
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize effect studies in catalysis: a simple surfactant-free synthesis of sub 3 nm Pd nanocatalysts supported on carbon†
Jonathan Quinson *a,
Søren B. Simonsenb,
Luise Theil Kuhnb,
Sebastian Kunzc and
Matthias Arenz*d
*a,
Søren B. Simonsenb,
Luise Theil Kuhnb,
Sebastian Kunzc and
Matthias Arenz*d
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark. E-mail: jonathan.quinson@chem.ku.dk
bDepartment of Energy Conversion and Storage, Technical University of Denmark, Frederiksborgvej 399, 4000 Roskilde, Denmark
cInstitute for Applied and Physical Chemistry, University of Bremen, Leobenerstraße, 28359 Bremen, Germany
dDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: matthias.arenz@dcb.unibe.ch
First published on 4th October 2018
Abstract
Supported Pd nanoparticles are prepared under ambient conditions via a surfactant-free synthesis. Pd(NO3)2 is reduced in the presence of a carbon support in alkaline methanol to obtain sub 3 nm nanoparticles. The preparation method is relevant to the study of size effects in catalytic reactions like ethanol electro-oxidation.
A key achievement in the design of catalytic materials is to optimise the use of resources. This can be done by designing nanomaterials with high surface area due to their nanometre scale. A second achievement is to control and improve catalytic activity, stability and selectivity. These properties are also strongly influenced by size.1–3 To investigate ‘size effects’ it is then important to develop synthesis routes that ensure well-defined particle size distribution, especially towards smaller sizes (1–10 nm).
Metal nanoparticles are widely studied catalysts. In several wet chemical syntheses, NP size can be controlled using surfactants. These additives are, however, undesirable for many applications4,5 since they can block active sites and impair the catalytic activity. They need to be removed in ‘activation’ steps which can negatively alter the physical and catalytic properties of the as-produced NPs. Surfactant-free syntheses are well suited to design catalysts with optimal catalytic activity6 but their widespread use is limited by a challenging size control.3
Palladium (Pd) NPs are important catalysts for a range of chemical transformations like selective hydrogenation reactions and energy applications.7–9 It is however challenging to obtain sub 3 nm Pd NPs, in particular without using surfactants.2 Surfactant-free syntheses are nevertheless attracting a growing interest due to the need for catalysts with higher performances.10–14
Promising surfactant-free syntheses of Pd NPs were recently reported.8,15 The NPs obtained in these approaches are in the size range of 1–2 nm and show enhanced activity for acetylene hydrogenation8 and dehydrogenation of formic acid.15 Enhanced properties are attributed to the absence of capping agents leading to readily active Pd NPs. The reported syntheses consist in mixing palladium acetate, Pd(OAc)2, in methanol and the reduction of the metal complex to NPs occurs at room temperature. The synthesis is better controlled in anhydrous conditions to achieve a fast reaction in ca. 1 hour. Another drawback is that the synthesis must be stopped to avoid overgrowth of the particles. Therefore, a support material needs to be added after the synthesis has been initiated and no simple control over the NP size is achieved.8,15
In this communication a more straightforward surfactant-free synthesis leading to sub 3 nm carbon-supported Pd NPs in alkaline methanol at ambient conditions is presented. A solution of Pd(OAc)2 in methanol undergoes a colour change from orange to dark, indicative of a reduction to metallic Pd, after ca. a day. However, only ca. 1 hour is needed with Pd(NO3)2, Fig. 1 and UV-vis data in Fig. S1.† The fast reduction of the Pd(NO3)2 complex in non-anhydrous conditions is a first benefit of the synthesis presented as compared to previous approaches.
 | ||
| Fig. 1 Pictures of 4 mM Pd metal complexes in methanol without or with a base (as indicated). | ||
For particle suspensions prepared with Pd(OAc)2 or Pd(NO3)2 the NPs agglomerate and quickly sediment leading to large ‘flake-like’ materials. When the reduction of Pd(NO3)2 in methanol is performed in presence of a carbon support and after reduction the solution is centrifuged and washed in methanol, a clear supernatant is observed indicating that no significant amount of NPs are left in methanol. Transmission electron microscope (TEM) analysis confirms that NPs are formed and well-dispersed on the carbon support surface and no unsupported NPs are observed, Fig. 2a. Likely, the reduction of the NPs proceeds directly on the carbon support. However, the size of the NPs is in the range 5–25 nm, which is still a relatively large particle size and broad size distribution.
 | ||
| Fig. 2 TEM micrographs of Pd NPs obtained by stirring 4 mM Pd(NO3)2 in methanol and a carbon support for 3 hours, (a) without NaOH and (b) with 20 mM NaOH. Size distribution histograms are reported in Fig. S4.† The same samples after electrochemical treatments are characterised in (c) and (d) respectively. Size distribution histograms are reported in Fig. S7.† | ||
Assuming a ‘nucleation and growth’ mechanism, the NPs should become larger over time.16 But the reaction is so fast that by stopping the reaction before completion, size control is not achieved and unreacted precious metal is observed, Fig. S2.† To achieve a finer size control and more efficient use of the Pd resources, a base was added to the reaction mixture, e.g. NaOH.3 In alkaline media, the formation of Pd NPs is slower; it takes ca. 60 minutes to observe a dark colour for a 5 mM Pd(NO3)2 solution with a base/Pd molar ratio of 10 in absence of a support, Fig. 1.
Also in alkaline methanol, the NPs agglomerate over time in absence of a support material. However, if the alkaline solution of Pd(NO3)2 is left to stir in presence of a carbon support the desired result is achieved, i.e. Pd NPs with a significantly smaller size and size distribution of ca. 2.5 ± 1.0 nm, Fig. 2b. The NPs homogeneously cover the carbon support and no unsupported NPs are observed by TEM suggesting that the NPs nucleate directly on the carbon surface. Furthermore, the supernatant after centrifugation is clear, indicating an efficient conversion of the Pd(NO3)2 complex to NPs, Fig. S3.† Furthermore, there is no need for an extra reducing agent as in other approaches, for instance in alkaline aqueous solutions.9
The benefits of surfactant-free syntheses of Pd NPs for achieving improved catalytic activity have been demonstrated for heterogeneous catalysis.8,15 Surfactant-free syntheses are also well suited for electrochemical applications where fully accessible surfaces are required for fast and efficient electron transfer. Several reactions for energy conversion benefit from Pd NPs. An example is the electro-oxidation of alcohols,7 in particular ethanol17 (see also Table S1†).
Previous studies optimised the activity of Pd electrocatalysts by alloying,18–20 by using different supports17,21–23 or crystal structures.24,25 Investigating NPs with a diameter less than 3 nm was challenging.2,26,27 The surfactant-free synthesis method presented here allows to further study the size effect on Pd NPs supported on carbon (Pd/C) for electrocatalytic reactions.
In Fig. 3, results for ethanol electro-oxidation in 1 M ethanol solution mixed with 1 M KOH aqueous electrolyte are reported based on cyclic voltammetry (CV) and chronoamperometry (CA) with Pd/C catalysts exhibiting 2 significantly different size distributions. The electrode preparation, the measurement procedure and the sequence of electrochemical treatments are detailed in the ESI.† In order to highlight size effects, we compare geometric and Pd mass normalized currents (Fig. 3a and c) as well as the oxidation currents normalized to the Pd surface area (Fig. 3b).
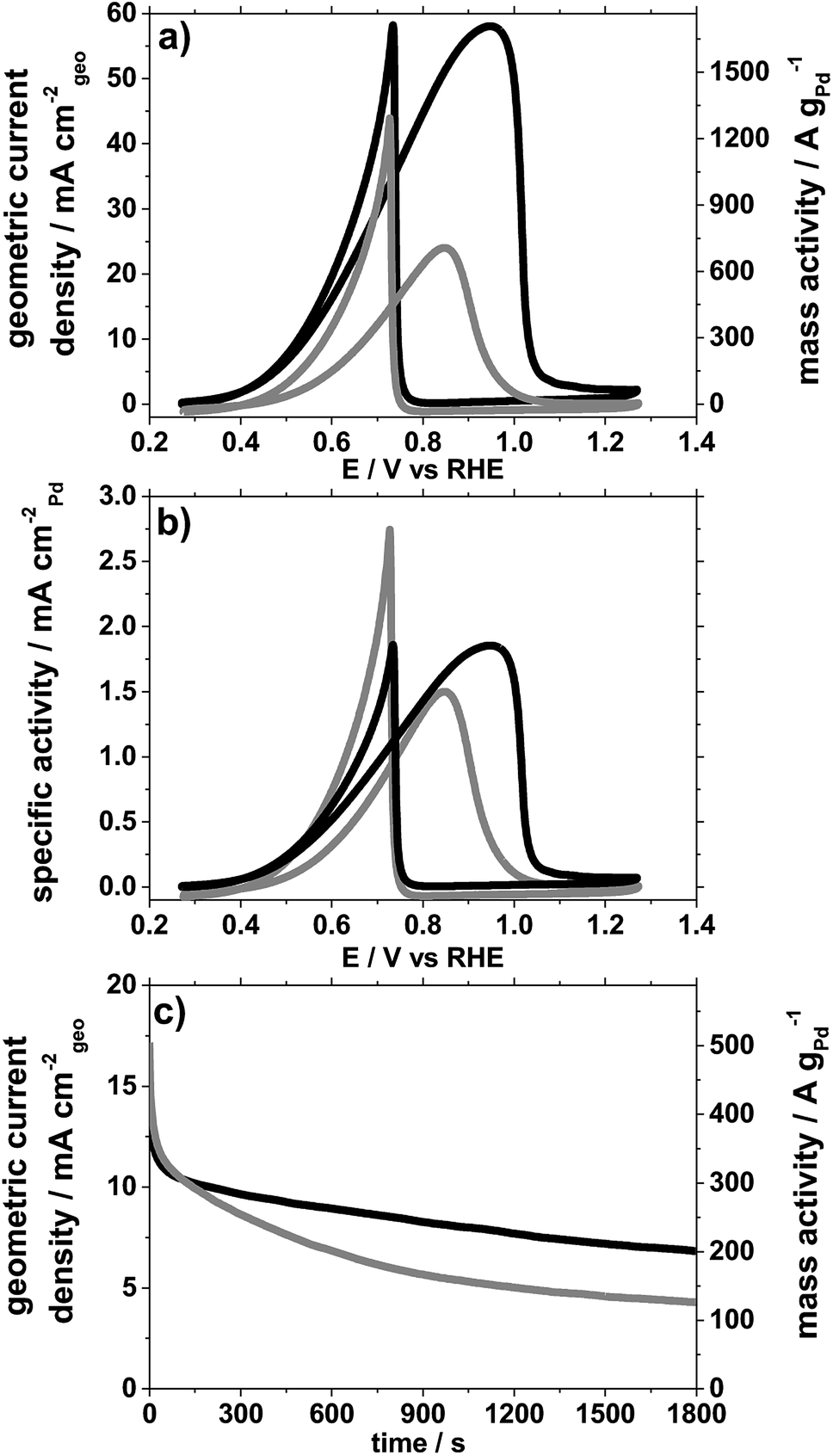 | ||
| Fig. 3 Electrochemical characterisation of carbon supported Pd NPs with 5–25 nm (grey) and 2.5 nm (dark) size in 1 M KOH + 1 M ethanol aqueous electrolyte. (a) 2nd CV before chronoamperometry (CA), (b) current normalised by the electrochemically active surface area of Pd, (c) CA recorded at 0.71 V vs. RHE after 50 cycles between 0.27 and 1.27 V. | ||
It is clearly seen that based on the geometric current density, the smaller Pd NPs exhibit significantly higher currents for ethanol oxidation than the larger NPs. To differentiate if this observation is a sole consequence of the different surface area, the electrochemically active surface area (ECSA) has been estimated based on “blank” CVs (without ethanol) recorded between 0.27 and 1.27 V vs. RHE in pure 1 M KOH aqueous electrolyte and integrating the area of the reduction peak at ca. 0.68 V, Fig. S5.† As conversion factor, 424 μC cm−2 was used.28
Using this method, the smaller NPs with a size around 2.5 nm exhibit an ECSA of 92 m2 g−1 whereas the larger NPs with a size in the range 5–25 nm exhibit an ECSA of 47 m2 g−1, consistent with a larger size. Normalising the ethanol electro-oxidation to these ECSA values instead of the geometric surface area, Fig. 3b, still indicates a size effect. It is clearly seen that the smaller Pd NPs exhibit higher surface specific ethanol oxidation currents, in particular at low electrode potentials. Furthermore, a clear difference in the peak ratios in the CVs is observed. The ratio in current density of the forward anodic peak (jf, around 0.9 V) and the backward cathodic peak (jb, around 0.7 V vs. SCE) is around one for the smaller NPs, whereas it is about 0.5 for the larger NPs. The forward scan corresponds to the oxidation of chemisorbed species from ethanol adsorption. The backward scan is related to the removal of carbonaceous species not fully oxidised in the forward scan. The higher jf/jb ratio therefore confirms that the smaller NPs are more active for ethanol electro-oxidation and less prone to poisoning, e.g. by formation of carbonaceous species that accumulate on the catalyst surface.29,30 This observation is further supported by a chronoamperometry (CA) experiment, Fig. 3c, at 0.71 V performed after continuous cycling (50 cycles between 0.27 and 1.27 V at a scan rate of 50 mV s−1). In the CA testing of the thus aged catalysts at 0.71 V, the ethanol oxidation current on the two catalysts starts at around the same values, however, its decay rate is significantly different. The Pd mass related oxidation currents for the smaller NPs are after 30 minutes almost twice as high (ca. 200 A gPd−1) as for the larger ones (ca. 130 A gPd−1), confirming that the small Pd NPs are less prone to poisoning. In particular a factor up to 4 in the Pd mass related ethanol oxidation currents after 1800 s of continuous operation is achieved compared to a recently characterised commercial Pd catalyst on carbon,20 Table S1.† Despite different testing procedure reported in the literature, it can be concluded from these investigations that the surfactant-free synthesis presented shows promising properties for electrocatalytic ethanol oxidation even after extended cycling.
The extended cycling, however, has different consequences for the two catalysts. For the small (2.5 nm) NPs of the Pd/C catalyst, a massive particle loss, but only moderate particle growth is observed as highlighted in Fig. 2 (see also Fig. S6†). TEM micrographs of the two Pd/C samples recorded before and after the complete testing (CVs and CAs, for details see Fig. S7†) show that the catalyst with small Pd NPs exhibits a pronounced particle loss as well as a particle growth to ca. 6 nm probably due to sintering. By comparison, for the Pd/C catalyst with the large Pd NPs, no significant influence of the testing on particle size or particle density is apparent.
Conclusions
Using alkaline methanol and Pd(NO3)2 a fast, simple, surfactant-free method is presented to prepare at ambient conditions sub 3 nm carbon supported Pd NPs. It is shown that the NPs uniformly cover the carbon support. Furthermore, the preparation method leads to readily active nanocatalysts without the need to remove surfactants. The NPs show promising catalytic activity for electro-oxidation of ethanol when compared with larger nanoparticles obtained without using NaOH. The sub 3 nm particles are more active and more important they are less prone to contamination by unreacted carbonaceous species. Interestingly, even if normalised to the electrochemical surface area, the activity of the small Pt NPs is still higher.Further studies of the effect of NaOH concentration and nature of the carbon support could bring more control on the size and possibly morphology of the nanoparticles. It is general challenge to follow the reduction of Pd2+ to Pd0 and the effect of a base on the nucleation and growth.2 However the simplicity of the method proposed should allow in situ studies to get a deeper understanding of the role and influence of the Pd(NO3)2 precursor on the reaction mechanism and kinetics.
The simple and relatively fast synthesis presented is promising to design, study and optimise a range of Pd based catalysts for electrochemical purposes (alcohol oxidation, O2 reduction, etc.) but also heterogeneous catalysis (hydrogenation, coupling reactions, etc.).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the Villum Foundation in form of a block stipend. J. Q. has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 703366 (SELECTRON).Notes and references
- E. Antolini, Appl. Catal., B, 2016, 181, 298–313 CrossRef CAS.
- S. Zhang, B. Jiang, K. Jiang and W.-B. Cai, ACS Appl. Mater. Interfaces, 2017, 9, 24678–24687 CrossRef CAS PubMed.
- J. Quinson, M. Inaba, S. Neumann, A. Swane, J. Bucher, S. Simonsen, L. Theil Kuhn, J. Kirkensgaard, K. Jensen, M. Oezaslan, S. Kunz and M. Arenz, ACS Catal., 2018, 8, 6627–6635 CrossRef CAS.
- Z. Q. Niu and Y. D. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- W. X. Huang, Q. Hua and T. Cao, Catal. Lett., 2014, 144, 1355–1369 CrossRef CAS.
- J. Quinson, S. Neumann, T. Wannmacher, L. Kacenauskaite, M. Inaba, J. Bucher, F. Bizzotto, S. B. Simonsen, L. T. Kuhn, D. Bujak, A. Zana, M. Arenz and S. Kunz, Angew. Chem., Int. Ed., 2018, 57, 12338–12341 CrossRef CAS PubMed.
- H. Meng, D. Zeng and F. Xie, Catalysts, 2015, 5, 1221–1274 CrossRef CAS.
- P. D. Burton, T. J. Boyle and A. K. Datye, J. Catal., 2011, 280, 145–149 CrossRef CAS.
- Q. L. Zhu, N. Tsumori and Q. Xu, Chem. Sci., 2014, 5, 195–199 RSC.
- H. H. Duan, D. S. Wang and Y. D. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- C. Jimenez-Gonzalez, P. Poechlauer, Q. B. Broxterman, B. S. Yang, D. A. Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorrnan, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900–911 CrossRef CAS.
- C. A. Charitidis, P. Georgiou, M. A. Koklioti, A.-F. Trompeta and V. Markakis, Manuf. Rev., 2014, 1, 11 Search PubMed.
- S. Olveira, S. P. Forster and S. Seeger, J. Nanotechnol., 2014, 324089 Search PubMed.
- D. S. Zhang, B. Goekce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- Q. L. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 11743–11748 CrossRef CAS PubMed.
- M. Harada, N. Tamura and M. Takenaka, J. Phys. Chem. C, 2011, 115, 14081–14092 CrossRef CAS.
- C. Xu, L. Cheng, P. Shen and Y. Liu, Electrochem. Commun., 2007, 9, 997–1001 CrossRef CAS.
- S. Arulmani, S. Krishnamoorthy, J. J. Wu and S. Anandan, Electroanalysis, 2017, 29, 433–440 CrossRef CAS.
- M. D. Obradovitc, Z. M. Stancic, U. C. Lacnjevac, V. V. Radmilovic, A. Gavrilovic-Wohlmuther, V. R. Radmilovic and S. L. Gojkovic, Appl. Catal., B, 2016, 189, 110–118 CrossRef.
- L. Chen, L. Lu, H. Zhu, Y. Chen, Y. Huang, Y. Li and L. Wang, Nat. Commun., 2017, 8, 14136 CrossRef CAS PubMed.
- Y. H. Qin, H. H. Yang, X. S. Zhang, P. Li and C. A. Ma, Int. J. Hydrogen Energy, 2010, 35, 7667–7674 CrossRef CAS.
- H. Yang, X. Zhang, H. Zou, Z. Yu, S. Li, J. Sun, S. Chen, J. Jin and J. Ma, ACS Sustainable Chem. Eng., 2018, 6, 7918–7923 CrossRef CAS.
- Z.-R. Yang, S.-Q. Wang, J. Wang, A.-J. Zhou and C.-W. Xu, Sci. Rep., 2017, 7, 15479 CrossRef PubMed.
- R. C. Cerritos, M. Guerra-Balcazar, R. F. Ramirez, J. Ledesma-Garcia and L. G. Arriaga, Materials, 2012, 5, 1686–1697 CrossRef CAS.
- X.-Y. Ma, Y. Chen, H. Wang, Q.-X. Li, W.-F. Lin and W.-B. Cai, ChemComm, 2018, 54, 2562–2565 RSC.
- C. N. Wen, Z. P. Li, C. Y. Cao, Y. Q. Wang, P. Z. Guo and X. S. Zhao, RSC Adv., 2016, 6, 91991–91998 RSC.
- J. W. Ma, J. Wang, G. H. Zhang, X. B. Fan, G. L. Zhang, F. B. Zhang and Y. Li, J. Power Sources, 2015, 278, 43–49 CrossRef CAS.
- M. Lukaszewski, M. Soszko and A. Czerwinski, Int. J. Electrochem. Sci., 2016, 11, 4442–4469 CrossRef CAS.
- L. J. Zhang and D. G. Xia, Appl. Surf. Sci., 2006, 252, 2191–2195 CrossRef CAS.
- R. Manoharan and J. B. Goodenough, J. Mater. Chem., 1992, 2, 875–887 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06912c |
| This journal is © The Royal Society of Chemistry 2018 |