
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solventless, selective and catalytic oxidation of primary, secondary and benzylic alcohols by a Merrifield resin supported molybdenum(VI) complex with H2O2 as an oxidant†
Jeena Jyoti Boruah‡
*ab and
Siva Prasad Das‡ *a
*a
aDepartment of Chemistry, School of Science, RK University, Bhavnagar Highway, Kasturbadham, Rajkot-360020, Gujarat, India. E-mail: siva.spd@gmail.com; siva.das@rku.ac.in; jeena.jyoti@gmail.com; jeena.jyoti@rku.ac.in; Tel: +91-9678084296
bDepartment of Chemistry, Moridhal College, Moridhal, Dhemaji-787057, Assam, India
First published on 8th October 2018
Abstract
Here, we have described the synthesis, characterization and catalytic activity of a dioxo-molybdenum(VI) complex supported on functionalized Merrifield resin (MR-SB-Mo). The functionalization of Merrifield resin (MR) was achieved in two-steps viz. carbonylation (MR-C) and Schiff base formation (MR-SB). The compounds, MR-C, MR-SB and MR-SB-Mo, were characterized at each step of the synthesis by elemental, SEM, EDX, thermal, BET and different spectroscopic analysis. The catalyst, MR-SB-Mo, efficiently and selectively oxidized a wide variety of alcohols to aldehydes or ketones using 30% H2O2 as an oxidant with reasonably good TOF (660 h−1 in case of benzyl alcohol). The catalyst acted heterogeneously under solventless reaction conditions and did not lead to over oxidized products under optimized conditions. The catalyst afforded regeneration and can be reused for at least five reaction cycles without loss of efficiency and product selectivity. A reaction mechanism for the catalytic activity of MR-SB-Mo was proposed and a probable reactive intermediate species isolated.
1. Introduction
The catalytic oxidation of alcohols is a basic transformation reaction, in synthetic organic chemistry in particular, used in the chemical and pharmaceutical industries, where aldehydes find wide application.1 Besides these, biologically the oxidation of alcohol to carbonyl offers a good contribution towards the degradation of fats during metabolism in humans (e.g. L-malate to oxaloacetate) which is part of the citric acid cycle.2 Due to the presence of a carbonyl group as an intermediate and final product in several natural products,1 fine chemicals1 and medicinally important components,1 there has been an upsurging interest in developing a variety of reagents and catalytic systems which has led to an improvement in the economic and environmentally benign synthetic methodology.Traditional protocol for the oxidation of alcohol carried out by using stoichiometric inorganic oxidant such as permanganate, bromate, or Cr(VI) based reagents3 suffer from drawback such as large amount of heavy metal waste, generation of toxic by-products, difficulty in work up and requirement of larger amount of oxidizing agents.3,4 Moreover, in some cases, the oxidation reactions are performed under severe reaction conditions, such as high temperature and high oxygen pressure, in presence of environmentally undesired solvents, typically chlorinated hydrocarbons which are very toxic, corrosive and environmentally hazardous.3,4a,5 Therefore, search for alternative environmentally benign and safe protocols for the synthesis of different organic carbonyl compounds and their derivatives by alcohol oxidation continues unabated.
Although myriads of traditional user-friendly oxygen sources such as molecular oxygen, hydrogen peroxide, tert-butylhydroperoxide (TBHP) are used to eradicate such harmful waste for alcohol oxidation,5 but aqueous H2O2 constitutes a clean, waste avoiding, potentially green, and environmental-friendly ideal oxidant due to its cost effectiveness, easy availability, ecologically acceptable and it generates only water as the by-product.6 Also, the reaction parameters can be highly tuned by using H2O2 which play a vital role in oxidation reaction. We foresee that H2O2 and O2 (or air) will be complementary useful clean oxidants in practical chemical synthesis. Though molecular oxygen is an ideal oxidant, however, aerial oxidation has one or more of the major impacts such as difficult to control which result combustion and occasionally the reaction should have to be performed with a low conversion to circumvent from over oxidation limit their synthetic applications.6a Besides these, although both the oxygen atom present in O2 may be employed for oxidation to meet the metrices for measuring the ‘greenness’ i.e. 100% atom efficiency, but in most of the reactions, only one oxygen atom is used and show only 50% atom efficiency, consequently to meet the requirement, the oxidation reactions often need certain reducing agents to arrest the extra oxygen atom during the reaction.6a,7 The rate of oxidation towards H2O2 induced oxidation reaction including sulfide, olefin and alcohol oxidation is relatively slow or negligible owing to its weak oxidative nature and as a consequence, it has to be activated by using a suitable catalysts.1b,6b,8 As a result, immense number of transition metal based catalytic systems such as vanadium,9 osmium,10 palladium,11 ruthenium,12 manganese,13 tungsten,9f,g,14 molybdenum,9f,14a,e,f,15 rhenium,16 cobalt,11c,17 copper,11c,18 and iron19 have been developed towards oxidation of alcohols. Though the transition metal ion complexes are reported to have their catalytic activity with high selectivity, good efficiency and reproducibility, however several catalytic processes associate with one or more of the disadvantages such as requirement of additives, use of halogenated solvents, homogeneous in nature, production of huge waste materials, corrosion to the industrial materials and some of them are deposited on the reactor wall as well as disrupting the environmental and ecological stability. These shortcomings are fetching increasingly conspicuous in the light of growing ecological awareness in recent years.20 Interestingly, over the recent years, a new class of catalyst has been developed which utilizes light for alcohol oxidation.21 These photocatalysts absorbs light (visible or UV) and coming under “Greener” approaches. However, majority of these protocols also suffered from drawbacks of the requirement of organic solvents and higher reaction time.
Therefore, constructing a safer, efficient and highly selective heterogeneous catalyst towards alcohol oxidation with H2O2 as the oxidant is considered to be the better choice. Apart from these, heterogenization of homogeneous catalysts22 by immobilization of active soluble catalysts on insoluble polymeric matrix offers improved stability, increased product selectivity, easy separation and recycling to the precious metal complexes.22a,23 In this regard, the use of Merrifield resin, which is a chloromethylated polystyrene cross-linked with divinyl benzene,24 is quite reasonable as the resin is readily availability, easy to handle, cheap, mechanically and chemically robust and most importantly able to undergo facile functionalization as well as provide high-loading capacity.23a,25
Keeping these facts into our account, we presented here the synthesis and characterization of a Merrifield resin supported dioxomolybdenum(VI) compound which acts as a heterogenous catalyst for the efficient and selective oxidation of alcohols to aldehydes or ketones. Interestingly, the protocol worked under solventless conditions with aqueous H2O2 as an oxidant and achieved the product selectivity with a wide range of substrates viz. primary, secondary and benzylic alcohols.
2. Experimental
2.1 Materials
The source of chemicals are given below: Merrifield resin (2% crosslinked, 2–4 mmol Cl per g of polymeric support), tetraethylammoniumchloride, 2-(aminomethyl)pyridine, 4-fluorobenzyl alcohol, 4-chlorobenzyl alcohol, 4-bromobenzyl alcohol, 4-methoxybenzyl alcohol, 4-hydroxybenzyl alcohol, diphenylmethanol, 1-phenylethanol, borneol, 2-octanol, 2-ethyl-1-hexanol, 1-butanol (Alfa Aesar, Mumbai, India), hydrogen peroxide (30%, 50% and 6%), sodium bicarbonate, dimethyl sulfoxide (DMSO), tetrahydrofurane (THF), methanol (MeOH), benzyl alcohol, 4-nitrobenzyl alcohol, cyclohexanol, cyclopentanol, menthol, 2-butanol, 1-octanol, 1-pentanol, 1-decanol (S D Fine-Chem Limited, Mumbai, India), silica gel (60–120 mesh) (Molychem, Mumbai, India). TLC plates (TLC Silica gel 60 F254) were purchased from Merck Limited (India). [MoO2(acac)2] was prepared by following a reported procedure.26 The other reagents and solvents were of commercially available reagent quality, unless otherwise stated.2.2 Physical measurements
Elemental analysis of the compounds for C, H and N were carried out with the help of Perkin-Elmer 2400 series II CHN analyzer. The molybdenum content was determined by using atomic absorption spectroscopy (Thermo iCE 3000 series Atomic absorption spectrophotometer model analyst 200) and EDX analysis. Chlorine content was determined by EDX analysis. C, H and N content were also determined with the help of EDX analysis. IR spectra of the compounds were recorded in KBr pellet (4000–400 cm−1) using a Nicolet model 410 FTIR spectrophotometer. The UV-visible diffuse reflectance analysis of the samples were carried out with the help of a Hitachi U-3400 spectrophotometer equipped with an integrating sphere of 60 mm inner diameter using spectroscopic grade BaSO4 as a reference in the range of 240–800 nm. The SEM images of the samples were recorded by using the JEOL JSM-6390LV Scanning Electron Micrograph with an attached energy-dispersive X-ray detector. The dinitrogen adsorption/desorption measurements were carried out on a Quantachrome model Nova 4200e porosimeter at 77.3 K. The thermogravimetric analysis of the samples was done by using a SHIMADZU TGA-50 system under an atmosphere of nitrogen using an aluminium pan at a heating rate of 10 °C min−1. The powder X-ray diffraction (XRD) patterns were carried out by a Rigaku X-ray diffractometer (Miniflax, UK) using Cu Kα (λ = 0.154 nm) radiation over the 2θ range of 10–70°. The XPS measurements were conducted in a multi-probe system (Omicron Nanotechnology, Germany) equipped with a dual Mg/Al X-ray source and a hemispherical analyzer operating in constant analyzer energy (CAE) mode. The X-ray source (Mg Kα) was operated at 15 kV and 300 W. Charging effects were corrected by setting the binding energies of the adventitious C ls line at 284.8 eV. GC analysis was carried out on a CIC, Gas Chromatograph model 2010 using a SE-52 packed column (length 2 m, 1/8′′ OD) with a Flame Ionization Detector (FID), and nitrogen as the carrier gas (30 mL min−1). The 1H NMR spectra of the substrates were recorded with the help of Bruker, AVANCE 400 MHz spectrophotometer in CDCl3. Tetramethylsilane (TMS) or residual solvent peak was used as an internal standard.2.3 Functionalization of the Merrifield resin (MR)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 24 h at room temperature. Subsequently, the carbonyl functionalized polystyrene, MR-C was synthesized by mixing 1.0 g of MR with 0.50 g, 5.95 mmol sodium bicarbonate (NaHCO3) in 50 mL DMSO under reflux for 24 h. After cooling, the resultant polymer was filtered off, washed with water (3 × 10 mL) and methanol (3 × 10 mL), then dried under high vacuum.
:1) for 24 h at room temperature. Subsequently, the carbonyl functionalized polystyrene, MR-C was synthesized by mixing 1.0 g of MR with 0.50 g, 5.95 mmol sodium bicarbonate (NaHCO3) in 50 mL DMSO under reflux for 24 h. After cooling, the resultant polymer was filtered off, washed with water (3 × 10 mL) and methanol (3 × 10 mL), then dried under high vacuum.2.4 Synthesis of the Merrifield resin supported molybdenum(VI) complex (MR-SB-Mo)
The supported molybdenum(VI) complex was synthesized by reacting the MR-SB, and MoO2(acac)2 in MeOH-THF (1:1) under reflux condition. In the typical procedure, 1.0 g of MR-SB was swallowed in 25 mL MeOH-THF (1:1) for 24 h at room temperature. Subsequently, 0.82 g, 2.51 mmol of MoO2(acac)2 (1.5 equiv. of Schiff base loading) and 1.66 g, 10.02 mmol of tetraethylammoniumchloride (Et4NCl, 6 equiv. of Schiff base loading) in 5 mL MeOH was added and stirred the reaction mixture under reflux for 36 h. The reaction mixture was filtered and the solid was washed with MeOH (5 × 10 mL). The slightly brownish solid products were dried under high vacuum for 6 h and stored in dry place.
2.5 Catalytic activity of MR-SB-Mo
:H2O2:Mo = 1000:1100:1. The reaction mixture was stirred at 65 °C and progress was monitored by TLC and GC. After completion of the reaction, the solid catalyst was separated by filtration and the crude filtrate was extracted with ethyl acetate (3 × 5 mL). The combined extract was dried over anhydrous magnesium sulfate, filtered and removed the excess ethyl acetate under reduced pressure. This followed by column chromatography of the crude product on silica gel. The pure products were subjected to 1H NMR analysis for identification.:H2O2:Mo at 1000:1100:1. The progress of the reaction was monitored by TLC and GC. After completion, the process was repeated so on for upto fifth reaction cycle.3. Results and discussion
3.1 Synthesis
The synthetic pathway for the synthesis of the polymer-supported molybdenum(VI) compound, MR-SB-Mo is shown in Scheme 1. The carbonyl functionalization of MR to form MR-C was achieved by adopting a previously reported procedure.27 In the next step, the carbonyl group of MR-C was condensed with 2-aminomethylpyridine under reflux condition in methanol to form MR-SB. The molybdenum complex was formed by refluxing MR-SB, Et4NCl and MoO2(acac)2 in MeOH-THF (1:1) for 36 h. The Et4NCl provides the counter anion required for the formation of the molybdenum(VI) complex. In order to maximized the molybdenum loading, a series of reactions conducted under different reaction condition such as variation of solvent, reaction temperature and time, molar ratio of molybdenum:Schiff base, etc. The experiments revealed that 1.5 equivalent of MoO2(acac)2 with respect to Schiff base loading, MeOH/THF as solvent, under reflux condition and reaction time of 36 h were optimum for giving maximum molybdenum loading on MR-SB-Mo.
 | ||
Scheme 1 Synthesis of MR-SB-Mo. “ ” represents polymeric support. ” represents polymeric support. | ||
3.2 Characterization
The elemental analysis data for the compounds at each step of functionalization are shown below in Table 1. It is seen from the table that the compound MR-SB showed 4.68% of nitrogen which were absent in MR or MR-C. This indicates the formation of Schiff base by the reaction of carbonyl group of MR-C and 2-aminomethylpyridine. This corresponds to ligand (i.e., 2-aminomethylpyridine) loading of 1.67 mmol g−1 of the polymer support. The value is consistent with the loss of the chlorine from the MR as evident from EDX analysis. From EDX analysis, it was also found that the amount of chlorine was decreased from 9.82% to 1.99% during carbonyl functionalization which indicates that nearly 80% of the chloromethylated groups were converted to carbonyl group in the process. The molybdenum loading in MR-SB-Mo was found to be 0.45 mmol g−1 of the polymeric support. This indicates that nearly half of the Schiff base ligands were coordinated with molybdenum (as two Schiff base molecules coordinated with one molybdenum center). Besides these, after metal incorporation, the chlorine content was increased from 1.69% (for MR-SB) to 4.60% (for MR-SB-Mo), which is due to the presence of the counter anion, Cl− for charge neutralization as shown in the structure for MR-SB-Mo (Scheme 1). The magnetic susceptibility measurements of MR-SB-Mo showed the diamagnetic nature of the compound. This confirmed the +6 oxidation state of molybdenum centers.| Compound | Data obtained from elemental analysis (%) (data obtained from EDX analysis (%)) | Metal loadinga (mmol g−1 of polymer) | ||||
|---|---|---|---|---|---|---|
| C | H | N | Cl | Mo | ||
| a Metal loading = (Observed molybdenum % × 10)/(atomic weight of molybdenum).b Data obtained from AAS. “—” stands for not determined.c Molybdenum content determined by AAS after 5th reaction cycle. | ||||||
| MR | 83.20 | 6.98 | — | — | — | — |
| (83.13) | — | (9.82) | ||||
| MR-C | 87.10 | 7.31 | — | — | — | — |
| (87.04) | — | (1.99) | ||||
| MR-SB | 86.02 | 7.22 | 4.68 | — | — | — |
| (68.14) | — | (4.66) | (1.69) | |||
| MR-SB-Mo | 78.07 | 6.20 | 4.41 | — | 4.32b | 0.45 |
| (78.21) | — | (4.36) | (4.60) | (4.28) | ||
| 4.29c | ||||||
The scanning electron micrographs (SEM) of the compounds obtained at different stages in the preparation of MR-SB-Mo are shown in Fig. 1. It is revealed from the micrograph that the virgin polymer beads, MR undergoes striking morphological changes to form MR-SB-Mo. The surface of the smooth and spherical beads of MR became lightly rough after carbonyl functionalization (MR-C, Fig. 1(b)) as well as Schiff base formation (MR-SB, Fig. 1(c)). The even roughening as seen in the images also suggest the uniform covalent functionalization on the surface of the MR beads. However, after molybdenum coordination, randomly oriented depositions were observed causing further roughening on the surface of the polymeric beads (Fig. 1(d)). The EDX spectra of MR-SB-Mo (Fig. 1(e)) showed the presence of carbon, nitrogen, oxygen, chlorine and molybdenum on the support.
 | ||
| Fig. 1 Scanning electron micrographs of (a) MR, (b) MR-C, (c) MR-SB, and (d) MR-SB-Mo. EDX spectra of (e) MR-SB-Mo. | ||
The powder XRD patterns of MR, MR-C, MR-SB and MR-SB-Mo are shown in Fig. 2 and were recorded at 2θ values between 5 and 70°. A broad peak cantered at 2θ value of ca. 20° was observed in pristine MR. This type of diffraction pattern is characteristic for the PS-DVB resin.28 Similar to MR, almost identical diffraction patterns were observed in MR-C, MR-SB and MR-SB-Mo. This type of broad peak indicates that the developed catalyst and its precursors, i.e., MR, MR-C and MR-SB are almost amorphous in nature. Moreover, the absence of sharp diffraction peaks in MR-SB-Mo indicated the amorphous nature of the attached MoO22− moiety. It is notable that during the synthesis of the MR-SB-Mo via different functionalization steps, there is no change in amorphousness of MR.
 | ||
| Fig. 2 The XRD patterns of (a) MR, (b) MR-C, (c) MR-SB and (d) MR-SB-Mo. | ||
In order to gain the electronic properties for the surface of MR-SB-Mo, XPS spectrum of the compound was recorded and presented in Fig. 3. From the figure it is seen that the spectrum displayed two well resolved peaks at 232.5 and 235.7 eV in the Mo (3d) region.29 On the basis of available literature reports,29 these peaks correspond to Mo (3d3/2) and Mo (3d5/2), respectively which is attributable to the molybdenum centers in +6 oxidation state, i.e., diamagnetic. This diamagnetic behavior of MR-SB-Mo shown by XPS analysis is in agreement with the results obtained from magnetic susceptibility analysis.
 | ||
| Fig. 3 XPS Mo (3d3/2) and Mo (3d5/2) spectra of (a) MR-SB-Mo and (b) MR-SB-Mo after the 5th reaction cycle. | ||
The surface area, pore volume, and pore size of the synthesized compounds viz. MR, MR-C, MR-SB, and MR-SB-Mo, were investigated by N2 absorption and desorption measurements at liquid nitrogen temperature. The surface areas were measured by following the Brunauer–Emmett–Teller (BET) method30 and the pore volume was determined by following the Barrett–Joyner–Halenda (BJH) model in the nitrogen isotherms.31 The data are presented in Table 2. It is seen from the data that the surface areas, pore volumes and pore radius were decreased with increasing functionalization steps. This is because the functionalization may as well as metal loading blocked the pore of the polymeric beads. Similar types of observation were reported earlier with MR based catalysts.32 The nitrogen adsorption/desorption isotherms showed typical TYPE II adsorption (Fig. S1, ESI†) of an IUPAC standard33 which is the characteristics of macroporous or nonporous material.34
Vital information can be derived by comparing the infrared (IR) spectral data for the compounds at each synthetic step. After each synthetic step, characteristic new peaks may appear or shifted to a new position which confirmed the chemical transformations. The FT-IR spectra for the compounds are shown in Fig. 4 and important peaks are assigned in Table 3. On the basis of available reports, the change in intensity of the ν(C–Cl) peak is found to be the indicator of the functionalization of chloromethyl group.32a,35 It is evident by comparing the IR spectra of MR and MR-C that the strong peak appeared at 1262 cm−1 in MR which is attributed to ν(C–Cl)32a,35a,b was disappeared (or significantly decreased the intensity) after carbonyl functionalization in MR-C. It is pertinent here to mention that after carbonyl functionalization the concentration of chloromethyl group in MR was dropped nearly 80%. Thus, this observation is in consistent with the data obtained from elemental analysis. Besides this, a strong peak appeared at 1728 cm−1 which is due to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the aldehyde group.36 Additionally, two new weak intensity peaks appeared for ν(C–H)aldehydic at 2826 and 2721 cm−1.36b,c This further confirmed the conversion of chloromethyl group to aldehyde group.
O) of the aldehyde group.36 Additionally, two new weak intensity peaks appeared for ν(C–H)aldehydic at 2826 and 2721 cm−1.36b,c This further confirmed the conversion of chloromethyl group to aldehyde group.
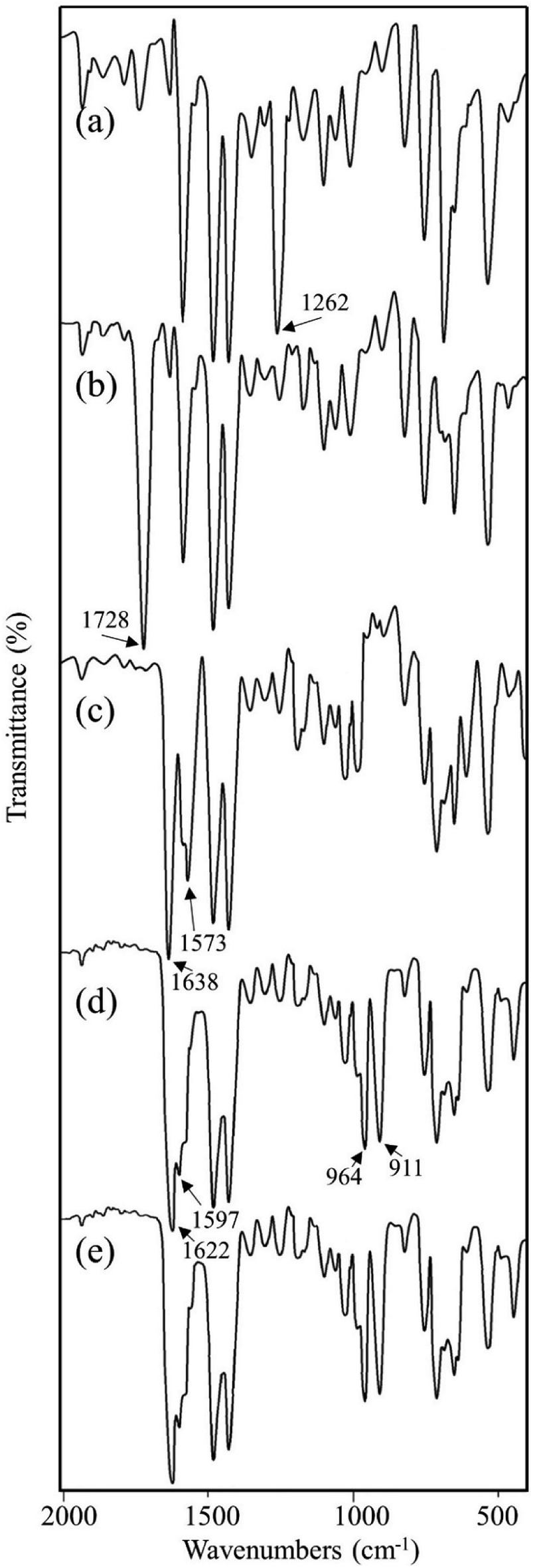 | ||
| Fig. 4 FTIR spectra for (a) MR, (b) MR-C, (c) MR-SB, (d) MR-SB-Mo, and (e) MR-SB-Mo after 5th reaction cycle. | ||
| Compound | Peak position (cm−1) | Peak assignment | |
|---|---|---|---|
| IR | Raman | ||
| a Py, pyridine; vs, very strong; s, strong; m, medium; w, weak; vw, very weak; sh, shoulder; “—” stands for not seen. | |||
| MR | 1262 (s) | 1260 (w) | ν(C–Cl) |
| MR-C | 1261 (vw) | — | ν(C–Cl) |
| 1728 (vs) | 1730 (vw) | ν(CO) |
|
| 2826 (m) | 2822 (w) | ν(C–H)aldehydic | |
| 2721 (m) | 2726 (w) | ||
| MR-SB | 1261 (vw) | — | ν(C–Cl) |
| 1638 (vs) | 1640 (m, sh) | ν(CN)imine |
|
| 1573 (s) | 1570 (m, sh) | ν(CN)py |
|
| 609 (m) | 610 (m) | Py(in-plane ring deformation) | |
| 408 (m) | 407 (m) | Py(out-of-plane ring deformation) | |
| MR-SB-Mo | 1263 (vw) | — | ν(C–Cl) |
| 1622 (vs) | 1625 (m, sh) | ν(CN)imine |
|
| 1597 (s) | 1596 (m, sh) | ν(CN)py |
|
| 964 (s) | 962 (s) | ν(OMoO)asym |
|
| 911 (s) | 910 (s) | ν(OMoO)sym |
|
| 640 (m) | 645 (m) | Py(in-plane ring deformation) | |
| 440 (m) | 441 (m) | Py(out-of-plane ring deformation) | |
In MR-SB, a new strong peak appeared at 1638 cm−1 which is attributable to ν(CN)imine.36a,37 However, the peak at 1728 cm−1 due to ν(CO) in MR-C was disappeared in MR-SB. This confirmed the condensation of aldehyde group with the amine group of 2-aminomethylpyridine to form an imine. Additionally, new medium to strong intensity peaks were appeared at 1573 (s), 609 (m), and 408 (m) cm−1 which can be assigned to ν(CN)pyridine, Py(in-plane ring deformation), and Py(out-of-plane ring deformation), respectively of a pyridine moiety.37,38 Thus the IR spectral analysis clearly confirmed the formation of MR-SB as shown in Scheme 1. Interestingly, in MR-SB-Mo, new and strong peaks appeared at 964 and 911 cm−1 which were assigned for ν(OMoO)asym and ν(OMoO)sym, respectively.39 Further the peak due to ν(CN)imine appeared at 1638 cm−1 of MR-SB was shift to 1622 cm−1 in MR-SB-Mo. In view of the existing literature, such lower shifting of ν(CN)imine peak confirmed the coordination of imine group with molybdenum(VI) center.36a Apart from this, the peaks due to ν(CN)pyridine, Py(in-plane ring deformation) and Py(out-of-plane ring deformation) were shifted to higher wave no after molybdenum(VI) coordination. This type of shifting suggest the coordination of pyridine with the metal center.37,38a Thus the FTIR spectral analysis clearly showed the formation of MR-C, MR-SB and MR-SB-Mo.
The Raman spectral analysis further confirmed the formation of MR-C, MR-SB and MR-SB-Mo that shown by FTIR analysis. The spectral data of the samples are given in Table 3 and the spectrum for MR-SB-Mo is shown in Fig. 5. The strong peaks appeared commonly in each of the spectrum at ca. 1600 and 1000 cm−1 are attributed to the benzene skeletal vibrations arising from the polymeric backbone.32a,40 The weak peak appeared at 1260 cm−1 due to ν(C–Cl) mode of –CH2Cl group in MR was absent in MR-C.40a This observation is in consistent with the results obtained from IR spectral analysis. The very weak peak appeared in the spectrum of MR-C at 1730 cm−1 is attributed to the ν(CO) mode of aldehydic group. Similar to IR spectral analysis, this peak was vanished in the spectrum of MR-SB due to the Schiff base formation. The ν(CN)imine and ν(CN)pyridine peaks were appeared as medium intensity shoulder in the spectrum of MR-SB. However, after complex formation in MR-SB-Mo, the former peak was shifted to lower wave number and the latter was shifted to higher wave number that confirmed the coordination of the imine and pyridine moieties with the molybdenum(VI) center (through nitrogen atom). Further, the Raman spectrum of MR-SB-Mo displayed two strong peaks at 962 and 910 cm−1 for ν(OMoO)asym and ν(OMoO)sym, respectively. These two peaks are characteristic of dioxomolybdenum(VI) moiety.41 Thus, the Raman spectral analysis confirmed the formation of MR-C, MR-SB and MR-SB-Mo.
 | ||
| Fig. 5 Raman spectrum for MR-SB-Mo. | ||
The diffuse reflectance UV–visible spectra of MR-SB and MR-SB-Mo were recorded by taking BaSO4 as reference. The spectrum of MR-SB display two well resolved peaks at ca. 264 and 345 nm. The relatively higher energy peaks at 264 nm corresponds to the π → π* transition of the pyridyl ring, benzene ring of polymeric support and imine functional group.37,42 On the other hand, the lower energy peak at 345 nm may be assigned to n → π* transition of the pyridyl ring and imine functional group.37,42 Interestingly, in MR-SB-Mo, the peak for n → π* transition was shifted to ca. 331 nm. But the peaks for π → π* transition were remained unaffected after complex formation. This blue shift of n → π* transition indicate the coordination of molybdenum(VI) via the pyridyl and imine nitrogen.37,42 Beside these, there was no LMCT bands present in the UV-Vis spectrum of MR-SB-Mo. This is because those bands generally appear at higher concentration of metal complexes.
The thermal stability of the compounds was studied with the help of TGA-DTG analysis. The thermograms are shown in Fig. 6 and data are presented in Table 4. The thermograms showed multiple stage of thermal degradation for each of the compounds. The pristine polymer, MR showed three weight loss steps in the temperature range of 291–366, 366–477, and 477–700 °C with corresponding weight loss of 14.23, 39.53, and 46.24%, respectively. On the basis of available literatures data, the first step of decomposition is attributed to the loss of chloromethylated group and the subsequent steps are attributed to the decomposition of the cross-linked polystyrene backbone.43 The MR was completely decomposed at ca. 700 °C. Similar to MR, MR-C also showed three stage thermal decomposing. The thermogram of MR-SB provided vital information regarding the Schiff base loading on the support. The decomposition at ca. 177–230 °C with weight loss of 20.77% matched exactly with the data obtained from elemental analysis for Schiff base loading (20.53% Schiff base, based on N content). Further evidence about the loss of Schiff base ligand at this stage was confirmed by recording IR spectrum of the sample at this temperature which did not show the presence of characteristic peak for ν(CN)imine. The subsequent decomposition steps are attributed to the decomposition of the polystyrene backbone. The thermogram of MR-SB-Mo showed four steps of thermal degradation at the temperature range of 195–234, 299–350, 350–486, and 486–700 °C with the corresponding weight loss of 18.86, 2.34, 29.90, and 39.27%, respectively. Interestingly, the compound never decomposed completely upto 700 °C which was due to the residual oxo-molybdenum species formed after complete decomposition of polymeric materials.32a,44 The thermal stability of upto 195 °C for MR-SB-Mo also provide additional evidence about the stability of the catalyst under different reaction temperatures.
 | ||
| Fig. 6 The TGA thermograms of MR (black), MR-C (red), MR-SB (blue), and MR-SB-Mo (green). | ||
| Compound | Temperature, oC | Weight loss (%) |
|---|---|---|
| MR | 291–366 | 14.23 |
| 366–477 | 39.53 | |
| 477–700 | 46.24 | |
| MR-C | 296–344 | 3.27 |
| 344–480 | 46.74 | |
| 480–700 | 48.92 | |
| MR-SB | 177–230 | 20.77 |
| 294–340 | 2.46 | |
| 340–480 | 32.08 | |
| 480–700 | 43.47 | |
| MR-SB-Mo | 195–234 | 18.86 |
| 299–350 | 2.34 | |
| 350–486 | 29.90 | |
| 486–700 | 39.27 |
On the basis of the above analysis, a structure for MR-SB-Mo is proposed as shown in Scheme 1 where a dioxomolybdenum(VI) species is anchored to the support via coordination with the nitrogen atom of imine and pyridine group.
3.3 Catalytic activity
In order to gain a standard reaction condition for product selectivity and better yield for oxidation of alcohols to ketones or aldehydes several reaction parameters such as solvents, amount of catalyst and oxidant, reaction temperature, etc. were screened using benzyl alcohol as model substrate.
In the initial investigation, the oxidation of benzyl alcohol was carried out with varying amount of 30% aqueous H2O2 as oxidant. We have conducted the reactions by keeping the molar ratios of benzyl alcohol:H2O2 at 1:0.5, 1:1.1, 1:1.5 and 1:2 and Mo: benzyl alcohol at 1:1000. The results are summarized in Table 5. The reaction at molar ratio of 1:0.5 was not completed even after 5 h [Table 5, entry 1]. So, we have increased the amount of H2O2 to 1:1.1, i.e., slightly excess than the substrate. At this molar ratio, the reaction comfortably completed in 90 min with benzaldehyde as the sole product [Table 5, entry 2]. With a view to decrease the reaction time, we have further conducted the reactions at 1:1.5 and 1:2, but product selectivity was lost and formed 6% and 9% of benzoic acid, respectively [Table 5, entries 3 and 4]. Thus, the molar ratio of substrate:H2O2 at 1:1.1 is found to be the optimum condition for selectively oxidize benzyl alcohol to benzaldehyde.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Sl. no. | Molar ratio | Solvent | Temperature (°C) | Time (min) | Isolated yield (%) | Selectivity (%) a:b |
TOFb (h−1) | |
| Mo:Sc |
S:H2O2c |
|||||||
| a All reactions were carried out with benzyl alcohol as substrate (2.5 mmol), MR-SB-Mo (5.6 mg for 0.0025 mmol of Mo) and 5 mL solvent (unless otherwise indicated).b TOF = (mmol of product)/[(mmol of catalyst) × (time)].c ‘S’ stands for substrate.d Reaction with 6% aqueous H2O2 as oxidant.e Reaction with 50% aqueous H2O2 as oxidant.f Reaction with 70% aqueous TBHP as oxidant.g Reaction conducted with MR-SB-Mo but no added oxidant.h Reaction conducted without MR-SB-Mo or blank reaction.i Reaction conducted under optimum condition with SB-Mo (1.5 mg, 0.0025 mmol) as catalyst. | ||||||||
| 1 | 1:1000 |
1:0.5 |
Solventless | 65 | 300 | 47 | 100:0 |
94 |
| 2 | 1:1000 |
1:1.1 |
Solventless | 65 | 90 | 99 | 100:0 |
660 |
| 3 | 1:1000 |
1:1.5 |
Solventless | 65 | 70 | 97 | 91:6 |
831 |
| 4 | 1:1000 |
1:2.0 |
Solventless | 65 | 60 | 98 | 89:9 |
980 |
| 5 | 1:1000 |
1:1.1 |
Water | 65 | 90 | 87 | 100:0 |
580 |
| 6 | 1:1000 |
1:1.1 |
Acetonitrile | 65 | 90 | 89 | 100:0 |
593 |
| 7 | 1:1000 |
1:1.1 |
Chloroform | 65 | 90 | 24 | 100:0 |
160 |
| 8 | 1:1000 |
1:1.1 |
Dichloromethane | 65 | 90 | 23 | 100:0 |
153 |
| 9 | 1:1000 |
1:1.1 |
Toluene | 65 | 90 | 20 | 100:0 |
133 |
| 10 | 1:500 |
1:1.1 |
Solventless | 65 | 80 | 98 | 100:0 |
368 |
| 11 | 1:100 |
1:1.1 |
Solventless | 65 | 70 | 99 | 100:0 |
84 |
| 12 | 1:1000 |
1:1.1d |
Solventless | 65 | 150 | 96 | 100:0 |
384 |
| 13 | 1:1000 |
1:1.1e |
Solventless | 65 | 70 | 94 | 92:2 |
806 |
| 14 | 1:1000 |
1:1.1f |
Solventless | 65 | 300 | 97 | 100:0 |
194 |
| 15 | 1:1000 |
In air | Solventless | 65 | 90 | 0 | — | 0 |
| 16 | 1:1000 |
In O2 | Solventless | 65 | 90 | 0 | — | 0 |
| 17 | 1:1000 |
1:1.1 |
Solventless | RT | 300 | 98 | 100:0 |
196 |
| 18 | 1:1000 |
1:1.1 |
Solventless | 50 | 120 | 99 | 100:0 |
495 |
| 19 | 1:1000 |
1:1.1 |
Solventless | 90 | 90 | 91 | 100:0 |
607 |
| 20g | 1:1000 |
— | Solventless | 65 | 90 | 0 | — | 0 |
| 21h | — | 1:1.1 |
Solventless | 65 | 90 | 0 | — | 0 |
| 22i | 1:1000 |
1:1.1 |
Solventless | 65 | 90 | 53 | 91:9 |
353 |

Keeping the molar ratio of substrate:H2O2 at 1:1.1, we have screened the oxidation of benzyl alcohol by using different solvents such as water, acetonitrile, chloroform, dichloromethane, toluene, etc. as well as under solventless condition. The highest activity in terms of TOF was found in solventless condition [Table 5, entry 2]. The reaction conducted in water decreased the TOF [Table 5, entry 5]. This may be due to dilution of the reaction mixture by the added water. In acetonitrile, the reactivity is not improved [Table 5, entry 6]. Further, reactions in chloroform and dichloromethane are very slow with poor TOF [Table 5, entries 7 and 8]. This may be due to the immiscibility of the oxidant with the solvents. Moreover, we do not intend to use halogenated solvents. Similar type of reactivity was also found while doing reaction in toluene [Table 5, entry 9]. The use of methanol and ethanol were avoided as these solvents may compete for oxidation. Indeed, the reactions were very slow and not completed (data not shown). Thus, solventless condition was found to be optimum for selectively oxidize benzyl alcohol to benzaldehyde.
The catalyst amount has a great impact on TOF. We have conducted reactions by keeping molar ratio of Mo:benzyl alcohol at 1:100, 1:500 and 1:1000. Each of the reactions were conducted by keeping benzyl alcohol: H2O2 at 1:1.1 and under solventless condition at 65 °C. There was decrease in reaction time with increased amount of catalyst but the TOF was significantly decreased [Table 5, entries 2, 10 and 11].
Reactions were conducted with 6%, 30% and 50% of aqueous H2O2 solutions [Table 5, entries 2, 12 and 13] at molar ratio of benzyl alcohol: H2O2 and Mo:benzyl alcohol at 1:1.1 and 1:1000, respectively under solventless condition at 65 °C. The slowest reaction and lowest TOF was found with 6% H2O2 whereas fastest was found with 50% H2O2 [Table 5, entry 13]. However, trace amount of carboxylic acid was formed with 50% H2O2, i.e., loss of product selectivity. The slowest reaction with 6% H2O2 may be due to the dilution of the oxidant in the reaction mixture as was found in case of reactions in water as solvent [Table 5, entry 5]. Therefore, we have chosen 30% H2O2 as optimum for this reaction.
We have also screened the reaction with different types of oxidant such as 30% H2O2 (aqueous), t-butyl hydroperoxide (70% aqueous solution, TBHP), air and O2 balloon. The highest TOF was found with 30% H2O2 [Table 5, entry 2] and no reaction with air or O2 balloon under identical reaction conditions [Table 5, entries 15 and 16]. There were 3 to 4-fold increase in reaction time with TBHP [Table 5, entry 14] as an oxidant. Moreover, it was reported that TBHP generates butanol as byproduct.45 Interestingly, the use of H2O2 is advantageous as it is cheap, environmentally clean, easy to handle and water is the only byproduct.6a–c,e,g
The reaction temperature has a remarkable impact on rate of reaction. So, we have conducted reactions at different temperatures viz. room temperature, 50, 65, and 90 °C [Table 5, entries 2, 17–19]. The reactions were conducted under solventless condition by keeping molar ratio of benzyl alcohol:H2O2 (30%):Mo at 1000:1100:1. It was found that with increasing reaction temperature, the TOF increases and reached a highest value at 65 °C. Thereafter, the TOF decreases which indicates that 65 °C is the optimum reaction temperature.
Thus the optimum reaction condition for the selective oxidation of benzyl alcohol to benzaldehyde was found to be substrate:H2O2 (30%):Mo at 1000:1100:1 under solventless condition at 65 °C as shown in Scheme 2.
 | ||
| Scheme 2 Optimum reaction condition for oxidation of alcohol catalyzed by MR-SB-Mo using 30% H2O2 as oxidant. | ||
Having gained the optimal conditions, we explored the substrate scope of the newly developed catalyst, MR-SB-Mo under the optimum condition, for the selective oxidation of a wide range of alcohols such as primary, secondary and benzyl alcohols to their corresponding aldehydes or ketones. The results are summarized in Table 6. It is seen from the table that each of the substrate oxidized in high yields with reasonably good TOF. Besides this, under same reaction conditions, benzylic [Table 6, entries 1–9] and secondary alcohols [Table 6, entries 10–15] were found to be oxidize relatively at faster rate than the primary alcohols [Table 6, entries 16–20]. The beauty of the protocol is that no overoxidation to carboxylic acid took place with all the studied substrates. In case of substituted benzyl alcohols, different types of substituents such as –F, –Cl, –Br, –OMe, –OH and –NO2 well-tolerate during the oxidation process [Table 6, entries 2–7], some of which could be utilized for further derivation. One of the notable aspect of the developed catalytic system is its ability to oxidize benzyl alcohol to benzaldehyde at relatively higher scale (10 g scale) without losing the catalytic efficiency and product selectivity [Table 6, entry 1d] which provides its potential application towards commercial processes. It is pertinent here to mentioned that in a separate blank reaction using benzyl alcohol as substrate, i.e., under identical optimum conditions without the added catalyst, the reaction did not progress within the stipulated time which indicate the active role of the catalyst in the oxidation processes (Table 5, entry 21). Similarly, the reaction was not successful without the added H2O2 (Table 5, entry 20).
| Sl. no. | Substrate | Time (min) | Product | Isolated yield (%) | TOFb (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: unless otherwise stated, all reactions were performed solventless at 65 °C using 2.5 mmol of substrate, 5.6 mg of MR-SB-Mo (contain 0.0025 mmol of Mo) and 2.75 mmol of 30% aqueous H2O2.b TOF=(mmol of product)/[(mmol of catalyst) × (time)].c Yield at 5th reaction cycle.d Yield at 10 g scale reaction.e Reaction conducted with 2 mL acetonitrile. | |||||
| 1 |  |
90 |  |
99 | 660 |
| 653 | |||||
| 646 | |||||
| 98c | |||||
| 97d | |||||
| 90 | |||||
| 90 | |||||
| 2e |  |
100 |  |
97 | 582 |
| 3e |  |
100 |  |
96 | 576 |
| 4e |  |
100 |  |
97 | 582 |
| 5e |  |
105 |  |
97 | 554 |
| 6e |  |
100 |  |
99 | 594 |
| 7 |  |
100 |  |
98 | 588 |
| 8 |  |
120 |  |
98 | 490 |
| 9 |  |
105 |  |
97 | 554 |
| 10 |  |
150 |  |
97 | 388 |
| 11 |  |
135 |  |
96 | 426 |
| 12 |  |
165 |  |
98 | 356 |
| 13e |  |
180 |  |
98 | 327 |
| 14 |  |
165 |  |
96 | 349 |
| 15 |  |
180 |  |
97 | 323 |
| 16 |  |
240 |  |
99 | 248 |
| 17 |  |
270 |  |
96 | 213 |
| 18 | CH3(CH2)3CH2OH | 240 | CH3(CH2)3CHO | 97 | 243 |
| 19 | CH3(CH2)8CH2OH | 270 | CH3(CH2)8CHO | 98 | 218 |
| 20 | CH3(CH2)2CH2OH | 240 | CH3(CH2)8CHO | 96 | 240 |
In order to check the advantage of synthesizing the heterogeneous catalyst, MR-SB-Mo, over homogeneous catalyst, a neat dioxomolybdenum complex, SB-Mo was synthesized (detailed synthetic procedure is in ESI†). The ligand for the neat complex was designed in such a way that it provides almost identical coordination environment that present in MR-SB-Mo. Moreover, the catalytic oxidation reaction was conducted under identical optimum condition using benzyl alcohol as substrate. From the reaction it was seen that the SB-Mo could reached upto TOF = 353 h−1 within the stipulated time period (Table 5, entry 22) and produced benzaldehyde along with benzoic acid (9%). Thus, under identical optimum condition the heterogeneous catalyst, MR-SB-Mo showed superior catalytic activity in terms of product yield as well as product selectivity over the homogeneous catalyst, SB-Mo.
In order to compare the catalytic activity of MR-SB-Mo over the reported catalyst, a separate comparison table containing catalytic activity of reported molybdenum-based catalyst towards oxidation of benzyl alcohol is given in ESI (Table S1†). From the table it is seen that MR-SB-Mo exhibit superior activity over the reported catalysts.
 | ||
| Fig. 7 Catalyst recycling. | ||
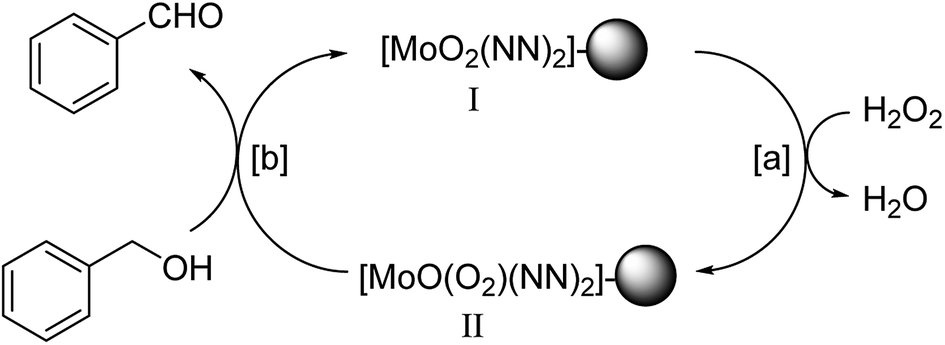 | ||
Scheme 3 Proposed reaction mechanism for oxidation of alcohols using benzyl alcohol as representative. “NN” stands for the nitrogen coordination site for imine and pyridine group and “ ” represents polymeric support. ” represents polymeric support. | ||
The change of substituents in the ligand environment of the metal complexes lead to a dramatic change in their catalytic activity. In this regard, the Hammett relation47 can be used to predict the catalytic activity of MR-SB-Mo. The substituent constant, σ of Hammett equation, for the substituents with (+)ve and (−)ve values indicate the electron withdrawing and electron releasing group, respectively. Thus, choosing substituents from the series could help to design a better catalyst for their activity. From the available literature report,14f,47,48 it is found that oxidation of alcohol by the peroxometal complexes take place by electrophilic attack of the peroxo moiety on the oxygen atom of the alcohol group (–OH). Thus, adding substituents with (+)ve σ-value in the ligand environment of MR-SB-Mo will increase the electrophilicity of the peroxo moiety which in return increase the catalytic activity of the catalyst. The catalyst, MR-SB-Mo can be substituted in its both the aromatic rings viz. phenyl or pyridyl ring. So, the synthesis of MR-SB-Mo with electron withdrawing substituents anticipated a higher reaction rate.
4. Conclusions
In summary, we have developed a heterogeneous molybdenum(VI) based catalyst using Schiff base functionalized Merrifield resin as support. The compound, MR-SB-Mo, served as an efficient catalyst for the selective oxidation of alcohols to aldehydes or ketones using 30% aqueous H2O2 as an oxidant, which is considered as a Green oxidant, under solvent-less condition. The catalyst showed recyclability atleast upto 5th reaction cycles without loss of activity and product selectivity. The developed protocol for the oxidation of alcohols is simple and the products were isolated in highly pure form. On the basis of available literature reports (Table S1, ESI†), the catalyst is found to offer reasonably good catalytic activity and higher TOF. Thus, the protocol complies with the principles of “Green Chemistry” which is very important in concern with the current environmental prospects.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
J. J. B. acknowledges University Grants Commission, New Delhi, India for providing financial support under Minor Research Project (No. F. 5–30/2013–14/MRP/NERO/427). We are also thankful to Sophisticated Analytical Instrumentation Centre (SAIC), Tezpur University, Sonitpur, Assam, India and Centre of Excellence, NFDD Centre, Saurashtra University, Rajkot, Gujarat, India for recording the FTIR and UV-Vis spectra, TGA-DTG thermogram and BET analysis.References
- (a) M. Hudlicky, Oxidation in Organic Chemistry, ACS Monograph Series no. 186, American Chemical Society, Washington DC, 1990 Search PubMed; (b) R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, London, 1981 Search PubMed; (c) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed; (d) M. Beller, Adv. Synth. Catal., 2004, 346, 107–108 CrossRef CAS; (e) J. Cai, J.-Y. Lu, Q.-Y. Chen, L.-L. Qu, Y.-Q. Lub and G.-F. Gao, New J. Chem., 2017, 41, 3882–3886 RSC; (f) Z. Hao, N. Li, X. Yan, Y. Li, S. Zong, H. Liu, Z. Han and J. Lin, New J. Chem., 2018, 42, 6968–6975 RSC; (g) S. Farhadi, M. Hakimib and M. Maleki, RSC Adv., 2018, 8, 6768–6780 RSC; (h) W. C. de Abreu, M. A. S. Garcia, S. Nicolodi, C. V. R. de Moura and E. M. de Moura, RSC Adv., 2018, 8, 3903–3909 RSC.
- K. Tornheim and N. B. Ruderman, Intermediary Metabolism of Carbohydrate, Protein, and Fat, in Metabolic Basis of Obesity, ed. R. S. Ahima, Springer, New York, 2011, pp 25–51 Search PubMed.
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons: New York, 4 edn, 1992 Search PubMed.
- (a) N. Bhati, K. Sarma and A. Goswami, Synth. Commun., 2008, 38, 1416–1424 CrossRef CAS; (b) S. Biella and M. Rossi, Chem. Commun., 2003, 378–379 RSC; (c) H. Ünver and I. Kani, Polyhedron, 2017, 134, 257–262 CrossRef.
- M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, Catalytic Oxidation of Alcohols: Recent Advances in Advances in Organometallic Chemistry, ed. P. J. Pérez, 2015, 63, pp 91–174 Search PubMed.
- (a) R. Noyori, M. Aokib and K. Sato, Chem. Commun., 2003, 1977–1986 RSC; (b) K. Kaczorowska, Z. Kolarska, K. Mitkab and P. Kowalski, Tetrahedron, 2005, 61, 8315–8327 CrossRef CAS; (c) K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS PubMed; (d) B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed; (e) C. W. Jones, Applications of Hydrogen Peroxide and Derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed; (f) K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310–8311 CrossRef CAS PubMed; (g) B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS PubMed.
- (a) L. I. Simándi, Catalytic Activation of Dioxygen by Metal Complexes, Kluwer Academic, Dordrecht, The Netherlands, 1992 CrossRef; (b) The Activation of Dioxygen and Homogeneous Catalytic Oxidation, ed. D. H. R. Barton, A. E. Bartell and D. T. Sawyer, Plenum, New York, 1993; (c) M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS; (d) R. Meiers, U. Dingerdissen and W. F. Holderich, J. Catal., 1998, 176, 376–386 CrossRef CAS.
- N. D. Gillitt, J. Domingos and C. A. Bunton, J. Phys. Org. Chem., 2003, 16, 603–607 CrossRef CAS.
- (a) A. Butler, M. J. Clague and G. E. Meister, Chem. Rev., 1994, 94, 625–638 CrossRef CAS; (b) U. R. Pillai and E. Sahle-Demessie, Appl. Catal., A, 2004, 276, 139–144 CrossRef CAS; (c) H. Veisi, P. Safarimehr and S. Hemmati, J. Taiwan Inst. Chem. Eng., 2018, 88, 8–17 CrossRef CAS; (d) S. D. Kurbah, M. Asthana, I. Syiemlieh and R. A. Lal, Appl. Organomet. Chem., 2018, 32, e4299 CrossRef; (e) A. L. Cánepa, V. R. Elías, V. M. Vaschetti, E. V. Sabre, G. A. Eimer and S. G. Casuscelli, Appl. Catal., A, 2017, 545, 72–78 CrossRef; (f) R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS; (g) B. Eftekhari-Sis, M. Akbari, A. Akbari and M. Amini, Catal. Lett., 2017, 147, 2106–2115 CrossRef CAS.
- (a) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina and P. V. Petrovskiy, Appl. Organomet. Chem., 2010, 24, 464–472 Search PubMed; (b) L. S. Shul'pina, D. Veghini, A. R. Kudinov and G. B. Shul'pin, React. Kinet. Catal. Lett., 2006, 88, 157–163 CrossRef.
- (a) B. A. Steinhoff, S. R. Fix and S. S. Stahl, J. Am. Chem. Soc., 2002, 124, 766–767 CrossRef CAS PubMed; (b) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Science, 2000, 287, 1636–1639 CrossRef CAS PubMed; (c) P. Saxena and R. Murugavel, ChemistrySelect, 2017, 2, 3812–3822 CrossRef CAS.
- (a) V. Kogan, M. M. Quintal and R. Neumann, Org. Lett., 2005, 7, 5039–5042 CrossRef CAS PubMed; (b) G. Barak, J. Dakka and Y. Sasson, J. Org. Chem., 1988, 53, 3553–3555 CrossRef CAS.
- (a) R. V. Ottenbacher, E. P. Talsi and K. P. Bryliakov, Chem. Rec., 2018, 18, 78–90 CrossRef CAS PubMed; (b) C. Miao, X.-X. Li, Y.-M. Lee, C. Xia, Y. Wang, W. Nam and W. Sun, Chem. Sci., 2017, 8, 7476–7482 RSC; (c) G. B. Shulpin, G. Süss-Fink and L. S. Shulpina, J. Mol. Catal. A: Chem., 2001, 170, 17–34 CrossRef CAS.
- (a) M. H. Dickman and M. T. Pope, Chem. Rev., 1994, 94, 569–584 CrossRef CAS; (b) K. Sato, M. Aoki, J. Takagi and R. Noyori, J. Am. Chem. Soc., 1997, 119, 12386–12387 CrossRef CAS; (c) B. S. Chhikara, R. Chandra and V. Tandon, J. Catal., 2005, 230, 436–439 CrossRef CAS; (d) K. Sato, J. Takagi, M. Aoki and R. Noyori, Tetrahedron Lett., 1998, 39, 7549–7552 CrossRef CAS; (e) N. Gharah, S. Chakraborty, A. K. Mukherjee and R. Bhattacharyya, Inorg. Chim. Acta, 2009, 362, 1089–1100 CrossRef CAS; (f) S. E. Jacobson, D. A. Muccigrosso and F. Mares, J. Org. Chem., 1979, 44, 921–924 CrossRef CAS.
- (a) Y. Ishii, K. Yamawaki, T. Yoshida, T. Ura and M. Ogawa, J. Org. Chem., 1987, 52, 1868–1870 CrossRef CAS; (b) S. D. Kurbah, A. Kumar, I. Syiemlieh, M. Asthana and R. A. Lal, Inorg. Chem. Commun., 2017, 86, 39–43 CrossRef CAS.
- C. Crestini, M. C. Caponi, D. S. Argyropoulos and R. Saladino, Bioorg. Med. Chem., 2006, 14, 5292–5302 CrossRef CAS PubMed.
- (a) R. Behling, G. Chatel and S. Valange, Ultrason. Sonochem., 2017, 36, 27–35 CrossRef CAS PubMed; (b) P. B. Bhat, R. Rajarao, V. Sahajwalla and B. R. Bhat, J. Mol. Catal. A: Chem., 2015, 409, 42–49 CrossRef CAS.
- H. Ünver and I. Kani, J. Chem. Sci., 2018, 130, 33 CrossRef.
- (a) É. Balogh-Hergovich and G. Speier, J. Mol. Catal. A: Chem., 2005, 230, 79–83 CrossRef; (b) B. Stanje, P. Traar, J. A. Schachner, F. Belaja and N. C. Mösch-Zanetti, Dalton Trans., 2018, 47, 6412–6420 RSC.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- (a) S. Liang, L. Wen, S. Lin, J. Bi, P. Feng, X. Fu and L. Wu, Angew. Chem., Int. Ed., 2014, 53, 2951–2955 CrossRef CAS PubMed; (b) Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS; (c) K. Jing, J. Xiong, N. Qin, Y. Song, L. Li, Y. Yu, S. Liang and L. Wu, Chem. Commun., 2017, 53, 8604–8607 RSC; (d) X. Wang, D. Baiyil and X. Li, RSC Adv., 2016, 6, 107233–107238 RSC; (e) T. Li, S. Zhang, S. Meng, X. Ye, X. Fu and S. Chen, RSC Adv., 2017, 7, 6457–6466 RSC; (f) N. Gogoi, G. Borah, P. K. Gogoi and T. R. Chetia, Chem. Phys. Lett., 2018, 692, 224–231 CrossRef CAS; (g) X. Xiao, C. Zheng, M. Lu, L. Zhang, F. Liu, X. Zuo and J. Nan, Appl. Catal., B, 2018, 228, 142–151 CrossRef CAS; (h) S. Yurdakala, B. S. Tek, Ç. Değirmenci and G. Palmisano, Catal. Today, 2017, 281, 53–59 CrossRef; (i) M. Zhang, Q. Wang, C. Chen, L. Zang, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081–6084 CrossRef CAS PubMed; (j) S. Higashimoto, K. Okada, T. Morisugi, M. Azuma, H. Ohue, T.-H. Kim, M. Matsuoka and M. Anpo, Top. Catal., 2010, 53, 578–583 CrossRef CAS; (k) T. W. Goh, C. Xiao, R. V. Maligal-Ganesh, X. Li and W. Huang, Chem. Eng. Sci., 2015, 124, 45–51 CrossRef CAS.
- (a) A. E. C. Collis and I. T. Horváth, Catal. Sci. Technol., 2011, 1, 912–919 RSC; (b) D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed.
- (a) K. C. Gupta, A. K. Sutar and C.-C. Lin, Coord. Chem. Rev., 2009, 253, 1926–1946 CrossRef CAS; (b) C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275–3300 CrossRef CAS PubMed; (c) P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515–529 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- S. B. Roscoe, C. Gong, J. M. J. Fréchet and J. F. Walzer, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2979–2992 CrossRef CAS.
- G. J.-J. Chen, J. W. McDonald and W. E. Newton, Inorg. Chem., 1976, 15, 2612–2615 CrossRef CAS.
- J. M. Frechet and C. Schuerch, J. Am. Chem. Soc., 1971, 93, 492–496 CrossRef.
- (a) B. Yan and M. Guo, J. Photochem. Photobiol., A, 2013, 257, 34–43 CrossRef CAS; (b) L. Sen, W. Kuan, C. K.-C. Alan, Z. Ming, W. Jing, C. Y. Gang, B. YuHua, W. TaiMing and Z. ZhiRen, Sci. China: Chem., 2012, 55, 1134–1139 Search PubMed; (c) J.-W. Kim, K.-J. Kim, S. Park, K.-U. Jeong and M.-H. Lee, Bull. Korean Chem. Soc., 2012, 33, 2966–2970 CrossRef CAS.
- (a) J.-G. Choi and L. T. Thompson, Appl. Surf. Sci., 1996, 93, 143–149 CrossRef CAS; (b) Y. Li, X. Fu, B. Gong, X. Zou, X. Tu and J. Chen, J. Mol. Catal. A: Chem., 2010, 322, 55–62 CrossRef CAS; (c) P.-S. E. Dai and J. H. Lunsford, J. Catal., 1980, 64, 173–183 CrossRef CAS; (d) J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of XPS, PerkinElmer Corporation, Eden Prairie, MN, 1992 Search PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- (a) J. J. Boruah, S. P. Das, S. R. Ankireddy, S. R. Gogoi and N. S. Islam, Green Chem., 2013, 15, 2944–2959 RSC; (b) D. T. Gokak and R. N. Ram, J. Mol. Catal., 1989, 49, 285–298 CrossRef CAS.
- (a) K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CAS; (b) S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, London, 1982 Search PubMed.
- S.-J. Park, D.-I. Seo and C. Nah, J. Colloid Interface Sci., 2002, 251, 225–229 CrossRef CAS PubMed.
- (a) V. B. Valodkar, G. L. Tembe, M. Ravindranathan and H. S. Rama, React. Funct. Polym., 2003, 56, 1–15 CrossRef CAS; (b) V. B. Valodkar, G. L. Tembe, M. Ravindranathan, R. N. Ram and H. S. Rama, J. Mol. Catal. A: Chem., 2003, 202, 47–64 CrossRef CAS.
- (a) Md. M. Islam, A. S. Roy and Sk. M. Islam, Catal. Lett., 2016, 146, 1128–1138 CrossRef CAS; (b) D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introduction to Spectroscopy, 5th edn, Cengage Learning, USA, 2015 Search PubMed; (c) R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley and Sons, New York, 7th edn, 2005 Search PubMed.
- P. F. Rapheal, E. Manoj and M. R. P. Kurup, Polyhedron, 2007, 26, 818–828 CrossRef CAS.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Co-ordination Compounds, Part B, Wiley and Sons, New York, 5th edn, 1997 Search PubMed; (b) K. H. Reddy, M. R. Reddy and K. M. Raju, Indian J. Chem., 1999, 38A, 299–302 CAS; (c) B. S. Garg, M. R. P. Kurup, S. K. Jain and Y. K. Bhoon, Transition Met. Chem., 1991, 16, 111–113 CrossRef CAS.
- (a) M. R. Maurya, S. Sikarwar, T. Joseph, P. Manikandan and S. B. Halligudi, React. Funct. Polym., 2005, 63, 71–83 CrossRef CAS; (b) M. R. Maurya, U. Kumar and P. Manikandan, Dalton Trans., 2006, 3561–3575 RSC.
- (a) B. Altava, M. I. Burguete, E. Garcia-Verdugo, S. V. Luis and M. J. Vincent, Tetrahedron, 2001, 57, 8675–8683 CrossRef CAS; (b) D. Lin-Ven, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of the Organic Molecules, Academic, London, 1991 Search PubMed; (c) J. R. Ferraro and K. Nakamoto, Introductory Raman Spectroscopy, Academic, London, 1994 Search PubMed; (d) M. J. Pelletier, Analytical Applications of Raman Spectroscopy, Blackwell, Oxford, 1999 Search PubMed.
- (a) F. E. Kuhn, M. Groarke, E. Bencze, E. Herdtweck, A. Prazeres, A. M. Santos, M. J. Calhorda, C. C. Romao, I. S. Goncalves, A. D. Lopes and M. Pillinger, Chem. - Eur. J., 2002, 8, 2370–2383 CrossRef PubMed; (b) L. J. Willis, T. M. Loehr, K. F. Miller, A. E. Bruce and E. I. Stiefel, Inorg. Chem., 1986, 25, 4289–4293 CrossRef CAS.
- (a) A. O. Sobola, G. M. Watkins and B. V. Brecht, S. Afr. J. Chem., 2014, 67, 45–51 Search PubMed; (b) A. O. Sobola, G. M. Watkins and B. V. Brecht, J. Serb. Chem. Soc., 2018, 83, 1–12 CrossRef.
- (a) G. Lisa, E. Avram, G. Paduraru, M. Irimia, N. Hurduc and N. Aelenei, Polym. Degrad. Stab., 2003, 82, 73–79 CrossRef CAS; (b) W. Mo, H. Liu, H. Xiong, M. Li and G. Li, Appl. Catal., A, 2007, 333, 172–176 CrossRef CAS.
- J. J. Boruah, D. Kalita, S. P. Das, S. Paul and N. S. Islam, Inorg. Chem., 2011, 50, 8046–8062 CrossRef CAS PubMed.
- (a) M. Uyanik, D. Suzuki, T. Yasui and K. Ishihara, Angew. Chem., 2011, 123, 5443–5446 CrossRef; (b) M. Uyanik and K. Ishihara, ChemCatChem, 2012, 4, 177–185 CrossRef CAS.
- (a) K. Amakawa, Y. V. Kolen'ko, A. Villa, M. E. Schuster, L.-I. Csepei, G. Weinberg, S. Wrabetz, R. N. d'Alnoncourt, F. Girgsdies, L. Prati, R. Schlögl and A. Trunschke, ACS Catal., 2013, 3, 1103–1113 CrossRef CAS; (b) C. J. Carrasco, F. Montilla, E. Álvarez, C. Mealli, G. Mancac and A. Galindo, Dalton Trans., 2014, 43, 13711–13730 RSC; (c) B. Trost and Y. Masuyam, Isr. J. Chem., 1984, 24, 134–143 CrossRef CAS; (d) M. R. Maurya, M. Kumar and U. Kumar, J. Mol. Catal. A: Chem., 2007, 273, 133–143 CrossRef CAS; (e) M. R. Maurya, A. K. Chandrakar and S. Chand, J. Mol. Catal. A: Chem., 2007, 274, 192–201 CrossRef CAS.
- (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS; (b) H. Szatylowicz, O. A. Stasyuk and T. M. Krygowski, Adv. Heterocycl. Chem., 2015, 116, 137–192 CrossRef CAS; (c) H. H. Jaffe and G. O. Doak, J. Am. Chem. Soc., 1955, 77, 4441–4444 CrossRef CAS.
- (a) R. Malakooti and A. Feghhi, New J. Chem., 2017, 41, 3405–3413 RSC; (b) A. V. Biradar, M. K. Dongare and S. B. Umbarkar, Tetrahedron Lett., 2009, 50, 2885–2888 CrossRef CAS; (c) S. Campestrini and F. D. Furia, Tetrahedron, 1994, 50, 5119–5130 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: N2 adsorption–desorption isotherms, IR spectra of the isolated peroxomolybdate(VI) intermediate, comparison table, and detailed synthetic procedure and characterization of SB-Mo. See DOI: 10.1039/c8ra05969a |
| ‡ Both the authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |