
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic-propelled Fe3O4–chitosan carriers enhance L-asparaginase catalytic activity: a promising strategy for enzyme immobilization
Burhan Ates *a,
Ahmet Ulu*a,
Suleyman Köytepea,
Samir Abbas Ali Nomaa,
Veli Serkan Kolatb and
Tekin Izgib
*a,
Ahmet Ulu*a,
Suleyman Köytepea,
Samir Abbas Ali Nomaa,
Veli Serkan Kolatb and
Tekin Izgib
aDepartment of Chemistry, Faculty of Science & Arts, Inonu University, Malatya, 44280, Turkey. E-mail: burhan.ates@inonu.edu.tr; Fax: +90-422 341 0037; Tel: +90-422 377 3888
bDepartment of Physics, Faculty of Science & Arts, Inonu University, Malatya, 44280, Turkey. E-mail: ahmet.ulu@inonu.edu.tr; Fax: +90-422 341 0037; Tel: +90-422 377 3774
First published on 23rd October 2018
Abstract
Magnetic-propelled carriers comprising magnetic Fe3O4–chitosan nanoparticles were immobilized with L-asparaginase (L-ASNase). The enzyme displayed enhanced catalytic activity in a weak magnetic field, and thermal and pH stabilities. The conjugated L-ASNase presented higher thermostability and wider range of pH stability in comparison with those of free L-ASNase. Moreover, the reusability of conjugated L-ASNase significantly improved after immobilization and it retained 60.5% of its initial activity after undergoing 16 cycles. The conjugated L-ASNase maintained more than 50% and 48% initial activity after 4 weeks of storage at 4 °C and room temperature, respectively. Furthermore, we reveal that the activity of conjugated L-ASNase onto magnetic Fe3O4–chitosan particles increased by about 3-fold in the weak magnetic field at certain frequencies and flux density compared with that of free L-ASNase. Considering these excellent attributes, the magnetic-propelled mechanism in the transporting and activation of L-ASNase can be used by enhancing the catalytic activity, stability, and efficiency in vital implications for medicinal biotechnology.
1. Introduction
L-Asparagine (L-Asn) is an essential amino acid for the growth of tumor cells. Cancer cells must absorb this amino acid from the bloodstream in order to synthesize protein and grow. L-ASNase (asparagine amidohydrolase, EC 3.5.1.1) catalyzes the hydrolysis of L-Asn to aspartic acid and ammonia.1 At the end of this enzymatic hydrolysis, the cancer cells grow slowly and they die. L-ASNase is one of the most widely used chemotherapeutic agent in the treatment of many cancer types.2,3 It is also an important enzyme in the food industry since it reduces the formation of acrylamide, a carcinogenic compound. Since this enzyme is commonly used in two important industries, it is essential to enhance its half-life, reusability and catalytic activity. To date, to the best of our knowledge, a few studies have been reported to evaluate the catalytic efficiency of enzymes after exposure to magnetic fields. For instance, Mizuki et al. reported the activity of immobilized α-amylase, lipase and chitinase activity increases in a rotational magnetic field and reaches maximum at a certain frequency.4 According to Prando et al.,5 since the increase in activity due to magnetic treatment may be related to an increase in the average particle size and a reduction of 25% in α-helix content, the substrate facilitates access to the active site of lysozyme. Based on these results, the activities of some enzymes can be efficiently improved owing to exposure to magnetic field, which may make a great contribution to nanoscience and biotechnology.In recent years, magnetic nanoparticles have received considerable interest and attention due to their versatile characteristics such as high surface area, superparamagnetism, stability, low toxicity, small and regular shape, inexpensiveness, and ease of synthesis.6,7 Therefore, they have been used for a broad range of applications including drug delivery,8 biosensors,9 catalysis,10 and magnetic resonance imaging.11 Among them, enzyme immobilization is probably one of the most widely used applications. In particular, the separation of magnetic particles with an external magnet contributes to enhanced reusability of enzyme. However, magnetic nanoparticles tend to aggregate in aqueous medium due to the strong magnetic dipole–dipole attractions between particles. In addition, there are not many functional reactive groups, except a small amount of hydroxyl groups located on its surface, which will react with other molecules such as enzymes and proteins.12 Perhaps, these drawbacks may hinder the applications of enzymes in industrial areas.13,14 To overcome these drawbacks, modification of Fe3O4 nanoparticles is an efficient strategy using different molecules such as precious metals,15 carbon,16 SiO2,17 MCM-41,18 and polymers.19 At this point, biocompatible and biodegradable polymers are generally preferred because they diminish unwanted toxicity of nanoparticles and offer active groups.14 Chitosan, a natural polymer derived from chitin, is found in the membrane of many organisms including lobsters, crabs, and shrimp. It is widely used as a biomaterial due to its features such as inexpensiveness, stability, biocompatibility, hydrophilicity, biodegradability, flexibility, versatility, and environment friendliness.20–22 Additionally, the amino groups in chitosan are very useful for the covalent attachment of enzyme onto the surface of chitosan.14 Therefore, chitosan can be considered as a promising support because of the aforementioned properties. Thus far, there are many reports about immobilization of enzyme on Fe3O4–chitosan particles.23–27
The main novelty of this study is that for the first time, we determined the effect of magnetic excitation caused by a weak magnetic field on the catalytic activity of the conjugated L-ASNase. Therefore, L-ASNAse activity improved with magnetic-propelled mechanism in a weak magnetic field, and may be used with improved efficiency in treatment of leukemia. In addition, for the first time, L-ASNase was immobilized on Fe3O4–chitosan particles via covalent attachment and the optimum parameters of the conjugated enzyme were investigated.
For this purpose, magnetic Fe3O4 nanoparticles were synthesized via co-precipitation technique. The Fe3O4 core was coated with chitosan to obtain core–shell structure magnetic nanoparticles. The thermal behavior, morphology and chemical composition of Fe3O4–chitosan magnetic nanoparticles were characterized by FTIR, TGA, DTA, DSC, SEM and EDX analysis. In addition, the hydrodynamic particle size, surface charge, structure and magnetic property of Fe3O4–chitosan nanoparticles were characterized by DLS, Zetasizer Nano ZS, XRD and VSM, respectively. L-ASNase was covalently immobilized onto the Fe3O4–chitosan magnetic particles using glutaraldehyde as an activating reagent (Fig. 1). The characterization of conjugated L-ASNase was performed through several technics including FTIR, SEM and XRD. In addition, the optimum pH and temperature, pH and thermal stability, kinetic parameters, reusability and storage capability of the conjugated enzyme were investigated and were compared with those of free enzyme. Moreover, we examined and discussed the activity of conjugated L-ASNase exposed to an external weak magnetic field using different frequency or flux density.
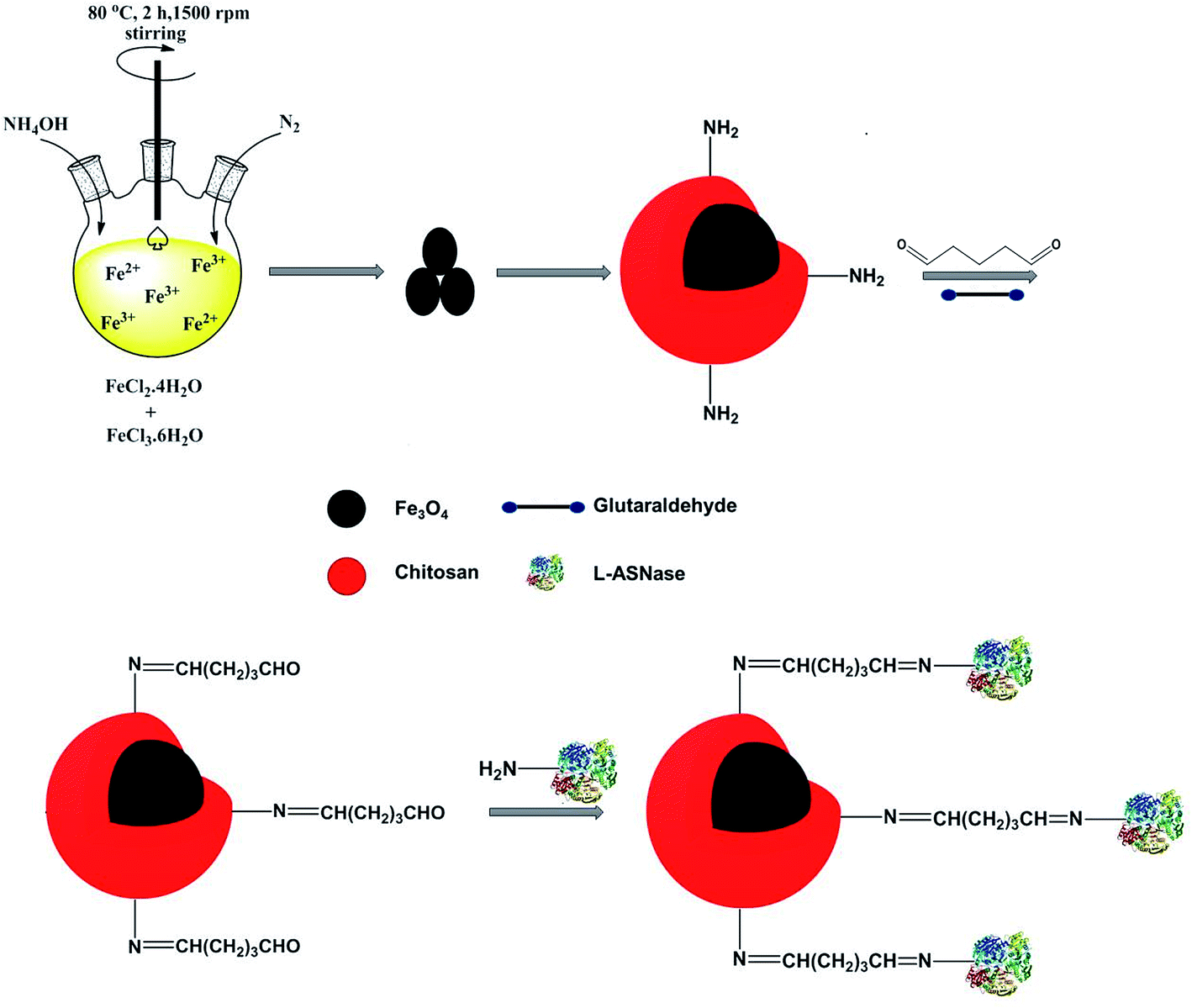 | ||
| Fig. 1 Immobilization of L-ASNase onto the magnetic Fe3O4–chitosan core–shell particles. | ||
2. Experimental
2.1 Materials
Iron(II) chloride tetrahydrate (FeCl2·4H2O) and iron(III) chloride hexahydrate (FeCl3·6H2O) were purchased from Merck (Darmstadt, Germany). L-Asn, chitosan (medium molecular weight, deacetylation: 75-85%), acetic acid, glutaraldehyde (GDA, 25%, v/v), sodium hydroxide (NaOH), and ammonium hydroxide (NH4OH, 26 wt%) were supplied by Sigma-Aldrich (St. Louis, MO). L-ASNase from Escherichia coli was purchased from ProSpec-Tany TechnoGene Ltd. Trichloroacetic acid (TCA) was obtained from Riedel de Haen (Deelze, Germany). Unless otherwise noted, all reagents and chemicals were of analytical or biological grade. Ultra-pure water (Milli Q, Millipore) was used to prepare all aqueous solutions used in this study.2.2 Preparation of Fe3O4 and Fe3O4–chitosan carriers
Magnetic Fe3O4 nanoparticles were synthesized by a co-precipitation method using ferric and ferrous salts.24 FeCl2·4H2O (1.99 g) and FeCl3·6H2O (5.4 g) were dissolved in 50 mL of deionized water. The pH of this mixture was adjusted by adding ammonia solution (26 wt%). The reaction was allowed to continue for 2 h at 80 °C under the protection of nitrogen. The obtained Fe3O4 nanoparticles were separated by using an external magnet and washed with distilled water to remove excess ammonia. Finally, the precipitate was dried at 60 °C for 6 h. In order to prepare Fe3O4–chitosan carrier, chitosan (0.3 g) was completely dissolved in 2% (v/v) acetic acid solution. Then, dry Fe3O4 nanoparticles (0.15 g) were added to chitosan solution. The solution was dispersed homogeneously by ultrasonication for 20 min. Subsequently, 50 mL of 1 M NaOH solution was added to the mixture and the Fe3O4–chitosan particles were obtained. The obtained particles were washed with deionized water until neutral pH was obtained. Finally, the particles were dried again in the vacuum oven at 50 °C for 12 h and stored at 4 °C for immobilization.2.3 Characterization of magnetic Fe3O4–chitosan carriers
The structural characterization of magnetic Fe3O4–chitosan particles was performed by FTIR spectroscopy (Perkin Elmer). Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) studies were performed using a Shimadzu Thermal Analyzer at room temperature to 700 °C a rate of 10 °C min−1 in air atmosphere. Differential Scanning Calorimetry (DSC) was performed on a Shimadzu DSC-60 at a heating rate of 20 °C min−1 from 0 to 450 °C. Crystalline phases of the samples were verified using powder X-ray diffraction (XRD, Rigaku RadB X-ray diffractometer). Morphology of the as-synthesized nanomaterial was determined using by scanning electron microscope (SEM, LEO Evo-40 VPX). Magnetization of the as-prepared nanoparticles was measured on a VSM (PPMS) at room temperature. EDX analysis was performed using a Ronteck Xflash detector analyzer equipped with Leo-Evo 40xVP. The average hydrodynamic particle size was measured using dynamic light scattering (DLS). Zeta potential measurements were performed using Malvern Zetasizer Nano ZS. L-ASNase activity was observed by an ELISA plate reader (BioTek) at 480 nm.2.4 Immobilization of L-ASNase onto Fe3O4–chitosan carriers
L-ASNase was covalently immobilized onto Fe3O4–chitosan carriers by the following procedure. First, 1.0 g support was reacted with 30 mL of 2.5% (v/v) GDA solution with stirring at 25 °C for 2 h to activate the support by offering aldehyde groups.24 Then, the modified support was rinsed with deionized water three times to remove excess unreacted GDA. L-ASNase was dissolved in Tris–HCl buffer and mixed with an appropriate amount of Fe3O4–chitosan particles. The mixture was shaken in Eppendorf tube for immobilization by chemical bonding at 4 °C for 24 h. Subsequently, the particles were collected by an external magnet and then washed with 2 times Tris–HCl buffer to remove unconjugated L-ASNase. The solid particles were dried at room temperature and stored in the refrigerator at 4 °C until further use. In addition, the supernatant and washing solutions were stored at 4 °C to calculate immobilization efficiency (IE). The protein concentration of the reaction mixture was measured by the Bradford method using bovine serum albumin (BSA) as standard.28 The following formula was used to determine immobilization efficiency:
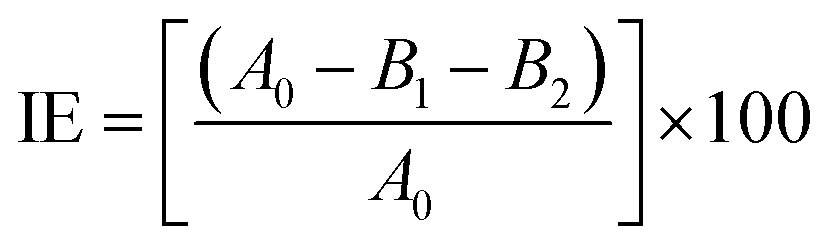 | (1) |
2.5 Assay of L-ASNase activity
The assay of free and conjugated enzyme activity was determined by the Nesslerization method, as previously reported.29 The method is based on colorimetric measurement of the amount of ammonia released during the hydrolysis of L-Asn. The reaction mixture contains 1.0 mL of 0.01![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M L-Asn (prepared in 0.05M Tris–HCl buffer at pH 8.6) and free (20 μL) or conjugated enzyme (5 mg). The mixture was incubated at 37 °C for 15 minutes. The enzymatic reaction was terminated by adding 0.1mL of 1.5M TCA. The conjugated enzyme was removed by centrifugation at 5000 rpm for 5min. The supernatant (100 μL) was added into the well containing Nessler's reagent (100 μL). The well-plate was incubated at room temperature for 10min. The observed yellow color was read using an ELISA microplate reader (Biotek, EON) at 480nm. One unit (U) of L-ASNase is defined as the amount of enzyme that catalyzed the formation of 1μmol of ammonia from L-Asn per minute under the standard assay conditions.30 All the experiments were repeated three or more times. The highest L-ASNase activity was considered 100% while calculating the relative activities.
M L-Asn (prepared in 0.05M Tris–HCl buffer at pH 8.6) and free (20 μL) or conjugated enzyme (5 mg). The mixture was incubated at 37 °C for 15 minutes. The enzymatic reaction was terminated by adding 0.1mL of 1.5M TCA. The conjugated enzyme was removed by centrifugation at 5000 rpm for 5min. The supernatant (100 μL) was added into the well containing Nessler's reagent (100 μL). The well-plate was incubated at room temperature for 10min. The observed yellow color was read using an ELISA microplate reader (Biotek, EON) at 480nm. One unit (U) of L-ASNase is defined as the amount of enzyme that catalyzed the formation of 1μmol of ammonia from L-Asn per minute under the standard assay conditions.30 All the experiments were repeated three or more times. The highest L-ASNase activity was considered 100% while calculating the relative activities.
2.6 Characterization of the conjugated L-ASNase
The influence of the temperature on the activities of free and conjugated L-ASNase was studied from 25 to 70 °C with interval of 5 °C at pH 8.0 and 8.5.
For thermal stability experiments, free and conjugated enzymes were incubated in a water bath at 55 °C for different durations ranging from 30 to 180 min. After incubation, the residual activities were determined using the abovementioned assay.
Storage stability was measured by determining the activity of free and conjugated L-ASNase every two days for a period of 30 days at 4 °C and 25 °C. The activity at the first day was taken to be 100%.
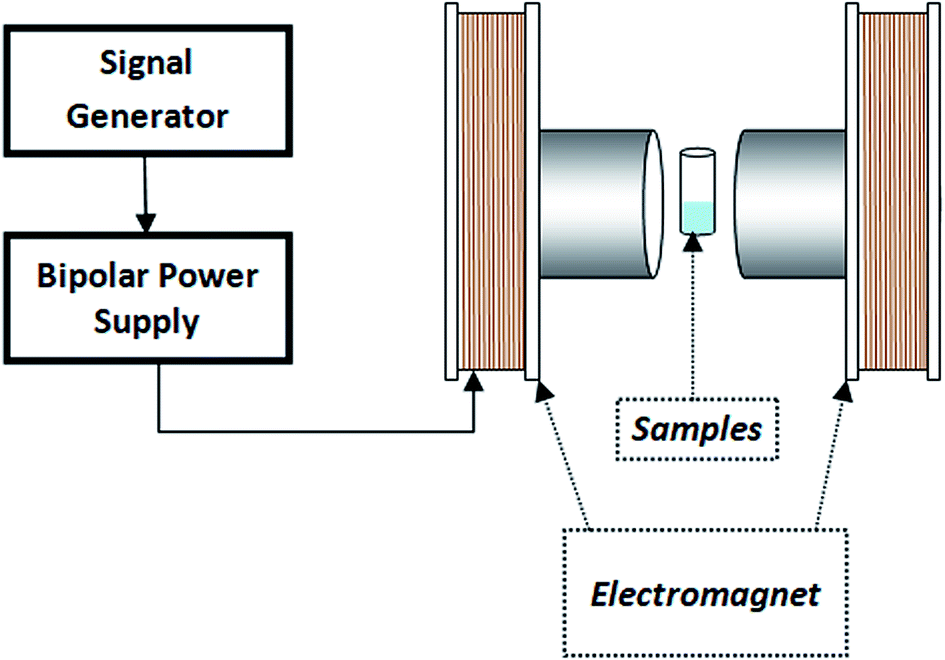 | ||
| Fig. 2 Schematic of magnetic field experiments(VSM). | ||
In addition, the influence of magnetic flux density on enzyme activity was examined at different flux densities (10, 20, 30, 40, 50 and 100 mT) with constant frequency (3 Hz). For this experiment, we repeated the above enzyme assay.
3. Result and discussion
3.1 Characterization of magnetic Fe3O4–chitosan carriers
In order to characterize the functional groups on the surface of the samples, FTIR analyses were performed, as shown in Fig. 3. The characteristic peak of pristine Fe3O4, ascribed to the stretching vibration of Fe–O bonds, appeared at 560 cm−1 (Fig. 3a).31 The other peaks at around 3400 cm−1 and 1630 cm−1 correspond to the adsorbed water in the sample.32 For chitosan (Fig. 3b), the FTIR spectra shows bands arising from stretching of –NH and –OH groups (3400 cm−1), C–H (2871 and 2929 cm−1) stretching vibrations, and from amide II bending (1569 cm−1).33 In addition, the bands at 1060, 1368 and 1400 cm−1 were assigned to the stretching vibrations of –C–O–C, –C–O and –COO groups, respectively.34–36 As expected, the FTIR spectrum of Fe3O4–chitosan showed characteristic bands of both Fe3O4 and chitosan. However, in the spectrum of Fe3O4–chitosan particles (Fig. 3c), the peak of Fe–O vibration at 560 cm−1 shifted to 440 cm−1 due to the interaction between Fe3O4 and chitosan compared with that observed in the spectrum of pristine Fe3O4.37,38 These results indicated that the Fe3O4 magnetic nanoparticles were coated by the chitosan. For Fe3O4–chitosan–GDA, the new absorption peaks at 1645 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, imine bond) and 1587 cm−1 (CC bond) confirmed that cross-linking occurred between chitosan and GDA (Fig. 3d). Similar absorption peaks were observed in some reports of chitosan cross-linked with GDA.39,40 The FTIR spectrum of Fe3O4–chitosan–L-ASNase is presented in Fig. 3e. In this spectrum, intense OH stretching band in the range of 3400–3600 cm−1 is attributed to the presence of enzyme.41 In addition, the increase in the intensity of two peaks at 1654 cm−1 (amide I bending) and 1553 cm−1 (amide II bending) is further evidence of the successful immobilization of L-ASNase.42
N, imine bond) and 1587 cm−1 (CC bond) confirmed that cross-linking occurred between chitosan and GDA (Fig. 3d). Similar absorption peaks were observed in some reports of chitosan cross-linked with GDA.39,40 The FTIR spectrum of Fe3O4–chitosan–L-ASNase is presented in Fig. 3e. In this spectrum, intense OH stretching band in the range of 3400–3600 cm−1 is attributed to the presence of enzyme.41 In addition, the increase in the intensity of two peaks at 1654 cm−1 (amide I bending) and 1553 cm−1 (amide II bending) is further evidence of the successful immobilization of L-ASNase.42
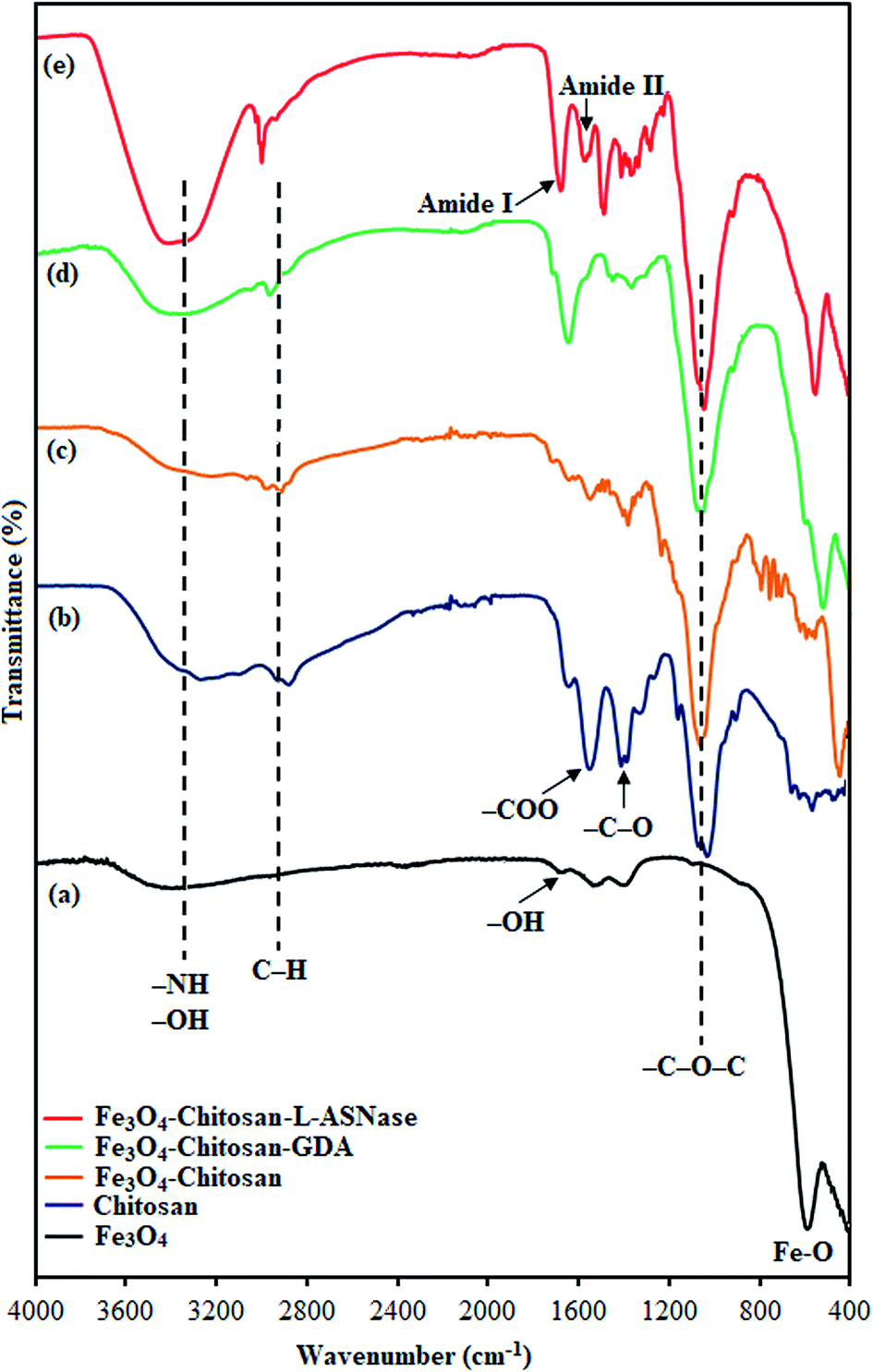 | ||
| Fig. 3 FTIR spectra of (a) Fe3O4, (b) chitosan, (c) Fe3O4–chitosan particles, (d) Fe3O4–chitosan–GDA, and (e) Fe3O4–chitosan–L-ASNase. | ||
TGA analysis of pure Fe3O4, chitosan and magnetic Fe3O4–chitosan carriers were performed between 0 °C and 700 °C for quantitative analysis. These thermograms are given in Fig. 4. In the thermogram of pure Fe3O4, weight loss is not observed up to 700 °C. Three weight losses are observed in the TGA thermogram of pure chitosan. The first thermal degradation of chitosan structure resulted from the loss of moisture from 60 to 110 °C. The second thermal decomposition was observed between 220 and 310 °C due to the decomposition of polymer with low molecular weight. The third thermal decomposition presented 40% weight loss between 310 °C and 590 °C. This weight loss stems from the decomposition of cyclic moiety (saccharide rings). The magnetic Fe3O4–chitosan particles show a similar thermal decomposition profile to that of chitosan i.e., there are three different weight losses in the TGA thermogram of the magnetic Fe3O4–chitosan carriers. These weight losses are due to the loss of physiosorbed water between 50–110 °C, the degradation of the chitosan polymeric structure between 200 °C and 320 °C and the saccharide ring breaking peak 320 °C and 550 °C. However, the weight loss of Fe3O4–chitosan is very different from that of chitosan. Fe3O4 and chitosan were connected together through the chelation between Fe3+ and chitosan in the course of the reaction. The conformational changes of chitosan and the additional bridging between Fe3O4 and chitosan enhanced the thermal stability of chitosan in Fe3O4–chitosan. Therefore, the final decomposition temperature of Fe3O4–chitosan was higher than that for pure chitosan. Similar TGA thermograms have also been reported previously.43,44 The TGA thermogram results of Fe3O4–chitosan structure indicate that Fe3O4 nanoparticles are successfully functionalized with chitosan groups. In addition, these thermal analysis results were confirmed by DTA and DSC techniques.
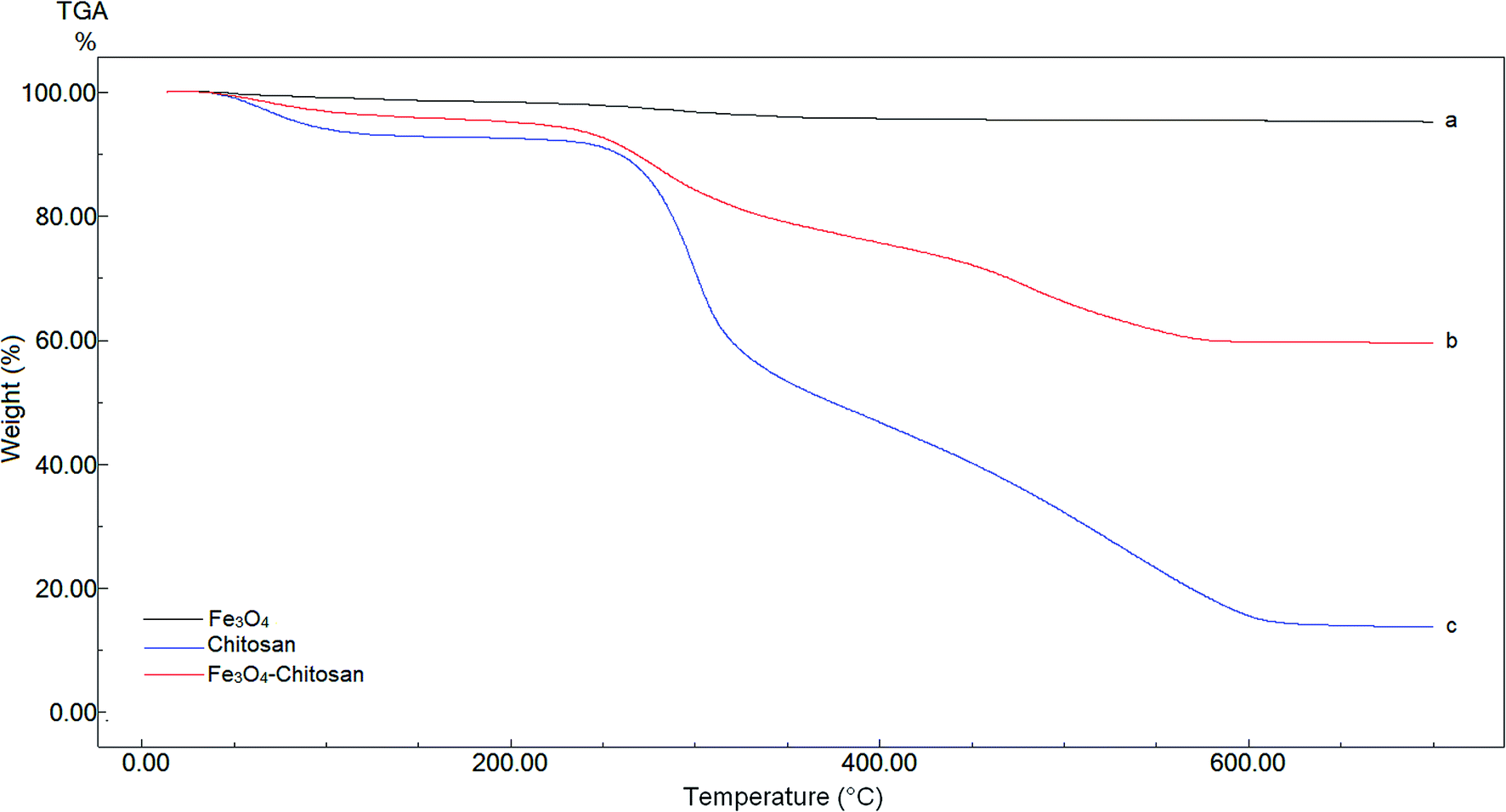 | ||
| Fig. 4 TGA curves of (a) Fe3O4, (b) chitosan, and (c) Fe3O4–chitosan particles. | ||
DTA thermograms of Fe3O4, chitosan and magnetic Fe3O4–chitosan carriers are shown in Fig. 5; the thermogram of the magnetic Fe3O4–chitosan carriers shows three exothermic peaks. The first exothermic peak at about 100 °C is due to loss of water and second peak at about 320 °C is related to the decomposition of polymeric structure of chitosan structure. The last exothermic peak at about 400 °C is due to the degradation of saccharide ring groups. Moreover, DSC thermograms of the Fe3O4, chitosan and magnetic Fe3O4–chitosan carriers are shown in Fig. 6. No significant peaks were detected in the DSC thermogram of Fe3O4 nanoparticles. However, the Tg was observed at 30 °C in the DSC thermogram of chitosan.45 In addition, this DSC thermogram shows that the decomposition of chitosan started at 220 °C.46,47 In the DSC thermogram of Fe3O4–chitosan, the exothermic peaks between 220 °C and 380 °C corresponds to the oxidative decomposition of organic group onto the Fe3O4 nanoparticle surface.
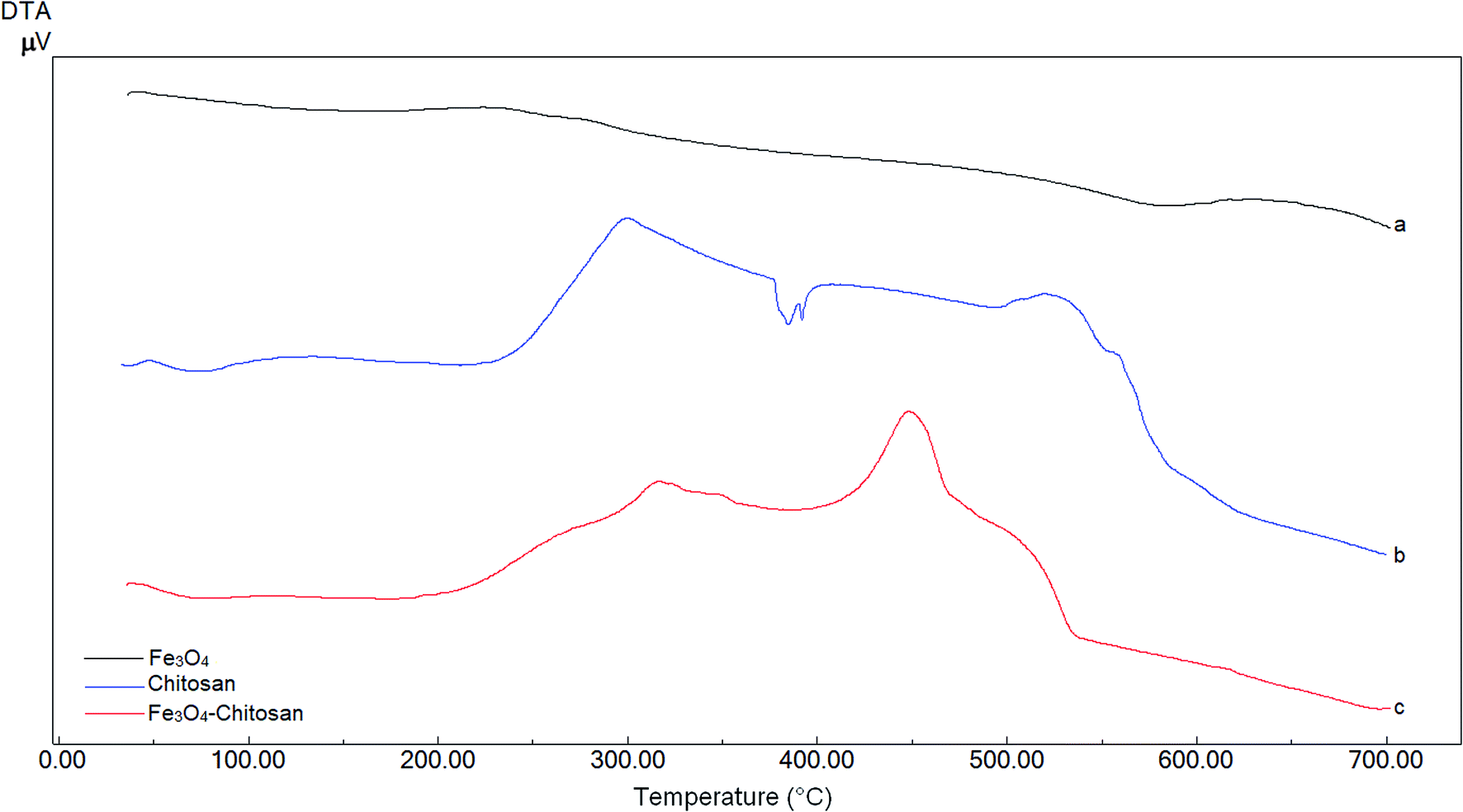 | ||
| Fig. 5 DTA curves of (a) Fe3O4, (b) chitosan, and (c) Fe3O4–chitosan particles. | ||
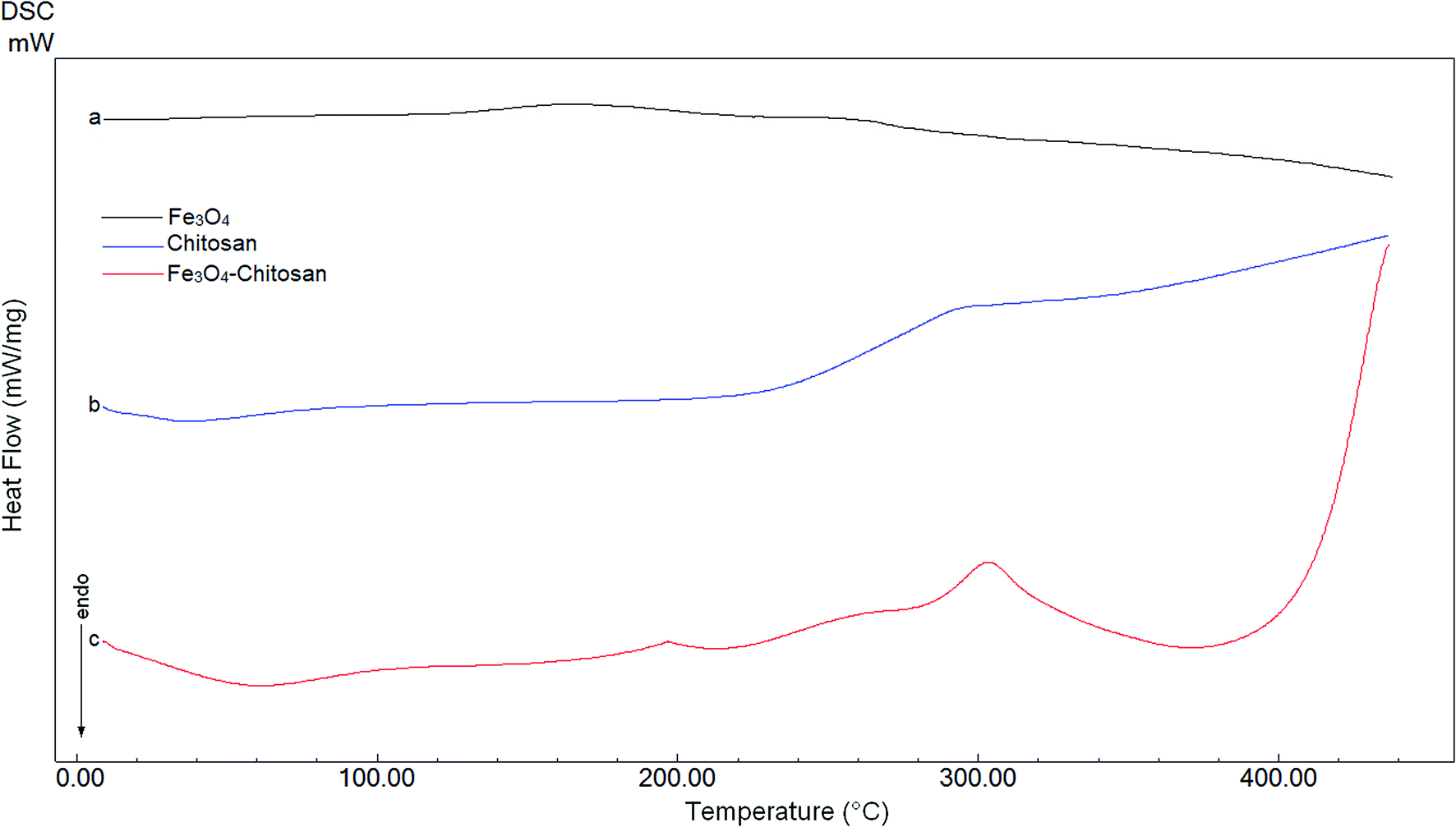 | ||
| Fig. 6 DSC curves of (a) Fe3O4, (b) chitosan, and (c) Fe3O4–chitosan particles. | ||
To verify the presence of crystalline Fe3O4 and Fe3O4–chitosan particles, the structure of the magnetic nanoparticles was characterized by XRD and the diffractogram is shown in Fig. 7. In both samples, six characteristic peaks were observed ((220), (311), (400), (422), (511), and (440)).48 These peaks are consistent with the database in JCPDS file (PDF No. 65-3107) and reveal that the resultant nanoparticles are pure Fe3O4 with a spinel structure.25 The weaker diffraction lines of magnetic Fe3O4–chitosan particles confirm that the Fe3O4 nanoparticles were coated by amorphous chitosan polymer.49 In addition, chitosan did not result in phase change of Fe3O4 nanoparticles. The conjugation of L-ASNase leads to a decrease in the intensity of all the characteristic peaks, which may be due to the interaction of the Fe3O4–chitosan particles with enzyme. XRD measurements further confirmed the abovementioned FTIR results.
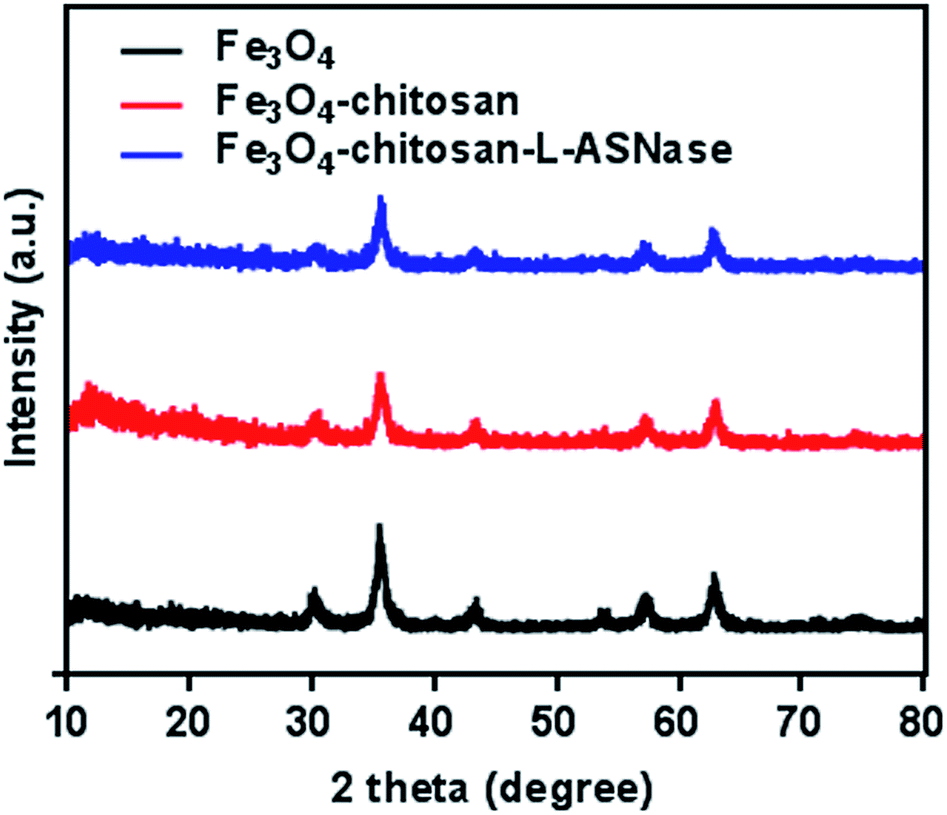 | ||
| Fig. 7 XRD patterns of the Fe3O4, Fe3O4–chitosan particles, and Fe3O4–chitosan–L-ASNase. | ||
The average hydrodynamic particle sizes of Fe3O4 and Fe3O4–chitosan are 100 nm and >200 nm, respectively (Fig. 8a). The increase in the hydrodynamic size of functionalized Fe3O4 nanoparticles is due to the interaction between amino groups of chitosan with hydroxyl groups onto the surface of the Fe3O4. It has also been reported in previous studies that the particle size increases after nanoparticles are coated with chitosan.35,36,50
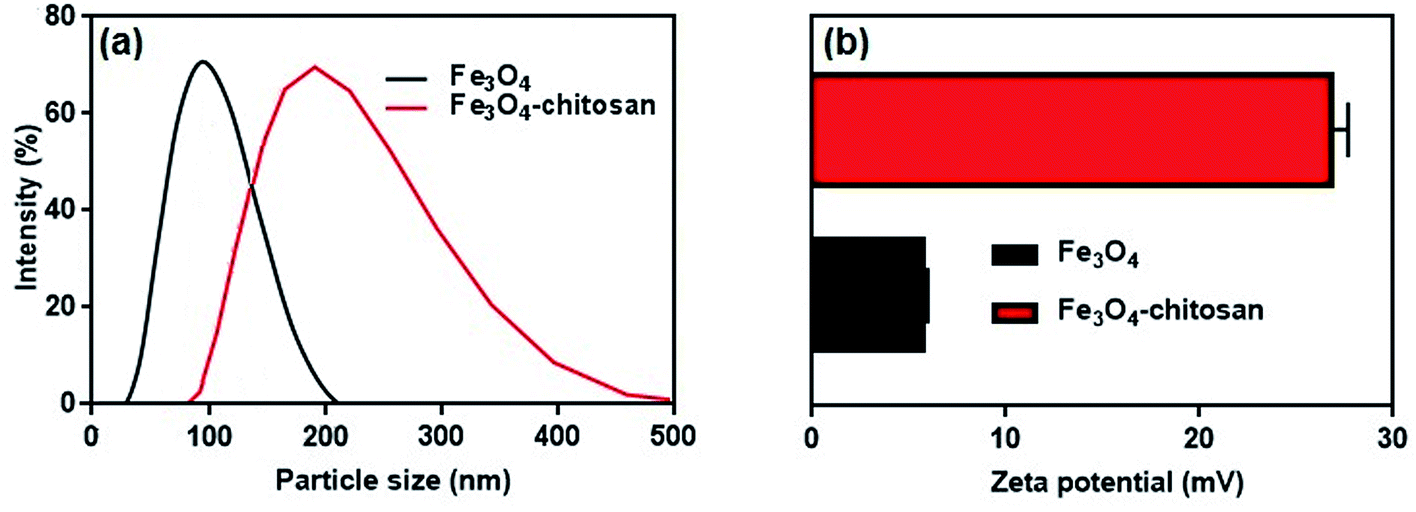 | ||
| Fig. 8 (a) Hydrodynamic particle size distribution and (b) zeta potential values of Fe3O4 nanoparticles and Fe3O4–chitosan particles. | ||
It is well known that zeta potential measurements might provide some additional insights into the magnitude of the repulsion or attraction between particles.51 Therefore, the successful coating of chitosan was confirmed by the zeta potential analysis of the resulting composite. The zeta potential results are shown in Fig. 8b. The zeta potential of unmodified magnetic Fe3O4 is 5.74 ± 0.22 mV. However, as shown in Fig. 8b, after coating of chitosan molecule, the zeta potential value of the Fe3O4–chitosan composite increased to 26.8 ± 0.87 mV. Chitosan can adsorb onto Fe3O4 nanoparticles easily by electrostatic attraction, and the amino groups generate positive charge on the Fe3O4 nanoparticles surface, which is similar to that reported previously.52,53
The surface morphology of Fe3O4, Fe3O4–chitosan particles and Fe3O4–chitosan–L-ASNase were confirmed by SEM (Fig. 9). The Fe3O4 nanoparticles were rough, nearly spherical in shape and aggregated (Fig. 9a–c). After the coating with chitosan, the surface morphology of Fe3O4 nanoparticles evidently changed. In Fig. 9(b–d), the SEM images confirm the coating process of Fe3O4 with increasing of the particles size. The results of this experiment were similar to those in previously published papers.54 Fig. 9(e and f) represents the image of Fe3O4–chitosan particles after bioconjugation. The SEM images revealed that there are differences in the surface micromorphology of Fe3O4–chitosan particles. As expected, the size of particles increased after the immobilization procedure.
 | ||
| Fig. 9 SEM analysis of magnetic (a and c) Fe3O4, (b and d) Fe3O4–chitosan particles and (e and f) Fe3O4–chitosan–L-ASNase at different magnifications. | ||
The surface composition of unmodified Fe3O4, and Fe3O4–chitosan particles was investigated by EDX, as shown in Fig. 10. The EDX map of Fe3O4 nanoparticles is mainly composed of only iron (Fe) and oxygen (O) elements (Fig. 10a). In addition, no peaks related to other elements were detected, which revealed the high purity of the sample. However, the new signals corresponding to carbon (C) and nitrogen (N) appeared for Fe3O4–chitosan (Fig. 10b). Therefore, it can be assumed that chitosan is coated onto the surface of Fe3O4 nanoparticles.
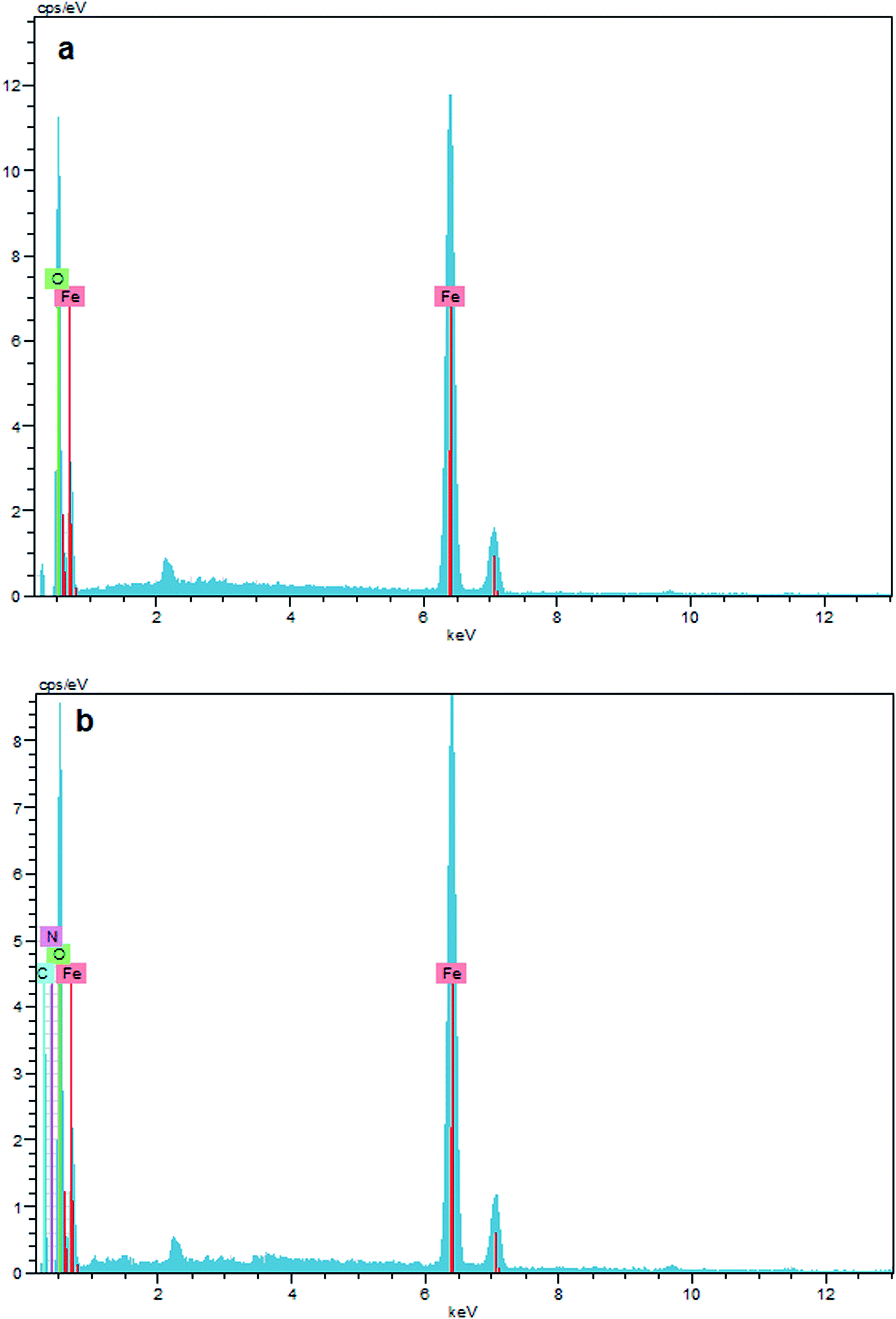 | ||
| Fig. 10 EDX result of (a) Fe3O4, and (b) Fe3O4–chitosan particles. | ||
The magnetic properties of the Fe3O4 and Fe3O4–chitosan particles were investigated by VSM analysis at room temperature. As shown in Fig. 11, there is no hysteresis in the magnetization for the two nanoparticles. The saturation magnetization of the bare Fe3O4 nanoparticles was about 67 emu g−1, while that for Fe3O4–chitosan particles was about 37 emu g−1 in this experiment. The existence of chitosan on the surface of Fe3O4 nanoparticles gave rise to the decrease in the uniformity due to quenching of surface moments, resulting in the reduction of magnetic moment in the nanoparticles.55 A similar decrease in saturation magnetization values were reported by other researchers. In addition, neither coercivity nor remanence was observed, implying that the nanoparticles are almost superparamagnetic. The inset of Fig. 11 shows the photographs of an aqueous solution of Fe3O4–chitosan particles in the absence of magnetic field and in the presence of externally applied magnetic field.
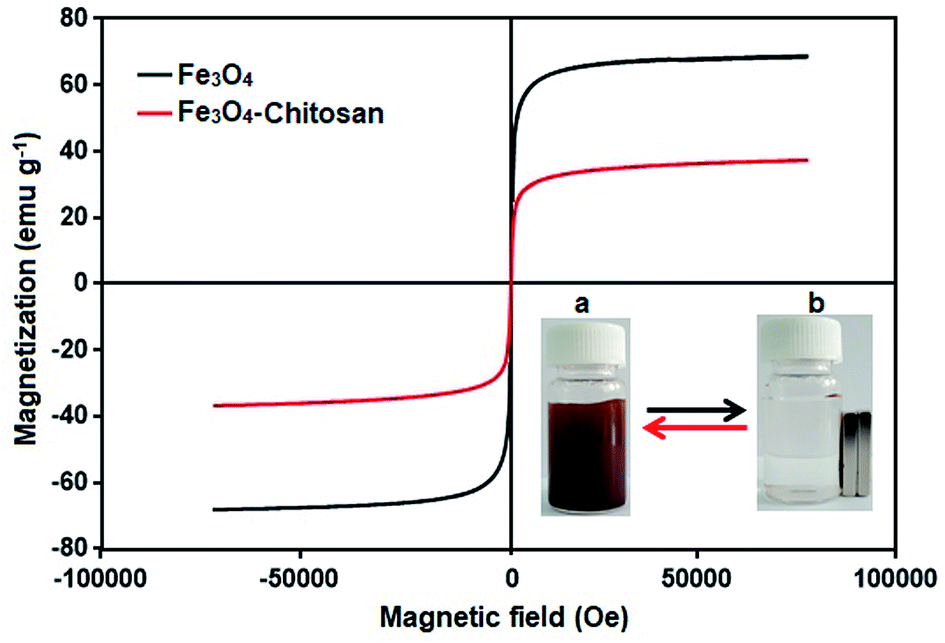 | ||
| Fig. 11 VSM measurements of the obtained Fe3O4 and Fe3O4–chitosan particles. The photographic image of Fe3O4–chitosan particles (a) without magnet and (b) with magnet. | ||
3.2 Optimization of conditions on the conjugated L-ASNase
The efficiency of L-ASNase immobilized on Fe3O4–chitosan was calculated as 73.2%. This high immobilization efficiency revealed that Fe3O4–chitosan particles can be a prominent carrier matrix for immobilization of L-ASNase. Moreover, the specific activity of free L-ASNase was 289 ± 8.4 U mg−1 protein, while the specific activity of conjugated L-ASNase was 179 ± 4.6 U mg−1 protein. This decrease in the specific activity after immobilization may be due to diffusion limitation. Additionally, immobilization of the enzyme by covalent binding would lead to a decrease in the flexibility of the enzyme molecule, which is commonly reflected by a decrease in catalytic activity.56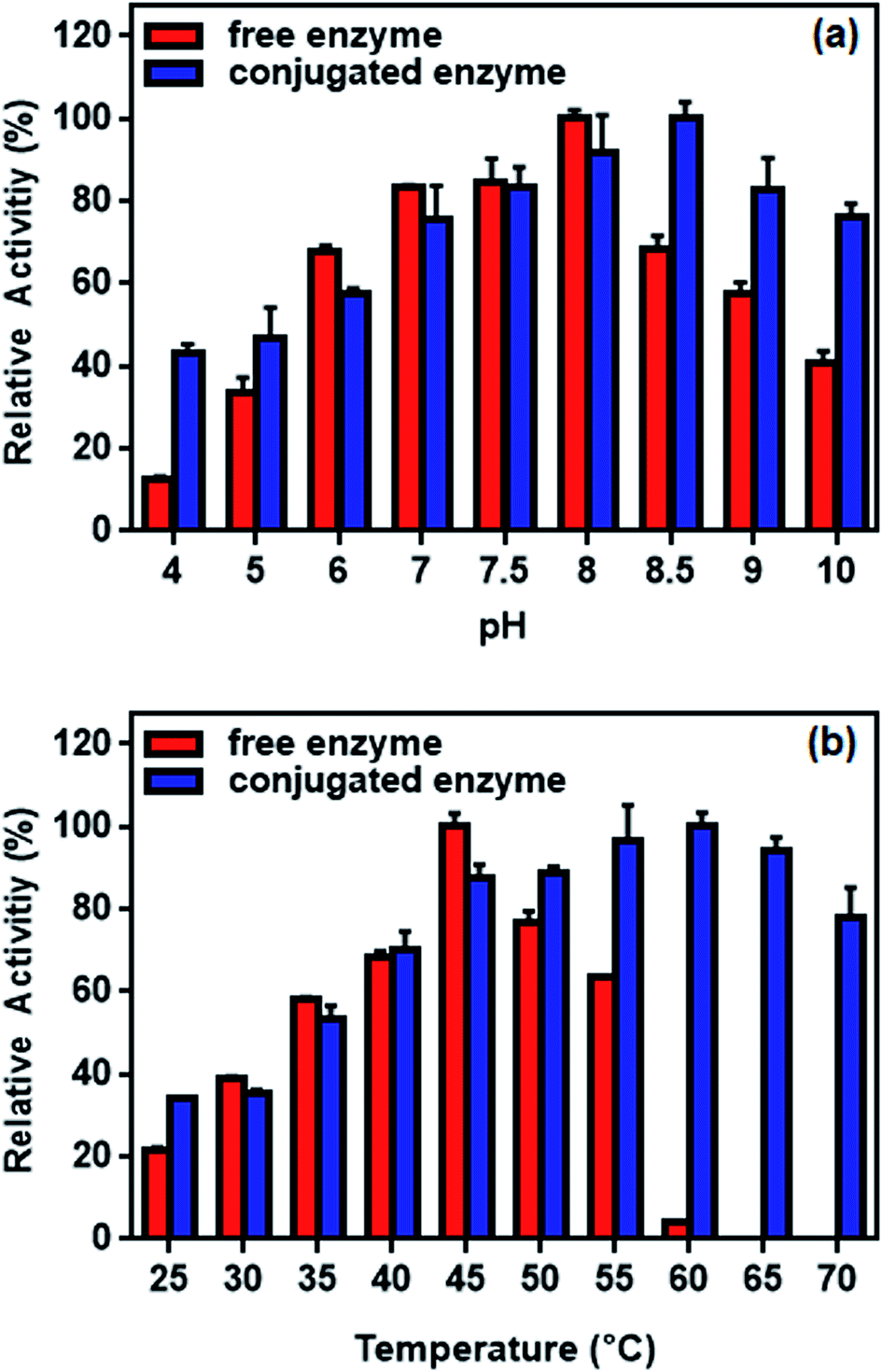 | ||
| Fig. 12 Relative activity of free and conjugated L-ASNase: (a) at different pH values and (b) at different temperature. | ||
The effect of temperature on the enzyme activity is shown in Fig. 12b. While the optimum temperature for free enzyme was 45 °C, the conjugated enzyme showed maximum activity at 60 °C. The activity of the free enzyme decreased more sharply than that of the conjugated enzyme at higher temperature. Although the free enzyme was readily inactivated at 60 °C, the conjugated L-ASNase maintains more than 60% of the initial activity even at 70 °C. This occurs probably because the immobilization of L-ASNase on the support limited the conformational mobility of the enzyme molecules at high temperature, preventing it from inactivation.57 Similar results of improved pH and temperature adaptability could be observed in other immobilization studies of L-ASNase.58,59
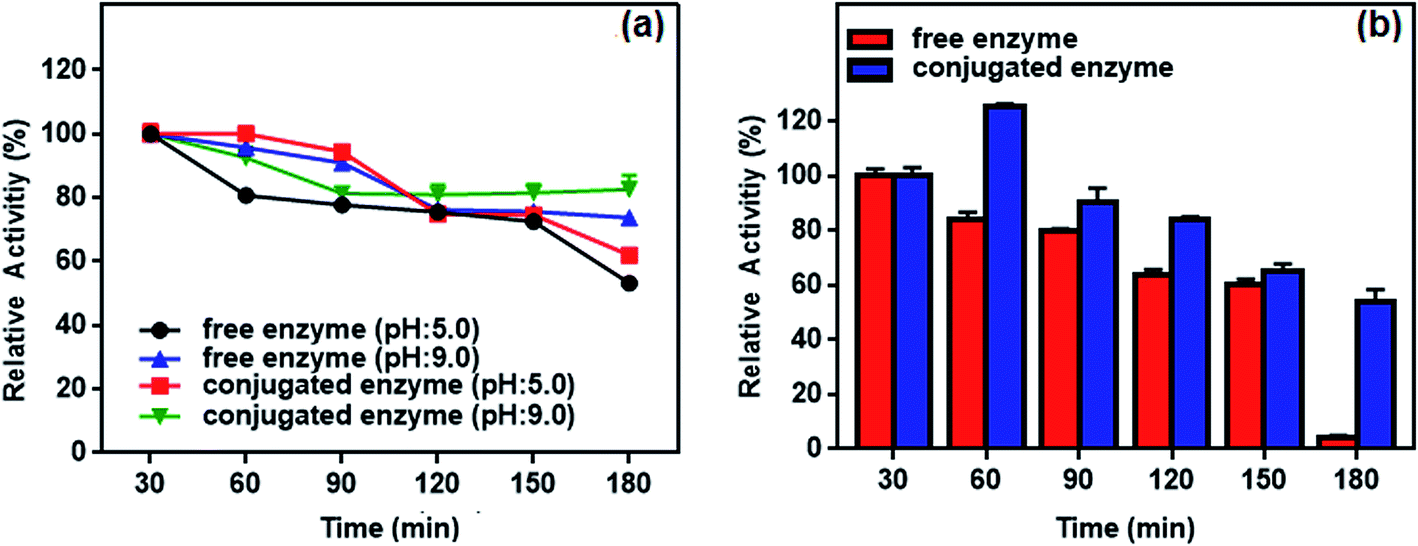 | ||
| Fig. 13 (a) pH and (b) thermal stability of the free and conjugated enzyme at different time intervals (30–180 min). | ||
Thermal stability of the conjugated enzyme is one of the most important criteria with respect to applications. Fig. 13b plots residual activity for the free and conjugated L-ASNase during 180 min test at 55 °C. The relative activity of conjugated L-ASNase remained higher than that of free L-ASNase at each time interval. The conjugated L-ASNase still retained 84% of its initial activity after 120 min incubation, whereas free L-ASNase retained 64% activity at the same conditions. When the incubation time is 180 min, conjugated enzyme holds approximately 54% of initial activity, while free enzyme lost almost all of its activity. Thus, the thermal stability of L-ASNase significantly improved after conjugation. Considering the results described above, the enzymatic activity of the conjugated L-ASNase was improved significantly, and it was more stable than the free L-ASNase, particularly under harsh conditions, as described above.
| Km (mM) | Vmax (U mg protein−1) | |
|---|---|---|
| Free L-ASNase | 0.65 ± 0.078 | 578.3 ± 38.0 |
| Conjugated L-ASNase | 0.14 ± 0.007 | 284.7 ± 12.5 |
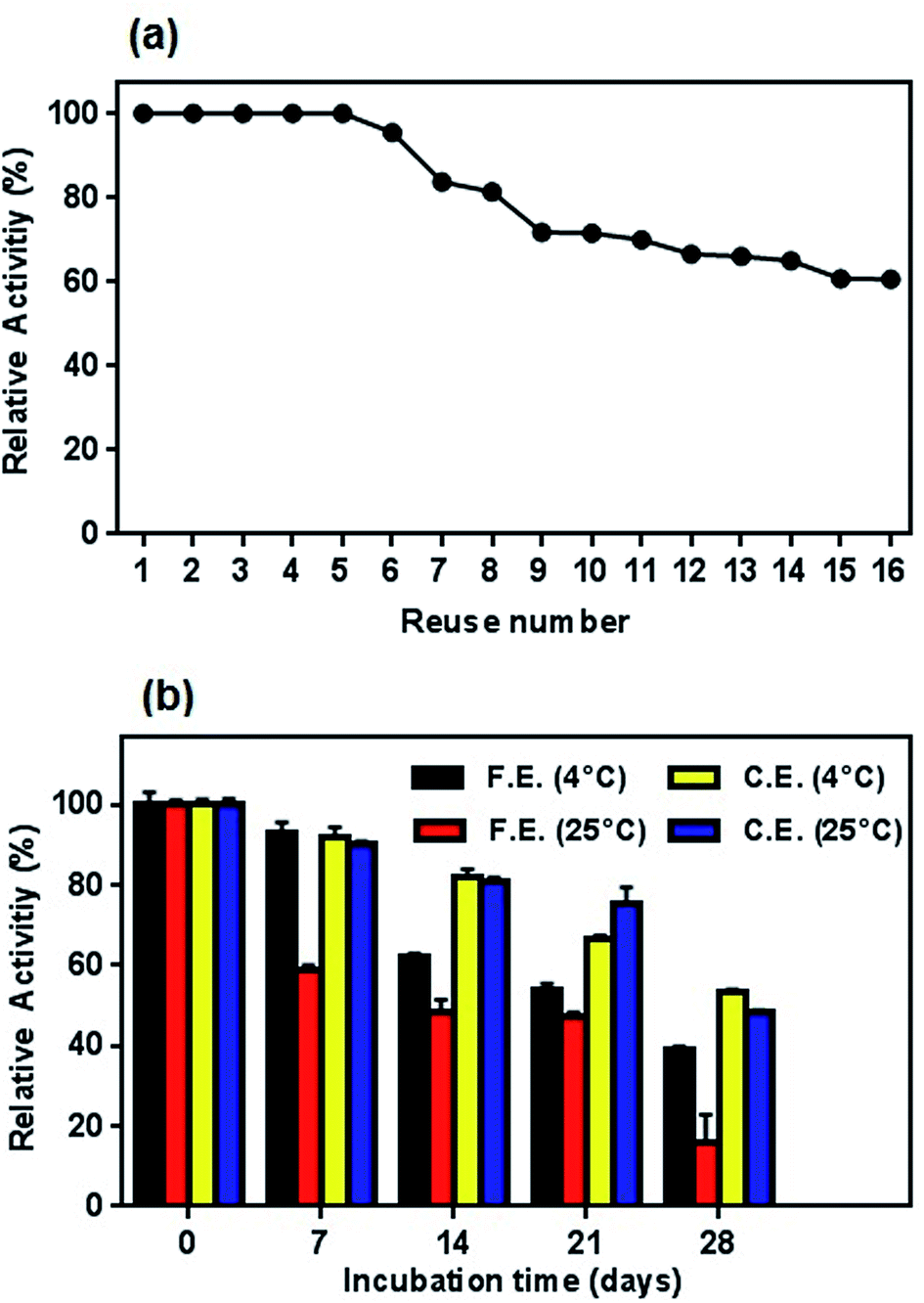 | ||
| Fig. 14 (a) Profile of reusability test for conjugated enzyme and (b) storage stability of free and conjugated L-ASNase on Fe3O4–chitosan at 4 °C and r.t. in dried state. | ||
Similar to reusability, long-time storage of L-ASNase is also a significant parameter to reduce the cost of the enzyme in the industrial applications since free enzyme denatures after a certain period of time. To test the long-term storage stability of enzymes, both the free and conjugated L-ASNase were preserved at 4 °C and room temperature for 4 weeks. The residual activities were measured as described in the assay section. As exhibited in Fig. 14b, the storage stability of conjugated L-ASNase was much higher than that of free L-ASNase at 4 °C and r.t. It could be observed that free L-ASNase retained 39.2% of the initial activity after 4 weeks, while the conjugated L-ASNase still maintained more than half (53.3%) of the initial activity after the same period. Moreover, it was found that the conjugated L-ASNase retained 48.4% of its initial activity at r.t. However, the free enzyme only retained 16% of the initial activity after 4 weeks at the same storage condition. These outcomes illustrated that the storage stability of L-ASNase fairly improved after immobilization with Fe3O4–chitosan magnetic particles due to increased stabilization of its active conformation by formation of a covalent bond between Fe3O4–chitosan and L-ASNase molecule.
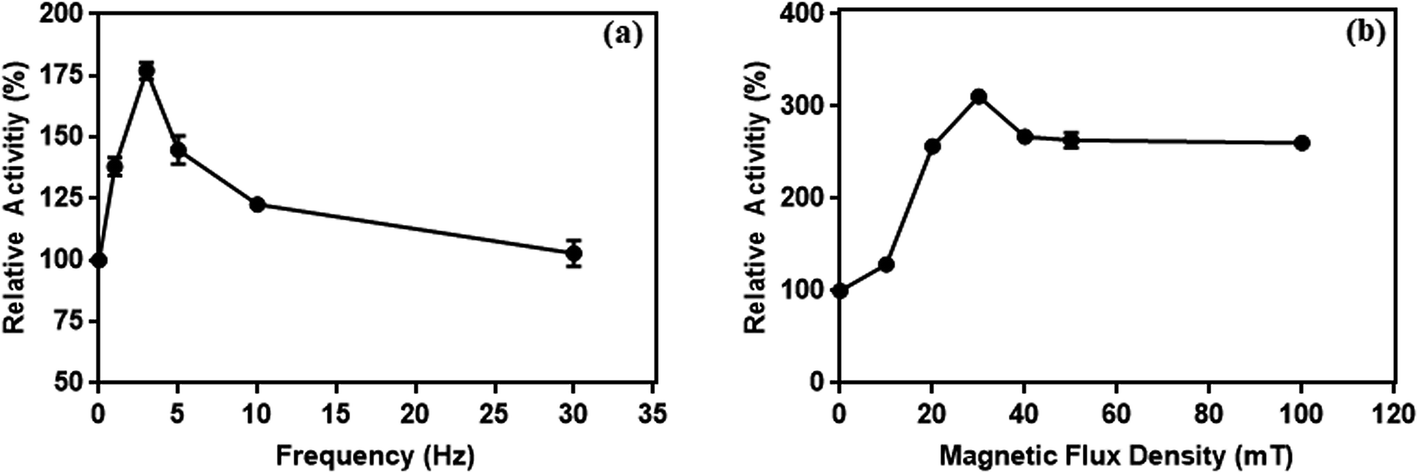 | ||
| Fig. 15 The effect of frequency at 10 mT (a) and (b) magnetic flux density at 3 Hz on the enzymes activity conjugated on magnetic particles. | ||
It has been reported that a relative increase in the interaction between enzymes and substrate molecules is associated with an increase in activity. In our study, we think that there are two important reasons for the increase in enzyme activity at certain frequency and flux density in the weak magnetic field. First is the effective prevention of the aggregation of L-ASNase-immobilized particles during the reaction with weak magnetic field. Second, magnetic-propelled enzymes more likely show enzyme–substrate interaction. We also believe that the enzymes are more stable in the covalent modification on the magnetic particle during these processes and this promotes the increase in activity of the immobilized L-ASNase.
4. Conclusions
L-ASNase is a unique enzyme for the enzymatic hydrolysis of L-Asn in medicine and food industry. Nevertheless, its instability, toxicity and poor reusability significantly limit the long-term applications. In order to overcome these disadvantages, for the first time, in this study, chitosan-modified magnetic particles were synthesized and employed for covalent immobilization of L-ASNase to improve its stability and reusability performance. Additionally, we developed a magnetic field platform and successfully studied the effect of magnetic field on conjugated L-ASNase activity. To demonstrate the superiority of the conjugated L-ASNase, we also compared the immobilization parameters when free enzyme was used as a control. The conjugated L-ASNase prepared in this study displayed excellent long-term storage and incubation stability, thermal stability, and reusability as compared with those of the free enzyme. More importantly, the magnetic field experiment results indicated that there was a considerable effect of magnetic exposure on the conjugated L-ASNase. Taking the abovementioned results into consideration, magnetic Fe3O4–chitosan particles formed with the combination of the inorganic material and natural polymer material could be used as an ideal carrier matrix for L-ASNase immobilization. Moreover, the application of a static magnetic field can be a promising strategy to increase the enzyme activity, particularly the activity of industrially important enzymes and their broad applications.Conflicts of interest
The authors report no declarations of interest.Acknowledgements
Authors are grateful to the Inonu University (FDK-2017-751).Notes and references
- A. Ulu and B. Ates, Bioconjugate Chem., 2017, 28, 1598–1610 CrossRef CAS PubMed.
- U. K. Narta, S. S. Kanwar and W. Azmi, Crit. Rev. Oncol. Hematol., 2007, 61, 208–221 CrossRef PubMed.
- R. Pieters, S. P. Hunger, J. Boos, C. Rizzari, L. Silverman, A. Baruchel, N. Goekbuget, M. Schrappe and C. H. Pui, Cancer, 2011, 117, 238–249 CrossRef CAS PubMed.
- T. Mizuki, N. Watanabe, Y. Nagaoka, T. Fukushima, H. Morimoto, R. Usami and T. Maekawa, Biochem. Biophys. Res. Commun., 2010, 393, 779–782 CrossRef CAS PubMed.
- L. T. Prando, P. R. De Lima, K. Rezzadori, J. V. De Oliveira and M. Di Luccio, Ind. Eng. Chem. Res., 2017, 56, 9065–9071 CrossRef CAS.
- A. Ali, H. Zafar, M. Zia, I. ul Haq, A. R. Phull, J. S. Ali and A. Hussain, Nanotechnol., Sci. Appl., 2016, 9, 49–67 CrossRef CAS PubMed.
- S. Sabale, P. Kandesar, V. Jadhav, R. Komorek, R. K. Motkuri and X. Y. Yu, Biomater. Sci., 2017, 5, 2212–2225 RSC.
- C. Yang, W. Guo, L. Cui, N. An, T. Zhang, H. Lin and F. Qu, Langmuir, 2014, 30, 9819–9827 CrossRef CAS PubMed.
- Y. Wang, J. Dostalek and W. Knoll, Anal. Chem., 2011, 83, 6202–6207 CrossRef CAS PubMed.
- M. An, J. Cui and L. Wang, J. Phys. Chem. C, 2014, 118, 3062–3068 CrossRef CAS.
- Y. K. Peng, C. N. P. Lui, Y. W. Chen, S. W. Chou, E. Raine, P. T. Chou, K. K. L. Yung and S. C. E. Tsang, Chem. Mater., 2017, 29, 4411–4417 CrossRef CAS.
- X. M. Ling, X. Y. Wang, P. Ma, Y. Yang, J. M. Qin, X. J. Zhang and Y. W. Zhang, J. Microbiol. Biotechnol., 2016, 26, 829–836 CrossRef CAS PubMed.
- Y. Liu, S. Jia, Q. Wu, J. Ran, W. Zhang and S. Wu, Catal. Commun., 2011, 12, 717–720 CrossRef CAS.
- X. Y. Wang, X. P. Jiang, Y. Li, S. Zeng and Y. W. Zhang, Int. J. Biol. Macromol., 2015, 75, 44–50 CrossRef CAS PubMed.
- C. H. Jun, Y. J. Park, Y. R. Yeon, J. Choi, W. Lee, S. Ko and J. Cheon, Chem. Commun., 2006, 0, 1619–1621 RSC.
- Z. Zhang, H. Duan, S. Li and Y. Lin, Langmuir, 2010, 26, 6676–6680 CrossRef CAS PubMed.
- S. A. Adams, J. L. Hauser, A. C. Allen, K. P. Lindquist, A. P. Ramirez, S. Oliver and J. Z. Zhang, ACS Appl. Nano Mater., 2018, 1, 1406–1412 CrossRef CAS.
- A. Ulu, I. Ozcan, S. Koytepe and B. Ates, Int. J. Biol. Macromol., 2018, 115, 1122–1130 CrossRef CAS PubMed.
- K. Manna and S. K. Srivastava, ACS Sustainable Chem. Eng., 2017, 5, 10710–10721 CrossRef CAS.
- Y. Lin, X. Liu, Z. Xing, Y. Geng, J. Wilson, D. Wu and H. Kong, Cellulose, 2017, 24, 5541–5550 CrossRef CAS.
- A. Mohseni-Bandpi, B. Kakavandi, R. R. Kalantary, A. Azari and A. Keramati, RSC Adv., 2015, 5, 73279–73289 RSC.
- A. Ulu, S. A. A. Noma, C. Gurses and S. Koytepe, Starch - Stärke, 2018 DOI:10.1002/star.201700303.
- J. Guo, Z. Zheng, C. Chen, X. Lu, Y. Zhang and B. Zheng, J. Agric. Food Chem., 2017, 65, 7934–7943 CrossRef CAS PubMed.
- L. Zang, J. Qiu, X. Wu, W. Zhang, E. Sakai and Y. Wei, Ind. Eng. Chem. Res., 2014, 53, 3448–3454 CrossRef CAS.
- X. Liu, X. Chen, Y. Li, X. Wang, X. Peng and W. Zhu, ACS Appl. Mater. Interfaces, 2012, 4, 5169–5178 CrossRef CAS PubMed.
- J. Long, Z. Wu, X. Li, E. Xu, X. Xu, Z. Jin and A. Jiao, J. Agric. Food Chem., 2015, 63, 3534–3542 CrossRef CAS PubMed.
- H. Suo, L. Xu, C. Xu, H. Chen, D. Yu, Z. Gao, H. Huang and Y. Hu, Int. J. Biol. Macromol., 2018, 119, 624–632 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- L. T. Mashburn and J. C. Wriston, Arch. Biochem. Biophys., 1964, 105, 450–452 CrossRef CAS PubMed.
- N. E. A. El-Naggar, H. Moawad, N. M. El-Shweihy and S. M. El-Ewasy, BioMed Res. Int., 2015, 2015, 627031 Search PubMed.
- Y. Haldorai, A. Rengaraj, T. Ryu, J. Shin, Y. S. Huh and Y. K. Han, J. Mater. Sci. Eng. B, 2015, 195, 20–29 CrossRef CAS.
- J. Qu, G. Liu, Y. Wang and R. Hong, Adv. Powder Technol., 2010, 21, 461–467 CrossRef CAS.
- H. Qin, C. M. Wang, Q. Q. Dong, L. Zhang, X. Zhang, Z. Y. Ma and Q. R. Han, J. Magn. Magn. Mater., 2015, 381, 120–126 CrossRef CAS.
- C. Yuwei and W. Jianlong, Chem. Eng. J., 2011, 168, 286–292 CrossRef.
- L. Y. Zhang, X. J. Zhu, H. W. Sun, G. R. Chi, J. X. Xu and Y. L. Sun, Curr. Appl. Phys., 2010, 10, 828–833 CrossRef.
- G. Y. Li, Y. R. Jiang, K. L. Huang, P. Ding and J. Chen, J. Alloys Compd., 2008, 466, 451–456 CrossRef CAS.
- N. F. M. Rodrigues, S. Y. Neto, R. de C. S. Luz, F. S. Damos and H. Yamanaka, Biosensors, 2018, 8(1), 16 CrossRef PubMed.
- C. Pan, B. Hu, W. Li, Y. Sun, H. Ye and X. Zeng, J. Mol. Catal. B: Enzym., 2009, 61, 208–215 CrossRef CAS.
- P. G. Ingole, N. R. Thakare, K. Kim, H. C. Bajaj, K. Singh and H. Lee, New J. Chem., 2013, 37, 4018–4024 RSC.
- Ö. Aybastier, S. Şahin, E. Işik and C. Demir, Anal. Methods, 2011, 3, 2289–2297 RSC.
- S. Ghosh, S. R. Chaganti and R. S. Prakasham, J. Mol. Catal. B: Enzym., 2012, 74, 132–137 CrossRef CAS.
- E. Bahreini, K. Aghaiypour, R. Abbasalipourkabir and A. R. Mokarram, Nanoscale Res. Lett., 2014, 9, 340 CrossRef PubMed.
- C. Cao, L. Xiao, C. Chen, X. Shi, Q. Cao and L. Gao, Powder Technol., 2014, 260, 90–97 CrossRef CAS.
- W. Li, L. Xiao and C. Qin, J. Macromol. Sci., Part A: Pure Appl.Chem., 2011, 48, 57–64 CrossRef CAS.
- J. A. Ratto, T. Hatakeyama and R. B. Blumstein, Polymer, 1995, 36, 2915–2919 CrossRef CAS.
- D. de Britto and S. P. Campana-Filho, Thermochim. Acta, 2007, 465, 73–82 CrossRef CAS.
- M. Mucha and A. Pawlak, Polimery, 2002, 47, 509–516 CAS.
- W. Jiang, W. Wang, B. Pan, Q. Zhang, W. Zhang and L. Lv, ACS Appl. Mater. Interfaces, 2014, 6, 3421–3426 CrossRef CAS PubMed.
- J. Safari and L. Javadian, Ultrason. Sonochem., 2015, 22, 341–348 CrossRef CAS PubMed.
- S. F. Shi, J. F. Jia, X. K. Guo, Y. P. Zhao, D. S. Chen, Y. Y. Guo, T. Cheng and X. L. Zhang, Int. J. Nanomed., 2012, 7, 5593–5602 CAS.
- Y. H. Chen, H. E. Tu and J. Leu, Microporous Mesoporous Mater., 2012, 162, 181–188 CrossRef CAS.
- G. Cheng and S. Y. Zheng, Sci. Rep., 2014, 4, 6947 CrossRef CAS PubMed.
- A. Zhu, L. Yuan and T. Liao, Int. J. Pharm., 2008, 350, 361–368 CrossRef CAS PubMed.
- Y. Liu, S. Jia, Q. Wu, J. Ran, W. Zhang and S. Wu, Catal. Commun., 2011, 12, 717–720 CrossRef CAS.
- S. Nasirimoghaddam, S. Zeinali and S. Sabbaghi, J. Ind. Eng. Chem., 2015, 27, 79–87 CrossRef CAS.
- M. A. Abdel-Naby, A. A. Sherif, A. B. El-Tanash and A. T. Mankarios, J. Appl. Microbiol., 1999, 87, 108–114 CrossRef CAS.
- J. Song, T. Lei, Y. Yang, N. Wu, P. Su and Y. Yang, New J. Chem., 2018, 42, 8458–8468 RSC.
- A. Ulu, S. Koytepe and B. Ates, J. Appl. Polym. Sci., 2016, 133, 19 CrossRef.
- A. Ulu, S. A. A. Noma, S. Koytepe and B. Ates, Artif. Cells, Nanomed., Biotechnol., 2018, 0, 1–11 CAS.
- S. A. Ahmed, F. A. Mostafa and M. A. Ouis, Int. J. Biol. Macromol., 2018, 112, 371–382 CrossRef CAS PubMed.
- A. Ulu, S. Koytepe and B. Ates, Polym. Bull., 2016, 73, 1891–1907 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |