
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe deactivation of a ZnO doped ZrO2–SiO2 catalyst in the conversion of ethanol/acetaldehyde to 1,3-butadiene†
Minhua Zhangab,
Xuechao Tanab,
Tong Zhangab,
Zheng Hanab and
Haoxi Jiang *ab
*ab
aKey Laboratory for Green Chemical Technology of Ministry of Education, R&D Center for Petrochemical Technology, Tianjin University, Tianjin 300072, China. E-mail: hxjiang@tju.edu.cn; Fax: +86-22-27406119; Tel: +86-22-27406119
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin, 300072, China
First published on 3rd October 2018
Abstract
A deactivation study on the ethanol/acetaldehyde conversion to 1,3-butadiene over a ZnO promoted ZrO2–SiO2 catalyst prepared by a sol–gel method was performed. The samples were characterized by N2 adsorption–desorption isotherms, scanning electron microscopy (SEM), NH3 temperature programmed desorption (NH3-TPD), X-ray powder diffraction characterization (XRD), thermogravimetric analyses (TGA), Fourier transform infrared resonance (FT-IR), 13C magic-angle spinning nuclear magnetic resonance (13C NMR) and X-ray photoelectron spectroscopy (XPS). The pore structure characteristics and surface acidity of Zn0.5–Zr–Si catalysts were largely decreased with time-on-stream and no crystal structure was formed in the used catalyst, indicating that the deactivation was caused by carbon deposition. Two main types of carbon deposition were formed, namely low-temperature carbon deposition with the oxidation temperature of around 400 °C and high-temperature carbon deposition with the oxidation temperature of 526 °C. The carbon species were mainly composed of graphitized carbon, amorphous carbon, carbon in C–O bonds and carbonyls. The deactivated catalyst could be regenerated by a simple oxidation process in air. Adding a certain amount of water into the feed had a positive effect on reducing the carbon deposition.
1 Introduction
1,3-Butadiene is an important chemical with extensive application in industry. It has been reported that about 80% of the production was used in synthetic rubber and elastomers. In the past century, the majority of 1,3-butadiene produced came from the petroleum route, by which 1,3-butadiene was treated as a by-product of ethylene production. In recent years, ethanol conversion into 1,3-butadiene has attracted attention from all over the world because of the shift to light feedstocks for steam crackers and the increase in production of bio-ethanol.Many literature studies on the mechanism of the reaction,1 active components of catalyst2–5 and the function of additives6–9 were reported previously. Good catalytic performances have been reported by Huang et al.10 and Jian et al.,11 but challenges also existed in the reaction, among which, the deactivation should be the most important problem of these highly active catalysts. As is known to all, the deactivation of catalyst may be resulted from three main reasons, i.e. catalyst agglomeration, carbon deposition and catalyst poisoning.12 For most organic reactions, coke, which is formed on the acid sites of the catalyst, may be the most likely cause of catalyst deactivation. Chae et al.13 reported a similar phenomenon in ethanol and acetaldehyde conversion into 1,3-butadiene process. And they pointed out that the pore size and the crystal size of the catalyst would play an important role in alleviating coke formation in the reaction process. However, no further studies were conducted to reveal the connection between the structural properties and the carbon deposition. Baylon et al.14 studied the stability and regenerability of Na doped ZnxZryOz catalyst. After 60 h of TOS, the selectivity to 1,3-butadiene reduced by 20% and could be recovered in 30 min at 450 °C in the presence of 12 vol% O2. It suggested that the reason of deactivation mainly was the coking of the active sites for secondary acetaldehyde to 1,3-butadiene reaction. Tripathi et al.15 reported the bimetallic Cu–Ag supported on MgO–SiO2 catalysts in the conversion of ethanol to 1,3-butadiene. The 5 wt%Ag/MgO–SiO2 showed higher deactivation compared with the 5 wt%Cu/MgO–SiO2. And the bimetallic 5 wt%CuAg/MgO–SiO2 catalyst showed less deactivation than 5 wt%Ag/MgO–SiO2, but higher than 5 wt%Cu/MgO–SiO2, suggesting that the addition of Cu could inhibit the carbon deposition.
In this paper, for the first time, the problems associated with the deactivation of 1,3-butadiene from ethanol/acetaldehyde catalyzed by ZnO–ZrO2–SiO2 were investigated. BET, XRD and SEM were used to ensure that carbon deposition was the reason of catalyst deactivation. The physicochemical properties of deactivated catalyst were examined by means of NH3-TPD, FT-IR, TGA, 13C NMR and XPS. In addition, the regenerability of the catalyst and the effect of water in feed were also investigated.
2 Experimental section
2.1 Chemicals
Zirconium oxynitrate (AR) and acetaldehyde (97%) were purchased from Aladdin Industrial Corporation. Neat ethanol (AR) was purchased from Tianjin Jiangtian Chemical Technology Co., Ltd. Nitric acid (AR) was purchased from Beijing Chemical Works. Tetraethyl orthosilicate (AR) and zinc nitrate hexahydrate (AR) were purchased from Tianjin Guangfu Chemical Reagent Co., Ltd.2.2 Catalyst preparation
The ZnO–ZrO2–SiO2 catalyst was prepared by hybrid sol–gel method as already described in ref. 6. Specifically, a certain amount of ZrO(NO3)2 and Zn(NO3)2·9H2O was dissolved in deionized water and then some amount of pure ethanol was added into the solution. The mixture was then subjected to the addition of diluted nitric acid solution under vigorously stirring. Some amount of ethyl orthosilicate was purged into the solution afterwards. The obtained gel was then allowed to stand at room temperature for 24 h. The colloidal sample was then dried at 110 °C for 6 h and subsequently calcined in a muffle furnace at 650 °C for 6 h with a temperature ramping rate of 5 °C min−1.2.3 Catalyst evaluation
The catalyst evaluation of ethanol and acetaldehyde converting into 1,3-butadiene was performed on a fixed bed reactor. The samples were ground and sieved into 20–40 mesh before experiment. The reaction conditions were chosen as follows: reaction temperature of 310 °C, mole ratio of ethanol to acetaldehyde of 3.5/1, WHSV of 1.8 h−1. In a typical experiment, the catalyst was loaded into the quartz tubular reactor, which was purged with N2 at reaction temperature for 1 h. Ethanol was fed into the vaporizer (120 °C) by using a syringe pump. All pipelines were kept 180 °C to prevent condensation. The composition of the exhaust gas was analyzed by Agilent 7890A gas chromatograph using a 30 m HP-PLOT-Q column. The main products contained 1,3-butadiene, ethylene, propylene, butylene, diethyl ether, ethyl acetate and unidentified compounds, which was represented as BD, EL, PL, BL, DE, EA and C6+, respectively. When the evaluation finished, the catalyst was purged by nitrogen at the reaction temperature for 1 h and then to the room temperature. The mixed conversion of ethanol and acetaldehyde as well as the selectivity towards the main products were calculated as follows:13

2.4 Catalyst characterization
X-ray powder diffraction characterization (XRD) was carried out using a Rigaku D/Max 2500 X-ray powder Diffractometer operated with Cu Kα radiation (λ = 1.5456 Å). The data was obtained in the 2θ range of 10–90° with a rate of 5° s−1.Scanning electron microscope (SEM) was recorded with a Hitachi S-4800 operated with the resolution of 1 nm. Before measurement the samples were ground thoroughly and pre-treated with conductive coating.
N2 adsorption–desorption isotherms were measured at −196 °C using a Micromeritics Tristar 3000 surface area and porosity analyzer. Before the analysis was conducted, all samples were pretreated at 300 °C for 6 h. The Brunauer–Emmett–Teller (BET) equation was used in the study.
Fourier transform infrared resonance (FT-IR) were obtained using a Nicolet 6700 spectrometer operated with the scanning range from 4000 to 400 cm−1 and at a resolution of 4 cm−1. All the catalysts were diluted by KBr before measurement.
X-ray photoelectron spectroscopy (XPS) were taken on a PerkinElmer PHI-1600 spectrometer using Mg Kα X-ray radiation source at a vacuum of 1.2 × 10−8 Mbar. All binding-energy values were referenced to the C 1s line at 284.6 eV.
13C magic-angle spinning nuclear magnetic resonance (13C-MAS-NMR) was recorded on a Varian Infinity plus 300 MHz spectrometer. The resonance frequency of 13C was 75.4 MHz, the spinning frequency of the rotor was 5 kHz and the pulse delay was 5 s. Adamantane was used as the secondary reference for the 13C spectrum.
The quantity and strength of acidity were obtained by NH3-temperature-programmed desorption (NH3-TPD) using a Micromeritic Autochem II 2920 equipment. All samples were pretreated at 310 °C for 2 h under flowing helium (50 mL min−1) and then cooled to 70 °C. After that the samples were saturated with ammonia (ammonia 1%, argon 99%) at a flow rate of 20 mL min−1 for 50 min, followed by flowing helium (30 mL min−1) for 1 h. Then the samples were heated to 400 °C with a ramping rate of 15 °C min−1 in helium (50 mL min−1). The signals of desorbed NH3 were recorded by a thermal conductivity detector.
The acidic properties were studied by FT-IR spectroscopy of adsorbed pyridine using a Nicolet 6700 FT-IR spectrometer with the optical resolution of 4 cm−1. The loading quantity of catalyst was about 20 mg. Before measurement, the sample was evacuated at 150 °C for 2 h. Breaking the vacuum with nitrogen and then getting the blank spectra at 450, 350, 250 and 150 °C. Pyridine was adsorbed at 150 °C and different spectra were obtained at 150, 250, 350 and 450 °C after subtracting the blank spectra of each temperature, respectively.
Thermogravimetric analyses (TGA) were carried out in a thermobalance unit using a METTLER DSTA851 analyzer. Weight data was automatically collected every 30 s with an accuracy of 0.001 mg. Air was taken as the reaction gas with a flow rate of 50 mL min−1. The temperature was linearly increased from 25 °C to 800 °C and the ramping rate was 10 °C min−1.
3 Results and discussion
3.1 Structural and acid properties of the deactivated catalysts
As reported in our previous work in ref. 6, the stability test of Zn0.5–ZrO2–SiO2 catalyst was performed. As shown in Fig. 1, the catalytic performances as to both BD selectivity and ethanol/acetaldehyde conversion decreased to some extent, indicating deactivation occurred during the test. | ||
| Fig. 1 Stability test of Zn0.5–ZrO–SiO2 catalyst. | ||
As listed in Table 1, the physical properties of used catalysts, i.e. SBET, average pore size and pore volume were much lower than those of fresh counterparts. This may be resulted from the deposition of carbon species on active sites of the catalyst, which, in return, blocked most pores of the catalyst. Notably, the specific surface area of catalyst decreased by 165 m2 g−1 after 3 h of TOS. Meanwhile, the specific surface area decreased by only 172 m2 g−1 from 3 h to 60 h. The pore structure properties of the catalysts showed that the initial specific surface area and pore volume reduction were high and then continued to decrease slowly. In addition, Fig. S1a and b† were drawn according to the data in Table 1. From the figures, we speculated that rapid decrease of specific surface area and pore volume, during the beginning 3 h, was due to block of some smaller pores by carbon species. When carbon species deposited on acid sites, it was not easy to escape from smaller pores, where there were more and more carbon species until the pores were blocked completely. The carbon species depositing in larger pores were more likely to diffuse out of pores due to the larger pore size.
| TOS (h) | SBET (m2 g−1) | d (Å) | V (cm3 g−1) | Quantity of acidity (mmol g−1) |
|---|---|---|---|---|
| 0 | 563 | 25.5 | 0.36 | 0.08 |
| 3 | 398 | 23.6 | 0.23 | 0.06 |
| 10 | 361 | 23.4 | 0.21 | 0.05 |
| 30 | 307 | 23.8 | 0.18 | 0.03 |
| 60 | 226 | 23.4 | 0.13 | 0.02 |
XRD characterization was used to study the structural changes of used catalysts with different TOS. As clearly revealed in Fig. 2, a broad halo was shown at the range of 2θ = 20–30°, which evidenced a relatively good dispersion of ZrO2 and ZnO on SiO2 support.16,17 In other words, the deactivation may have little connection with the change of the crystal structure of the catalyst. When comparing the diffraction peak intensity, it can be seen that upon TOS increasing, the intensity of the characteristic diffraction peak increased gradually, which evidenced that the amorphous property of the catalyst decreased. This may be due to carbon deposition on the catalyst surface or the prolonged high temperature (310 °C) treatment.
 | ||
| Fig. 2 XRD patterns of catalysts with different TOS. (a) 0 h; (b) 3 h; (c) 10 h; (d) 30 h; (e) 60 h. | ||
Further study by using SEM was conducted to observe the morphology of the used catalysts. The catalyst with 60 h of TOS was chosen as a representative sample. As we can see in Fig. 3c, the surface of fresh catalyst was relatively smooth at the scale of 20.0 μm. For the used catalyst, the agglomeration was evident and some obvious particles were formed on the surface of the catalyst, which was due to the carbon deposition, as shown in Fig. 3a and b.
 | ||
| Fig. 3 SEM images of catalysts. (a) Catalyst with 60 h of TOS (scale bar: 5 μm); (b) catalyst with 60 h of TOS (scale bar: 1 μm); (c) fresh catalyst. | ||
Scholars have done some research about carbon deposition on various reactions and catalysts. Castro et al.18 observed higher amount of carbon formed on the catalyst exhibiting the higher density of acid sites on the support, when studying steam reforming of toluene over CexZr1−xO2/Al2O3 catalysts. Li et al.19 studied coke formation on the surface of Ni/HZSM-5 and Ni–Cu/HZSM-5 catalysts, finding the first stage of carbon deposition was oxygenated hydrocarbons adsorbed Lewis acid sites in HZSM-5 support. Ni20 et al. studied carbon deposition on borated alumina supported nano-sized Ni catalysts for dry reforming of CH4 and believed carbon deposition was related to strong acid sites. In order to explore the influence of the carbon deposition on the acid property of the catalysts, NH3-TPD was conducted to analyse the quantity of acidity of the catalysts after different TOS. As shown in Fig. 4, the quantity of acidity of the catalysts with different TOS was much lower than that of the fresh sample. Moreover, as shown in Fig. S1c,† it is evident that the decreasing rate of the quantity of acidity tended to diminish with TOS increasing, which was in good accordance with the trend of the structural changes in XRD patterns.
 | ||
| Fig. 4 NH3-TPD profiles of catalysts with different TOS. (a) 0 h; (b) 3 h; (c) 10 h; (d) 30 h; (e) 60 h. | ||
The acidic property of the catalysts was also studied by Py-IR spectra. As shown in Fig. S2,† the adsorption at about 1446 and 1604 cm−1 were typical bands of pyridine adsorbed on Lewis acid sites21,22 and no Bronsted acid was found at 1540 cm−1. Thus, in ETB (ethanol to butadiene) reaction, we also suspect the carbon species deposited on Lewis acid sites. In addition, the carbon deposition of the catalyst tended to be a process, in which carbon species began to deposit on acid sites of the inner surface and gradually grew and spread around until filling the pore completely. The smaller pores were blocked quickly, while the larger ones slowly.
What's more, there was an interesting phenomenon, during the beginning 10–20 h on stream, the drop in acidity was pronounced, while no obvious effect on catalysis was observed. BTD is a very complicated reaction, which requires different active sites, including acid sites, basic sites23,24 and even redox sites.25 In this work, we believe the acid sites were excess relative to the other active sites and the key active sites of the catalyst were not acid sites. Of course, they were necessary. Key active sites mean the active sites are required for the rate control step, and their quantity and nature determine the rate of reaction. After 20 h of TOS, the performances began to decrease slowly, because the excess acid sites had been consumed by carbon deposition.
3.2 Quantity and composition of carbon deposition on the deactivated catalysts
TG analysis was used in order to specify the carbon type and quantify the carbon deposition of the used catalysts with different TOS. In Fig. 5, four maps were shown and each represented the used catalyst with 3 h, 10 h, 30 h and 60 h of TOS, respectively. As we can see from the figures, the major weight losses appeared with the temperature higher than 200 °C. It was reported in ref. 26 that the weight loss with the temperature lower than 200 °C may result from the desorption of molecular ethanol and water on the surface of the catalyst. In the present study, two major weight loss peaks were shown at the temperature of around 400 °C and 526 °C, which evidenced that two types of carbon species were formed during the ethanol and acetaldehyde conversion into BD process. But apparently, the integrated area of the two peaks varied, implying the change of the quantities of two carbon types upon TOS increasing. The quantities of two carbon types on the catalysts were calculated and the results were listed in Table 2. It is easy to draw that the quantities of the low temperature carbon and high temperature carbon were accumulating with TOS increasing. As shown in the Fig. S3† (drawn according to the data in Table 2), the quantity of the high temperature carbon was much higher than that of low temperature carbon and both type of carbons accumulated almost under the same increasing rate. Notably, the quantity of total carbon with 3 h of TOS was 3.78 wt% and with 60 h, the number reached 9.69 wt%. In other words, only 5.91 wt% of total carbon was formed during the 57 h reaction period. It is clear that the quantity of carbon deposition was relatively high during the first couple of hours which in return blocked most of the smaller pores of the catalyst. The TG results were in good accordance with the results of BET and NH3-TPD analysed above. Thus we can draw a conclusion that the deactivation of the catalyst was caused by the continuous increase of carbon deposition, which led to blockage of pores in catalysts and coverage of the acid sites on the surface. | ||
| Fig. 5 TG curves of catalysts with different TOS. (a) 3 h, (b) 10 h, (c) 30 h, (d) 60 h. | ||
| TOS (h) | Low temperature carbon (wt%) | High temperature carbon (wt%) | Total (wt%) |
|---|---|---|---|
| 3 | 1.55 | 2.23 | 3.78 |
| 10 | 1.55 | 3.14 | 4.69 |
| 30 | 2.27 | 3.75 | 6.02 |
| 60 | 3.58 | 6.11 | 9.69 |
Studies on the chemical structure of the carbon deposition during the reaction process were performed via FT-IR, 13C NMR and XPS. According to ref. 27, the bands at 3100–2800 cm−1 could be assigned to stretching vibration of C–H bonds. Specifically, the vibration of –CH in aromatic compounds was shown at 3050 cm−1, methyl hydrogen atoms was shown at 2970 cm−1 and symmetric stretching vibration of methylene could be detected at 2930 and 2860 cm−1. As revealed in Fig. 6, some typical bands were shown at 2981, 2937 and 2877 cm−1. The first band could be assigned to the vibration of the methyl hydrogen atoms, and the last two bands could be ascribed to the symmetric stretching vibration of methylene. Moreover, two additional bands were shown at 3452 and 1631 cm−1, which was assigned to the antisymmetric stretching vibration of hydroxyl groups of molecular water and physically adsorbed water, respectively. With TOS increasing, the absorbance of the methyl and methylene groups was enhanced, indicating the increasing quantity of those groups in carbon species. On the other hand, no aromatic or olefins C–H stretch (∼3050 cm−1) and aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch (∼1600 cm−1) were observed in the IR spectra,28 probably because there were no aromatic compounds in carbon species or aromatic compounds were too few to be detected.
C stretch (∼1600 cm−1) were observed in the IR spectra,28 probably because there were no aromatic compounds in carbon species or aromatic compounds were too few to be detected.
 | ||
| Fig. 6 FT-IR spectra of catalysts with different TOS. (a) 0 h; (b) 3 h; (c) 10 h; (d) 30 h; (e) 60 h. | ||
However, it has been reported in many literatures that aromatic compounds exist in the carbon deposition of organic reactions.29,30 To confirm whether there were aromatic compounds, 13C NMR was used to further study the carbon species. The catalyst with 60 h of TOS was chosen as a representative sample. The chemical shifted at the range of 0–50 ppm, 50–90 ppm and 90–150 ppm were correspond to saturated carbon species, C–O moieties and sp2 carbon (aromatic carbon, olefin carbon or graphitized carbon), respectively.31,32 As we can see from Fig. 7, two sharp peaks were shown at the chemical shift of 16.1 ppm and 59.6 ppm, which was assigned to the carbon species formed by saturated carbon and carbon species with C–O moieties, respectively. A broad peak found at the range of 115–150 ppm evidenced the formation of sp2 carbon during the reaction process. Together with the results of FT-IR spectra, it can be inferred that some amount of aromatic compounds was formed on the catalyst and most of them converted to graphitized carbon by dehydrocyclization.
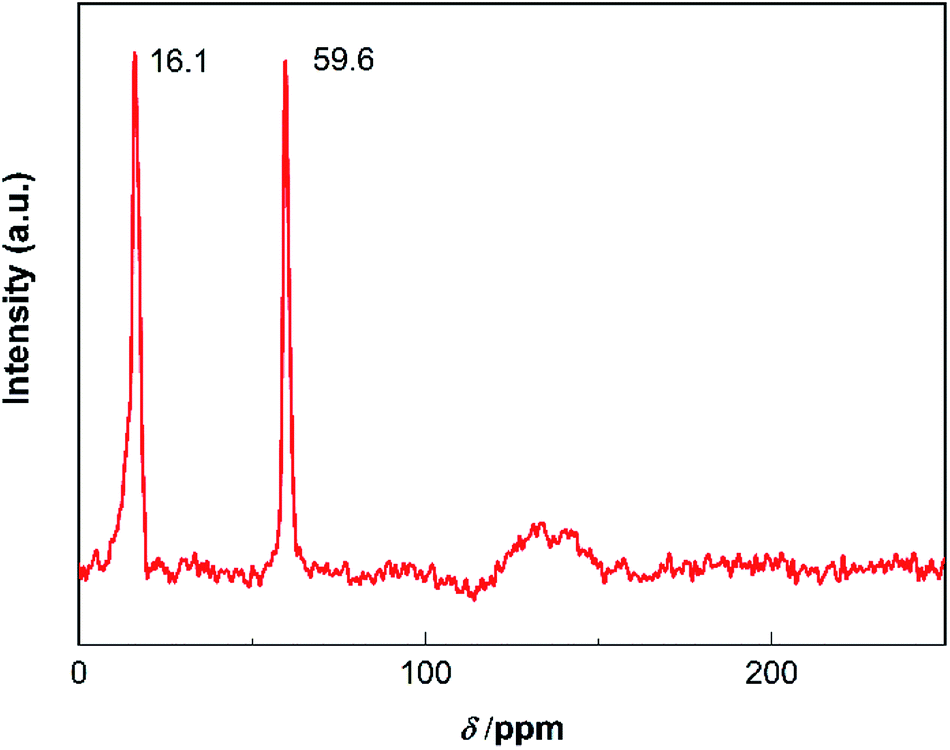 | ||
| Fig. 7 13C NMR spectra of the catalyst after 60 h of TOS. | ||
Further characterization by XPS was conducted to confirm the results as analysed above. The catalyst with 60 h of TOS was chosen as a representative sample. In Fig. 8, four banding energy values were shown at 284.2 eV, 285.2 eV, 286.4 eV and 288.1 eV, which was correspond to graphitized carbon, amorphous carbon formed by aliphatic hydrocarbon, C–O bond, carbonyl, respectively.33,34 To make a better insight of the comparison of the carbon species on the catalyst surface, the quantities of those four carbon species were listed in Table 3. As can be seen, the majority of carbon was graphitized carbon, which accounted for 52.2 wt% of total carbon. Amorphous carbon and carbon bonded to oxygen (most long-chain compounds containing C–O bonds, Fig. S4†) accounted for 28.0 wt% and 19.2 wt%, respectively. The rest was the CO bonded carbon. The result of XPS was in good agreement with the results of 13C-NMR and FT-IR characterization. Generally, the graphitized carbon formed in organic reactions was of high stability and difficult to remove, higher temperature was needed in thermal treatment.35 Thus, it can be concluded that the accumulation of graphitized carbon on the surface was the major cause of deactivation.
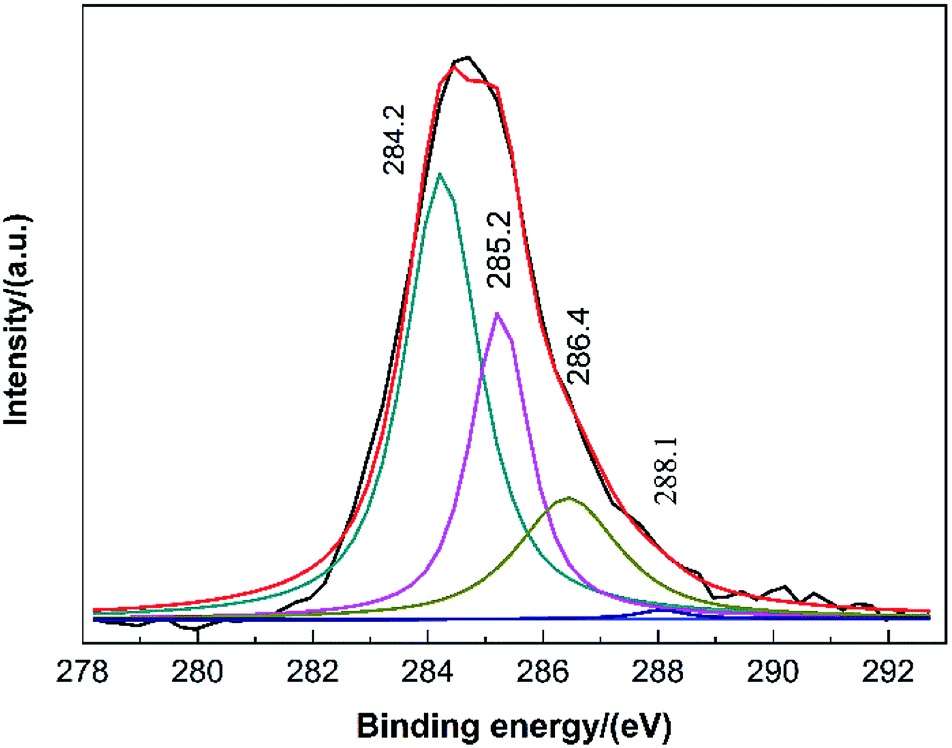 | ||
| Fig. 8 C 1s XPS spectrum of the catalyst with 60 h of TOS. | ||
| Binding energy (eV) | Chemical state of carbon | Quantity (wt%) |
|---|---|---|
| 284.2 | Graphitized carbon | 52.2 |
| 285.2 | Amorphous carbon | 28.0 |
| 286.4 | C–O bond | 19.2 |
| 288.1 | Carbonyl | 0.6 |
3.3 Regeneration of the deactivated catalysts
As analysed above, the blockage of pore structure and coverage of acid sites of the catalysts by carbon was the main reason of the deactivation. Generally, the deactivated catalysts caused by the carbon deposition can be regenerated by thermal-treatment. In this study, the used catalyst with 60 h of TOS was regenerated by calcining in air at 650 °C for 6 h with a temperature ramping rate of 5 °C min−1. The catalytic performances of the fresh and regenerated catalysts were listed in Table S1.† In addition, to better understand the process of regeneration, the catalysts were also regenerated in-suit in the reactor. Due to the change of conversion and BD selectivity was not obvious, such as in Fig. 1, we processed the data. As shown in Fig. S5,† both conversion and BD selectivity declined to some extent like regeneration in air. During 20 h of TOS, the conversion fell within 14%, while the selectivity ranged within 5%. Moreover, after 1st regeneration and 2nd regeneration in-suit, the catalytic performances of the deactivated catalyst were close to performances of the fresh one. Therefore, we believe that the heat treatment can basically restore the performances of the catalyst and there was not much difference between regeneration in air or regeneration in situ in the reactor.The pore parameters of fresh catalyst, catalyst with 60 h of TOS, catalysts after 1st regeneration and 2nd regeneration were listed in Table 4. As can be seen, the parameters of regenerated catalysts were largely improved compared with those of the used catalysts but still lower than those of the fresh catalyst, evidencing that the carbon species did block the pore structures and some structures had undergone permanent changes due to prolonged heat treatment.
| Catalyst | SBET (m2 g−1) | d (Å) | V (cm3 g−1) |
|---|---|---|---|
| Fresh | 563 | 25.5 | 0.36 |
| 60 h of TOS | 226 | 23.4 | 0.13 |
| 1st Regeneration | 514 | 24.6 | 0.32 |
| 2nd Regeneration | 530 | 25.1 | 0.33 |
In order to investigate whether the crystal structure of the regenerated catalyst changed during the coke oxidation process, the used catalysts were subjected to XRD characterization and the results were shown in Fig. 9. A broad halo at 20–30° existed in regenerated catalysts just as the fresh sample. No typical peaks for ZrO2 and ZnO crystals were found at the scan range of 2θ = 10–90°, evidencing a well-dispersion state of the ZrO2 and ZnO. Nevertheless, for the regenerated catalysts, a sharp peak was shown at 2θ = 27° and this peak could be assigned to the (011) crystal face of SiO2 (JCPDS 47-1144), which implied that some amount of silica crystal was formed through regeneration process. The conversion of amorphous silica to crystalline silica is not an easy task, which requires a high temperature.36,37 There may be two possible reasons. On the one hand, the introduction of Zr and Zn reduced the temperature, at which amorphous silica transformed into crystalline silica. Milonjic et al.38 concluded the transformation temperature decreased with increasing metal ion impurities in silica samples and Muroya et al.39 believed the growth of α-quartz was influenced strongly by cations such as Ca2+, Cd2+ and Zn2+. On the other hand, fresh catalyst did not show obvious crystalline state, so we speculate the second factor was heat treatment time. When the sharp XRD peak appeared, the catalyst had been heated at 650 °C for 12 h (catalyst calcination time + 1st regeneration time). With heat treatment time increasing (2nd regeneration time), the intensity of the peak at 2θ = 27° tended to be enhanced. Moreover, the amorphous phase of the silica was largely increased upon regeneration, but could not recover completely. This result confirmed the view that the decreased amorphous property of the deactivated catalyst may be the result of the coverage of the catalyst surface by carbon deposition and the prolonged high temperature treatment.
 | ||
| Fig. 9 XRD patterns for catalysts. (a) With 60 h of TOS; (b) 1st regeneration; (c) 2nd regeneration; (d) fresh catalyst. | ||
NH3-TPD was performed to analyze the acid property of the regenerated catalysts. As shown in Fig. 10, there was no significant difference between the fresh and regenerated catalysts on aspect of acidity. The acidity's quantities of the fresh, 1st regenerated and 2nd regenerated catalyst was 0.08 mmol g−1, 0.07 mmol g−1 and 0.06 mmol g−1, respectively. This result may be due to the heat treatment in air at 650 °C. From the pore parameters results in Table 4, the XRD results in Fig. 9 and the TPD results above, we can concluded that after prolonged heat treatment in regenerations, some structures undergone the permanent changes, while these small changes almost had none significant effect on performances of the regenerated catalysts.
 | ||
| Fig. 10 NH3-TPD profiles of fresh and regenerated catalysts. (a) Fresh catalyst; (b) 1st regeneration; (c) 2nd regeneration. | ||
3.4 The function of H2O in feed on the alleviation of carbon deposition
As we all know, the deactivation of the catalyst due to carbon deposition has brought many adverse effects to the industrial production and how to alleviate the catalyst carbon deposition has been a matter of great concern. As reported in ref. 40 and 41, some amount of H2O vapor in feed could alleviate carbon deposition on the catalyst to some extent. In addition, some literatures have illustrated the effect of water in feed in ethanol conversion. For example, Rahman et al.42 studied the effect of water on ethanol conversion on ZnO, concluding that molecular water could not only hinder ethanol dehydration by blocking strong Lewis acid sites but also inhibit acetaldehyde coupling to crotonaldehyde. Ochoa et al.43 also investigated the co-fed water on one-step ethanol to butadiene conversion over MgO/SiO2 catalysts, and unfortunately decrease of butadiene selectivity and rise of ethylene selectivity were observed, owing to the in situ formation of Bronsted acid sites by the adsorption of molecular water on Lewis acid sites. In this section, the effect of water in feed on the catalytic performance and carbon deposition in the conversion of ethanol/acetaldehyde to butadiene was studied and the amount of ethanol/acetaldehyde feed, regardless of the water content, was the same (WHSV (ethanol + acetaldehyde) = const).As shown in Table S2,† the result of water in feed in this study was beneficial as the addition of water increased the BD selectivity as well as ethanol/acetaldehyde conversion. With increasing the amount of H2O in feed, the production of EL and DE was prohibited to some extent, but excessive amount of water would lower the ethanol/acetaldehyde conversion. Zhu et al.44 studied the effect of water in the ethanol–acetaldehyde feed over MgO/SiO2 catalysts. They found that an appropriate amount of water (10 wt%) inhibited the formation of 1-butanol and heavier C6 compounds and to a certain extent decreased the selectivity to ethanol dehydration products (ethylene and ethyl ether), which was opposite to the results of Ochoa et al.,43 but similar to the results in this study. The authors concluded that after adding water in feed, different inhibition levels of ethanol conversion to acetaldehyde led to the increase of ethylene selectivity in the one-step process and acetaldehyde adding into the feed in two-step process led to avoid the hindrance of ethanol dehydrogenation. Therefore, after addition of some water (5–15 vol%) in feed, BD selectivity increased.
In order to investigate the function of water in feed to eliminate carbon deposition, TG analysis was applied by comparing the catalysts of the reaction with or without 10 vol% water in feed. There was a big difference on the distribution of carbon deposition between the two feeding methods. As shown in Fig. 11, except the peak occurred at 75 °C attributed to desorption of physically adsorbed water, three weight loss peaks were recorded when 10 vol% of water was added in feed and the temperature of each peak was 314 °C, 408 °C and 479 °C, respectively. While only two obvious peaks with 400 °C and 526 °C were found in the DTG curves of water free feeding. The high temperature carbon deposition peak shifted from the highest of 526 °C to 479 °C, which implied that the addition of water in feed could shift the high temperature carbon species to the high temperature carbon species with lower temperature. The quantity of carbon deposition of each catalyst was calculated and listed in Table 5. As shown in the table, the total quantity of carbon deposition dropped by 6.4%. The quantity of low temperature and high temperature carbon decreased by 5.8% and 39.5%, respectively. Quantity of lower temperature carbon increased from 0 wt% to 1.03 wt%. These results indicated that addition of water could decrease high temperature carbon species and increase carbon species with the lower temperature, including both lower temperature and low temperature carbon species. The conversion from high temperature carbon deposition to lower temperature and low temperature deposition was beneficial to alleviate the carbon deposition process.
 | ||
| Fig. 11 TG curves of catalysts with 10 h of TOS. (a) 10 vol% H2O in feed; (b) H2O free in feed. | ||
| Feed condition | Lower temperature carbon (wt%) | Low temperature carbon (wt%) | High temperature carbon (wt%) | Total (wt%) |
|---|---|---|---|---|
| H2O free | 0 | 1.55 | 3.14 | 4.69 |
| 10 vol% H2O | 1.03 | 1.46 | 1.90 | 4.39 |
4 Conclusions
In this work, different characterizations were performed to investigate the deactivation and the regeneration of the ZnO–ZrO2–SiO2 catalysts. Carbon deposition reduced the activity of the catalyst by blocking some pores and covering some acid sites and most of the carbon existed in the form of graphite. After regeneration, the performance of catalyst can almost completely restore. The addition of water into the feed could not only reduce the total amount of carbon deposition, but also shifted the high temperature carbon to low temperature or lower temperature carbon, which alleviated carbon deposition on the catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Key Laboratory for Green Chemical Technology of Ministry of Education of Tianjin University for technical support and the large precision instrument platform for XPS, XRD, 13C-MAS-NMR and SEM measurements.References
- V. L. Sushkevich and I. I. Ivanova, Appl. Catal., B, 2017, 215, 36–49 CrossRef CAS.
- Z. Han, X. Li, M. H. Zhang, Z. Z. Liu and M. X. Gao, RSC Adv., 2015, 5, 103982–103988 RSC.
- V. L. Sushkevich, I. I. Ivanova, V. V. Ordomsky and E. Taarning, ChemSusChem, 2014, 7, 2527–2536 CrossRef CAS PubMed.
- T. De Baerdemaeker, M. Feyen, U. Muller, B. Yilmaz, F. S. Xiao, W. P. Zhang, T. Yokoi, X. H. Bao, H. Gies and D. E. De Vos, ACS Catal., 2015, 5, 3393–3397 CrossRef CAS.
- M. D. Jones, C. G. Keir, C. Di Iulio, R. A. M. Robertson, C. V. Williams and D. C. Apperley, Catal. Sci. Technol., 2011, 1, 267–272 RSC.
- Y. Xu, Z. Liu, Z. Han and M. Zhang, RSC Adv., 2017, 7, 7140–7149 RSC.
- J. V. Ochoa, A. Malmusi, C. Recchi and F. Cavani, ChemCatChem, 2017, 9, 2128–2135 CrossRef CAS.
- W. Janssens, E. V. Makshina, P. Vanelderen, F. De Clippel, K. Houthoofd, S. Kerkhofs, J. A. Martens, P. A. Jacobs and B. F. Sels, Chemsuschem, 2015, 8, 994–1008 CrossRef CAS PubMed.
- S. Da Ros, M. D. Jones, D. Mattia, J. C. Pinto, M. Schwaab, F. B. Noronha, S. A. Kondrat, T. C. Clarke and S. H. Taylor, Chemcatchem, 2016, 8, 2376–2386 CrossRef CAS.
- X. Huang, Y. Men, J. Wang, W. An and Y. Wang, Catal. Sci. Technol., 2017, 7, 168–180 RSC.
- J. L. Cheong, Y. Shao, S. J. R. Tan, X. Li, Y. Zhang and S. S. Lee, ACS Sustainable Chem. Eng., 2016, 4, 4887–4894 CrossRef CAS.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- H.-J. Chae, T.-W. Kim, Y.-K. Moon, H.-K. Kim, K.-E. Jeong, C.-U. Kim and S.-Y. Jeong, Appl. Catal., B, 2014, 150, 596–604 CrossRef.
- R. A. L. Baylon, J. Sun and Y. Wang, Catal. Today, 2016, 259, 446–452 CrossRef CAS.
- A. Tripathi, K. Faungnawakij, A. Laobuthee, S. Assabumrungrat and N. Laosiripojna, Int. J. Chem. React. Eng., 2016, 14, 945–954 CAS.
- Q. Jiang, Z. Y. Wu, Y. M. Wang, Y. Cao, C. F. Zhou and J. H. Zhu, J. Mater. Chem., 2006, 16, 1536–1542 RSC.
- R. G. R. Avendano, J. A. de los Reyes, J. A. Montoya and T. Viveros, J. Sol-Gel Sci. Technol., 2005, 33, 133–138 CrossRef.
- T. P. de Castro, R. P. S. Peguin, R. C. R. Neto, L. E. P. Borges and F. B. Noronha, Top. Catal., 2015, 59, 292–302 CrossRef.
- Y. Li, C. Zhang, Y. Liu, S. Tang, G. Chen, R. Zhang and X. Tang, Fuel, 2017, 189, 23–31 CrossRef CAS.
- J. Ni, L. Chen, J. Lin and S. Kawi, Nano Energy, 2012, 1, 674–686 CrossRef CAS.
- O. V. Larina, P. I. Kyriienko and S. O. Soloviev, Theor. Exp. Chem., 2015, 51, 252–258 CrossRef CAS.
- V. V. Ordomsky, V. L. Sushkevich and I. I. Ivanova, J. Mol. Catal. A: Chem., 2010, 333, 85–93 CrossRef CAS.
- J. V. Ochoa, C. Bandinelli, O. Vozniuk, A. Chieregato, A. Malmusi, C. Recchi and F. Cavani, Green Chem., 2016, 18, 1653–1663 RSC.
- X. Huang, Y. Men, J. Wang, W. An and Y. Wang, Catal. Sci. Technol., 2017, 7, 168–180 RSC.
- C. Angelici, M. E. Velthoen, B. M. Weckhuysen and P. C. Bruijnincx, ChemSusChem, 2014, 7, 2505–2515 CrossRef CAS PubMed.
- V. V. Bokade and G. D. Yadav, Appl. Clay Sci., 2011, 53, 263–271 CrossRef CAS.
- P. E. Eberly Jr, C. N. Kimberlin Jr, W. M. Miller and H. V. Drushel, Ind. Eng. Chem. Process Des. Dev., 1966, 5, 193–198 CrossRef.
- J. P. Cain, P. L. Gassman, H. Wang and A. Laskin, Phys. Chem. Chem. Phys., 2010, 12, 5206–5218 RSC.
- A. Martinez, J. Rollan, M. A. Arribas, H. S. Cerqueira, A. F. Costa and E. F. S-Aguiar, J. Catal., 2007, 249, 162–173 CrossRef CAS.
- L. Pinard, K. Ben Tayeb, S. Hamieh, H. Vezin, C. Canaff, S. Maury, O. Delpoux and Y. Pouilloux, Catal. Today, 2013, 218, 57–64 CrossRef.
- J. D. Mao, A. Schimmelmann, M. Mastalerz, P. G. Hatcher and Y. Li, Energy Fuels, 2010, 24, 2536–2544 CrossRef CAS.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- H. Yang, F. Li, C. Shan, D. Han, Q. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- S. Pylypenko, T. S. Olson, N. J. Carroll, D. N. Petsev and P. Atanassov, J. Phys. Chem. C, 2010, 114, 4200–4207 CrossRef CAS.
- I. Rossetti, E. Bencini, L. Trentini and L. Forni, Appl. Catal., A, 2005, 292, 118–123 CrossRef CAS.
- T. Iwasaka, K. Inoue, R. Katayama and T. Uchino, J. Phys. Chem. C, 2012, 116, 6754–6761 CrossRef CAS.
- S. Tie and S. Zhang, Int. J. Mod. Phys. B, 2017, 31, 1744083 CrossRef CAS.
- S. K. Milonjic, L. S. Cerovic, D. M. Cokesa and S. Zec, J. Colloid Interface Sci., 2007, 309, 155–159 CrossRef CAS PubMed.
- M. Muroya and S. Kondo, Bull. Chem. Soc. Jpn., 1970, 43, 3453–3456 CrossRef CAS.
- T. Mendiara, J. M. Johansen, R. Utrilla, P. Geraldo, A. D. Jensen and P. Glarborg, Fuel, 2011, 90, 1049–1060 CrossRef CAS.
- R. Villa, C. Cristiani, G. Groppi, L. Lietti, P. Forzatti, U. Cornaro and S. Rossini, J. Mol. Catal. A: Chem., 2003, 204, 637–646 CrossRef.
- M. M. Rahman, S. D. Davidson, J. M. Sun and Y. Wang, Top. Catal., 2016, 59, 37–45 CrossRef.
- J. V. Ochoa, C. Bandinelli, O. Vozniuk, A. Chieregato, A. Malmusi, C. Recchi and F. Cavani, Green Chem., 2016, 18, 1653–1663 RSC.
- Q. Q. Zhu, B. Wang and T. W. Tan, ACS Sustainable Chem. Eng., 2017, 5, 722–733 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06757k |
| This journal is © The Royal Society of Chemistry 2018 |