
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrene-based metal–organic framework NU-1000 photocatalysed atom-transfer radical addition for iodoperfluoroalkylation and (Z)-selective perfluoroalkylation of olefins by visible-light irradiation†
Tiexin Zhang *a,
Pengfang Wanga,
Zirui Gaoa,
Yang Ana,
Cheng Hea and
Chunying Duan*ab
*a,
Pengfang Wanga,
Zirui Gaoa,
Yang Ana,
Cheng Hea and
Chunying Duan*ab
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China. E-mail: zhangtiexin@dlut.edu.cn; cyduan@dlut.edu.cn; Fax: +86-411-84986476; Tel: +86-411-84986476
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300071, China
First published on 24th September 2018
Abstract
The photocatalytic atom-transfer radical addition (ATRA) of perfluoroalkyl iodides onto olefins is of potential biointerest; the relatively negative reductive potential of perfluoroalkyl iodide makes it difficult to generate the perfluoroalkyl radical by the photoinduced single-electron transfer from the excited state of the photocatalyst. In the presence of the easily available well-known pyrene-based metal–organic framework (MOF) NU-1000, the ATRA was achieved for iodoperfluooralkylation of olefins in a heterogeneous mode upon 405 nm visible-light irradiation with LEDs. The investigation supports a mechanism whereby the pyrene-based chromophores within NU-1000 photochemically generate the reactive radical species by sensitisation of the perfluoroalkyl iodides through an energy-transfer (EnT) pathway. Besides the activation of singlet oxygen for oxidative application, it is the first time to directly utilise the photoinduced EnT process of MOFs in organic transformations. Compared with the photocatalysis using homogeneous free ligand or other pyrene-based MOFs, the spatial isolation of chromophores in NU-1000 is believed to hamper the destructive excited-state energy loss from self-quenching or interligand interactions, ensuring the efficiency of reaction. When employing conjugated arylalkenes as substrates, the photocatalytic ATRA reaction, the HI elimination of the ATRA product, and the EnT-mediated (E)/(Z)-isomerisation could be merged together in one-pot to afford highly (Z)-selective perfluoroalkyl styrenes, which might be attractive in the pharmaceutical field.
Introduction
Atom-transfer radical addition (ATRA)1 is known as an atom-economical and effective way to functionalise olefin substrates to construct a new C–C and C–X bond (X = halogen) in a single strike. The recently developed photoredox catalytic systems circumvented the use of stoichiometric amounts of toxic or explosive reagents, or high reaction temperatures to generate radicals for the initiation of the ATRA reaction,2,3 and further expanded the scope of ATRA to the perfluoroalkyl-halogenation of alkenes,4–7 which is of fundamental importance in pharmaceutical fields since the introduction of perfluoroalkyl (Rf, –CnF2n+1), into organic molecules leads to drastic enhancement of their solubility, metabolic stability, and bioavailability.8 However, in comparison to the facile generation of an alkyl radical from precursors such as the widely-used diethyl 2-bromomalonate through a photoinduced single-electron transfer (PET) process,9 the relatively negative oxidation potentials10–12 and the generally needed large enough over-potential for common perfluoroalkyl radical precursors perfluoroalkyl iodides (RfI) (Scheme 1b)13 made it challenging to afford perfluoroalkyl (Rf) radicals through PET routes10 in the presence of photoresponsive Ru/Ir multipyridyl complexes4,10,15,16 or organic dyes.17,18 Thus, exogenous electron donors (EDs),11,16 excess amounts of RfI,15 or Lewis acid activation11 were employed to enhance the reductive heterolytic cleavage of C–I bonds of RfI for the formation of Rf radicals (Scheme 1a, PET pathways). It should be noted that the remarkably lower bond dissociation energies (BDEs) of C–I bonds of RfI compared with the substantial BDE value of diethyl 2-bromomalonate implies the facile generation of perfluoroalkyl radicals by homolytic cleavage of C–I bonds (Scheme 1b). Note that the redox innocent nature of energy transfer (EnT) process provides the possibility of mediating direct homolytic C–I bonds cleavages to take advantage of their medium BDE values, and circumvents the severe dependency of the PET-induced heterolytic cleavages on reduction potentials of photocatalysts (Scheme 1a, EnT pathway). Melchiorre19 and Vincent20 successfully developed photocatalytic EnT mediated generation of perfluoroalkyl radicals by using ultraviolet (UV) responsive benzaldehyde or benzophenone derivatives as triplet photosensitisers and applied the methodologies in ATRA reactions of unsaturated C–C bonds. Considering the inevitable self-quenching and bleaching of organic dyes owing to their uncontrollable random collisions and other inter-chromophore interactions in solution phase and the destruction of labile moieties under the high energetic UV light, it is highly desirable to perform the EnT mediated ATRA of RfI to olefins under visible-light irradiation in a heterogeneous manner.21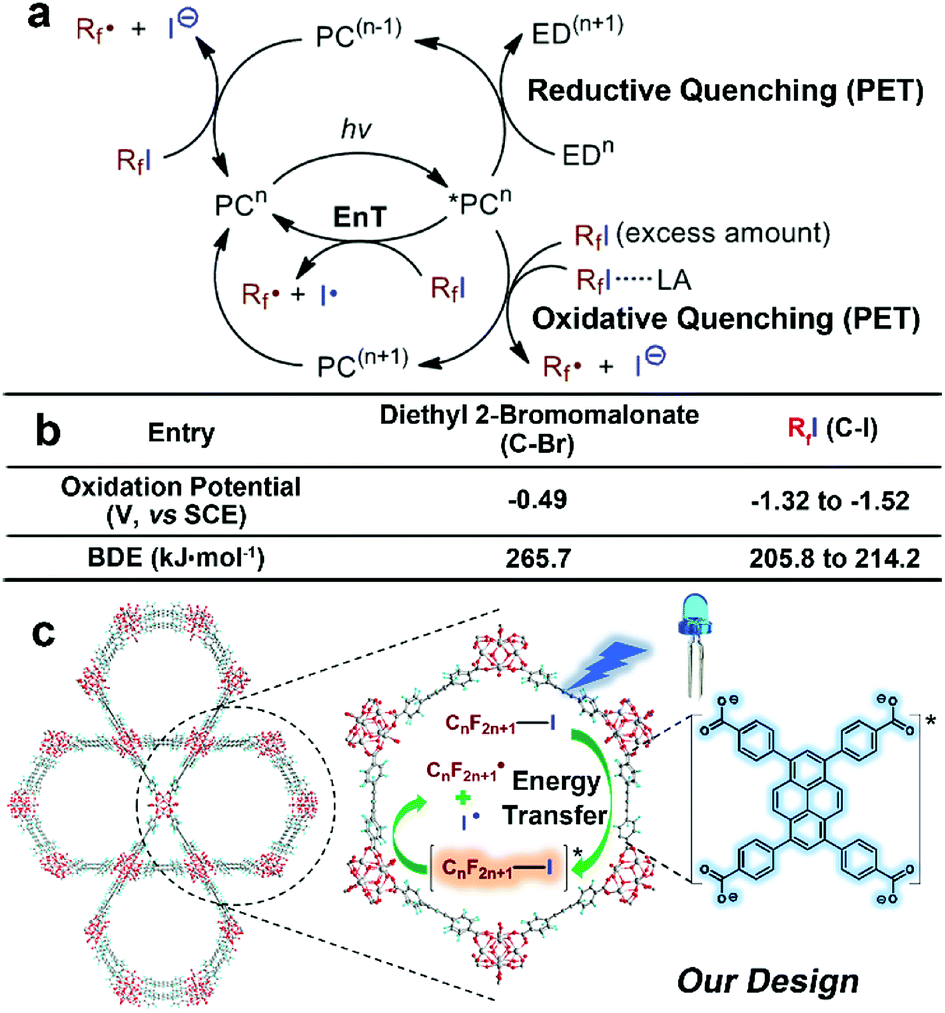 | ||
| Scheme 1 (a) Mechanistic diagram of formation of perfluoroalkyl (Rf) radicals via different photocatalytic routes. (PC = photocatalyst, LA = Lewis acid, EnT = energy transfer) (b) comparison of oxidative potentials9–12 and bond dissociation energies (BDEs)14 of diethyl 2-bromomalonate and RfI, favouring different strategies for generation of radicals. (c) Schematic illustration of EnT induced homolytic cleavage of RfI to generate perfluoroalkyl radical for ATRA by using pyrene-based MOF NU-1000 under visible-light irradiation of 405 nm LEDs. | ||
Due to the good visible-light harvesting22–26 and powerful triplet photosensitising ability,27–29 the organic dye pyrene-based chromophores mediated EnT processes have been involved in the singlet oxygen generation,30 and other hot topics like fluorescence resonance energy transfer (FRET)31 or up-conversions,32,33 and those processes were widely applied in the bioinspired applications such as protein labelling,34 and biosensing.35 The aggregation state of pyrene-derivatives imposed great impact on light-harvesting, EnT, or other aspects of their photochemical/physical performances.36–40 Thus, this aggregation state-induced modulation was envisioned to be more controllable when assembling pyrene-based chromophores in a highly uniform manner within crystalline materials like metal–organic frameworks (MOFs).41 As a well-known representative of pyrene-based MOF,42–44 NU-1000 is constructed from 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPy) and zirconium ions,45 The open channels of NU-1000 facilitate the diffusion of substrates/reagents for the efficient conversion, the spatial isolation of TBAPy moieties within framework avoids the self-quenching or photobleaching of chromophores, and the strong coordination between zirconium ions and carboxylate groups of ligands endows NU-1000 with excellent durability for its recycle after use. Note that the photoreductive potential of excited-state NU-1000+/NU-1000* couple (ca. −1.12 eV, Fig. 1) is not negative enough to drive the PET-induced reduction of RfI to form the Rf radical (Scheme 1b), thus the EnT process mediated by NU-1000 might be the alternative way to homolytic cleavage C–I of RfI for generating Rf radicals to initiate ATRA reactions (Scheme 1c). In the pioneering works of Farha,46–48 Kim, and Lee,49 the EnT processes of NU-1000 and other pyrene-based MOFs have been successfully utilised in the activation of singlet oxygen for oxidative applications. In comparison, the direct use of photoinduced EnT of MOF for organic transformations is still in the infancy stage, and the EnT-induced formation of carbon-centred radicals by using MOFs have never been reported before. Herein, we report the EnT-mediated ATRA reaction of RfI onto olefins for the first time by using pyrene-based MOF NU-1000 under visible-light irradiation.
 | ||
| Fig. 1 (a) Normalized absorption and emission spectra of NU-1000, and the reductive potential of excited state of NU-1000 was determined to be −1.12 V (vs. SCE) on the basis of a free energy change of 2.62 eV (vs. SCE) and (b) the ground state oxidative potential of 1.50 V (vs. SCE). Solid-state cyclic voltammogram (CV) of NU-1000 with a scan rate of 200 mV s−1 in the scan range 0.0–2.0 V. | ||
Results and discussion
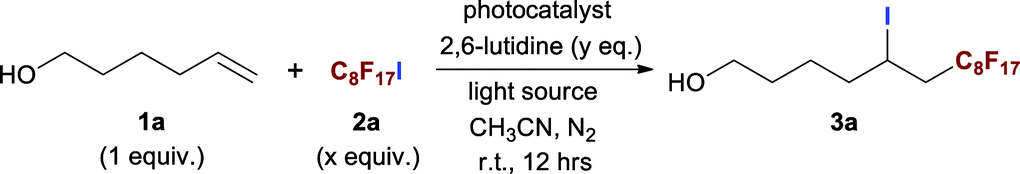
To evaluate our design, the terminal olefin 5-hexenol 1a was chosen as the model substrate, the ATRA reaction was carried out with 1a (0.25 mmol, 1.0 equiv.), perfluorooctyl iodide C8F17I 2a (2.0 equiv.), and the additive base 2,6-lutidine (2.0 equiv.) in the presence of 2.5 mol% of NU-1000 in acetonitrile at room temperature, by using Xe light as the light source. Gratifyingly, the desired product of ATRA reaction 7,7,8,8,9,9,10,10,11,12,12,13,13,14,14,14-hexadecafluoro-5-iodo-11-methyltetradecan-1-ol 3a was afforded in an isolated yield of 37% after 12 h (Table 1, entry 1). After filtering off the UV wavelength range below 400 nm of Xe light, the yield of 3a was remarkably increased to 59% (entry 2), implying the destructive effect of UV towards the labile product and reflecting the importance of visible-light irradiation. For the subtle tune of visible-light source, the narrow band LEDs with different wavelengths were screened, the LEDs with a centred wavelength of 405 nm (93% isolated yield, entry 4) were more favoured than the blue light ones with longer wavelength of 455 nm (63% isolated yield, entry 3), which could be easily rationalised by the stronger absorption of NU-1000 at 405 nm (Fig. 1a). After reaction, the brown colour of reaction mixture was attributed to the formation of I2, which was easily confirmed by using the starch test paper (Scheme 3d and e). The I2 was estimated to be generated from self-coupling of iodine radicals, corroborating the homolytic cleavage route of RfI. From another perspective, the evidence of self-coupling of radical intermediates revealed the necessity of using excess amount (2.0 equiv.) of radical source 2a, and decreasing the amount of 2a lead to lower yields (entries 5 and 6). By the way, the broad absorption of I2 covering 400–500 nm range also should be responsible for the inferior catalytic efficiency at 455 nm owing to the stronger photo-shielding effect there compared with the weaker absorption at more marginal 405 nm. Furthermore, different loading amounts of photocatalyst were examined, and 2.5 mol% of NU-1000 was proved to be more practical (entries 7 and 8). The following control experiments demonstrated that the photocatalyst, visible-light irradiation, inert gas atmosphere, and additive base are indispensable factors of this reaction (entries 9 to 12).
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst (mol%) | Light source | C8F17I/lutidine (x/y equiv.) | Yieldb (%) |
| a Reaction conditions: 1a (0.25 mmol, 1.0 equiv.), RfI 2a and additive base 2,6-lutidine (specified amounts), photocatalyst (specified amount calculated based on pyrene moiety), degassed acetonitrile (1.0 mL), room temperature (r.t.) and N2 atmosphere, 12 h.b Isolated yields.c NMR yield is shown in parenthesis.d Calculated based upon the amount of 2a. | ||||
| 1 | NU-1000 (2.5) | Xe light | 2/2 | 37 |
| 2 | NU-1000 (2.5) | Xe light (>400 nm) | 2/2 | 59 |
| 3 | NU-1000 (2.5) | LED (455 nm) | 2/2 | 63 |
| 4 | NU-1000 (2.5) | LED (405 nm) | 2/2 | 93(98)c |
| 5 | NU-1000 (2.5) | LED (405 nm) | 1.2/1.2 | 66 |
| 6 | NU-1000 (2.5) | LED (405 nm) | 0.5/0.5 | 57d |
| 7 | NU-1000 (5.0) | LED (405 nm) | 2/2 | 94 |
| 8 | NU-1000 (1.25) | LED (405 nm) | 2/2 | 61 |
| 9 | NU-1000 (2.5) | Dark | 2/2 | 0 |
| 10 | NU-1000 (2.5) | LED (405 nm) (in air) | 2/2 | Trace |
| 11 | None | LED (405 nm) | 2/2 | 0 |
| 12 | NU-1000 (2.5) | LED (405 nm) | 2/0 | 0 |
| 13 | Pyrene (2.5) | LED (405 nm) | 2/2 | Trace |
| 14 | H4TBAPy (2.5) | LED (405 nm) | 2/2 | 16 |
| 15 | ZrCl4 (7.5) + H4TBAPy (2.5) | LED (405 nm) | 2/2 | 21 |
| 16 | NU-901 (2.5) | LED (405 nm) | 2/2 | 81 |
The photocatalyst plays a vital role in this ATRA reaction, switching MOF to the homogeneous pyrene molecule gave ignorable amount of conversion (entry 13). The integrity of MOF catalyst is also important, only using the free ligand 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPy) or the simply mixed free ligand and zirconium salt as photocatalyst resulted in sharp falls in conversions (entries 14 and 15). Next, NU-901,50,51 another MOF constructed from H4TBAPy and zirconium salt but featured with different topology and stronger interchromophoric interaction than NU-1000,52 was also amenable to driving this reaction but in lower efficiency (entry 16), reflecting the advantage for NU-1000 in reducing the undesirable energy loss of excited state by its weaker inter-ligand interactions.
Then, the heterogeneity of this photocatalytic system was explored, as shown in Fig. 2. Removal of MOF particles by hot-filtration after 0.5 h shut down the reaction immediately (Fig. 2a), and zirconium ions could not be detected from the supernatant of reaction mixture by Inductively coupled plasma mass spectrometry (ICP-MS), excluding the possible decomposition of NU-1000. After the reaction, the MOF solids could be easily recovered by simple filtration, and then reused for at least 5 cycles without remarkable deterioration in activity (Fig. 2b). There were not significant changes of powder X-ray diffraction (XRD) patterns (Fig. 2c) and infrared (IR) spectrum of photocatalyst NU-1000 (Fig. 2d) before and after the reactions, which disclosed the maintenance of the crystallinity and composition of MOF NU-1000, furtherly demonstrating the important role of structural rigidity for the durable catalytic performance.
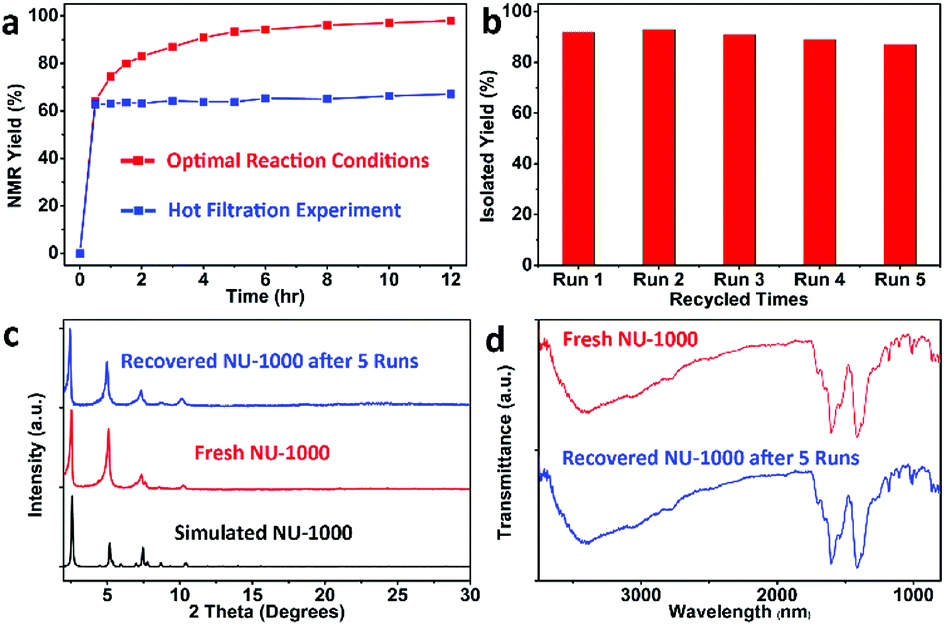 | ||
| Fig. 2 (a) The time-course reaction of 1a under the optimal condition and the hot filtration experiment in the presence of NU-1000. (b) Histograms of reuse experiments of MOF NU-1000. (c) X-ray powder diffraction (XRD) patterns of freshly prepared NU-1000 (middle), the recycled catalyst after 5 runs (up), and its simulated pattern based on the reported single-crystal data (bottom). (d) IR spectrum of the freshly prepared NU-1000 (up) and the recovered one after 5 runs (bottom). | ||
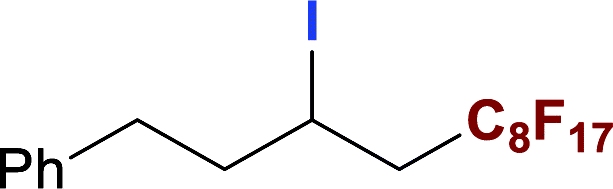
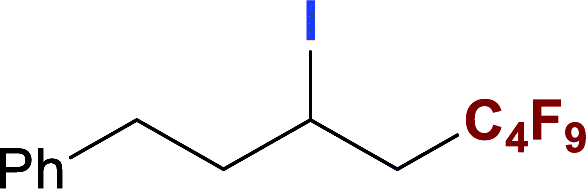
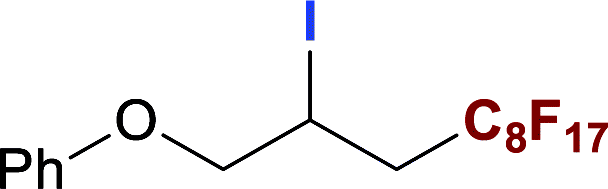
With optimized conditions in hand, we explored the substrate scope of the ATRA of RfI 2 onto olefins 3. The relative results were summarized in Table 2. In the presence of 2.5 mol% of NU-1000, the aliphatic terminal alkenes 1a to 1c bearing hydrophilic hydroxyl or hydrophobic phenyl/phenoxy substituents on the other ends of olefins were smoothly transformed into the corresponding difunctionalized products in moderate to high yields (3a to 3g). The perfluoroalkyl radical precursor 2 bearing longer perfluoroalkyl chains gave higher reaction efficiency than the ones with shorter perfluoroalkyl chains (entries 1 to 4 and entries 5 and 6). Judging from the trend of conversions, the reactivities of RfI 2 might be negatively correlated with the BDEs of their C–I bonds.10,14,53 It should be noted that the 2,2,2-trifluoroethyl iodide 2d, which was difficult to be reduced owing to its considerably high reductive potential (ca. −1.80 V vs. SCE),53 could also engage in this reaction to give a 67% isolated yield of desired product 3d.
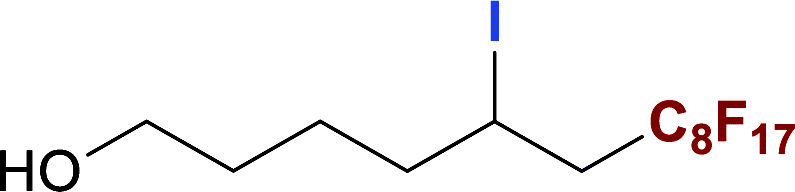
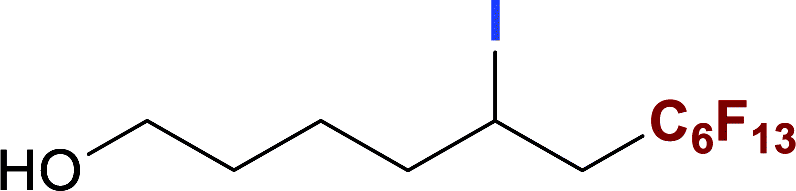
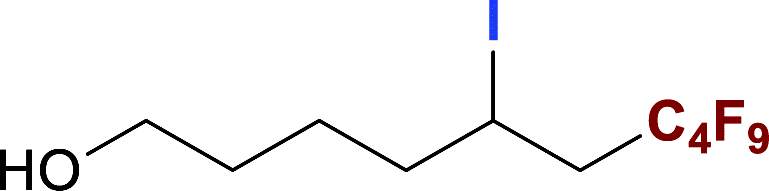
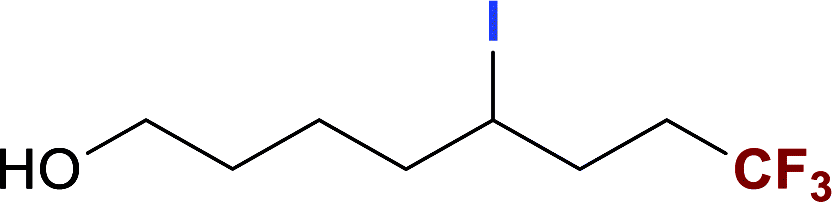
The naturally occuring compound bearing terminal olefin like (−)-β-pinene 1h, was also examined in this photocatalytic system (Scheme 2), and the four-membered spiroring of 1h was found to undergo a ring-opening process at the bridgehead carbon position near the alkene moiety to release the ring strain of the intermediate with a neighboring carbon centered radical, which was an evidence for the addition of perfluoroalkyl radical towards olefin moiety. However, the desired product 3h was subject to a base-assisted elimination of HI after the ATRA step, yielding a mixture of 4h and 4h′ with a ratio of ca. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50.
:50.
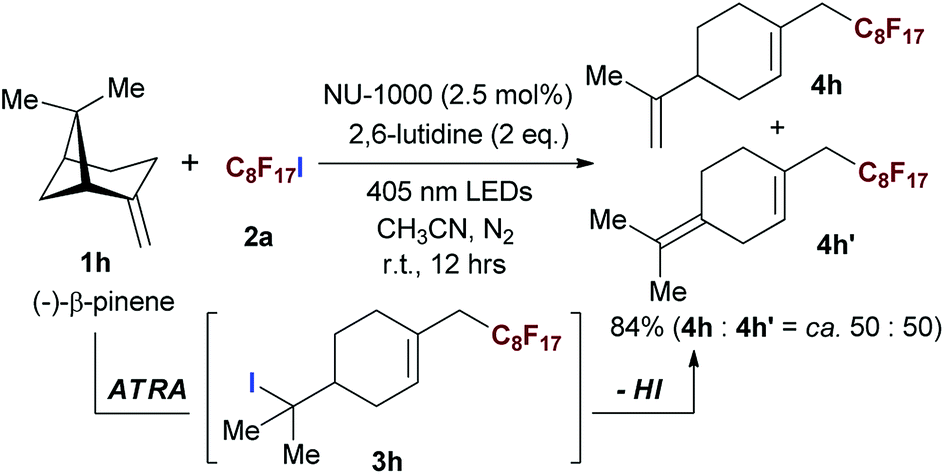 | ||
| Scheme 2 Evaluating the scope of the photocatalytic ATRA using aliphatic olefin bearing naturally occurring compound. Isolated yields. | ||
To investigate the mechanism of this photocatalytic ATRA reaction, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was employed as the radical scavenger. When 1.2 equiv. of TEMPO was added, the photocatalytic ATRA reaction was totally suppressed (Scheme 3a). Then, a radical clock experiment was conducted by using an intramolecular diallyl containing substrate 1i, furnishing 5-exo-trig radical cyclization in 86% yield (Scheme 3b). Those results together with the detection of I2 (Scheme 3c–e) well demonstrated the generation of perfluoroalkyl and iodine radical, implying the homolytic cleavage of C–I bonds of RfI.
 | ||
| Scheme 3 (a) The photocatalytic ATRA in the presence of radical scavenger. (b) The radical clock experiment. Isolated yields. The comparison of colour of reaction mixture (c) before and (d) after the photocatalytic process. (e) Detecting I2 from the reaction mixture by starch testing paper (left) before and (right) after photocatalysis. | ||
It is known that the outer-sphere single-electron transfer reduction of alkyl halides depends on their reductive potentials. In comparison, the inner-sphere electron transfer or EnT process are sensitive to the BDEs of carbon–halogen bonds of the alkyl halides and the nature of the halogen.54–56 The conversion plots of photocatalytic ATRA of various alkyl halides 2 onto 1a vs. the BDEs of carbon–halogen bonds,14 and vs. the oxidative potentials of the alkyl halides,57–59 were shown in Scheme 4. It was evident from the two sets of plots that a better correlation of the reaction conversions was obtained with BDEs (Scheme 4a) than with reductive potentials (Scheme 4b), indicative of an inner-sphere electron transfer or EnT pathway. It was shown that the alkyl halides with lower BDEs roughly underwent more efficient ATRA reactions in this photocatalytic system, and nearly no conversion was observed in the case of using carbon tetrachloride (2l) which was featured with a moderate oxidative potential (−0.64 V vs. SCE) but a high BDE (298 kJ mol−1). Although the presence of electron transfer pathways were still difficult to be excluded, it was theoretically less possible for a pyrene-based Zr-MOF to undergo an inner-sphere electron transfer process without the assistance of transition metal ions.60
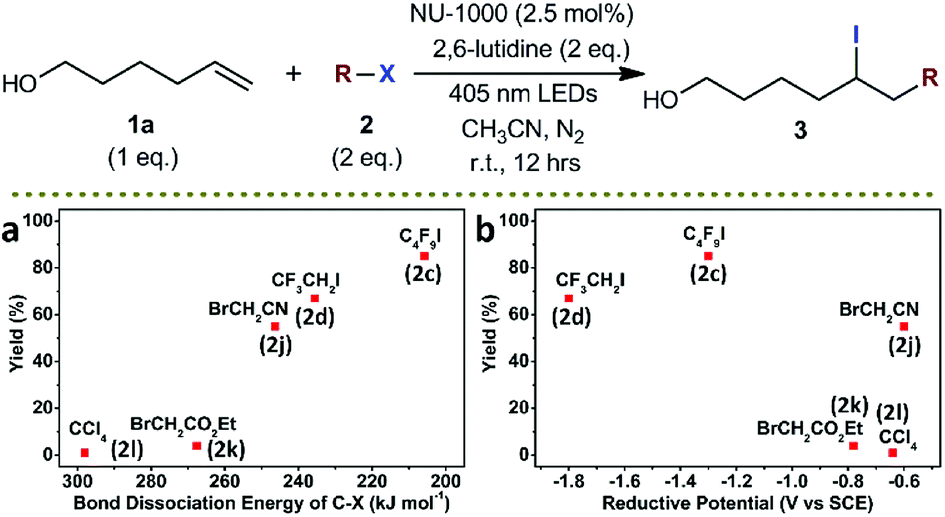 | ||
| Scheme 4 Conversion plots of photocatalytic ATRA of various alkyl halides 2 as a function of (a) BDEs of carbon–halogen bonds and (b) reductive potentials of alkyl halides. X refers to halogen atom. NMR yields. Compound labels are shown in parenthesis. | ||
The very recent computational works of Deria and coworkers revealed the correlation between the topological structures of pyrene-based MOFs and their photoinduced excited-state property,52,61 and the energy levels of singlet excited state S1 of NU-1000 and the isomeric NU-901 were caculated to be ca. 294 kJ mol−1 (ca. 3.05 eV) and ca. 256 kJ mol−1 (ca. 2.65 eV), repectively, endowing NU-1000 with theoretical possibility of driving the more efficient EnT mediated homolytic bond cleavage of RfI with medium BDE values in comparison to alkyl halides with much higher BDEs (Schemes 1b and 4a). Noticeably, the luminescence intensity of NU-1000 was significantly quenched upon the addition of C4F9I (2c) (Fig. 3a), and the luminescence lifetime of NU-1000 also decreased from 7.59 ns to 6.92 ns after addition of 2c (Fig. 3b), both phenomena evidenced the photoinduced singlet–singlet EnT process from excited-state NU-1000 to the perfluoroalkyl iodide molecules.62–65
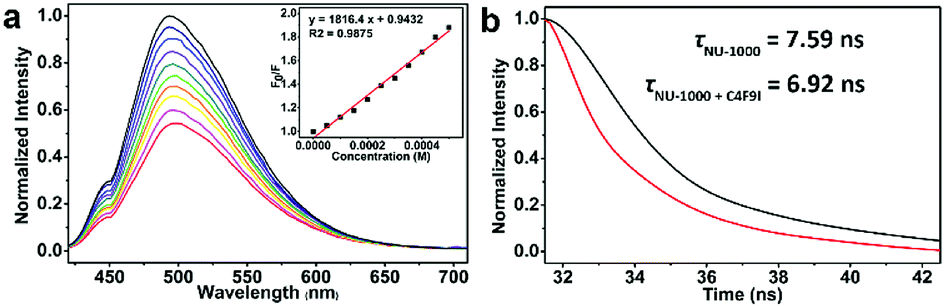 | ||
| Fig. 3 (a) Luminescence spectra of NU-1000 upon the addition of C4F9I (2c) and the corresponding simulated Stern–Volmer curve (inset). (b) The decreased luminescence lifetime of the NU-1000 suspension upon addition of C4F9I (2c). | ||
As previously mentioned, the ATRA reaction hugely diminished in the presence of O2 (ΔET1→S1 = ca. 95 kJ mol−1) as an effective triplet energy quencher of excited state (Table 1, entry 10).66 Then, the ATRA reaction of 2c and 1a was performed in the presence of triplet quenchers with different triplet state energies, and the triplet quencher 2,5-dimethyl hexa-2,4-diene, which triplet-state energy (ET = ca. 243 kJ mol−1) was much lower than that of another triplet quencher pyridazine (ET = ca. 297 kJ mol−1),19 was found to depress the conversion more effectively than pyridazine (Table 3, entries 1 and 2),67 the results of these triplet quenching experiments together with the reported works on photocatalytic singlet oxygen generation by using pyrene-based MOFs46–49 further implied the possible existence of triplet excited state of NU-1000 generated from singlet-to-triplet intersystem crossing during the photocatalysis.68 According to the computational results of Deria and coworkers, the topology-determined inter-ligand interactions within MOFs facilitated the transitions among different excited states (such as transitions among optically relevant S1, S2, and Sn states), lengthened their lifetime, and expedited their spatial shifts across the ligands in comparison to the free ligand H4TBAPy,61 providing more possible routes to the formation of triplet excited states of MOFs for EnT processes during photocatalysis. Moreover, compared with stronger inter-ligand interactions within MOF NU-901, the weaker inter-ligand interactions within NU-1000 were helpful to retain the energy level of excited state by avoiding the excited-state energy losses owing to stronger inter-ligand interactions,61 benefiting the more effective EnT process for homolytic bond break of RfI (Table 1, entries 4 and 16).
The application of this photocatalytic strategy was further expended to ATRA of aliphatic and aromatic terminal alkynes, furnishing iodoperfluoroalkylation of carbon–carbon triple bonds in good yields (Scheme 5). Interestingly, although the (E)-geometries of ATRA products were still more favoured, the corresponding ratios of (Z)-/(E)-isomers were higher than most of the reported results in literatures.20,69–71 These phenomena gave a strong hint of triplet EnT induced enrichment of (Z)-alkenes, and the (E)- to (Z)-conversion was confirmed by exposing (E)-isomer of 6b to the reaction condition (Scheme 5). The heavy-atom effect of iodo group of product 6b was believed to facilitate the conversion from (E)- to (Z)-geometry.74–78 Simultaneously, the EnT-induced intersystem crossing behaviour of conjugated arylalkene also imposed an competitive driving force to pull the perfluoroalkyl group to the ‘(Z)’-side of aryl moiety (caution: here, the (Z)-geometry relative to aryl is equivalent to the (E)-geometry relative to iodine atom). The ratio of (Z)-/(E)-isomers should represent the balance status of ‘tug-of-war’ between iodo and aryl group, and it was interesting to examine the (Z)-/(E)-ratio if iodine atom of 6b was absent, since it might provide a pathway for the facile preparation of (Z)-enriched perfluoroalkyl styrenes (caution: the (Z)-geometry here refers to the case of aryl moiety) which was of potential interest in pharmaceutical field and generally needed multi-step synthesis.79,80
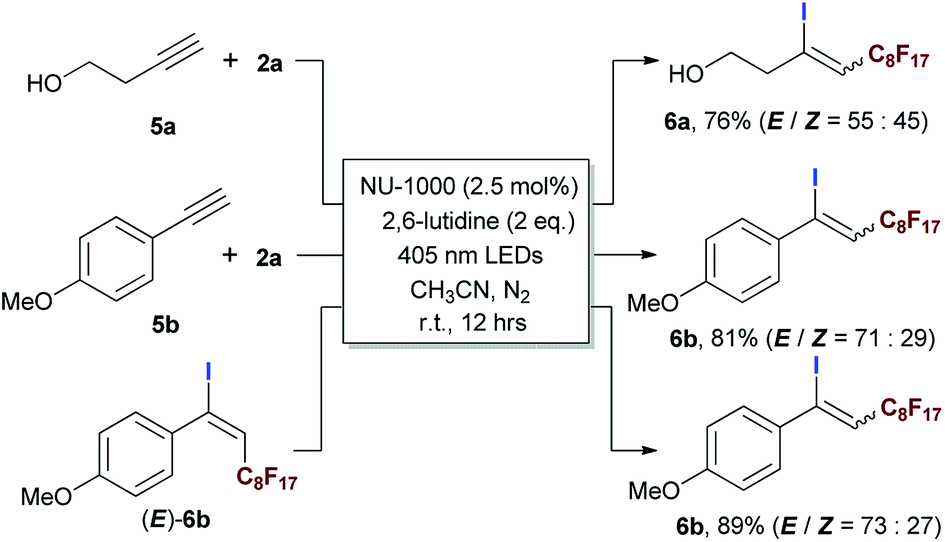 | ||
| Scheme 5 Evaluating the scope of the photocatalytic ATRA using aliphatic and aromatic terminal alkynes. Isolated yields. | ||
By merging photocatalytic ATRA of RfI onto styrene and subsequent elimination of HI together in one pot, we successfully developed a strategy for the highly stereoselective preparation of (Z)-perfluoroalkyl styrene derivatives 7 in moderate to good yields (Scheme 6). To the best of our knowledge, it was the first time to develop the direct transformation from styrene to (Z)-selective perfluoroalkyl styrene. The biointeresting fragment such as estrone derivative could be tolerated in this heterogeneous photocatalytic tandem reaction (7c), showing the potential value of this method in pharmaceutical field. The ATRA step was proven to be first stage in this one pot process, the ATRA product 3m was successfully isolated in a yield of 64% when ceasing the reaction at 4 h, and re-subjecting 3m into reaction condition steadily gave rise to the formation of desired 7a. The (E)-isomer of product could not be detected either from the reaction mixture of one pot conversion or that of stepwise process, implying that the (Z)-geometry product was rapidly formed once upon elimination of HI. In comparison, this reaction gave much lower efficiency and inferior stereoselectivity in the presence of free ligand H4TBAPy instead of using NU-1000 (Scheme 6 and 7c‡), showing the significance of structure of NU-1000 in conducting this tandem process and enriching the (Z)-isomers.
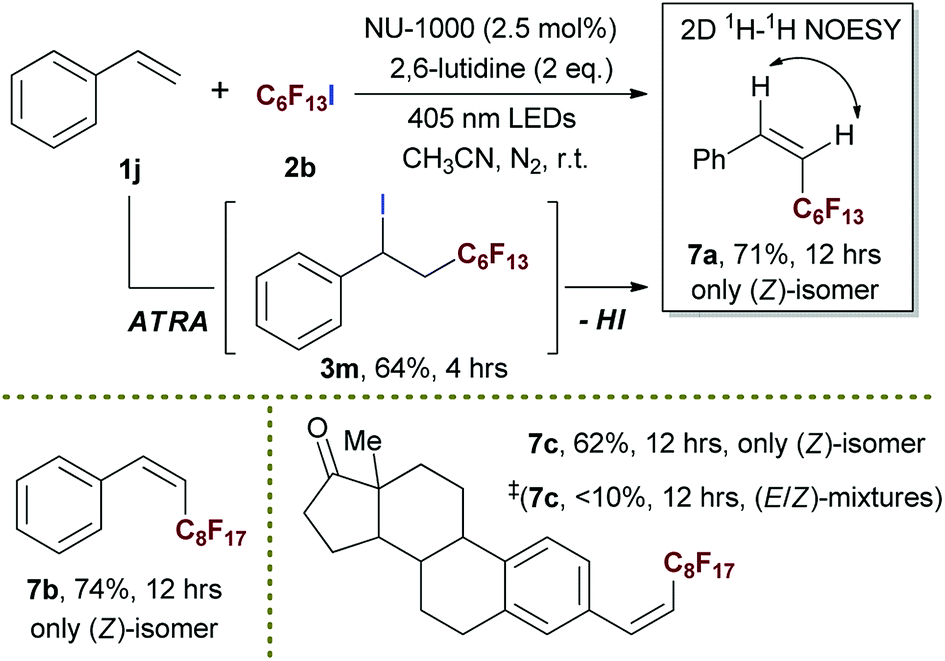 | ||
| Scheme 6 Highly stereoselective preparation of (Z)-perfluoroalkyl styrenes via tandem photocatalytic ATRA and subsequent HI elimination in one-pot. Isolated yields. ‡Using free ligand H4TBAPy as photocatalyst instead of MOF NU-1000, and the ratio of (E)/(Z)-isomer is not assignable due to the overlaps of peaks in NMR spectra. | ||
Based upon our experimental results and the previous experimental19,20 and computational52,61 works, a possible mechanism was outlined in Scheme 7. The singlet–singlet EnT from the excited-state NU-1000 to RfI triggered the homolytic cleavage of carbon–iodine bond of RfI to generate perfluoroalkyl and iodine radicals which were added towards the β- and α-positions of olefin, repectively, furnishing the ATRA reaction (Scheme 7, Pathway A). In the case of employing aromatic alkenes as substrates, the ATRA products easily underwent the base-assisted elimination of HI, then the obtained (E)/(Z)-isomeric mixtures were imposed with another EnT process from the triplet excited state of NU-1000,81 forging the (E)/(Z)-isomerisation to enrich the (Z)-isomer (Scheme 7, Pathway B).72,73
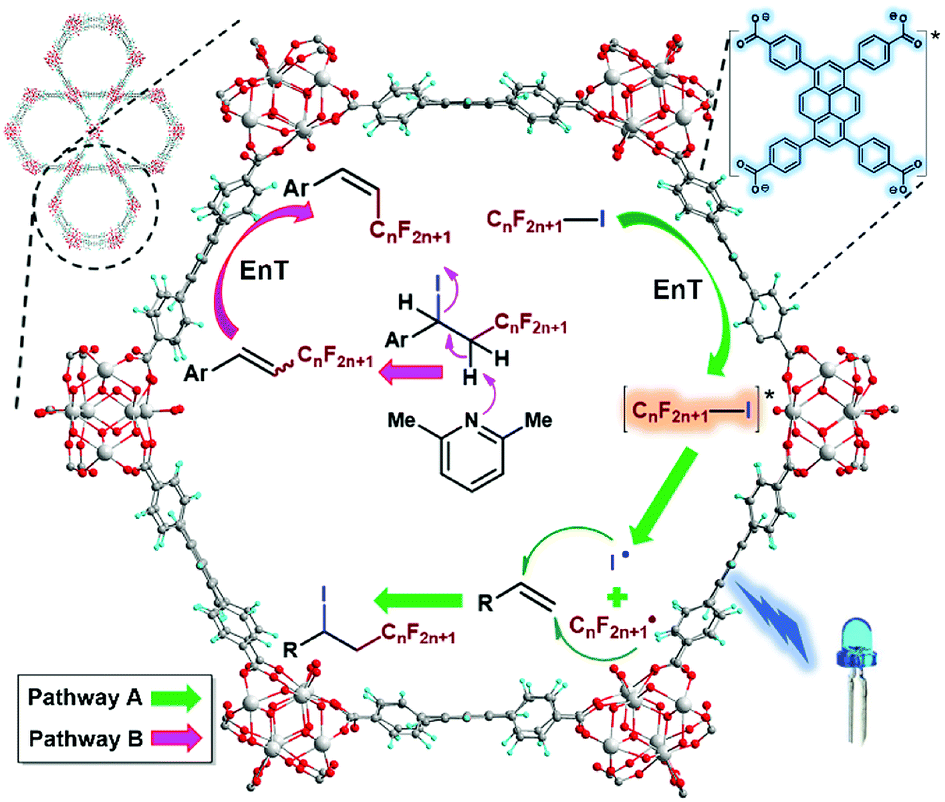 | ||
| Scheme 7 Proposed mechanisms of (Pathway A) photocatalytic ATRA reaction of perfluoroalkyl iodids onto olefins by using NU-1000, and (Pathway B) the formation of (Z)-enriched perfluoroalkyl alkene products. | ||
Experimental section
A. Materials and methods
All reagents were obtained from commercial sources and used without further purification. All the solvents involved were distilled before use. NU-1000 (ref. 21) and NU-901(ref. 22) were prepared, and activated according to literature procedures.NMR spectra were measured on a Bruker Avance 500 WB or Bruker Avance 400 WB spectrometers, and the relative chemical shifts were recorded in parts per million (ppm, δ). Powder X-ray diffractograms (PXRD) experiments were performed with PANalytical Empyrean X-ray powder diffractometer (Cu Kα radiation, 40 kV, 40 mA). Elemental analysis with inductively coupled plasma-atomic emission spectroscopy (ICP-MS) was measured on a Shimadzu ICPE-9000. FT-IR spectra were recorded as KBr pellets on JASCO FT/IR-430. High-resolution mass spectrum (EI) were recorded on a Micromass GC-TOF mass spectrometer.
Solid state cyclic voltammograms were conducted by using a carbon-paste working electrode; a well-ground mixture of each bulk sample and carbon paste (graphite and mineral oil) was set in the channel of a glass tube and connected to a copper wire. A platinum-wire counter electrode and an Ag/AgCl reference electrode were utilised. Measurements were conducted by using a three-electrode system in an aqueous solution of KNO3 (0.1 M) at a scan rate of 200 mV s−1.
The solid and liquid fluorescent spectrum were measured on an Edinburgh FS920 instrument. The excitation and emission slits were both 3 nm wide, and the wavelengths of excitation and emission were chosen according to specific cases. The time resolved luminescence spectrum were measured in an Edinburgh FLS920 spectrometer.
B. Experimental procedures and characterization of data
The reactions catalyzed by other kinds of (or other specified amounts of) pyrene-based heterogeneous/homogeneous species were conducted by similar manners.
1.2 equiv. of radical scavenger TEMPO (Scheme 3a) or triplet quencher (2,5-dimethylhexa-2,4-diene/pyridazine, Table 3) was also added to the reaction mixture besides the other components in the case of corresponding quenching experiments.
In case of the hot filtration experiment, the photocatalyst solid particles were filtered off by a syringe filter at 0.5 h under positive pressure of N2, and the filtrate was transferred to another pre-dried Pyrex tube, stirred, and irradiated by visible light of 405 nm LEDs under N2 atmosphere. The timely-monitoring of conversion of 1a was performed in the same way, as shown above.
7,7,8,8,9,9,10,10,11,12,12,13,13,14,14,14-Hexadecafluoro-5-iodo-11-methyltetradecan-1-ol (3a)82. This compound was synthesized according to the general procedure (GP) and isolated by column chromatography as sticky oil (150.1 mg, 93%) using petroleum ether/ethyl acetate (40
:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 4.39–4.29 (m, 1H), 3.69 (t, J = 6.0 Hz, 2H), 3.03–2.68 (m, 2H), 1.94–1.78 (m, 2H), 1.72–1.47 (m, 4H), 1.30 (br s, 1H). 13C NMR (126 MHz, CDCl3) δ 62.7, 41.9 (t, J = 20.9 Hz), 40.3 (d, J = 1.8 Hz), 31.8, 26.2, 20.6. 19F NMR (470 MHz, CDCl3) δ −80.77 (t, J = 9.8 Hz, 3F), −111.13 to −115.08 (m, 2F), −121.56 (virt. s, 2F), −121.91 (virt. s, 4F), −122.72 (virt. s, 2F), −123.57 (virt. s, 2F), −126.10 (virt. s, 2F).
7,7,8,8,9,9,10,10,11,11,12,12,12-Tridecafluoro-5-iodododecan-1-ol (3b)83. This compound was synthesized according to the general procedure (GP) and isolated by column chromatography as sticky oil (121.5 mg, 89%) using petroleum ether/ethyl acetate (40
:1 v/v) as the eluent system. 1H NMR (500 MHz, CDCl3) δ 4.34 (ddd, J = 13.4, 8.8, 5.0 Hz, 1H), 3.69 (t, J = 6.0 Hz, 2H), 3.01–2.86 (m, 1H), 2.79 (ddt, J = 31.3, 15.7, 7.9 Hz, 1H), 1.92–1.78 (m, 2H), 1.68–1.47 (m, 4H), 1.31 (br s, 1H). 13C NMR (126 MHz, CDCl3) δ 62.7, 41.9 (t, J = 20.8 Hz), 40.3 (d, J = 1.8 Hz), 31.8, 26.2, 20.6. 19F NMR (470 MHz, CDCl3) δ −79.47 to −82.07 (m, 3F), −110.36 to −115.76 (m, 2F), −121.75 (virt. s, 2F), −122.72 to −123.00 (m, 2F), −123.59 (virt. s, 2F), −126.05 to −126.27 (m, 2F).
7,7,8,8,9,9,10,10,10-Nonafluoro-5-iododecan-1-ol (3c)83. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (94.8 mg, 85%) using petroleum ether/ethyl acetate (40
:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 4.39–4.27 (m, 1H), 3.69 (t, J = 5.6 Hz, 2H), 2.95–2.71 (m, 2H), 1.95–1.77 (m, 2H), 1.71–1.47 (m, 4H), 1.33 (br s, 1H). 13C NMR (126 MHz, CDCl3) δ 62.7, 41.8 (t, J = 20.8 Hz), 40.3 (d, J = 2.1 Hz), 31.8, 26.2, 20.6. 19F NMR (470 MHz, CDCl3) δ −80.98 to −81.05 (m, 3F), −111.42 to −115.38 (m, 2F), −124.53 (virt. s, 2F), −125.75 to −126.05 (m, 2F).
8,8,8-Trifluoro-5-iodooctan-1-ol (3d)84. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (51.9 mg, 67%) using petroleum ether/ethyl acetate (40
:1 v/v) as the eluent system. 1H NMR (500 MHz, CDCl3) δ 4.07 (dq, J = 13.2, 4.4 Hz, 1H), 3.68 (t, J = 6.0 Hz, 2H), 2.48–2.34 (m, 1H), 2.28–2.14 (m, 1H), 2.12–1.98 (m, 2H), 1.98–1.89 (m, 1H), 1.75 (ddd, J = 14.2, 9.9, 5.3 Hz, 1H), 1.68–1.59 (m, 3H), 1.55–1.48 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 127.0 (d, J = 276.3 Hz), 62.8, 40.6, 35.7, 34.4 (q, J = 28.9 Hz), 33.0 (q, J = 2.9 Hz), 31.9, 26.0. 19F NMR (470 MHz, CDCl3) δ −65.89 (t, J = 10.6 Hz).
(5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-Heptadecafluoro-3-iodododecyl)benzene (3e)20. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (150.9 mg, 89% yield) using petroleum ether/ethyl acetate (100
:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 7.33–7.28 (m, 2H), 7.24–7.20 (m, 3H), 4.31–4.22 (m, 1H), 3.04–2.84 (m, 2H), 2.75 (m, 2H), 2.19–2.08 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 140.1, 128.8, 128.7, 126.6, 42.0 (d, J = 2.0 Hz), 41.9 (d, J = 21.1 Hz), 35.9, 20.3. 19F NMR (470 MHz, CDCl3) δ −80.77 (t, J = 9.9 Hz, 3F), −110.92 to −114.85 (m, 2F), −121.55 (virt. s, 2F), −121.90 (virt. s, 4F), −122.71 (virt. s, 2F), −123.58 (virt. s, 2F), −126.07 to −126.16 (virt. m, 2F).
(5,5,6,6,7,7,8,8,8-Nonafluoro-3-iodooctyl)benzene (3f)85. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (99.0 mg, 83% yield) using petroleum ether/ethyl acetate (100
:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 7.34–7.28 (m, 2H), 7.24–7.20 (m, 3H), 4.32–4.21 (m, 1H), 3.04–2.84 (m, 2H), 2.82–2.69 (m, 2H), 2.21–2.05 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 140.1, 128.8, 128.7, 126.6, 42.0 (d, J = 2.1 Hz), 41.8 (d, J = 20.4 Hz), 35.9, 20.2. 19F NMR (470 MHz, CDCl3) δ −80.16 to −81.82 (m, 3F), −110.43 to −116.34 (m, 2F), −123.72 to −125.38 (m, 2F), −125.38 to −126.67 (m, 2F).
((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-Nonadecafluoro-2-iodododecyl)oxy)benzene (3g). This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (137.5 mg, 81% yield) using petroleum ether/ethyl acetate (100
:1 v/v) as the eluent system. 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.9 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.92 (d, J = 7.9 Hz, 2H), 4.56–4.48 (m, 1H), 4.30 (dd, J = 10.3, 4.8 Hz, 1H), 4.18 (dd, J = 10.2, 6.9 Hz, 1H), 3.19 (ddd, J = 25.6, 16.2, 8.6 Hz, 1H), 2.80 (ddd, J = 33.5, 16.6, 8.7 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 157.9, 129.9, 122.1, 115.2, 73.0, 38.0 (t, J = 21.2 Hz), 13.0. 19F NMR (470 MHz, CDCl3) δ −80.79 (t, J = 9.9 Hz, 3F), −112.70 to −114.34 (m, 2F), −121.53 (virt. s, 2F), −121.88 (virt. s, 4F), −122.70 (virt. s, 2F), −123.51 (virt. s, 2F), −126.04 to −126.20 (m, 2F). HRMS (EI) for C17H10F17IO [M]+: calculated: 679.9505; found: 679.9504.
1-(2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-Heptadecafluorononyl)-4-(prop-1-en-2-yl)cyclohex-1-ene (4h) & 1-(2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-heptadecafluorononyl)-4-(propan-2-ylidene)cyclohex-1-ene (4h′). A pair of inseparable mixture of regioisomers (ca. 50
:50) was synthesized according to the GP and isolated by column chromatography as sticky oil (116.0 mg, 84% yield) using petroleum ether/ethyl acetate (100:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 5.71 (virt. s, 1H; 4h), 5.67 (virt. s, 1H; 4h′), 4.76–4.73 (m, 1H; 4h), 4.72 (virt. s, 1H; 4h), 2.82 (s, 2H), 2.79–2.65 (m, 4H), 2.34 (t, J = 6.2 Hz, 2H), 2.25–2.12 (m, 6H), 2.04–1.98 (m, 1H), 1.87–1.78 (m, 1H), 1.74 (s, 3H), 1.70 (s, 3H), 1.66 (s, 3H), 1.55–1.47 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 149.6, 130.03, 129.98, 127.1, 126.7, 126.3, 122.9, 109.0, 40.6, 39.0 (t, J = 21.9 Hz), 31.2, 30.9, 30.1, 30.0, 29.9, 27.9, 26.7, 21.0, 20.4, 20.0. 19F NMR (377 MHz, CDCl3) δ −80.96 (t, J = 10.0 Hz, 6F; overlapped), −111.27 to −113.80 (m, 4F; overlapped), −121.72 to −121.74 (m, 4F; overlapped), −122.02 (virt. s, 8F; overlapped), −122.83 (virt. s, 4F; overlapped), −123.30 (virt. s, 4F; overlapped), −126.24 (td, J = 14.1, 3.3 Hz, 4F; overlapped). HRMS (EI) for C18H15F17 [M]+: calculated: 554.0902; found: 554.0896.
Diethyl 3-(2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-heptadecafluorononyl)-4-(iodomethyl)cyclopentane-1,1-dicarboxylate (3i)4. A pair of diastereoisomers (d.r. = 86
:14) was synthesized according to the GP and isolated by column chromatography as sticky oil (169.0 mg, 86% yield) using petroleum ether/ethyl acetate (40:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 4.25–4.17 (m; overlapped 4H of minor diastereomer and 4H of major diastereomer), 3.35 (dd, J = 10.2, 3.8 Hz, 1H; minor), 3.17 (dd, J = 9.8, 5.6 Hz, 1H; major), 3.14–3.10 (m, 1H; minor), 3.05 (t, J = 9.7 Hz, 1H; major), 2.76 (q, J = 11.8 Hz, 1H; minor), 2.66–2.48 (m; overlapped 4H of major and 1H of minor), 2.35–1.99 (m; overlapped 4H of major and 6H of minor), 1.30–1.23 (m; overlapped 6H of major and 6H of minor). 13C NMR (101 MHz, CDCl3) δ 172.5 (major), 172.2 (major), 171.9 (minor), 62.1 (major), 62.04 (major), 62.01 (minor), 61.97 (minor), 58.5 (major), 58.3 (minor), 46.8 (minor), 45.6 (major), 41.1 (d, J = 2.7 Hz; minor), 40.7 (minor), 40.0 (major), 38.6 (d, J = 2.3 Hz; major), 37.9 (minor), 35.6 (major), 29.9 (t, J = 21.7 Hz; major), 14.20 (overlapped major and minor), 14.18 (overlapped major and minor), 8.56 (minor), 5.73 (major). 19F NMR (470 MHz, CDCl3) δ −81.52 (t, J = 9.9 Hz, 3F), −111.25 to −116.20 (m, 2F), −122.48 to −122.66 (m, partially overlapped 6F), −123.46 (virt. s, 2F), −124.34 (virt. s, 2F), −126.57 to −127.29 (m, 2F).
4-Bromo-8-hydroxyoctanenitrile (3j)19. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (29.4 mg, 53% yield) using petroleum ether/ethyl acetate (4
:1 v/v) as the eluent system. 1H NMR (500 MHz, CDCl3) δ 4.13–3.99 (m, 1H), 3.68 (t, J = 5.8 Hz, 2H), 2.68–2.56 (m, 2H), 2.23–2.16 (m, 1H), 2.12–2.06 (m, 1H), 1.96–1.85 (m, 2H), 1.72–1.52 (m, 5H). 13C NMR (126 MHz, CDCl3) δ 118.9, 62.73, 54.9, 38.8, 34.8, 32.1, 24.1, 16.3.
(E/Z)-5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,12-Heptadecafluoro-3-iodododec-3-en-1-ol (6a)20. The (E)- and (Z)-stereoisomer of 6a were synthesized according to the GP and purified by column chromatography as white solids (total 117.2 mg, 76% yield) using petroleum ether/ethyl acetate (10
:1 v/v) as the eluent system. ((E)-isomer, 64.3 mg, 42%) 1H NMR (500 MHz, CDCl3) δ 6.49 (t, J = 14.3 Hz, 1H), 3.87 (virt. s, 2H), 2.94 (t, J = 6.0 Hz, 2H), 1.49 (br. s, 1H). 13C NMR (126 MHz, CDCl3) δ 129.2 (t, J = 23.8 Hz), 117.3 (t, J = 6.2 Hz), 62.1, 43.8. 19F NMR (470 MHz, CDCl3) δ −80.77 (t, J = 9.9 Hz, 3F), −105.03 (q, J = 13.5 Hz, 2F), −121.44 (virt. s, 2F), −121.89 (virt. s, 4F), −122.72 (virt. s, 2F), −123.08 (virt. s, 2F), −126.07 to −126.18 (m, 2F). ((Z)-isomer, 52.9 mg, 34%) 1H NMR (500 MHz, CDCl3) δ 6.41 (t, J = 13.0 Hz, 1H), 3.85 (t, J = 5.8 Hz, 2H), 2.92 (t, J = 5.2 Hz, 2H), 1.55 (br. s, 1H). 13C NMR (126 MHz, CDCl3) δ 124.5 (t, J = 23.8 Hz), 111.8 (t, J = 6.6 Hz), 60.8, 51.2. 19F NMR (470 MHz, CDCl3) δ −80.82 (t, J = 9.9 Hz, 3F), −108.82 (q, J = 12.6 Hz, 2F), −121.49 (virt. s, 2F), −121.93 (virt. s, 4F), −122.77 (virt. s, 2F), −122.89 (virt. s, 2F), −126.13 to −126.19 (m, 2F).
1-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-Heptadecafluoro-1-iododec-1-en-1-yl)-4-methoxybenzene (6b)20. The (E)- and (Z)-stereoisomer of 6b were synthesized according to the GP and purified by column chromatography as white solids (total 137.5 mg, 81% yield) using petroleum ether/ethyl acetate (10
:1 v/v) as the eluent system. ((E)-isomer) 1H NMR (500 MHz, CDCl3) δ 7.26 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 6.55 (t, J = 13.5 Hz, 1H), 3.82 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 160.4, 133.8, 129.0, 126.6 (t, J = 21.6 Hz), 113.6, 55.5. 19F NMR (470 MHz, CDCl3) δ −80.75 (t, J = 9.9 Hz, 3F), −104.84 (q, J = 13.1 Hz, 2F), −121.47 (virt. s, 2F), −121.89 (virt. s, 4F), −122.72 (virt. s, 2F), −122.83 (virt. s, 2F), −126.06 to −126.15 (m, 2F). ((Z)-isomer could not be totally separated from the (E)-isomer, and its pure NMR spectrum were not obtained.)
(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluoro-1-iodooctyl)benzene (3m). This compound was synthesized according to the GP with a decreased reaction time of 4 h, and isolated by column chromatography as sticky oil (88.0 mg, 64% yield) using petroleum ether/ethyl acetate (100
:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 7.45–7.40 (m, 2H), 7.35–7.24 (m, 3H), 5.44 (dd, J = 9.7, 5.2 Hz, 1H), 3.39–3.08 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 143.0, 129.1, 128.8, 127.0, 42.8 (t, J = 20.6 Hz), 16.7. 19F NMR (470 MHz, CDCl3) δ −80.68 to −81.19 (m, 3F), −111.85 to −115.16 (m, 2F), −121.83 (virt. s, 2F), −122.75 to −123.13 (m, 2F), −123.58 (virt. s, 2F), −126.04 to −126.44 (m, 2F). HRMS (EI) for C14H8F13 [M−I]+: calculated: 423.0413; found: 423.0423.
(Z)-(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooct-1-en-1-yl)benzene (7a)69. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (74.6 mg, 71% yield) using petroleum ether as the eluent system. 1H NMR (500 MHz, CDCl3) δ 7.48 (dd, J = 6.4, 2.9 Hz, 2H), 7.40 (dd, J = 5.1, 1.7 Hz, 3H), 7.18 (d, J = 16.1 Hz, 1H), 6.20 (dt, J = 15.8, 12.2 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 141.06–138.87 (m), 134.86–133.04 (m), 130.43 (virt. s), 129.20 (virt. s), 127.88 (virt. s), 115.53–112.98 (m). 19F NMR (470 MHz, CDCl3) δ −80.79 (t, J = 10.0 Hz, 3F), −110.88 to −111.37 (m, 2F), −121.60 (virt. s, 2F), −122.86 (virt. s, 2F), −123.12 to −123.32 (m, 2F), −126.14 (ddd, J = 17.8, 10.8, 5.3 Hz, 2F).
(Z)-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-Heptadecafluorodec-1-en-1-yl)benzene (7b)86. This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (96.4 mg, 62% yield) using petroleum ether as the eluent system. 1H NMR (500 MHz, CDCl3) δ 7.51–7.45 (m, 2H), 7.41 (dd, J = 9.7, 5.5 Hz, 3H), 7.18 (d, J = 16.1 Hz, 1H), 6.20 (dd, J = 28.2, 12.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 140.0 (t, J = 9.4 Hz), 133.8, 130.4, 129.2, 127.9, 114.6 (t, J = 23.0 Hz). 19F NMR (377 MHz, CDCl3) δ −80.80 (t, J = 10.0 Hz, 3F), −111.09 (q, J = 12.5 Hz, 2F), −121.37 to −121.39 (m, 2F), −121.83 to −122.07 (m, 4F), −122.62 to −122.80 (m, 2F), −123.11 to −123.27 (m, 2F), −126.04 to −126.19 (m, 2F).
(Z)-3-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-Heptadecafluorodec-1-en-1-yl)-13-methyl-7,8,9,11,12,13,15,16-octahydro-6H-cyclopenta[a]phenanthren-17(14H)-one (7c). This compound was synthesized according to the GP and isolated by column chromatography as sticky oil (92.5 mg, 74% yield) using petroleum ether/ethyl acetate (40
:1 v/v) as the eluent system. 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.2 Hz, 1H), 7.27 (d, J = 7.8 Hz, 1H), 7.22 (s, 1H), 7.12 (td, J = 16.1 and 2.0 Hz, 1H), 6.16 (dd, J = 28.2, 12.3 Hz, 1H), 2.97–2.91 (m, 2H), 2.57–2.40 (m, 2H), 2.37–2.27 (m, 1H), 2.22–1.95 (m, 4H), 1.71–1.42 (m, 6H), 0.92 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 220.8, 142.6, 139.7 (t, J = 9.6 Hz), 137.5, 131.4, 128.5, 126.2, 125.2, 113.8 (t, J = 23.0 Hz), 50.8, 48.2, 44.8, 38.2, 36.0, 31.8, 29.5, 26.5, 25.9, 21.8, 14.0. 19F NMR (377 MHz, CDCl3) δ −80.78 (t, J = 9.9 Hz, 3F), −110.88 (q, J = 12.3 Hz, 2F), −121.33 (virt. s, 2F), −121.90 (virt. s, 4F), −122.70 (virt. s, 2F), −123.11 to −123.32 (m, 2F), −125.99 to −126.29 (m, 2F). HRMS (ESI) for C28H23F17O [M]+: calculated: 698.1477; found: 698.1470.
Conclusions
We reported a photocatalytic strategy which simultaneously integrated the perfluoroalkyl and iodo groups into olefins by using a pyrene-based metal–organic framework NU-1000 as heterogeneous photocatalyst under irradiation with LEDs (405 nm). The smoothly undergoing radical clock experiment and the detected formation of I2 as a by-product confirmed the in situ generation of perfluoroalkyl and iodine radicals, respectively, supporting the homolytic cleavage of carbon–iodine bond of radical precursor. The positive correlation between BDE of alkyl halide and conversion of ATRA reaction and the triplet quenching experiments supported the EnT process from the pyrene-based chromophore of MOF to RfI as a plausible driving force for the homolytic bond dissociation to generate the Rf and iodine radicals and trigger the ATRA process. The NU-1000 photocatalytic system was more efficient than that of homogeneous counterparts or other pyrene-based MOF with chromophores in closer π–π proximity, implying that either the uncontrollable thermal collisions in solution phase or the stronger inter-ligand interactions within framework impaired the effectiveness of photoinduced EnT from the pyrene-based chromophore to RfI. It is the first time to utilise the photoinduced EnT process of MOF in organic transformations other than photosensitising the oxygen. The competent visible-light harvesting ability of this system effectively prohibited the possible decomposition or over-reaction of products which were the common ailments of the high energetic UV light promoted reactions. When extending the substrate scope to styrene derivatives, the photocatalytic ATRA reaction of styrene derivatives was found to be concomitant with an HI elimination of ATRA product and an (E)/(Z)-isomerisation in one-pot, affording highly stereoselective (Z)-perfluoroalkyl styrenes, which was applicable to the biointeresting molecules like estrone derivative and might show the power in the late-stage modification of drug derivatives.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the financial support for this work from the National Natural Science Foundation of China (21402020, U1608224, and 21531001), the Fundamental Research Funds for the Central Universities (DUT16RC(4)09 and DUT18LK50), and the 111 Project (B16008).Notes and references
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC.
- O. Reiser, Acc. Chem. Res., 2016, 49, 1990 CrossRef PubMed.
- T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 CrossRef PubMed.
- T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, ACS Catal., 2018, 8, 3950 CrossRef.
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem.–Eur. J., 2012, 18, 7336 CrossRef PubMed.
- X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246 CrossRef PubMed.
- D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef PubMed.
- P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity, Applications, Wiley, New York, 2nd edn, 2013 Search PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef PubMed.
- E.g. CF3I, Ered = -1.52 V vs. SCE, see the reference, C. P. Andrieux, L. Gélis, M. Medebielle, J. Pinson and J.-M. Savéant, J. Am. Chem. Soc., 1990, 112, 3509 CrossRef.
- E.g. C8F17I, Ered = -1.32 V vs. SCE, see the reference, C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef PubMed.
- V. G. Koshechko and L. A. Kiprianova, Theor. Exp. Chem., 1999, 35, 18 CrossRef.
- Generally, the substantial overpotential as large as 1.0 V is necessary for the reduction cleavage of C-I bond of RfI, see also ref. 10.
- Y.-R. Luo, Handbook of bond dissociation energies in organic compounds, CRC Press LLC, 2003 Search PubMed.
- J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef PubMed.
- N. Iqbal, S. Choi, E. Kim and E. J. Cho, J. Org. Chem., 2012, 77, 11383 CrossRef PubMed.
- G. Magagnano, A. Gualandi, M. Marchini, L. Mengozzi, P. Ceroni and P. G. Cozzi, Chem. Commun., 2017, 53, 1591 RSC.
- T. Yajima and M. Ikegami, Eur. J. Org. Chem., 2017, 2126 CrossRef.
- E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064 CrossRef PubMed.
- R. Beniazza, R. Atkinson, C. Absalon, F. Castet, S. A. Denisov, N. D. McClenaghan, D. Lastécouères and J.-M. Vincent, Adv. Synth. Catal., 2016, 358, 2949 CrossRef.
- P. Riente and M. A. Pericàs, ChemSusChem, 2015, 8, 1841 CrossRef PubMed.
- O. A. Khakhel, J. Appl. Spectrosc., 2001, 68, 280 CrossRef.
- G. B. Ray, I. Chakraborty and S. P. Moulik, J. Colloid Interface Sci., 2006, 294, 248 CrossRef PubMed.
- J. Xiao, C. D. Malliakas, Y. Liu, F. Zhou, G. Li, H. Su, M. G. Kanatzidis, F. Wudl and Q. Zhang, Chem.–Asian J., 2012, 7, 672 CrossRef PubMed.
- G. Li, H. M. Duong, Z. Zhang, J. Xiao, L. Liu, Y. Zhao, H. Zhang, F. Huo, S. Li, J. Ma, F. Wudl and Q. Zhang, Chem. Commun., 2012, 48, 5974 RSC.
- G. Li, Y. Wu, J. Gao, J. Li, Y. Zhao and Q. Zhang, Chem.–Asian J., 2013, 8, 1574 CrossRef PubMed.
- A. Yildiz and C. N. Reilley, Spectrosc. Lett., 1968, 1, 335 CrossRef.
- M. A. Slifkin and A. O. Al-Chalabi, Chem. Phys. Lett., 1975, 31, 198 CrossRef.
- C. Rullière, E. C. Colson and P. C. Roberge, Can. J. Chem., 1975, 53, 3269 CrossRef.
- A. Abdel-Shafi, D. R. Worrall and F. Wilkinson, J. Photochem. Photobiol., A, 2001, 142, 133 CrossRef.
- T. Ogoshi, D. Yamafuji, T.-a. Yamagishia and A. M. Brouwer, Chem. Commun., 2013, 49, 5468 RSC.
- Y. Lu, J. Wang, N. McGoldrick, X. Cui, J. Zhao, C. Caverly, B. Twamley, G. M. Ó Máille, B. Irwin, R. Conway-Kenny and S. M. Draper, Angew. Chem., Int. Ed., 2016, 55, 14688 CrossRef PubMed.
- M. Marchini, G. Bergamini, P. Giorgio Cozzi, P. Ceroni and V. Balzani, Angew. Chem., Int. Ed., 2017, 56, 12820 CrossRef PubMed.
- G. Ulrich, C. Goze, M. Guardigli, A. Roda and R. Ziessel, Angew. Chem., Int. Ed., 2005, 44, 3694 CrossRef PubMed.
- M. Sasmal, R. Bhowmick, A. S. Musha Islam, S. Bhuiya, S. Das and M. Ali, ACS Omega, 2018, 3, 6293 CrossRef.
- P. Ensslen and H.-A. Wagenknecht, Acc. Chem. Res., 2015, 48, 2724 CrossRef PubMed.
- C. B. Winiger, S. Li, G. R. Kumar, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2014, 53, 13609 CrossRef PubMed.
- S. Cicchi, P. Fabbrizzi, G. Ghini, A. Brandi, P. Foggi, A. Marcelli, R. Righini and C. Botta, Chem.–Eur. J., 2009, 15, 754 CrossRef PubMed.
- S. A. Denisov, Q. Gan, X. Wang, L. Scarpantonio, Y. Ferrand, B. Kauffmann, G. Jonusauskas, I. Huc and N. D. McClenaghan, Angew. Chem., Int. Ed., 2016, 55, 1328 CrossRef PubMed.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352 CrossRef PubMed.
- L. Zeng, X. Guo, C. He and C. Duan, ACS Catal., 2016, 6, 7935 CrossRef.
- K. C. Stylianou, R. Heck, S. Y. Chong, J. Bacsa, J. T. A. Jones, Y. Z. Khimyak, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2010, 132, 4119 CrossRef PubMed.
- K. C. Stylianou, J. Rabone, S. Y. Chong, R. Heck, J. Armstrong, P. V. Wiper, K. E. Jelfs, S. Zlatogorsky, J. Bacsa, A. G. McLennan, C. P. Ireland, Y. Z. Khimyak, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, J. Am. Chem. Soc., 2012, 134, 20466 CrossRef PubMed.
- R.-J. Li, M. Li, X.-P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047 RSC.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294 CrossRef PubMed.
- Y. Liu, C. T. Buru, A. J. Howarth, J. J. Mahle, J. H. Buchanan, J. B. DeCoste, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 13809 RSC.
- A. Atilgan, T. Islamoglu, A. J. Howarth, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 24555 CrossRef PubMed.
- A. J. Howarth, C. T. Buru, Y. Liu, A. M. Ploskonka, K. J. Hartlieb, M. McEntee, J. J. Mahle, J. H. Buchanan, E. M. Durke, S. S. Al-Juaid, J. F. Stoddart, J. B. DeCoste, J. T. Hupp and O. K. Farha, Chem.–Eur. J., 2017, 23, 214 CrossRef PubMed.
- K. C. Park, C. Seo, G. Gupta, J. Kim and C. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 38670 CrossRef PubMed.
- C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012 CrossRef.
- H. Noh, C.-W. Kung, T. Islamoglu, A. W. Peters, Y. Liao, P. Li, S. J. Garibay, X. Zhang, M. R. DeStefano, J. T. Hupp and O. K. Farha, Chem. Mater., 2018, 30, 2193 CrossRef.
- P. Deria, J. Yu, T. Smith and R. P. Balaraman, J. Am. Chem. Soc., 2017, 139, 5973 CrossRef PubMed.
- Y. Xu, M. Fletcher and W. R. Dolbier Jr, J. Org. Chem., 2000, 65, 3460 CrossRef PubMed.
- C. L. Wong and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 5593 CrossRef.
- J. K. Kochi, Acc. Chem. Res., 1992, 25, 39 CrossRef.
- R. S. Miller, J. M. Sealy, M. Shabangi, M. L. Kuhlman, J. R. Fuchs and R. A. Flowers, J. Am. Chem. Soc., 2000, 122, 7718 CrossRef.
- A. A. Isse and A. Gennaro, J. Phys. Chem. A, 2004, 108, 4180 CrossRef.
- D. A. Lutterman, N. N. Degtyareva, D. H. Johnston, J. C. Gallucci, J. L. Eglin and C. Turro, Inorg. Chem., 2005, 44, 5388 CrossRef PubMed.
- A. A. Isse, C. Y. Lin, M. L. Coote and A. Gennaro, J. Phys. Chem. B, 2011, 115, 678 CrossRef PubMed.
- A. Haim, Acc. Chem. Res., 1975, 8, 264 CrossRef.
- J. Yu, J. H. Park, A. Van Wyk, G. Rumbles and P. Deria, J. Am. Chem. Soc., 2018, 140, 10488 CrossRef PubMed.
- M. T. Indelli, M. Ghirotti, A. Prodi, C. Chiorboli, F. Scandola, N. D. McClenaghan, F. Puntoriero and S. Campagna, Inorg. Chem., 2003, 42, 5489 CrossRef PubMed.
- C. V. Kumar and M. R. Duff Jr, Photochem. Photobiol. Sci., 2008, 7, 1522 RSC.
- A. Chaudhary and S. P. Rath, Chem.–Eur. J., 2011, 17, 11478 CrossRef PubMed.
- Y. Ishida, R. Kulasekharan, T. Shimada, S. Takagi and V. Ramamurthy, Langmuir, 2013, 29, 1748 CrossRef PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef.
- The experimental and theoretical data of triplet excited-state energies of 1,3,6,8-tetraphenylpyrene were reported, see ref. 22.
- N. I. Nijegorodov and W. S. Downey, J. Phys. Chem., 1994, 98, 5639 CrossRef.
- N. J. W. Straathof, S. E. Cramer, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2016, 55, 15549 CrossRef PubMed.
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., 2014, 126, 549 CrossRef.
- T. Xu, C. W. Cheung and X. Hu, Angew. Chem., Int. Ed., 2014, 53, 4910 CrossRef PubMed.
- J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254 CrossRef PubMed.
- J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040 CrossRef PubMed.
- K. A. Muszkat, D. Gegiou and E. Fischer, J. Am. Chem. Soc., 1967, 89, 4814 CrossRef.
- J. Saltiel, D. W. L. Chang and E. D. Megarity, J. Am. Chem. Soc., 1974, 96, 6521 CrossRef.
- W. I. Ferree Jr, B. F. Plummer and W. W. Schloman Jr, J. Am. Chem. Soc., 1974, 96, 7741 CrossRef.
- A. R. Gutierrez and D. G. Whitten, J. Am. Chem. Soc., 1974, 96, 7128 CrossRef.
- J. Saltiel, A. Marinari, D. W. L. Chang, J. C. Mitchener and E. D. Megarity, J. Am. Chem. Soc., 1979, 101, 2982 CrossRef.
- T. Konno, J. Chae, T. Tanaka, T. Ishihara and H. Yamanaka, J. Fluorine Chem., 2006, 127, 36 CrossRef.
- Y. Zhao, Y. Zhou, C. Zhang, H. Wang, J. Zhao, K. Jin, J. Liu, J. Liu and J. Qu, Org. Biomol. Chem., 2017, 15, 5693 RSC.
- According to the report of Gilmour and co-worker (ref.72 and 73), the efficacy of (E)/(Z)-isomerization of β-substituted styrene derivatives has obvious correlation with the triplet state energy (ET) of photocatalysts, and the photocatalysts with ET in a range from 179 kJ mol−1 to 255 kJ mol−1 could give rise to highly (Z)-selective isomerization.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef PubMed.
- A. Kohlmeier and D. Janietz, Chem. Mater., 2006, 18, 59 CrossRef.
- T. Baba, T. Takagi and T. Kanamori, Jpn. Kokai Tokkyo Koho, JP2009209251A, 2009.
- D. P. Tiwari, S. Dabral, J. Wen, J. Wiesenthal, S. Terhorst and C. Bolm, Org. Lett., 2017, 19, 4295 CrossRef PubMed.
- J. Feng and C. Cai, J. Fluorine Chem., 2013, 146, 6 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06181e |
| This journal is © The Royal Society of Chemistry 2018 |