 5 min turned into a clog. Then a white precipitate formed which was placed into a tightly closed bottle in a drying oven at 80 °C for 20 h. After that the powder was filtered and left to dry in air. The template was separated by boiling in an acidified ethanol (30 cm3 of ethanol per 1 g of mesophase) for 3 h, the procedure was repeated 4 times. The product was dried in vacuum for 30 min without heating, for 30 min with heating at 50 °C., and for 2 h at 90 °C.
:1 (mol)). Firstly, a template solution (1.6 g P123 plus 26 cm3 water and 14.7 cm3 concentrated HCl) was prepared (solution no. 1). Separately, 3.4 g of sodium metasilicate was dissolved in 10 cm3 of water (solution no. 2). Then 0.28 cm3 of MPTMS dissolved in 2 cm3 of 0.0024 M HCl was added to the transparent template solution. Slowly growing clouding of the solution was observed. In 5 min, the solution no. 2 in the form of a thin jet was poured into the solution no. 1 with MPTMS. At once the mixture became transparent, later opalescence appeared which in 5 min turned into a clog. Then a white precipitate formed which was placed into a tightly closed bottle in a drying oven at 80 °C for 20 h. After that the powder was filtered and left to dry in air. The template was separated by boiling in an acidified ethanol (30 cm3 of ethanol per 1 g of mesophase) for 3 h, the procedure was repeated 4 times. The product was dried in vacuum for 30 min without heating, for 30 min with heating at 50 °C., and for 2 h at 90 °C.
DSH2 (SM/MPTMS = 15:1 (mol)). The synthesis was the same as for DSH1 except for the fact that the applied amount of MPTMS was 0.42 cm3.
To modify the surface of these materials by sulfonic groups, DSH1 and DSH2 were treated with an excess of concentrated nitric acid (2 mL 20% HNO3 and 10 mL HNO3conc per 0.5 g of the sample), which oxidized the surface thiol groups. We applied a simple approach (5) that gives quantitative oxidation of –SH groups into SO3H groups. The oxidized samples were named DSH1SOH and DSH2SOH, accordingly.
Intensive oxidation of thiols with nitric acid, permanganate, or hydrogen peroxide leads to formation of sulfonic acids, possibly due to the intermediate formation of disulfide.
Based on knowledge of organic transformations, we supposed the intermediate formation of sulfenic or sulfinic acids, which are also easily oxidized, so that they can barely be isolated (Fig. 1). The oxidation can be traced qualitatively, i.e. the process is accompanied by the release of a brown gas – NO2. According to authors,5 approximately 80% of SH groups can be converted into sulfonic groups.
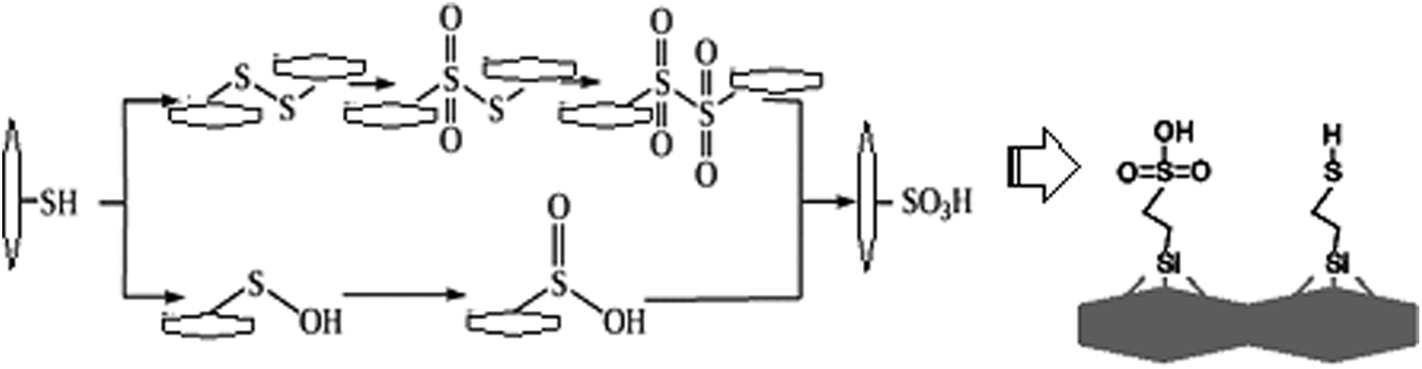 | ||
| Fig. 1 Possible schemes of oxidation of the surface thiol groups and expected surface layer. | ||
Discussion and conclusions
We considered the option of introducing oxidized mercapto-propyltrimethoxysilane into the reaction mixture. The synthesis of MPTMSOH: hydrolysis of MPTMS in the excess amount of HNO3was performed in the following way: 0.95 cm3 of MPTMS was hydrolyzed in 3.1 cm3 of HNO3conc. (1:10) at room temperature. A precipitate appeared immediately. When heated, a rapid reaction occurred with a release of brown gas NO2, and the entire volume of the vessel was filled with a white foamy substance of creamy structure. The effectiveness of such method for transformation of thiol groups into sulfonic ones was confirmed by means of IR spectroscopy (Fig. 2). We observed a disappearance of the band at 2566 cm−1, which corresponds to the stretching vibrations of the thiol groups and an appearance of bands of the stretching symmetric and asymmetric oscillations of –SOH group. The difference in the intensities of bands in the IR spectra of silane before and after the treatment with acid can be explained by different modes of registration of spectra. Since MPTMS is liquid, its drop was placed between NaCl plates and the IR spectrum was recorded in the transmission mode. Meanwhile, MPTMSOH is a powdery product; therefore, it was ground with KBr at sample/KBr ratio of 1/20 and the spectrum was recorded in the reflection mode. Thus, it is not rational to compare the intensities of bands in these spectra. However, the resulting product was insoluble in dilute hydrochloric acid or alcohol and could not be used in sol–gel synthesis. Consequently, such technique is not appropriate to incorporate the functional groups under study into the structure of silica.
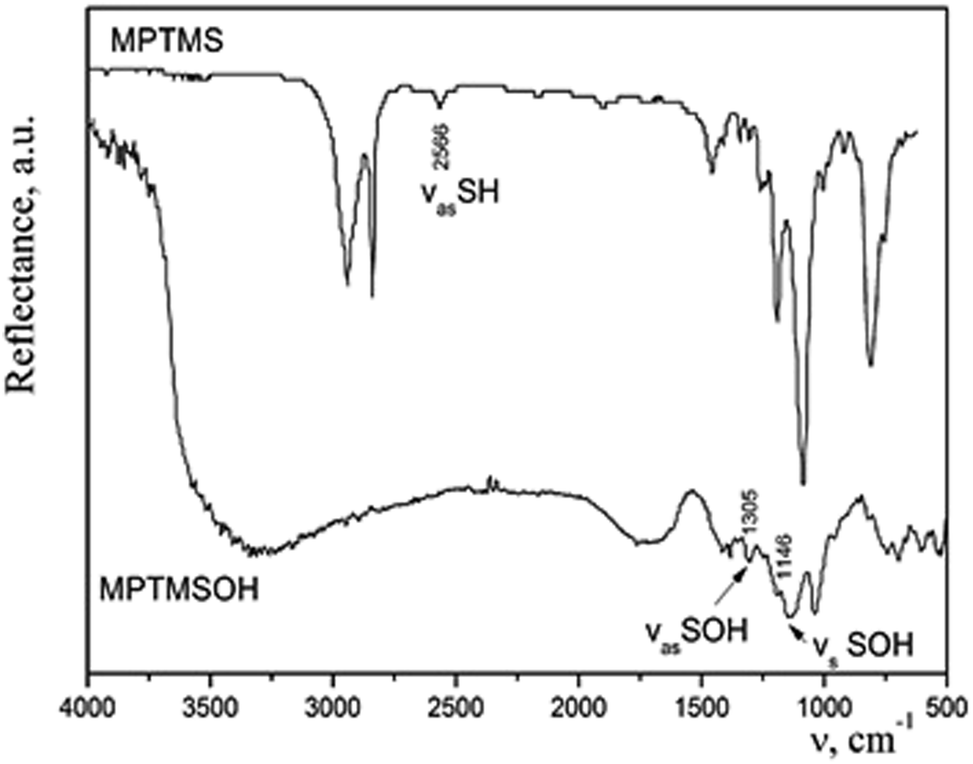 | ||
| Fig. 2 IR spectra of MPTMS before and after treatment with concentrated nitric acid. | ||
Therefore, we used the previously described approach of synthesizing the SBA-15 type silicates with thiol functional groups via traditional template method. Afterwards, the thiol groups were oxidized to sulfonic ones with concentrated nitric acid as described above.
All the synthesized samples had typical look for such a class of materials: white finely dispersed powders consisted mostly of particles of spherical shape, but also a small (appr. 10%) amount of particles of irregular shape (dish-shaped morphology or short rod) were registered. The size of spheres before and after treatment with acid was different. The DSH1 (Fig. 3a) sample before processing consisted of small 1.5 μm size oval particles, which were combined into agglomerates. After processing with nitric acid, the share of small particles of irregular shape in this sample increased, the spherical agglomerates became smaller and their diameter reached 4.2 μm, see sample DSH1SOH (Fig. 3c). The sample DSH2 (Fig. 3b) contained spherical particles with a diameter of about 6 μm, whereas after acidic treatment their diameter reduced to 5 μm (DSH2SOH Fig. 3d).
 | ||
| Fig. 3 SEM photographs of functionalized SBA-15 samples (a – DSH1, b – DSH2, c – DSH1SOH, d – DSH2SOH). | ||
Typically, P123 forms spherical micelles,40 but there are special conditions at which micelles of different types (rod-shaped, cylindrical, and scaly) are formed. This was observed in those cases when the formation of micelles occurred quickly enough due to the weak electrostatic repulsion between the polyethylene oxide (PEO) links. Previously, it was found that silicon dioxide and a part of the PEO interact with each other at the early stage of forming the cylindrical micelles. An increase in the concentration of the silica component in the reaction mixture (as in case DSH1) affected the radius of the micelle. Further interaction of silica units led to the formation of large cylindrical aggregates, and at this stage, hexagonal ordering was observed. At low pH values, protonated PEO chains were bound to silica cations by weak electrostatic interactions via negatively charged chlorine ions.41,42 We supposed that an increase of the MPTMS concentration in the DSH2 sample could change the pH of the reaction mixture only in the little extent but it was sufficient to initiate the structure-forming processes.
An X-ray diffraction analysis of samples was performed to identify the spatial arrangement of synthesized materials with thiol groups (Fig. 4a) and to clarify whether or not such orderliness is maintained after the acidic treatment (Fig. 4b).
