
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSonodynamic therapy induces oxidative stress, DNA damage and apoptosis in glioma cells
Yue Sun,
Haiping Wang,
Kun Zhang,
Jingfei Liu,
Pan Wang ,
Xiaobing Wang and
Quanhong Liu*
,
Xiaobing Wang and
Quanhong Liu*
National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest China, The Key Laboratory of Medicinal Resources and Natural Pharmaceutical Chemistry, The Ministry of Education, College of Life Sciences, Shaanxi Normal University, Xi'an, Shaanxi 710119, People's Republic of China. E-mail: lshaof@snnu.edu.cn; Tel: +86-029-85310275
First published on 25th October 2018
Abstract
Malignant glioma remains one of the most challenging diseases to treat because of the invasive growth of glioma cells and the existence of the blood–brain barrier (BBB), which blocks drug delivery to the brain. New strategies are urgently needed to overcome these shortcomings and improve the outcomes. Ultrasound represents a promising noninvasive and reversible BBB opening approach and the related sonodynamic therapy (SDT) is rapidly emerging. This study aims to explore the ultrasound parameters for BBB opening and the cell killing effect of SDT in human glioma U373 cells by using a recently reported sonosensitizer, sinoporphyrin sodium (DVDMS). The in vitro BBB model indicated that SDT caused a time-dependent permeability increase, which peaked at 2 h post treatment and then recovered gradually. The results of toxicology tests showed significant U373 cell viability loss and apoptosis increase after DVDMS-SDT, accompanied by enhanced cleaved-caspase-3 level and DNA fragmentation, in which reactive oxygen species (ROS) were a major triggering intermediate during DVDMS-SDT. Furthermore, DVDMS-SDT produced DNA damage and the underlying mechanisms were evaluated, in order to provide a fundamental basis for DVDMS-SDT application in glioma therapy. The findings indicated that the DNA molecules could be temporarily regulated by SDT and DNA double-strand breaks (DSBs), which increased the difficulty of cellular self-repair, thus aggravating cell apoptosis and inhibiting glioma cell invasive growth. Therefore, this study supports the use of SDT as an alternative approach for glioma therapy.
1. Introduction
Malignant glioma, the most aggressive brain cancer, is generally recalcitrant to current chemotherapeutic and radiotherapeutic treatment. The primary predicament is that the blood–brain barrier (BBB) has a strict selectivity against foreign substances, which prevents over 98% of small molecule drugs and almost 100% of macromolecular drugs from penetrating into the brain.1,2 Therefore, disruption of BBB without any damage to normal brain tissues is very challenging in glioma treatment. Ultrasound refers to a mechanical wave form which can propagate through deep tissue and can focus its energy into a small volume to increase vessel/tissue/cell permeability.3 Opening the BBB by using ultrasound has recently attracted many researchers' attention, because this process is reversible and nondestructive, minimizing damage to surrounding normal brain tissues.4–6 Leinenga et al. have reported that scanning ultrasound can transiently open the BBB in order to remove amyloid-β peptide and restore the memory of a mouse model of Alzheimer's disease.7 Landhuis proposed the technique of transcranial ultrasound modulation, which has brought new hope to the treatment of many brain diseases.8It is worth mentioning that opening the BBB with ultrasound also enables a new strategy for glioma treatment. The combination of ultrasound and a sonosensitizer, called sonodynamic therapy (SDT), may combine the distinct merits of ultrasound and nontoxic sonosensitizers, providing a non-invasive, repeatable, effective and targeted approach for glioma therapy. It has been reported that low frequency ultrasound has a strong penetrability through the animal skull and can focus into a small volume of the brain, localizing at the precise position of a tumor.9–11 Ohmura et al. have investigated the tumor destruction and growth inhibition using ultrasound combined with a traditional endogenous sensitizer, 5-ALA, in rat C6 gliomas.12 Dai et al. reported that the sensitizer HMME (hematoporphyrinmonomethyl ether) facilitated obvious sonodynamic effects on C6 glioma cells.13 Xu's study suggested that glioma stem-like cells could be damaged by using Photofrin-mediated SDT, in which reactive oxygen species (ROS) generation triggered cell apoptosis under ultrasound irradiation.14 Although these studies suggest sonodynamic therapy has a great potential for glioma treatment through an undefined mechanism, more realistic trials of sonosensitizers and human glioma models should be conducted, and concurrently the parameters for reversible BBB opening using ultrasound and the mechanistic basis of the glioma killing effects of SDT should be explored.
We previously reported that the sensitizer sinoporphyrin sodium (also called DVDMS, Fig. 1) elicited more efficient sonodynamic efficacy than HMME, Photofrin and protoporphyrin IX.15,16 The efficacy of combined SDT varies depending on the sensitizer properties, ultrasound parameters, cell type features and so on. In this study, we aimed to investigate DVDMS-mediated SDT (DVDMS-SDT) in the human glioma U373 cell line, which represents the highly invasive, apoptosis-resistant (P53 mutation), abnormal DNA-repair properties of clinical malignant glioma.17,18 Inspired by the possibility of DVDMS-SDT in glioma, we optimized the reversible BBB opening parameters by establishing an in vitro BBB model and compared the cellular response to sonodynamic stimulus between malignant cells and normal cells. More importantly, we focused on the structural and functional DNA damage of glioma cells after DVDMS-SDT treatment.
 | ||
| Fig. 1 Molecular structure of DVDMS (sinoporphyrin sodium). | ||
DNA, as a crucial genetic substance, determines cell proliferation, migration, and all the important activities of cell life. In cancer therapy, several approaches have been developed to target DNA molecules to induce DNA damage and tumor cell apoptosis. DNA damage includes single-strand break, double-strand break (DSB), tautomeric shift, base-transitions and so on. DSB is probably the most severe type of DNA damage, and can cause irreversible effects.19–21 Wieczorek reported that DSBs could activate checkpoint kinases such as ATM and CHK2, which then trigger cell apoptosis.22 As is widely known, ROS are generally formed during SDT and closely interact with cellular components.23–25 Excessive amounts of ROS will initiate oxidative stress in many cellular biomacromolecules, which then culminates in subcellular organelle dysfunction and cell death.26 Dizdaroglu et al. reported that DNA base modifications could be induced by ROS, especially ·OH.27 ROS-induced DNA damage can occur through oxidative modification of DNA bases or by spontaneous hydrolysis of nucleosides. Oxidative damage of DNA includes base damage, single- or double-strand break and other serious disruptions.28 Therefore, SDT-triggered excessive ROS may closely interact with cellular DNA molecules via some undefined mechanism, and the induced DNA damage level and cell fate would be determined by the SDT doses. However, what types of DNA damage would be induced by SDT, and whether it could be influenced by the specific sensitizer, have not been established. Here we show that the mechanisms of oxidative stress, DSB-derived DNA damage, and apoptotic response occur as a result of DVDMS-SDT in U373 cells, and these effects synergistically enhance each other. These findings will provide a foundation for SDT application in glioma therapy.
2. Materials and methods
2.1. Reagents
DVDMS (Fig. 1) was obtained as previously described.16 It was dissolved in a physiological saline solution to a final storage concentration of 1 mM and was stored in the dark at −20 °C.3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Hoechst 33258, propidium iodide (PI), ethidium bromide (EB), fluorescein sodium and calf thymus DNA were purchased from Sigma Chemical Company (St. Louis, MO, USA). Anti-GAPDH monoclonal antibody was supplied by EarthOx Inc. (San Francisco, CA, USA). N-Acetylcysteine (NAC) and 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH-DA) were pursed from Invitrogen (Carlsbad, CA, USA). Caspase-3 antibody and phosphorylation histone family 2A variant (γH2A.X) were acquired from Cell Signaling Technology Inc. (Beverly, MA, USA). An Annexin V-PI Apoptosis Detection Kit was obtained from Keygen Technology Co., Ltd. (Nanjing, China). All other reagents were commercial products of analytical grade.
2.2. Cell culture
Human glioma U373 cells and murine fibroblastic NIH 3T3 cells were obtained from the cell bank of the Chinese Academy of Science, Shanghai, China. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U mL−1 penicillin–streptomycin solution and 1 mM L-glutamine at 37 °C in a humidified atmosphere with 5% CO2.2.3. Ultrasound setup and SDT protocol
The ultrasound exposure system used in this experiment consisted of a power amplifier (AG1020, T&C Power Conversion Inc., USA) and a planar transducer with frequency of 1.0 MHz (ndtXducer®, Northborough, MA, USA). The ultrasound intensity was calibrated before it was displayed on the screen of the power amplifier. The transducer was submerged in an acrylic tank filled with distilled degassed water. An intensity of 0.45 W cm−2 and duration of 1 min were used for ultrasound exposure. Human glioma cancer U373 cells were cultured on a 35 mm culture dish at a density of 2 × 105 cells per mL. After reaching around 70% confluence, the cells were divided randomly into four groups: (i) control (no treatment), (ii) DVDMS, (iii) ultrasound (Us), (iv) DVDMS plus ultrasound (SDT), each group having three repetitions. For the DVDMS and SDT groups, cells were incubated with 2 μM DVDMS over a 24 h drug-loading time in culture medium. Cells in the Us and SDT groups were exposed to continuous-wave ultrasound at a frequency of 1.0 MHz and an intensity of 0.45 W cm−2 for 1 min. For the inhibitory assays, the general ROS scavenger NAC (5 mM) was added to the culture medium 1 h prior to loading DVDMS.2.4. BBB model in vitro
To determine the ultrasound-mediated disruption of the BBB, the in vitro BBB model was established using a murine brain microvascular endothelial (bEnd.3) cell monolayer in a transwell.29–31 In brief, as shown in Fig. 2A, 1 × 105 bEnd.3 cells were seeded onto the upper chamber of the transwell in 24-well plates, in a medium that also contained FBS, and cultured for 5 days. Then, the permeability change of the BBB model in vitro was analyzed by a fluorescein sodium leakage test. 1 mg mL−1 fluorescein sodium was added to the upper chamber, then exposed to 0.45 W cm−2 ultrasonic irradiation for 1 min. The fluorescence intensity of the medium in the lower chamber at different time points was analyzed by a fluorescence photometer.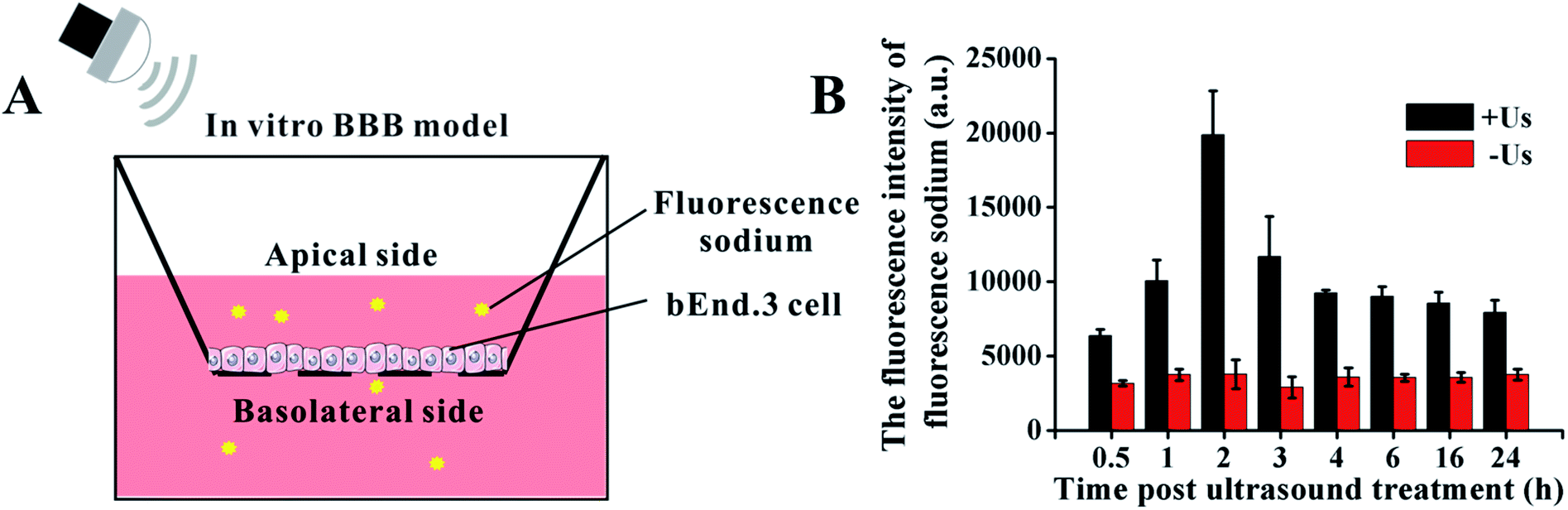 | ||
| Fig. 2 The model for studying the BBB in vitro. (A) Schematic illustration of BBB model. (B) The changes of BBB permeability after ultrasonic treatment with 0.45 W cm−2. | ||
2.5. Cytotoxicity assay
Cytotoxicity was evaluated with the conventional MTT reduction assay at 4 h and 24 h after different treatments in U373 cells and NIH 3T3 cells. The cell viability was determined by adding 10 μL MTT solution (5 mg mL−1 in PBS) to each sample (100 μL) in 96-well plates, followed by incubation at 37 °C for 4 h in a 5% CO2 incubator. Then, we added dimethyl sulfoxide (DMSO, Sigma) to dissolve formazan crystals and the absorbance (OD value) at 570 nm was recorded by using a microplate reader (ELX800, Bio-Tek, USA). Cell survival was calculated using the following equation:| Cell survival (%) = OD treatment group/OD control group × 100%. |
2.6. Morphologic study
Cells were seeded on a coverslip for 24 h, and then incubated with 2 μM DVDMS at 37 °C for 24 h prior to ultrasound treatment. After ultrasound treatment at a frequency of 1.0 MHz and an intensity of 0.45 W cm−2 for 1 min, the changes of cell morphology were observed using a light microscope (Axio Imager A2, Zeiss, Germany).2.7. Determination of intracellular ROS
Intracellular ROS generation was measured by using DCFH-DA as previously described.32 Briefly, to detect the intracellular ROS production, at 24 h after different treatments, cells were incubated with 10 μM DCFH-DA solution at 37 °C for 30 min, then washed with PBS and immediately analyzed by flow cytometry (NovoCyte, ACEA Biosciences Inc., USA). Data were analyzed using De Novo Software (De Novo Software, Los Angeles, CA, USA).2.8. Analysis of cell membrane integrity
The membrane-impermeable dye PI was used to stain cell nuclei. PI enters dead cells because the cell membranes become permeable. PI is widely used in cell death detection to evaluate the integrity of the plasma membrane. Different groups of cells were treated as per the protocol described above, then measured by flow cytometry after co-incubation with 5 μg mL−1 PI for 5 min.2.9. Assessment of DNA fragmentation
The DNA fragmentation was evaluated by flow fluorocytometric detection of DNA hypoploidy after adding PI and permeabilization using freeze-thawing.33 At 24 h post-treatment, U373 cells and NIH 3T3 cells were mixed with 200 μL PI solution (5 μg mL−1). The tubes were immediately placed in liquid nitrogen for 30 s and thawed at 37 °C for 5 min. Then, samples were analyzed using flow cytometry. Histograms were analyzed using De Novo Software.2.10. Fluorescence microscopy for nuclear morphological detection
Hoechst 33258 is a commercially available fluorescent probe that binds with DNA and reflects the nuclear morphology. The nuclei of dead cells can also be simultaneously stained with PI. After different treatments, cells were stained with Hoechst 332585 and PI for 15 min at 37 °C and imaged under a fluorescence microscope.2.11. Analysis of cell apoptosis
Cell apoptosis was determined at 24 h post treatment through Annexin V-FITC/PI double staining. Briefly, 100 μL of sample was mixed with 100 μL binding buffer containing Annexin V-FITC and PI. After incubation at room temperature for 30 min, samples were analyzed by a flow cytometer.2.12. Immunofluorescence staining
Histone γH2A.X was employed as a marker to reflect the DNA double-stranded damage in this study. An immunofluorescence test was performed at 24 h after different treatments. Cells were fixed with 4% paraformaldehyde for 15 min, followed by permeabilization with 0.1% Triton X-100 and blocking with 5% BSA. Cells were incubated with γH2A.X antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :800 dilution) at 4 °C overnight. After washing with PBS, samples were incubated with the corresponding secondary antibodies at 37 °C for 1 h, and images were recorded using a fluorescence microscope.
:800 dilution) at 4 °C overnight. After washing with PBS, samples were incubated with the corresponding secondary antibodies at 37 °C for 1 h, and images were recorded using a fluorescence microscope.
2.13. Single cell gel electrophoresis
DNA damage was measured by single cell gel electrophoresis (comet electrophoresis). Briefly, the samples were mixed with 0.75% low melting point agarose and poured into a microscope slide quickly. After lysing for 30 min, the slides were placed in an electrophoresis tank, allowing the DNA to unwind for 25 min in alkaline solution. Electrophoresis was performed at 25 V (300 mA) for 25 min in the same alkaline solution at room temperature. The samples were then neutralized and gradient eluted using ethanol and stained with EB. The slides were observed by a fluorescence microscope. Data were analyzed with Comet Assay Software Project (CASP).2.14. Western blotting
Western blotting was performed to analyze the expression levels of apoptosis-related protein and γH2A.X. Briefly, equal amounts of proteins from different samples quantified with a BCA kit were added to 10% SDS-polyacrylamide gel to separate distinct molecular weights of proteins. The proteins on the gel were then transferred to PVDF membrane, followed by blocking and incubation with primary antibody (caspase-3, γH2A.X). After reaction with the corresponding secondary antibodies, samples were imaged using an Odyssey infrared imaging system (Li-Cor Bioscience, Lincoln, USA).2.15. Assessment of DNA damage in cell-free system
DNA DSBs confer an increase of the characteristic absorption at 260 nm, which is called the hyperchromic effect and can be utilized to evaluate the DNA damage level.34 Using an in vitro cell-free system, ultraviolet spectrophotometry was applied to test the DVDMS-induced calf thymus DNA damage with or without the assistance of ultrasound exposure. 10 μM DVDMS was added into 0.05 μg mL−1, 0.1 μg mL−1 and 0.2 μg mL−1 of calf thymus DNA solution, then samples were detected by ultraviolet spectrophotometry at 260 nm. Additionally, 0.1 μg mL−1 DNA solution was mixed with 10 μM DVDMS and exposed to ultrasound for 1 min, 2 min and 3 min, respectively. Simultaneously, samples without DVDMS were also used as references to determine the absorption values after different ultrasound treatments.In electrophoresis assay, samples in different groups were loaded in 1% neutral agarose gel, and then electrophoresized at 150 V for 1.0 h in electrophoresis apparatus. The gel was stained with a solution of 0.5 μg mL−1 EB for 30 min and visualized by a gel image analysis system.
2.16. Statistical analysis
All data are expressed as mean ± standard deviation (S.D.) of three independent experiments. Differences between the treatment groups were assessed with one-way analysis of variance (ANOVA). A value of p < 0.05 was considered significant.3. Results
3.1. BBB model in vitro
The in vitro BBB model was utilized to investigate the cross-BBB transportation at different times under ultrasonic irradiation of 0.45 W cm−2 (Fig. 2B). Ultrasound assisted the penetration of fluorescein sodium across the bEnd.3 monolayer, and the evidence suggested that the greatest extent of BBB opening occurred at 2 h post irradiation and gradually recovered soon afterwards. This result indicates that the BBB can be temporarily and reversibly opened by ultrasound exposure at specific conditions.3.2. Cell damage induced by DVDMS-SDT
The results in Fig. 3A show that 2 μM DVDMS and ultrasound alone did not produce obvious cytotoxicity, with cell viability of about 95.03% and 89.93% at 4 h after treatment, but the cell survival rate decreased to 41.82% (p < 0.01) in the SDT group. When the incubation time was prolonged to 24 h, the cell viability was measured as 87.27%, 90.81% and 37.76% (p < 0.01) in the 2 μM DVDMS alone, 0.45 W cm−2 ultrasound and SDT group, respectively. We also investigated the viability of NIH 3T3 cells after DVDMS-SDT treatment (2 μM DVDMS, 0.45 W cm−2 ultrasound). In contrast with tumor cells, no significant changes were observed in the viability of normal cells (Fig. 3C). Based on this, it is speculated that DVDMS can selectively kill tumor cells, which may be due to DVDMS' preferential accumulation in tumor cells. Furthermore, microscopy observation showed that the cells were normal fusiform shape in the control group, whereas the morphology of the U373 cells was seriously disturbed after DVDMS-SDT treatment, with a decrease of cell numbers (Fig. 3B).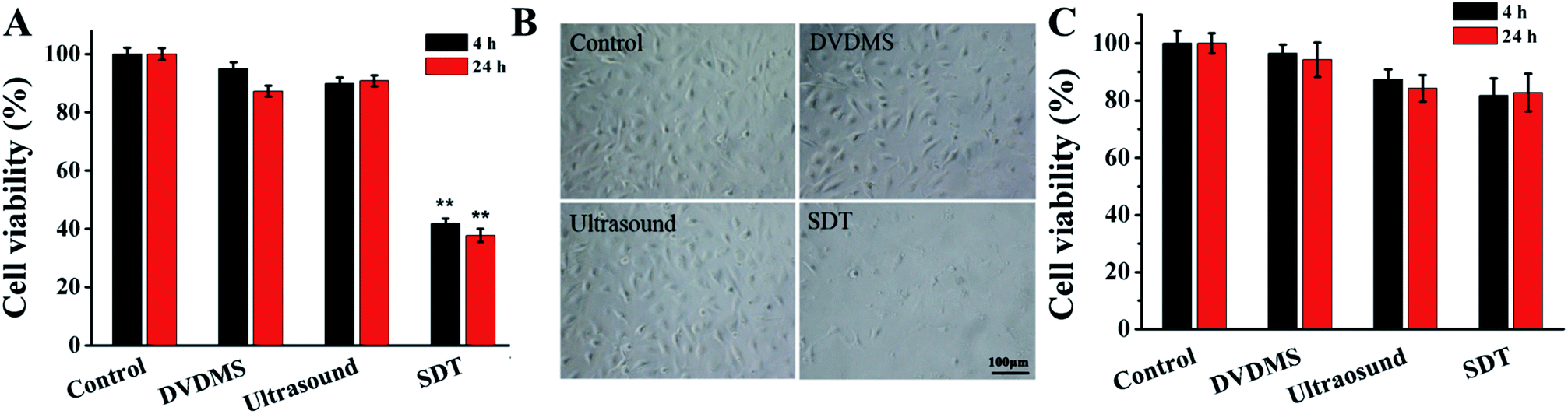 | ||
| Fig. 3 Cell damage triggered by DVDMS-SDT treatment. (A) Evaluation of cell viability by MTT assay. Error bars represent the S.D. from three independent experiments. **p < 0.01 versus control. (B) Observation of cell morphology with light microscope after SDT treatment (the scale bar is 100 μm). Control, without any treatment; DVDMS, 2 μM DVDMS alone; Ultrasound, 0.45 W cm−2 ultrasound for 1 min; SDT, 2 μM DVDMS combined with 0.45 W cm−2 ultrasound for 1 min. (C) Detection of cell viability of NIH 3T3 cells by MTT assay at 24 h after different treatments. | ||
3.3. Integrity of cell membranes
PI staining combined with flow cytometry was used to determine cell membrane integrity. Fig. 4 shows that there were no significant changes observable by PI positive staining in the 2 μM DVDMS alone (6.80%) and control groups, and a slight increase of cell membrane integrity in the ultrasound treatment group (11.30%, p < 0.01). Compared with the other groups, the SDT group exhibited a significant increase of membrane permeability (25.75%, p < 0.01). | ||
| Fig. 4 Effects of DVDMS on the cell membrane integrity of U373 cells. After different treatments, cells were stained with PI and analyzed by flow cytometry. Error bars represent S.D. of the means from three independent experiments. **p < 0.01 versus untreated cells. | ||
3.4. Intracellular ROS generation
Quantitative measurements of intracellular ROS generation in different samples were performed using flow cytometry (Fig. 5A). The DVDMS alone group showed a minuscule ROS increase, with 11.92% of U373 cells emitting high dichlorofluorescein (DCF) fluorescence. However, the intracellular ROS generation of DVDMS was greatly enhanced upon ultrasound exposure, representing up to four-fold increased potency compared with no ultrasound trigger. The ROS production caused by SDT reduced to 13.45% when cells were pre-incubated with the free radical scavenger NAC. Moreover, as shown in Fig. 5B, NAC pretreatment also rescued the cell viability rate from 37.76% to 82.17% (p < 0.01) when ROS generation was triggered by SDT. These results demonstrate that ROS formation was a major factor for the SDT-induced cell killing effect in vitro.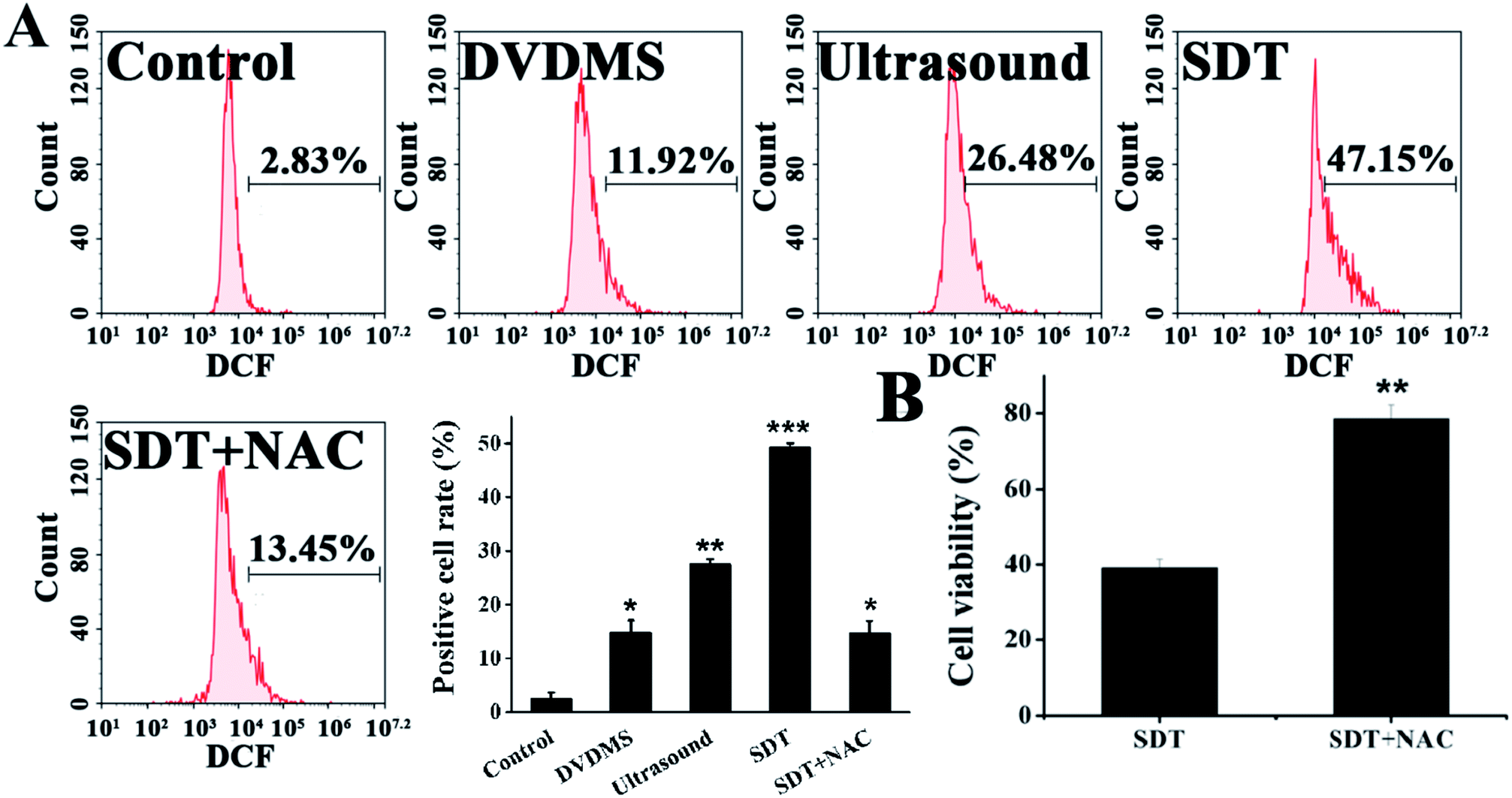 | ||
| Fig. 5 Intracellular ROS generation in U373 cells at 0.5 h post treatment. The cells were labeled with DCFH-DA and the mean fluorescence intensity of the oxidized product DCF in cells was detected by flow cytometry (A). The changes of cell viability after NAC treatment was detected by MTT assay (B). Error bars represent the S.D. from three independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 versus control. | ||
3.5. DNA damage caused by DVDMS-SDT
Hoechst 33258-PI double staining was used to detect the DNA damage after SDT treatment. As shown in Fig. 6A, the untreated cells of the control group retained their normal nucleus morphology with homogeneous blue staining. DVDMS alone did not show any difference compared with the control. Ultrasound treatment slightly enhanced Hoechst 33258 staining, and a few cells were stained with bright PI fluorescence. In the DVDMS-SDT group, the cell nuclei were more condensed and emitted bright Hoechst 33258 fluorescence and most of the cells were stained with PI, showing altered nucleus morphology, apoptotic features and decreased cell numbers.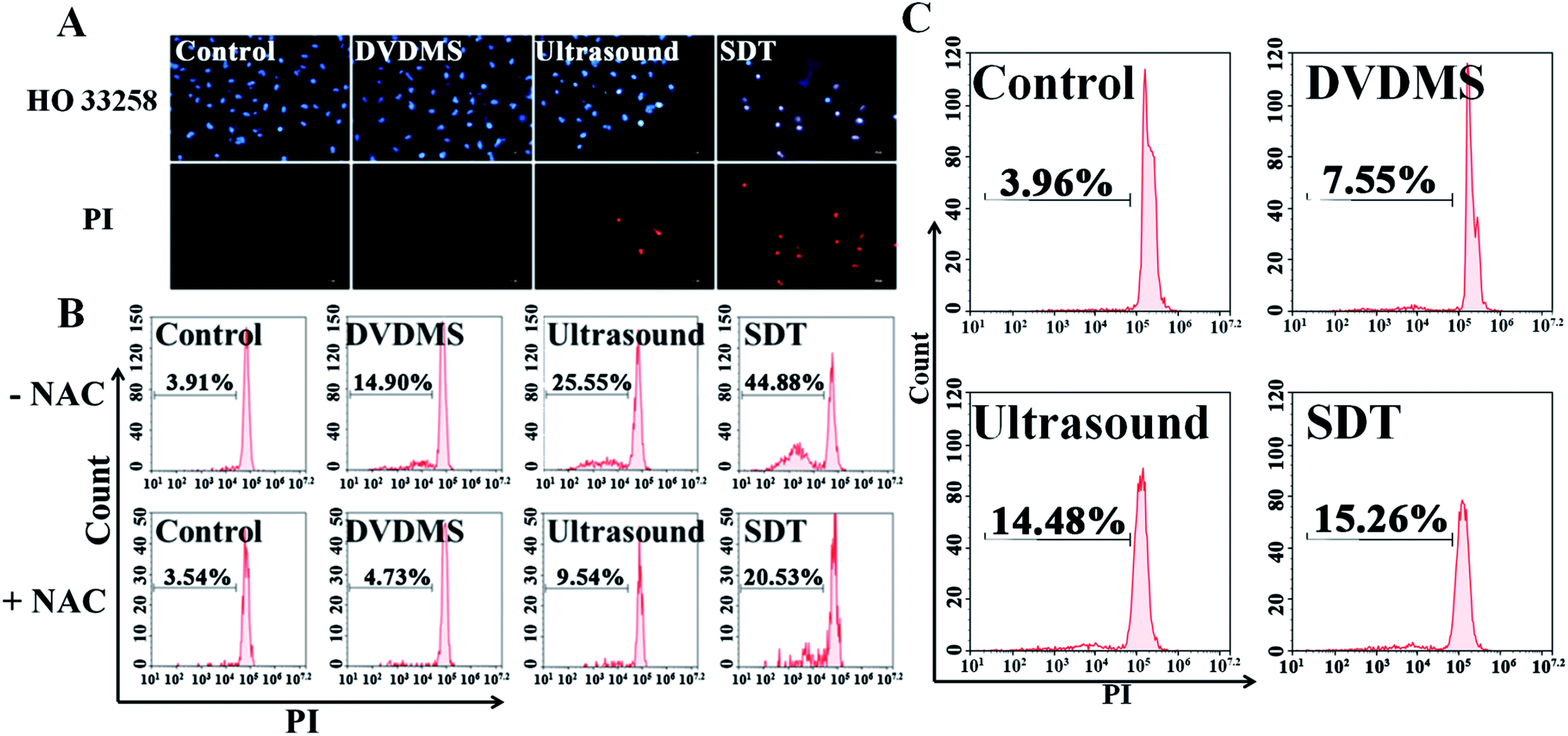 | ||
| Fig. 6 Detection of nuclear damage and DNA fragmentation of differently treated U373 cells. The apoptotic nuclear changes and cell membrane integrity detected by PI/Hoechst staining using fluorescence microscopy (A) and the analysis of DNA fragmentation after staining with PI for U373 cells (B) and NIH 3T3 cells (C). Error bars represent the S.D. from three independent experiments. **p < 0.01 versus control, **p < 0.01 SDT group versus SDT + NAC group. | ||
Following Hoechst 33258-PI observation, we also designed a PI staining method with freeze-thawing to quantitatively analyze DNA fragmentation. Fig. 6B shows that the level of DNA fragmentation is 3.91% in the control group, no significant difference was measured in the DVDMS alone group (14.90%), ultrasound alone increased the DNA fragmentation to 25.55% (p < 0.05), whereas the most serious DNA damage was observed in the SDT group with 44.88% DNA fragmentation (p < 0.01). These results suggest that DNA damage was aggravated when ultrasound was combined with DVDMS. With NIH 3T3 cells serving as normal controls, the results in Fig. 6C indicate no significant changes were observed in the DNA damage of the normal cells.
Comet assay is based on the fact that smaller DNA fragments migrate faster in an electric field, and the pattern of migration produces a profile resembling the shape of a comet. The results in Fig. 7 show that the untreated cells in the control maintained the regular morphology of the nucleus. Meanwhile, minor DNA damage was observed in some of the cells treated with 2 μM DVDMS and ultrasound alone, with slight comet tailing. Cells in the SDT group produced a large number of comet tails consisting of low molecular weight DNA (p < 0.001).
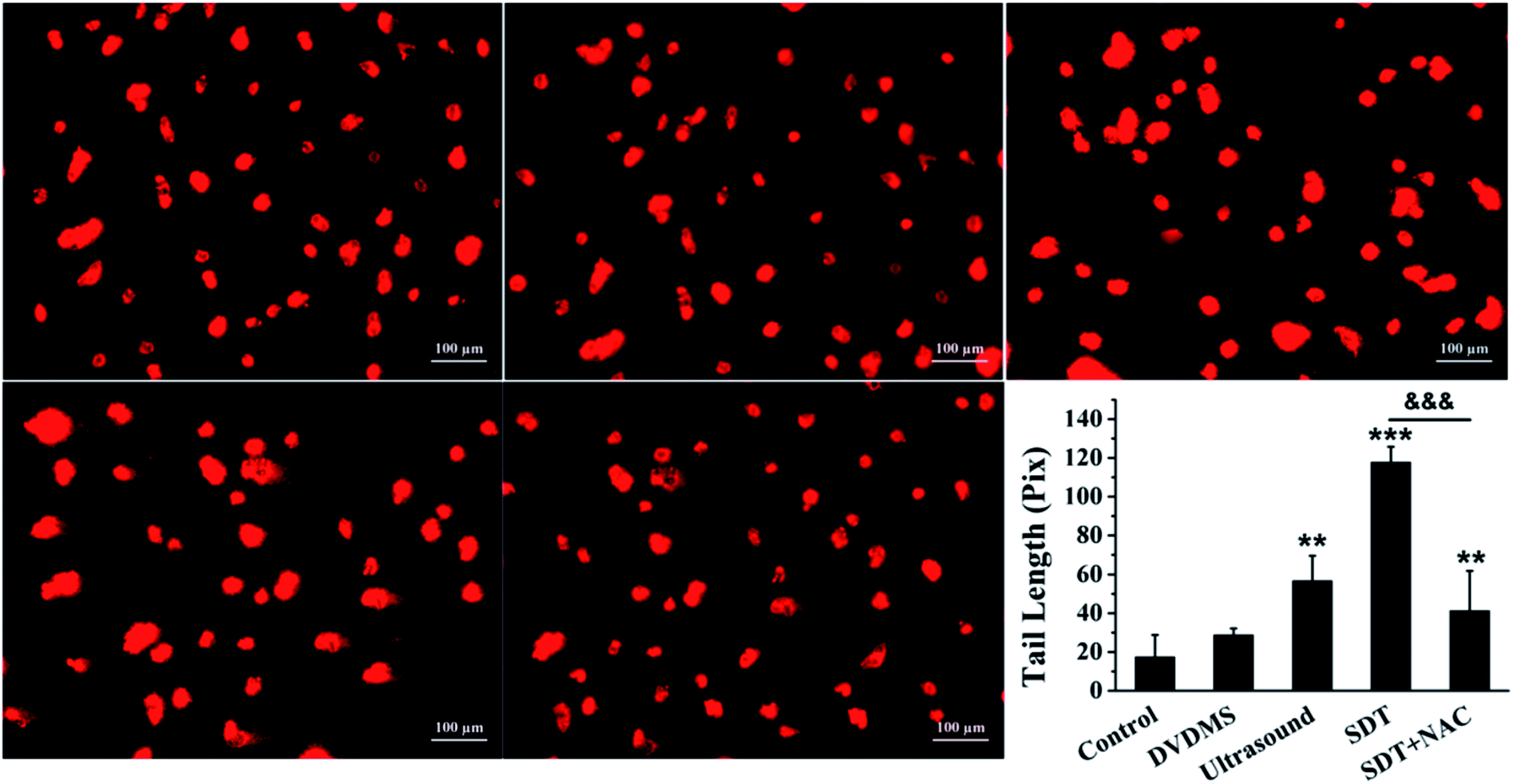 | ||
| Fig. 7 Examination of DNA damage by comet assay in U373 cells. The DNA damage was evaluated by gel electrophoresis and analyzed by CASP software, and the DNA tail length was calculated after different treatments. The data are represented as means ± S.D (n = 3). *p < 0.05, **p < 0.01 and ***p < 0.001 versus the untreated control, ***p < 0.001 SDT group versus SDT + NAC group. | ||
To explore the ROS effect on DNA damage in SDT treatment, cells in each group were pre-treated with NAC. As seen in Fig. 6B and 7, U373 cells treated with NAC showed a prominent decrease of DNA fragmentation in the ultrasound and SDT groups (9.54% and 20.53%, respectively), and comet tailing was obviously alleviated in the SDT + NAC group (p < 0.001), suggesting that the elimination of ROS decreased the oxidative damage of DNA induced by DVDMS-SDT.
3.6. DNA double-strand break triggered by DVDMS-SDT
The above data indicate that much severe DNA injury occurred after DVDMS-SDT treatment, but the specific types of DNA damage remain unclear. Histone γH2A.X, the marker of DSB, was detected by immunofluorescence and western blot to investigate the possible classes of DNA damage. As illustrated in Fig. 8 and 9B, treatment with 2 μM DVDMS or 0.45 W cm−2 ultrasound alone slightly increased the γH2A.X expression level compared with the control. However, γH2A.X production was particularly pronounced in the DVDMS-SDT group. After addition of NAC to scavenge ROS, the expression level of γH2A.X was significantly lower than that of the SDT group. These results suggest that DVDMS-SDT induced DNA DSB in U373 cells via ROS production.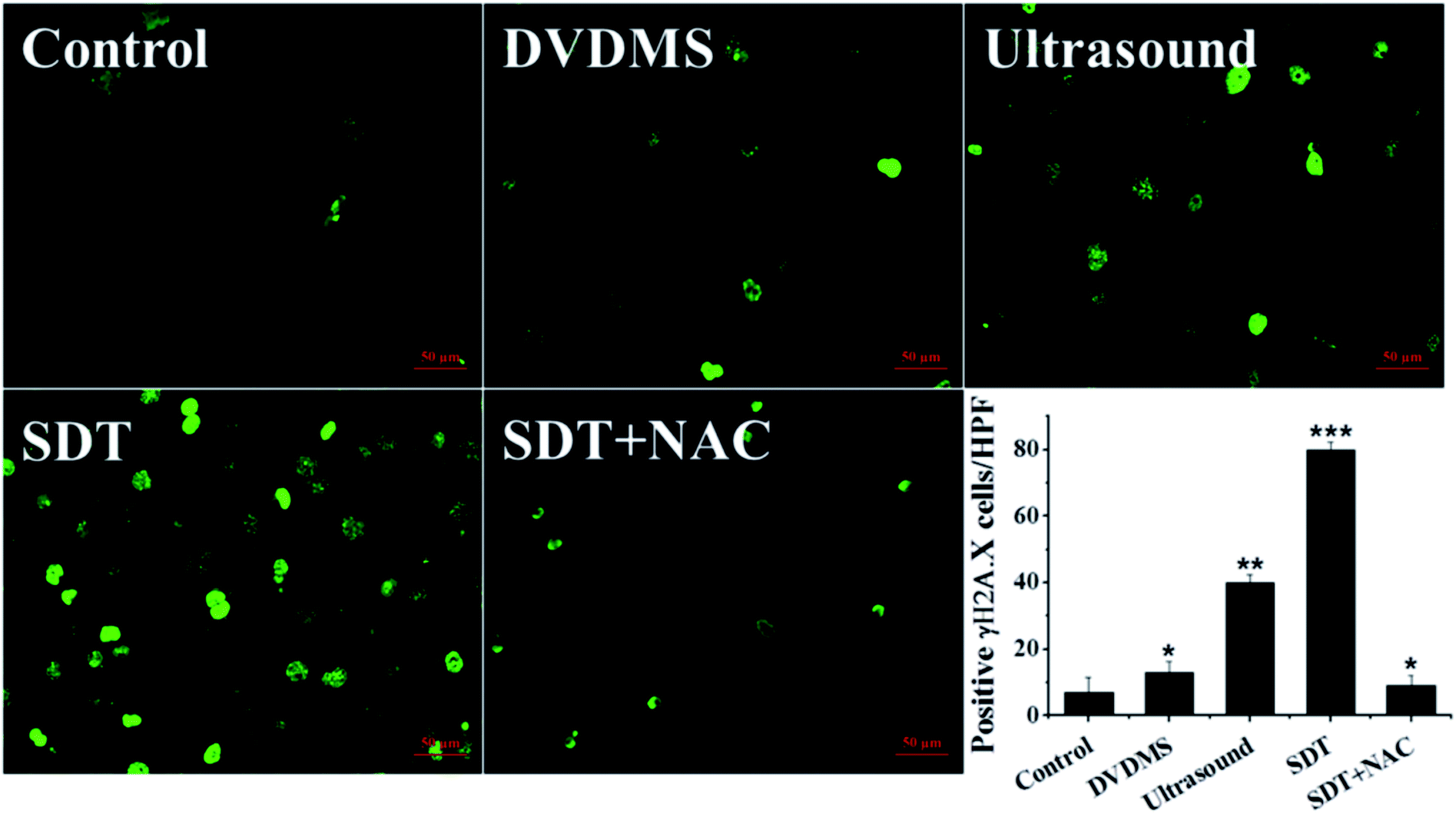 | ||
| Fig. 8 Measurement of histone γH2A.X expression level in U373 cells after different treatments. Cells were fixed and processed for immunofluorescence, γH2A.X was visualized using FITC-labeled secondary antibody (green). The numbers of γH2A.X-positive cells per high power field were calculated. Representative images are shown and the quantitative result is presented. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with control group. | ||
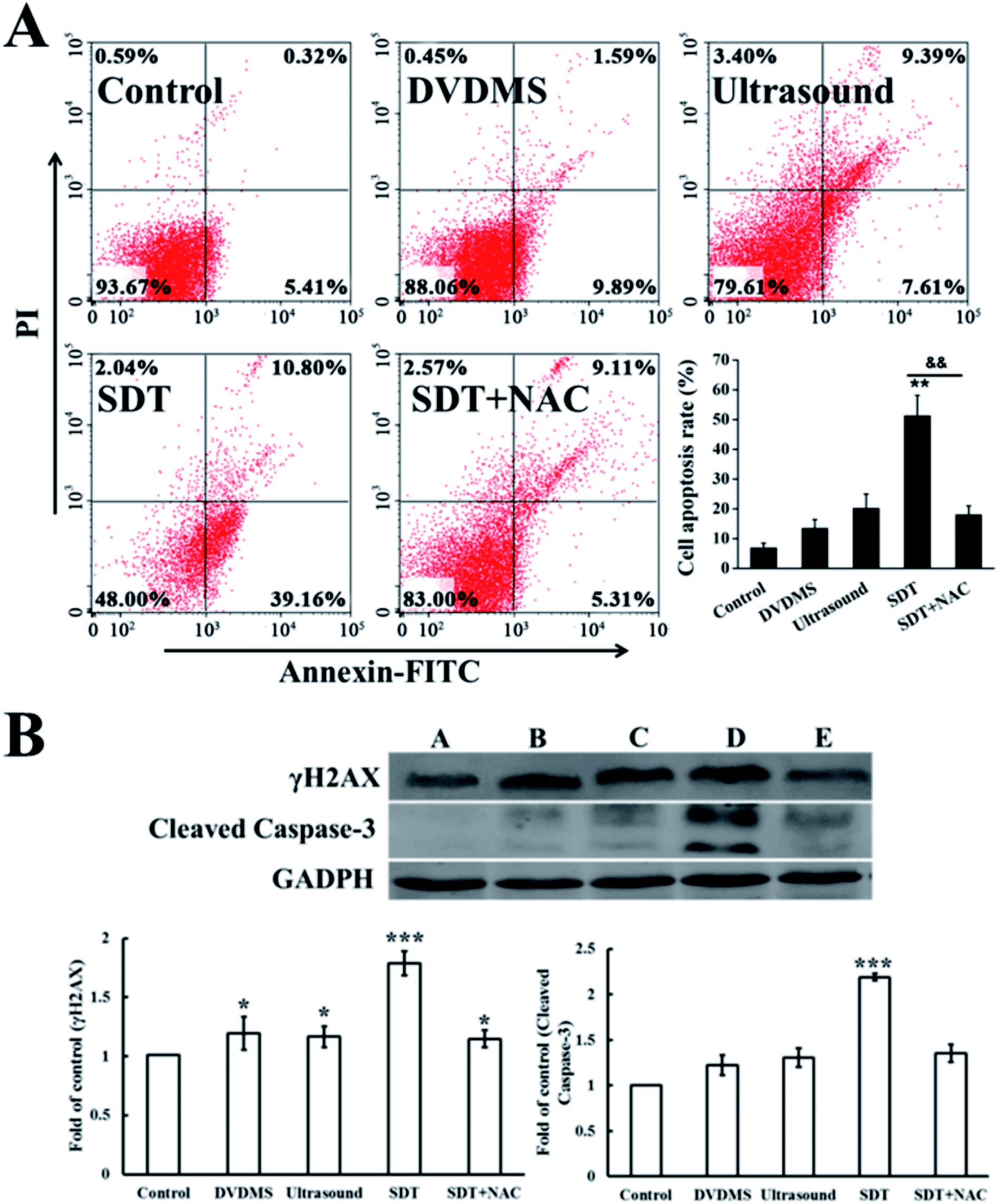 | ||
| Fig. 9 DVDMS-SDT-induced apoptosis in U373 cells. (A) Dot plots of uptake of Annexin V and PI by differently treated cells 24 h post-treatment as determined by flow cytometry. (B) Western blot analysis of the expression level of histone γH2A.X and apoptosis-related protein cleaved-caspase-3 after different treatments. Significant differences between the control and SDT groups are indicated: *p < 0.05, **p < 0.01 and ***p < 0.001 versus control, **p < 0.01 represents SDT group versus SDT + NAC group. | ||
3.7. Cell apoptosis induced by DVDMS-SDT
Cell apoptosis was determined through Annexin V-FITC/PI double staining with flow cytometry and western blotting of activated caspase-3. Fig. 9A shows the quantification of cell apoptosis. The proportions of apoptotic cells in the DVDMS alone, ultrasound and SDT groups were 11.94%, 20.39% and 52.00% (p < 0.01), respectively. Compared with the control group (8.09%), SDT greatly enhanced the level of cell apoptosis (p < 0.01). However, cell apoptosis decreased to 20.21% in the SDT + NAC group, showing a significant difference from the SDT group (p < 0.01), which was consistent with the cell viability assay. Moreover, procaspase-3 is usually activated and cleaved in the apoptotic process. The expression level of cleaved-caspase-3 in the DVDMS-SDT group was 2.8 times that of the control (Fig. 9B), and NAC effectively inhibited cleaved-caspase-3 expression, indicating ROS played a vital role in SDT-induced cell apoptosis.3.8. DNA damage induced by DVDMS- SDT in cell-free system
Because the studies in vitro suggested serious DNA damage after SDT treatment, we next investigated the direct interaction between DNA and DVDMS in the presence or absence of ultrasound. As shown in Fig. 10A and 10B, DNA displayed a characteristic absorption peak at 260 nm, which showed a DVDMS dose-dependent variation, and increased together with the ultrasound duration-time. More obviously, the intensity of DNA absorption at 260 nm was enhanced by increasing the duration time in SDT (Fig. 10C), indicating that the DNA structure was seriously disrupted in the sonodynamic process.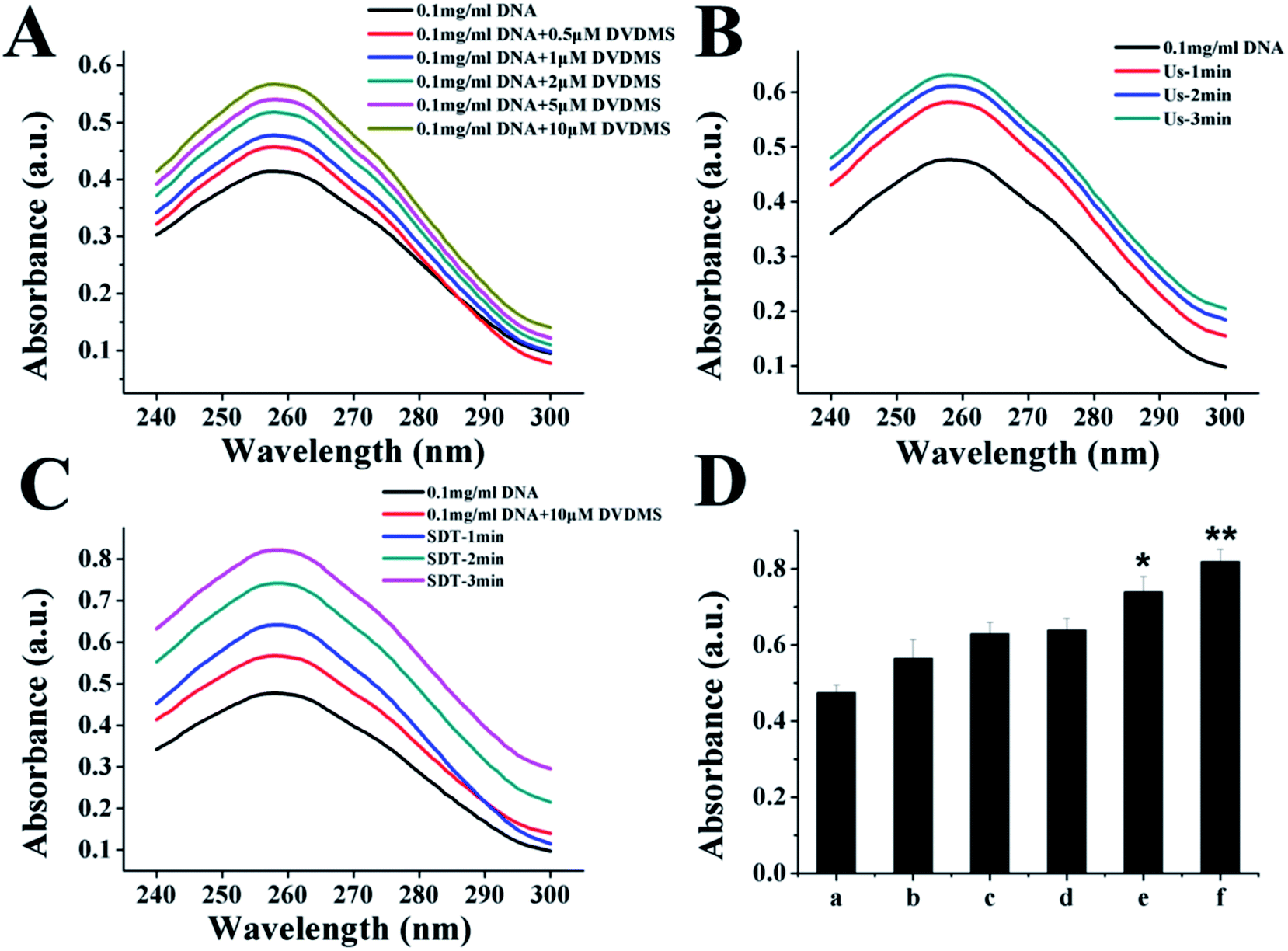 | ||
| Fig. 10 UV-vis spectra of DNA after different treatments. (A) Effect of different concentrations of DVDMS on DNA damage. (B) Effects of ultrasound exposure time on DNA damage. (C) DNA damage (0.1 mg mL−1 DNA) after DVDMS-SDT was detected by ultraviolet spectrophotometry. (D) Quantification of absorbance value of DNA at 260 nm under different DVDMS-SDT. (a), 0.1 mg mL−1 DNA; (b), 0.1 mg mL−1 DNA+10 μM DVDMS; (c), ultrasonic irradiation for 3 min; (d), SDT-1 min; (e), SDT-2 min; (f), SDT-3 min. Data expressed as mean ± standard deviation of three batches. *p < 0.05, **p < 0.01 compared with control. | ||
Further, agarose gel electrophoresis was performed to confirm DNA fragmentation and DNA degradation. The results in Fig. 11 exhibit that the untreated cells of the control and DVDMS alone groups showed little DNA damage with very short DNA tailing. Slight DNA tailing was captured after ultrasound treatment. Meanwhile, DNA tailing and DNA degradation were gradually aggravated after DVDMS-SDT treatment, in an ultrasound-intensity-dependent manner. The DNA damage tests in cell-free systems and cell experiments demonstrate that DVDMS-SDT interfered with DNA molecules, and the induced DSB might have contributed to the cell viability loss of the U373 cells.
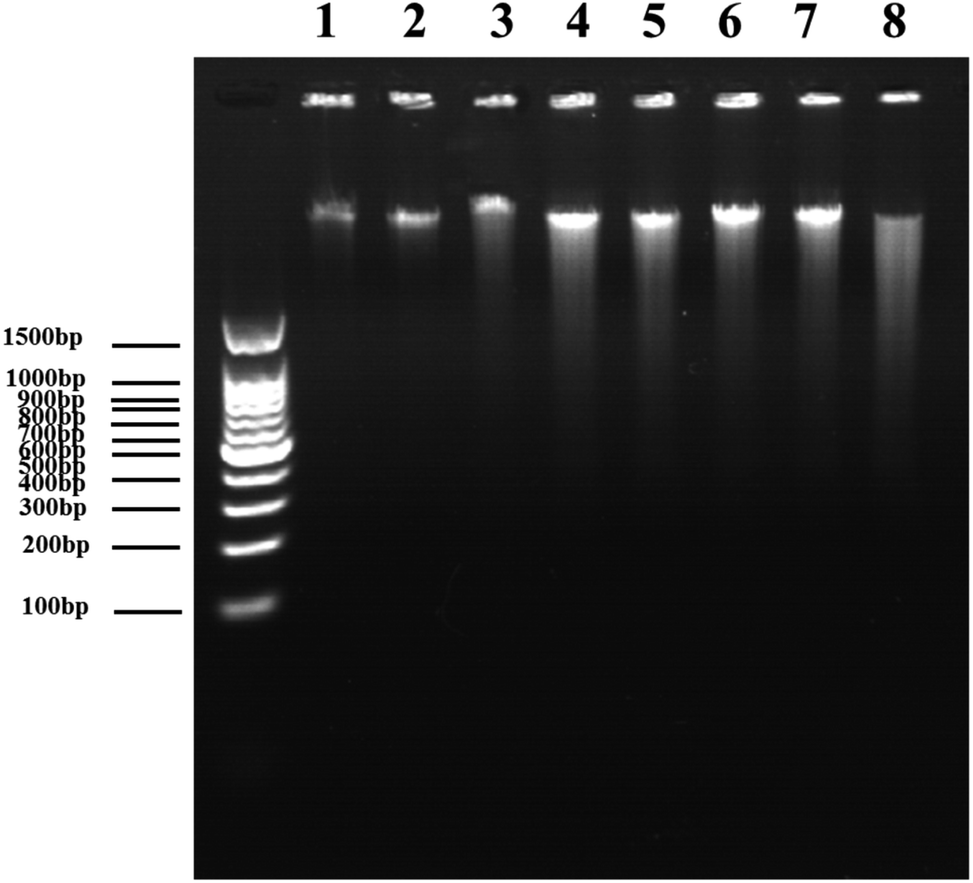 | ||
| Fig. 11 Agarose gel electrophoretic patterns of DNA treated with SDT. lane 1:0.1 mg mL−1 DNA, lane 2:0.1 mg mL−1 DNA + 10 μM DVDMS, lane 3:0.1 mg mL−1 DNA + ultrasound for 1 min, lane 4:0.1 mg mL−1 DNA + ultrasound for 2 min, lane 5:0.1 mg mL−1 DNA + ultrasound for 3 min, lane 6:0.1 mg mL−1 DNA + 10 μM DVDMS + ultrasound for 1 min, lane 7:0.1 mg mL−1 DNA + 10 μM DVDMS + ultrasound for 2 min, lane 8:0.1 mg mL−1 DNA + 10 μM DVDMS + ultrasound for 3 min. | ||
4. Discussion
Recently, the effects of sonocatalytic reagents in combination with ultrasound have been reported in glioma cells,14,35 which involves a complex and diverse SDT process. The possible SDT target sites remain unclear, but they are determined by the features of sonosensitizers and can greatly influence the cellular responses and SDT outcomes. In this study, the emphasis on the sonodynamic efficacy in human glioma U373 cells by utilizing a sonosensitizer, DVDMS, aimed to explain the mechanistic basis of BBB opening, DNA oxidative damage, and potential glioma therapy.The topic of BBB disruption using ultrasound has aroused considerable attention in brain diseases because it is noninvasive and reversible.2 In this study, we chose therapeutic ultrasound to optimize the BBB opening parameters, which indicated that the in vitro established BBB model could be temporarily and reversibly opened by ultrasound exposure at frequency of 1.0 MHz and intensity of 0.45 W cm−2 for 1 min duration, which did not produce any viability loss in normal cells. Controllable BBB opening facilitates subsequent drug delivery at specific time points and local sites.36 It is reported that ultrasound can induce a disintegration of the tight-junctional complexes in brain microvessels, involving the redistribution and loss of immunosignals for occludin, claudin-5 and reduction in the expression of the submembranous junction-related proteins ZO-1 and claudin-1.29,30 Therefore, the ultrasound-assisted drug penetration into BBB and accumulation in the brain also pave a way for increased SDT anti-glioma efficiency.
Using human glioma U373 cells, the toxic assays in vitro indicated a significant cell viability loss and apoptosis increase after the combined DVDMS-SDT treatment, whereas either alone did not cause obvious cell damage at the chosen doses. Additionally, there was no significant cytotoxicity in normal cells after DVDMS-SDT treatment. This suggests that DVDMS-mediated SDT had a selective lethal effect on cancer cells without affecting healthy cells. Our previous studies showed that DVDMS was taken up quickly and preferentially accumulated in tumor cells, compared with normal cells.37,38 This might be due to the high expression of low-density lipoprotein receptors in tumor cell membranes, which promote the internalization of sonosensitizers with high binding affinity via endocytosis pathways.39 SDT has been suggested as an activatable approach for tumor therapy, and the oxidative radicals produced during the process contribute to cellular toxicity. In agreement with previous studies,40 DVDMS-SDT stimulated obvious ROS generation in U373 cells and pretreatment with NAC significantly rescued the cell viability loss and apoptosis induction. Although the ROS generation sites, which determine the cellular targets of SDT, usually occur in the cytosol, the nuclear genetic material may also be greatly influenced by an undefined mechanism. Furusawa reported that nuclear membranes can be permeabilized by ultrasound and the DNA strands could be directly exposed to the ultrasound wave, which produced distinct breaks or cross-linking.41 Excess ROS can activate nuclear factor E2-related factor 2 (Nrf2), nuclear transcription factor-κB (NF-κB), and mitogen-activated protein kinases (MAPKs) to regulate expression of oxidative stress-related genes,42 which then further disrupt cellular DNA molecules. Therefore, we subsequently focused on the structural and functional DNA damage after DVDMS-SDT treatment. Both flow cytometry and comet electrophoresis showed severe oxidative DNA damage (Fig. 6B and 7) post SDT in U373 cells. DVDMS-SDT produced little damage to DNA of normal cells. DNA damage involves not only base pair substitution and point mutation, but also serious DNA single-strand or double-strand breaks. Phosphorylation of γH2A.X is currently known as a DNA marker for DSB, and can be catalyzed by ATM and ATR at the Ser139 site and in turn leads to recruitment of DNA-related proteases to repair the damaged sites.43 γH2A.X is dephosphorylated when the DNA repair process is completed.44 In the present study, γH2A.X phosphorylation following DVDMS-SDT was confirmed by immunofluorescence and western blot, and the results suggest that the most serious DNA damage type – DSB – was induced by SDT. In various tumor types including glioma, several proteins with particular mutations endow tumor cells with distinct DNA damage-repair ability. In this case, however, U373 cells lack the ability to activate the p53-dependent apoptotic pathway due to p53 mutation, which may trigger other factors at the S or G2 checkpoint to lead to cell cycle arrest under external stimuli.45 Ruiz-Magana et al. reported that the cell checkpoint can be activated in response to therapeutics in p53 mutant cells.46 As the time of DNA repair was lengthened, unrepaired DNA strand breaks then easily led to cell death. For these reasons, DNA DSBs following DVDMS-SDT hindered DNA repair in U373 cells and caused serious toxicity.
Cells struggle to maintain stable functioning when exposed to various stimuli, which then initiate complicated cellular damage-repair responses and compensation mechanisms, the ultimate outcome being a synergistic combination of several responses. In order to investigate whether DNA could be directly influenced by DVDMS-SDT, a cell-free system was established and showed evidence for ultrasound-intensity- and DVDMS-concentration-dependent DNA damage and fragmentation after SDT treatment.
5. Conclusions
Taken together, our findings indicate that glioma therapy using DVDMS-SDT has potential benefits derived from the reversible BBB opening and significant sonochemical effects. DVDMS-SDT may directly regulate DNA molecules and induce DSBs through ROS generation, which provides some new insights into SDT's biological mechanism for glioma therapy. Nevertheless, an orthotopic glioma model is also now underway, which involves several ultrasound treatment cycles, BBB opening windows, DNA damage in situ, and so on. In short, the present study represents an alternative SDT strategy for glioma therapy.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81472846, 81571834), the Fundamental Research Funds for the Central Universities (Grant No. 2017CSY022) and the Shaanxi Normal University Training Programs of Innovation and Entrepreneurship for Undergraduates (No. 201710718015).References
- N. A. Bush, S. M. Chang and M. S. Berger, Neurosurg. Rev., 2017, 40, 1–14 CrossRef PubMed.
- X. Wang, Y. Jia, P. Wang, Q. Liu and H. Zheng, Ultrason. Sonochem., 2017, 37, 592–599 CrossRef CAS PubMed.
- P. Wu, Y. Jia, F. Qu, Y. Sun, P. Wang, K. Zhang, C. Xu, Q. Liu and X. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 25706–25716 CrossRef CAS PubMed.
- M. A. O'Reilly, R. M. Jones, E. Barrett, A. Schwab, E. Head and K. Hynynen, Theranostics, 2017, 7, 3573–3584 CrossRef PubMed.
- C. H. Fan, C. Y. Lin, H. L. Liu and C. K. Yeh, J. Controlled Release, 2017, 261, 246–262 CrossRef CAS PubMed.
- M. Aryal, C. D. Arvanitis, P. M. Alexander and N. McDannold, Adv. Drug Delivery Rev., 2014, 72, 94–109 CrossRef CAS PubMed.
- G. Leinenga and J. Gotz, Sci. Transl. Med., 2015, 7, 278ra233 Search PubMed.
- E. Landhuis, Nature, 2017, 551, 257–259 CrossRef PubMed.
- P. Huang, X. Qian, Y. Chen, L. Yu, H. Lin, L. Wang, Y. Zhu and J. Shi, J. Am. Chem. Soc., 2017, 139, 1275–1284 CrossRef CAS PubMed.
- X. Qian, Y. Zheng and Y. Chen, Adv. Mater., 2016, 28, 8097–8129 CrossRef CAS PubMed.
- I. Rosenthal, J. Z. Sostaric and P. Riesz, Ultrason. Sonochem., 2004, 11, 349–363 CAS.
- T. Ohmura, T. Fukushima, H. Shibaguchi, S. Yoshizawa, T. Inoue, M. Kuroki, K. Sasaki and S. Umemura, Anticancer Res., 2011, 31, 2527–2533 CAS.
- S. Dai, C. Xu, Y. Tian, W. Cheng and B. Li, Oncol. Lett., 2014, 8, 1675–1681 CrossRef CAS PubMed.
- Z. Y. Xu, K. Wang, X. Q. Li, S. Chen, J. M. Deng, Y. Cheng and Z. G. Wang, Ultrasonics, 2013, 53, 232–238 CrossRef CAS PubMed.
- H. Wang, P. Wang, L. Li, K. Zhang, X. Wang and Q. Liu, Int. J. Biol. Sci., 2015, 11, 1401–1409 CrossRef CAS PubMed.
- H. Wang, X. Wang, S. Zhang, P. Wang, K. Zhang and Q. Liu, Int. J. Nanomed., 2014, 9, 3077–3090 CrossRef CAS PubMed.
- C. S. Djuzenova, V. Fiedler, S. Memmel, A. Katzer, S. Hartmann, G. Krohne, H. Zimmermann, C. J. Scholz, B. Polat, M. Flentje and V. L. Sukhorukov, Exp. Cell Res., 2015, 330, 346–357 CrossRef CAS PubMed.
- S. W. Lee, H. K. Kim, N. H. Lee, H. Y. Yi, H. S. Kim, S. H. Hong, Y. K. Hong and Y. A. Joe, Cancer Lett., 2015, 360, 195–204 CrossRef CAS PubMed.
- A. Khalil, R. N. Morgan, B. R. Adams, S. E. Golding, S. M. Dever, E. Rosenberg, L. F. Povirk and K. Valerie, Cell Cycle, 2011, 10, 481–491 CrossRef CAS PubMed.
- K. K. Khanna and S. P. Jackson, Nat. Genet., 2001, 27, 247–254 CrossRef CAS PubMed.
- T. Rich, R. L. Allen and A. H. Wyllie, Nature, 2000, 407, 777–783 CrossRef CAS PubMed.
- S. A. Wieczorek, F. Breitenbuecher, A. Soni, K. Paul-Konietzko, S. Ziegler, A. Sak, G. Iliakis and M. Schuler, J. Cancer Res. Clin. Oncol., 2017, 143(9), 1733–1744 CrossRef CAS PubMed.
- Y. Li, Q. Zhou, Z. Deng, M. Pan, X. Liu, J. Wu, F. Yan and H. Zheng, Sci. Rep., 2016, 6, 25968 CrossRef CAS PubMed.
- Y. Liu, P. Wang, Q. Liu and X. Wang, Ultrason. Sonochem., 2016, 31, 437–448 CrossRef CAS PubMed.
- X. Su, P. Wang, X. Wang, L. Guo, S. Li and Q. Liu, Int. J. Radiat. Biol., 2013, 89, 915–927 CrossRef CAS PubMed.
- S. Das, C. J. Neal, J. Ortiz and S. Seal, Nanoscale, 2018 10.1039/c8nr04640a.
- M. Dizdaroglu, R. Olinski, J. H. Doroshow and S. A. Akman, Cancer Res., 1993, 53, 1269–1272 CAS.
- F. Salehi, H. Behboudi, G. Kavoosi and S. K. Ardestani, Sci. Rep., 2018, 8, 13902 CrossRef PubMed.
- Y. Liu, L. Mei, C. Xu, Q. Yu, K. Shi, L. Zhang, Y. Wang, Q. Zhang, H. Gao, Z. Zhang and Q. He, Theranostics, 2016, 6, 177–191 CrossRef CAS PubMed.
- N. Sheikov, N. McDannold, S. Sharma and K. Hynynen, Ultrasound Med. Biol., 2008, 34, 1093–1104 CrossRef PubMed.
- J. Xue, Z. Zhao, L. Zhang, L. Xue, S. Shen, Y. Wen, Z. Wei, L. Wang, L. Kong, H. Sun, Q. Ping, R. Mo and C. Zhang, Nat. Nanotechnol., 2017, 12, 692–700 CrossRef CAS PubMed.
- F. Caldefie-Chezet, S. Walrand, C. Moinard, A. Tridon, J. Chassagne and M. P. Vasson, Clin. Chim. Acta, 2002, 319, 9–17 CrossRef CAS.
- D. V. Krysko, T. Vanden Berghe, K. D'Herde and P. Vandenabeele, Methods, 2008, 44, 205–221 CrossRef CAS PubMed.
- A. E. Gasperini, S. Sanchez, A. L. Doiron, M. Lyles and G. K. German, Sci. Rep., 2017, 7, 6631 CrossRef PubMed.
- T. Kujawska, W. Secomski, K. Bilmin, A. Nowicki and P. Grieb, Ultrasonics, 2014, 54, 1366–1372 CrossRef CAS PubMed.
- M. T. Burgess, I. Apostolakis and E. E. Konofagou, Phys. Med. Biol., 2018, 63, 065009 CrossRef CAS PubMed.
- W. Xiong, P. Wang, J. Hu, Y. Jia, L. Wu, X. Chen, Q. Liu and X. Wang, Sci. Rep., 2015, 5, 17485 CrossRef PubMed.
- J. Hu, X. Wang, K. Zhang, P. Wang, X. Su, Y. Li, Z. Huang and Q. Liu, Anticancer Drugs, 2014, 25, 174–182 CrossRef CAS PubMed.
- M. Korbelik, J. Photochem. Photobiol., B, 1992, 12, 107–109 CrossRef CAS.
- Y. Li, P. Wang, X. Wang, X. Su and Q. Liu, Ultrasound Med. Biol., 2014, 40, 990–1000 CrossRef PubMed.
- Y. Furusawa, M. A. Hassan, Q. L. Zhao, R. Ogawa, Y. Tabuchi and T. Kondo, Ultrason. Sonochem., 2014, 21, 2061–2068 CrossRef CAS PubMed.
- H. Zhang, K. J. Davies and H. J. Forman, Free Radical Biol. Med., 2015, 88, 314–336 CrossRef CAS PubMed.
- M. S. Siddiqui, M. Francois, M. F. Fenech and W. R. Leifert, Mutat. Res., Rev. Mutat. Res., 2015, 766, 1–19 CrossRef CAS PubMed.
- J. Fillingham, M. C. Keogh and N. J. Krogan, Biochem. Cell Biol., 2006, 84, 568–577 CrossRef CAS PubMed.
- J. Zhang, M. Hummersone, C. S. Matthews, M. F. Stevens and T. D. Bradshaw, Oncology, 2015, 88, 28–48 CrossRef CAS PubMed.
- M. J. Ruiz-Magana, J. M. Rodriguez-Vargas, J. C. Morales, M. A. Saldivia, K. Schulze-Osthoff and C. Ruiz-Ruiz, Int. J. Cancer, 2012, 130, 1195–1207 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |