
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective and sensitive spectrofluorimetric quantification of velpatasvir in presence of sofosbuvir. Application to their co-formulated tablet
Rania M. El-Gamal†
*ab,
Sherif A. Abdel-Gawad†ac,
Fathalla F. Belal†b and
Moustapha E. Moustapha†d
aPharmaceutical Chemistry Department, College of Pharmacy, Prince Sattam Bin-Abdul Aziz University, P. O. Box 173, Al-Kharj, 11942, Kingdom of Saudi Arabia. E-mail: r_m_elgamal@yahoo.com; Fax: +966 115886832; Tel: +966 561411680
bDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt
cAnalytical Chemistry Department, Faculty of Pharmacy, Cairo University, Cairo, ET-11562, Egypt
dDepartment of Chemistry, College of Science and Humanities, Prince Sattam Bin-Abdul Aziz University, Al-Kharj, 11942, Kingdom of Saudi Arabia
First published on 24th September 2018
Abstract
A new sensitive, rapid and simple spectrofluorimetric method was utilized for the assessment of velpatasvir (VPS) in its bulk form as well as in its combined tablet with sofosbovir (SFV). The technique relies on measuring the native fluorescence of VPS in methanol at 385 nm and 400 nm after excitation at 295 nm. The fluorescence–concentration plots were rectilinear through the range of 2.0–20.0 ng mL−1 at both emission maxima with lower detection limits of 0.146 ng mL−1 and 0.378 ng mL−1, and lower quantification limits of 0.444 ng mL−1 and 1.147 ng mL−1 at 385 nm and 400 nm, respectively. The proposed method was appropriately used for the analysis of VPS in its commercial tablet formulation and the results were in good agreement with those achieved with the applied comparison method.
1. Introduction
Hepatitis C virus (HCV) infection can be considered as a dangerous disease, affecting about three to five million people in the United States (US) and about one hundred and seventy million people worldwide. This disease has no symptoms in the early stages but if it becomes chronic, it may lead to dangerous life-threatening complications, including liver failure, hepatocellular carcinoma and death.1 Velpatasvir (VPS) is methyl {(2S)-1-[(2S,5S)-2-(9-{2-[(2S,4S)-1-{(2R)-2-[(methoxycarbonyl)amino]-2-phenylacetyl}-4-(methoxymethyl)pyrrolidin-2-yl]-1H-imidazol-4-yl}-1,11-dihydro[2]benzopyrano[4′,3′:6,7]naphtho[1,2-d]imidazol-2-yl)-5-methylpyrrolidin-1-yl]-3-methyl-1-oxobutan-2-yl}carbamate, Fig. 1.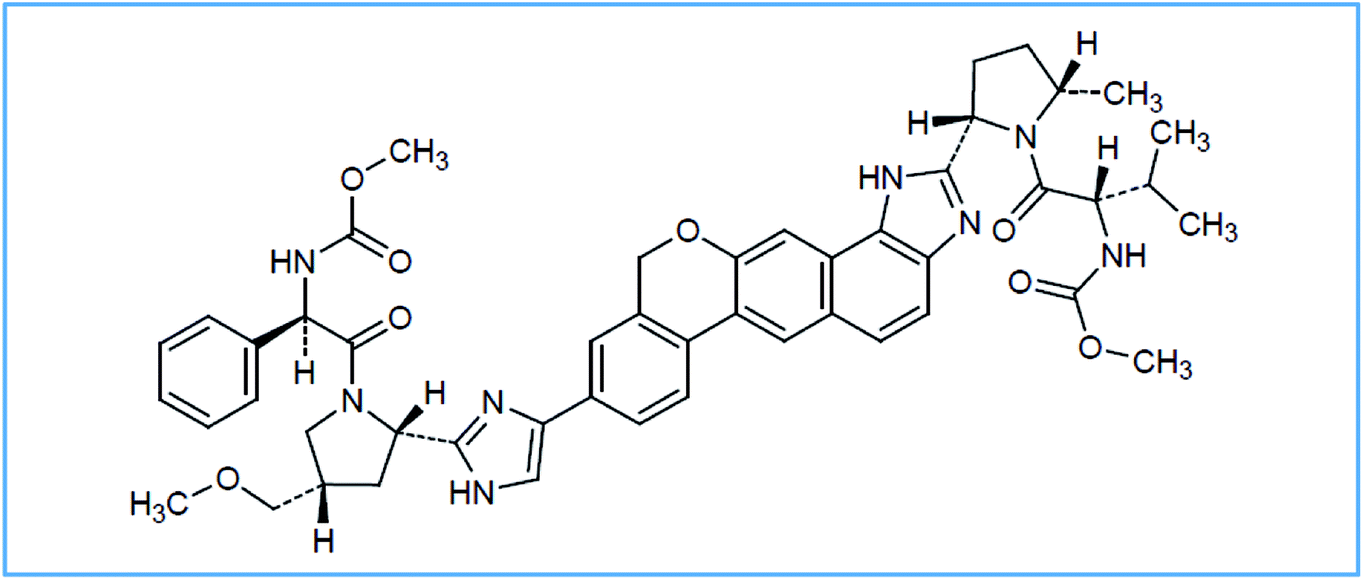 | ||
| Fig. 1 The structural formulae of VPS. | ||
