
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of newly emerging psychoactive substances using Raman spectroscopy and chemometrics†
Jesus Calvo-Castro‡
 ,
Amira Guirguis‡,
Eleftherios G. Samaras,
Mire Zloh,
Stewart B. Kirton* and
Jacqueline L. Stair*
,
Amira Guirguis‡,
Eleftherios G. Samaras,
Mire Zloh,
Stewart B. Kirton* and
Jacqueline L. Stair*
Department of Pharmacy, Pharmacology and Postgraduate Medicine, School of Life and Medical Sciences, University of Hertfordshire, Hatfield, AL10 9AB, UK. E-mail: s.b.kirton3@herts.ac.uk; j.stair@herts.ac.uk
First published on 12th September 2018
Abstract
A novel approach for the identification of New Psychoactive Substances (NPS) by means of Raman spectroscopy coupled with Principal Components Analysis (PCA) employing the largest dataset of NPS reference materials to date is reported here. Fifty three NPS were selected as a structurally diverse subset from an original dataset of 478 NPS compounds. The Raman spectral profiles were experimentally acquired for all 53 substances, evaluated using a number of pre-processing techniques, and used to generate a PCA model. The optimum model system used a relatively narrow spectral range (1300–1750 cm−1) and accounted for 37% of the variance in the dataset using the first three principal components, despite the large structural diversity inherent in the NPS subset. Nonetheless, structurally similar NPS (i.e., the synthetic cannabinoids FDU-PB-22 & NM-2201) grouped together in the PCA model based on their Raman spectral profiles, while NPS with different chemical scaffolds (i.e., the benzodiazepine flubromazolam and the cathinone α-PBT) were well delineated, occupying markedly different areas of the three-dimensional scores plot. Classification of NPS based on their Raman spectra (i.e., chemical scaffolds) using the PCA model was further investigated. NPS that were present in the initial dataset of 478 NPS but were not part of the selected 53 training set (validation set) were observed to be closely aligned to structurally similar NPS within the generated model system in all cases. Furthermore, NPS that were not present in the original dataset of 478 NPS (test set) were also shown to group as expected in the model (i.e., methamphetamine and N-ethylamphetamine). This indicates that, for the first time, a model system can be applied to potential ‘unknown’ psychoactive substances, which are new to the market and absent from existing chemical libraries, to identify key structural features to make a preliminary classification. Consequently, it is anticipated that this study will be of interest to the broad scientific audience working with large structurally diverse chemical datasets and particularly to law enforcement agencies and associated scientific analytical bodies worldwide investigating the development of novel identification methodologies for psychoactive substances.
Introduction
The market for New Psychoactive Substances (NPS) has been characterised by its huge variety and diversity. In the last decade there has been an increasingly large and rapid appearance of newly emerging drugs, with more than 700 currently monitored.1–3 In most cases, this can be attributed to efforts by suppliers to evade their detection and circumvent existing legislation.1,4 This rapid appearance, poor detectability, increased potency and availability of newly emerging drugs is often accompanied by an increase in reported health harms, emergency department visits and even fatalities, making NPS a major public health concern.1,2 In an attempt to restrict the production, supply and abuse of NPS, countries such as New Zealand, and more recently the United Kingdom, have approved psychoactive substances Acts, often referred to as ‘blanket bans’.5,6 Current NPS legislation however represents an even greater challenge for law enforcement units and associated scientific bodies trying to develop more efficient, sensitive and selective detection methodologies, particularly for the front line (i.e. customs), that could possibly match the rapid and continuous surge of newly abused substances.To date, the majority of spectroscopic and chromatographic analytical techniques employed in the detection of NPS rely on, and are restricted to, reference standard availability.1 This often precludes the application of such approaches to the identification of newly appearing substances, which is crucial in reducing the potential harms caused by NPS. Subsequently, efforts have been devoted to the development of selective and sensitive detection methodologies that build upon the modulation of analyte response.1,7–9 However, it is acknowledged that such approaches are currently limited to a discrete number of traditional drugs of abuse or well established NPS, requiring time-consuming development and optimisation of techniques for each new substance. Accordingly, there remains a need for more universal in-field identification methodologies that can not only effectively identify existing NPS substances, but more importantly be applicable for newly emerging drugs. To address these shortcomings, a number of studies have engaged in the rational design and realisation of novel methodologies and algorithms for the in-field detection of NPS.8,10–19 Along these lines, a number of vibrational spectroscopy techniques (i.e. IR, NIR and Raman) which can be realised in portable, handheld instruments have been successfully utilised for the in-field identification of NPS. Among these, Raman spectroscopy shows great potential due to desirable properties such as non-contact/non-destructive analysis; and low sensitivity to cutting agents/adulterants, moisture and the physical properties of the sample.18,20–24
An added complexity in the development of novel approaches for the identification of NPS arises from the fact that existing NPS are often classified using a pragmatic approach (i.e., based on pharmacological activity,25,26 chemical structure, etc.), which causes difficulty in interpreting the control status of an NPS. Also, NPS classification is constantly being modified and changed based on popularity, current trends, new evidence and legislation.15,27 As a result, our recent work28 attempts to address the existing issues with regards to the classification of NPS, where a dataset of 478 NPS was systematically categorised according to their chemical structural features. Using a software based on a technique called hierarchical clustering, the NPS investigated were divided into 21 categories based on all compounds in the category sharing a common structural core referred to as the maximum common substructure (MCS).27 These top-level categories were broken down further into 79 subcategories (13 of which contained only a single molecule i.e. compounds that were significantly structurally distinct from all other known NPS, also referred to as singletons). As such, it was hypothesised an adequately selected subset of NPS compounds could be used to represent the entire structural diversity of the dataset (478 NPS) and that furthermore, this subset could aid in the identification and/or prediction of key structural features of both known and newly emerging NPS.
Thus, the aim of this study was to develop a model that can be used to identify chemical structural features of NPS, including newly emerging NPS, using Raman spectral profiles and Principal Component Analysis (PCA).
Experimental
Selection of training, validation and test set
In our previous work,28 79 NPS were identified as representatives of the chemical space inherent in the 478 NPS dataset. Fifty-three (SI.1†) of these 79 NPS were selected and purchased to be used as the training set following the exclusion of singletons (i.e., a molecule that did not share significant structural similarity with any other NPS in the chemical space studied) and availability. An additional 21 NPS were used to further validate and test the generated model. These were split into two different groups, the validation set (17) and test set (4), where validation set NPS (SI.2†) were compounds that were part of the initial 478 compound dataset but not used as training set, and test set substances, that were psychoactive drugs external to the initial dataset of 478 compounds (SI.3†).Dissimilarity calculations
Structural similarity between all NPS investigated in this work was quantified by calculating the pairwise dissimilarity values (i.e. 1 – tanimoto similarity value, determined from a chemical fingerprint) using the ChemAxon JChem software suite.29New psychoactive substances
Reference standard materials (purity ≥ 98%) for training, validation and test sets were purchased in powder form from Chiron AS (Trondheim, Norway) and LGC Group (Teddington, UK) in all cases unless otherwise stated and used as supplied. Controlled substances were purchased under UK Home Office License.Raman spectroscopy
Raman analyses were carried out utilising a benchtop Renishaw inVia Raman microscope equipped with a high sensitivity ultra-low noise RenCam CCDC detector and ultra-high precision diffraction grating of 1200 lines per mm. The instrument was operated using the WiRE software supplied by the manufacturer. All reported spectra were measured employing a 785 nm excitation wavelength with ca 5.8 mW power at sample. NPS standards were analysed on Al plates and interrogated a total of 10 times focusing on different regions of the powder to account for anisotropic effects.Subsequently, the experimentally acquired Raman spectra (100–3200 cm−1) were pre-processed prior to multivariate analysis. Spectra were smoothed, and baseline subtracted by means of the Savitzky–Golay algorithm as implemented in OriginPro 2016 software to reduce shot/residual noise and raised baseline respectively. Maximum normalisation was then applied to each spectrum individually.
Principal component analysis
Pre-processed Raman spectra were initially critically assessed using PCA, a multivariate technique that has previously been employed in the analysis of spectroscopic data.16,30–32 For this study, the NIPALS (Non-linear Iterative Projections by Alternating Least Squares) algorithm was implemented in the Unscrambler X 10.4 software (CAMO, Oslo, Norway). Each generated model was full crossed validated (one sample per segment). Validation and test-set representatives were tested against the generated model system by means of projection PCA.Results and discussion
Three-dimensional model system using NPS training set
In order to develop a model system, the Raman spectra of the NPS training set were examined using PCA. Given the large size of data matrix (530 spectra × 3642 variables), a systematic step-wise data reduction methodology was explored to identify the optimum spectral region of interest whilst maximising the amount of explained variance, particularly by the first three principal components. In short, it was observed that a decrease in the number of spectral replicates from 10 to 5, then 3 and finally 1 had a negative impact with respect to the model robustness. In turn, a systematic and rational selection of spectral regions of interest underpinned by the in-depth analysis of line loading plots for the generated PCA models resulted in a consistent improvement in the amount of explained variance for the dataset (i.e., 11/9/6 and 16/12/9% explained variance for PC1/PC2/PC3 using the 250–3200 and 1300–1750 cm−1 spectral ranges respectively).A PCA model system was developed (Fig. 1) comprising 10 replicate spectra for each of the (53) NPS in the training set and a narrow33 spectral region of interest (from 1300 to 1750 cm−1), that still contained the majority of the spectral information for all investigated NPS and yielded the largest explained variance from all investigated spectral regions (vide supra). Moreover, the optimised model accounted for 37% of the variance using the first three principal components. This arguably low amount of explained variance can be attributed to the intrinsic chemical diversity of the training set, a selected subset of 53 representatives from a dataset of 478 NPS. Nevertheless, as illustrated in Fig. 1, it is important to note the discriminative ability of the generated model. NPS previously shown to assemble together in hierarchical clustering experiments28 based on chemical connectivity (Categories 1–13), largely group together and furthermore occupy distinctly different areas of the three-dimensional scores plot with respect to Raman active chemical scaffolds. Again, the structural similarity between NPS in the training set drawn from each different structural category can be seen in SI.1.† Examination of different regions of the scores plot arising from generating the PCA model will be discussed further below by focusing on particular categories of compounds from Fig. 1.
 | ||
| Fig. 1 A three-dimensional scores plot generated using Raman spectra for the NPS training set (ten replicate spectra per substance, spectral region: 1300–1750 cm−1). Refer to SI.1† for details on the members belonging to each of the categories identified by hierarchical clustering. | ||
Fig. 2 shows Category 1 compound data whose members share an indole core (depicted in black on the chemical structure) isolated with example spectra. Via judicious analysis of the three-dimensional scores plot, it was observed that training set NPS bearing indole cores occupy two distinct regions, primarily delineated by PC2, which agrees with current pragmatic classifications.2 For example, the compounds FDU-PB-22 and NM-2201 group together and are known synthetic cannabinoids, whilst 5-MeO-DALT, 5-MeO-MiPT and 4-HO-DET group together in a different area of the scores plot and are known tryptamines (Fig. 2).
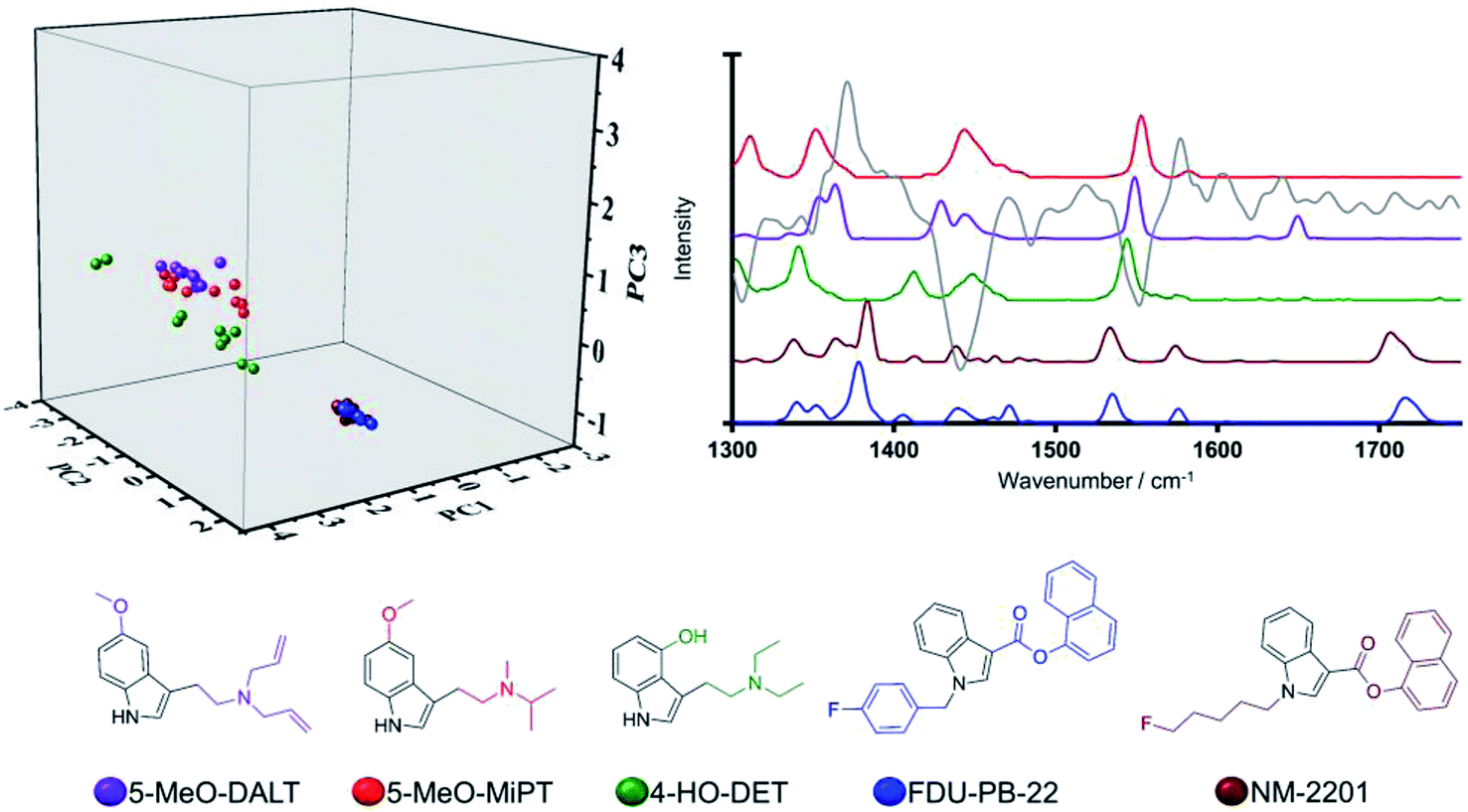 | ||
| Fig. 2 Three-dimensional scores plot (left), Raman spectral profile (right) (grey solid line denotes PC2 loadings) and chemical structures of training set NPS sharing an indole core (illustrated in black on their chemical structures). | ||
This finding can be ascribed to the different substitution patterns on position 3 of the indole cores in these synthetic cannabinoids and tryptamines, leading to distinct spectral profiles in the region of interest as illustrated in Fig. 2. The different locations observed on the scores plot can be furthermore accounted for by the loading plot for PC2 (grey solid line in Fig. 2), exhibiting high loading at a vibrational frequency that coincides with vibrational bands uniquely observed in the Raman profiles for the three tryptamines at ca. 1560 cm−1 and attributed to quadrant stretching vibrational motions of the indole core,33,34 bearing substitution on the N atoms, which are not present in the synthetic cannabinoids counterparts. This example demonstrates that although all these substances share an indole core, their specific substitution patterns were distinguished via their Raman profile and delineated by the PCA scores plot. Thus, there is ability to predict not only a core structure but also to suggest the unique substitution pattern of a previously ‘unknown’ NPS. Another key example in the model system is illustrated by the compounds in Category 5. In our previous work,28 the largest number (n = 18/53) of NPS representatives were drawn from Category 5. The large number of compounds in this category is a result of the MCS being a simple benzene ring. Thus, in the initial dataset of 478 compounds Category 5 contains more members than any other category of compounds. Despite the diversity in this category (see SI.1†), all 18 representative NPS were observed to closely group along the two first principal components in the PCA model, with delineation amongst the group occurring due to separation of the molecules along PC3 (see Fig. 1). This is demonstrated by the only two substances belonging to the quinazoline class in the training set; afloqualone and mebroqualone, (Fig. 3). The delineation of these two molecules along PC3 can be ascribed to large similarities between the PC3 loading plot and the spectral profile of afloqualone in the region of interest, i.e. the vibrational band associated with the carbonyl stretching motion at ca. 1670 cm−1. A higher frequency (ca. 1690 cm−1) is observed for mebroqualone, which can be attributed to an intramolecular H-bonding interaction, precluded in afloqualone upon o-methyl substitution a difference, which allows for the separation of these molecules along PC3.
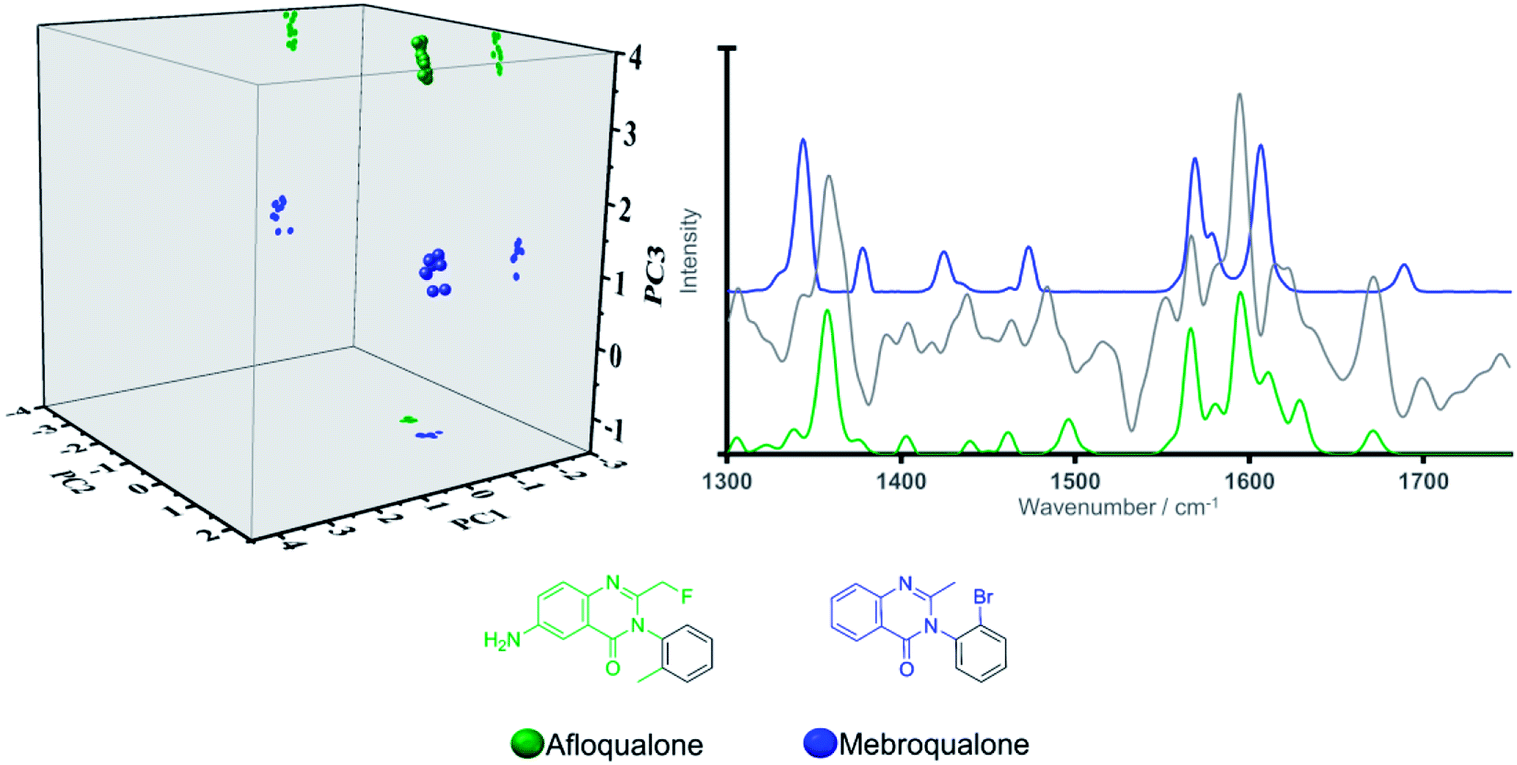 | ||
| Fig. 3 Three-dimensional scores plot (left) (projections illustrated for ease of visualization), Raman spectral profiles (right) (grey solid line denotes PC3 loadings) and chemical structures of the quinazoline containing structures, afloqualone and mebroqualone. | ||
The third example is focused on Category 9, whose representative structures are two thiophenyl containing structures (depicted in black on the chemical structure in Fig. 4), which is not present in the MCS of any other of the 21 categories identified by hierarchical clustering. The two substances, MPA (arylalkylamine) and α-PBT (cathinone), were evaluated with respect to the PCA model scores plot, and their Raman spectra. Although these are the only two thiophenyl containing structures in the training set, a detailed understanding of Category 9 is anticipated to play a crucial role in accounting for newly emerging NPS with a unique scaffold, such as thiophene-based compounds. In the three-dimensional scores plot (Fig. 4), MPA and α-PBT are closely grouped along PC1. In turn, clear delineation between the two is achieved along PC2 and to a lesser extent PC3. In this regard, delineation along PC2 can be readily understood as a consequence of the high negative loading of this principal component at ca. 1440 cm−1, often associated with aliphatic stretching vibrational motions, which coincides with the highest intensity vibrational band of MPA in the selected spectral region. In addition, neither PC1 nor PC3 are characterised by particularly high loadings that coincide with vibrational frequencies of significant intensity for MPA or α-PBT, hence the lack of significant delineation along these two principal components is not unexpected. Thus, based on spectral region selected, delineation occurs based on the functionalities substituted on the thiophene groups (i.e. carbonyl group present in α-PBT and absent in MPA).
 | ||
| Fig. 4 Three-dimensional scores plot (left) (projections illustrated for ease of visualization), Raman spectral profile (right) (grey solid line denotes PC2 loadings) and chemical structures of the thiophenyl containing structures, α-PBT and MPA. | ||
Evaluation of model system using validation set NPS
The model system was then tested by using NPS from the original 478 substance dataset28 that were not designated as training set compounds (henceforth referred to as the “validation set”). Contrary to the training set, validation set scaffolds (SI.2†) were selected to exhibit larger dissimilarity with respect to their category medoids i.e. the members of a given category that are structurally most similar, on average, to all other members in that category. Dissimilarities greater than 0.200 were calculated for 10 out of 17 (59%) validation set NPS, as opposed to 2 out of 53 (4%) for the training set,28 thus posing a genuine challenge for the evaluation of the PCA model. It was of interest to investigate the performance (amount of explained variance) of the PCA model with respect to the validation set. To achieve this, the generated model system was used to calculate the amount of explained variance within the validation set without altering the generated model system (i.e. via projection of the validation set onto the PCA model). In this regard, percentages of explained variance for the validation subset that approach that of the training set would indicate a robust model system able to account for newly emerging NPS. Performance of the model, with the validation set (15/11/7% for PC1/PC2/PC3 respectively regarding the percentage of explained variance) was comparable to that achieved by the model for the training set (16/12/9 for PC1/PC2/PC3 respectively). Hence, the model was considered robust and predictive.The next step used to evaluate the performance of the model with respect to the validation set was individual comparisons of each substance in the validation set compared to the category representative used in the 53-molecule training set (MCS for the compounds in each category are shown in black). Each substance in the validation set had a pre-determined category designation (see SI.2†),28 which was used as the basis for determining model success (i.e. if the model predicted the compound to be in the same category as the pre-determined categorisation, it was deemed successful). It was observed that, in all cases, validation set substances were closely grouped with structurally similar training set NPS when projected onto the model. Key examples to demonstrate this are highlighted below.
Due to its scaffold being present in a wide-range of NPS, it was considered of particular interest to examine validation set NPS with the phenylethylamine backbone (again illustrated in black) from Category 2, namely 5-APB, 6-APB and βk-2C-B. Related NPS from the training set were N-Me-2-CB and 2,5-dimethoxy-4-methylamphetamine (DOM or STP). This allowed for an in-depth analysis of the relative position of the validation samples in the three-dimensional scores plot. Examination of the three-dimensional scores plot illustrated in Fig. 5, illustrated close grouping of the arylalkylamines from the validation set (5-APB and 6-APB) with STP from the validation set. This can be ascribed to the presence of a medium-intensity peak in all three Raman spectra at ca. 1620 cm−1 and the high loadings registered for PC1 and PC3 in this spectral region.
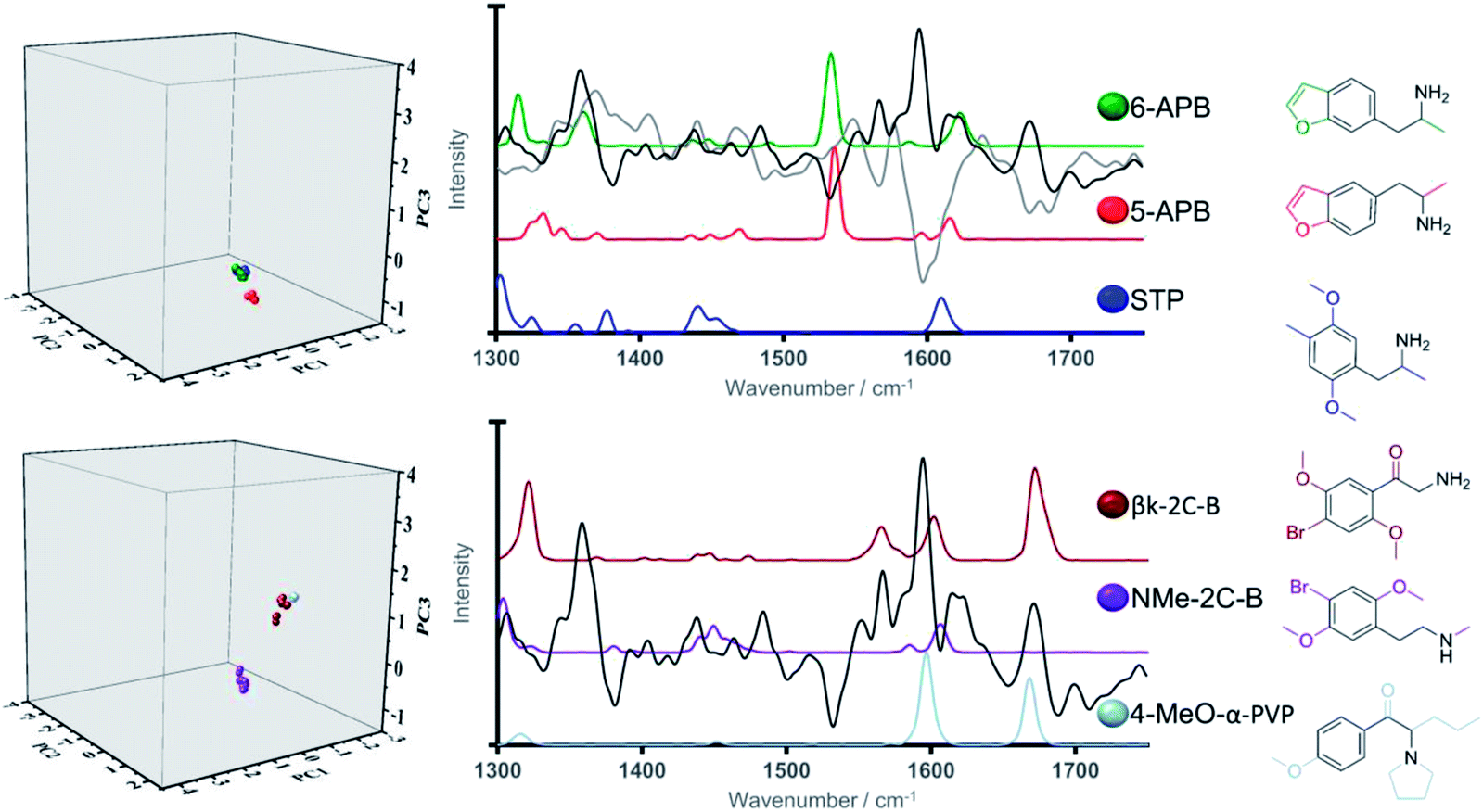 | ||
| Fig. 5 Three-dimensional score plots (left), Raman spectral profiles (right) (solid grey and black lines denote PC1 and PC3 loadings respectively) and chemical structures for training and validation set NPS bearing phenylmethanamine core. | ||
In turn, greater delineation between βk-2C-B and its training set counterpart, NMe-2C-B was illustrated, particularly along PC3. Closer analysis shows βk-2C-B to exhibit close alignment to one of the other phenylethylamine containing structures within the training set, 4-MeO-α-PVP. This finding is validation for the model system and also reinforces its discriminative power which can be ascribed structurally to the presence of carbonyl groups (absent in NMe-2C-B) in both βk-2C-B and 4-MeO-α-PVP, i.e. the latter two are both cathinones. This carbonyl functionality is represented spectrally by a vibrational band at ca. 1650 cm−1, which coincides with a region of high loading for PC3. Therefore, we anticipate that particular functional groups and associated substitution patterns that exhibit distinct Raman active bands can play a crucial role in the identification and correct classification of newly emerging NPS.
The second key example compares the popular synthetic cannabinoid 5F-PB-22 from the validation set which is most similar to phenyl acetates from the training set (Category 6 members). Classification of 5F-PB-22 via the PCA model system (Fig. 6) revealed a closer alignment to its non-fluorinated training set analogue, PB-22, than to the other phenyl acetate containing structures within the training set, namely N-PB-22, a synthetic cannabinoid and 4-AcO-DMT, a tryptamine. PB-22 and its fluorinated structural analogue, 5F-PB-22, exhibited very close alignment in the three-dimensional scores plot along all three principal components in line with their closely related chemical structures and Raman spectral profiles. This further validates the strength of the model system. The Raman spectral profile throughout the region of interest featured medium to high intensity vibrational bands centred at ca. 1382, 1425, 1531 and 1711 cm −1. The lack of significant spectral differences between these two synthetic cannabinoids is ascribed to the fluorine substitution carried out on the terminal position of the long aliphatic chain, hence precluding strong polarizability changes that would result in distinct spectral features in the region of 1300–1750 cm−1. Along these lines, terminal halogen substitutions have become popular strategies in the design of novel psychoactive substances.35–37 Herein, we have demonstrated that their negligible impact on the Raman spectral properties facilitates their identification based on previously existing non-halogenated analogues.
 | ||
| Fig. 6 Three-dimensional scores plot (left), Raman spectral profile (right) (solid light grey line denote PC2 loadings) and structures for phenyl acetate containing systems, N-PB-22, PB-22, 4-AcO-DMT (training set) and 5F-PB-22 (validation set). | ||
All three synthetic cannabinoids in Fig. 6, (5F-PB-22, PB-22 and N-PB-22) were observed to be closely aligned in three-dimensional scores plot, which can be accounted for by means of the vibrational bands at ca. 1382, 1425 and 1577 cm−1 all of which are present in the Raman profiles of the three compounds and that are ascribed to vibrational motions within their common quinoline motif.
In turn, the tryptamine, 4-AcO-DMT, bears an indole core instead, which was observed to result in small shifts to these vibrational bands (centred at ca. 1390, 1438 and 1550 cm−1). Thus, the delineation in the three-dimensional scores plot observed between these phenyl-acetate containing synthetic cannabinoids and tryptamine representative, particularly along PC1, can be accounted for on the basis of the observed PC1 high loadings at ca. 1438 and 1550 cm−1, which strikingly coincide with strong vibrational bands of 4-AcO-DMT and absent in the spectral profile of the three synthetic cannabinoids.
The third example selected from the validation set is the benzodiazepine, pyrazolam, as it possesses a unique fused herterocycle ring system. Fig. 7 looks at pyrazolam with regards to the structurally related training set benzodiazepines (Category 8), etizolam and flubromazolam. All three structures bear the 3,7-dimethyl-9H-[1,2,4]triazolo[4,3-a][1,4]diazepine core (depicted in black on the chemical structure). In most cases, benzodiazepines include a 7-membered ring, an additional benzene ring and an electron attracting group at position 7 of the fused heterocyclic rings to ensure biological activity.38 Along these lines, benzodiazepines are commonly sub-categorised according to the functional group attached to the 7-membered ring, which may include keto, hydroxyl, imidazo or triazolo groups. The three NPS in Fig. 7 have a triazolo group as the functional group. In-depth analysis of the validation set projection on the PCA model reveals clear delineation between pyrazolam and etizolam (along PC2 and PC3) and in turn close alignment of the validation compound pyrazolam with flubromazolam along PC1 and PC3 and, to a lesser extent, along PC2.
 | ||
| Fig. 7 Three-dimensional scores plot (left), Raman spectral profile (right) (solid black and grey lines denote PC3 and PC2 line loadings plots respectively) and chemical structures for flubromazolam, etizolam and pyrazolam. | ||
Close examination of their Raman spectral profiles in the region of interest and associated line loadings reveals coinciding strong Raman active bands at ca. 1440 and 1593 cm−1 in flubromazolam and pyrazolam (absent in etizolam) with high intensity loadings along PC2 and PC3 respectively (Fig. 7). Structurally, this can be attributed to the fused thiophene ring which is present in etizolam and absent in its counterparts which bear a fused benzene ring to the pyrimidine ring instead. Thus, the pyrazolam was positioned closely in the model to the most structurally similar benzodiazepine training structure which provides validation, but once again reinforces the delineation capability of the generated model system.
Evaluation of the model system using the test set
The performance of the model system was then evaluated using ‘unknown’ psychoactive substances external to the initial 478 NPS dataset (in the following denoted as test set, SI.3†). The purpose of this section is to evaluate the model's capability to propose chemical scaffolds for previously unknown substances (i.e., unknown with respect to the dataset used to create the initial model). To do this, the dissimilarity scores (vide supra) of the test set compounds, namely MDMA, methamphetamine, S-cathinone and methylphenidate, to all cluster medoids were calculated and corresponding spectra investigated using PCA.In this regard, it was observed that methylphenidate represents a complex scenario with a lowest dissimilarity value (0.540) computed against training set representatives 4-MeO-PCP, 4-MeO-α-PVP and 4-Me-N-ethylnorpentedrone, which further illustrates the complexity posed by newly emerging NPS. In light of this arguably high dissimilarity scores calculated for methylphenidate, we anticipate that this derivative would have been classified as a singleton in our original 478 NPS dataset.
Interestingly, projections of MDMA, methamphetamine and S-cathinone on the three-dimensional scores plot (Fig. 8) exhibit close alignment to the training set molecules that have the closest structural similarity (or lowest dissimilarity scores) i.e., methylone (0.190), N-ethyl-amphetamine (0.090) and mephedrone (0.170) respectively. These findings were further explored by the analysis of their Raman spectral profiles in the region of interest. It was observed that the ‘pair’ exhibiting the lowest dissimilarity scores (N-ethylamphetamine/methamphetamine) was characterised by the closest alignment in the three-dimensional scores plot and that furthermore, the ‘pair’ methylone/MDMA were the furthest apart in the three-dimensional scores plot, in line with their computed highest dissimilarity score (0.190) for these three investigated ‘pairs’. The low dissimilarity score (0.090) and close location of N-ethylamphetamine and methamphetamine can be ascribed to their high structural similarity, solely differing in the ethyl/methyl substitution on the nitrogen, which was accounted for in the Raman spectra. In turn, we observed larger distances within the pairs S-cathinone/mephedrone and methylone/MDMA, in line with their calculated larger dissimilarity score (0.170 and 0.190 respectively).
 | ||
| Fig. 8 Three-dimensional scores plot, Raman spectral profiles (grey solid line denotes line loadings plot for PC3) and structures for test set representatives and associated training set compounds. | ||
In the case of the pair formed by S-cathinone and mephedrone, both synthetic cathinones, their delineation along PC3 in the three-dimensional scores plot can be ascribed to close alignment of the peak centered at ca 1590 cm−1 with a high PC3 loading. Discriminating between the different structural analogues of synthetic cathinones has been reported to be often afforded by means the position of the two high intensity Raman active bands at ca 1600 and 1700 cm−1 and their relative intensities.14,17,19,39 However, successful identification is limited by the availability of reference standard materials in the chemical libraries used. In addressing this limitation, it is anticipated the ability of our generated model system in delineating structural analogues of synthetic cathinones by means of the high intensity loadings of PC1 and PC3 at ca 1600 cm−1 (SI.4†).
MDMA projection is delineated with respect to its least dissimilar training set representative (methylone) along the third principal component. We observed this to be largely associated to the strong vibrational peak at ca 1680 cm−1 from the carbonyl group in methylone, which is absent in MDMA and that importantly coincides with a region of high loadings in PC3 (Fig. 8). Along these lines, it is of interest that our findings agree with previous reports whereby samples containing MDMA were differentiated from those containing cocaine based on the absence of a carbonyl group in MDMA, which is present in the chemical structure of cocaine.40
Accordingly, it has been demonstrated the optimum performance of the selected training set representatives in accounting for the large structural diversity of NPS and the subsequent associated ability of the proposed model system to account for newly emerging architectures.
Conclusions
A three-dimensional model system was successfully demonstrated for an NPS dataset possessing an inherently large structural diversity, using Raman spectroscopy with PCA. Due to this diversity, a systematic optimisation process was required (i.e., spectral pre-processing and reduction of the spectral range) to achieve the maximum explained variance (37% for the three first principal components), whilst retaining model robustness. The predictive potential was evaluated using both a validation and a test set, which in all cases these substances were closely aligned in the three-dimension scores plots with respect to their structurally related NPS training set. This demonstrates the utility of combining Raman spectroscopy (where the signal generated is restricted to Raman active functional groups) with a multivariate approach for model generation for a structurally diverse dataset. Future work should focus on implementing models such as these in Raman handheld instruments for use in-field. Thus, the results presented herein will be invaluable to a wide chemistry audience working with large and structurally complex datasets as well as to the growing scientific community developing novel identification methodologies for NPS and the targeted end-users of this technology (e.g., health care professionals, law enforcement and border control).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the European Commission for funding under the Drug Prevention and Information Programme 2014–16, contract no. JUST/2013/DPIP/AG/4823, EU-MADNESS project and JUST/ISEC/DRUGS/AG/6428, EPSNPS project.Notes and references
- P. I. Dargan and D. M. Wood, Novel Psychoactive Substances: Classification, Pharmacology and Toxicology, Elsevier, 2013 Search PubMed.
- E. Cuypers, A.-J. Bonneure and J. Tytgat, Drug Test. Anal., 2016, 8, 136–140 CrossRef PubMed.
- P. M. Geyer, M. C. Hulme, J. P. B. Irving, P. D. Thompson, R. N. Ashton, R. J. Lee, L. Johnson, J. Marron, C. E. Banks and O. B. Sutcliffe, Anal. Bioanal. Chem., 2016, 408, 8467–8481 CrossRef PubMed.
- D. Mainali and J. Seelenbinder, Appl. Spectrosc., 2016, 70, 916–922 CrossRef PubMed.
- S. D. Brandt, L. A. King and M. Evans-Brown, Drug Test. Anal., 2014, 6, 587–597 CrossRef PubMed.
- M. Paillet-Loiler, A. Cesbron, R. Le Boisselier, J. Bourgine and D. Debruyne, Subst. Abuse Rehabil., 2013, 5, 37–52 Search PubMed.
- L. E. Regester, J. D. Chmiel, J. M. Holler, S. P. Vorce, B. Levine and T. Z. Bosy, J. Anal. Toxicol., 2015, 39, 144–151 CrossRef PubMed.
- M. Philp, R. Shimmon, M. Tahtouh and S. Fu, Forensic Chem., 2016, 1, 39–50 CrossRef.
- K. Kellett, J. H. Broome, M. Zloh, S. B. Kirton, S. Fergus, U. Gerhard, J. L. Stair and K. J. Wallace, Chem. Commun., 2016, 52, 7474–7477 RSC.
- Scientific Working Group for the Analysis of Seized Drugs (SWGDRUG), SWGDRUG recommendations, edn 7.1, 2016 Search PubMed.
- J. Lobo Vicente, H. Chassaigne, M. V. Holland, F. Reniero, K. Kolář, S. Tirendi, I. Vandecasteele, I. Vinckier and C. Guillou, Forensic Sci. Int., 2016, 8(265), 107–115 CrossRef PubMed.
- W. W. Y. Lee, V. A. D. Silverson, L. E. Jones, Y. C. Ho, N. C. Fletcher, M. McNaul, K. L. Peters, S. J. Speers and S. E. J. Bell, Chem. Commun., 2016, 52, 493–496 RSC.
- S. E. J. Bell, D. T. Burns, A. C. Dennis, L. J. Matchett and J. S. Speers, Analyst, 2000, 125, 1811–1815 RSC.
- S. P. Stewart, S. E. J. Bell, N. C. Fletcher, S. Bouazzaoui, Y. C. Ho, S. J. Speers and K. L. Peters, Anal. Chim. Acta, 2012, 711, 1–6 CrossRef PubMed.
- L. Elie, M. Elie, G. Cave, M. Vetter, R. Croxton and M. Baron, J. Raman Spectrosc., 2016, 47, 1343–4350 CrossRef.
- B. Li, A. Calvet, Y. Casamayou-Boucau, C. Morris and A. G. Ryder, Anal. Chem., 2015, 87, 3419–3428 CrossRef PubMed.
- R. Christie, E. Horan, J. Fox, C. O'Donnell, H. J. Byrne, S. McDermott, J. Power and P. Kavanagh, Drug Test. Anal., 2014, 6, 651–657 CrossRef PubMed.
- S. Assi, A. Guirguis, S. Halsey, S. Fergus and J. L. Stair, Anal. Methods, 2015, 7, 736–746 RSC.
- L. E. Jones, A. Stewart, K. L. Peters, M. McNaul, S. J. Speers, N. C. Fletcher and S. E. J. Bell, Analyst, 2016, 141, 902–909 RSC.
- M. D. Hargreaves, A. D. Burnett, T. Munshi, J. E. Cunningham, E. H. Linfield, A. G. Davies and H. G. M. Edwards, J. Raman Spectrosc., 2009, 40, 1974–1983 CrossRef.
- J. M. Chalmers, H. G. M. Edwards and M. D. Hargreaves, in Infrared and Raman Spectroscopy in Forensic Science, 2012, pp. 45–86 Search PubMed.
- S. Christesen, B. Maciver, L. Procell, D. Sorrick, M. Carrabba and J. Bello, Appl. Spectrosc., 1999, 53, 850–855 CrossRef.
- A. Guirguis, S. Girotto, B. Berti and J. L. Stair, Forensic Sci. Int., 2017, 273, 113–123 CrossRef PubMed.
- J. Calvo-Castro, A. Guirguis, M. Zloh and J. L. Stair, in Light in Forensic Science: Issues and Applications, The Royal Society of Chemistry, 2018, pp. 257–278 Search PubMed.
- F. Schifano, L. Orsolini, G. Duccio Papanti and J. M. Corkery, World Psychiatry, 2015, 14, 15–26 CrossRef PubMed.
- United Nations Office on Drugs and Crime (UNODC), 2018, Understanding the synthetic drug market: the NPS factor Search PubMed.
- R. B. Cody, J. A. Laramée and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef PubMed.
- M. Zloh, E. G. Samaras, J. Calvo-Castro, A. Guirguis, J. L. Stair and S. B. Kirton, RSC Adv., 2017, 7, 53181–53191 RSC.
- J. W. Godden, L. Xue, D. B. Kitchen, F. L. Stahura, E. J. Schermerhorn and J. Bajorath, J. Chem. Inf. Comput. Sci., 2002, 42, 885–893 CrossRef PubMed.
- J. Welter-Luedeke and H. H. Maurer, Ther. Drug Monit., 2016, 38, 4–11 CrossRef PubMed.
- J. Mounteney, I. Giraudon, G. Denissov and P. Griffiths, Int. J. Drug Policy, 2015, 26, 626–631 CrossRef PubMed.
- Z. Han, H. Liu, J. Meng, L. Yang, J. Liu and J. Liu, Anal. Chem., 2015, 87, 9500–9506 CrossRef PubMed.
- P. Larkin, Infrared and Raman Spectroscopy. Principles and Spectral Interpretation, Elsevier Academic Press, 2011 Search PubMed.
- F. A. Settle, Handbook of instrumental techniques for analytical chemistry, Prentice-Hall PTR, 1997 Search PubMed.
- C. McKenzie, O. B. Sutcliffe, K. D. Read, P. Scullion, O. Epemolu, D. Fletcher, A. Helander, O. Beck, A. Rylski, L. H. Antonides, J. Riley, S. A. Smith and N. Nic Daeid, Forensic Toxicol., 2018, 36, 359–374 CrossRef PubMed.
- S. D. Banister, A. Olson, M. Winchester, J. Stuart, A. R. Edington, R. C. Kevin, M. Longworth, M. Herrera, M. Connor, I. S. Mcgregor, R. R. Gerona and M. Kassiou, Drug Test. Anal., 2018, 10, 1099–1109 CrossRef PubMed.
- M. Kusano, K. Zaitsu, K. Taki, K. Hisatsune, J. Nakajima, T. Moriyasu, T. Asano, Y. Hayashi, H. Tsuchihashi and A. Ishii, Drug Test. Anal., 2018, 10, 284–293 CrossRef PubMed.
- P. A. Borea, T. A. Hamor and I. L. Martin, Structure activity relationships at the benzodiazepine receptor, in Analysis of Psychiatric Drugs. Neuromethods, ed. A. A. Boulton, G. B. Baker and R. T. Coutts, Humana Press, 1988, vol 10 Search PubMed.
- C. R. Maheux and C. R. Copeland, Drug Test. Anal., 2012, 4, 17–23 CrossRef PubMed.
- M. D. Hargreaves, K. Page, T. Munshi, R. Tomsett, G. Lynch and H. G. M. Edwards, J. Raman Spectrosc., 2008, 39, 873–880 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05847d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |