
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing local structural disruption of Bid protein during thermal denaturation by absorption-mode ESR spectroscopy†
Chien-Lun Hung ,
Yu-Ying Lin,
Hsin-Ho Chang and
Yun-Wei Chiang*
,
Yu-Ying Lin,
Hsin-Ho Chang and
Yun-Wei Chiang*
Department of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: ywchiang@gapp.nthu.edu.tw
First published on 9th October 2018
Abstract
Bid is a requisite protein that connects death receptors to the initiation of mitochondrial-dependent apoptosis. Its structure was determined more than a decade ago, but its structure–function relationship remains largely unexplored. Here we investigate the thermostability of Bid protein and explore how the death-promoting function of Bid is affected by thermally-induced unfolding. First, we show by circular dichroism (CD) spectroscopy that Bid remains partially folded at high temperatures (350–368 K), and that the thermal unfolding of Bid is irreversible and accompanied with intermolecular associations that lead to protein aggregation. In 3 M GdnHCl, the onset of unfolding can, however, be conveniently observed at much lower temperatures around 320 K. We employ pulsed ESR dipolar spectroscopy to show that the structure of Bid remains almost unchanged between 0 and 3 M GdnHCl before thermal denaturation. More than 30 single-labeled Bid mutants are studied using the peak-height analysis method based on ESR absorption spectroscopy, in the temperature range of 300–345 K. The ESR results provide site-specific information about the temperature dependence of the local environment of Bid, thus enabling the discrimination between the onsets of unfolding and aggregation for respective sites. Consequently, we map out the local thermostability over the Bid structure and identify a new interface between helices 3, 6, and 8 as the beginning of structural unfolding. This study also investigates the apoptotic activity of the thermally-induced Bid aggregates and shows that Bid retains the death-promoting function even when unfolded and aggregated. The applicability of the new ESR absorption peak-height method is demonstrated for protein thermostability.
Introduction
The BCL-2 (B-cell lymphoma 2) protein family plays a crucial role in mitochondria-mediated apoptosis.1,2 The common feature of this family is that they contain one or more BCL-2 homology (BH) domains. Depending on the role they play in opposing or promoting cell death, the family members are divided into anti- and pro-apoptotic groups. Upon receipt of apoptotic stimuli, proteins in the pro-apoptotic group, which can be sub-divided into multi-BH domain proteins (such as BAX, BOK, and BAK) and BH3-only proteins (such as Bid, PUMA, Bad, and Bim), are post-translationally modified to trigger the apoptotic pathways. For example, full-length Bid (BH3 interacting domain death agonist) is cleaved by caspase-8 at site D59 (for mouse Bid) to produce cleaved Bid (cBid) comprising two fragments (p7 and p15), in which the latter is termed truncated Bid (tBid).3–5 tBid is known to directly or indirectly interact with BAX and BAK proteins and induce conformational changes leading to the activation of BAX and BAK, hence promoting mitochondrial outer membrane (MOM) permeabilization (MOMP).6,7 Interestingly, while many studies find that truncation of Bid is essential for Bid-induced MOMP, there are also reports suggesting a pro-apoptotic role of full-length Bid.8–12 It remains a mystery as to by what means a full-length Bid can be triggered to propagate apoptotic signaling. Studies on the structure–function relationships are urgently needed.Structures of (mouse and human) Bid proteins have been determined using NMR spectroscopy for more than a decade.13,14 Notably, of all the BH3-only proteins studied until now, only Bid shows a defined structure while others are intrinsically unstructured in the absence of binding partners.15 However, what has received little attention is the stability and folding/unfolding of Bid. Studies on the stability of Bid have been sparsely reported.16 A previous result shown by circular dichroism (CD) spectroscopy that full-length Bid is a highly stable protein with a thermal denaturation midpoint difficult to be determined; its secondary structure remains largely unchanged until temperature is increased to the range of 80–90 °C. The onset temperature of thermal denaturation is so high that revealing a clear sigmoidal curve in the CD measurements has been challenging. Cleavage of Bid was reported to reduce the melting temperature to 77 °C. It is only when tBid (i.e., the p15 fragment of Bid) was used that a clear sigmoidal curve with a midpoint temperature approximately 60 °C was observed.16 It has been long desired to develop a biophysical methodology for exploring molecular details of folding/unfolding of such a highly thermostable protein.
This study aims to explore the thermostability of full-length Bid protein using site-directed spin-labeling (SDSL) electron spin resonance (ESR) spectroscopy. The SDSL-ESR approach has been demonstrated as a powerful tool for biophysical chemistry.17–24 The advantage of the ESR approach lies in site specificity as the denaturation of local domain can be followed independently of global proteins. In the present report, more than 30 cysteine variants of Bid mutant have been studied using continuous-wave (cw) and pulsed ESR techniques during thermal denaturation. An improvement in the analysis of cw-ESR spectra is introduced to resolve the difficulty of studying thermostability of Bid protein at varying temperature. The ESR results provide site-specific information on temperature dependence of local environment in Bid protein, onset of transition between native and denatured states, long range interspin distance measurements that allow one to reveal the conformational changes of Bid in response to thermal and chemical denaturation effects. This study, therefore, improves our understanding about the stability of Bid on a molecular level and, moreover, reveals new insights into how the intermolecular interactions in thermally unfolded Bid proteins are induced to promote protein aggregation and affect the death-promoting function of Bid. All of the information is valuable to the therapeutic use and storage of this apoptotic Bid protein.
Results
To study the stability of Bid protein (Fig. 1A), we collected CD spectra in the far-UV region (Fig. 1B) in the temperature range of 298–368 K (25–95 °C). At low temperatures, CD spectra of wild-type (wt) Bid are characterized by two distinct minimum bands at 208 and 222 nm, in line with the literature spectrum of Bid protein.16,20 Interestingly, at the highest temperature studied (368 K) the CD spectrum is by no means similar to the spectrum of random coil, indicating that Bid retains some secondary structure and remains partially folded at 368 K. Fig. 1C shows the temperature dependence of CD signals at 222 nm, recorded in a heating and cooling cycle (298–368 K, with intervals of 5 K). In the heating process, the mean residual ellipticity (MRE) signals at 222 nm (Fig. 1C) decrease gradually with increasing temperature and diminish abruptly when the temperature is (approximately) >350 K (>77 °C). The temperature dependence of the MRE signal is consistent with the expectation for a protein unfolding during thermal denaturation, though the onset temperature of unfolding is unusually high. In the cooling process (following the heating), the magnitude of the MRE signals at 222 nm is not restored when temperature is decreased from 368 K to 298 K (Fig. 1C), indicating that the thermally-induced unfolding in Bid proteins is irreversible. The majority of the thermally-denatured Bid proteins failed to refold to the native structure when temperature was recovered to the initial temperature of 298 K.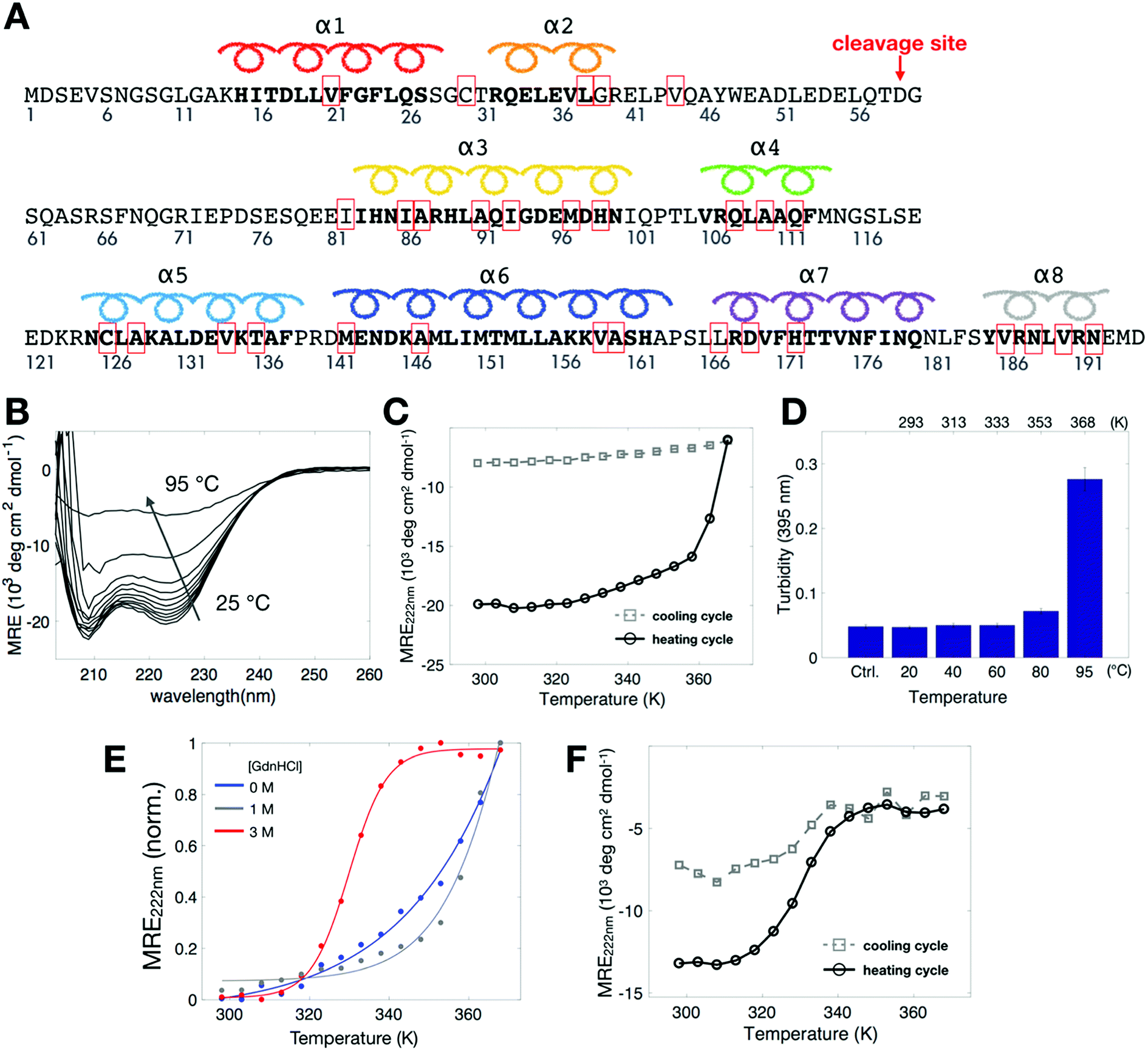 | ||
| Fig. 1 Thermal denaturation study of Bid by CD spectroscopy. (A) The sequence of mouse wt-Bid protein. The sites, which were mutated to cysteines to form disulfide bonds with the MTSSL probe (designated as R1), are highlighted with red boxes. D59 in mouse Bid is the cleavage site for caspase-8. (B) Thermal denaturation (298–368 K, or 25–95 °C) study of wt-Bid by CD in the far-UV region. Bid is not completely unfolded at high temperatures. (C) MRE at 222 nm for wt-Bid in the heating and then cooling cycle (298–368 K, with intervals of 5 K) in 0 M GdnHCl. (D) Wt-Bid samples (10 μM) were incubated at the indicated temperatures for 1 h. The extent of aggregation was determined by turbidity (absorbance at 395 nm). Buffer without proteins serves as control. Values represent means ± S.D. (n = 5). (E) MRE at 222 nm in the heating process when wt-Bid was incubated in different concentrations of GdnHCl. (F) MRE at 222 nm in the heating and cooling cycle for wt-Bid in 3 M GdnHCl. Measurements were repeated to verify the irreversibility of the thermal unfolding. | ||
During the heating process we observed that the protein solution tends to become opaque after heated up to temperatures >90 °C. The opaque solution suggests that at high temperatures the unfolding of Bid proteins takes place along with intermolecular associations, which consequently leads to protein aggregation. To investigate the aggregation in the opaque solutions, we measured turbidity of the solutions after heat treatments of different temperature for 1 h (Fig. 1D). The result shows that Bid proteins aggregate, which leads to large increase in turbidity, only when incubated at temperatures >90 °C (363 K). Taken together the results of CD and turbidity measurements, we found that Bid is a thermally stable protein, which remains largely folded at temperatures below 350 K (approximately), and that the thermally-induced unfolding begins to occur at temperatures above 350 K and is accompanied with intermolecular interactions that lead to protein aggregation at elevated temperatures. It is desirable to seek an approach to weaken the stability of Bid, to better characterize the transition between folded and unfolded states, and to distinguish the onset of unfolding from the onset of protein aggregation.
To weaken the stability of Bid proteins, we prepared Bid proteins in solutions containing different concentrations of denaturant GdnHCl (guanidine hydrochloride) and performed CD measurements. A clear sigmoidal-like curve (Fig. 1E) was gradually revealed in the CD spectra when the concentration of GdnHCl was increased from 0 to 3 M. In 3 M GdnHCl, the MRE signal at 222 nm begins to decrease drastically at temperature around 320 K (i.e., the onset of unfolding) and reaches a plateau around 340 K. The reversibility of the thermal denaturation for Bid proteins in 3 M GdnHCl is examined in a heating and cooling cycle. It (Fig. 1F) basically shows that the thermally-denatured Bid in 3 M GdnHCl fails to refold to the native state in the cooling process, indicating that the intermolecular association between the unfolded proteins remains somewhat at high temperatures. The presence of GdnHCl in protein solution is mainly useful to weaken the stability of Bid, which consequently reduces the onset temperature of unfolding from >350 K to 320 K. The CD studies reveal the onset of unfolding during heat denaturation and show that the unfolding is irreversible as it is followed by intermolecular association (hence aggregation, evidenced by apparent precipitation and turbidity) at high temperatures.
Study of protein conformation by DEER spectroscopy
To investigate what changes in the conformation of Bid are caused by the presence of 3 M GdnHCl, we performed double electron-electron resonance (DEER)25 experiment to measure distances in spin pairs so as to evaluate the structural differences in Bid between 0 and 3 M GdnHCl. Basically, spin-labeled Bid proteins at 300 K were plunge-cooled to liquid nitrogen (ca. 80 K) following a standard procedure for DEER measurement, as detailed in Experimental. Previously, we showed that the partially unfolded structures of cytochrome c protein corresponding to different concentrations of GdnHCl (0, 0.7, and 1.5 M) can be obtained using this DEER method.26 Here, three double-labeled Bid mutants (30/126R1, 30/82R1, and 82/126R1; shown in Fig. 2A) were examined by DEER spectroscopy. (Note that 30/126R1 represents the doubly spin-labeled wt-Bid as wt-Bid has two native cysteine residues C30/C126.) Strikingly, the DEER time-domain raw experimental data (Fig. 2B) are highly similar between the results in 0 M versus 3 M GdnHCl. The time-domain data were analyzed to yield the inter-spin distance distributions (Fig. 2C) using the approach combining the Tikhonov regularization and the maximum entropy methods.18,26,27 The average distances of the distance distributions are consistent with the structural model of Bid (PDB: 1DDB).14 Predications of the distance distributions (Fig. S1A†) from MtsslWizard28 analysis of the PDB structure are in a good agreement with the DEER results. Most importantly, it demonstrates that Bid protein retains its conformation in the presence of 3 M GdnHCl. It suggests that although the presence of 3 M GdnHCl weakens the structural stability (as evidenced in Fig. 1E), it affects insignificantly the structural conformation of Bid. | ||
| Fig. 2 Exploring structural differences between 0 and 3 M GdnHCl with DEER spectroscopy. (A) Cartoon model of Bid protein (PDB: 1DDB) highlighting helices 1, 2, 3, and 5. Three pairs of cysteine variants of Bid were engineered to obtain three double-labeled mutants, 30/126R1, 30/82R1, and 82/126R1. As displayed, the global structure of Bid is well represented by the three sites. (B) Raw experimental data (blue) of the DEER measurements for the indicated mutants in 0 M versus 3 M GdnHCl. Fits to DEER background signals are shown in red. (C) Interspin distance distributions and average distances obtained from analyzing the DEER data in (B). Results of 0 M and 3 M GdnHCl are shown in black and gray lines, respectively. The distance distributions are highly similar between the two GdnHCl conditions. | ||
Cw-ESR reveals local structural changes due to temperature and denaturant
To reveal site-specific information about local stability in Bid, we collected cw-ESR spectra of various single-labeled Bid mutants, one at a time, at 300 K for 0 and 3 M GdnHCl (Fig. 3A). For this study, 30 single-cysteine substitution mutants (denoted by red boxes in Fig. 1A) over the entire protein were engineered and subsequently spin labeled with the R1 side chain. CD measurements were performed to verify that the cysteine variants of Bid retain the same structure as wt-Bid (Fig. S1B†). Importantly, we found that most of the cw-ESR spectra are similar between the solutions containing 0 versus 3 M GdnHCl. Exceptions are the results of 21R1, 147R1, 159R1, 167R1, and 186R1, whereas the spectral differences are reasonably small reporting subtle changes in the local environments. Cw-ESR is a common biophysical approach for studying protein dynamics.24,29–33 The cw-ESR spectrum of a spin-labeled protein encodes information on the overall motion of the nitroxide ring on the nanosecond time scale with contributions mainly from the internal motion of the R1 side chain, which can be modulated by interactions of the spin probe with the environment, and rotational diffusion of the protein. The spectral lineshapes obtained (Fig. 3A) all reflect distinct variations in detail from site-to-site attributable to differences in amplitude and/or rate of motions on the ns time scale. The local environments over the Bid protein structure can therefore be characterized by the lineshape changes. Also, this site-specific cw-ESR study supports that little difference occurs in the local environment of Bid between 0 and 3 M GdnHCl, in a good agreement with the conclusion drawn from the DEER measurements (Fig. 2). | ||
| Fig. 3 Studying local structural changes in Bid using cw-ESR. Cw-ESR spectra of various single-labeled Bid mutants in 0 M and 3 M GdnHCl conditions, reordered at temperatures (A) 300 K and (B) 345 K. All spectra are scaled to have the same height in the central peak for a clear presentation. Spectra of individual sites in the two [GdnHCl] conditions are reasonably similar at the lower temperature 300 K, supporting a view that Bid structure remains undisturbed. Whereas, many spectra of individual sites show clear dissimilarity between the two [GdnHCl] conditions at high temperature 345 K. At 345 K, the spectra of 0 M GdnHCl are generally characterized by a greater linewidth and spectral anisotropy as compared to the spectra of 3 M GdnHCl. This indicates that some local structures in Bid are not completely disrupted in 0 M GdnHCl at 345 K. | ||
We also repeated the above cw-ESR measurements at an elevated temperature 345 K (Fig. 3B). All of the spectra for 3 M GdnHCl at 345 K exhibit a similar lineshape, characterized by three sharp peaks indicating of a highly mobile dynamic. It indicates that at 345 K and 3 M GdnHCl the local environments in Bid are mostly unrestricted and the local structures are disrupted. As opposed to the 3 M GdnHCl result (Fig. 3B), the majority of the spectra for 0 M GdnHCl at 345 K are characterized by relatively broadened linewidths representing anisotropy. Several spectra (e.g., 91R1, 126R1, 159R1, 167R1, 186R1) retain the characteristics of restricted and immobile lineshape. It indicates that some local structures in Bid remain folded at 345 K.
Spectroscopic evidence for the T-induced structural changes of Bid
To investigate the temperature dependence of the local stability in Bid proteins, we recorded cw-ESR spectra of single-labeled Bid proteins in the temperature range of 300–345 K in the absence and presence of 3 M GdnHCl. (The whole collection of the temperature-dependent spectra is given in Fig. S2.†) Fig. 4A shows two typical examples demonstrating the sensitivity of cw-ESR to the local stability in Bid. The spectra of 39R1 between 0 and 3 M GdnHCl are alike at low temperatures, but they change differently with increasing temperature, from the slow-motional-like lineshape (featured by broadened linewidth) to the fast-motional-like lineshape (characterized by three sharp peaks) at high temperatures. In 3 M GdnHCl, the 39R1 spectra display a clear transition from the broadened to the sharp linewidths in the temperature range around 320–330 K (as highlighted by blue dashed lines in Fig. 4A). This is important spectroscopic evidence warranting more rigorous spectral analysis.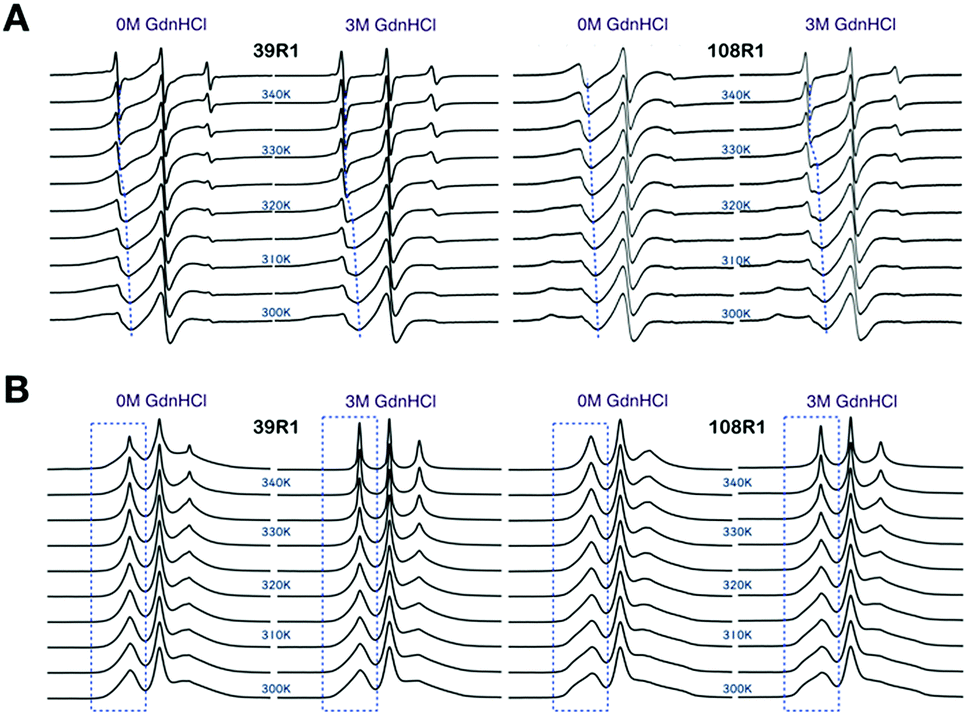 | ||
| Fig. 4 Cw-ESR spectra of 39R1 and 108R1 in 0 M versus 3 M GdnHCl, recorded in the temperature range of 300–345 K (in increments of 5 K). (A) Spectra displayed in the first-derivative mode. Blues lines are guides to the eye indicating the shift in position of lower-field peak, important spectroscopic evidence, with temperature. (B) Spectra converted to the absorption mode. Blue boxes highlight that when displayed in the absorption mode, the lower-field peak is more sensitive to present the temperature-dependent effect (which includes sharp and broadened spectral components) on spin label mobility. Totally, 30 single-labeled Bid mutants were studied. See Fig. S2† for the temperature-dependent ESR spectra of the all Bid mutants. | ||
The spectra of 108R1 (Fig. 4A) represents another typical example of the spectra in this study. The spectra of 108R1 are similar between 0 and 3 M GdnHCl at low temperatures, both of which display lineshape reflecting anisotropic slow motion. Likewise, the spectral lineshape changes distinctly and continuously with increasing temperature. At 345 K, the spectra for 0 M GdnHCl exhibit an increase in the spectral mobility, but the corresponding linewidth remains large compared to the spectra of 39R1 at the same temperature, which indicates that the local environment of 108R1 is relatively less exposed and unfolded in 0 M GdnHCl. As denoted by dashed guide line (Fig. 4A), the spectral characteristic peak of 108R1 in 0 M GdnHCl changes smoothly with temperature, showing no sign of discontinuity. However, the spectra of 108R1 in 3 M GdnHCl display a typical fast-motional isotropic lineshape at high temperatures, which indicates of the local environment being highly mobile and unstructured. A clear discontinuity in the spectra (3 M GdnHCl) is observed around 330 K. Their counterpart spectra presented in the absorption mode are shown in Fig. 4B. When presented in the absorption mode, the lower-field peak is found to be more sensitive to exhibit the temperature-dependent changes (highlighted by blue boxes in Fig. 4B). The lower-field peak height (in the absorption mode) is therefore used to yield a convenient measure of the ESR lineshape variations with temperature. It is based on the strong spectroscopic evidence discussed above that we develop the peak-height analysis method, as detailed in Experimental section, to determine the onset of local disruption of Bid protein during thermal denaturation.
Fig. S3† provides another comparison highlighting the spectral differences between 300 K and 345 K. In 0 M GdnHCl, many of the spectra recorded at 345 K exhibit some similarity to the spectra of 300 K and display lineshapes reflecting anisotropy. This indicates that the local environments in Bid remain somewhat structured and folded even at 345 K. In 3 M GdnHCl, spectra are highly dissimilar between 300 K and 345 K; all of the spectra recorded at 345 K exhibit lineshape of highly mobile and unstructured characteristics, indicating that in 3 M GdnHCl, Bid is completely unfolded at temperatures >345 K.
Results of the ESR absorption peak-height analysis
To systematically extract the mobility information from the recorded temperature-dependent spectra, we analyze the spectra in the normalized absorption mode (see Experimental section) and plot lower-field peak heights of the spectra as a function of temperature (Fig. 5). The analysis principles are given in the following. The peak-height data provide two important pieces of information about local environment: (1) in the lower temperature range, in which protein remains structured and folded, they provide the site-specific information about temperature dependence of the local environment in a native protein fold; (2) when there is a changeover in the slope of the peak-height plot, it provides the site-specific information about the onset of local structural disruption due to thermal denaturation. Considering a globular protein freely tumbling in a solution, the peak-height data of its ESR absorption spectra would increase linearly with increasing temperature as demonstrated in Fig. S4.† Basically, the mobility of the spin label increases with increasing temperature, which consequently leads to a decrease in the linewidth of ESR spectrum, hence an increase in the peak height. An abrupt change in the local environment of the spin-labeled site (e.g., an exposure to solvent due to protein structural changes, an increase in the mobility of spin-label side-chain due to disruption of local structure, etc.) would lead to a distinct change in the slope of the temperature-dependent plot of the ESR peak height. By determining the changeover in the peak-height plot, we can, therefore, identify the onset of local structural change during thermal denaturation.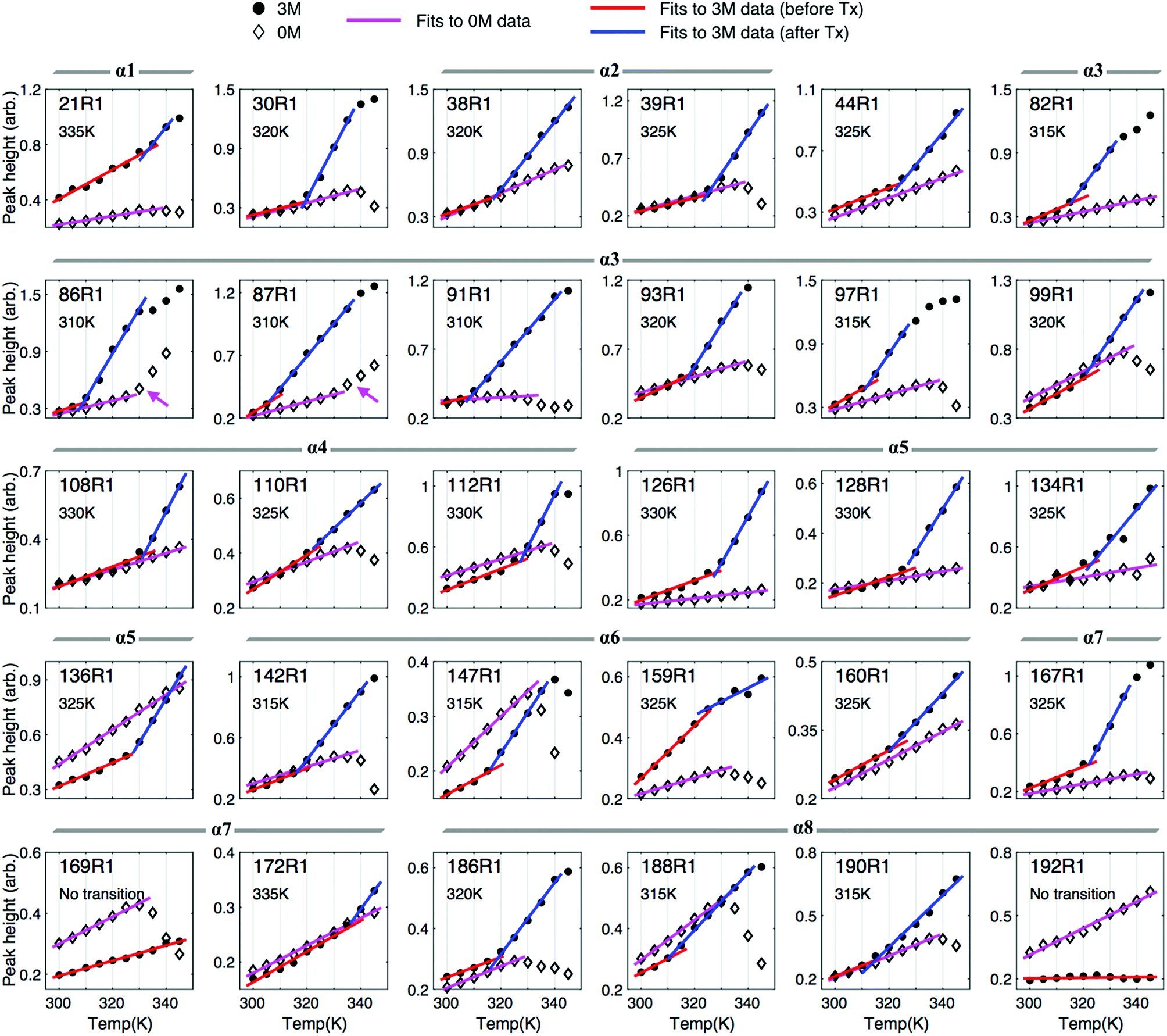 | ||
| Fig. 5 Monitoring the onset of local unfolding in Bid during thermal denaturation with the peak height of the spectra in the absorption mode. The peak-height data were analyzed in a linear regression fit with criteria given in Experimental section. Basically, the peak height increases with increasing temperature, consistent with the general expectation for temperature dependence of ESR lineshape, whereas at high temperatures it drops largely with increasing temperature due to the increased intermolecular associations in the thermally unfolded proteins. For the results of 3 M GdnHCl, we can easily identify a changeover in the slope of the peak-height data. The changeover temperature (Tx), denoted in each of the subplots, indicates the onset of local unfolding in Bid during thermal denaturation. | ||
First, we describe the results of the peak-height analysis for 3 M GdnHCl in the following (Fig. 5). In contrast to the data for 0 M GdnHCl, we can easily identify a drastic increase in the slope of the data for most of the 3 M GdnHCl results. The peak-height data were analyzed (cf. Experimental section) to determine the first changeover temperature (Tx, as denoted in each subplot) encountered when increasing temperature from the lower temperature side. Linear regression fits to the data of lower temperatures (<Tx) and higher temperatures (>Tx) are plotted in red and blue colors, respectively. Among the 30 mutation sites studied, 28 of them display a similar trend with increasing temperature; the slope of the red lines, which corresponds to the data < Tx, is distinctly less than the slope of the blue lines. The increase in the slope with increasing temperature (from red to blue lines) indicates that the local environment of the spin-labeled sites becomes more unrestricted and mobile upon the changeover, consistent with the expectation that Bid proteins become largely unfolded at high temperatures in 3 M GdnHCl. Specifically, the averages (and standard deviations) of the obtained slopes are 0.0067 (0.0033) and 0.0236 (0.0124) for red and blue lines, respectively. The small variation in the slopes of the red lines indicates that all of the spin-labeling sites experience a similar temperature-dependent effect. The difference in the slope between the blue and red lines is distinct. The values of Tx are thus determined by the intersection of the blue and red lines. The determination of Tx provides site-specific information about the thermally-induced effect that causes the local disruption of Bid structure.
Note that few data points at high temperatures are excluded from the analysis because they are clearly deviated from the blue lines. The deviation occurs since at high temperatures the intermolecular association in the (partially) unfolded Bid proteins is increased, which leads to protein aggregation even in the presence of 3 M GdnHCl. As a result, the population of the immobile spectral component is increased, hence causing a linewidth broadening and, consequently, a reduction in the peak height. As a result, data points would deviate from the trend of blue line when the aggregation-induced broadened component is present. As these excluded data points show a decrease with increasing temperature, they also provide support for the protein aggregation at high temperatures.
Below, we describe the peak-height plot for 0 M GdnHCl (Fig. 5). In contrast to the results of 3 M GdnHCl, the data for 0 M GdnHCl vary (relatively) insignificantly (in magnitude) with temperature. Except for the results of 86R1 and 87R1 (denoted by magenta arrows in the subplots), an unambiguous determination of Tx for the 0 M GdnHCl data is hardly possible. The data points for 0 M GdnHCl from lower temperatures are analyzed by linear regression fits (magenta color in Fig. 5), yielding 0.0051 and 0.0024 for average slope and derivation, respectively. Basically, all of the peak-height data increase gradually with increasing temperature, consistent with the expectation for the temperature dependence of ESR spectra. At higher temperatures (ca. >335 K), most of the peak-height data points, however, decrease largely with increasing temperature, suggesting that strong intermolecular interactions in the thermally-unfolded proteins take place, which leads to an increase in the population of broadened component, hence a decrease in the peak height. Therefore, data points at the higher temperatures cannot be analyzed using the linear regression method. Totally, 18 out of the 30 samples studied were observed to have strong intermolecular associations at high temperatures (for 0 M GdnHCl).
Mapping local structural disruptions of Bid during thermal denaturation
Fig. 6 shows a plot of the onset temperatures of unfolding (Tx) determined from the peak-height analysis for 3 M GdnHCl. The red dashed line (Fig. 6A) indicates the onset temperature (320 K) of wt-Bid determined from the CD study, which is also close to the average of Tx (321 K) determined in the ESR study. These onset temperatures of unfolding allow one to explore how each of the sites studied responds to temperature variations. Strikingly, we found that sites with relatively lower Tx (≤ 315 K) are spatially separated from those with relatively higher Tx (≥ 320 K; Fig. 6B). For a comparison, a cartoon model of Bid (colored by helix) is shown (Fig. 6C) to indicate that the sites of the lower Tx are at the interfaces between helices 3, 6, and 8, providing new insights into the molecular interface in Bid protein. Based on the ESR results, a model for the thermally-induced unfolding of Bid is proposed (Fig. 6D) to uncover the sequence of unfolding events. Our results suggest that the thermally-induced unfolding of Bid initiates from the interfaces between helices 3, 6, and 8, followed by the disruption of the associated helices 4 and 5 at elevated temperatures. The first event occurs during thermal unfolding of Bid is the exposure of BH3 domain (i.e., residues 90–98 within α3 facing the association interface with α6 and α8) rather than the separation of the p7 and p15 segments. | ||
| Fig. 6 Mapping thermal unfolding events in Bid protein. (A) The onset temperatures (Tx) of Bid unfolding (for 3 M GdnHCl) determined from the peak-height data, with estimated errors of ±2 K. The average of the Tx temperatures is 321 K, denoted by red dashed line, and is close to the onset temperature (ca. 321 ± 2 K) in the sigmoidal curve of CD spectra shown in Fig. 1E. This observation provides some support to the suitability of the ESR method for protein stability. (B) A cartoon model illustrating the positions of the studied sites in Bid protein. They are colored according to Tx values. (C) A cartoon model exhibiting the 8 helices of Bid protein, for a convenient comparison with the presentation in B. (D) A cartoon model illustrating that the disruption of the interface between helices 3, 6, and 8, which leads to the exposure of the BH3 domain, is the initial event during thermal denaturation. | ||
Native structure of Bid is not important to the death-promoting function
To explore the biological significance of the thermally-induced unfolding and aggregation, we performed assays of cytochrome c release from mitochondria (Fig. 7). Bid is known to promote the apoptotic activity of BAX and its activity is enhanced when cleaved by caspase-8.4,9,34 Our result shows that in the presence of BAX and caspase-8, wt-Bid is most potent for inducing the release of cytochrome c from mitochondria (cf. lanes 1–5 in Fig. 7), consistent with the expectation for Bid activity. After different heat treatments (70 or 95 °C incubation for 1 h; Fig. 7) were applied to Bid proteins, the activity of Bid to promote the BAX-induced cytochrome c release seemed to change insignificantly (cf. lanes 6–8 in Fig. 7). Interestingly, the heating treatment brings about structural unfolding and subsequent Bid aggregations, but it causes insignificant change to the death-promoting function of Bid protein.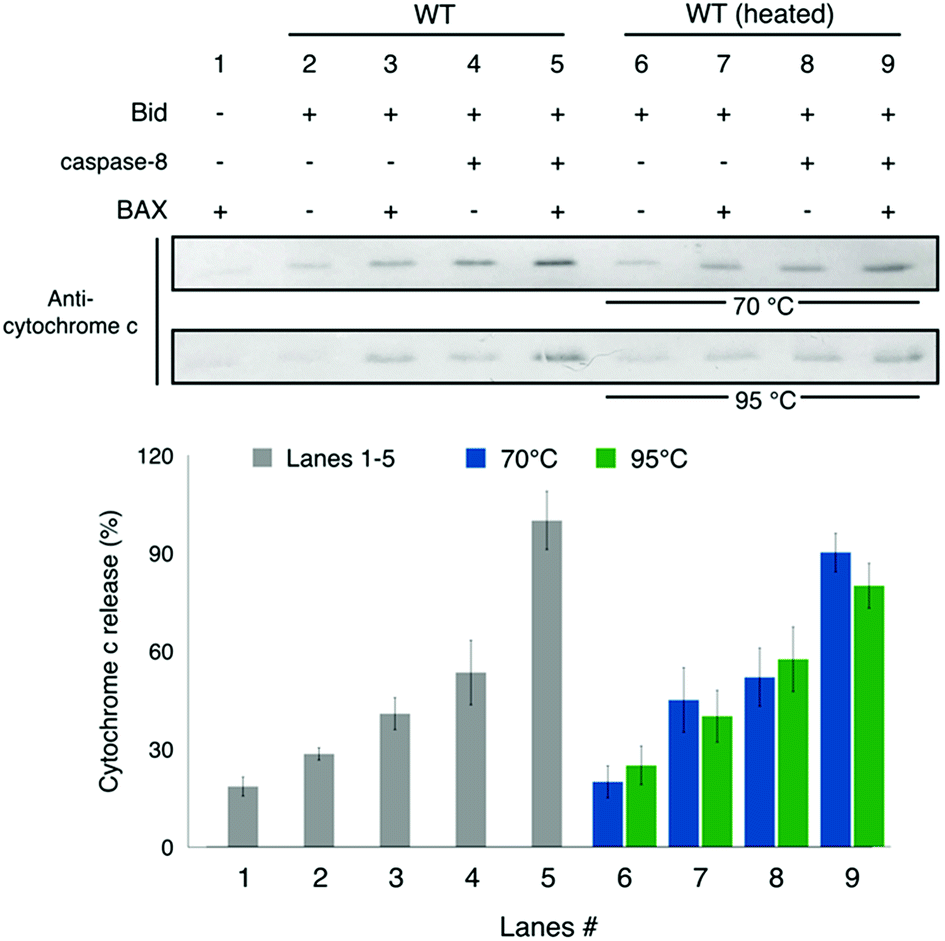 | ||
| Fig. 7 Assay for cytochrome c release from mitochondria. Lanes 1–5 show that the incubated Bid is most potent for promoting the BAX-induced cytochrome c release from mitochondria when caspase-8 is present that causes the cleavage of Bid, consistent with the understanding of Bid. Lanes 6–9 show that after the heat treatments (70 or 95 °C incubation for 1 h) to Bid, the activity of Bid to promote the BAX-induced cytochrome c release is barely affected. It suggests that the heat-treated Bid proteins retain the death-promoting function. Error bar represents the mean ± SD (n = 5). See also Fig. S8.† | ||
Discussion
Differences in stabilizing force within the structures of BAX and Bid
Although Bid belongs to the BH3-only subgroup in the Bcl-2 family, structural and phylogenetic evidence suggests that it is both structurally and functionally related to proteins in the multi-BH subgroup such as BAX and Bcl-XL.5,35–39 Therefore, Bid is also known as BAX-like BH3 protein.5 Both Bid and BAX were found to proceed with membrane association by similar mechanisms driving the proteins to mitochondrial membrane.5,40–42 Despite the similarities, Bid and BAX actually participate differently in the regulation of mitochondria-dependent apoptosis. While both of them are considered as “direct activator” for the induction of apoptosis, BAX participates to form membrane pores in the MOM and subsequently induce MOMP, whereas Bid only promotes the BAX-mediated apoptosis rather than merges into the pores with other members. It remains unclear as to what leads to the difference in the behavior of theses structurally-alike proteins.Previously, BAX was found to be highly stable against thermal and chemical denaturations.43 As evidenced by the strong CD signals of BAX in the near-UV region, aromatic interaction in side chains was demonstrated to play an important role in stabilizing the structure of BAX protein against the denaturing effects. The aromatic interaction in the C-terminal region (CTR) of BAX (helices 4–9) is so strong that the CTR of BAX remains folded even in 6 M GdnHCl or at temperature 95 °C. This present study has performed the same turbidity measurements (as done in Fig. 1D for Bid) with heat-treated BAX and found no sign of aggregation after the heat treatments (40–95 °C). Contrary to what was found for the stability of BAX, the present study shows that Bid proteins unfold and subsequently associate