It is a Direct-Acting Antiviral (DAA) drug that can be used in combination with sofosbuvir (SFV) for HCV treatment. It acts as a defective substrate for NS5A and so it plays an important role in preventing viral replication.2 SFV is a nucleotide analog NS5B polymerase inhibitor. It is a prodrug which can be utilized for HCV treatment, either alone or in combination with other drugs like, ribavirin, ledipasvir and VPS.3 In June 2016, The FDA (Food and Drug Administration) approved a fixed dose combination of SFV and VPS for the treatment of adult patients with chronic HCV infection (genotypes 1, 2, 3, 4, 5, or 6).4 Referring to the literature, SFV alone was determined using chromatographic and spectrophotometric techniques.5,6 Also, SFV forced degradation was investigated using liquid chromatography-tandem mass spectrometry (LC-MS/MS).7 On the other hand, simultaneous determination of SFV and VPS was performed using different RP-HPLC methods.8–11
A single spectroflurimetric method had been recently reported for the assay of VPS in pharmaceutical tablets and body fluids.12 The developed method was based on dispersive solid phase extraction (dSPE) utilizing the synergistic effect of reduced graphene oxide (RGO) and cobalt hydroxide nanoparticles (CHNPs) in addition to cloud point extraction (CPE) using polyethylene glycol 6000 (PEG 6000) as non-ionic surfactant. Despite the sensitivity of the method it is highly complicated with time consuming procedures that outweighs its merits.
The goal of this work was to adopt a novel simple, rapid, sensitive and economic spectroflurimetric method for the selective quantification of VPS in presence of SFV either in bulk or dosage forms.
2. Experimental
2.1. Apparatus
A spectrofluorophotometer with a xenon lamp, model FP-8200 JASCO; Japan was utilized. A quartz cell of 1 cm path length was used for all measurements. The slit widths were 10 nm for both excitation and emission, the sensitivity was adjusted at low sensitivity. pH measurements were determined using a Consort NV P-901 pH meter (Belgium). An ultrasonic bath, model Branson 2800 was utilized as well. A&D GR300 analytical balance.2.2. Materials and reagents
All chemicals utilized in this work were of analytical grade and the solvents were of HPLC grade. Deionized water was used for all preparations.- VPS (Catakog #B1194-5, 25 and Lot #2L30B11940) was purchased from BioVision, Milpitas Boulevard, Milpitas, CA 95035 USA. Its purity percentage was certified to be ≥ 98%.
- SFV (PSI-7977) was purchased from Cayman chemical company, Ann Arbor, USA. Its purity percentage was 99.98 ± 0.741.8
- Sodium dodecyl sulphate (SDS) and sodium acetate trihydrate, were purchased from Lobachemie, Mumbai, India.
- Methanol, acetonitrile, acetone and n-propanol were obtained from Sigma-Aldrich (Germany).
- Glacial acetic acid was obtained from BDH laboratory supplies, England.
- Sodium hydroxide pellets and boric acid were obtained from central drug house (CDH), New Delhi, India.
- Tween-80, methyl cellulose, borax were all obtained from Assagaf pharma, Saudi Arabia – acetate buffer (0.2 mol L−1) solution was used. It was of pH range 3.0–5.5. Borate buffer (0.2 mol L−1) solution was used. It was of pH range 6.0–8.7. Aqueous solutions (1.0% w/v) of Tween-80, methylcellulose and SDS were prepared and used.
2.3. Dosage form
Epclusa® extended-release tablets (NDC 51267-890-99) labeled to contain 400 mg SFV and 100 mg VPS per tablet. It was manufactured by Gilead Sciences International, Cambridge, UK.2.4. Standard solutions
Stock solution of concentration 100.0 μg mL−1 of VPS was prepared by dissolving 10 mg of pure drug in 100 mL methanol with the aid of an ultrasonic bath. Working standard solution of 0.1 μg mL−1 was prepared by appropriate dilution of the stock solutions with methanol. Solutions were properly stored in refrigerator to ensure their stability.3. Results and discussion
VPS was found to exhibit an intense native fluorescence in methanolic solution at 385 and 400 nm after excitation at 295 nm (Fig. 2). Hence, we aimed to utilize these emission bands to develop a new approach for accurate and selective VPS quantification in its bulk form as well as in its tablet.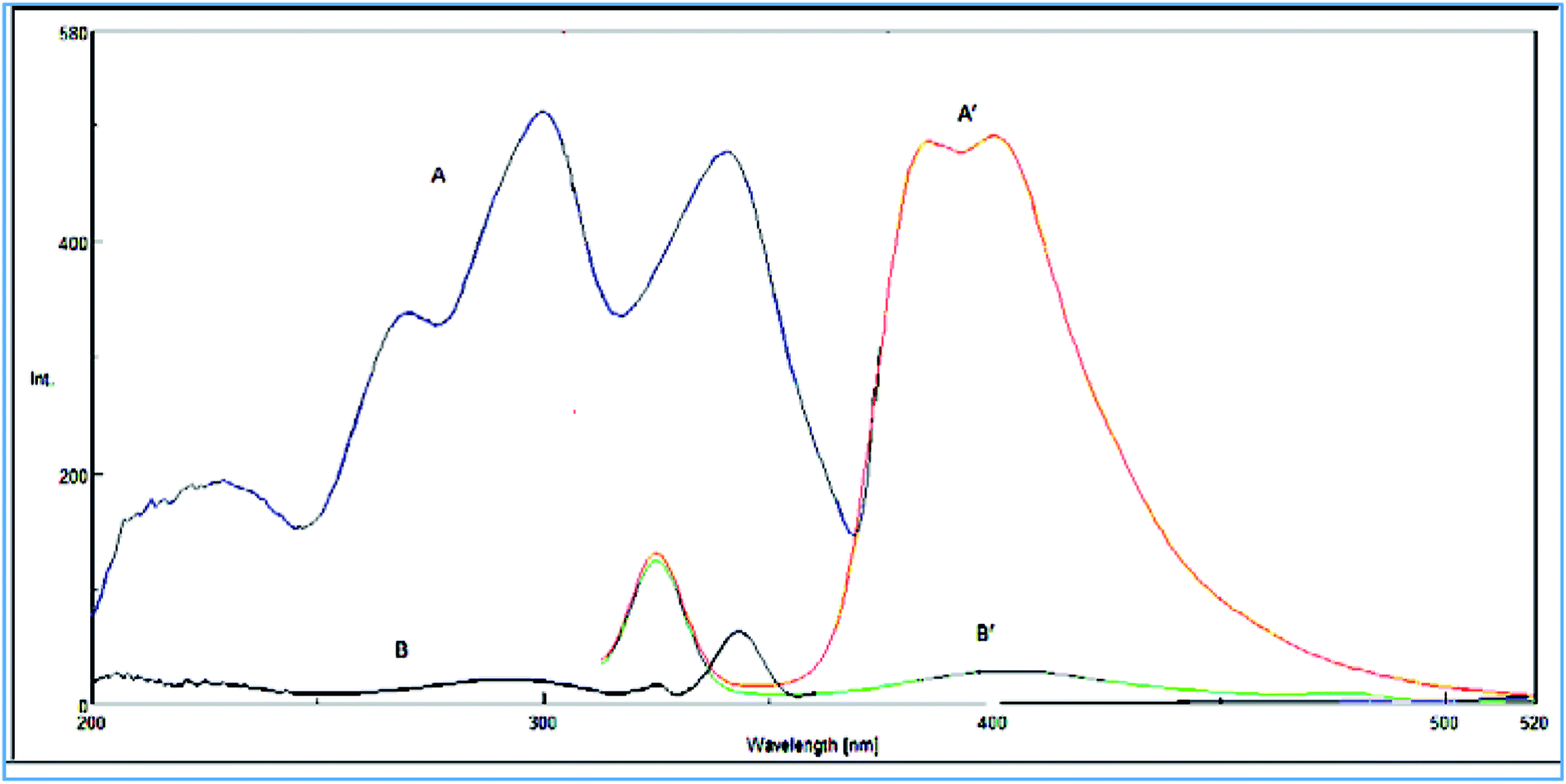 | ||
| Fig. 2 (A) Excitation and (A′) emission spectra of (15 ng mL−1) of VPS in methanol. (B) Excitation and (B′) emission spectra of methanol. | ||
3.1. Factors affecting the fluorescence properties
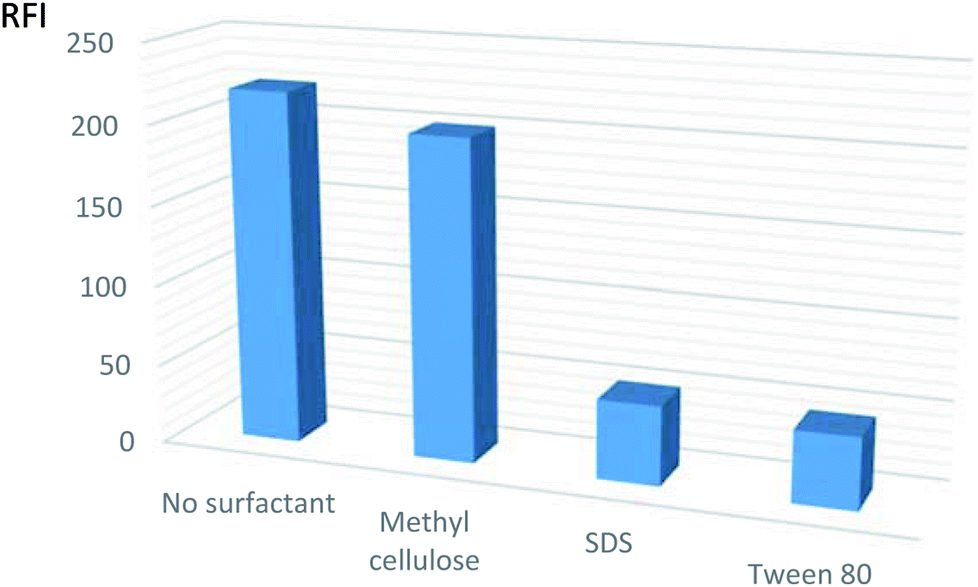 | ||
| Fig. 3 Effect of different organized media on the relative fluorescent intensity (RFI) of VPS (5 ng mL−1 in methanol). | ||
The results depicted that the use of surfactants caused no appreciable effect or may reduce the fluorescence intensity. Subsequently, the procedures were adapted without the use of any of the mentioned surfactants.
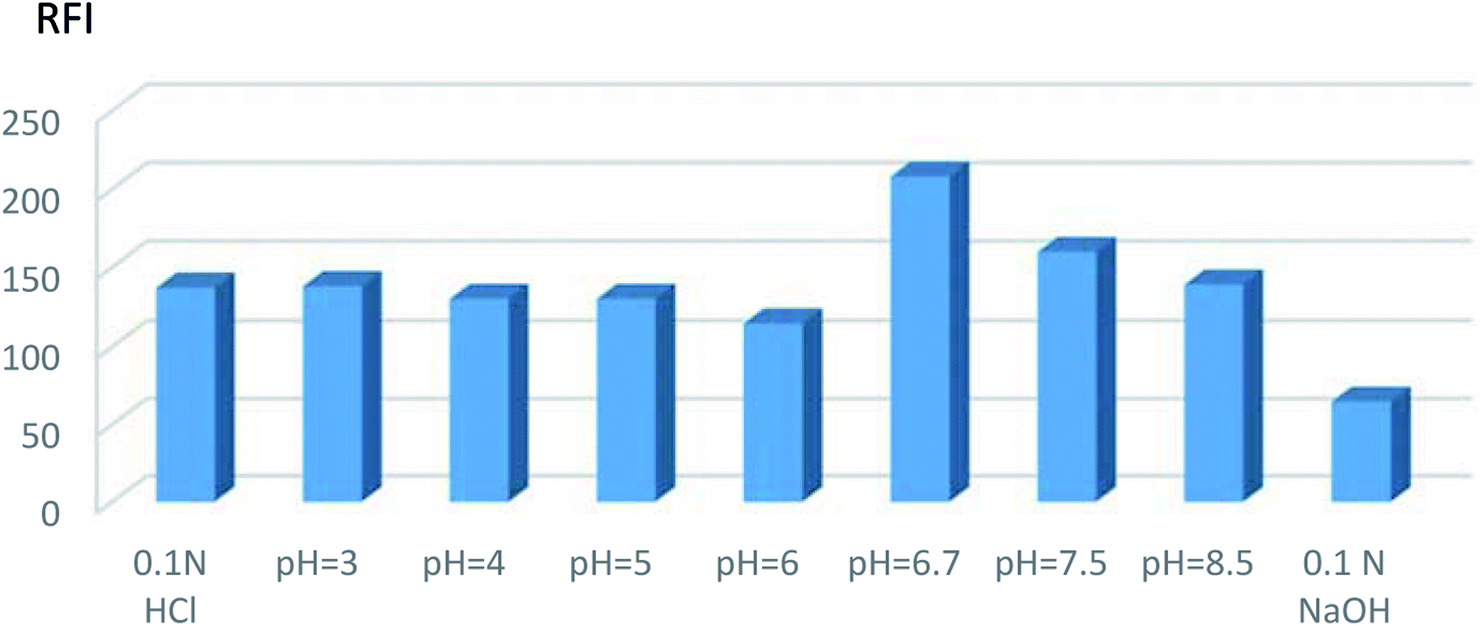 | ||
| Fig. 4 Effect of different pH on the relative fluorescence intensity (RFI) of VPS (5 ng mL−1 in methanol). | ||
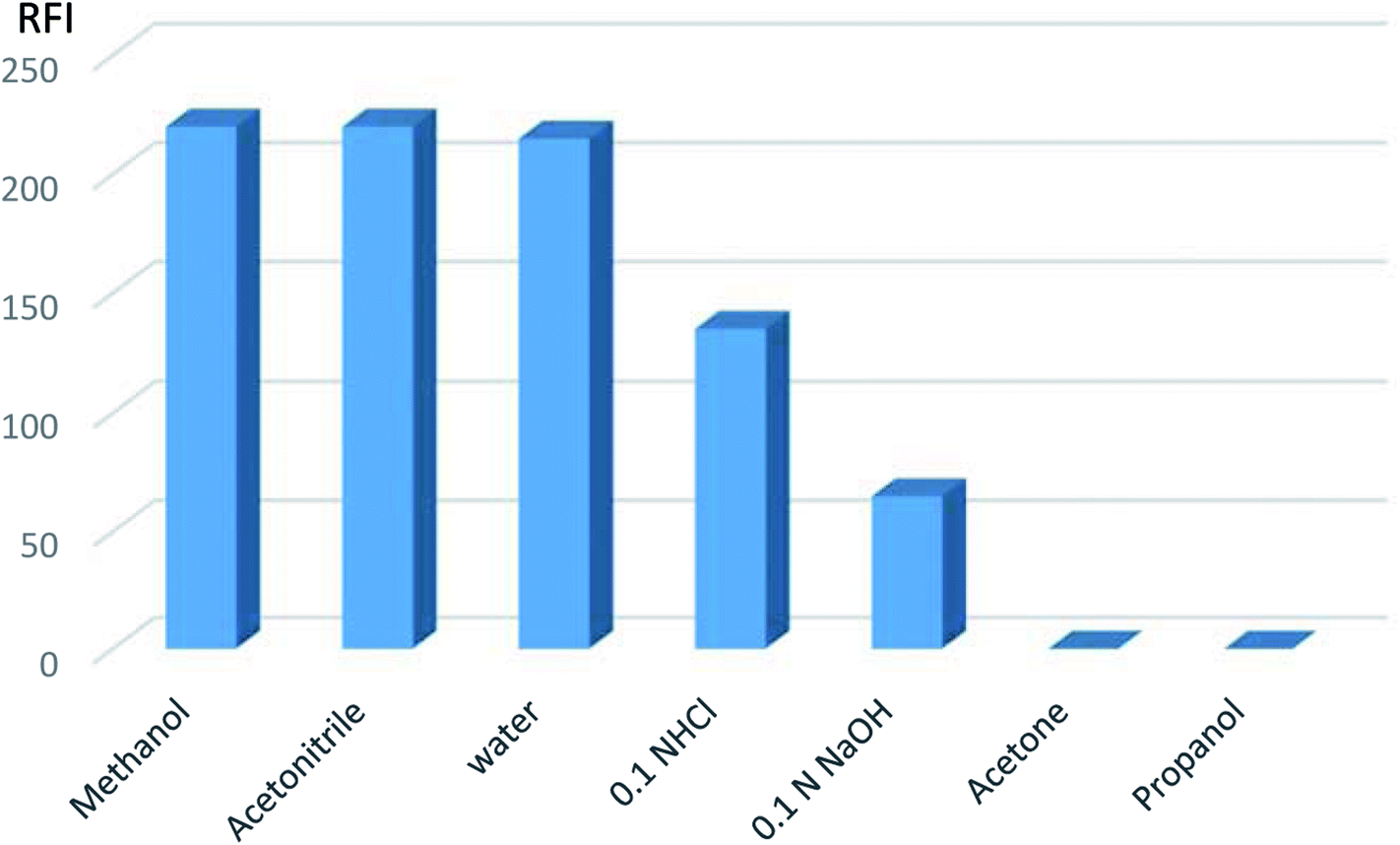 | ||
| Fig. 5 Effect of different diluting solvents on the relative fluorescent intensity of VPS (5 ng mL−1). | ||
Hence, methanol was the diluting solvent of choice producing a more symmetric peak that increased quantitatively with the change in VPS concentration.
3.2. Method validation
The method was validated in accordance to ICH recommendations.14| RFI = 28.976 + 40.406C (r = 0.9999) at 385 nm |
| RFI = 14.805 + 39.980C (r = 0.9999) at 400 nm |
Statistical assessment15 of the results shows acceptable high correlation coefficient (r) values and small values of standard deviation of residuals (Sy/x). It also, reveals small values of standard deviation of intercept (Sa), small values of the percentage relative standard deviation and the percentage relative error (Table 1).
| Parameter | Value | |
|---|---|---|
| At 385 nm | At 400 nm | |
| a Percentage relative standard deviation.b Percentage relative error.c Limit of detection.d Limit of quantitation. | ||
| Linearity and range (ng mL−1) | 2.0–20.0 | 2.0–20.0 |
| Correlation coefficient (r) | 0.9999 | 0.9999 |
| Slope | 40.406 | 39.980 |
| Intercept | 28.976 | 14.805 |
| Sy/x, S.D. of the residuals | 2.131 | 5.450 |
| Sa,S.D. of the intercept | 1.792 | 4.584 |
| Sb, S.D. of the slope | 0.146 | 0.373 |
| S.D. | 0.60 | 0.89 |
| % RSDa | 0.599 | 0.892 |
| % errorb | 0.267 | 0.399 |
| LODc (ng mL−1) | 0.146 | 0.378 |
| LOQd (ng mL−1) | 0.444 | 1.147 |