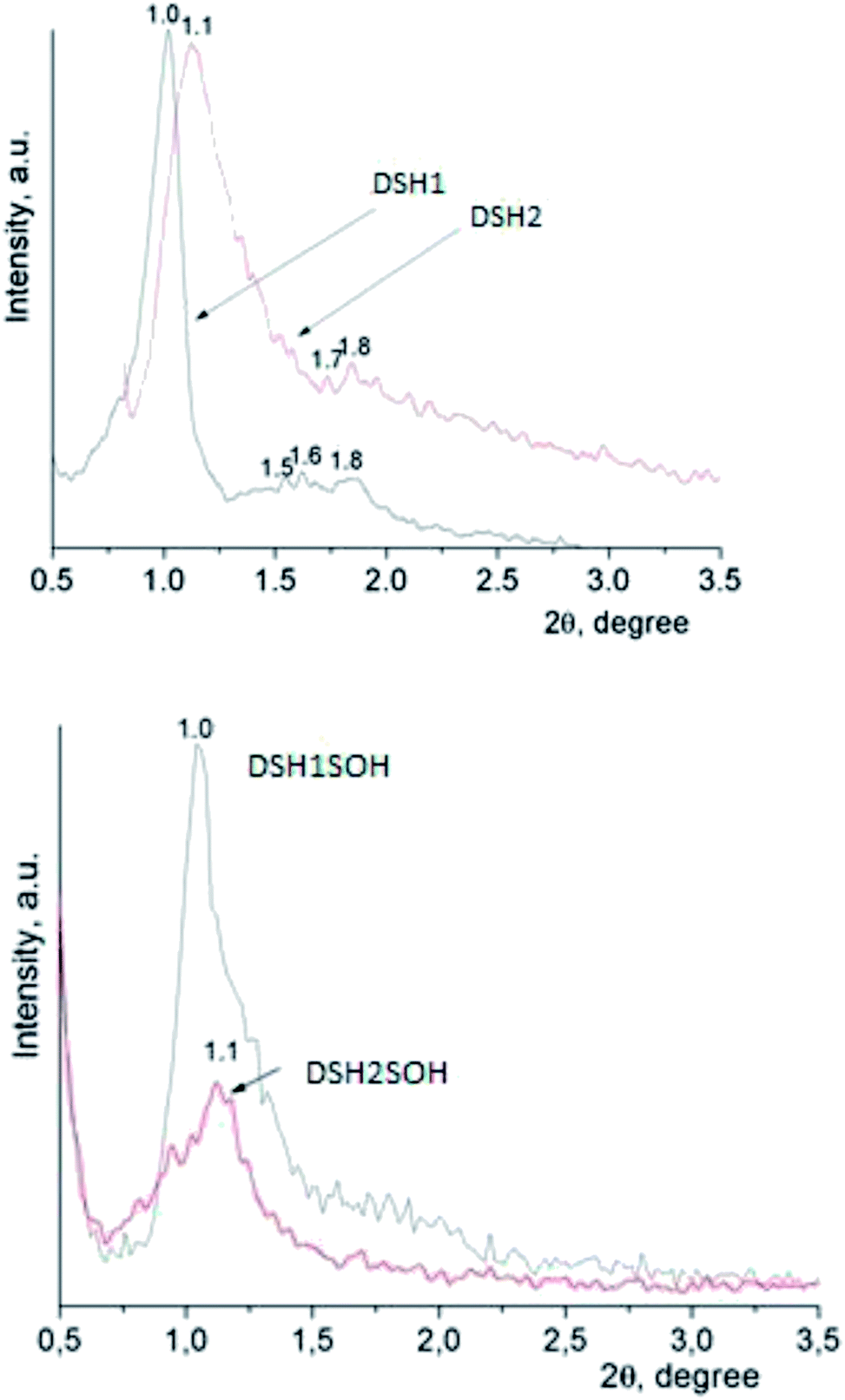 | ||
| Fig. 4 The diffraction patterns of samples of mesoporous silica. | ||
The presence of reflections in the small-angle region, as a rule, testifies the formation of mesoporous structure, as well as its type. The DSH1, DSH2 (Fig. 4a) diffraction patterns are characterized by the presence of one reflex at 1.0 and 1.1 2θ/°, respectively, and also additional reflections at higher values of 2θ/° were identified.
The latter is typical for the ordered structures with hexagonal lattice type, but these values did not correspond to the parameters of the spatial group p6mm √1:√3:√4,33 they are more similar in their data to the cubic Im3m ([110] – 1.0, [211] – 1.6, [220] – 1.8). This picture is not characteristic for functionalized mesoporous materials of SBA-15 type, but it is typical for samples obtained using other templates.43 With an increase in the concentration of MPTMS in the reaction mixture, the share of ordered structure was decreased. Treatment of samples with concentrated nitric acid gives a decrease in the intensity of reflections (Fig. 4b). We can assume that the partway destruction of them is a result of the sample treatment at these conditions.
To confirm or refute this hypothesis, a study of the synthesized materials was performed using TEM technique. It is interesting that the DSH1SOH photo clearly shows rather a cubic structure but not a hexagonal one (Fig. 5). Also an amorphous phase can be detected in the sample, which may be the result of the acidic treatment.
 | ||
| Fig. 5 TEM images of the sulphonic-containing samples (a – DSH1SOH, b – DSH2SOH). | ||
Investigation of textural parameters of the synthesized and treated materials by low-temperature adsorption–desorption of nitrogen showed that there was a change in parameters and, first of all, changes in the number of micropores (see Table 1).
| Samples | Ratio SM/MPTMS | Ssp, m2 g−1 | Vsp, cm3 g−1 | Vmicropores, cm3 g−1 | deff, nm |
|---|---|---|---|---|---|
| a Ssp – Specific surface area calculated from desorption data in the relative pressures range 0.05–0.20; Vsp – single point pore volume calculated at relative pressure of 0.98; deff – pore width calculated from the isotherms by DFT method. | |||||
| DSH1 | 20:1 |
675 | 0.75 | 0.00 | 2.2, 6.4 |
| DSH1SOH | 20:1 |
420 | 0.55 | 0.00 | 2.0, 6.5 |
| DSH2 | 15:1 |
320 | 0.22 | 0.04 | 3.8, 11.5 |
| DSH2SOH | 15:1 |
385 | 0.23 | 0.08 | 2.3, 4.7 |
Acidic treatment caused more changes in the textural parameters of mesoporous samples DSH1 and DSH1SOH, as can be seen from the Fig. 6a and c.
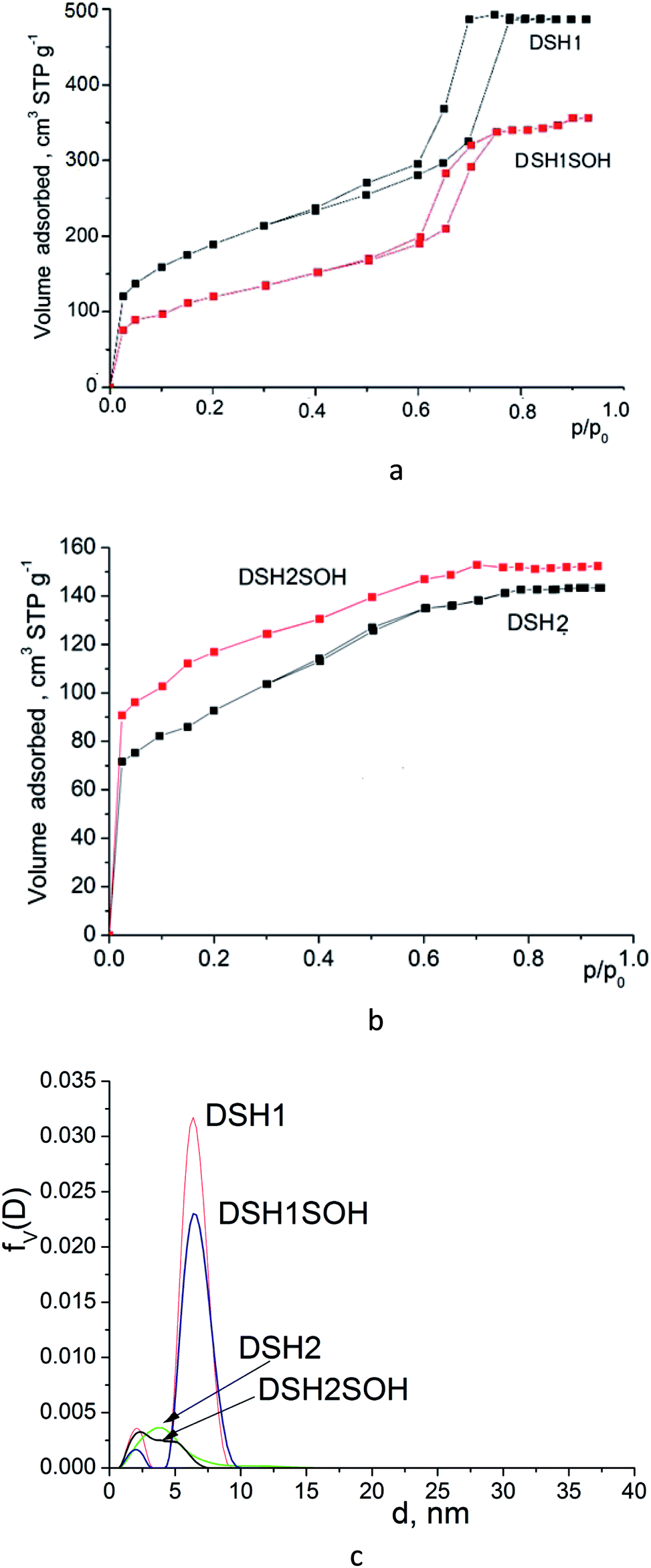 | ||
| Fig. 6 Nitrogen adsorption/desorption isotherms for mesoporous silicas (a and b) and pore size distribution curves calculated from desorption branches of the isotherms (c) by DFT method. | ||
A substantial decrease in the surface area was observed (Table 1) which might be the result both of the decrease in the number of micropores, and the possible restructuring of the constituent elements of the structure. Reducing the effective pore diameter may be resulted from an increase in the size of functional groups, which can create steric barrier for the sorption of nitrogen molecules. When we considered the samples DSH2 and DSH2SOH (Fig. 6b), it was noticeable that the form of the isotherm did not change. It belongs to type I according to the classification.44 Moreover, the value of the specific surface area after treatment increased due to the contribution of micropores, since neither the effective diameter of the pores nor the sorption volume changed (see Table 1). On the other hand, presence of pores in the SBA-15 walls has been supposed45,46 which also contributes to the effective diameter of pores and sorption volume. These pores could be either blocked or destroyed under the nitric acid treatment. Introduction of large amounts of thiol groups during the synthesis probably changes the pH of the reaction mixture, and therefore the structure-formation does not follow the SBA-15 type (DSH2). Only an increase in the ratio of the reactive components of SS:MPTMS up to 20:1 allows formation of a well-ordered structure (DSH1). Calculations for the DFT (Fig. 6c) show that for the DSH2 and DSH2SOH samples, micropores with a small amount of mesopores are identified. At the same time, for other samples (DSH1 and DSH1SOH), the dominant structure is mesoporous. The presence of a small number of micropores can be explained by the formations of plugs in mesopore channels in addition to the complimentary fine pores existing in the SBA-15.
We have used potentiometric titration to determine the concentration of active acidic centres on the surface of the oxidized materials (DSH2SOH and DSH1SOH). The measurements were carried out both in pure water and in 0.1 mol L−1 NaNO3 solution. Fig. 7a demonstrates the difference between curves for DSH2SOH in different media as an example to clarify which environment is better for the dissociation of functional groups. Lower values of the surface charge density in NaNO3 solution compared to that in water (Fig. 7b) is explained by the charge screening effect of the salt.
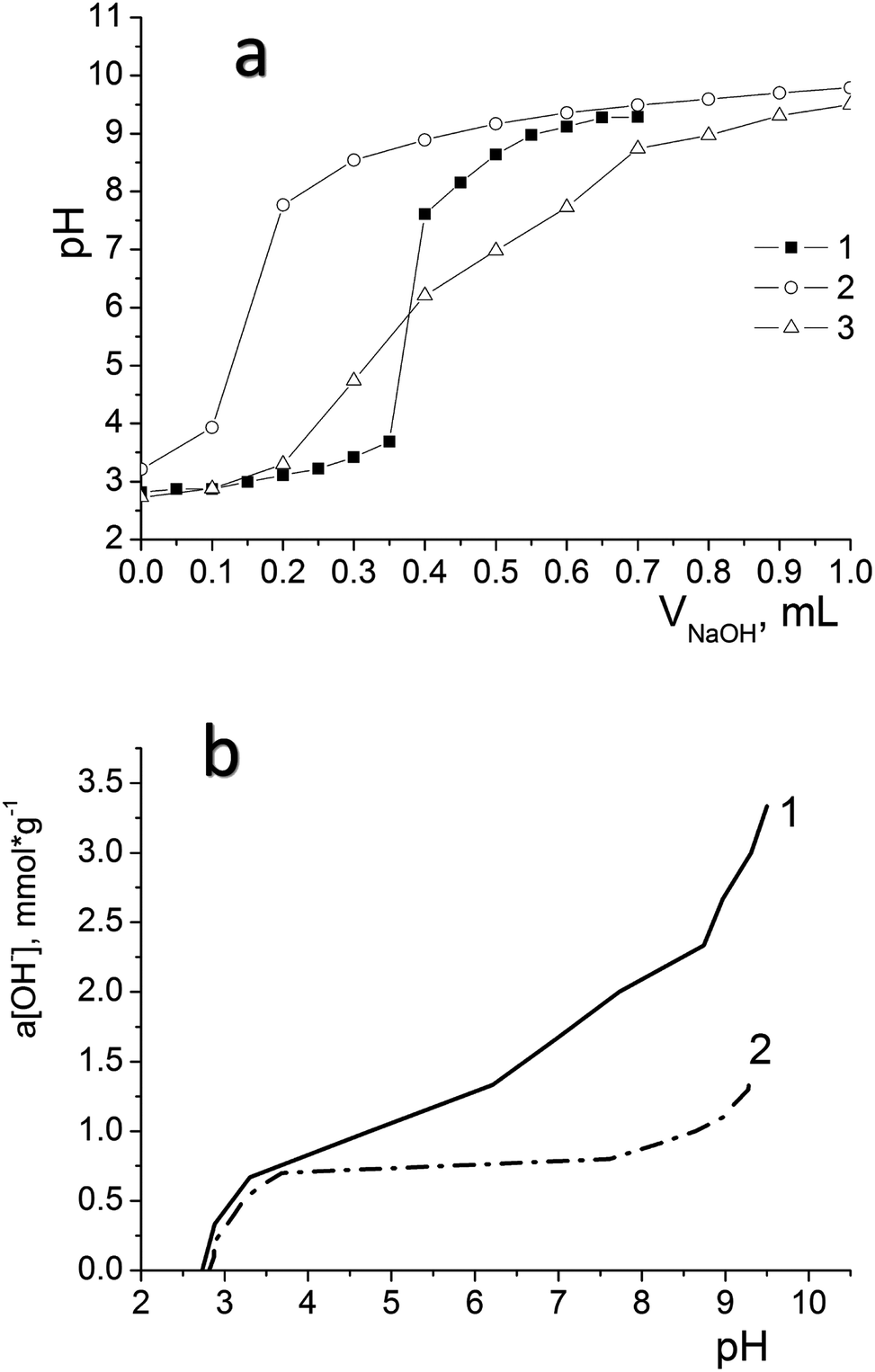 | ||
| Fig. 7 Curves of potentiometric titration of samples after their treatment with nitric acid (a) 1 – DSH1SOH. 2 – DSH2SOH in 0.1 N NaNO3 and 3 – DSH2SOH in water; pH dependence of the content of surface groups calculated from titration data for DSH2SOH (1 – in water, 2 – in 0.1 N NaNO3) (b) Vsusp = 25 mL, CNaOH = 0.1 mL. | ||
Analysis of the acid–base titration curves confirmed that the sample DSH2SOH contains higher concentration of functional groups, compared to those predicted because of theoretical calculation starting from the ratio of reagents in the reaction mixture. The number of available groups is lower than that theoretically calculated by the reaction equation, but the sorption equilibrium is reached quickly, within 15 minutes. That is why they have an advantage over disordered materials with higher content of functional groups. So, for the sample DSH1SOH, the concentration of acidic groups available for adsorption was found 0.3 mmol g−1 (the theoretical value calculated from the ratio of silanes content is 0.5 mmol g−1 SH) and 0.76 mmol g−1 for the sample DSH2SOH (at 0.75 mmol g−1 SH theoretical value).
Starting from the results of conductometric titration, the concentration of the sulfonic groups available for interaction with alkaline groups was determined. In addition, from these data the degree of transformation of the thiol groups into the sulfonic groups has been established. We have to keep in mind that the thiol groups are not titrated with alkali, so that the difference in surface structures (the presence of functional groups of different acidity) can be estimated using conductometric titration. As an example, let us inspect the results of titration of DSH1SOH sample with attached alkylsulfonic acid groups (Fig. 8a). It is seen that the curve of conductometric titration is V-shaped, which is characteristic for titration of strong acids with strong bases in solution. This indicates the high acidity of the corresponding functional groups (Fig. 8a). With a decrease in the strength of immobilized acid (DSH1), the curve of direct conductivity titration (Fig. 8a) became less informative. The titration curve of the sample with the attached thiol groups showed a marked weakness, due to the ionization of thiol groups.
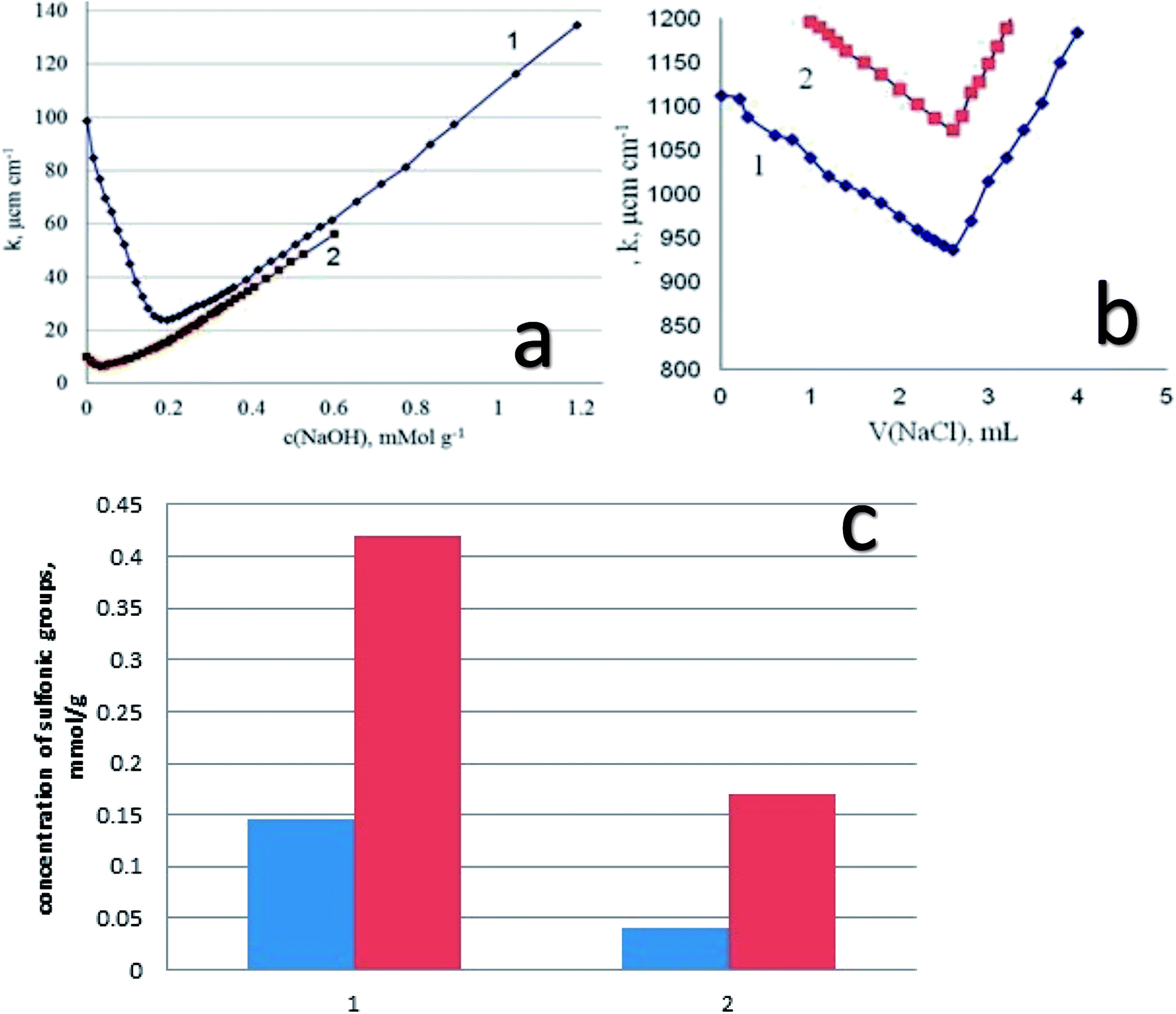 | ||
| Fig. 8 Results of acid–based conductometric titration (a) and the inverse conductometric titration after contact with AgNO3 (b) for DSH1SOH(1) and DSH1 (2); concentration of sulfonic groups for DSH2SOH (1 red) and DSH2 (1 blue), DSH1SOH (2 red) and DSH1 (2 blue) (c). | ||
In order to increase the reliability of determination of the concentration of thiol groups, fixed on the surface, the method of inverse conductometric titration of the excess of Ag ions after their interaction with the samples (the reaction in Scheme 1) with NaCl solution was also applied.
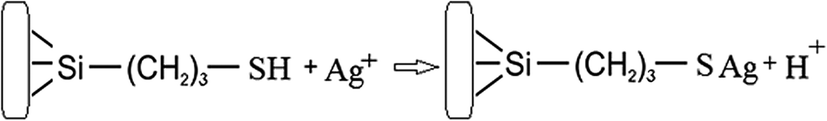 | ||
| Scheme 1 | ||
Thus, a combination of methods of direct and inverse conductometric titration allowed to identify and determine the concentration of thiol and sulfonic groups on the surface of the samples at their joint presence (Fig. 8b).
Based on the results of conductometric titration (Fig. 8c) we have found the amount of functional groups in the synthesized samples for DSH2 – 0.145 mmol g−1 and for sample DSH2SOH – 0.42 mmol g−1 sulfonic groups. For the DSH1 surface 0.04 mmol g−1 sulfonic groups, and 0.60 mmol g−1 thiol groups, and on the surface of DSH1SOH 0.17 mmol g−1 of sulfonic groups and 0.44 mmol g−1 of thiol groups were detected.
These results testify that a part of thiol groups was converted in the course of template synthesis. The lower value of theoretically calculated concentration of sulfonic groups compared to the experimentally determined one can be explained by partial inaccessibility of groups within the structure for oxidation. At the same time, the measured concentration of functional groups is high enough to recommend the produced samples for application in catalytic processes, especially if one takes into account the possibility of reusing of these types of the material. There is a good reason to recommend them for application in catalytic processes.
Since it is planned to apply these materials as catalysts in liquid medium, it was interesting to study the electric surface behavior of the samples through electrokinetic potential measurements. Fig. 9a and b illustrates the dependencies of the electrokinetic potential of samples on the pH value.
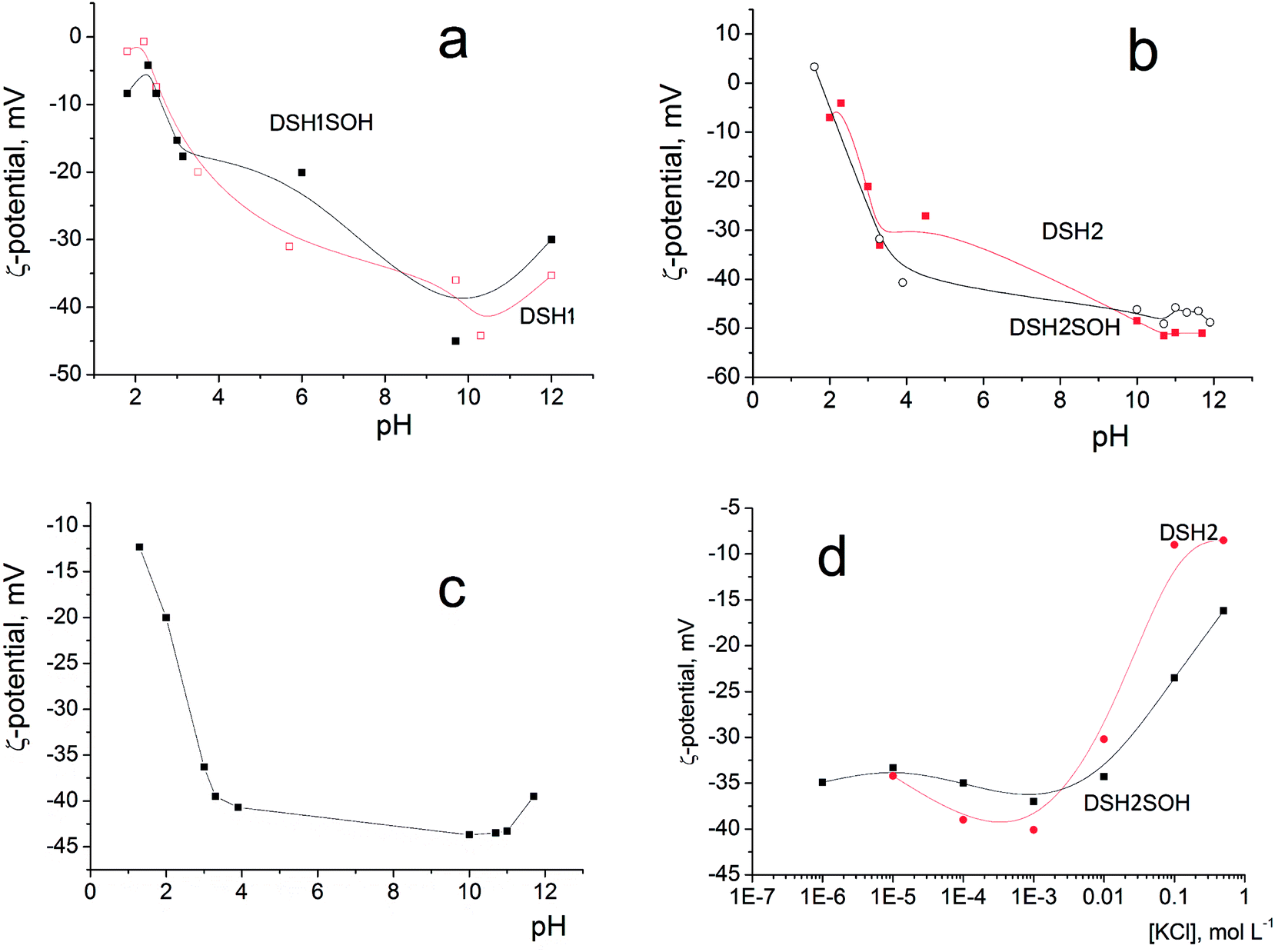 | ||
| Fig. 9 Dependence of the electrokinetic potential of DSH2SOH particles on pH in aqueous solution (a and b), on pH in the presence of 10–3 M KCl (c), and on the concentration of KCl solution (d). | ||
The general trend is a substantial rise in the absolute (negative) value of zeta potential with increasing pH, from about −10 mV at pH 2 to −40–50 mV at pH 8–10. This is due to increasing dissociation of different kinds of functional groups having different degree of dissociation: initial Si–OH groups, weakly charged –SH and moderately charged –SOH groups. A decrease in zeta potential at pH <2 and pH >10 is explained by an increase in the ionic strength of the solution (>10−2 M of HCl or KOH, correspondingly) accompanied by the electrical double layer compression. Addition of 10−3 M KCl does not change the character of ζ (pH) dependencies (example: Fig. 9c), only steeper rise in ζ at pH >11 is observed as a result of the impact of increased electrolytes concentration in the system (mixture of KOH + KCl). This is also an evidence of the absence of considerable preferential adsorption of cations or anions by the surfaces studied.
The effect of increasing amounts of KCl on the electrokinetic potential of modified samples (Fig. 9d) is characterized by a slight increase of the negative ζ with subsequent substantial decrease of its absolute value. An appearance of the maximum on ζ(CKCl) curves is usually explained by the impact of two effects acting in opposite directions: (i) polarization of the electrical double layer which gives a rise to ζ with increasing of the ionic strength and (ii) compression of this layer which leads to a decrease of the potential (see in detail).47,48
Note that high zeta-potential values (−40–50 mV) were observed in a wide range of pH values, between 4 and 11. The reason is the strong acidic character of adsorbed functional –SOH groups. Thiol groups have lower acidity than silanol, about pK = 4. Sulfo-groups remain strongly acidic and when attached to the surface, as a rule, their pK was not indicated.
These figures provide additional confirmation of the change in the acidity of the surface layer, especially as shown in Fig. 9d, and how the zeta-potential values of the sample before and after treatment with concentrated acid changed.
Mesoporous materials functionalized with sulfonic acids with different content of sulfur-containing groups were successfully synthesized by a one-pot method of condensation of silica precursors in acidic media. The obtained samples are characterized by developed specific surface area (Ssp = 320–675 m2 g−1) and porous structure, with effective pore diameter of 3.5–5.7 nm. The acidic-catalyzed synthesis allows for the slow and even condensation of silica units and facilitates the ordering of mesoporous materials. The main requirements that apply to these materials as catalysts are not only their performance, mechanical, hydrolytic and thermal stability, but also their ability to be reused and environmental safety. Hybrid materials based on silica, obtained by template methods almost completely satisfy all these needs. The materials produced are relatively cheap, have competitive characteristics compared to existing sulfur-containing SBA-15. The structural parameters and physical–chemical characteristics of the products make them promising for practical use as catalysts supporters or as catalysts for acid–based organic syntheses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was carried out in the framework of the GINOP-2.3.2-15-2016-00010 “Development of enhanced engineering methods with the aim at utilization of subterranean energy resources” project of the Research Institute of Applied Earth Sciences of the University of Miskolc in the framework of the Széchenyi 2020 Plan, funded by the European Union, co-financed by the European Structural and Investment Funds. The authors would like to thank the partial financial support of this study from the project “Establishment of collaboration between the University of Miskolc and industry (FIEK) for advanced materials and intelligent technologies” Program (GINOP-2.3.4-15-2016-00004), the cooperation project between National Academy of Sciences of Ukraine and Hungarian Academy of Sciences and the cooperation project between National Academy of Sciences of Ukraine and Polish Academy of Sciences “Synthesis of novel organic–inorganic hybrid materials to use as catalysts for fine organic synthesis”(2018–2020).Notes and references
- I. K. Mbaraka, D. R. Radu, V. S.-Y. Lin and B. H. Shanks, J. Catal., 2003, 219, 329 CrossRef CAS.
- B. Rác, A. ́. Molnár, P. Forgo, M. Mohai and I. Bertóti, J. Mol. Catal. A: Chem., 2006, 244, 46 CrossRef.
- O. A. Dudarko, Bulletin of the Precarpathian National University named after VasylStefanyk. Chemistry Series. 2016, vol. XX, p. 72 Search PubMed.
- W. D. Bossaert, D. E. De Vos, W. M. Van Rhijn, J. Bullen, P. J. Grobet and P. A. Jacobs, J. Catal., 1999, 182, 156 CrossRef CAS.
- Q. Yang, M. P. Kapoor, N. Shirokura, M. Ohashi, S. Inagaki, J. N. Kondo and K. Domen, J. Mater. Chem., 2005, 15, 666 RSC.
- X. Liu, L. Zhu, T. Zhao, J. Lan, W. Yan and H. Zhang, Microporous Mesoporous Mater., 2011, 142, 614 CrossRef CAS.
- R. K. Nagarale, G. S. Gohil, V. K. Shahi and R. Rangarajan, Macromolecules, 2004, 37, 10023 CrossRef CAS.
- I. D́ıaz, F. Mohino, T. Blasco, E. Sastre and J. Ṕerez-Pariente, Microporous Mesoporous Mater., 2005, 80, 33 CrossRef.
- C. Li, J. Yang, P. Wang, J. Liu and Q. Yang, Microporous Mesoporous Mater., 2009, 123, 228 CrossRef CAS.
- Z. Tai, M. A. Isaacs, C. M. A. Parlett, A. F. Lee and K. Wilson, Catal. Commun., 2017, 92, 56 CrossRef CAS.
- A. Galarneau, M. Nader, F. Guenneau, F. Di Renzo and A. Gedeon, J. Phys. Chem. C, 2007, 111, 8268 CrossRef CAS.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448 CrossRef CAS.
- F. Alrouh, A. Karam, A. Alshaghel and S. El-Kadri, Arabian J. Chem., 2017, 10, 281 CrossRef.
- J. A. Melero, R. l van Grieken and G. l Morales, Chem. Rev., 2006, 106, 3790 CrossRef CAS PubMed.
- Q. Yang, M. P. Kapoor, S. Inagaki, N. Shirokura, J. N. Kondo and K. Domen, J. Mol. Catal. A: Chem., 2005, 230, 85 CrossRef CAS.
- G. Morales, G. Athens, B. F. Chmelka, R. van Grieken and J. A. Melero, J. Catal., 2008, 254, 205 CrossRef CAS.
- R. VanGrieken, J. A. Melero and G. Morales, J. Mol. Catal. A: Chem., 2006, 256, 29 CrossRef CAS.
- B. Schäfgen, O. D. Malter, E. Kaigarula, A. Schüßler, S. Ernst and W. R. Thiel, Microporous Mesoporous Mater., 2017, 251, 122 CrossRef.
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2006, 244, 78 CrossRef CAS.
- M. Alvaro, A. Corma, D. Das, V. Fornes and H. Garcia, Chem. Comxmun., 2004, 956 RSC.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403 CrossRef CAS PubMed.
- C. Li, J. Yang, P. Wang, J. Liu and Q. Yang, Microporous Mesoporous Mater., 2009, 123, 228 CrossRef CAS.
- J. M. Kim and G. D. Stucky, Chem. Commun., 2000, 1159 RSC.
- M. Choi, W. Heo, F. Kleitz and R. Ryoo, Chem. Commun., 2003, 1340 RSC.
- K. Kosuge, T. Sato, N. Kikukawa and M. Takemori, Chem. Mater., 2004, 16, 899 CrossRef CAS.
- P. F. Fulvio, S. Pikus and M. Jaroniec, J. Colloid Interface Sci., 2005, 287, 717 CrossRef CAS PubMed.
- W. Wang, W. Shana and H. Ru, J. Mater. Chem., 2011, 21, 17433 RSC.
- Y. Ding, G. Yin, X. Liao, Z. Huang, X. Chen and Y. Yao, Mater. Lett., 2012, 75, 45 CrossRef CAS.
- N. Rahmat, F. Hamzah, N. Sahiron, M. Mazlan and M. M. Zahari, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 133, 012011 Search PubMed.
- W. Wang, W. Shan and H. Ru, J. Mater. Chem., 2011, 21, 17433 RSC.
- S. Saravanamurugan, D. S. Han, J. B. Koo and S. E. Park, Catal. Commun., 2008, 9, 158 CrossRef CAS.
- S. E. Park and E. A. Prasetyanto, Top. Catal., 2009, 52, 91 CrossRef CAS.
- V. V Sliesarenko, O. A. Dudarko, Y. L Zub, G. A Seisenbaeva, V. G. Kessler, P. Topka and O. Šolcová, J. Porous Mater., 2013, 20, 1315 CrossRef.
- O. A. Dudarko, C. Gunathilake, V. V. Sliesarenko, Y. L. Zub and M. Jaroniec, Colloids & Surfaces A, 2014, 459, 4 CrossRef CAS.
- O. A. Dudarko, C. Gunathilake, N. P. Wickramaratne, V. V. Sliesarenko, Y. L. Zub, J. Gorka, S. Dai and M. Jaroniec, Colloids & Surfaces A, 2015, 460, 1 CrossRef.
- P. F. Siril, N. R. Shiju, D. R. Brown and K. Wilson, Appl. Catal., A, 2009, 364, 95 CrossRef CAS.
- J. S. Brunauer, P. H. Emmet and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef.
- V. M. Gun'ko and D. D. Do, Colloids Surf., A, 2001, 193, 71 CrossRef.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, London, 1982 Search PubMed.
- M. Imperor-Clerc, P. Davidson and A. Davidson, J. Am. Chem. Soc., 2000, 122, 11925 CrossRef CAS.
- M. A. Shenashen, S. A. El-Safty and E. A. Elshehy, Analyst, 2014, 139, 6393 RSC.
- A. Sundblom, C. L. P. Oliveira, A. E. C. Palmqvist and J. S. Pedersen, J. Phys. Chem. C, 2009, 113, 7706 CrossRef CAS.
- X. Fang and L. Wu, Handbook of Innovative Nanomaterials: From Syntheses to Applications, CRC Press. 2012 Search PubMed.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids. principles, methodology and application. London: Academic Press, 1999, p. 467 Search PubMed.
- P. Van-Der-Voort, P. I. Ravikovitch, K. P. De Jong, A. V. Neimark, A. H. Janssen, M. Benjelloun, E. Van Bavel, P. Cool, B. M. Weckhuysen and E. F. Vansant, Chem. Commun., 2002, 1010 RSC.
- P. Van Der Voort, P. I. Ravikovitch, K. P. De Jong, M. Benjelloun, E. Van Bavel, A. H. Janssen, A. V. Neimark, B. M. Weckhuysen and E. F. Vansant, J. Phys. Chem. B, 2002, 106, 5873 CrossRef CAS.
- S. S. Dukhin and B. V. Derjaguin, Electrokinetic Phenomena, Surface and Colloid Science, ed. E.Matijevic, John Willey & Sons, NY, 1974 Search PubMed.
- S. Barany, Adv. Colloid Interface Sci., 1998, 75(1), 45 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |