
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocked nucleic acid modified primer-based enzymatic polymerization assay: towards allele-specific genotype detection of human platelet antigens†
Bao T. Le ab,
Quintin Hughesc,
Shilpa Rakeshc,
Ross Bakerc,
Per T. Jørgensend,
Jesper Wengel*d and
Rakesh N. Veedu*abd
ab,
Quintin Hughesc,
Shilpa Rakeshc,
Ross Bakerc,
Per T. Jørgensend,
Jesper Wengel*d and
Rakesh N. Veedu*abd
aCentre for Comparative Genomics, Murdoch University, Perth, Australia-6150
bPerron Institute for Neurological and Translational Science, Perth, Australia-6009
cPerth Blood Institute, Nedlands, Perth, WA, Australia
dBioNEC, Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Odense M, 5231, Denmark. E-mail: R.Veedu@murdoch.edu.au; jwe@sdu.dk
First published on 21st September 2018
Abstract
Accurate detection of single nucleotide polymorphisms (SNPs) is paramount for the appropriate therapeutic intervention of debilitating diseases associated with SNPs. However, in some cases current nucleic acid probes fail to detect allele-specific mutations, for example, human platelet antigens, HPA-15a (TCC) and HPA-15b (TAC) alleles associated with neonatal alloimmune thrombocytopenia. Towards this, it is necessary to develop a novel assay for detection of allele-specific mutations. In this study, we investigated the potential of unlocked nucleic acid (UNA)-modified primers in SNP detection utilising an enzymatic polymerisation-based approach. Our results of primer extension and asymmetric polymerase chain reaction by KOD XL DNA polymerase revealed that UNA-modified primers achieved excellent allele-specificity in discriminating the human platelet antigen DNA template, whereas the DNA control primers were not able to differentiate between the normal and mutant alleles, demonstrating the scope of this novel UNA-based enzymatic approach as a robust methodology for efficient detection of allele-specific mismatches. Although further evaluation is required for other disease conditions, we firmly believe that our findings offer a great promise for the diagnosis of neonatal alloimmune thrombocytopenia and other SNP-related diseases.
Introduction
Chemically-modified nucleic acid analogues have drawn significant interest for the development of novel therapeutic and diagnostic approaches towards combatting diseases. A number of chemically-modified nucleic acid analogues with excellent physicochemical properties have been reported in the last two decades, allowing the construction of oligonucleotides with high resistance to nuclease degradation, and increased binding affinity to complementary DNA or RNA targets compared to natural nucleotide monomers.1,2 Unlocked nucleic acid (UNA) nucleotide has recently emerged as a useful chemical modification with potential applicability in both diagnostics and therapeutic development.3 UNA is considered to be an RNA mimic in which the C2′ and C3′ bond is cleaved in the ribose ring (Fig. 1). Structurally, UNA is highly flexible4,5 unlike other conformationally constrained analogues such as locked nucleic acid (LNA).6,7 Previous studies demonstrated the potential of UNA in siRNA-based gene-silencing applications by rendering high silencing potency, reduced off-target effects and high resistance to nuclease degradation.8 The scope of UNA nucleotides was also investigated in aptamer development by selective incorporation of UNA nucleotides mainly into the loops of known aptamers that showed improvements in target binding affinity.9,10 In addition, UNA nucleotides can also be recognised by polymerases, offering great potential in developing novel polymerase-based diagnostic assays involving UNA.11 | ||
| Fig. 1 Structural representations of RNA, UNA and LNA nucleotide monomers. | ||
Single nucleotide polymorphisms (SNPs) refer to a specific genetic variation occurring in the whole genome, and there are several genetic disorders associated with SNPs.12 Thus, SNPs detection or mismatch discrimination technology plays a key role in identifying the root cause of such diseases for the appropriate therapeutic interventions. There are several approaches available currently to detect SNPs such as primer extension, hybridization, ligation, and enzymatic cleavage-based assays.13 Nonetheless, in some cases, the existing technologies involving natural nucleotide monomers (including TaqMan™ probes) fail miserably. To improve the efficiency, an effective method must be developed. One approach is to incorporate novel chemically-modified nucleotides into detection probe to enhance binding affinity and mismatch recognition capabilities. Towards this, LNA, 2′-O-methyl, peptide nucleic acid (PNA) and UNA have been utilised and reported previously, but these studies were based on non-enzymatic approach.14–16 Herein we report, for the first time, an efficient enzymatic primer-extension approach for mismatch recognition using UNA-modified primers towards allele-specific SNPs detection.
Results and discussion
First, we designed and synthesised a set of 19-mer UNA-modified 5′-FAM (6-carboxyfluorescein)-labelled primers and a set of 57-mer DNA templates for initial primer extension experiments. Primers included P1 – DNA primer composed of natural nucleotides (Fig. 2A), P2-UNA-A5 containing a UNA-A nucleotide at position 5 (in adjacent to 3′-end, Fig. 2B), and P3-UNA-C7 containing a UNA-C nucleotide at position 7 (in adjacent to 3′-end, Fig. 2C). A fully matched DNA template T1, and two DNA templates with mismatches (‘A’ instead of ‘T’) located at position 15 (T2) and position 17 (T3) from the 3′-end were also synthesised (Table S1†). It is worth noting that P2-UNA-A5 was designed to fully anneal with template T1, while template T2 containing ‘A’ mutation that directly opposite to the UNA-A nucleotide of the primer when annealed (Fig. 2B). In addition, template T3 was designed with an ‘A’ mutation at position 17 from the 3′-end, i.e. one nucleotide away from the UNA-A nucleotide and closer to the 3′-end of P2-UNA-A5 upon annealing. | ||
| Fig. 2 Primer extension assay of (A) P1 annealed to templates T1, T2 and T3; (B) P2-UNA-A5 annealed to templates T1, T2 and T3; (C) P3-UNA-C7 annealed to templates T1, T2 and T3; (D) gel images of extension products from (A), (B) and (C) respectively; lane 1: P1 alone, lane 2: P2-UNA-A5 alone, lane 3: P3-UNA-C7 alone. UNA nucleotides are represented in underlined letters with superscript “U”; mismatched nucleotides are represented in lower-case underlined italic letters. All primers are labelled with 5′-FAM. Full sequence of template T1, T2 and T3 are shown in ESI Table S1.† Gel images were cropped and demonstrated as (D), for original gel images please see ESI Fig. S1.† | ||
Primer extension assays were performed to assess the ability of different polymerases to read through the UNA nucleotides and recognise mismatches. Initially, we tested three different enzymes including KOD XL DNA polymerase, Q5® High-Fidelity DNA polymerase and Platinum Taq DNA polymerase. Briefly, the primer was annealed to the template and then subjected to the polymerases-based extension step by following the manufacturer recommendations. Samples were subsequently removed from the reaction at 30 minutes time point, and the products were analysed by denaturing polyacrylamide gel electrophoresis (PAGE).
The results showed that only KOD XL polymerase was able to extend the UNA nucleotides efficiently. In the case of DNA primer P1, it is not surprising that the primer yielded full-length extension products when annealed to both matched and mismatched templates T1, T2, T3 (Fig. 2D, lanes 4, 5 and 6) due to poor efficiency of DNA nucleotides in recognising mismatches.17 But, the primer P2-UNA-A5 containing a UNA-A did not yield any extension product when annealed to the templates T2 and T3 that contain mismatch “A:A” at either directly opposite or one nucleotide away from UNA-A nucleotide of P2-UNA-A5, respectively (Fig. 2B and D, lanes 8 and 9). When annealed to a fully matched template T1, as expected, P2-UNA-A5 successfully yielded the full-length extension product (Fig. 2D, lane 7). These initial results clearly suggested that KOD XL DNA polymerase and UNA-modified primers could yield efficient mismatch discrimination.
Next, we investigated the effect of UNA nucleotide positioning in the primer. P3-UNA-C7 was designed for this purpose by incorporating an UNA-C nucleotide at position 7 (in adjacent to 3′-end), and performed the same experiment as above. Once annealed to the templates T2 and T3 (Fig. 2C), the mismatches (“A:A”) were positioned one and two nucleotides away from the 3′-end of the primer, respectively. The experiment revealed that P3-UNA-C7 did not yield any extension product suggesting that the change in positioning of UNA nucleotide did not impact the mismatch discrimination potential (Fig. 2D, lanes 11 and 12). Not surprisingly, P3-UNA-C7 yielded the full-length extension product when annealed to the matched template T1 (Fig. 2D, lane 10). It was also noted that the non-elongated primers were not homogenous and exhibited different bands. We speculate that this is due to the exonuclease activity of KOD XL polymerase enzyme, in line with previous report.18
Encouraged by the initial results, we further evaluated the efficacy of our UNA-modified primer-based enzymatic assay to detect mutations using a disease relevant DNA template specific to neonatal alloimmune thrombocytopenia (NAIT). NAIT is a serious complication affecting new-borns who express human platelet antigens (HPAs) inherited from the father which reacts with maternally-derived antibodies that can result in severe haemorrhage, particularly intracranial haemorrhage, and death.19,20 Early and rapid diagnosis of NAIT is essential for therapeutic intervention. To confirm NAIT, HPAs genotyping is often performed for both the maternal and neonate, and the current gold standard for HPA genotyping utilises TaqMan™ probes to differentiate between allele-specific variants for all common HPA genotypes, except HPA-15. This is because the significant degree of cross-reactivity between the two TaqMan™ probes designed to detect the HPA-15a and HPA-15b genotypes. Therefore, an alternative approach that could overcome this problem would be of great interest to help save the life of new-borns suffering NAIT. Thus, we evaluated UNA primer-based enzymatic approach for the detection of HPA-15a (TCC) and HPA-15b (TAC) genotypes (c.2108A>C, SNP ID: rs10455097).21
First, we designed and synthesised two 87-mer DNA templates T4 and T5 mimicking the HPA-15b and HPA-15a allele-specific variants (with ‘A’ nucleotide and ‘C’ nucleotide at position 19 from 3′-end, respectively).22 To differentiate between the “C” and the “A” alleles of the two genotypes, we then designed and synthesised two 21-mer DNA control primers, P4 and P5 that complement to templates T4 and T5 (Fig. 3A and C), respectively. In parallel, six UNA-modified primers containing one UNA nucleotide systematically positioned at 7, 6, 5, 4, 3 and 2 in relative to the 3′-end of the primers (P6-UNA-C7–P11-UNA-A2; Fig. 3A and C) were also constructed. Primer extension experiments were performed using KOD XL polymerase as described above but with 45 minutes of incubation time. It was also noted that all UNA-modified primers were designed to be fully complement to template T4 (containing “A” allele), and therefore, full-length extension products should be observed. On the other hand, P4 should not yield any product with T4 but T5 (containing “C” allele). Likewise, all UNA-modified primers should not show any full-length extension products upon annealing to T5, as it contains a ‘T/U’ nucleotide that miss-match to “C” allele of T5.
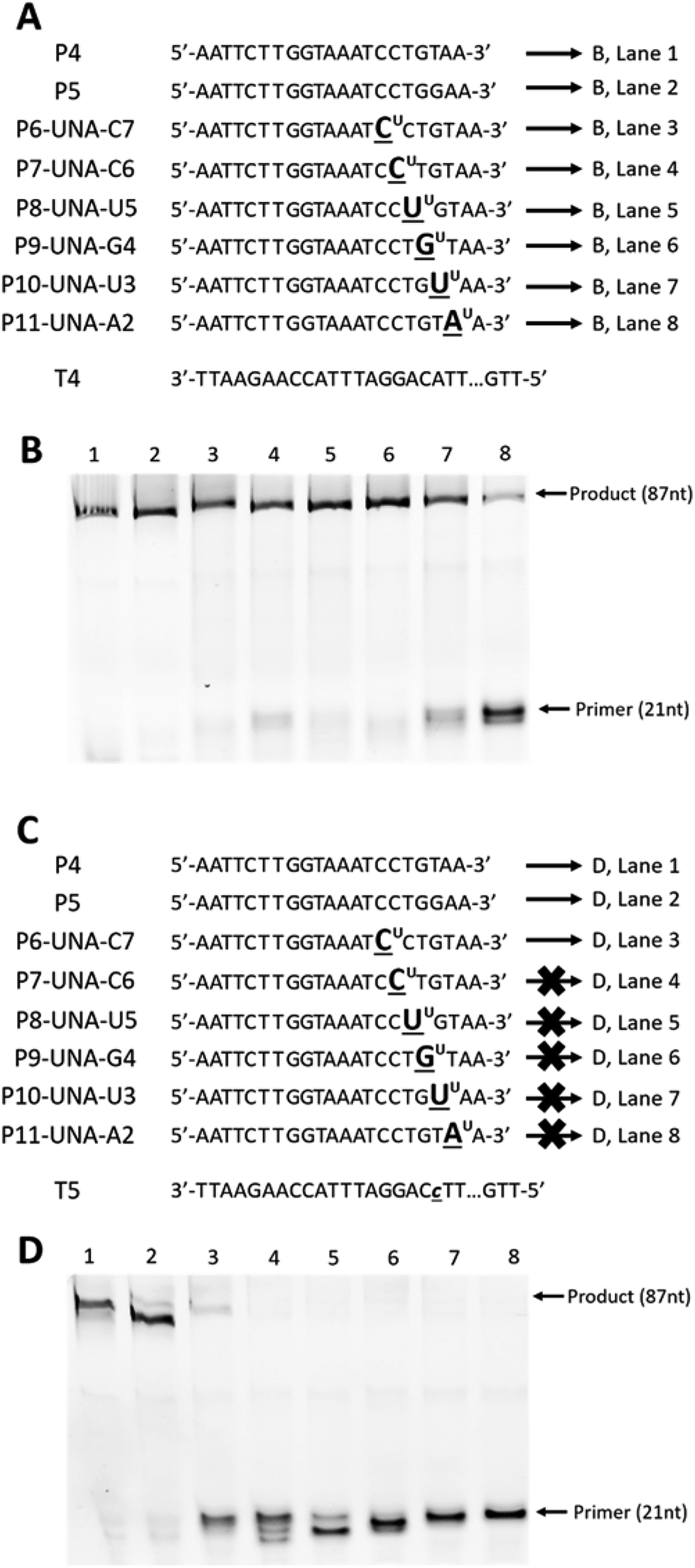 | ||
| Fig. 3 Primer extension assays of (A): DNA (P4 and P5) and UNA-modified primers (P6-UNA-C7–P11-UNA-A2) annealed to template T4; (C) DNA (P4 and P5) and UNA-modified primers (P6-UNA-C7–P11-UNA-A2) annealed to template T5; (B) and (D) gel images from (A) and (C), respectively. UNA nucleotides are represented in underlined letters with superscript “U”; mismatched nucleotides are represented in lower-case underlined italic letters. All primers are labelled with 5′-FAM. Full sequence of template T4 and T5 are shown in ESI Table S1.† | ||
The results showed that all primers yielded full-length extension products (87 nt) when annealed to template T4 (Fig. 3B, lane 1–8). Not surprisingly, P5, which designed to discriminate the “A” mutation, could not achieve the mismatch recognition due to the poor capability of the natural nucleotide monomer (Fig. 3B, lane 2). In contrast, most UNA-modified primers, when annealed to template T5 (containing “C” allele), did not yield any full-length 87-mer extension products, demonstrating the potential of UNA nucleotides and KOD XL polymerase for excellent mismatch recognition ability (Fig. 3D, lanes 4–8).
It is worth mentioning that P6-UNA-C7 primer yielded the extension product in very low yield which may be due to the positioning of the UNA nucleotide that is six nucleotides away from the 3′-end of the primer. Not surprisingly, both the control DNA primers P4 and P5 yielded full-length products underlining their poor mismatch discrimination ability (Fig. 3D, lanes 1 and 2).
Next, we utilised asymmetric polymerase chain reaction (PCR) to further evaluate our UNA primer-based enzymatic assay for allele-specific mismatch recognition. This assay was similar to primer extension, but with repeating cycles of denaturation, annealing, and extension. Briefly, PCR was performed using KOD XL polymerase in 20 cycles of denaturation at 94 °C for 30 seconds, annealing at 50 °C for 10 seconds, extension at 72 °C for 30 seconds. The experimental design using templates T4, T5 and the DNA (P4 and P5) and UNA primers (P6-UNA-C7–P1-UNA-A2) as above. Hence, we expected the full-length 87-mer products in the case of T4 extension whereas T5 ideally should not yield any full-length extension products due to the “C” mutation.
The experiment revealed that KOD XL DNA polymerase yielded full-length extension products in all reactions where the primers annealed to the fully-matched template T4 (Fig. 4A, lanes 1–8). In line with previous observations, the DNA primer P4 containing mismatch also yielded the full-length product (Fig. 4A, lane 2). Remarkably, the UNA-modified primers did not generate any products when tested with mismatched template T5, demonstrating its high efficiency for allele-specific recognition (Fig. 4B; lanes 3–8). On the other hand, the DNA primers (P4 and P5) failed to achieve the mismatch recognition as full-length products were found in both cases (Fig. 4B, lanes 1 and 2).
 | ||
| Fig. 4 Asymmetric PCR assays using DNA and UNA-modified primers. (A): template T4 and (B): template T5. For both (A and B): lane 1: P4; lane 2: P5; lane 3: P6-UNA-C7; lane 4: P7-UNA-C6; lane 5: P8-UNA-U5; lane 6: P9-UNA-G4; lane 7: P10-UNA-U3; lane 8: P11-UNA-A2. | ||
Experimental
Design and synthesis of UNA-modified primers
All UNA-modified primers were prepared in-house on an Expedite 8909 DNA synthesizer via standard phosphoramidite chemistry in 1 μmol scale. Synthesised oligonucleotides were deprotected and cleaved from the solid support by treatment with NH4OH at 55 °C overnight. The crude oligonucleotides were then purified, desalted and verified by MALDI-ToF MS analysis (ESI Fig. S2–S9†). All the templates and DNA primers were obtained from Integrated DNA Technologies (Iowa, USA).General procedure for primer extension assay
Primer extension assay was performed by following a 2-step procedure. Firstly, 5′-FAM (6-carboxyfluorescein)-labelled primers were annealed to the templates by mixing the primer and template at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (primer:template) plus 5× TSC buffer and H2O. The mixture was then heated to 82 °C for 4 min, followed by slow cooling to 37 °C and subjected to the extension step. Briefly, the extension reaction was set up as followed: 1× reaction buffer (specific to each polymerase), 2.5 mM MgCl2, 5 mM dNTPs, 1:10 (v/v) of the annealed primer–template complex, KOD XL DNA polymerase (Merck Millipore, Victoria, Australia) (amount as recommended by the manufacturer) and top up with H2O to reach 25 μL. The reaction was then gently vortexed and incubated at 74 °C. Samples were subsequently removed at 30 or 45 minutes, quenched by added with formamide loading buffer and run on 15% denaturing polyacrylamide gel in Tris–borate–EDTA buffer (13 W for 30 minutes). Results were visualized on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). All reagents were purchased form Sigma Aldrich, New South Wales, Australia unless specify.
:2 (primer:template) plus 5× TSC buffer and H2O. The mixture was then heated to 82 °C for 4 min, followed by slow cooling to 37 °C and subjected to the extension step. Briefly, the extension reaction was set up as followed: 1× reaction buffer (specific to each polymerase), 2.5 mM MgCl2, 5 mM dNTPs, 1:10 (v/v) of the annealed primer–template complex, KOD XL DNA polymerase (Merck Millipore, Victoria, Australia) (amount as recommended by the manufacturer) and top up with H2O to reach 25 μL. The reaction was then gently vortexed and incubated at 74 °C. Samples were subsequently removed at 30 or 45 minutes, quenched by added with formamide loading buffer and run on 15% denaturing polyacrylamide gel in Tris–borate–EDTA buffer (13 W for 30 minutes). Results were visualized on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). All reagents were purchased form Sigma Aldrich, New South Wales, Australia unless specify.
Asymmetric polymerase chain reaction
The asymmetric PCR reaction was prepared in a total volume of 25 μL containing 1× reaction buffer, 50 μM of dNTPs, 5 μM of primer, 4 μM of template and 1.5 U of KOD XL DNA polymerase. The mixture was gently vortexed and then amplified for 20 cycles of denaturation at 94 °C for 30 seconds, annealing at 50 °C for 10 seconds, extension at 72 °C for 30 seconds using Bio-Rad S-1000 thermal cycler (Hercules, California, United States). PCR products were then run on a 15% denaturing polyacrylamide gel in Tris–borate–EDTA buffer (13 W for 30 minutes). Results were visualized on a Fusion Fx gel documentation system.Conclusions
In summary, we have developed a novel and robust enzymatic allele-specific mismatch recognition methodology by performing primer-extension and asymmetric-PCR assays using UNA-modified primer oligonucleotides. The efficacy of UNA-modified primers was evaluated using synthetic templates based on the gene sequences of human platelet antigens, i.e. HPA-15a (TCC) and HPA-15b (TAC) alleles associated with NAIT. UNA-modified primers showed excellent allele-specificity whereas the corresponding DNA-primers failed to display allele-specificity in the enzymatic extension assays using KOD XL DNA polymerase. Although additional detailed evaluation of this assay for other disease conditions are necessary for further validation, we firmly believe that our findings offer a great potential in the diagnosis of SNPs-related diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RNV acknowledges funding from the McCusker Foundation and Perron Institute for Neurological and Translational Science. JW acknowledges the funding support from the WILLUM Foundation for funding the Biomolecular Nanoscale Engineering Center (BioNEC), grant number VKR022710. BTL thanks the MIPS funding scheme of Murdoch University.References
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef PubMed.
- T. L. Bao, R. N. Veedu, S. Fletcher and S. D. Wilton, Expert Opin. Orphan Drugs, 2015, 4, 139–152 CrossRef.
- A. Pasternak and J. Wengel, Org. Biomol. Chem., 2011, 9, 3591–3597 RSC.
- N. Langkjær, A. Pasternak and J. Wengel, Bioorg. Med. Chem., 2009, 17, 5420–5425 CrossRef PubMed.
- M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680–5689 RSC.
- R. N. Veedu and J. Wengel, Chem. Biodiversity, 2010, 7, 536–542 CrossRef PubMed.
- R. N. Veedu and J. Wengel, RNA Biol., 2009, 6, 321–323 CrossRef PubMed.
- M. B. Laursen, M. M. Pakula, S. Gao, K. Fluiter, O. R. Mook, F. Baas, N. Langkjær, S. L. Wengel, J. Wengel, J. Kjems and J. B. Bramsen, Mol. BioSyst., 2010, 6, 862–870 RSC.
- A. Pasternak, F. J. Hernandez, L. M. Rasmussen, B. Vester and J. Wengel, Nucleic Acids Res., 2011, 39, 1155–1164 CrossRef PubMed.
- L. J. Aaldering, V. Poongavanam, N. Langkjær, N. A. Murugan, P. T. Jørgensen, J. Wengel and R. N. Veedu, ChemBioChem, 2017, 18, 755–763 CrossRef PubMed.
- C. Dubois, M. A. Campbell, S. L. Edwards, J. Wengel and R. N. Veedu, Chem. Commun., 2012, 48, 5503–5505 RSC.
- R. Chaudhary, B. Singh, M. Kumar, S. K. Gakhar, A. K. Saini, V. S. Parmar and A. K. Chhillar, Drug Metab. Rev., 2015, 47, 281–290 CrossRef PubMed.
- S. Kim and A. Misra, Annu. Rev. Biomed. Eng., 2007, 9, 289–320 CrossRef PubMed.
- S. Fontenete, J. Barros, P. Madureira, C. Figueiredo, J. Wengel and N. F. Azevedo, Appl. Microbiol. Biotechnol., 2015, 99, 3961–3969 CrossRef PubMed.
- Y. You, B. G. Moreira, M. A. Behlke and R. Owczarzy, Nucleic Acids Res., 2006, 34, e60 CrossRef PubMed.
- R. Owczarzy, Y. You, C. L. Groth and A. V. Tataurov, Biochemistry, 2011, 50, 9352–9367 CrossRef PubMed.
- K. Kubota, A. Ohashi, H. Imachi and H. Harada, Appl. Environ. Microbiol., 2006, 72, 5311–5317 CrossRef PubMed.
- R. N. Veedu, B. Vester and J. Wengel, Org. Biomol. Chem., 2009, 7, 1404–1409 RSC.
- M. Simsek and H. Adnan, J. Sci. Res. Med. Sci., 2000, 2, 11–14 Search PubMed.
- J. A. Peterson, J. G. McFarland, B. R. Curtis and R. H. Aster, Br. J. Haematol., 2013, 161, 3–14 CrossRef PubMed.
- A. C. Schuh, N. A. Watkins, Q. Nguyen, N. J. Harmer, M. Lin, J. Y. Prosper, K. Campbell, D. R. Sutherland, P. Metcalfe, W. Horsfall and W. H. Ouwehand, Blood, 2002, 99, 1692–1698 CrossRef PubMed.
- P. Moncharmont, S. Courvoisier, A. Pagnier, L. Cotta, T. Debillon and D. Rigal, Acta Paediatr., 2007, 96, 1701–1703 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra06050a |
| This journal is © The Royal Society of Chemistry 2018 |