| Studied drug | Proposed method | Comparison method8 | ||||
|---|---|---|---|---|---|---|
| Conc. taken (ng mL−1) | At 385 nm | At 400 nm | % found | |||
| Conc. found (ng mL−1) | % found | Conc. found (ng mL−1) | % found | |||
| a Figures between parentheses are the tabulated t and F values at P = 0.05.15 Each result is the average of three separate determinations. | ||||||
| At 385 nm | 2.0 | 1.9805 | 99.03 | 2.006 | 100.30 | 101.12 |
| 5.0 | 4.9751 | 99.50 | 5.007 | 100.15 | 102.45 | |
| 10.0 | 10.0486 | 100.49 | 9.885 | 98.85 | 99.78 | |
| 15.0 | 15.0478 | 100.32 | 15.187 | 101.25 | 101.77 | |
| 20.0 | 19.9480 | 99.74 | 19.915 | 99.57 | 99.14 | |
| X− ± SD | 99.82 ± 0.60 | 100.02 ± 0.890 | 100.85 ± 1.37 | |||
| t-test | 1.55 (2.31)a | 1.13 (2.31)a | ||||
| F-test | 5.27 (6.39)a | 2.37 (6.39)a | ||||
The intraday precision was assessed by determining three different VPS concentrations on the same day. On the other hand, interday precision was determined by quantifying three different concentrations over three consecutive days (Table 3).
| Parameters | At 385 nm | At 400 nm | |||||
|---|---|---|---|---|---|---|---|
| Concentration (ng mL−1) | Concentration (ng mL−1) | ||||||
| 5.0 | 10.0 | 15.0 | 5.0 | 10.0 | 15.0 | ||
| N. B. each result is the average of three separate determinations. | |||||||
| Intraday | % found | 99.50 | 100.49 | 100.32 | 100.15 | 98.85 | 101.25 |
| 100.11 | 98.88 | 101.13 | 99.74 | 99.45 | 100.84 | ||
| 101.22 | 98.45 | 98.94 | 100.98 | 98.73 | 99.32 | ||
(![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) ± S.D. ) ± S.D. |
100.28 ± 0.87 | 99.27 ± 1.08 | 1001.3 ± 1.11 | 100.29 ± 0.63 | 99.01 ± 0.39 | 100.47 ± 1.02 | |
| % RSD | 0.87 | 1.08 | 1.11 | 0.63 | 0.39 | 1.01 | |
| % error | 0.50 | 0.63 | 0.64 | 0.36 | 0.23 | 0.58 | |
| Inter-day | % found | 99.50 | 100.49 | 100.32 | 100.15 | 98.85 | 101.25 |
| 98.76 | 99.21 | 99.50 | 98.86 | 100.43 | 100.55 | ||
| 100.37 | 98.65 | 100.68 | 99.02 | 98.15 | 99.68 | ||
| () ± S.D. |
99.54 ± 0.81 | 99.54 ± 0.94 | 100.17 ± 0.61 | 99.34 ± 0.70 | 99.14 ± 1.17 | 100.49 ± 0.79 | |
| % RSD | 0.81 | 0.95 | 0.60 | 0.71 | 1.18 | 0.78 | |
| % error | 0.47 | 0.55 | 0.35 | 0.41 | 0.68 | 0.45 | |
Acceptable repeatability and intermediate precision of the cited method were assured by the very small values of relative standard deviations.
3.3. Analysis of VPS and SFV in a laboratory prepared mixture of their pharmaceutical ratio
The cited procedure was applied to the determination of VPS in a laboratory prepared mixture with SFV at their pharmaceutical ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4). The results obtained are summarized in Table 4. The results were in good agreement with those obtained using the reference method.8
:4). The results obtained are summarized in Table 4. The results were in good agreement with those obtained using the reference method.8
| Combination | Proposed method | Comparison method8 | ||||
|---|---|---|---|---|---|---|
| Amount taken (ng mL−1) | At 385 nm | At 400 nm | ||||
| Amount found (ng mL−1) | % found | Amount found (ng mL−1) | % found | % found | ||
| a N. B. each result is the average of three separate determinations. The figures between parentheses are the tabulated t and F values at P = 0.05.15 | ||||||
| SFV/VPS mixture 4/1 | 5.0 | 5.018 | 100.35 | 5.035 | 100.69 | 101.45 |
| 10.0 | 9.813 | 98.13 | 10.166 | 101.66 | 100.47 | |
| 15.0 | 14.889 | 99.26 | 14.909 | 99.39 | 99.12 | |
| Mean | 99.25 | 100.58 | 100.35 | |||
| ±S.D. | 1.11 | 1.14 | 1.17 | |||
| t-Test | 1.18 (2.78)a | −0.25 (2.78)a | ||||
| F-Test | 1.11 (19.0)a | 1.05 (19.0)a | ||||
3.4. Application to dosage form
The cited method was applied to determine VPS in its commercially available tablet form (Table 5). The results were in good agreement with those achieved using the reference method.8| Preparation | Proposed method | Comparison method8 | ||||
|---|---|---|---|---|---|---|
| Amount taken (ng mL−1) | At 385 nm | At 400 nm | ||||
| Amount found (ng mL−1) | % found | Amount found (ng mL−1) | % found | % found | ||
| The figures between parentheses are the tabulated t and F values at P = 0.05.15 | ||||||
| Epclusa® tablet | 5.0 | 4.929 | 98.59 | 4.984 | 99.67 | 102.45 |
| 10.0 | 10.045 | 100.45 | 9.956 | 99.56 | 101.78 | |
| 15 | 15.002 | 100.01 | 14.696 | 97.97 | 98.91 | |
| Mean | 99.68 | 99.07 | 101.05 | |||
| ±S.D. | 0.97 | 0.95 | 1.88 | |||
| t-Test | 1.12 (2.78) | 1.63 (2.78) | ||||
| F-Test | 3.74 (19.0) | 3.91 (19.0) | ||||
4. Conclusion
A new simple rapid and highly sensitive spectrofluorimetric method was clarified for the selective quantification of VPS in presence of its co-formulated drug (SFV) either in bulk or dosage forms. The cited method is rapid, economic and does not require the elaborate treatment associated with chromatographic methods or previously reported spectrofluorimetric method. In addition, the proposed method shows high sensitivity, with no need for derivatization procedures. This method can be applied for the sensitive and selective determination of VPS in the routine quality control work.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project was supported by the Deanship of Scientific Research at Prince Sattam Bin Abdulaziz University under the research project # 2017/03/7249.References
- H. S. Te and D. M. Jensen, Epidemiology of hepatitis B and C viruses: a global overview, Clin. Liver Dis., 2010, 14, 1–21 CrossRef PubMed.
- PubChem (2018), https://pubchem.ncbi.nlm.nih.gov/compound/Velpatasvir#section=Top, accessed, 12 February 2018.
- Wikipedia (2018), https://en.wikipedia.org/wiki/Sofosbuvir, accessed, 12 February 2018.
- Drugs.com (2018), https://www.drugs.com/history/epclusa.html, accessed, 12 February 2018.
- S. A. Abdel-Gawad, Simple chromatographic and spectrophotometric determination of sofosbuvir in pure and tablet forms, Eur. J. Chem., 2016, 7, 375–379 CrossRef.
- M. Miraghaei, B. Mohammadi, A. Babaei, S. Keshavarz and G. Bahrami, Development and validation of a new HPLC-DAD method for quantification of sofosbuvir in human serum and its comparison with LC–MS/MS technique: application to a bioequivalence study, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1063, 118–122 CrossRef PubMed.
- M. Nebsen and E. S. Elzanfaly, Stability-Indicating method and LC–MS-MS characterization of forced degradation products of sofosbuvir, J. Chromatogr. Sci., 2016, 54, 1631–1640 Search PubMed.
- J. Uppalapati and U. DrParimi, Analytical method development and validation for the simultaneous estimation of sofosbuvir and velpatasvir drug product by RP-HPLC method, Indo Am. J. Pharm. Res., 2017, 7, 401–409 Search PubMed.
- J. Bandla and S. Ganapaty, Stability indicating RP-HPLC method development and validation for the simultaneous determination of Sofosbuvir and Velpatasvir in tablet dosage forms, Indian J. Pharm. Biol. Res., 2017, 5, 10–16 Search PubMed.
- N. Sarath and J. V. L. N. Seshagiri Rao, A stability indicating RP-HPLC method for simultaneous estimation of velpatasvir and sofosbuvir in combined tablet dosage forms, World J. Pharm. Pharm. Sci., 2017, 6, 1596–1611 Search PubMed.
- G. K. Swamy, K. Pranay, M. Rajkumar and D. Sudheer Kumar, Novel stability indicating RP-HPLC method simultaneous determination of sofosbuvir and velpatasvir in bulk and combined tablet dosage forms, Asian J. Res. Biol. Pharm. Sci., 2017, 5, 143–151 Search PubMed.
- M. E. Mohamed, R. H. A. Hassan, A. M. Adel and A. Ramadan, Enhanced dispersive solid phase extraction assisted by cloud point strategy prior to fluorometric determination of anti-hepatitis C drug velpatasvir in pharmaceutical tablets and body fluids, RSC Adv., 2018, 8, 13292–13300 RSC , https://pubs.rsc.org/en/content/articlelanding/2018/ra/c7ra13719b#!divAbstract.
- G. G. Guilbault, Fluorescence, theory, instrumentation and practice, Marcel Dekker, Inc., New York, USA, 1967 Search PubMed.
- ICH Harmonized Tripartite Guidelines, validation of analytical procedures: Text and Methodology, Q2(R1), current Step 4 Version, arent Guidelines on Methodology Dated November 6 1996, Incorporated in November 2005, through (https://www.ich.org/LOB/media/MEDIA417.pdf), (accessed February 9, 2018).
- J. N. Miller, and J. C. Miller, Statistics and chemometrics for analytical chemistry, Pearson Education Limited, Harlow, 2005 Search PubMed.
Footnote |
| † Current address: College of Pharmacy, Prince Sattam Bin-Abdul Aziz University, King Abdullah Road, Al-Kharj, Kingdom of Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2018 |