to form aggregates at high temperatures, and moreover they are largely unfolded in 6 M GdnHCl (Fig. S5A†). In measuring CD signals of Bid in the near-UV region, the recorded signals are insignificant, indicating that Bid has hardly any aromatic interaction in structural stabilization. To further verify this finding, we performed calculations using the protein interaction calculator (PIC) and the CaPTURE program to evaluate the contribution of aromatic interactions to the structure of Bid.44,45 As opposed to the previous finding (using the same simulation programs) that a total of 12 aromatic pairs exist within BAX, we found that aromatic interactions barely exist in Bid; specifically, the calculations indicate that the total number of cation–pi interactions (with energy less than −1.0 kcal mol−1) in Bid structure is zero. This study, therefore, suggests that Bid is primarily stabilized by hydrophobic interactions in side chains (cf. Fig. S5B†), in line with previous studies of Bid that the hydrophobic face of the BH3 domain must remain buried to prevent Bid from activation.13,46 Also, this study reports that the unfolding of Bid is susceptible to aggregation. We conclude that the stabilization forces in BAX versus Bid are different.
Bid retains its death-promoting function even when it is largely unfolded and aggregated
This study has revealed that at high temperatures (>350 K, in a regular buffer without GdnHCl) Bid proteins unfold partially, followed by intermolecular associations which tend to cause protein aggregation. To explore the biological significance of the thermally-induced aggregation, we have performed the assays of cytochrome c release from mitochondria. We found that although the heat treatment to Bid proteins causes structural unfolding and subsequent aggregation, it brings about insignificant change to the death-promoting function of Bid proteins; upon the cleavage by caspase-8, Bid proteins, even in an aggregated form, retain the function of promoting BAX-induced cytochrome c release from mitochondria. Despite that Bid loses largely its secondary structure after the thermal denaturation (as indicated by the CD study in Fig. 1C), the thermally-induced Bid aggregates have the functional activity similar to native Bid proteins. An important implication of the result is that the death-promoting function of Bid is not strictly dependent on the native structure of Bid.Proteins are polypeptides formed from a sequence of amino acids (i.e., the primary structure) which can fold into three additional levels of structural order (i.e., secondary, tertiary, and quaternary). The higher orders of protein structure involve mainly non-covalent interactions and often contain key information in determining protein function. As the structural folds of BAX are critical to the assembly of a BAX oligomer that induces MOMP,18,47 BAX evolves to possess a core structural domain able to resist extreme denaturing effects (e.g., 6 M GdnHCl and high temperatures such as 95 °C) so as to maintain its pro-apoptotic activity. As for Bid, which is responsible for propagating apoptotic signals within a cell rather than participating into the pore-forming complex, it is, therefore, reasonable to find that the higher orders of Bid structure are not important to the death-promoting function of Bid. Our results suggest that the exposure of the BH3 domain sequence in Bid, which can be induced upon the association with MOM as suggested in literature,13,14,20,42,46 is sufficient for Bid to propagate apoptotic signaling, and the death-promoting function of Bid is enhanced in the presence of caspase-8. Accordingly, Bid evolves to have only the high thermostability to maintain itself in the inactive state in solution. As the extreme conditions (e.g., high T and [GdnHCl]) required to unfold Bid are nearly impossible to occur under physiological conditions in cytosol, this study may also imply that the association of cBid with membrane is key to initiating the death-promoting function of cBid, warranting further investigations.
Conclusions
This study has reported an investigation of thermostability of Bid protein using spin-label ESR techniques. Bid is a thermally stable protein as it remains folded at temperatures up to 350 K. It is partially folded at temperatures up to 368 K (95 °C) and the thermally-induced unfolding is accompanied with intermolecular associations that lead to protein aggregation, as verified by the CD and ESR studies. To weaken the stability of this highly thermostable protein, the use of denaturant GdnHCl was introduced. We found that in 3 M GdnHCl, the conformational structure of Bid is barely affected, whereas the structural stability of Bid is weakened such that its onset temperature of unfolding is decreased more than 30 K from the result in a regular buffer without GdnHCl. With the use of the peak-height analysis method, our result has provided site-specific information about the temperature dependence of local structure of Bid and thus enabled the discrimination between the onsets of local unfolding and aggregation of Bid. As a result, we map out the local stability over Bid and identify the new interface between helices 3, 6, and 8 as the beginning of the structural unfolding. This study also investigates the apoptotic activity of the thermally-induced aggregates and shows that Bid retains the death-promoting function even when unfolded and aggregated. The local thermostability of the 30 individual sites in Bid reported herein is valuable information for further biophysical investigations of protein folding and storage. The ESR absorption peak-height analysis method is carefully demonstrated and proven in practice. While other biophysical tools (e.g., CD and DSC) provide an overall estimation of protein thermostability, our ESR method is distinguished by the site specificity. The methodology established provides a convenient approach to studying protein folding/unfolding events at varying temperature.Experimental
Protein expression and purification
Unless specified otherwise, all chemicals used in this study were from Sigma-Aldrich, Inc. The full-length mouse Bid was cloned into NdeI/XhoI site of pET28a vector (New England Biolabs, Inc.). Wild-type (wt) Bid has two native cysteine residues (C30 and C126). Cysteine-free construct (C30S/C126S) was used to prepare single-cysteine variants of Bid mutant for spin labeling study. All point mutations were generated using the QuikChange site-directed mutagenesis kit (Stratagene) and verified by DNA sequencing. The recombinant pET28a vector was transformed into the E. coli BL21(DE3) expression strain (Novagen). Recombinant proteins fused with six histidines at the N-terminal of Bid were expressed and purified by an affinity Ni column, as previously described.20,42 Briefly, bacterial culture was grown at 37 °C in Luria–Bertani medium containing kanamycin (30 μg mL−1) until OD600 reached 0.6–0.8. Protein expression was induced by addition of 1 mM IPTG (isopropyl 1-thio-β-D-galactopyranoside) at 30 °C for 4–6 h. The cell pellet was collected by centrifugation and resuspended in ice-cold lysis buffer (20 mM Tris, 100 mM NaCl, 20 mM imidazole, pH 7.4 and protease inhibitor tablet (cOmplete)). The resuspended pellet was sonicated on ice for 5 min, followed by centrifugation at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 40 min. The supernatant was filtrated through a 0.22 μm filter and then loaded onto an affinity Ni column using HisTrap HP (GE Healthcare) at a flow rate about 1 mL min−1. The column was washed with 10 column volumes of wash buffer (50 mM Tris, 500 mM NaCl and 40 mM imidazole, pH 7.4). Bid fraction was eluted with 10 column volumes of elution buffer-1 (20 mM Tris, 100 mM NaCl and 100 mM imidazole, pH 7.4) and elution buffer-2 (20 mM Tris, 100 mM NaCl and 500 mM imidazole, pH 7.4), respectively. Purified protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie blue staining. Imidazole was removed using a PD-10 desalting column (GE Healthcare) equilibrated with storage buffer (20 mM Tris and 100 mM NaCl, pH 7.4), and the protein concentration was estimated via absorption spectroscopy at 280 nm.
000g for 40 min. The supernatant was filtrated through a 0.22 μm filter and then loaded onto an affinity Ni column using HisTrap HP (GE Healthcare) at a flow rate about 1 mL min−1. The column was washed with 10 column volumes of wash buffer (50 mM Tris, 500 mM NaCl and 40 mM imidazole, pH 7.4). Bid fraction was eluted with 10 column volumes of elution buffer-1 (20 mM Tris, 100 mM NaCl and 100 mM imidazole, pH 7.4) and elution buffer-2 (20 mM Tris, 100 mM NaCl and 500 mM imidazole, pH 7.4), respectively. Purified protein was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie blue staining. Imidazole was removed using a PD-10 desalting column (GE Healthcare) equilibrated with storage buffer (20 mM Tris and 100 mM NaCl, pH 7.4), and the protein concentration was estimated via absorption spectroscopy at 280 nm.
Purified Bid proteins were labeled with a 10-fold excess of (1-oxy-2,2,5,5-tetramethyl-3-pyrroline-3-methyl)methanethiosulfonate spin label (MTSSL) (Alexis Biochemicals, San Diego, CA) per cysteine residue in the dark. The reaction was performed at 4 °C for overnight for solvent-accessible sites and at room temperature overnight for buried sites. Excess MTSL reagent was removed using a PD-10 desalting column equilibrated with storage buffer.
The procedure for expression and purification of caspase-8 is given below. The truncated human caspase-8 (ΔDEDs-Casp-8, amino acids 217–496) was cloned into NdeI/BamHI site of pET15b vector (New England Biolabs, Inc.). The recombinant pET15b vector was transformed into the E. coli BL21(DE3) pLysS expression strain (Novagen). Recombinant proteins fused with six histidines at the N-terminal of caspase-8 were expressed and purified by an affinity Ni column followed by size exclusion chromatography. We basically followed the literature procedure.48 Briefly, bacterial culture was grown at 37 °C in 2xYT medium containing ampicillin (100 μg mL−1) until OD600 reached 0.6–0.8. Protein expression was induced by addition of 0.2 mM of IPTG at 37 °C for 4–6 h. The cell pellet was collected by centrifugation and resuspended in ice-cold lysis buffer (50 mM Tris, 100 mM NaCl, 20 mM imidazole, pH 7.4 and 1 mM PMSF). The resuspended pellet was sonicated on ice for 5 min, followed by centrifugation at 13000g for 40 min. The supernatant was filtrated through a 0.22 μm filter and then loaded onto an affinity Ni column using HisTrap HP (GE Healthcare) at a flow rate about 1 mL min−1. The column was washed with 10 column volumes of wash buffer (50 mM Tris, 500 mM NaCl and 40 mM imidazole, pH 7.4). Caspase-8 fraction was eluted with elution buffer (50 mM Tris, 100 mM NaCl and 500 mM imidazole, pH 7.4). Protein was then purified with storage buffer (20 mM Tris and 100 mM NaCl, pH 7.4) by a size exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare). The purified caspase-8 was confirmed by SDS-PAGE with Coomassie blue staining and western blot. Protein concentration was estimated via absorption spectroscopy at 280 nm.
The pseudo-wild type T4 lysozyme (T4L) construct containing the substitutions C54T and C97A was subcloned into NdeI/HindIII site of pET28a vector (New England Biolabs, Inc.), and then used to prepare single-cysteine construct K65C. The expression and purification of T4L were performed as previously described in our publication.31
Cw-ESR experiment
A Bruker ELEXSYS E580 cw/pulsed spectrometer was used. The ESR experiment was performed at an operating frequency of 9.4 GHz with incident microwave power 1.5 mW. Approximately 40 μL solution volume, containing 30% (v/v) glycerol as a cryoprotectant, was added into the quartz ESR tube. The concentration of Bid in the ESR measurements was ca. 0.3–0.5 mM. For thermal denaturation (300–345 K) study, each spectrum was recorded with a magnetic width of 150 G, 1024 points, and 10 scans. Equilibrium time was 5 min for each temperature variation.Peak-height analysis of ESR absorption spectra
ESR absorption-mode spectrum was obtained from the conversion of the experimental first-derivation lineshape by integration. The temperature-dependent peak-height plot was obtained by analyzing the stacked plots of ESR absorption spectra (i.e., all spectra in the absorption mode are normalized by double integration). This absorption-based analysis was previously used to study protein intermediates induced by GdnHCl at a fixed temperature,22,49,50 and also to investigate the phase diagram of ternary lipid mixtures at room temperature and determine transitions between the lipid phases,51 but it has yet to be developed for studying ESR spectra at varying temperatures. The analysis of temperature-dependent spectra is more challenging as both the changes in local environment of a protein (e.g., in folded versus denatured states) and in mobility of spin label with varying temperature contribute to the spectral differences. The present study has further developed the method with the goal to study the spectra of a spin-labeled protein during thermal denaturation.Basically, we record cw-ESR spectra of singly labeled protein at varying temperatures. We then convert the spectra to the normalized absorption mode and plot the height of the lower-field peak as a function of temperature. The lower-field peak is selected for the analysis because its change between the two temperature extremes is greater than the changes in the central- and higher-field peaks (Fig. 4 and S4†). The peak-height data were basically fit to two linear regression lines, provided that some data points, which deviate largely from the regression due to protein aggregation, at higher temperatures were excluded from the analysis (Fig. 5). This study has also demonstrated using EasySpin software package52 that the peak-height data of ESR spectra from a freely tumbling protein would change linearly with temperature (Fig. S4†), providing a foundation for the linear regression analysis. Temperature corresponding to the onset of thermally-induced unfolding (denoted by Tx) is determined by the intersection of the two linear regression lines. R2 values of the fits were all greater than 0.95. Random sample consensus (RANSAC) algorithm was used to choose the inliers for linear regression in the respective temperature ranges.53 In this algorithm, two random points were selected for each iteration and the number of iteration steps is set to 500 to generate a reproducible and reliable result. The threshold was set to 0.032 to rule out the outliers. The probability of choosing inliers in the selected temperature range was >90% as criteria to define the range in which the chosen inliers follow the same linear behavior.
The peak-height plot provides a convenient measure of how the respective local environments respond to temperature changes. At lower temperatures where protein structure remains folded, the slope (i.e., the ratio of the peak-height change between two temperatures) changes little between spin-labeled sites, reporting only temperature effect on the spectra. At higher temperatures, where protein local folds are disrupted, the local dynamics and ordering of spin label is certainly changed and, consequently, the corresponding slope of the peak-height curve is different from those at lower temperatures. The temperature dependences of the site-specific local environment in protein before and after local structure disruption can be clearly distinguished by the slopes of the peak-height curve. This method allows one to conveniently access the temperature-dependent behaviors of individual sites and identify respective onset temperatures of local disruption during thermal denaturation. One key to the success of this methodology is that the spectral difference between the maximum and minimum temperatures should be sufficiently great and the spectral lineshapes of the two extremes must span over the fast-to-slow motional regime (ca. rotational correlation time τc ≈ 10−11 to 10−7 s). Using a model protein T4L whose thermal denaturation is well studied,54 we demonstrated that the onset of local disruption temperature of T4L determined by our absorption-based ESR peak-height method is consistent with the result by CD (Fig. S6†).
The absorption-based peak-height plot is a semi-empirical analysis of the nitroxide spectrum. There are other semi-empirical analysis methods available in literature, such as the inverse linewidth of the center-field ESR resonance (ΔH0−1) and the empirical equation for obtaining the rotational correlation time τc (as given below), to provide a convenient measure of relative mobilities in spin-label side-chain. An approximate value of τc for the rotational motions of spin label can be obtained using the equation, τc = 6.5 × 10−10ΔH0[(A0/A−1)1/2 − 1], where A0 and A−1 are the peak-to-peak heights of the center- and high-field resonances, respectively, and τc is in seconds.55 Fig. S7† shows a comparison of the results from the above two methods as well as the peak-height analysis. It shows that the absorption-based peak-height analysis does a better job in distinguishing the temperature-dependent effect on spin label motion from the onset of local structural disruption of protein. The peak-height analysis brings out the temperature-dependent effect on the spin label spectra that can be clearly characterized by slopes of the curve. Importantly, the result about the thermal denaturation of Bid is in a good agreement with the CD data as discussed in the main text. It demonstrates the suitability of the peak-height method for the reported spectra spanning over the fast-to-slow motional regime (ca. τc ≈ 10−11 to 10−7 s). As the other two methods are known to be sensitive to molecular motions in (a shorter time scale) the fast-to-intermediate motional regime (τc ≈ 10−11 to 10−9 s, where one observes three distinct resonance lines),55 it is reasonable that they appear inappropriate to analyze the spectra of the present study. While the two methods (ΔH0−1 and τc) focus on the spectral characteristics (i.e., the center-field linewidth and the ratio A0/A−1) that dominate the sharp-line components in amplitude (cf. Fig. S7†), the peak-height analysis takes into consideration the all components in the lower-field peak such that it retains the sensitivity to the broader components in the spectra, thus revealing the linear dependence of the lower-field peak-height on temperature.
DEER experiment and analysis
Approximately, 40 μL solution volume, containing 30% (v/v) d8-glycerol as cryoprotectant, was added into quartz ESR tube. All buffers in the DEER experiments were deuterated. The concentration of Bid in the DEER measurements was ca. 0.3–0.5 mM. A Bruker ELEXSYS E580-400 CW/Pulsed spectrometer, with a split-ring resonator (EN4118X-MS3) and a helium gas flow system (4118CF and 4112HV), was used. The ESR probe head was precooled to 80 K prior to the transfer of the ESR sample tube into the cavity. DEER experiments were performed using the typical four-pulse constant-time DEER sequence as previously described.18,25 The detection pulses were set to 32 and 16 ns for π and π/2 pulses, respectively, and the pump frequency was set to approximately 70 MHz lower than the detection pulse frequency. The pulse amplitudes were chosen to optimize the refocused echo. The π/2-pulse was employed with +x/−x phase cycles to eliminate receiver offsets. The duration of the pumping pulse was about 32 ns, and its frequency was coupled into the microwave bridge by a commercially available setup (E580-400U) from Bruker. All pulses were amplified via a pulsed traveling wave tube (TWT) amplifier (E580-1030). The field was adjusted such that the pump pulse is applied to the maximum of the nitroxide spectrum, where it selects the central mI = 0 transition of Azz together with the mI = ±1 transitions. The accumulation time for each set of data was about 10 h at a temperature of 80 K. A common cooling approach was used.17,18 The sample tube was plunge-cooled in liquid nitrogen and then transferred into the ESR probe head, which was precooled to 80 K using a helium flow system. The determination of interspin distance distribution of the DEER spectroscopy was performed using time-domain analysis by Tikhonov regularization based on the L-curve method,27 followed by a data refinement process using the maximum entropy method (MEM)26 to obtain the non-negative probability density P(r).Mitochondria isolation and cytochrome c release assays
Mouse liver tissue (0.2 mg) was ground using a douce homogenizer with 15–20 strokes, and the mitochondria were separated using centrifugation according to Mitochondria Isolation Kit (Thermo Scientific). The mitochondria were then washed with Mitochondria Assay Buffer (MAB: 200 mM mannitol, 68 mM sucrose, 10 mM HEPES-KOH pH 7.4, 110 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1% bovine serum albumin, protease inhibitor) and centrifuged at 12000 × g, and the supernatant was removed. The final mitochondria fraction was placed on ice for further downstream processing. For cytochrome c release assay, the mitochondria (50 μg of protein) were incubated with recombinant Bid (100 nM) and caspase-8 (100 nM), or BAX (100 nM) at 37 °C for 30 min. For the heat-treated samples, Bid was heated to 70 °C or 95 °C for 1 h and cooled down to room temperature prior to the incubation with mitochondria for another 30 min. The sample was then centrifuged at 12000 × g for 5 min separating the supernatant and pellets fractions. The supernatant was subjected to subsequent immunoblotting with mouse anti-cytochrome c antibodies (clone 7H8.2C12, MAB1800, Millipore, MA, USA). Quantification of immunoblots was done using ImageJ (version 1.47, NIH).
Circular dichroism (CD) spectroscopy
CD measurements were performed with an Aviv Model 410 circular dichroism spectrometer, using quartz cuvettes with a path length of 1 mm. Protein concentration was 10 μM. Equilibrium time was 5 min at each temperature. All spectra shown were corrected by subtracting corresponding blanks. In the absence of GdnHCl, some protein precipitation was observed on the inner surface of cuvette after thermal denaturation. CD measurements were repeated several times to confirm that the CD signal at 222 nm is not restored after the heating and cooling cycle, validating that the thermal unfolding is irreversible. In the studies with GdnHCl, proteins were incubated with GdnHCl for >0.5 h prior to the CD measurements.Turbidity measurement
Turbidity was used to assess the extent of protein aggregation by measuring absorbance at 395 nm, as previously followed.56,57 To study the thermally-induced aggregation, samples (10 μM Bid protein) were incubated at desired experimental temperatures (e.g., 40, 60, 80 and 95 °C) for 1 h prior to the turbidity measurement. The data was recorded using Tecan Infinite 200 PRO plate reader. The estimation of errors was based on five independent measurements.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by grants from the Ministry of Science and Technology of Taiwan (105-2628-M-007-005 and 106-2627-M-007-009) and the Frontier Research Center on Fundamental and Applied Sciences of Matters at NTHU. All of the CW/pulse ESR measurements were conducted in the Research Instrument Center of Taiwan located at NTHU.Notes and references
- A. R. D. Delbridge and A. Strasser, The BCL-2 protein family, BH3-mimetics and cancer therapy, Cell Death Differ., 2015, 22, 1071–1080 CrossRef CAS PubMed.
- P. E. Czabotar, G. Lessene, A. Strasser and J. M. Adams, Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy, Nat. Rev. Mol. Cell Biol., 2014, 15, 49–63 CrossRef CAS PubMed.
- A. Shamas-Din, H. Brahmbhatt, B. Leber and D. W. Andrews, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 508–520 CrossRef CAS PubMed.
- C. Kantari and H. Walczak, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 558–563 CrossRef CAS PubMed.
- L. P. Billen, A. Shamas-Din and D. W. Andrews, Bid: a Bax-like BH3 protein, Oncogene, 2008, 27, S93–S104 CrossRef CAS PubMed.
- R. F. Gahl, P. Dwivedi and N. Tjandra, Bcl-2 proteins bid and bax form a network to permeabilize the mitochondria at the onset of apoptosis, Cell Death Dis., 2016, 7, e2424 CrossRef CAS PubMed.
- T. Moldoveanu, C. R. Grace, F. Llambi, A. Nourse, P. Fitzgerald, K. Gehring, R. W. Kriwacki and D. R. Green, BID-induced structural changes in BAK promote apoptosis, Nat. Struct. Mol. Biol., 2013, 20, 589–597 CrossRef CAS PubMed.
- K. Wang, X. M. Yin, D. T. Chao, C. L. Milliman and S. J. Korsmeyer, BID: a novel BH3 domain-only death agonist, Genes Dev., 1996, 10, 2859–2869 CrossRef CAS PubMed.
- R. Sarig, Y. Zaltsman, R. C. Marcellus, R. Flavell, T. W. Mak and A. Gross, BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts, J. Biol. Chem., 2003, 278, 10707–10715 CrossRef CAS PubMed.
- A. J. Valentijn and A. P. Gilmore, Translocation of full-length bid to mitochondria during anoikis, J. Biol. Chem., 2004, 279, 32848–32857 CrossRef CAS PubMed.
- H. Li, H. Zhu, C. J. Xu and J. Yuan, Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis, Cell, 1998, 94, 491–501 CrossRef CAS PubMed.
- A. Gross, X. M. Yin, K. Wang, M. C. Wei, J. Jockel, C. Milliman, H. Erdjument-Bromage, P. Tempst and S. J. Korsmeyer, Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-X(L) prevents this release but not tumor necrosis factor-R1/Fas death, J. Biol. Chem., 1999, 274, 1156–1163 CrossRef CAS PubMed.
- J. J. Chou, H. Li, G. S. Salvesen, J. Yuan and G. Wagner, Solution Structure of BID, an Intracellular Amplifier of Apoptotic Signaling, Cell, 1999, 96, 615–624 CrossRef CAS PubMed.
- J. M. McDonnell, D. Fushman, C. L. Milliman, S. J. Korsmeyer and D. Cowburn, Solution Structure of the Proapoptotic Molecule BID: A Structural Basis for Apoptotic Agonists and Antagonists, Cell, 1999, 96, 625–634 CrossRef CAS PubMed.
- M. G. Hinds, C. Smits, R. Fredericks-Short, J. M. Risk, M. Bailey, D. C. S. Huang and C. L. Day, Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets, Cell Death Differ., 2007, 14, 128–136 CrossRef CAS PubMed.
- S. Bleicken, A. J. García-Sáez, E. Conte and E. Bordignon, Dynamic interaction of cBid with detergents, liposomes and mitochondria, PLoS One, 2012, 7, e35910 CrossRef CAS PubMed.
- C.-J. Tsai, S. Liu, C.-L. Hung, S.-R. Jhong, T.-C. Sung and Y.-W. Chiang, BAX-induced apoptosis can be initiated through a conformational selection mechanism, Structure, 2015, 23, 139–148 CrossRef CAS PubMed.
- T.-C. Sung, C.-Y. C.-Y. Li, Y.-C. Y.-C. Lai, C.-L. Hung, O. Shih, Y.-Q. Y.-Q. Yeh, U.-S. U.-S. Jeng and Y.-W. Chiang, Solution Structure of Apoptotic BAX Oligomer: Oligomerization Likely Precedes Membrane Insertion, Structure, 2015, 23, 1878–1888 CrossRef CAS PubMed.
- A. L. Lai, E. M. Clerico, M. E. Blackburn, N. A. Patel, C. V. Robinson, P. P. Borbat, J. H. Freed and L. M. Gierasch, Key features of an Hsp70 chaperone allosteric landscape revealed by ion-mobility native mass spectrometry and double electron-electron resonance, J. Biol. Chem., 2017, 292, 8773–8785 CrossRef CAS PubMed.
- K. J. Oh, S. Barbuto, N. Meyer, R.-S. Kim, R. J. Collier and S. J. Korsmeyer, Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding. A site-directed spin labeling study, J. Biol. Chem., 2005, 280, 753–767 CrossRef CAS PubMed.
- C. S. Klug, W. Su, J. Liu, P. E. Klebba and J. B. Feix, Denaturant unfolding of the ferric enterobactin receptor and ligand-induced stabilization studied by site-directed spin labeling, Biochemistry, 1995, 34, 14230–14236 CrossRef CAS PubMed.
- M. Lindgren, M. Svensson, P. O. Freskgård, U. Carlsson, P. Jonasson, L. G. Mårtensson and B. H. Jonsson, Characterization of a folding intermediate of human carbonic anhydrase II: probing local mobility by electron paramagnetic resonance, Biophys. J., 1995, 69, 202–213 CrossRef CAS PubMed.
- Z. C. Liang, Y. Lou, J. H. Freed, L. Columbus and W. L. Hubbell, A multifrequency electron spin resonance study of T4 lysozyme dynamics using the slowly relaxing local structure model, J. Phys. Chem. B, 2004, 108, 17649–17659 CrossRef CAS.
- Z. Zhang, M. R. Fleissner, D. S. Tipikin, Z. Liang, J. K. Moscicki, K. A. Earl, W. L. Hubbell and J. H. Freed, Multifrequency Electron Spin Resonance Study of the Dynamics of Spin Labeled T4 Lysozyme, J. Phys. Chem. B, 2010, 114, 5503–5521 CrossRef CAS PubMed.
- G. Jeschke, DEER Distance Measurements on Proteins, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- Y.-W. Chiang, P. P. Borbat and J. H. Freed, Maximum entropy: a complement to Tikhonov regularization for determination of pair distance distributions by pulsed ESR, J. Magn. Reson., 2005, 177, 184–196 CrossRef CAS PubMed.
- Y.-W. W. Chiang, P. P. Borbat and J. H. Freed, The Determination of Pair Distance Distributions by Pulsed ESR Using Tikhonov Regularization, J. Magn. Reson., 2005, 172, 279–295 CrossRef CAS PubMed.
- G. Hagelueken, D. Abdullin, R. Ward and O. Schiemann, mtsslSuite: in silico spin labelling, trilateration and distance-constrained rigid body docking in PyMOL, Mol. Phys., 2013, 111, 2757–2766 CrossRef CAS PubMed.
- T.-C. Sung and Y.-W. Chiang, Identification of complex dynamic modes on prion protein peptides using multifrequency ESR with mesoporous materials, Phys. Chem. Chem. Phys., 2010, 12, 13117–13125 RSC.
- Y.-W. Chiang, Y. Otoshima, Y. Watanabe, O. Inanami and Y. Shimoyama, Dynamics and Local Ordering of Spin-Labeled Prion Protein: An ESR Simulation Study of a Highly PH-Sensitive Site, J. Biomol. Struct. Dyn., 2008, 26, 355–365 CrossRef CAS PubMed.
- K.-J. Chang, Y.-H. Kuo and Y.-W. Chiang, Study of Protein Dynamics under Nanoconfinement by Spin-Label ESR: A Case of T4 Lysozyme Protein, J. Phys. Chem. B, 2017, 121, 4355–4363 CrossRef CAS PubMed.
- W. L. Hubbell, H. S. Mchaourab, C. Altenbach and M. A. Lietzow, Watching proteins move using site-directed spin labeling, Structure, 1996, 4, 779–783 CrossRef CAS PubMed.
- C. J. López, S. Oga and W. L. Hubbell, Mapping molecular flexibility of proteins with site-directed spin labeling: A case study of myoglobin, Biochemistry, 2012, 51, 6568–6583 CrossRef PubMed.
- A. Shamas-Din, D. Satsoura, O. Khan, W. Zhu, B. Leber, C. Fradin and D. W. Andrews, Multiple partners can kiss-and-run: Bax transfers between multiple membranes and permeabilizes those primed by tBid, Cell Death Dis., 2014, 5, e1277 CrossRef CAS PubMed.
- A. M. Petros, E. T. Olejniczak and S. W. Fesik, Structural biology of the Bcl-2 family of proteins, Biochim. Biophys. Acta, 2004, 1644, 83–94 CrossRef CAS PubMed.
- N. Volkmann, F. M. Marassi, D. D. Newmeyer and D. Hanein, The rheostat in the membrane: BCL-2 family proteins and apoptosis, Cell Death Differ., 2014, 21, 206–215 CrossRef CAS PubMed.
- A. Gross, J. M. McDonnell and S. J. Korsmeyer, Genes Dev., 1999, 13, 1899–1911 CrossRef CAS PubMed.
- M. Suzuki, R. J. Youle and N. Tjandra, Structure of Bax: coregulation of dimer formation and intracellular localization, Cell, 2000, 103, 645–654 CrossRef CAS PubMed.
- S. W. Muchmore, M. Sattler, H. Liang, R. P. Meadows, J. E. Harlan, H. S. Yoon, D. Nettesheim, B. S. Chang, C. B. Thompson, S.-L. Wong, S.-C. Ng and S. W. Fesik, X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death, Nature, 1996, 381, 335–341 CrossRef CAS PubMed.
- S. Shivakumar, M. Kurylowicz, N. Hirmiz, Y. Manan, O. Friaa, A. Shamas-Din, P. Masoudian, B. Leber, D. W. Andrews and C. Fradin, The proapoptotic protein tbid forms both superficially bound and membrane-inserted oligomers, Biophys. J., 2014, 106, 2085–2095 CrossRef CAS PubMed.
- A. J. García-Sáez, I. Mingarro, E. Pérez-Payá and J. Salgado, Membrane-insertion fragments of Bcl-xL, Bax, and Bid, Biochemistry, 2004, 43, 10930–10943 CrossRef PubMed.
- A. Shamas-Din, S. Bindner, W. Zhu, Y. Zaltsman, C. Campbell, A. Gross, B. Leber, D. W. Andrews and C. Fradin, tBid undergoes multiple conformational changes at the membrane required for bax activation, J. Biol. Chem., 2013, 288, 22111–22127 CrossRef CAS PubMed.
- C.-H. Chan, C.-J. Tsai and Y.-W. Chiang, Side-chain packing interactions stabilize an intermediate of BAX protein against chemical and thermal denaturation, J. Phys. Chem. B, 2015, 119, 54–64 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Cation-π interactions in structural biology, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
- K. G. Tina, R. Bhadra and N. Srinivasan, PIC: Protein Interactions Calculator, Nucleic Acids Res., 2007, 35, W473–W476 CrossRef CAS PubMed.
- Y. Wang and N. Tjandra, Structural insights of tBid, the caspase-8-activated bid, and its BH3 domain, J. Biol. Chem., 2013, 288, 35840–35851 CrossRef CAS PubMed.
- S. Bleicken, G. Jeschke, C. Stegmueller, R. Salvador-Gallego, A. J. García-Sáez and E. Bordignon, Structural Model of Active Bax at the Membrane, Mol. Cell, 2014, 56, 496–505 CrossRef CAS PubMed.
- H. R. Stennicke and G. S. Salvesen, Caspases: preparation and characterization, Methods, 1999, 17, 313–319 CrossRef CAS PubMed.
- M. Svensson, P. Jonasson, P.-O. Freskgaard, B.-H. Jonsson, M. Lindgren, L.-G. Maartensson, M. Gentile, K. Boren and U. Carlsson, Mapping the Folding Intermediate of Human Carbonic Anhydrase II. Probing Substructure by Chemical Reactivity and Spin and Fluorescence Labeling of Engineered Cysteine Residues, Biochemistry, 1995, 34, 8606–8620 CrossRef CAS PubMed.
- V. M. Grigoryants, K. A. DeWeerd and C. P. Scholes, Method of rapid mix EPR applied to the folding of bi-spin-labeled protein as a probe for the dynamic onset of interaction between sequentially distant side chains, J. Phys. Chem. B, 2004, 108, 9463–9468 CrossRef CAS.
- Y. W. Chiang, Y. Shimoyama, G. W. Feigenson and J. H. Freed, Dynamic molecular structure of DPPC-DLPC-cholesterol ternary lipid system by spin-label electron spin resonance, Biophys. J., 2004, 87, 2483–2496 CrossRef CAS PubMed.
- S. Stoll, in Multifrequency Electron Paramagnetic Resonance, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 69–138 Search PubMed.
- M. A. Fischler and R. C. Bolles, Random Sample Consensus: A Paradigm for Model Fitting with Applications to Image Analysis and Automated Cartography, Commun. ACM, 1981, 24, 381–395 CrossRef.
- H. S. Mchaourab, M. A. Lietzow, K. Hideg and W. L. Hubbell, Motion of spin-labeled side chains in T4 lysozyme, correlation with protein structure and dynamics, Biochemistry, 1996, 35, 7692–7704 CrossRef CAS PubMed.
- C. S. Klug and J. B. Feix, Methods and applications of site-directed spin labeling EPR spectroscopy, Methods Cell Biol., 2008, 84, 617–658 CAS.
- Z. Sun, Z. Diaz, X. Fang, M. P. Hart, A. Chesi, J. Shorter and A. D. Gitler, Molecular determinants and genetic modifiers of aggregation and toxicity for the als disease protein fus/tls, PLoS Biol., 2011, 9, e1000614 CrossRef CAS PubMed.
- B. S. Johnson, D. Snead, J. J. Lee, J. M. McCaffery, J. Shorter and A. D. Gitler, TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity, J. Biol. Chem., 2009, 284, 20329–20339 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06740f |
| This journal is © The Royal Society of Chemistry 2018 |