
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem one-pot synthesis of 2-arylcinnolin-6-one derivatives from arylhydrazonopropanals and acetoacetanilides using sustainable ultrasound and microwave platforms†
Hamad M. Al-Matar *a,
Kamal M. Dawood*b and
Wael M. Tohamyc
*a,
Kamal M. Dawood*b and
Wael M. Tohamyc
aChemistry Department, Faculty of Science, University of Kuwait, P. O. Box 5969, Safat 13060, Kuwait. E-mail: h.almatar@ku.edu.kw; Fax: +965 24816482; Tel: +965 24987559
bChemistry Department, Faculty of Science, Cairo University, Giza 12613, Egypt. E-mail: dr_dawood@yahoo.com; Fax: +20 235727556; Tel: +20 235676602
cOrganometallic and Organometalloid Chemistry Department, National Research Centre, Cairo, Egypt
First published on 8th October 2018
Abstract
Several 2-arylcinnolin-6(2H)-one derivatives were synthesized via tandem annulation of a large number of 3-oxo-2-arylhydrazonopropanals with acetoacetanilide under three different heating modes (conventional heating, ultrasound and microwave irradiation) using triethylamine in ethanol. The factors affecting the optimization of the annulation process were thoroughly studied. The annulated structures were established on the basis of 1H and 13C NMR and MALDI-TOF/MS spectral data as well as single crystal X-ray analysis.
Introduction
Cinnolines are a class of aromatic nitrogen-heterocyclic compounds that are also known as benzo[c]pyridazines. Cinnoline derivatives have attracted considerable attention as drug candidates against several diseases due to their broad spectrum biological activities.1 They are patented as bactericides2–4 and fungicides.5 Cinnolin-3-one and cinnolin-4-one derivatives have also been reported to display potent antitumor,6–8 and antifungal,9 as well as human neutrophil elastase and STAT3 inhibitory activities10,11 and cannabinoid CB2 agonists.12,13 Cinnolin-5-ones were reported as atypical antipsychotic,14 and antitumor15 activities. In addition, cinnolin-3-carboxamides displayed ATM kinase inhibitors and were used in treating cancer16 and as BTK (Bruton's tyrosine kinase) inhibitors.17 Such potent biological importance of the cinnoline derivatives make them excellent templates for medicinal chemists to develop lead compounds for drug targets.Thus, several reaction protocols have been reported for the synthesis of the cinnoline building blocks, among those protocols are the cyclization of the arylhydrazono-cyanoacetanilide,18 Richter cyclization of ortho-ethynyldiazonium salts,19–22 acid catalyzed cyclization of 3-oxo-3-aryl-2-arylhydrazonopropanals,23 diazotization of ortho-substituted anilines.24 The cinnolin-3(2H)-one derivatives were also synthesized by rhodium or gold catalyzed reactions of azobenzene derivatives.25,26 To date, the only reported cinnolinone ring skeletons included cinnolin-3-one, cinnolin-4-one, cinnolin-5-one, and cinnoline-5,6-dione derivatives,6–15,27,28 however, synthesis of cinnolin-6(2H)-one structures are not reported so far.
The environmentally sustainable ultrasound technique has pronounced applications in organic synthesis, medicinal chemistry, and in materials science.29–31 The ultrasound is reported to be superior over conventional heating in organic synthesis, where formation of pure products in high yields, high selectivity, shorter reaction time and waste-minimization are among the most advantages of ultrasound techniques.32–39 In addition, a growing interest is focused on the use of microwave irradiation methodology because it markedly assists in achieving rapid incorporation of organic synthesis into broad industrial diversities.40–43 In continuation of our research work employed ultrasound and microwave irradiations in heterocyclic synthesis,44–51 we envisaged in the current work a feasible simple and straightforward approach for the construction of 2-arylcinnolin-6(2H)-one derivatives, which would be assembled by reaction of 3-oxo-2-arylhydrazonopropanals with double equivalents of acetoacetanilide via aldol-condensation followed by Michael-type addition reaction. For comparison with the conventional heating, two green and energy-saving platforms; ultrasound and microwave irradiation, are applied for achieving the goals of this work. Structures of the products are examined by measuring the X-ray crystallography along with all possible spectral analyses (IR, 1H and 13C NMR and MALDI-TOF/MS spectral data).
Results and discussion
At first, 3-oxo-2-arylhydrazonopropanal derivatives 1a–o were synthesized using reported procedure in the literature.52,53 Then, initial attempts to perform the annulation of the 3-oxo-2-phenylhydrazonopropanal 1a with acetoacetanilide 2 in ethanol in the presence of triethylamine were conducted under conventional thermal heating. The expected pyridazine 4 or phenylazophenol 6 structures, according to the previously published protocols54,55 could not be detected, however the assigned product was established as 2-arylcinnolin-6(2H)-one derivative 3a (Scheme 1). To our delight, phenylhydrazonopropanal 1a satisfactorily reacted with double equivalent of acetoacetanilide 2 to give the aldol-condensation followed by Michael-type addition product 3a, through in situ elimination of three molecules of water during the reaction path (Scheme 1). The structure 3a was established on the basis of IR, 1H and 13C NMR spectra, MALDI-TOF/MS spectrum, and X-ray diffraction data. Positive ion mode MALDI-TOF mass spectrum of 3a exhibited molecular ion peak of the form [M + K]+ at m/z 624.317. Optimization of the reaction of 1a with 2 was thoroughly investigated under various reaction conditions (different solvents, bases and heating modes) and the reaction path was monitored by TLC till full conversion of the starting substrates, and the results are summarized in Table 1. The results demonstrated that these factors greatly affected the productivity of this reaction. Among several solvents used (EtOH, MeOH, i-PrOH, n-hexane, DMF, 1,4-dioxane), ethanol was the proper one and the reaction was also much influenced by the type of base, where various organic and inorganic bases e.g. Et3N, pyridine, DBU, DABCO, NaHCO3, K2CO3 and KOH were tested. Using ethanol as solvent and triethylamine (10 mol%) as base, the reaction of the hydrazonal 1a with 2 under conventional heating at reflux for 4 hours resulted in 66% isolated yield, however, 70% isolated yield was obtained after 7 min of ultrasound irradiation and the yield was 75% after only 7 min of microwave irradiation (as examined by TLC) (entry 1, Table 1). The use of 20 mol% instead of 10 mol% of triethylamine led to production of 74%, 79% and 87% isolated yields under conventional heating (2 hours), US irradiation (50 min) and MW irradiation (5 min), respectively (entry 2, Table 1). Minor shift to lower yields were observed when Et3N was used in a higher ratio (30 mol%), where the isolated yields became 70%, 73% and 80% under thermal heating (3 hours), US (60 min) and MW (6 min), respectively (entry 3, Table 1). Very close product yields with low efficiency were obtained when MeOH or i-PrOH were employed as reaction solvent in the presence of 20 mol% of Et3N under all heating modes (entries 4 and 5, Table 1). Replacing ethanol with aprotic solvents (e.g. n-hexane, 1,4-dioxane, DMF and toluene) led to a sharp decrease in the product yields (varied from 28–55%) after longer reaction times regardless the type of heating mode (thermal, US or MW) (entries 6–9, Table 1). Keeping ethanol solvent and replacing Et3N by pyridine resulted in 55%, 60% and 75% isolated yields under thermal heating (6 hours), US (80 min) and MW (15 min), respectively (entry 10, Table 1). Use of other bases such as DBU, DABCO, NaHCO3, K2CO3 and KOH could not provide satisfactory yields (varied from 30–46% yields) under all the three heating modes (entries 11–15, Table 1). From the above findings, the benefit of microwave irradiation was confirmed by high efficiency with high conversion rate compared with the low conversion rate of the reactions that were carried out under conventional heating or ultrasound. The structure of the obtained reaction product 3a was unambiguously established by spectral analyses along with X-ray crystallography (Fig. 1).56 Mechanistically, polar solvents absorb microwave irradiation and convert it into heat that causes bulk heating and simultaneous rising of the temperature of the reaction mixture, in sharp contrast with conventional conductive heating.57 However, ultrasound waves cause cavitation phenomenon, that is, the creation, growth, and collapse of bubbles releasing enough energy to perform chemical reactions.31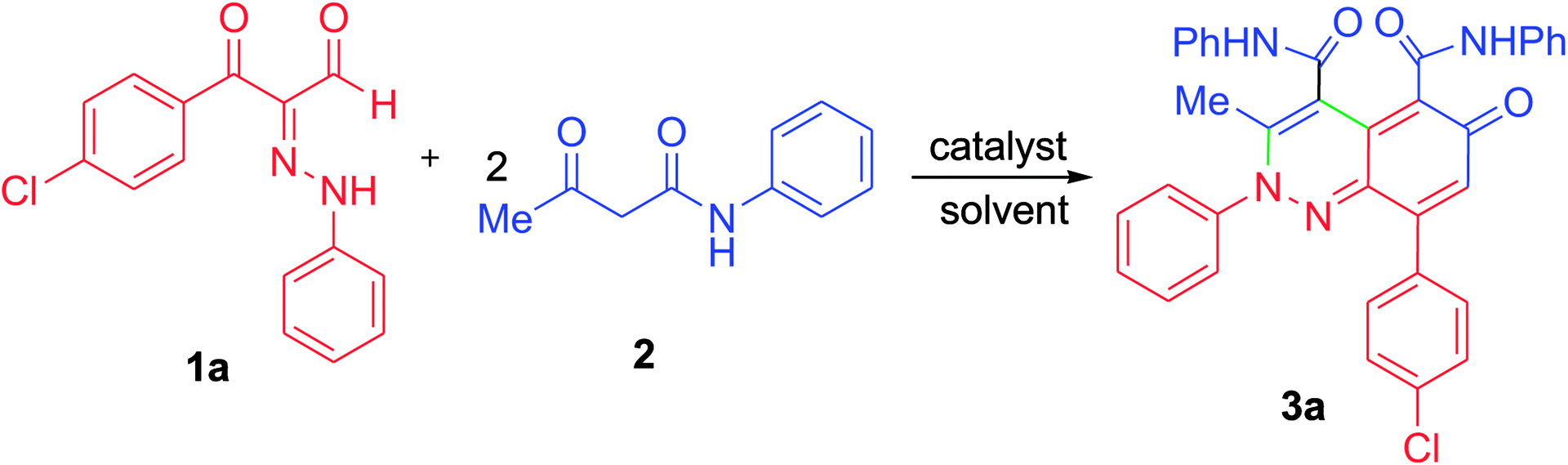 | ||
| Scheme 1 Annulation of phenylhydrazonopropanal 1a with acetoacetanilide 2. | ||
| Entry | Base catalyst | Solvent | Conv. heating | Sonication | MW irradiation | |||
|---|---|---|---|---|---|---|---|---|
| Yielde % | Time (h) | Yielde % | Time (min) | Yielde % | Time (min) | |||
| a Reaction conditions: phenylhydrazonopropanal 1a (2 mmol), acetoacetanilide 2 (4 mmol), solvent (15 mL) and base (10–30 mol%), conventional heating at reflux for 2–11 h, ultrasound at 80 °C for 50–110 min, microwave irradiation at 80 °C (200 W) for 5–20 min.b Base 10 mol%.c Base 20 mol%.d Base 30 mol%.e Isolated yields.f Yield was 12% after 20 min and 50% after 80 min.g Yield was 30% after 20 min and 62% after 40 min.h Yield was 53% after 3 min.i Yield was 17% after 2 h.j Yield was 38% after 50 min.k Yield was 25% after 5 min. | ||||||||
| 1 | Et3Nb | EtOH | 66 | 4 | 70 | 70 | 75 | 7 |
| 2 | Et3Nc | EtOH | 74f | 2 | 79g | 50 | 87h | 5 |
| 3 | Et3Nd | EtOH | 70 | 3 | 73 | 60 | 80 | 6 |
| 4 | Et3Nc | MeOH | 55 | 6 | 60 | 80 | 70 | 10 |
| 5 | Et3Nc | i-PrOH | 68 | 7 | 72 | 80 | 80 | 10 |
| 6 | Et3Nc | n-Hexane | 28 | 9 | 30 | 100 | 40 | 15 |
| 7 | Et3Nc | 1,4-Dioxane | 38 | 8 | 40 | 100 | 45 | 15 |
| 8 | Et3Nc | DMF | 42 | 9 | 45 | 110 | 55 | 20 |
| 9 | Et3Nc | Toluene | 34 | 8 | 37 | 100 | 42 | 15 |
| 10 | Pyridinec | EtOH | 55i | 6 | 60j | 80 | 75k | 15 |
| 11 | DABCOc | EtOH | 30 | 11 | 35 | 110 | 41 | 20 |
| 12 | DBUc | EtOH | 30 | 11 | 36 | 110 | 46 | 20 |
| 13 | NaHCO3c | EtOH | 34 | 9 | 36 | 90 | 42 | 12 |
| 14 | K2CO3c | EtOH | 38 | 7 | 40 | 90 | 45 | 15 |
| 15 | NaOHc | EtOH | 30 | 7 | 30 | 100 | 40 | 15 |
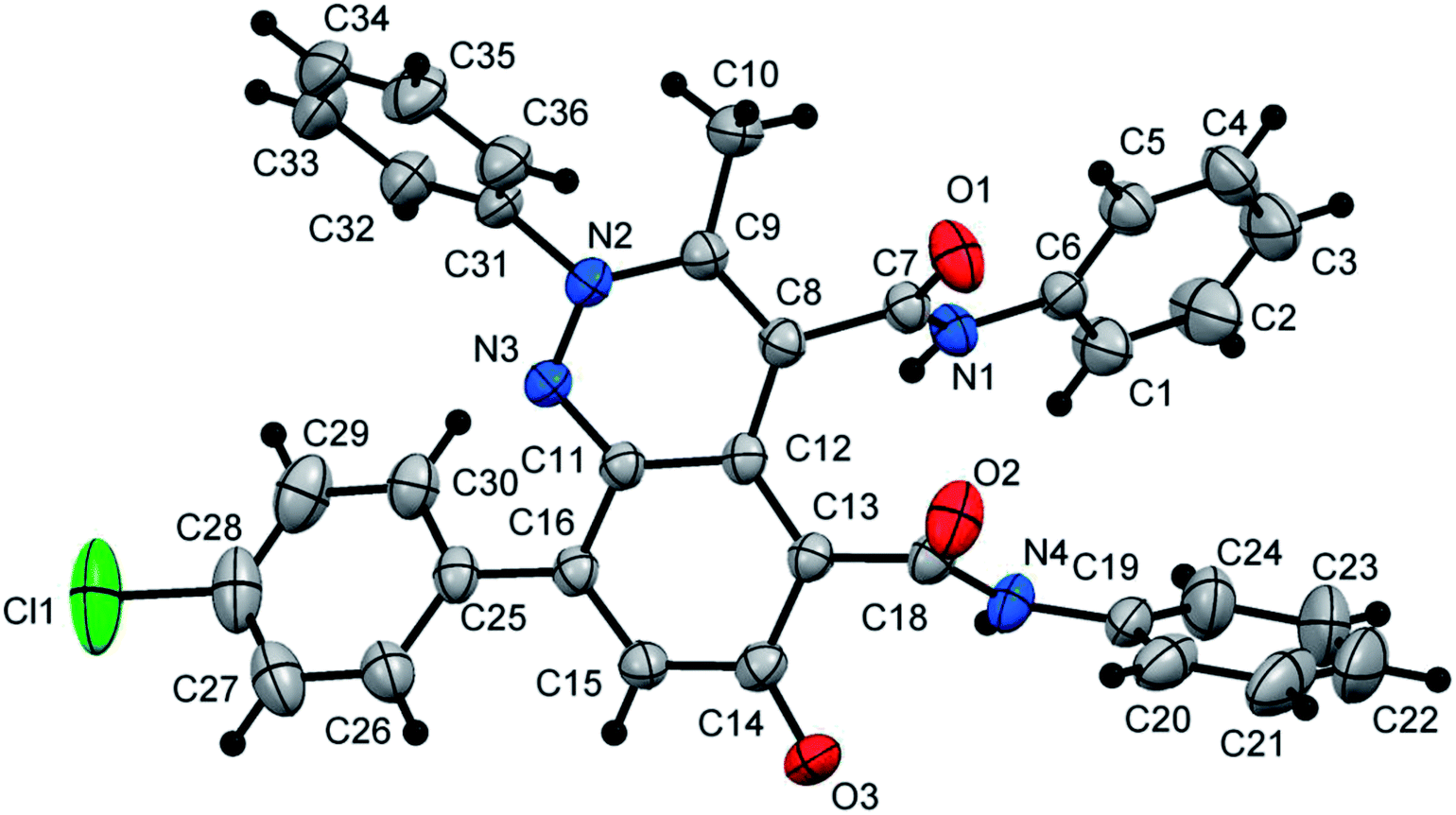 | ||
| Fig. 1 ORTEP plot of the X-ray crystallographic data determined for 3a. | ||
Prompted by the above interesting results and under the optimized reaction conditions, generalization and scope of the reaction using different substrates were investigated as shown in Table 2. The procedure was found to be applicable to a wide range of the arylhydrazonopropanal derivatives 1a–o with acetoacetanilide 2 (Scheme 2). Thus, annulation reaction of arylhydrazonopropanals 1a–o with acetoacetanilide 2 was conducted using EtOH (6 mL) and Et3N (20 mol%) under the three heating modes; conventional heating, ultrasound irradiation and microwave irradiation, afforded the corresponding 2-arylcinnolin-6(2H)-one derivatives 3a–o, Scheme 2. The reaction was monitored by TLC till full conversions of the starting substrates and the isolated yields of the corresponding annulated products 3a–o ranged from 45 to 75% (after 2–4 hours under conventional heating), from 50 and 85% yields (after 40–80 min under ultrasound), and from 60 to 90% yields (after 3–10 min of microwave irradiation) as depicted in Table 2, entries 1–15. From the structure–activity relationship point of view, it would be established, from the obtained results in Table 2, that the best yields within the shortest reaction times were obtained when R = 4-FC6H4 for the products 3f and 3g among all the tested derivatives under all heating techniques (Table 2, entries 6 and 7). In contrast, the yields of the products 3n and 3o (where R = CH3) were minimum and the reaction times were longest among all the derivatives under all applied heating modes (Table 2, entries 14 and 15). The 2-arylcinnolin-6(2H)-one structures 3a–o were determined from their all possible spectral data (IR, MALDI-TOF, 1H and 13C NMR spectra) and elemental analyses as mentioned in the Experimental section.
| Entry | Product | R | Ar | Conv. heating | Sonication | MW | |||
|---|---|---|---|---|---|---|---|---|---|
| Yieldb % | Time (h) | Yieldb % | Time (min) | Yieldb % | Time (min) | ||||
| a Reaction condition: 1a–o (1 mmol), acetoacetanilide (2 mmol), triethylamine (20 mol%) in EtOH (6 mL), heated under reflux for 2–4 h, ultrasound irradiation at 80 °C for 40–80 min, microwave irradiations at 80 °C (200 W) for 3–10 min.b Isolated yields.c Yield was 55% after 40 min.d Yield was 51% after 3 min.e Yield was 52% after 3 min.f Yield was 28% after 2 h.g Yield was 31% after 40 min.h Yield was 21% after 3 min. | |||||||||
| 1 | 3a | 4-ClC6H4 | C6H5 | 74 | 2 | 79 | 50 | 87 | 5 |
| 2 | 3b | C6H5 | 4-ClC6H4 | 70 | 3 | 74 | 60 | 75 | 8 |
| 3 | 3c | 4-ClC6H4 | 4-BrC6H4 | 72 | 3 | 75 | 50 | 84 | 5 |
| 4 | 3d | 4-BrC6H4 | 4-ClC6H4 | 75 | 2 | 82 | 40 | 87 | 3 |
| 5 | 3e | 4-BrC6H4 | 4-BrC6H4 | 73 | 2 | 80c | 60 | 84d | 5 |
| 6 | 3f | 4-FC6H4 | 4-ClC6H4 | 73 | 2 | 85 | 40 | 90 | 3 |
| 7 | 3g | 4-FC6H4 | 4-BrC6H4 | 70 | 3 | 78 | 40 | 85e | 5 |
| 8 | 3h | 4-MeOC6H4 | C6H5 | 63 | 4 | 68 | 70 | 74 | 8 |
| 9 | 3i | 4-MeOC6H4 | 4-ClC6H4 | 62 | 4 | 65 | 80 | 72 | 10 |
| 10 | 3j | 4-MeOC6H4 | 4-BrC6H4 | 65 | 3 | 70 | 60 | 80 | 8 |
| 11 | 3k | 4-MeOC6H4 | 2-NO2C6H4 | 58 | 4 | 62 | 80 | 72 | 10 |
| 12 | 3l | 4-NO2C6H4 | 4-ClC6H4 | 55f | 4 | 60g | 80 | 68h | 10 |
| 13 | 3m | 4-NO2C6H4 | 4-BrC6H4 | 52 | 4 | 62 | 70 | 69 | 10 |
| 14 | 3n | CH3 | 4-BrC6H4 | 50 | 4 | 58 | 80 | 64 | 10 |
| 15 | 3o | CH3 | 2-NO2C6H4 | 45 | 4 | 50 | 80 | 60 | 10 |
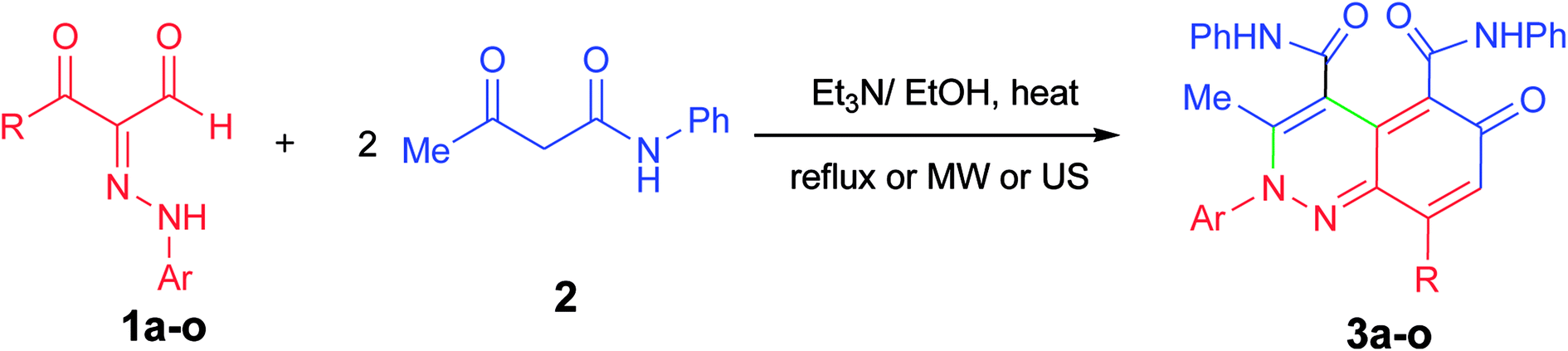 | ||
| Scheme 2 Annulation of arylhydrazonopropanals 1a–o with acetoacetanilide 2. | ||
A plausible mechanistic pathway for the annulation of the hydrazonals 1a–o with acetoacetanilide 2 was suggested as depicted in Scheme 3. First, base catalyzed aldol condensation of the active methylene of 2 with the aldehyde function of 1 followed by loss of one molecule of water led to the formation of the α,β-unsaturated carbonyl adduct A. Then, a second molecule of 2 attacks the adduct A via a Michael-type addition to give the intermediate B, which underwent cyclization via nucleophilic attack of the hydrazone-NH to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O function followed by loss of water molecule from C to give the pyridazine intermediate D. Further annulations took place by base catalyzed cyclization through C–C bond formation followed by loss of water molecule from E to give the intermediate F. Loss of hydrogen molecule through air oxidation of F led to the formation of the novel 6-oxo-2,6-dihydrocinnoline-4,5-dicarboxamide derivatives 3a–o.
O function followed by loss of water molecule from C to give the pyridazine intermediate D. Further annulations took place by base catalyzed cyclization through C–C bond formation followed by loss of water molecule from E to give the intermediate F. Loss of hydrogen molecule through air oxidation of F led to the formation of the novel 6-oxo-2,6-dihydrocinnoline-4,5-dicarboxamide derivatives 3a–o.
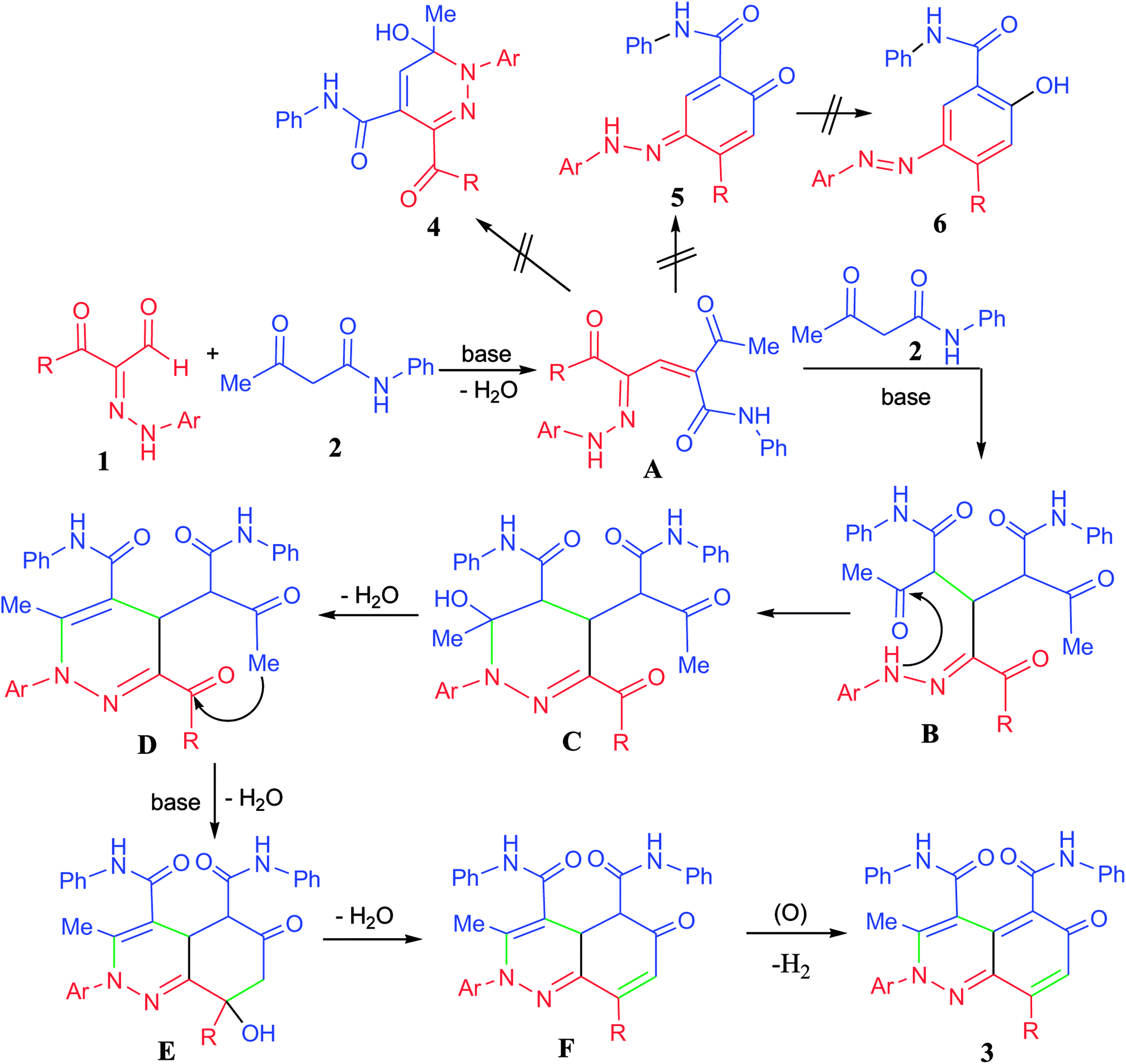 | ||
| Scheme 3 Proposed mechanism for the annulation pathway. | ||
Experimental section
General
Melting points were recorded on a Griffin melting point apparatus and are reported uncorrected. IR spectra were recorded using KBr disks using a Perkin-Elmer System 2000 FT-IR spectrophotometer. 1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra were recorded at 25 °C using DMSO-d6 as solvent with TMS as internal standard on a Bruker DPX 400 or 600 super-conducting NMR spectrometer. Chemical shifts are reported in ppm. Low-resolution electron impact mass spectra [MS (EI)] was performed using a high resolution GC-MS (DFS) thermo spectrometer at 70.1 eV using magnetic sector mass analyzer. Follow up of the reactions was made by using thin layer chromatography (TLC). Microwave experiments were carried out using a CEM Discover LabMate® microwave apparatus (300 W with ChemDriver software; Matthews, NC). Reactions were conducted under microwave irradiation in heavy-walled Pyrex tubes fitted with PCS caps (closed vessel under pressure). The X-ray crystal structures were determined by using a Rigaku R-AXIS RAPID diffractometer and Bruker X8 Prospector and the collection of single crystal data was made at room temperature by using Cu-Kα radiation. The data were collected at room temperature. The structures were solved by using direct methods and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. The structures were solved and refined using the Bruker SHELXTL Software Package (Structure solution program-SHELXS-97 and Refinement program-SHELXL 97).58 Data were corrected for the absorption effects using the multi-scan method (SADABS). Sonication was performed in MKC6, Guyson ultrasonic bath (Model-MKC6, operating frequency 38 kHz ± 10% and an output power of 110 watts) with digital timer (6 s to 100 min) and heater allows solution heating to be set from 20 to 80 °C in 1 °C increments. The inside tank dimensions are 150 × 300 × 150 mm (length × width × depth) with a fluid capacity of 6 L.Molecular weight determination using MALDI-TOF/MS
Determination of molecular weight was achieved using MALDI-TOF/MS (Bruker, model ultraflexXtreme). All the analyses were examined in positive mode using a reflectron time of flight mass spectrometer. Data processing was performed using Bruker flexAnalysis. The instrument was calibrated with peptide calibration standard.The sample was prepared as following: the matrix α-cyano-4-hydroxycinnamic acid (HCCA) was purchased from Sigma Aldrich. All reagents were used without any further purification. HCCA was dissolved in mixture of acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water:trifluoroacetic acid (70:30:0.1). All samples were dissolved in chloroform. Deposition of the samples on target pate was done as follows: 5 L of the sample was mixed with 5 L of the matrix (1:1 ratio). The solution mixture was placed on the target plate and allowed to dry at room temperature.
:water:trifluoroacetic acid (70:30:0.1). All samples were dissolved in chloroform. Deposition of the samples on target pate was done as follows: 5 L of the sample was mixed with 5 L of the matrix (1:1 ratio). The solution mixture was placed on the target plate and allowed to dry at room temperature.
Synthesis of 6-oxo-2,6-dihydrocinnoline-4,5-dicarboxamide derivatives 3a–o
8-(4-Chlorophenyl)-3-methyl-6-oxo-N4,N5,2-triphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3a). Orange crystals, mp 268–270 °C; IR (KBr): ν/cm−1 3259 (NH), 1683, 1654 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.95 (t, J = 6.9 Hz, 1H, Ar–H), 7.06 (t, J = 6.6 Hz, 1H, Ar–H), 7.11 (m, 3H, Ar–H), 7.24 (t, J = 7.8 Hz, 2H, Ar–H), 7.46–7.47 (m, 2H, Ar–H), 7.49–7.50 (m, 2H, Ar–H), 7.55–7.62 (m, 3H, Ar–H), 7.64 (d, J = 4.2 Hz, 4H, Ar–H), 7.66–7.68 (m, 2H, Ar–H), 10.48 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.35 (CH3), 115.17, 119.48, 119.79, 123.50, 124.20, 125.94, 127.33, 127.97, 128.09, 128.36, 129.78, 131.90, 133.46, 134.43, 135.41, 138.74, 139.43, 141.73, 142.56 (Ar–C) 162.59, 164.20, 178.96 (CO). MALDI-TOF: calcd for [M + K]+ m/z = 624.128, found m/z = 624.317. Anal. calcd for C35H25ClN4O3: C, 71.85; H, 4.31; N, 9.58 found: C, 71.77; H, 4.39; N, 9.61. Crystal data, C35H25ClN4O3, M = 585.04, monoclinic, a = 15.5696(6) Å, b = 19.6736(7) Å, c = 9.4182(3) Å, α = 90°, β = 95.241(3)°, γ = 90°, V = 2872.83(18) Å3, T = 296(2) K, space group: P121/c1, Z = 4, calculated density = 1.353 g cm−3, no. of reflection measured 24182, θmax = 66.51°, R1 = 0.0800.56
2-(4-Chlorophenyl)-3-methyl-6-oxo-N4,N5,8-triphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3b). Yellow crystals, mp 270–271 °C; IR (KBr): ν/cm−1 3249 (NH), 1677, 1660 (C
O); 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3); 6.97 (t, J = 7.2 Hz, 1H, Ar–H), 7.07–7.15 (m, 2H, Ar–H), 7.13 (t, J = 7.8 Hz, 2H, Ar–H), 7.25 (t, J = 8.1 Hz, 2H, Ar–H), 7.42–7.49 (m, 5H, Ar–H), 7.56 (d, J = 7.8 Hz, 2H, Ar–H), 7.65 (d, J = 8.4 Hz, 2H, Ar–H), 7.63–7.72 (m, 4H, Ar–H), 10.48 (s, 1H, NH), 10.56 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.33(CH3), 115.41, 119.49, 119.79, 122.54, 123.53, 123.95, 127.23, 127.92, 127.98, 128.11, 128.39, 128.59, 129.79, 130.11, 134.49, 135.23, 135.58, 138.75, 139.47, 140.59, 141.32, 141.59, 143.32 (Ar–C), 162.55, 164.23, 178.80 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 607.151, [M + K]+ m/z = 623.128, found m/z = 607.50 and 623.458; anal. calcd for C35H25ClN4O3: C, 71.85; H, 4.31; N, 9.58. Found: C, 71.71; H, 4.40; N, 9.51.
2-(4-Bromophenyl)-8-(4-chlorophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3c). Pale yellow crystals, mp 274–276 °C; IR (KBr): ν/cm−1 3251 (NH), 1676 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3); 6.95 (t, J = 7.25 Hz, 1H, Ar–H), 7.06 (t, J = 7.2 Hz, 1H, Ar–H), 7.10–7.13 (m, 3H, Ar–H), 7.24 (t, J = 7.8 Hz, 2H, Ar–H), 7.45 (d, J = 7.2 Hz, 2H, Ar–H), 7.50 (d, J = 8.4 Hz, 2H, Ar–H), 7.53 (d, J = 7.8 Hz, 2H, Ar–H), 7.61 (d, J = 8.4 Hz, 2H, Ar–H), 7.67 (d, J = 8.4 Hz, 2H, Ar–H), 7.85 (d, J = 8.4 Hz, 2H, Ar–H), 10.44 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 115.56, 119.52, 119.82, 122.59, 123.56, 123.16, 123.58, 124.04, 127.22, 128.03, 128.13, 128.15, 128.41, 131.94, 132.80, 133.55, 134.36, 135.39, 138.74, 139.45, 140.40, 141.63, 141.73, 142.03 (Ar–C), 162.55, 164.18, 178.68 (CO). MALDI-TOF: calcd for [M + K]+ m/z = 703.034, found 703.713; anal. calcd for C35H24BrClN4O3: C, 63.31; H, 3.64; N, 8.44. Found: C, 63.28; H, 3.72; N, 8.49.
8-(4-Bromophenyl)-2-(4-chlorophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3d). Orange crystals, mp 276–278 °C; IR (KBr): ν/cm−1 3247 (NH), 1674 (C
O); 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 6.95 (t, J = 7.2 Hz, 1H, Ar–H), 7.06 (t, J = 7.2 Hz, 1H, Ar–H), 7.11 (m, 3H, Ar–H), 7.24 (t, J = 8.1 Hz, 2H, Ar–H), 7.46 (d, J = 7.8 Hz, 2H, Ar–H), 7.54 (d, J = 7.8 Hz, 2H, Ar–H), 7.60–7.65 (m, 4H, Ar–H), 7.67–7.73 (m, 4H, Ar–H), 10.44 (s, 1H, NH), 10.54 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.37 (CH3), 115.54, 119.49, 119.79, 122.24, 122.56, 123.55, 124.00, 127.22, 128.11, 129.85, 130.94, 132.21, 134.55, 134.73, 135.36, 138.73, 139.44, 140.31, 141.29, 141.67, 142.07 (Ar–C), 162.53, 164.15, 179.43 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 686.937, [M + K]+ m/z = 703.045, found m/z = 687.294 and 703.261; anal. calcd For C35H24BrClN4O3: C, 63.31; H, 3.64; N, 8.44. Found: C, 63.38; H, 3.60; N, 8.41.
2,8-Di(4-bromophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3e). Pale yellow solid, mp 268–270 °C; IR (KBr): ν/cm−1 3250 (NH), 1676 (C
O); 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 6.95 (t, J = 7.5 Hz, 1H, Ar–H), 7.06 (t, J = 7.5 Hz, 1H, Ar–H), 7.10–7.13 (m, 3H, Ar–H), 7.24 (t, J = 7.8 Hz, 2H, Ar–H), 7.46 (d, J = 7.8 Hz, 2H, Ar–H), 7.54 (d, J = 7.8 Hz, 2H, Ar–H), 7.59–7.65 (m, 6H, Ar–H), 7.86 (d, J = 9 Hz, 2H, Ar–H), 10.44 (s, 1H, NH), 10.54 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.37 (CH3), 115.55, 119.49, 119.79, 122.24, 122.56, 123.15, 123.55, 124.01, 127.20, 128.11, 128.14, 128.39, 130.94, 132.20, 132.79, 134.72, 135.35, 138.72, 139.44, 140.31, 141.60, 141.71, 142.05 (Ar–C), 162.52, 164.15, 178.65 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 729.011, found 729.03; anal. calcd for C35H24Br2N4O3: C, 59.34; H, 3.41; N, 7.91. Found: C, 59.22; H, 3.49; N, 7.84.
2-(4-Chlorophenyl)-8-(4-fluorophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3f). Yellow solid, mp 271–273 °C; IR (KBr): ν/cm−1 3277 (NH), 1659 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.95 (t, J = 7.5 Hz, 1H, Ar–H), 7.06 (t, J = 7.2 Hz, 1H, Ar–H), 7.09 (s, 1H, Ar–H), 7.12 (t, J = 7.8 Hz, 2H, Ar–H), 7.22–7.29 (m, 4H, Ar–H), 7.45 (d, J = 7.2 Hz, 2H, Ar–H), 7.55 (d, J = 7.2 Hz, 2H, Ar–H), 7.67–7.72 (m, 6H, Ar–H), 10.46 (s, 1H, NH), 10.55 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.38 (CH3), 114.86, 115.00, 115.44, 119.51, 119.80, 122.57, 123.56, 124.01, 127.25, 127.92, 128.12, 128.41, 129.83, 131.87, 131.89, 132.26, 132.31, 134.51, 135.26, 138.75, 139.46, 140.54, 141.33, 141.61, 142.20 (Ar–C), 162.59, 164.20, 178.75 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 626.031, found 626.317; anal. calcd for C35H24ClFN4O3: C, 69.71; H, 4.01; N, 9.29. Found: C, 69.65; H, 4.15; N, 9.23.
2-(4-Bromophenyl)-8-(4-fluorophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3g). Yellow crystals, mp 267–269 °C; IR (KBr): ν/cm−1 3274 (NH), 1676 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3); 6.95 (t, J = 7.2 Hz, 1H, Ar–H), 7.06 (t, J = 7.5 Hz, 1H, Ar–H), 7.09 (s, 1H, Ar–H), 7.12 (t, J = 7.8 Hz, 2H, Ar–H), 7.22–7.29 (m, 4H, Ar–H), 7.47 (d, J = 7.2 Hz, 2H, Ar–H), 7.55 (d, J = 7.2 Hz, 2H, Ar–H), 7.61 (d, J = 8.4 Hz, 2H, Ar–H), 7.68–7.71 (m, 2H, Ar–H), 7.85 (d, J = 9 Hz, 2H, Ar–H), 10.46 (s, 1H, NH), 10.57 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.38 (CH3), 114.85, 114.99, 115.45, 119.50, 122.55, 123.10, 123.53, 124.02, 127.24, 128.11, 128.14, 128.39, 131.87, 132.24, 132.29, 132.76, 135.25, 138.75, 139.45, 140.54, 141.54, 141.74, 142.19, 161.54, 162.27 (Ar–C) 163.17, 164.19, 178.75 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 669.091, found 669.316; anal. calcd for C35H24BrFN4O3: C, 64.92; H, 3.74; N, 8.65. Found: C, 64.85; H, 3.66; N, 8.77.
8-(4-Anisyl)-3-methyl-6-oxo-N4,N5,2-triphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3h). Orange crystals, mp 242–244 °C; IR (KBr): ν/cm−1 3260 (NH), 1687, 1653 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 6.96 (t, J = 7.5 Hz, 1H, Ar–H), 7.00 (d, J = 9 Hz, 1H, Ar–H), 7.07–7.14 (m, 4H, Ar–H), 7.24 (t, J = 7.8 Hz, 2H, Ar–H), 7.38–7.48 (m, 4H, Ar–H), 7.57–7.61 (m, 4H, Ar–H), 7.64–7.82 (m, 3H, Ar–H), 7.95 (d, J = 7.8 Hz, 2H, Ar–H), 11.84 (br. s, 2H, 2NH); 13C NMR (DMSO-d6): δ = 19.45 (CH3), 55.77 (OCH3), 113.43, 113.56, 114.24, 114.91, 115.37, 116.61, 119.57, 119.87, 122.59, 124.18, 125.83, 126.07, 127.90, 128.46, 129.85, 131.57, 132.42, 138.89, 139.29, 141.63, 142.70 (Ar–C), 162.67, 164.42, 188.79 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 603.201, found 603.327; anal. calcd for C36H28N4O4: C, 74.47; H, 4.86; N, 9.65. Found: C, 74.40; H, 4.74; N, 9.77.
8-(4-Anisyl)-2-(4-chlorophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3i). Orange crystals, mp 225–227 °C; IR (KBr): ν/cm−1 3313 (NH), 1705, 1642 (C
O); 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 6.95 (t, J = 7.8 Hz, 1H, Ar–H), 7.00 (d, J = 8.4 Hz, 2H, Ar–H), 7.04 (s, 1H, Ar–H), 7.07 (d, J = 7.2 Hz, 1H, Ar–H), 7.12 (t, J = 7.8 Hz, 2H, Ar–H), 7.24 (t, J = 7.8 Hz, 2H, Ar–H), 7.47 (d, J = 7.2 Hz, 2H, Ar–H), 7.56 (d, J = 7.8 Hz, 2H, Ar–H), 7.60 (d, J = 8.4 Hz, 2H, Ar–H), 7.68–7.73 (m, 4H, Ar–H), 10.50 (s, 1H, NH), 10.53 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.35 (CH3), 55.18 (OCH3), 113.50, 115.18, 119.49, 119.79, 122.53, 123.51, 123.91, 127.34, 127.73, 127.93, 128.12, 128.39, 129.82, 131.50, 134.45, 138.78, 139.46, 140.68, 141.36, 141.46, 142.93, 159.70 (Ar–C), 162.66, 164.28, 178.95 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 637.162, [M + K]+ m/z = 653.136, found 637.488 and 653.447; anal. calcd For C36H27ClN4O4: C, 70.30; H, 4.42; N, 9.11. Found: C, 70.40; H, 4.55; N, 9.18.
8-(4-Anisyl)-2-(4-bromophenyl)-3-methyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3j). Orange crystals, mp 252–254 °C; IR (KBr): ν/cm−1 3268 (NH), 1678, 1659 (C
O); 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 3.79 (s, 3H, OCH3), 6.97 (t, J = 7.5 Hz, 1H, Ar–H), 7.02 (d, J = 8.4 Hz, 2H, Ar–H), 7.07 (t, J = 7.5 Hz, 2H, Ar–H), 7.12 (t, J = 7.8 Hz, 2H, Ar–H), 7.24 (t, J = 7.8 Hz, 2H, Ar–H), 7.46 (d, J = 7.8 Hz, 2H, Ar–H), 7.55–7.62 (m, 6H, Ar–H), 7.86 (d, J = 8.4 Hz, 2H, Ar–H), 10.49 (s, 1H, NH), 10.54 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 55.20 (OCH3), 113.52, 115.24, 119.51, 119.80, 122.56, 123.08, 123.54, 123.92, 127.32, 127.74, 128.14, 128.19, 128.42, 131.52, 132.78, 134.41, 138.80, 139.49, 140.70, 141.42, 141.80, 142.95, 159.72 (Ar–C), 162.68, 164.30, 178.96 (CO). MALDI-TOF: calcd for [M + H]+ m/z = 661.127, found 661.456; anal. calcd for C36H27BrN4O4: C, 65.56; H, 4.13; N, 8.49. Found: C, 65.40; H, 4.23; N, 8.58.
8-(4-Anisyl)-3-methyl-2-(4-nitrophenyl)-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3k). Orange crystals, mp 248–250 °C; IR (KBr): ν/cm−1 3274 (NH), 1661 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 3.78 (s, 3H, OCH3), 6.92–7.02 (m, 5H, Ar–H), 7.13 (t, J = 7.8 Hz, 2H, Ar–H), 7.22–7.31 (m, 4H, Ar–H), 7.47 (m, 2H, Ar–H), 7.54–7.58 (m, 3H, Ar–H), 7.89 (t, J = 7.8 Hz, 1H, Ar–H), 8.01 (s, 1H, Ar–H), 8.36 (d, J = 7.8 Hz, 1H, Ar–H), 10.40 (br. s, 1H, NH), 10.64 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 55.19 (OCH3), 113.40, 113.61, 119.43, 119.52, 119.78, 119.93, 122.46, 122.69, 123.59, 123.54, 126.68, 127.54, 128.07, 128.10, 128.32, 128.41, 128.73, 130.91, 131.52, 131.94, 134.53, 134.85, 135.37, 138.63, 139.54, 141.43, 141.95, 142.95, 144.05, 159.58, 159.64 (Ar–C), 162.20, 164.64, 179.15(CO). MALDI-TOF: calcd for [M + Na]+ m/z = 648.186, found 648.439; anal. calcd for C36H27N5O6: C, 69.11; H, 4.35; N, 11.19. Found: C, 69.22; H, 4.42; N, 11.26.
2-(4-Chlorophenyl)-3-methyl-8-(4-nitrophenyl)-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3l). Orange crystals, mp 249–251 °C; IR (KBr): ν/cm−1 3251 (NH), 1668 (C
O); 1H NMR (DMSO-d6): δ = 2.35 (s, 3H, CH3), 6.95 (t, J = 7.5 Hz, 1H, Ar–H), 7.07 (t, J = 7.5 Hz, 1H, Ar–H), 7.12 (t, J = 7.5 Hz, 2H, Ar–H), 7.22–7.25 (m, 3H, Ar–H), 7.47 (d, J = 7.8 Hz, 2H, Ar–H), 7.55 (d, J = 7.8 Hz, 2H, Ar–H), 7.68–7.72 (m, 4H, Ar–H), 7.94 (d, J = 8.4 Hz, 2H, Ar–H), 8.26 (d, J = 8.4 Hz, 2H, Ar–H), 10.41 (s, 1H, NH), 10.56 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.87 (CH3), 116.44, 119.99, 120.30, 123.09, 123.47, 124.09, 124.59, 127.61, 128.40, 128.61, 128.89, 130.37, 131.98, 135.07, 136.70, 139.18, 139.92, 140.66, 141.72, 142.35, 142.64, 147.86 (Ar–C), 162.92, 164.55, 178.84 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 652.136, found 652.859; anal. calcd for C35H24ClN5O5: C, 66.72; H, 3.84; N, 11.12. Found: C, 66.66; H, 3.95; N, 11.29.
2-(4-Bromophenyl)-3-methyl-8-(4-nitrophenyl)-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3m). Orange solid, mp 302–304 °C; IR (KBr): ν/cm−1 3259 (NH), 1668 (C
O); 1H NMR (DMSO-d6): δ = 2.36 (s, 3H, CH3), 6.96–7.26 (m, 7H, Ar–H), 7.47 (d, J = 7.2 Hz, 2H, Ar–H), 7.56 (d, J = 7.8 Hz, 2H, Ar–H), 7.60–7.65 (m, 2H, Ar–H), 7.86–7.95 (m, 4H, Ar–H), 8.27 (d, J = 7.8 Hz, 1H, Ar–H), 8.34 (d, J = 7.8 Hz, 1H, Ar–H), 10.45 (s, 1H, NH), 10.57 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 19.39 (CH3), 115.70, 115.81, 119.52, 119.82, 121.76, 122.60, 122.98, 123.16, 123.21, 123.60, 124.03, 124.69, 127.20, 128.12, 128.40, 130.78, 131.08, 131.48, 132.81, 135.92, 136.85, 138.74, 139.44, 140.35, 140.45, 141.66, 142.28, 143.46, 147.41 (Ar–C), 162.28, 164.18, 178.68 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 698.038, found 698.295; anal. calcd For C35H24BrN5O5: C, 62.32; H, 3.59; N, 10.38. Found: C, 62.26; H, 3.46; N, 10.42.
2-(4-Bromophenyl)-3,8-dimethyl-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3n). Orange crystals, mp 222–223 °C; IR (KBr): ν/cm−1 3240 (NH), 1658, 1640 (C
O); 1H NMR (DMSO-d6): δ = 2.34 (s, 3H, CH3), 2.39 (s, 3H, CH3), 6.95 (t, J = 6.9 Hz, 1H, Ar–H), 6.99 (s, 1H, Ar–H), 7.06 (t, J = 7.5 Hz, 1H, Ar–H), 7.12 (t, J = 7.5 Hz, 2H, Ar–H), 7.23 (t, J = 7.8 Hz, 2H, Ar–H), 7.45 (d, J = 7.2 Hz, 2H, Ar–H), 7.55 (d, J = 7.2 Hz, 2H, Ar–H), 7.66 (d, J = 7.8 Hz, 2H, Ar–H), 7.91 (d, J = 7.8 Hz, 2H, Ar–H), 10.43 (s, 1H, NH), 10.47 (s, 1H, NH); 13C NMR (DMSO-d6): δ = 16.88 (CH3), 19.27 (CH3), 114.32, 119.49, 119.77, 122.50, 123.16, 123.49, 123.61, 127.44, 128.11, 128.38, 132.86, 135.14, 138.80, 139.47, 140.68, 141.58, 141.92, 142.27 (Ar–C), 162.66, 164.30, 179.51 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 589.085, found 589.513; anal. calcd For C30H23BrN4O3: C, 63.50; H, 4.09; N, 9.87. Found: C, 63.55; H, 4.14; N, 9.76.
3,8-Dimethyl-2-(2-nitrophenyl)-6-oxo-N4,N5-diphenyl-2,6-dihydrocinnoline-4,5-dicarboxamide (3o). Orange crystals, mp 176–178 °C; IR (KBr): ν/cm−1 3260 (NH), 1677 (C
O); 1H NMR (DMSO-d6): δ = 2.37 (s, 3H, CH3), 2.38 (s, 3H, CH3), 7.06 (t, J = 7.2 Hz, 2H, Ar–H), 7.11–7.14 (m, 2H, Ar–H), 7.31 (t, J = 8.1 Hz, 2H, Ar–H), 7.57 (d, J = 8.4 Hz, 2H, Ar–H), 7.73–7.75 (m, 2H, Ar–H), 7.83 (s, 2H, Ar–H), 7.92–7.94 (m, 2H, Ar–H), 8.12 (d, J = 8.4 Hz, 2H, Ar–H), 11.52 (s, 2H, 2NH); 13C NMR (DMSO-d6): δ = 24.50 (CH3), 30.17 (CH3), 115.02, 116.84, 119.06, 119.50, 120.96, 121.22, 123.39, 125.70, 128.74, 130.02, 133.55, 135.22, 136.12, 138.87, 139.15, 141.13 (Ar–C) 165.02, 197.16, 202.83 (CO). MALDI-TOF: calcd for [M + Na]+ m/z = 556.159, found 556.233; anal. calcd For C30H23N5O5: C, 67.53; H, 4.35; N, 13.13. Found: C, 67.59; H, 4.28; N, 13.24.
Conclusions
A feasible and efficient one-pot tandem annulation of the hydrazonals 1a–o with acetoacetanilide 2 was performed under three different heating modes (conventional heating, ultrasound and microwave irradiation) and resulted in the formation of a series of novel class of the 2-arylcinnolin-6(2H)-one derivatives 3a–o. Microwave irradiation proved to be superior and efficient tool over ultrasound and conventional heating, for the promotion of such annulation reactions using ethanol as solvent and Et3N as a base.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was financially supported by the University of Kuwait through a research grant (SC07/13). The facilities of ANALAB and SAF supported by research grants GS01/01, GS01/05, GS01/03, and GS03/08 are greatly appreciated.References
- W. Lewgowd and A. Stanczak, Arch. Pharm. Chem. Life Sci., 2007, 340, 65–80 CrossRef CAS PubMed.
- S. Inoue, A. Yasaki, H. Mochizuki, H. Tutsumi, M. Murata and K. Sakane, Jpn. Pat., 05213951 A2 930824, 1993, Chem. Abstr., 1993, 120, 134503w.
- H. Tutsumi, T. Terasawa, D. Barret, M. Murata, K. Sakane, A. Yazaki and S. Inoue, Jpn. Pat., 9215584 A1 920217, 1992, Chem. Abstr., 1992, 118, 254944w.
- M. Yokomoto, A. Yazaki, N. Hayashi, S. Hatono, S. Ioue and Y. Kuramoto, Eur. Pat., 4700578 A1 920212, 1992, Chem. Abstr., 1992, 117, 7943c.
- M. J. Coghlan, B. A. Driekorn, R. G. Suhr and G. P. Jourdan, Eur. Pat., 0326328 A2 890802, 1989, Chem. Abstr., 1989, 112, 55907u.
- B. Stefanska, M. Arciemiuk, M. M. Bontemps-Gracz, M. Dzieduszycka, A. Kupiec, S. Martellib and E. Borowskia, Bioorg. Med. Chem., 2003, 11, 561–572 CrossRef CAS PubMed.
- S. Li, Y. Zhao, K. Wang, Y. Gao, J. Han, B. Cui and P. Gong, Bioorg. Med. Chem., 2013, 21, 2843–2855 CrossRef CAS PubMed.
- G. Zoidis, A. Sosic, S. Da Ros, B. Gatto, C. Sissi, F. Palluotto, A. Carotti and M. Catto, Bioorg. Med. Chem., 2017, 25, 2625–2634 CrossRef CAS PubMed.
- C.-K. Ryu and J. Y. Lee, Bioorg. Med. Chem. Lett., 2006, 16, 1850–1853 CrossRef CAS PubMed.
- M. P. Giovannoni, I. A. Schepetkin, L. Crocetti, G. Ciciani, A. Cilibrizzi, G. Guerrini, A. I. Khlebnikov, M. T. Quinn and C. Vergelli, J. Enzyme Inhib. Med. Chem., 2016, 31, 628–639 CrossRef CAS PubMed.
- D. Masciocchi, A. Gelain, F. Porta, F. Meneghetti, A. Pedretti, G. Celentano, D. Barlocco, L. Legnani, L. Toma, B.-M. Kwon, A. Asaid and S. Villa, Med. Chem. Commun., 2013, 4, 1181 RSC.
- C. Vernhet, E. Barbagallo, M. Rinaldi-Carmona and P. Roux, Fr. Demande, FR 2933697 A1 20100115, 2010.
- E. Barbagallo, M. Rinaldi-Carmona, P. Roux and C. Vernhet, PCT Int. Appl., WO 2010004215 A2 20100114, 2010.
- M. Alvarado, M. Barcelo, L. Carro, C. F. Masaguer and E. Ravina, Chem. Biodiversity, 2006, 3, 106–117 CrossRef CAS PubMed.
- Y. Sato, Y. Suzuki, K. Yamamoto, S. Kuroiwa and S. Maruyama, PCT Int. Appl., WO 2005121105 A1 20051222, 2005.
- K. G. Pike and B. C. Barlaam, PCT Int. Appl., WO 2017162605 A1 20170928, 2017.
- J. D. Lawson, M. Sabat, C. Smith, H. Wang, Y. K. Chen and T. Kanouni, Int. Appl., WO 2013148603 A1 20131003, 2013.
- C. Alhambra, C. Becker, T. Blake, A. Chang, J. R. Damewood, T. Daniels, B. T. Dembofsky, D. A. Gurley, J. E. Hall, K. J. Herzog, C. L. Horchler, C. J. Ohnmacht, R. J. Schmiesing, A. Dudley, M. D. Ribadeneira, K. S. Knappenberger, C. Maciag, M. M. Stein, M. Chopra, X. F. Liu, E. P. Christian, J. L. Arriza and M. J. Chapdelaine, Bioorg. Med. Chem., 2011, 19, 2927–2938 CrossRef CAS PubMed.
- A. Goeminne, P. J. Scammells, S. M. Devine and B. L. Flynn, Tetrahedron Lett., 2010, 51, 6882–6885 CrossRef CAS.
- O. V. Vinogradova, V. N. Sorokoumov, S. F. Vasilevsky and I. A. Balova, Tetrahedron Lett., 2007, 48, 4907–4909 CrossRef CAS.
- O. V. Vinogradova, V. N. Sorokoumov, S. F. Vasilevskii and I. A. Balova, Russ. Chem. Bull. Int. Ed., 2008, 57, 1725–1733 CrossRef CAS.
- R. Dey and B. C. Ranu, Tetrahedron, 2011, 67, 8918–8924 CrossRef CAS.
- N. A. Al-Awadi, M. H. Elnagdi, Y. A. Ibrahim, K. Kaul and A. Kumar, Tetrahedron, 2001, 57, 1609–1614 CrossRef CAS.
- K. W. Woods, J. P. Fischer, A. Claiborne, T. Li, S. A. Thomas, G. D. Zhu, R. B. Diebold, X. Liu, Y. Shi, V. Klinghofer, E. K. Han, R. Guan, S. R. Magnone, E. F. Johnson, J. J. Bouska, A. M. Olson, R. de Jong, T. Oltersdorf, Y. Luo, S. H. Rosenberg, V. L. Giranda and Q. Li, Bioorg. Med. Chem., 2006, 14, 6832–6846 CrossRef CAS PubMed.
- I. S. Kim and P. H. Lee, Repub. Korea, KR 1740155 B1 20170525, 2017.
- J. Yan, G. L. Tay, C. Neo, B. R. Lee and P. W. H. Chan, Org. Lett., 2015, 17, 4176–4179 CrossRef CAS PubMed.
- M. A. Berghot, E. M. Kandeel, A. H. Abdel-Rahman and A.-M. Marwa, Med. Chem., 2014, 4, 381–388 Search PubMed.
- A. D. T. Tuyet, L. Decuyper, T. P. Hoang, V. N. Doan, H. T. Nguyen, T. T. Nguyen, T. D. Huy, H. H. Nguyen, M. D'hooghe and T. V. Nguyen, Tetrahedron Lett., 2015, 56, 5855–5858 CrossRef CAS.
- T. J. Mason and D. Peters, Practical Sonochemistry, Ellis Horwood, New York, NY, USA, 1991 Search PubMed.
- J. L. Luche, Organic Sonochemistry, Plenum Press, New York, NY, USA, 1998 Search PubMed.
- T. J. Mason and J. P. Lorimer, Applied sonochemistry: uses of power ultrasound in chemistry and processing, Wiley-VCH, Weinheim, 2002 Search PubMed.
- A. Bazgir, S. Ahadi, R. Ghahremanzadeh, H. R. Khavasi and P. Mirzaei, Ultrason. Sonochem., 2010, 17, 447–452 CrossRef CAS PubMed.
- J. L. G. Ruano, A. Parra, L. Marzo, F. Yuste and V. M. Mastranzo, Tetrahedron, 2011, 67, 2905–2910 CrossRef.
- P. Cintas, Ultrason. Sonochem., 2016, 28, 257–258 CrossRef CAS PubMed.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 1–14 CrossRef CAS PubMed.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 15–35 CrossRef CAS PubMed.
- G. Chatel, Ultrason. Sonochem., 2018, 40, 117–122 CrossRef CAS PubMed.
- S. Puri, B. Kaur, A. Parmar and H. Kumar, Curr. Org. Chem., 2013, 17, 1790–1828 CrossRef CAS.
- J. L. G. Ruano, A. Parra, L. Marzo, F. Yuste and V. M. Mastranzo, Tetrahedron, 2011, 67, 2905–2910 CrossRef.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS PubMed.
- C. O. Kappe and A. Stadler, Microwave Theory. In Microwaves in Organic and Medicinal Chemistry, Wiley-Blackwell, 2005, pp. 9–28 Search PubMed.
- J. P. Tierney and P. Lidstrom, Microwave Assisted Organic Synthesis, Blackwell, Oxford, 2005 Search PubMed.
- A. Loupy, Microwaves in Organic Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2006 Search PubMed.
- H. M. Al-Matar, S. M. Riyadha and M. H. Elnagdi, J. Heterocycl. Chem., 2007, 44, 603–607 CrossRef CAS.
- K. D. Khalil and H. M. Al-Matar, Molecules, 2012, 17, 12225–12233 CrossRef CAS PubMed.
- K. M. Dawood and M. M. El-Deftar, Synthesis, 2010, 1030–1038 CrossRef CAS.
- H. A. Abdel-Aziz, H. S. A. El-Zahabi and K. M. Dawood, Eur. J. Med. Chem., 2010, 45, 2427–2432 CrossRef CAS PubMed.
- K. M. Dawood, A. M. Farag, M. M. El-Deftar, M. Gardiner and H. A. Abdelaziz, ARKIVOC, 2013,(iii), 210–226 Search PubMed.
- K. M. Dawood, M. B. Elamin and A. M. Farag, ARKIVOC, 2015,(vii), 50–62 Search PubMed.
- H. Behbehani, H. M. Ibrahim and K. M. Dawood, RSC Adv., 2015, 5, 25642–25649 RSC.
- H. Behbehani, K. M. Dawood, H. M. Ibrahim and N. S. Mostafa, Arabian J. Chem., 2017 DOI:10.1016/j.arabjc.2017.05.016 , published online.
- M. A. Al-Shiekh, H. Y. Medrassi, M. H. Elnagdi and E. A. Hafez, J. Chem. Res., 2007, 432–436 CrossRef CAS.
- H. M. E. Hassaneen and I. A. Abdelhamid, J. Heterocycl. Chem., 2017, 54, 1048–1053 CrossRef CAS.
- M. A. Al-Shiekh, H. Y. Medrassi, M. H. Elnagdi and E. A. Hafez, ARKIVOC, 2008,(xvii), 36–47 Search PubMed.
- F. Al-Qalaf, K. Almohammad, M. A. El-Apasery and H. Mahmoud, Eur. J. Chem., 2013, 4, 211–215 Search PubMed.
- The crystallographic data for compound 3a (ref. CCDC 1858311) can be obtained on request from the director, Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EW, UK.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1858311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06494f |
| This journal is © The Royal Society of Chemistry 2018 |