DOI:
10.1039/C8RA06854B
(Paper)
RSC Adv., 2018,
8, 31427-31439
Versiquinazolines L–Q, new polycyclic alkaloids from the marine-derived fungus Aspergillus versicolor†
Received
16th August 2018
, Accepted 2nd September 2018
First published on 7th September 2018
Abstract
Further chemical examination of a coral-associated fungus Aspergillus versicolor LZD-14-1 by the PHLC-DAD detection resulted in the isolation of six new polycyclic alkaloids, namely versiquinazolines L–Q (1–6). Their structures were determined by extensive analyses of spectroscopic data, including quantum ECD calculation and X-ray single crystal diffraction for the assignment of absolute configurations. Versiquinazoline L bearing a D-Ala residue and versiquinazoline M containing an L-serine residue are rarely found in the fumiquinazoline-type alkaloids, while versiquinazoline P displayed an unusual scaffold with a spiro-γ-lactone. Versiquinazolines P and Q exhibited significant inhibition against thioredoxin reductase (TrxR) with IC50 values of 13.6 ± 0.6 and 12.2 ± 0.7 μM, which showed higher activity than the positive control curcumin (IC50 = 25 μM). The weak cytotoxicity and potent inhibition toward TrxR suggested that versiquinazolines P and Q are potential for microenvironmental regulation of tumor progression and metastasis.
1. Introduction
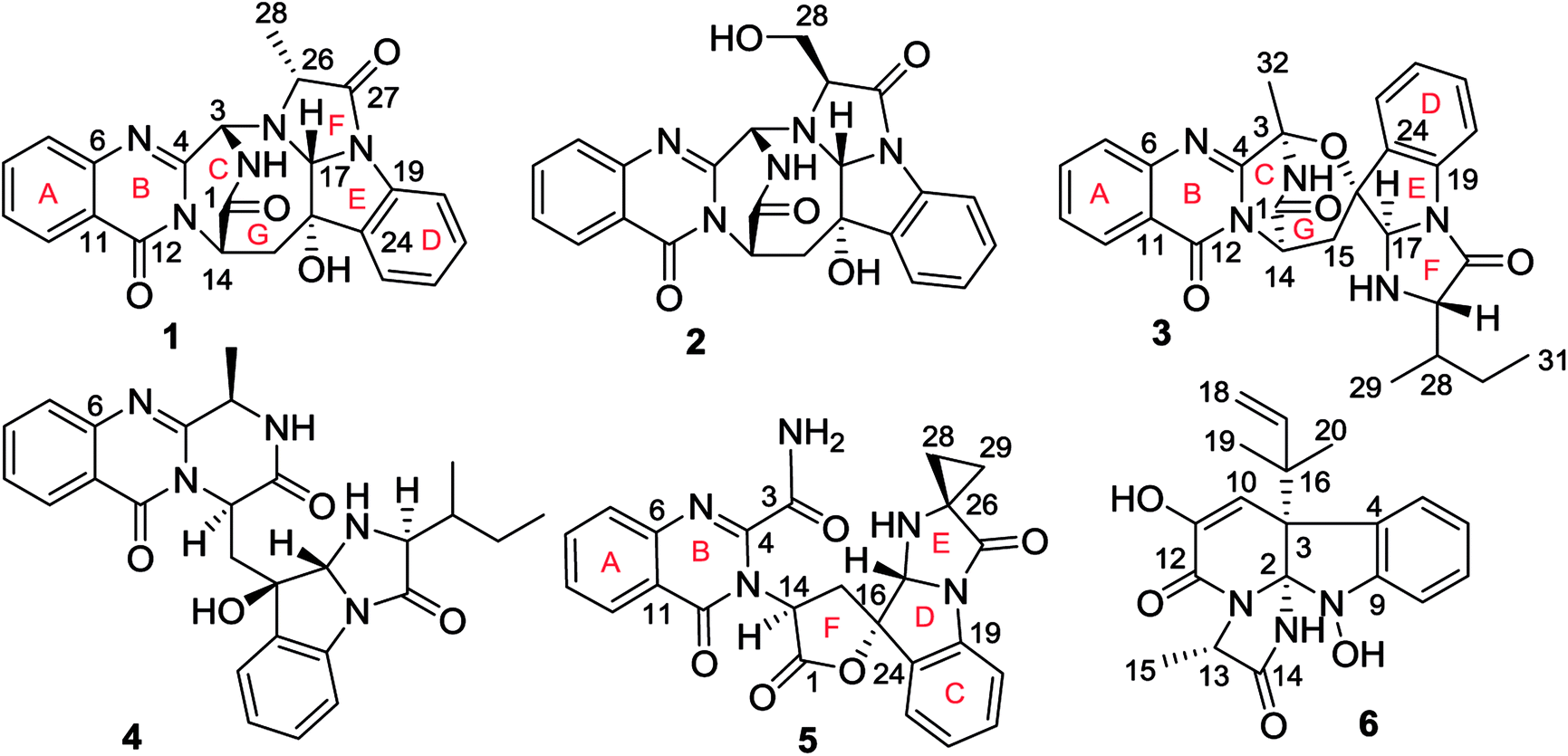
Fungal fumiquinazoline-type alkaloids are a group of structurally unique natural products with a diverse array of scaffolds that are widely distributed in fungi from terrestrial and marine origins.1–9 These typical compounds are nonribosomally assembled by a pyrazino[2,1-b]quinazoline-3,6-dione nucleus which incorporated various amino acids such as tryptophan and histidine to derive diverse scaffolds.10–12 Marine fungi are recognized to be a rich source to produce fumiquinazoline relevant structural diversity, while the marine hosts inhabited by the fungi that are able to produce the fumiquinazoline-type alkaloids covered a wide range of organisms, including fish, sponges, and corals, as well as marine mangrove plants. Since fumiquinazolines A–G were isolated from marine fish associated Aspergillus fumigatus in early 1990, a certain numbers of analogues including fumiquinazolines H–T were separated from diverse endophytic fungi, including Acremonium sp. in tunicate, A. fumigatus in soft coral, Scopulariopsis sp. in gorgonian, Aspergillus sp. in sponge and marine-submerged wood. Some fumiquinazoline derivatives displayed the cytotoxicity against human tumor cell lines, antibacterial and insecticidal effects as well as the interaction with protein targets such as breast cancer resistance protein inhibition. In previous work, we found that a coral-associated fungus A. versicolor LZD-14-1 is the source to produce fumiquinazoline-type alkaloids, while eleven new alkaloids namely versiquinazolines A–K were isolated.13 In present work, a HPLC-MS guided examination further isolated six minor fumiquinazoline-type alkaloids, namely versiquinazolines L–Q (1–6) (Fig. 1). Herein, we report the structural elucidation of the new alkaloids and their inhibition against thioredoxin reductase (TrxR).
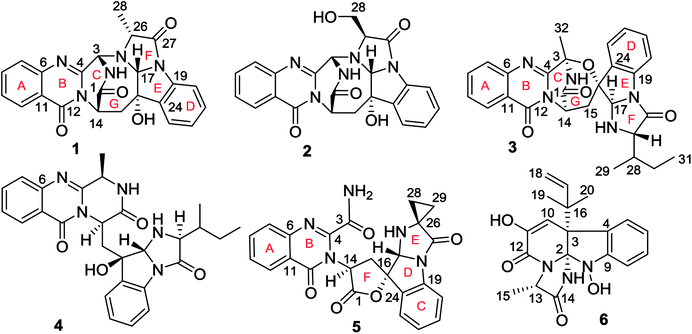 |
| | Fig. 1 Structures of versiquinazolines L–Q (1–6). | |
2. Experimental section
2.1. General procedure
Specific rotations were measured by an Autopol III automatic polarimeter (Rudolph Research Co., Ltd.). IR spectra were recorded on a Thermo Nicolet Nexus 470 FT-IR spectrometer. UV spectra were measured on a Cary 300 spectrometer. ECD spectra were measured on a JASCO J-810/J-815 spectropolarimeter. The 1H and 13C NMR spectra were recorded on a Bruker Avance-400FT NMR spectrometer. HRESIMS spectra were obtained on a Waters Xevo G2 Q-TOF spectrometer fitted with an ESI source. HF254 silica gel for TLC was obtained from Qingdao Marine Chemistry Co. Ltd. Sephadex LH-20 was purchased from Pharmacia. Semi-preparative HPLC was performed on an Alltech 426 pump using UV detector, and the Prevail C18 column (semipreparative, 5 μm) was used for separation.
2.2. Identification of fungal strain and fermentation
Fungus Aspergillus versicolor LZD-14-1 was isolated from the gorgonian Pseudopterogorgia sp. (LZD-14), which was collected from the South China Sea, in May 2015. The strain was identified by comparing the morphological characteristics and analysis of the ITS region of the rDNA sequence with those of standard records (GeneBank KX254916). The morphological examination was performed by scrutinizing the fungal culture, the mechanism of spore production, and the characteristics of the spores. For inducing sporulation, the fungal strains were separately inoculated onto potato dextrose agar. All experiments and observations were repeated at least twice leading to the identification of the strain LZD-14-1 as A. versicolor. The strain LZD-14-1 was deposited at the State Key Laboratory of Natural and Biomimetic Drugs, Peking University, China.
The large scale fermentation was carried out in Fernbach flasks (50 × 500 mL), each containing 80 g of rice. Distilled artificial seawater (NaCl 26.726 g, MgCl2 2.26 g, MgSO4 3.248 g, CaCl2 1.153 g, NaHCO3 0.198 g, KCl 0.721 g, NaBr 0.058 g, H3BO3 0.058 g, Na2SiO3 0.0024 g, Na2Si4O9 0.0015 g, H3PO4 0.002 g, Al2Cl6 0.013 g, NH3 0.002 g, LiNO3 0.0013 g, H2O 1 L) (100 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.
2.3. Extraction and isolation
The fermented material was extracted successively with EtOAc (3 × 500 mL). After evaporation under vacuum, the EtOAc extract (4.0 g) was subjected to a vacuum liquid chromatography (silica gel, 200–300 mesh) with PE/EtOAc (from 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 0:1, gradient) as an eluent to obtain four fractions (F1 to F4). Fraction F3 (2.0 g) was chromatographed on a Sephadex LH-20 column eluting with MeOH to collect five subfractions (SF3a–SF3e). SF3c (150 mg), which was not examined previously,13 was detected by the HPLC-DAD (UV) and ESIMS for the molecular profile, showing the HPLC peaks with UV absorptions ranging 215–320 nm and the molecular weights ranging m/z 356–486 [M + H]+. These data were characteristic of the fumiquinazoline-type alkaloids, relevant to versiquinazolines. Chromatographic separation of SF3c (150 mg) by semipreparative reversed phase (RP) HPLC using MeCN/H2O = 45:55 (2 mL min−1) as a mobile phase to obtain 2 (2.4 mg), 1 (3.5 mg), 6 (3.8 mg), 5 (3.5 mg), 3 (3.2 mg), and 4 (1.6 mg).
:1 to 0:1, gradient) as an eluent to obtain four fractions (F1 to F4). Fraction F3 (2.0 g) was chromatographed on a Sephadex LH-20 column eluting with MeOH to collect five subfractions (SF3a–SF3e). SF3c (150 mg), which was not examined previously,13 was detected by the HPLC-DAD (UV) and ESIMS for the molecular profile, showing the HPLC peaks with UV absorptions ranging 215–320 nm and the molecular weights ranging m/z 356–486 [M + H]+. These data were characteristic of the fumiquinazoline-type alkaloids, relevant to versiquinazolines. Chromatographic separation of SF3c (150 mg) by semipreparative reversed phase (RP) HPLC using MeCN/H2O = 45:55 (2 mL min−1) as a mobile phase to obtain 2 (2.4 mg), 1 (3.5 mg), 6 (3.8 mg), 5 (3.5 mg), 3 (3.2 mg), and 4 (1.6 mg).
Versiquinazoline L (1). Colorless crystal in acetone/MeOH, mp 286–287 °C, [α]20D + 252 (c 0.1, MeOH); UV (MeOH) λmax 206, 228, 258, 305, 316 nm; ECD (c 1.8 × 10−4 M, MeOH) λmax (Δε) 304 (+5.15), 257 (+6.95), 234 (+20.2), 206 (−34.0); IR (KBr) νmax 3385, 2920, 2851, 1686, 1610, 1471, 1207, 1140 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 430.1499 [M + H]+ (calcd for C23H20N5O4, 430.1510).
Table 1 1H NMR data of 1–6 in DMSO-d6a
| |
1 |
2 |
3 |
4 |
5 |
6 |
| Recorded at 400 MHz. Chemical shifts are in ppm, coupling constants J in Hz. |
| 3 |
5.44, d (4.5) |
5.67, d (4.5) |
|
4.94, q (6.5) |
|
|
| 5 |
|
|
|
|
|
7.45, d (7.6) |
| 6 |
|
|
|
|
|
6.91, t (7.6) |
| 7 |
7.67, d (8.1) |
7.67, d (8.1) |
7.76, d (8.1) |
7.66, d (8.0) |
7.82, d (8.1) |
7.17, t (7.6) |
| 8 |
7.83, dd (7.7, 8.1) |
7.82, dd (7.6, 8.1) |
7.90, dd (7.8, 8.1) |
7.83, dd (6.9, 8.0) |
7.97, dd (7.7, 8.1) |
6.80, d (7.6) |
| 9 |
7.55, dd (7.7, 8.0) |
7.54, dd (7.6, 8.0) |
7.63, dd (7.8, 8.1) |
7.53, dd (6.9, 8.0) |
7.69, dd (7.7, 8.0) |
|
| 10 |
8.16, d (8.0) |
8.16, d (8.0) |
8.20, d (8.1) |
8.12, d (8.0) |
8.25, d (8.0) |
5.16, s |
| 13 |
|
|
|
|
|
4.20, q (6.8) |
| 14 |
5.33, d (6.7) |
5.37, d (6.9) |
5.36, d (5.2) |
5.57, dd (4.4, 9.4) |
5.49, dd (9.4, 10.5) |
|
| 15 |
2.30, dd (1.4, 14.7) |
2.31, d (14.9) |
1.92, d (14.8) |
1.82, dd (4.4, 14.9) |
2.90, dd (9.4, 13.1) |
1.43, d (6.8) |
| |
3.11, dd (6.7, 14.7) |
3.12, dd (6.9, 14.9) |
2.98, dd (5.2, 14.8) |
2.58, dd (9.4, 14.9) |
3.44, dd (10.5, 13.1) |
|
| 17 |
4.98, d (1.3) |
5.17, s |
5.69, d (5.9) |
5.25, d (6.6) |
5.55, d (8.7) |
6.14, dd (10.8, 17.4) |
| 18 |
|
|
|
|
|
5.05, d (17.4); 5.00, d (10.8) |
| 19 |
|
|
|
|
|
1.21, s |
| 20 |
7.35, d (7.9) |
7.32, d (8.5) |
7.38, d (7.7) |
7.34, d (7.8) |
7.46, d (7.7) |
1.21, s |
| 21 |
7.31, dd (7.7, 7.9) |
7.32, dd (7.8, 8.5) |
7.31, dd (7.4, 7.7) |
7.27, dd (7.6, 7.8) |
7.57, dd (7.5, 7.7) |
|
| 22 |
7.09, t (7.7) |
7.14, dd (7.8, 8.5) |
7.04, dd (7.4, 7.7) |
7.09, dd (7.5, 7.6) |
7.40, dd (7.5, 8.0) |
|
| 23 |
7.36, d (7.7) |
7.31, d (8.5) |
7.26, d (7.7) |
7.80, d (7.5) |
7.66, d (8.0) |
|
| 26 |
4.25, dq (1.3, 6.5) |
4.11, dd (2.4, 4.6) |
3.66, dd (1.3, 4.5) |
3.60, dd (1.8, 4.3) |
|
|
| 28 |
1.43, d (6.5) |
3.79, ddd (2.4, 4.9, 12.2) |
1.83, m |
1.70, m |
0.93, m; 0.72, m |
|
| |
|
4.07, ddd (4.6, 4.9, 12.2) |
|
|
|
|
| 29 |
|
|
1.05, d (6.8) |
0.94, d (6.8) |
1.24, m; 1.06, m |
|
| 30 |
|
|
1.58, m; 1.40, m |
1.45, m; 1.26, m |
|
|
| 31 |
|
|
0.96, t (7.3) |
0.85, t (7.4) |
|
|
| 32 |
|
|
1.90, s |
1.58, d (6.5) |
|
|
| NH-2 |
9.19, d (4.5) |
9.00, d (4.5) |
9.74, s |
8.61, s |
8.30, s; 8.74, s |
|
| NH-25 |
|
|
2.61, dd (1.3, 5.9) |
3.46, dd (1.8, 6.6) |
4.80, d (8.7) |
9.11, s |
| OH-16 |
5.23, d (1.4) |
5.28, s |
|
5.67, s |
|
8.79, br s |
| OH-28 |
|
4.93, t (4.9) |
|
|
|
|
Table 2 13C NMR data of 1–6 in DMSO-d6a
| |
1 |
2 |
3 |
4 |
5 |
6 |
| Recorded at 100 MHz. |
| 1 |
170.4 |
170.0, C |
168.6, C |
169.0, C |
170.2, C |
|
| 2 |
|
|
|
|
|
101.9, C |
| 3 |
65.4, CH |
64.4, CH |
84.2, C |
48.6, CH |
163.2, C |
51.7, C |
| 4 |
150.2, C |
149.9, C |
151.5, C |
153.3, C |
148.9, C |
126.7, C |
| 5 |
|
|
|
|
|
124.2, CH |
| 6 |
147.5, C |
147.2, C |
146.6, C |
146.6, C |
145.7, C |
121.6, CH |
| 7 |
127.2, CH |
127.2, CH |
127.9, CH |
127.0, CH |
127.7, CH |
127.5, CH |
| 8 |
134.6, CH |
134.6, CH |
134.7, CH |
134.5, CH |
135.6, CH |
110.7, CH |
| 9 |
126.8, CH |
126.8, CH |
127.5, CH |
126.8, CH |
128.7, CH |
148.3, C |
| 10 |
126.1, CH |
126.2, CH |
126.4, CH |
126.5, CH |
126.5, CH |
101.9, CH |
| 11 |
120.0, C |
120.2, C |
120.7, C |
120.1, C |
121.1, C |
142.7, C |
| 12 |
159.8, C |
159.9, C |
158.9, C |
160.3, C |
160.1, C |
159.4, C |
| 13 |
|
|
|
|
|
52.8, CH |
| 14 |
53.7, CH |
53.3, CH |
52.8, CH |
51.8, CH |
54.9, CH |
173.7, C |
| 15 |
34.7, CH2 |
33.7, CH2 |
33.7, CH2 |
35.5, CH2 |
30.9, CH2 |
14.1, CH3 |
| 16 |
74.8, C |
77.4, C |
86.4, C |
80.1, C |
86.3, C |
42.1, C |
| 17 |
78.4, CH |
82.0, CH |
88.5, CH |
88.2, CH |
83.5, CH |
144.5, CH |
| 18 |
|
|
|
|
|
112.7, CH2 |
| 19 |
136.0, C |
139.0, C |
136.6, C |
137.3, C |
142.0, C |
24.4, CH3 |
| 20 |
113.9, CH |
116.6, CH |
114.4, CH |
115.1, CH |
117.5, CH |
23.6, CH3 |
| 21 |
129.6, CH |
129.4, CH |
129.8, CH |
128.9, CH |
131.6, CH |
|
| 22 |
124.6, CH |
125.4, CH |
125.2, CH |
124.7, CH |
126.0, CH |
|
| 23 |
124.7, CH |
123.9, CH |
126.3, CH |
125.5, CH |
125.3, CH |
|
| 24 |
139.0, C |
137.9, C |
137.4, C |
138.7, C |
131.6, C |
|
| 25 |
62.5, CH |
67.6, CH |
68.0, CH |
68.3, CH |
45.9, C |
|
| 26 |
165.7, C |
170.2, C |
171.6, C |
172.3, C |
177.8, C |
|
| 27 |
14.8, CH3 |
59.2, CH2 |
38.1, CH |
37.8, CH |
17.0, CH2 |
|
| 28 |
|
|
15.0, CH3 |
15.1, CH3 |
11.8, CH2 |
|
| 29 |
|
|
24.5, CH2 |
24.50, CH2 |
|
|
| 30 |
|
|
11.3, CH3 |
11.6, CH3 |
|
|
| 31 |
|
|
23.8, CH3 |
16.6, CH3 |
|
|
Versiquinazoline M (2). Colorless oil, [α]20D + 238 (c 0.1, MeOH); UV (MeOH) λmax 204, 227, 259, 303, 316 nm; ECD (c 2.0 × 10−4 M, MeOH) λmax (Δε) 304 (+6.36), 260 (+5.66), 234 (+22.04), 206 (−38.57); IR (KBr) νmax 3363, 2920, 2851, 1661, 1609, 1204, 1183, 1138 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 446.1455 [M + H]+ (calcd for C23H20N5O5, 446.1459).
Versiquinazoline N (3). White powder, [α]20D +6.7 (c 0.1, MeOH); UV (MeOH) λmax 203, 227, 303 nm; ECD (c 4.1 × 10−4 M, MeOH) λmax (Δε) 259 (−7.50), 237 (−8.79), 215 (+38.56); IR (KBr) νmax 2970, 1737, 1724, 1381, 1367, 1229, 1217, 1022 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 486.2136 [M + H]+ (calcd for C27H28N5O4, 486.2136).
Versiquinazoline O (4). White powder, [α]20D −37.2 (c 0.1, MeOH); UV (MeOH) λmax 204, 233, 306 nm; ECD (c 5.1 × 10−4 M, MeOH) λmax (Δε) 233 (−15.55), 211 (+20.15); IR (KBr) νmax 3399, 2923, 2852, 1714, 1228, 1018 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 470.2187 [M + H]+ (calcd for C27H30N5O4, 488.2292).
Versiquinazoline P (5). Colorless oil, [α]20D −116 (c 0.4, MeOH); UV (MeOH) λmax 202, 228, 277; ECD (c, 3.8 × 10−4 M, MeOH) λmax (Δε) 298 (−2.16), 274 (−3.33), 232 (−9.94), 212 (+9.27); IR (KBr) νmax 2921, 2851, 1686, 1467, 1377, 1218, 760 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 458.1447 [M + H]+ (calcd for C24H20N5O5, 458.1459); 480.1275 [M + Na]+ (calcd for C24H19N5O5Na, 480.1278).
Versiquinazoline Q (6). Colorless oil, [α]20D +53.7 (c 0.1, MeOH); UV (MeOH) λmax 207, 228, 294 nm; ECD (c 7.3 × 10−4 M, MeOH) λmax (Δε) 280 (−4.81), 253 (+3.78), 230 (−12.6); IR (KBr) νmax 3386, 2928, 1662, 1609, 1429, 1225, 1182 cm−1; 1H and 13C NMR data, see Tables 1 and 2; HRESIMS m/z 356.1601 [M + H]+ (calcd for C19H22N3O4, 356.1605).
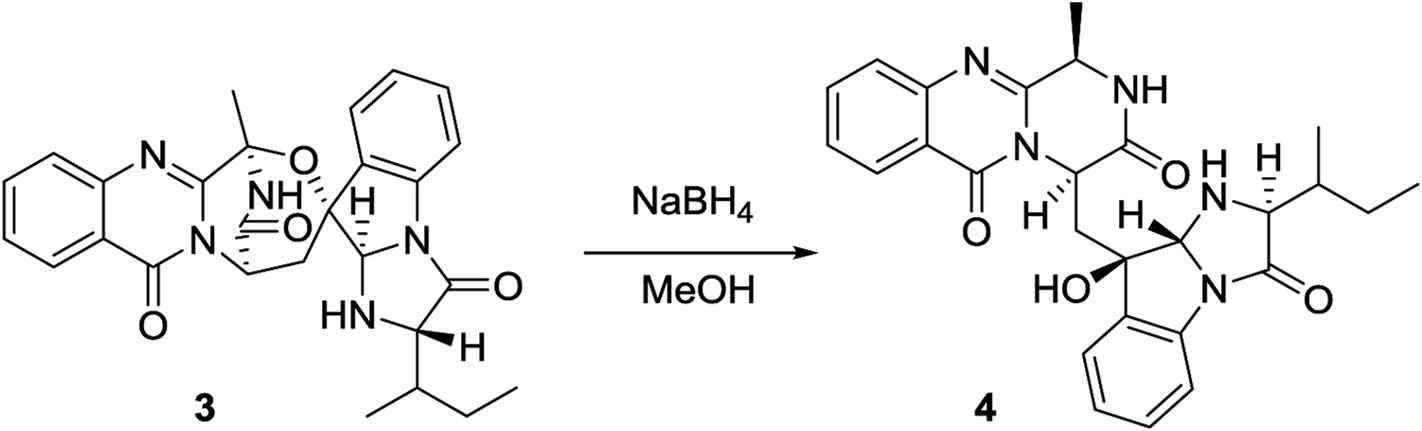
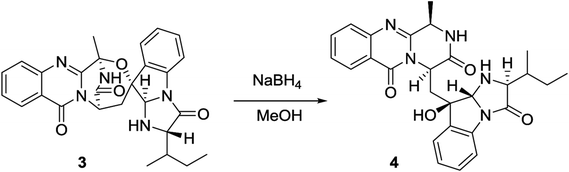
2.4. Hydrogenolysis of 3
To the MeOH (0.5 mL) solution of 3 (1.0 mg), NaBH4 (0.5 mg) was added to stir at rt for 5 min. The reacted mixture was subject to semi-preparative HPLC to afford a major product, which was identified to be 4 by the 1H NMR data, HPLC retention time, and specific rotation.
2.5. X-ray crystallographic analysis
Crystal data for 1: C23H19N5O4, M = 429.43 g mol−1, orthorhombic, space group P212121, a = 7.44940(7) Å, α = β = γ = 90.00°, b = 13.81570(14) Å, c = 18.11204(16) Å, V = 1864.07(3) Å3, Z = 4, T = 100 K, μ(CuKα) = 0.893 mm−1, Dcalc. = 1.530 g cm−3, 15719 reflections measured (8.048° ≤ 2Θ ≤ 137.254°), 3344 unique (Rint = 0.0239, Rsigma = 0.0129) which were used in all calculations. A crystal of dimensions 0.58 × 0.5 × 0.3 mm3 was selected for measurements. The final R1 was 0.0245 (I > 2σ(I)) and wR2 was 0.0625 (all data). The Flack parameter is 0.07(5). The crystal structures of 1 was solved using the program shelex-97 (G. M. Sheldrick, SHELXS 97, University of Gottingen, Germany, 1997) and subsequent Fourier difference techniques, and refined anisotropically by full matrix least-squares on F2 using SHELEX-97 (G. M. Sheldrick, SHELEX, version 6.10, Bruker AXS Inc., Madison, Wisconsin, USA, 2000). The crystallographic data for the structures of 1 has been deposited in Cambridge Crystallographic Data Center (CCDC numbers: 1813311).
2.6. Evaluation of the TrxR inhibitory activities
The TrxR inhibition assay was performed as described.13
2.7. Marfey's method
Compound 1 (0.5 mg) was placed in a 5 mL conical vial containing HCl (6 M, 1 mL), and the sealed vial was heated at 110 °C for 20 h. After evaporation of the solvent, H2O (100 μL) was added. Then NaHCO3 (1 M, 50 μL) and 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide (L-FDAA, 1%, 100 μL) in acetone were added to the hydrolysis solution, and the sealed vial was heated at 40 °C for 2 h. Then HCl (2 M, 20 μL) was added to the reaction mixture to stop the reaction. After evaporation, the reaction products were dissolved in MeOH for HPLC analysis on a Thermo BDS Hypersil C18 column (150 mm × 4.6 mm, 5 μm). MeOH–H2O (0.5% H3PO4) gradient: 0 min: 30% MeOH–H2O; 40 min: 70% MeOH–H2O, with a flow rate of 1 mL min−1. UV detection was performed at a wavelength of 340 nm. Compounds 2–4 were performed by the same protocol as 1.
2.8. Calculation of ECD data and 13C NMR chemical shifts
The ECD spectra were simulated by the overlapping Gaussian function (half the bandwidth at 1/e peak height, 0.16–0.3 eV). The simulated spectra of the two lowest energy conformers for each structure were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy (ΔG). Theoretical ECD spectra of the corresponding enantiomers were obtained by directly inversing the ECD spectra of compounds.
For 13C NMR calculation, the conformers were re-optimized using DFT at the B3LYP/6-31G* level in gas phase by the GAUSSIAN 09 program. The 13C NMR shielding constants of 1 and 5 were calculated with the GIAO method at MPW1PW91/6-31G(d,p) level in gas phase. The computational 13C NMR were finally obtained by linear regression analysis method.
3. Results and discussion
3.1. Structure elucidation of new alkaloids
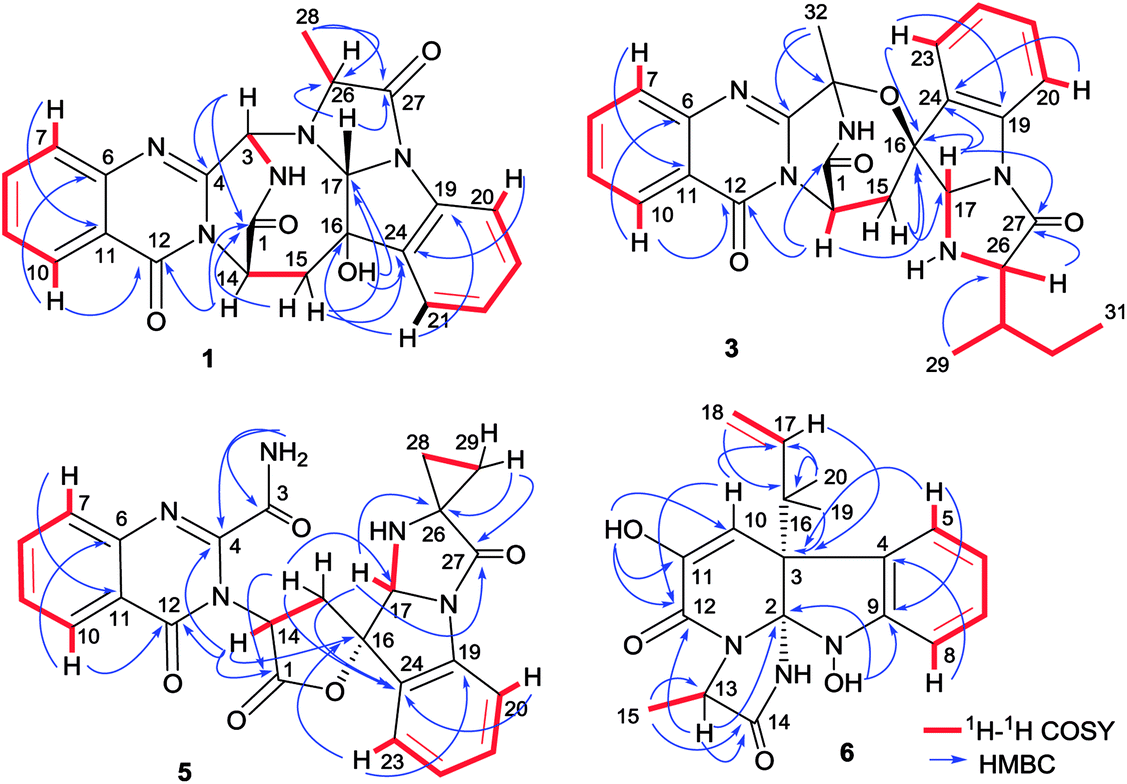
Versiquinazoline L (1) has the molecular formula of C23H19N5O4, as determined by the HRESIMS and 13C NMR data, indicating 17 degrees of unsaturation. The NMR data (Tables 1 and 2) featured a fumiquinazoline-type alkaloid, structurally related to the coexisted known compound cottoquinazoline B.13 Diagnostic 2D NMR (HMBC, HMQC and 1H–1H COSY) data (Fig. 2) established two substructures, involving a tricyclic pyrazinoquinazolinedione (from rings A to C) and an imidazoindolone segment (from rings D to F). Their linkage was determined by the COSY relationship between H2-15 (δH 2.30, 3.11) and H-14 (δH 5.33, d, J = 6.7 Hz) in addition to the HMBC correlations from H2-15 to the carbons C-1 (δC 170.4) and C-14 (δC 53.7) of pyrazinoquinazolinedione unit and to C-16 (δC 74.8), C-17 (δC 78.4), and C-24 (δC 139.0) of imidazoindolone unit. These data allowed the connection of C-14 and C-16 by a methylene bridge CH2-15. Moreover, the HMBC correlation from H-3 (δH 5.44, d, J = 4.5 Hz) to C-17 and C-26 (δC 62.5) (Fig. 2) linked C-3 (δC 65.4) and N-25 to form a C–N bond. Thus, the planar structure of 1 was identical to cottoquinazoline B.7 The NOESY correlations (Fig. 3) such as the interactions from H-17 to H-15b (δH 2.30) and H-26 and between OH-16 (δH 5.23) and H-15a (δH 3.11) indicated the opposite face of OH-16 toward H-17, while H-17 and H-26 were in cofacial orientation. The lactam ring C was oriented in the same face as H-17 due to the observation of the NOE correlations from NH (δH 9.19) to H-17 and H-26. In addition, calculations of the 13C NMR chemical shifts were performed using the Gaussian 03 software package. Geometry optimizations and frequency calculations were performed at the B3LYP/6-31G(d) DFT level. All geometries obtained were optimized to true minima as evidenced by the absence of imaginary frequencies. GIAO-based 13C NMR chemical shifts were calculated at the MPW1PW91/6-31G(d,p) level in gas phase.14,15 Reported shifts are based on a benzene reference (128.5 ppm) calculated at the same level of theory. To determine the accuracy of this computational model as applied to the analogues, 13C NMR chemical shifts were initially calculated for 1 and cottoquinazoline B, and were compared to the experimental data (Table 3). The calculated chemical shifts were in excellent agreement with those determined experimentally. For compound 1, the maximum absolute difference between predicted and observed shifts was 2.5 ppm (C-27), while that for cottoquinazoline B was 3.5 ppm. These data further supported the relative configuration of 1. Finally, the X-ray single-crystal diffraction using Flack parameter (0.07(5)) assigned the absolute configurations of 1 to be 3S, 14S, 16S, 17R, and 26R (Fig. 4). It is noted that the D-Ala residue assembled fumiquinazoline-type alkaloid is rarely found from nature.
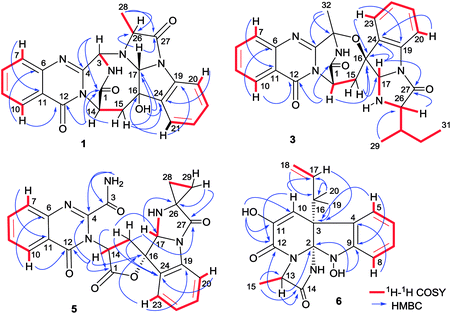 |
| | Fig. 2 Key 1H–1H COSY and HMBC correlations of 1, 3, 5 and 6. | |
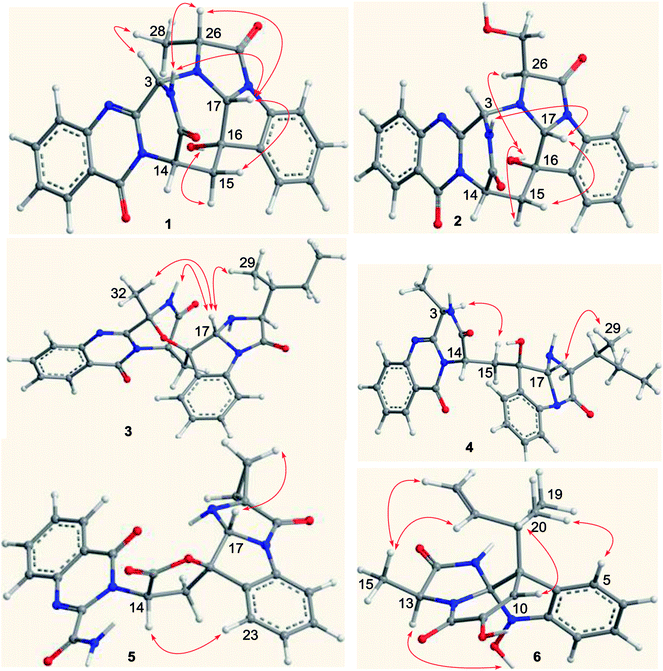 |
| | Fig. 3 Key NOE correlations of 1 to 6. | |
Table 3 Experiment and calculated 13C NMR data of 1, 5 and cottoquinazoline B
| No. |
1 |
5 |
Cottoquinazoline B |
| Exp. |
Cal. |
δΔ |
Exp. |
Cal. |
δΔ |
Exp. |
Cal. |
δΔ |
| 1 |
170.4 |
170.4 |
0.0 |
170.2 |
170.0 |
−0.2 |
169.9 |
169.1 |
−0.8 |
| 2 |
65.4 |
66.5 |
1.1 |
163.2 |
162.6 |
−0.6 |
64.5 |
65.2 |
0.7 |
| 3 |
150.2 |
149.5 |
−0.7 |
148.9 |
146.0 |
−2.9 |
149.4 |
150.0 |
0.6 |
| 4 |
147.5 |
147.7 |
0.2 |
145.7 |
145.4 |
−0.3 |
147.2 |
147.1 |
−0.1 |
| 5 |
127.2 |
127.8 |
0.6 |
127.7 |
128.0 |
0.3 |
127.4 |
127.7 |
0.3 |
| 6 |
134.6 |
134.0 |
−0.6 |
135.6 |
134.4 |
−1.2 |
134.7 |
133.9 |
−0.8 |
| 7 |
126.8 |
126.3 |
−0.5 |
128.7 |
128.3 |
−0.4 |
127.1 |
125.7 |
−1.4 |
| 8 |
126.1 |
128.7 |
2.6 |
126.5 |
129.1 |
2.6 |
126.4 |
127.9 |
1.5 |
| 9 |
120.0 |
121.1 |
1.1 |
121.1 |
122.4 |
1.3 |
120.4 |
120.6 |
0.2 |
| 10 |
159.8 |
158.4 |
−1.4 |
160.1 |
160.9 |
0.8 |
160.1 |
159.3 |
−0.8 |
| 11 |
53.7 |
53.4 |
−0.3 |
54.9 |
55.7 |
0.8 |
53.4 |
53.3 |
−0.1 |
| 12 |
34.7 |
33.3 |
−1.4 |
30.9 |
31.1 |
0.2 |
33.6 |
33.7 |
0.1 |
| 13 |
74.8 |
75.0 |
0.2 |
86.3 |
84.4 |
−1.9 |
78.6 |
81.6 |
3.0 |
| 14 |
78.4 |
77.2 |
−1.2 |
83.5 |
84.4 |
0.9 |
82.2 |
82.4 |
0.2 |
| 15 |
136.0 |
137.4 |
1.4 |
142.0 |
144.9 |
2.9 |
139.1 |
142.1 |
3.0 |
| 16 |
113.9 |
115.6 |
1.7 |
117.5 |
117.8 |
0.3 |
117.5 |
119.4 |
1.9 |
| 17 |
129.6 |
129.9 |
0.3 |
131.6 |
131.3 |
−0.3 |
129.5 |
130.7 |
1.2 |
| 18 |
124.6 |
124.8 |
0.2 |
126 |
124.9 |
−1.1 |
125.8 |
125.4 |
−0.4 |
| 19 |
124.7 |
124.2 |
−0.5 |
125.3 |
125.7 |
0.4 |
123.8 |
120.3 |
−3.5 |
| 20 |
139.0 |
138.8 |
−0.2 |
131.6 |
131.2 |
−0.4 |
138.7 |
137.7 |
−1.0 |
| 21 |
62.5 |
63.2 |
0.7 |
45.9 |
44.9 |
−1.0 |
59.2 |
58.1 |
−1.1 |
| 22 |
165.7 |
163.2 |
−2.5 |
177.8 |
177.6 |
−0.2 |
174.1 |
173.1 |
−1.0 |
| 23 |
14.8 |
14.0 |
−0.8 |
17.0 |
18.8 |
1.8 |
15.8 |
13.9 |
−1.9 |
| 24 |
|
|
|
11.8 |
9.9 |
−1.9 |
|
|
|
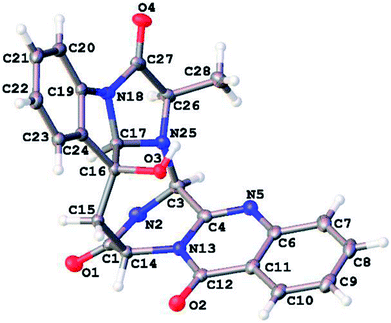 |
| | Fig. 4 X-ray crystallographic structure of 1. | |
Versiquinazoline M (2) has a molecular formula of C23H19N5O5 as determined by the HRESIMS and NMR data. Comparison of the NMR data (Tables 1 and 2) revealed the structure of 2 to be closely related to that of 1. The distinction was due to the imidazoindolone segment, in which the protons of a hydroxymethyl group H2-28 (δH 3.79, 4.07) showed the COSY relationship with H-26 (δH 4.11, dd, J = 2.4, 4.6 Hz) and a D2O exchangeable proton (δH 4.93, t, J = 4.9 Hz) and the HMBC correlations from H2-28 to C-26 (δH 67.6) and a carbonyl carbon C-27 (δC 170.2), indicating a serine unit instead of an alanine to incorporate ring F in 2. The NOE correlations from H-17 (δH 5.17) to H-15b (δH 2.31) and NH-2 (δH 9.00) and between OH-16 (δH 5.28, s) and H-15a (δH 3.12) as the cases in 1 were observed in the NOESY spectrum. Additional NOE correlation between OH-16 and H-26 indicated the distinct configuration at C-26 of 2 in comparison with that of 1 (Fig. 3), reflecting the hydroxymethylene group to be oriented in the same face as H-17. Based on the exciton chirality method,16 the positive Cotton effect (CE) at 234 nm (first Cotton effect) and the negative CE at 206 (second Cotton effect) suggested a clockwise orientation of the chromophores of aromatic rings A and D, assuming 3S and 14S configurations. Therefore, the remaining stereogenic centers were determined to be 16S, 17R, and 26S configurations. The similar specific rotation and ECD data (Fig. 5) of both 1 and 2 further confirmed the configurational assignment.
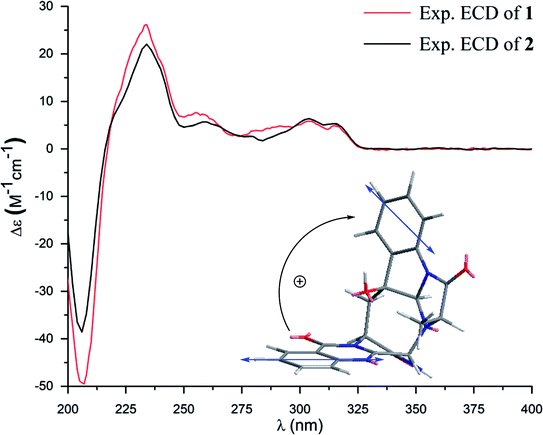 |
| | Fig. 5 Comparison of the Cotton effects between 1 and 2. | |
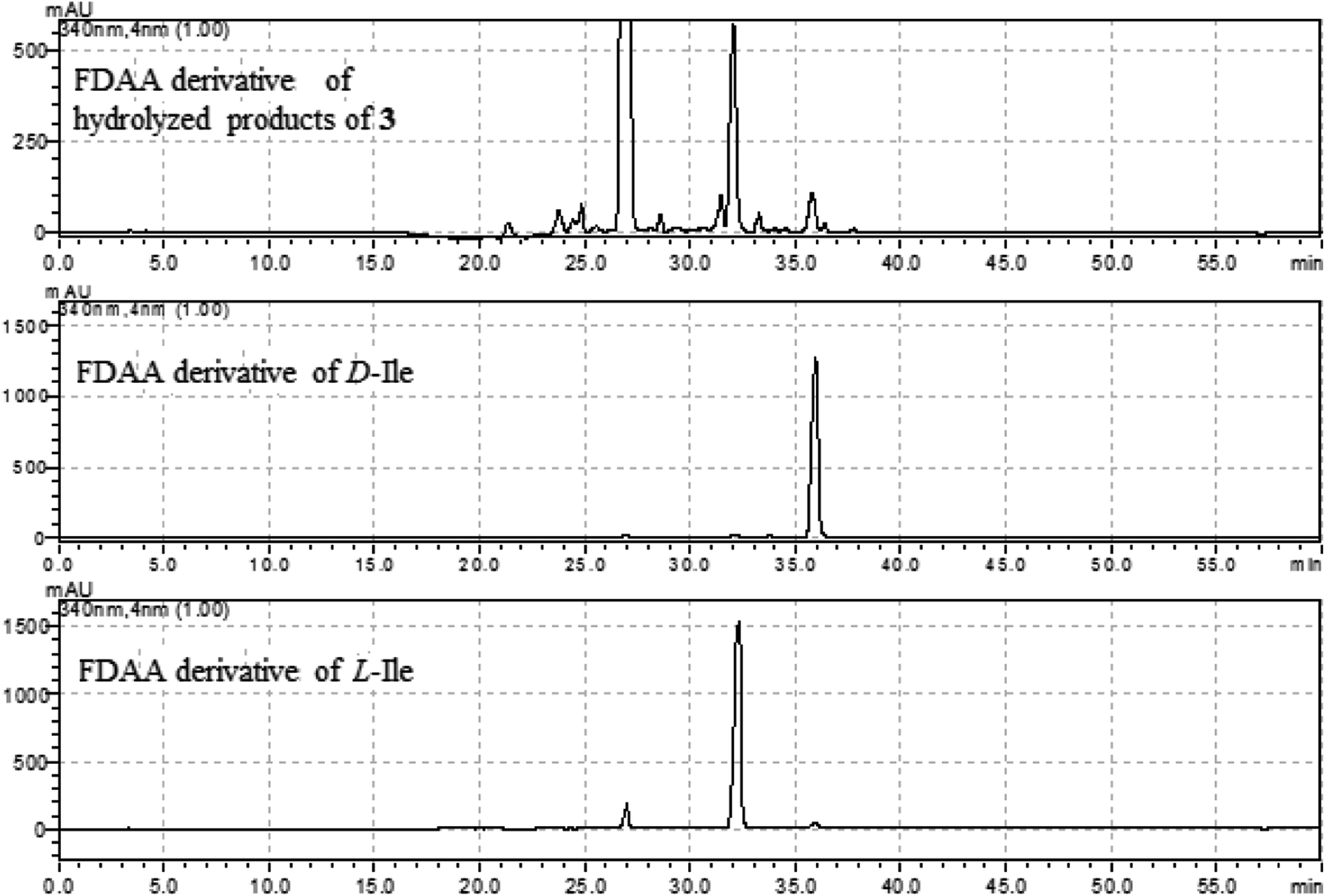
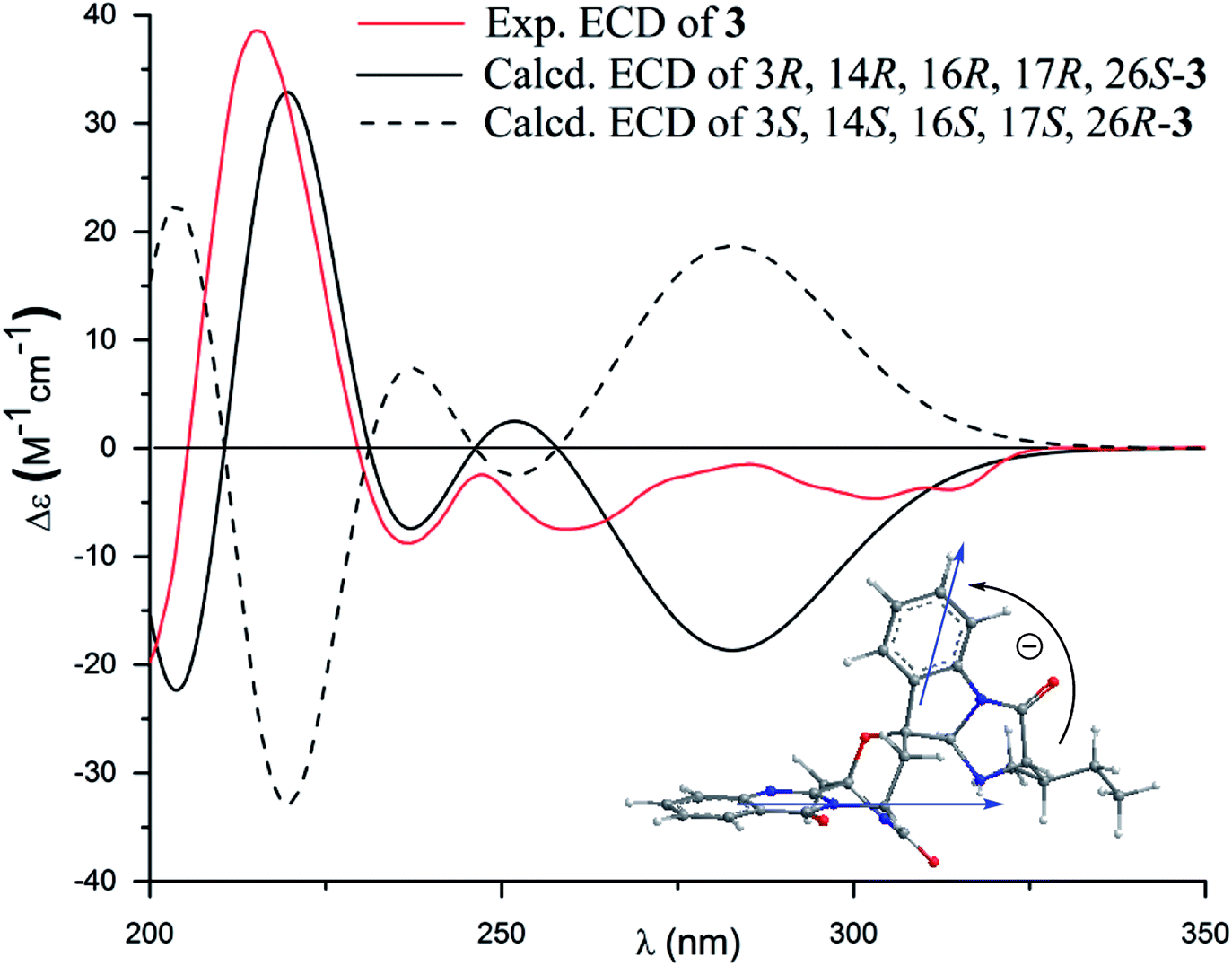
The molecular formula (C27H27N5O4) of versiquinazoline N (3) was established by the HRESIMS and NMR data, requiring 17 degrees of unsaturation. The 2D NMR data established the basic scaffold possessing two moieties, while a pyrazinoquinazolinedione moiety was identical to that of 1–2. In regard to the imidazoindolone unit, the COSY and HMBC correlations assigned an isoleucine unit to replace alanine of 1 for the incorporation of lactam ring F. The linkage of pyrazinoquinazolinedione to imidazoindolone unit by a methylene bridge across C-16 (δC 86.4) and C-14 (δC 52.8) was evident from the 1H–1H COSY correlation between H-14 (δH 5.36, d, J = 5.2 Hz) with H-15a (δH 2.98, dd, J = 5.2, 14.8 Hz), in association with the HMBC correlations from H2-15 to C-17 (δC 88.5) and C-24 (δC 137.4). The observation of HMBC correlation from a methyl singlet H3-32 (δH 1.90, s) to C-3 (δC 84.2) and C-4 (δC 151.5) conformed the methyl group to be substituted at C-3. The established moieties occupied 16 degrees of unsaturation, the remaining one site was suggested to form an additional ring, while the remarkably deshielded C-3 and C-16 in comparison with those of the respective carbons in 2 demonstrated an ether bond connected C-3 and C-16. The NOE correlation between H-17 (δH 5.69, d, J = 5.9 Hz) and H3-29 was indicative of the trans orientation of H-17 toward H-26 (Fig. 3). The isoleusine unit in 3 was assigned as L form based on the advanced Marfey's method,17–19 performing by acidic hydrolysis of 3 and subsequent generation of L-FDAA (1-fluoro-2,4-dinitrophenyl-5-L-alanine amide) amino acid derivative, whose HPLC retention time was excellent agreement with that of L-Ile (tR 32.2 min for L-Ile and tR 36.0 min for D-Ile) (Fig. 6). These findings resulted in 26S configuration, thus the stereogenic center C-17 was determined as 17R configuration. Additional NOE correlations from H-17 to H3-32 and NH-2 (δH 9.74, s) (Fig. 3) carried out the spiro form of ring E, of which H-17 was spatially approximated to NH-2. These assignments led to depict 3R and 14R configurations, which were distinguished to those of 1 and 2. In deed, the experimental ECD spectrum exhibited negative CE (first Cotton effect) at 237 nm and positive CE at 215 nm (second Cotton effect), reflecting counterclockwise twist of the aromatic chromophores (Fig. 7). Based on the TDDFT-ECD method,20,21 the ECD spectra of (3R, 14R, 16R, 17R and 26S)-3 and its enantiomer were calculated at the B31YP/6-311++G(2d,2p) level in the gas phase using the B3LYP/6-31G(d) optimized geometries after conformational searches via the MMFF94S force field. Comparison of the experimental ECD with the computed ECD data for the model molecules of 3 and its enantiomer further supported the configurational assignment.
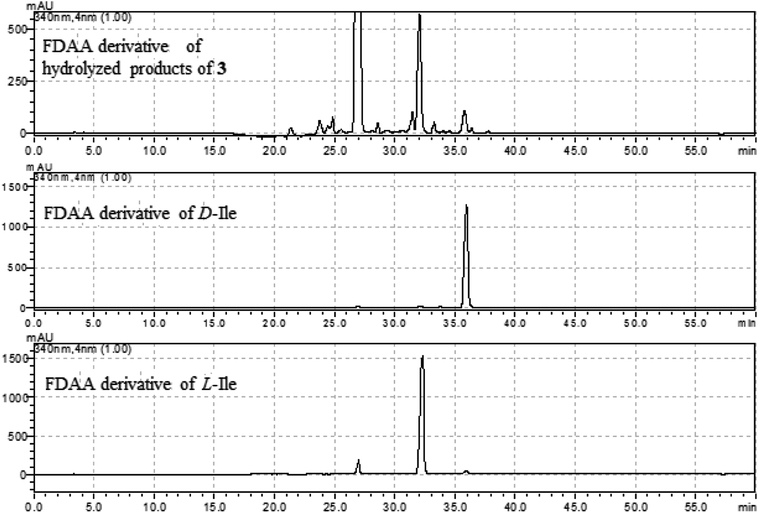 |
| | Fig. 6 HPLC chromatographs of the L-FDAA derivatives of hydrolyzed products of 3, and the L-FDAA derivatives of L-Ile and D-Ile. | |
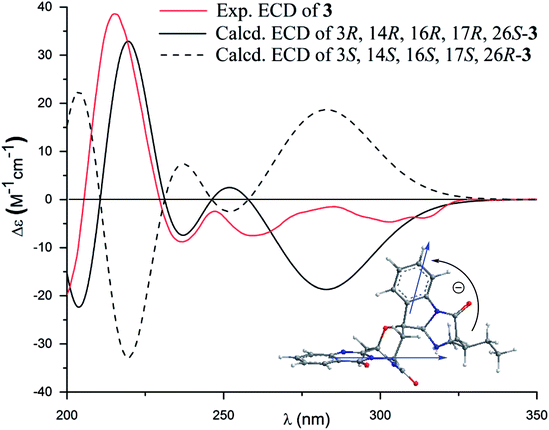 |
| | Fig. 7 Experimental and calculated ECD spectra of 3 and its enantiomer. | |
Versiquinazoline O (4) has a molecular formula (C27H29N5O4) with 2 amu more than that of 3 as determined by the HRESIMS data. The 1H and 13C NMR data featured a fumiquinazoline-type analogue,13 closely related to those of 3. Analyses of the 2D NMR data revealed that the structure of 4 possesses two moieties involving pyrazinoquinazolinedione and imidazoindolone. The connection of both units by a methylene group CH2-15 to link C-14 (δC 51.8) and C-16 (δC 80.1) was established by the COSY coupling of H2-15 (δH 1.82, 2.58) with H-14 (δH 5.57, dd, J = 4.4, 9.4 Hz) and the HMBC correlation of H2-15 with C-16, C-17 (δC 88.2), and C-24 (δC 138.7). A D2O exchangeable proton OH-16 (δH 5.67, s) correlating to C-15 (δC 35.5), C-16, C-17, and C-24 in the HMBC spectrum resided a OH group at C-16. In addition, the 1H–1H COSY relationship between H-3 (δH 4.94, q, J = 6.5 Hz) and H3-32 (δH 1.58, d, J = 6.5 Hz) and the HMBC correlations from H3-32 to C-3 (δC 48.6) and C-4 (δC 153.3) clarified 4 to be an analogue of 3 with the cleavage of C3–O ether bond. The NOE correlation between H-3 and H2-15 assigned the same orientation of these protons, while the NOE correlations from H-17 (δH 5.25) to OH-16 (δH 5.67) and H3-29 determined the relative configuration in imidazoindolone unit to be the same as that of 3. As the case of 3, the negative CE at 233 nm (first Cotton effect) and positive CE at 211 nm (second Cotton effect) (Fig. 8) reflected 14R configuration, while the configurations of the remaining stereogenic centers were assumed to be 3R, 16R, 17R, and 26S referred to the NOE data. Comparison of the experimental ECD data with those calculated for the model molecules of 4 and its enantiomer (Fig. 7) further confirmed the configurational assignment. In addition, a chemical conversion of 3 by NaBH4/MeOH13 (Fig. 9) produced a derivative, whose NMR data and specific rotation were corresponded to those of 4.
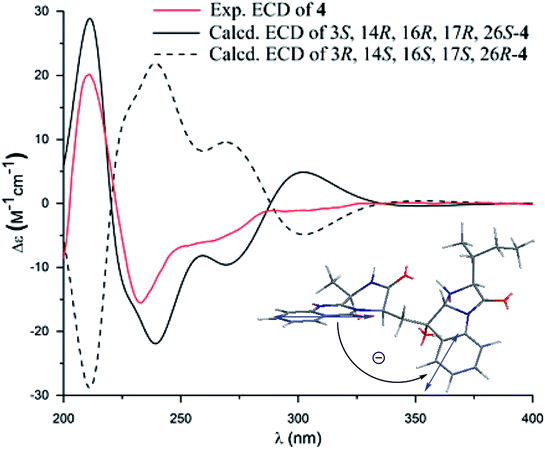 |
| | Fig. 8 Experimental and calculated ECD spectra of 4 and its enantiomer. | |
 |
| | Fig. 9 Chemical conversion of 3 to yield 4. | |
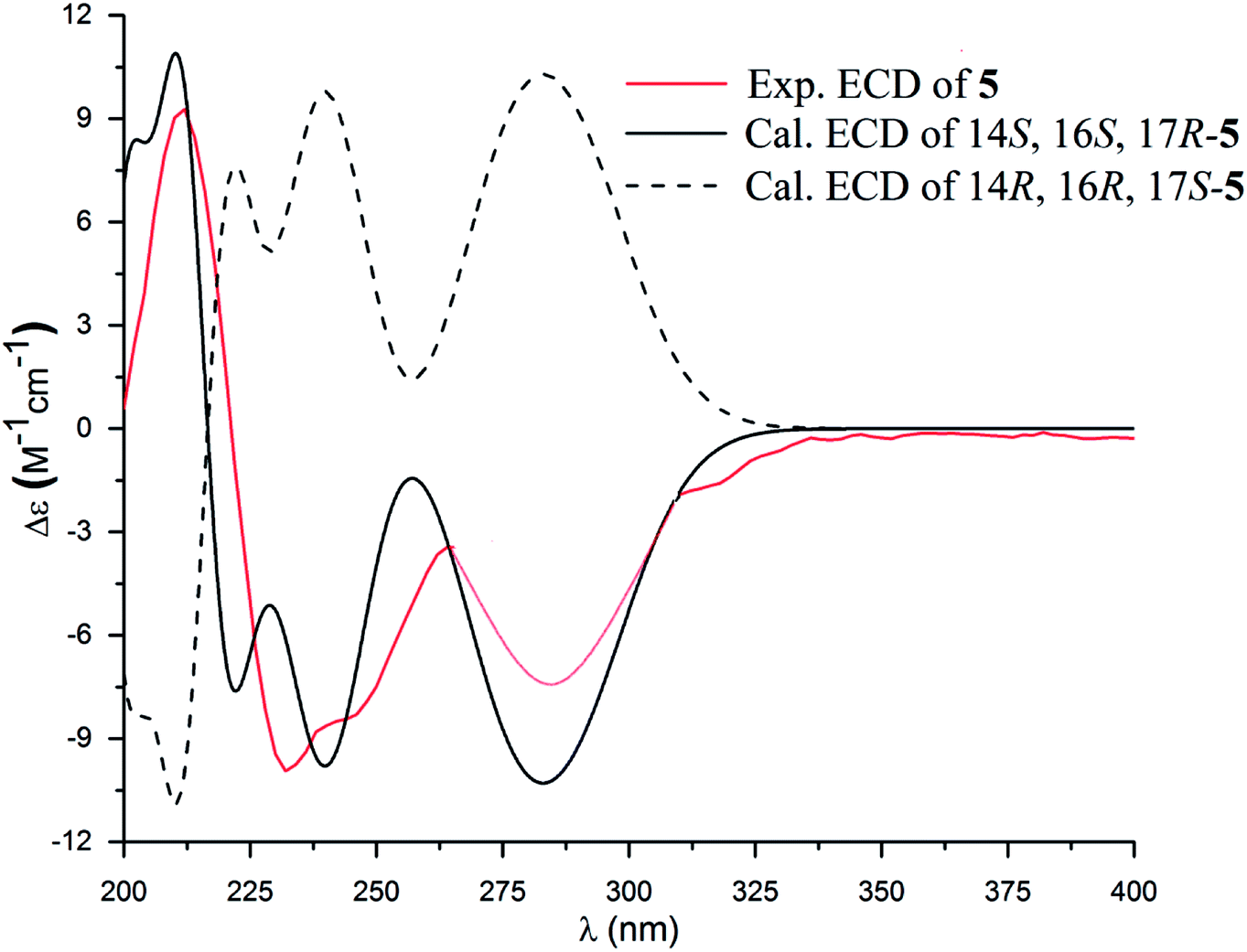
The molecular formula of versiquinazoline P (5) was determined to be C24H19N5O5N on the basis of the HMBC and NMR data, containing 18 degrees of unsaturation. The 1D and 2D NMR data established 5 to be a polycyclic alkaloid bearing a 2-quinazolinecarboxamide and an imidazo[1,2-a]indoline units, structurally related to versiquinazoline E.13 A formamide unit CONH2-3 was recognized by the NMR data for the NH2 (δH 8.4, 8.8) and a carbonyl carbon C-3 at δC 163.2, that was linked to C-4 (δC 148.9) of quinazolinone based on the HMBC correlation of the NH2 to C-3 and C-4. In addition, a γ-lactone ring fused to C-16 (δC 86.3) in a spiro form was uncovered by the COSY relationship between H-14 (δH 5.49, dd, J = 9.4, 10.5 Hz) and H2-15 (δH 2.90, 3.44) as well as the HMBC correlations from H2-15 to a carbonyl carbon C-1 (δC 170.2), C-16, C-17 (δC 83.5), as well as an aromatic carbon C-24 (δC 131.6). The remaining 2D NMR data were attributed to rings D–G in midazo[1,2-a]indoline unit, which was identical to that of versiquinazoline E. The connection of C-14 to the nitrogen atom of quinazolinone was evident from the HMBC correlation of H-14 with C-4 and C-12 (δC 160.1) (Fig. 2). The obvious NOE interaction between H-14 and the aromatic proton H-23 (δH 7.66, d, J = 8.0 Hz) clarified aromatic ring E to be oriented in the same face as H-14 (Fig. 3). The absence of NOE correlation between H-17 (δH 5.55) and H2-15 suggested H-17 to be oriented in opposite face toward H2-15 group in ring D. Calculation of the 13C NMR chemical shifts of the model molecule with 14S*, 16S*, 17R*-5 by the same method as for 1 resulted in the data virtually identical to the measured 13C NMR data of 5 (Table 3) with the maximum absolute difference between predicted and observed shifts was 3.0 ppm. Comparison of the experimental ECD data with those calculated for the model molecules (Fig. 10) further determined the absolute configurations to be 14S, 16S, and 17R.
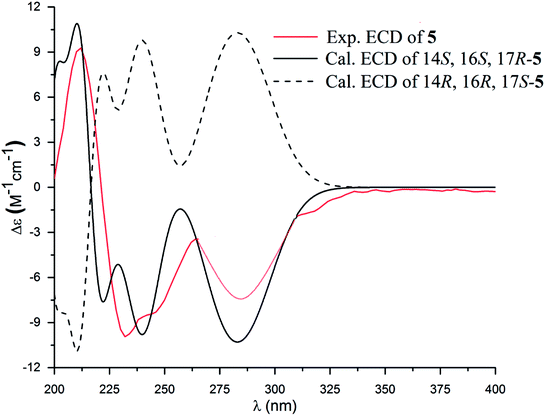 |
| | Fig. 10 Experimental and calculated ECD spectra of 5 and its enantiomer. | |
The structure of versiquinazoline Q (6) was determined to be a 11-hydroxylated analogue of versiquinazoline K,13 based on the similar NMR data of both compounds with the exception of a phenol proton (δH 8.79, OH-11) in 6 to replace a methoxy group of the known analogue. The HMBC correlation of OH-11 with C-10 (δC 101.9), C-11, and C-12 (δC 159.4) confirmed the phenol position. The similar specific rotation and ECD data of both 6 and versiquinazoline K (Fig. 11) assigned the absolute configuration of 6 to be the same as the known analogue.
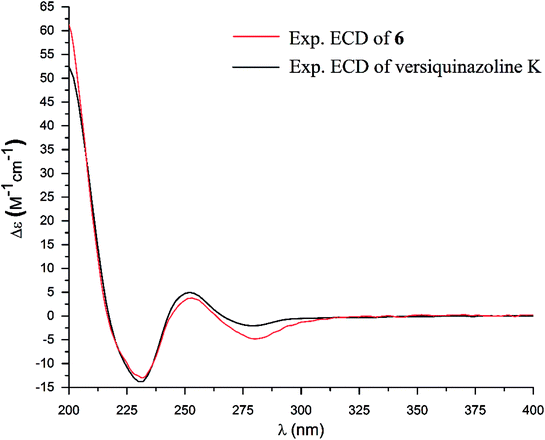 |
| | Fig. 11 Comparison of the Cotton effects between 6 and versiquinazoline K. | |
Table 4 TrxR inhibitory activity of compounds 1–6
| No. |
% Inhibition (50 μM) |
IC50 (μM) |
| Positive control. |
| 1 |
36 |
|
| 2 |
41 |
|
| 3 |
49 |
|
| 4 |
29 |
|
| 5 |
81 |
13.6 ± 0.6 |
| 6 |
84 |
12.2 ± 0.7 |
| Curcumina |
|
25 ± 2 |
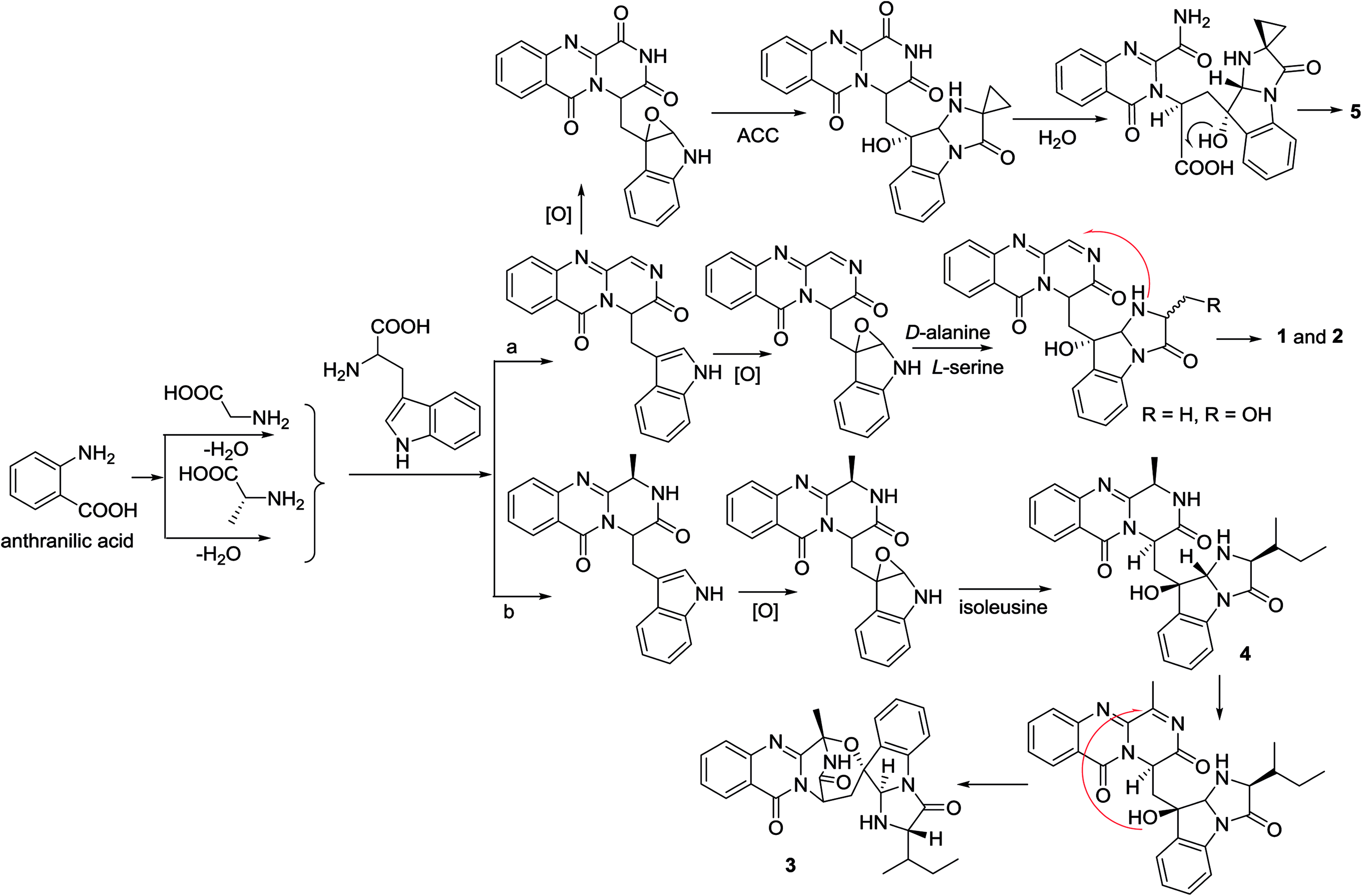
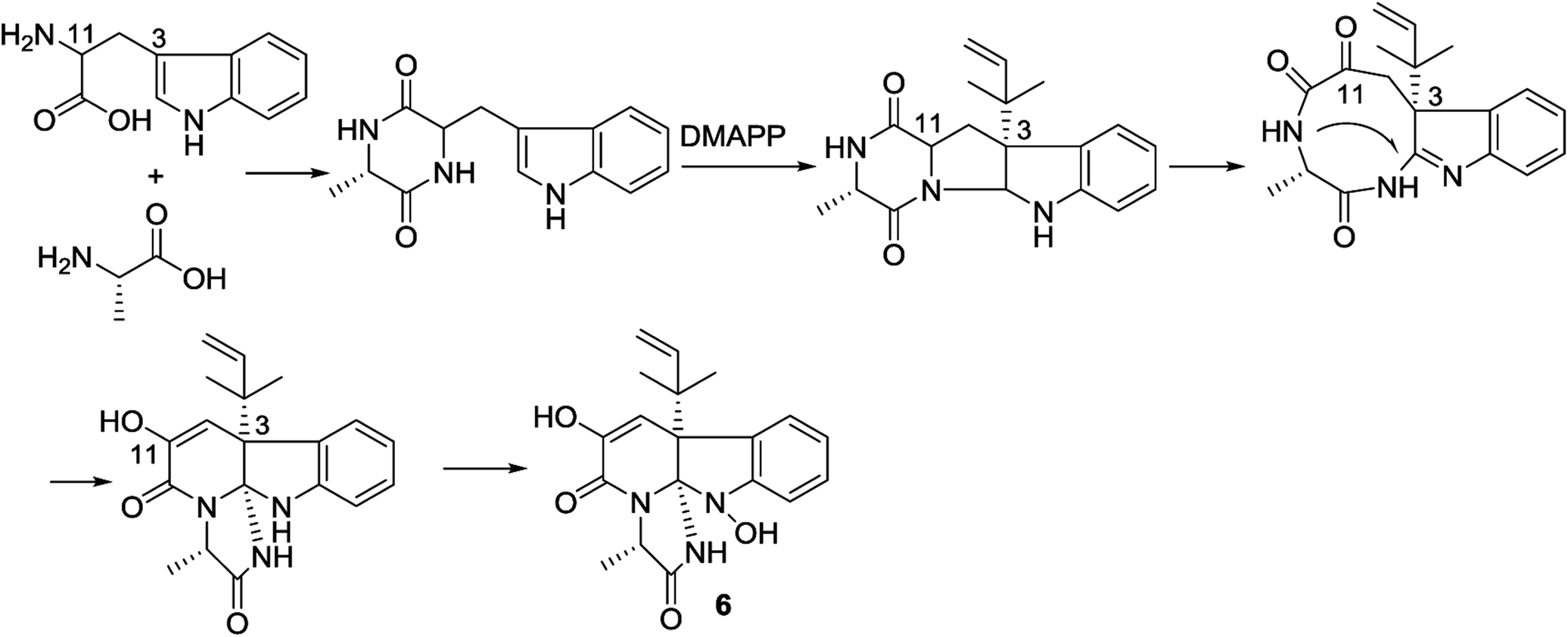
In the biogenetic consideration, anthranilic acid is speculated to be one of the precursors to generate the polycyclic alkaloids 1–5. Intermolecular condensation of anthranilic acid with glycine/alanine and tryptophan through a and b pathways (Scheme 1) yielded pyrazinoquinazolinedione scaffolds. Epoxidation at the indole ring resulted in the epoxy-bearing intermediates, which further incorporated with D-alanine or L-serine to afford imidazoindolone unit. A Mannich reaction is likely occurred to yield compounds 1 and 2 by the formation of an eight-membered ring. Pathway b followed the similar manner as that of pathway a, but the epoxy-bearing intermediate alternatively incorporated with L-isoleusine to derive compound 4. The latter compound via a Mannich reaction to generate an ether bond in 3. Compound 5 was assumed to share the same glyantrypine intermediate as that of 1 and 2 to undergo oxidation, epoxidation, 1-aminocyclopropanecarboxylic acid (ACC) incorporation, hydrolysis, and lactonization. Compound 6 was possible to be generated by the condensation of tryptophan and alanine to form a cyclodipeptide, which followed the dimethylallyl addition under dimethylallyl diphosphate (DMAPP) and ring cyclization. Hydroxylation at C-11 with a subsequent cleavage of the bond between C-11 and N-atom yielded a nine-membered ring with a keto-group at C-11. Further oxidation at nitrogen atom of the indole moiety led to the loss of water to generate an imine, which was attached by N-atom of amide to generate a tetracyclic nucleus. Finally, hydroxylation at nitrogen atom yielded compound 6 (Scheme 2).
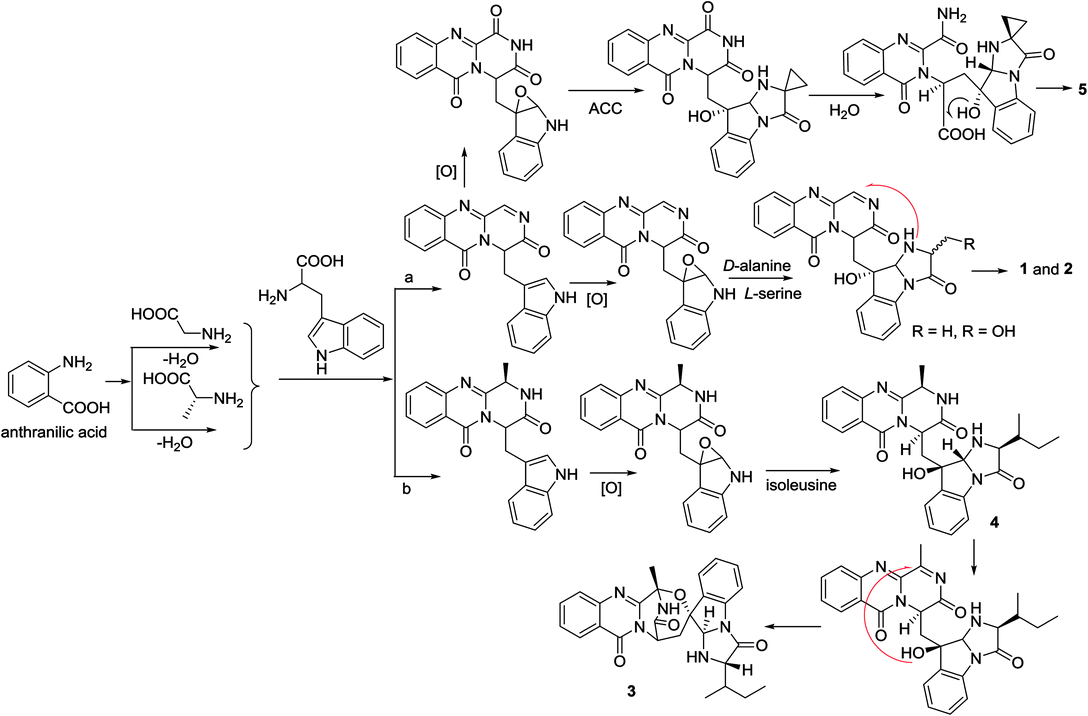 |
| | Scheme 1 Hypothesis of biosynthetic pathway of 1to 5. | |
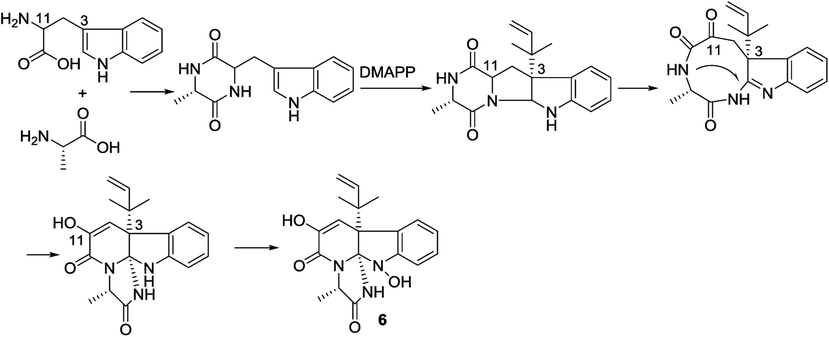 |
| | Scheme 2 Hypothesis of biosynthetic pathway of 6. | |
3.2. Bioassay
All compounds showed weak activity against human lung adenocarcinoma epithelial cell line A549 with IC50 > 10 μM. Additional examination revealed that compound 5 and 6 exhibited significantly inhibitory activities against thioredoxin reductase (TrxR) with IC50 values to be 13.6 ± 0.6 and 12.2 ± 0.7 μM (Table 4), which are more active than the positive control curcumin (IC50 = 25 μM). TrxR is a number of the Trx system and is regarded as a new target for anticancer drug development due to its overexpression in many aggressive tumors, while TrxR has been proved to be closely related to the tumor progression and metastasis in vivo.22,23 the weak cytotoxicity and potent inhibition toward TrxR suggested that compounds 1, 2, 7, and 11 may act as microenvironmental regulation of tumor progression and metastasis. The weak cytotoxicity and potent inhibition toward TrxR suggested that compounds 5 and 6 may act as microenvironmental regulation of tumor progression and metastasis.
4. Conclusion
In summary, this work reports six new fumiquinazoline-related alkaloids with diverse subtypes, while D-alanine and L-serine incorporated in the fumiquinazoline-based analogues was rarely found in nature. Compound 5 presented as a unusual scaffold which provided an additional subtype of fumiquinazoline alkaloids. The biogenetic relationships of the isolated alkaloids were postulated, whereas the biosynthetic method are required to prove the pathways. Although diverse secondary metabolites from the fungus Aspergillus versicolor have been reported in literature, present work indicated that the same fungal species from different environments provided the natural products with distinct chemical diversity due to each strain activating distinctive biosynthetic pathway. In addition, two compounds exhibited significantly inhibitory effect against thioredoxin reductases with low cytotoxic activities toward tumor cell lines, suggested that their inhibitory effects toward TrxR may be related to the microenvironmental regulation of tumor progression and metastasis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the grants of the National High Technology and Science 973 program (2015CB755906), and the National Natural Science Foundation of China (81630089, 41376127, U1606403).
References
- A. Numata, C. Takahashi, T. Matsushita, T. Miyamoto, K. Kawai, Y. Usami, E. Matsumura, M. Inoue, H. Ohishi and T. Shingu, Tetrahedron Lett., 1992, 33, 1621–1624 CrossRef.
- G. Yu, G. Zhou, M. Zhu, W. Wang, T. Zhu, Q. Gu and D. Li, Org. Lett., 2016, 18, 244–247 CrossRef PubMed.
- J. Liu, X. Wei, E. L. Kim, X. Lin, X. Yang, X. Zhou, B. Yang, J. H. Jung and Y. Liu, Tetrahedron, 2015, 71, 271–275 CrossRef.
- L. Liao, M. You, B. K. Chung, D. C. Oh, K. B. Oh and J. Shin, J. Nat. Prod., 2015, 78, 349–354 CrossRef PubMed.
- Y. Zhou, A. Debbab, A. Mandi, V. Wray, B. Schulz, W. E. G. Mueller, M. Kassack, W. Lin, T. Kurtan, P. Proksch and A. H. Aly, Eur. J. Org. Chem., 2013, 894–906 CrossRef.
- C. Shao, R. Xu, M. Wei, Z. She and C. Wang, J. Nat. Prod., 2013, 76, 779–782 CrossRef PubMed.
- Y. Zhuang, X. Teng, Y. Wang, P. Liu, G. Li and W. Zhu, Org. Lett., 2011, 13, 1130–1133 CrossRef PubMed.
- J. Peng, T. Lin, W. Wang, Z. Xin, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2013, 76, 1133–1140 CrossRef PubMed.
- C. An, X. Li, C. Li, M. Wang, G. Xu and B. Wang, Mar. Drugs, 2013, 11, 2682–2694 CrossRef PubMed.
- S. W. Haynes, B. D. Ames, X. Gao, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 5668–5679 CrossRef PubMed.
- C. T. Walsh, S. W. Haynes, B. D. Ames, X. Gao and Y. Tang, ACS Chem. Biol., 2013, 8, 1366–1382 CrossRef PubMed.
- F. Y. Lim, B. Ames, C. T. Walsh and N. P. Keller, Cell. Microbiol., 2014, 16, 1267–1283 CrossRef PubMed.
- Z. Cheng, L. Lou, D. Liu, X. Li, P. Proksch, S. Yin and W. Lin, J. Nat. Prod., 2016, 79, 2941–2952 CrossRef PubMed.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef PubMed.
- St. Thomas, I. Brühl, D. Heilmann and E. Kleinpeter, J. Chem. Inf. Comput. Sci., 1997, 37, 726–730 CrossRef.
- H. Uzawa, Y. Nishida, H. Ohrui and H. Meguro, J. Org. Chem., 1990, 55, 116–122 CrossRef.
- K. Fujii, Y. Ikai, H. Oka, M. Suzuki and K. Harada, Anal. Chem., 1997, 69, 3346–3352 CrossRef.
- K. Fujii, Y. Ikai, H. Oka, M. Suzuki and K. Harada, Anal. Chem., 1997, 69, 5146–5151 CrossRef.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef.
- G. Mazzeo, E. Santoro, A. Andolfi, A. Cimmino, P. Troselj, A. G. Petrovic, S. Superchi, A. Evidente and N. Berova, J. Nat. Prod., 2013, 76, 588–599 CrossRef PubMed.
- Y. Ding, X. Li and D. Ferreira, J. Org. Chem., 2007, 72, 9010–9017 CrossRef PubMed.
- D. Mustacich and G. Powis, Biochem. J., 2000, 346, 1–8 CrossRef PubMed.
- J. Lu, E. H. Chew and A. Holmgren, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12288–12293 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1813311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06854b |
| ‡ equal contribution |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a