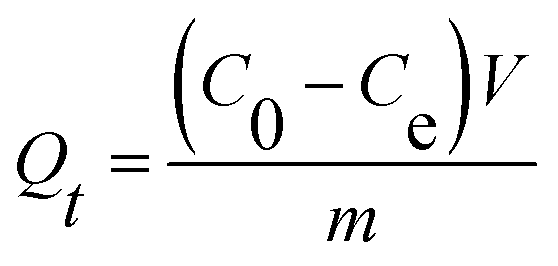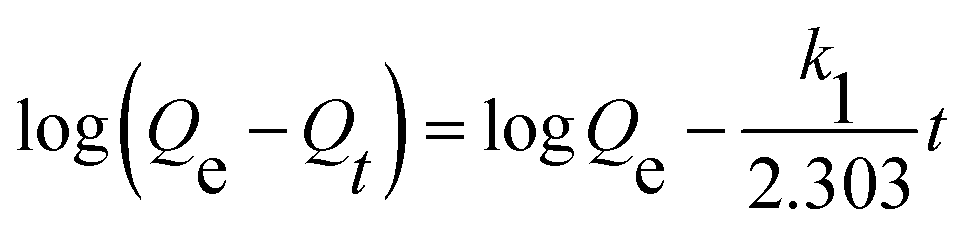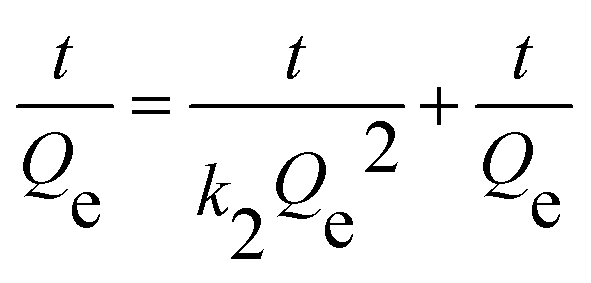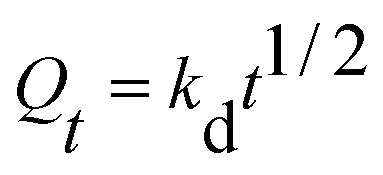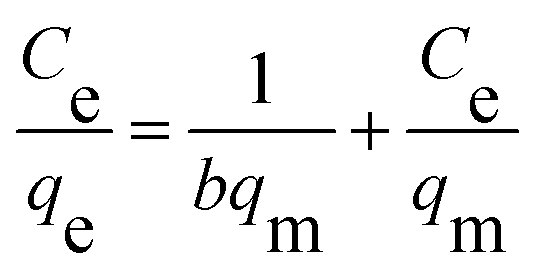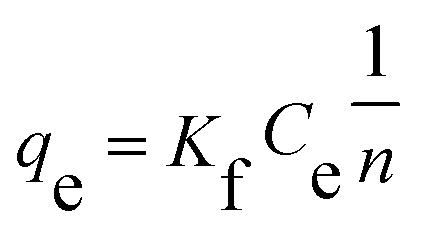DOI:
10.1039/C8RA05809A
(Paper)
RSC Adv., 2018,
8, 32358-32367
Facile preparation of UiO-66 /PAM monoliths via CO2-in-water HIPEs and their applications
Received
8th July 2018
, Accepted 10th September 2018
First published on 19th September 2018
Abstract
A novel clean method to synthesis a composite monolith was developed. Given its amphiphilic property, UiO-66 can emulsify water and CO2 to format a high internal phase emulsion (HIPE) under certain conditions. These UiO-66-emulsified Pickering HIPEs can be used as templates to prepare interconnected macroporous MOF/polymer composite monoliths. The effects of UiO-66 amount, cross-linking agent concentration, and CO2–water ratio on UiO-66/PAM structures were investigated. Then, the as-synthesized MOF/PAM composites were characterized by TGA, XRD, SEM, and FT-IR analyses, as well as rheological and DMA measurements. The results indicated that the composites are interconnected with hierarchical pores, and the diameter of the voids is 10–50 μm. The directly prepared monoliths exhibited relatively high stresses at 82% strain and recovered their shape quickly. The adsorption capacity of the composites for methylene blue (MB) is 50 mg g−1 at a faster adsorption rate. The monoliths also exhibit underlying applications in edible oil–water separation.
1 Introduction
Metal–organic frameworks (MOFs) are a type of crystalline coordination polymer that consist of metal or metal clusters and organic ligands. The MOFs displayed extremely high surface areas, controllable structures, porosity and steerable chemical properties through modified precursors or post-synthetic treatment procedures.1 The MOFs are easily synthesized and displayed excellent physical and chemical properties, so they have been widely used in catalysis,2–4 sensing,5–7 drug delivery,8,9 gas storage,10–12 and carbon dioxide (CO2) capture.13 Furthermore, MOFs can be adsorbed at the interface of two immiscible phases and format emulsions due to the blended constituents.14 UiO-66 is one of the most common MOFs with remarkable thermostability and chemical stability, it is comprised of a Zr6-cluster core has 12-coordination and contains multiple hierarchical metal structures.15 UiO-66 is stable up to 550 °C in an air atmosphere and possesses prominent mechanical stabilities under high stress compared with other MOFs.16
Emulsion templated method is common for the preparation of porous material. Generally, high internal phase emulsion (HIPE) is the emulsions which with volume fraction of internal phase at least 74.05% of the whole emulsion. These merits of HIPEs have attracted scientists' attention in various fields and have been applied in cosmetics,17 food,18 separation,19–21 carrier22 and other areas. Conventional HIPEs usually required large amounts of surfactant (more than 20 vol%) to format stable emulsions. But particles emulsified Pickering HIPEs can be stabilized only with a little emulsifier. Pickering emulsifiers are a kind of particles with partial dual wettability that can adsorb at interface of two immiscible phases to form a barrier against flocculation of emulsion drops. Such as functionalized silica,23 cellulose nanocrystals,24,25 and modified ferric oxide,26 and so on. Oil-in-water (O/W) HIPEs are one of the most studied emulsions that have employed plenty of oil phase. The oil phase often is organic solvent, which causes environmental contamination, difficulty to remove, organic residual, and toxicity. However, the organic solvent can be replaced by CO2 because of nontoxic, nonflammable, low price, environment-friendly and easily available properties.27–31 Accordingly, the destabilization of CO2-in-water (C/W) emulsion appeared in the presence of polar monomers such as AM and MBAM,32 this instability can be counteracted by the addition of cosurfactant, such as PVA.33 CO2-in-water emulsion stabilized by MOF was first reported by Zhang.34
In this work, supermacroporous UiO-66/polyacrylamide (UiO-66/PAM) monolith was first readily synthesized using UiO-66 emulsified CO2-in-water (C/W) HIPE templating. The effects of UiO-66 content, N,N′-methylenebisacrylamide concentration, co-stabilizer PVA amount, and the densities of CO2 on the monolith were investigated. Robust mechanical behavior was determined by the rheological and DMA measurements. The application performances of the porous composites were evaluated in oil and water separation and azo cationic dye adsorption.
2 Experimental
2.1 Reagents
1,4-Benzenedicarboxylic acid (H2BDC, ≥99.0% purity, Ourchem), zirconium(IV) chloride (ZrCl4, 98.0% purity, J&K), methanol (Tianjin ZhiYuan company), acetic acid (Tianjin ZhiYuan company), acrylamide (AM, ≥98.0% purity, Tianjin yongcheng company), cyclohexane (Tianjin bailian chemistry company), N,N′-methylene (bisacrylamide) (MBAM, ≥98.0% purity, Tianjin chemistry company), potassium persulfate (KPS, ≥99.5% purity, Tianjin tianda company), polyvinyl alcohol (PVA, Mw ∼ 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, DH = 87–89% Macklin), CO2 (>99.995% purity), methylene blue (MB, aladdin), edible oil (Jing Long yu, Jiali grain and oil (China) Co., Ltd.), deionized water. The reagents mentioned above were used as received.
000, DH = 87–89% Macklin), CO2 (>99.995% purity), methylene blue (MB, aladdin), edible oil (Jing Long yu, Jiali grain and oil (China) Co., Ltd.), deionized water. The reagents mentioned above were used as received.
2.2 Synthesis of UiO-66
UiO-66 was synthesized by hydrothermal method according to the literature.35 Briefly, ZrCl4 (0.357 g) and terephthalic acid (0.254 g) were dissolved into mixture of DMF (21 mL) and acetic acid (8.6 mL), the solution were transferred into Teflon lined autoclave and heated to 120 °C for 24 hours. After cooling to ambient temperature, the obtained powder was collected by centrifugation and washed with methanol for three times, then vacuum dried at 60 °C for 8 hours.
2.3 Cyclohexane-in-water HIPE emulsion stabilized by UiO-66
The stability of UiO-66 was verified by using an “inverted tube” method.34 A certain amount of pre-synthesized UiO-66 was added into 1 mL of water. Subsequently, solid dispersions of 1 w/v%, 3 w/v%, 5 w/v% were formed respectively. 4 mL of cyclohexane was then added separately and shake it violently by hand to form a gel-like emulsion.
2.4 Preparation of poly(acrylamide) HIPEs stabilized by UiO-66
Poly(acrylamide) HIPEs were synthesized by HIPE method in high pressure stainless steel reactor (100 cm3). Typically, stabilizer PVA (3 w/v% equivalent to water) was added in 15 mL of deionized water and magnetic stirring for 30 minutes to ensure completely dissolved. The monomer AM (46 w/v% equivalent to water), crosslinker MBAM (20% w/w relative to monomer), a certain amount of UiO-66 and initiator K2S2O8 (2% w/w relative to monomer) were added into reactor. The system was charged-vented by certain CO2 three times and a desired amount of CO2 was charged into the vessel (50 ± 5 bar) and commenced to stir vigorously for 30 min at 30 °C. Then, heated to 60 °C gradually and the polymerization was conducted for 12 h. After reaction, the vessel was cooled to room temperature, the internal phase removed via simple depressurization. Other samples were prepared according to the above method with different contents of UiO-66, cross-linking agent and different density of internal phase (CO2). The specific dosage is shown in Table 1. Furthermore, in order to keep the pore structure, the as-prepared UiO-66/PAM monolith composites were freeze drying.
Table 1 List of recipes for different variables and the density of UiO-66/PAM monolithsa
| |
PVAb w/v% |
UiO-66b wt% |
AMb wt% |
MBAMc wt% |
KPSc wt% |
CO2 (MPa) |
True densityd (g cm−3) |
| Reaction condition: the continuous water phase (V = 15 mL), contains PVA, AM, MBAM, KPS. w/v based on H2O. w/w relative to monomer weight. Density was measured by true density analyzer. |
| 1 |
8.0 |
0 |
46 |
20 |
5 |
12 ± 2 |
1.49 |
| 2 |
1.5 |
3 |
46 |
20 |
5 |
12 ± 2 |
1.35 |
| 3 |
1.5 |
5 |
46 |
20 |
5 |
12 ± 2 |
1.45 |
| 4 |
1.5 |
9 |
46 |
20 |
5 |
12 ± 2 |
1.52 |
| 5 |
1.5 |
6 |
46 |
5 |
5 |
16 ± 6 |
1.36 |
| 6 |
1.5 |
6 |
46 |
8 |
5 |
16 ± 6 |
1.41 |
| 7 |
1.5 |
6 |
46 |
20 |
5 |
16 ± 6 |
1.44 |
| 8 |
1.5 |
5 |
32 |
20 |
5 |
17 ± 7 |
1.49 |
| 9 |
1.5 |
5 |
32 |
20 |
5 |
12 ± 2 |
1.46 |
2.5 Characterization and instruments
As-prepared UiO-66 and UiO-66/PAM composites were analyzed by X-ray powder diffraction (XRD) patterns (Bruker D8 Advance, Cu-Kα radiation, λ = 1.5418 Å, 2θ = 5–50°). The UiO-66 and UiO-66/PAM were analyzed by a field-emission scanning electron microscope (SU-8000) and a low power scanning electron microscopy (TM 3030). It was operated at 5.0 kV to examine the structure and crystallite morphology of UiO-66 and UiO-66/PAM. The samples were outgassed at 120 °C, before to measurement. The nitrogen adsorption/desorption isotherms of the UiO-66 were measured with a surface area and porosity analyzer (Kubo-X1000). As prepared UiO-66 and UiO-66/PAM was analyzed by a thermogravimetric analyzer which was used from room temperature to 800 °C at 10 °C min−1 under an N2 flow of 100 mL min−1. FT-IR spectra of the UiO-66 and UiO-66/PAM were performed on a Bruker Vertex 70 spectrometer.
The densities of the samples were determined using the True Density Analyzer (TD-2200, Builder, China) at ambient temperature. A weighed sample was placed in the sample cell and degassed by pumping at a steady speed. An average of 5 measurements per sample was used to calculate the average density.
Compression tests were performed on an DMA Q800 at room temperature. The different MOFs contents of cylindrical UiO-66/PAM were compressed at a constant rate of 5 N min−1 to 15 N. The UiO-66/PAM composite with different of cross-linking agent contents were performed by oscillatory shear measurement. A dynamic shear rheometer (DHR-1) was used with parallel plate geometry (25 mm diameter). The parallel plate was needed to keep an interval of 2 mm during the measurement. To determine a linear viscoelastic regime, we group selected the varying strain% from 0.001% to 100% to monitor the storage and loss moduli at a frequency of ω = 1 s−1. The storage and loss moduli of the composites were measured by using the dynamic frequency sweeps from 0.1 s−1 to 100 s−1. All experiments were performed in isothermal conditions of 25 °C. The oil contact angle (OCA) of the samples underwater was determined with a contact angle analyzer (Powereach JJ2000B2, China). The average CA values were obtained with five parallel measurements.
2.6 Direction of material application
2.6.1 Oil–water separation test. A simple experiment was performed as follows: the oil–water device was shown in Fig. 11, as-prepared UiO-66/PAM composite materials were cut into slices with a diameter of 40 mm and a thickness of 2 mm and the slices was mounted between a centrifuge tube cover and a support base with a stainless steel mesh. The oil and water mixture (volume ratio = 1:3) was used in the gravity-driven separation produces were made immiscible and layered. The separations were performed by adding 80 mL oil and water mixture into the device.
2.6.2 Adsorption performance. The adsorption kinetics were investigated by adding 10 mg UiO-66/PAM composites into 10 mL MB solutions (10 mg L−1) for a different contact time. Then, the concentration of MB solutions was analyzed by UV-vis spectrophotometer (Shimadzu UV-2550) at 664 nm, before a calibration curve obtained from the known concentration MB solutions. The adsorption capacity Qt was calculated using the following equation:| |
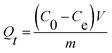 | (1) |
where V is the volume of solution, m is the mass of the UiO-66/PAM composite. C0 and Ce are the MB concentrations before and after adsorption, separately.The adsorption process with time changes was investigated by using the pseudo-first-order and pseudo-second-order kinetic model, which was:
| |
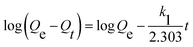 | (2) |
| |
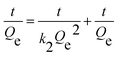 | (3) |
where
Qe and
Qt were the adsorption capacities equilibrium and at time
t, respectively, and
k1,
k2 were the rate constant of the pseudo-first-order and pseudo-second-order model.
3 Results and discussion
3.1 Characterization of UiO-66
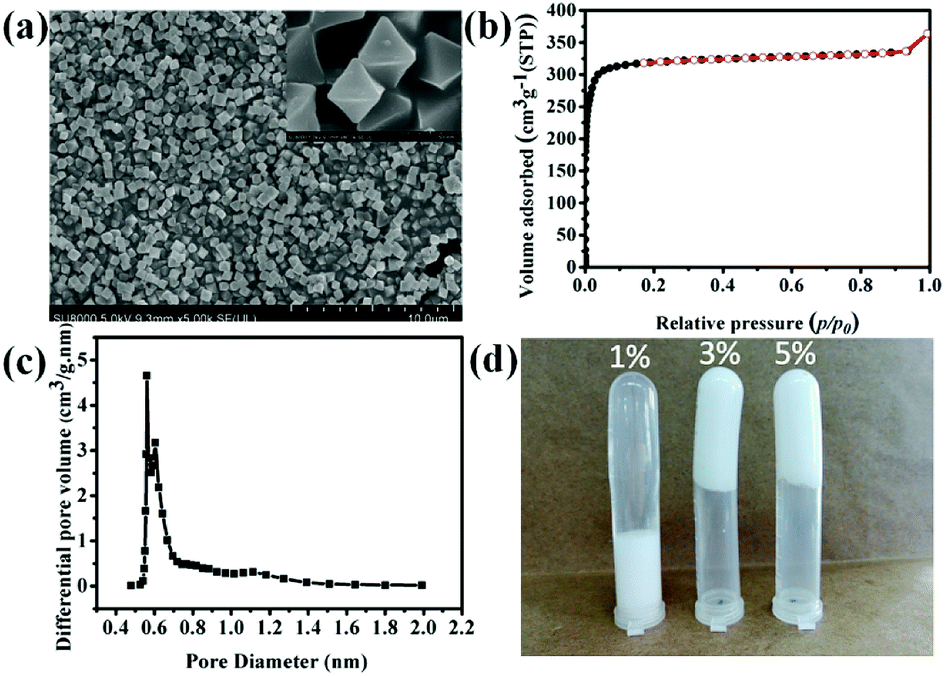
To prepared HIPEs, UiO-66 was used as Pickering emulsifier to prepare HIPEs. Fig. 2d shows Pickering HIPEs stabilized by 1 wt%, 3 wt% and 5 wt% UiO-66, respectively. The UiO-66 particles absorbed at the interface of water and cyclohexane (the volume ratio of 1:4) and formed a barrier to against the flocculation of emulsion drops. The amount of UiO-66 considerably influenced the stability of emulsion. The HIPE emulsified by 1 wt% UiO-66 are unstable, and the internal phase was expelled after one day. This may be ascribed to relatively low viscosity of emulsion. Meanwhile, the emulsions stabilized by 3 wt% and 5 wt% UiO-66 also have the same phenomenon (expelling of internal phase), however, these emulsions are more stable. The exceeded UiO-66 existed in the continuous phase, which has increased the viscosity of emulsion and hindered flocculation and phase inverse.
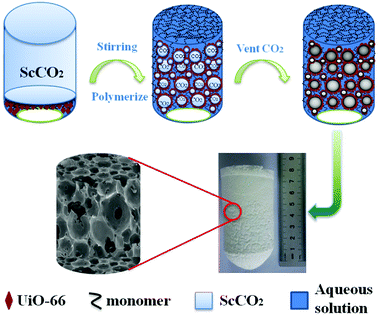 |
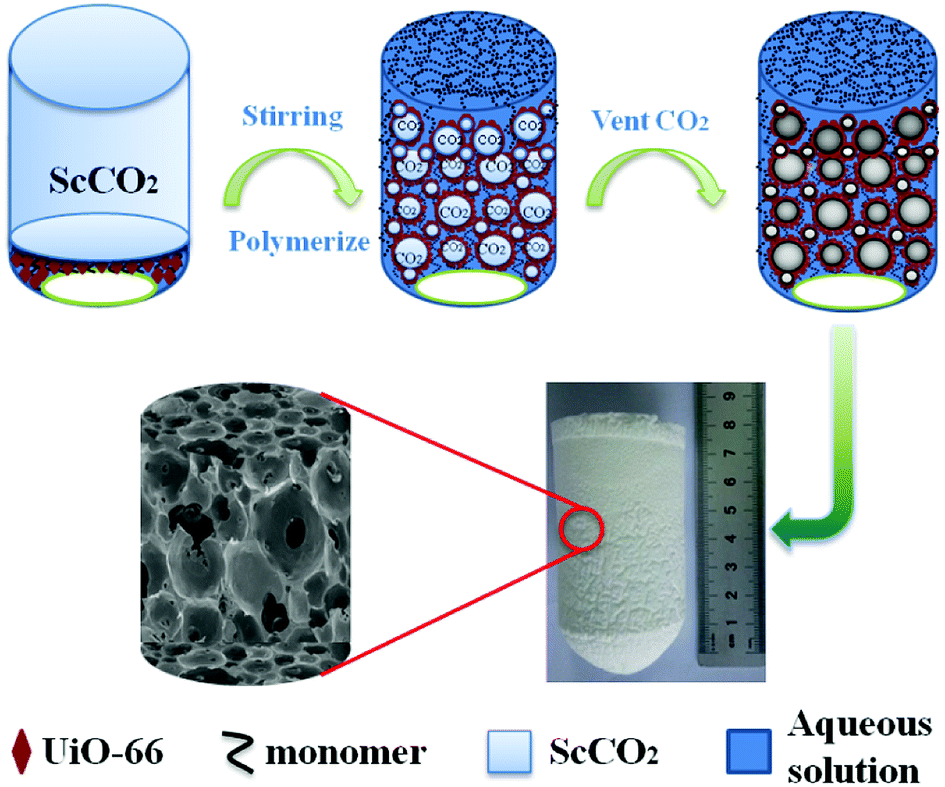
| | Fig. 1 Schematic illustration for the preparation of UiO-66/polyacrylamide. | |
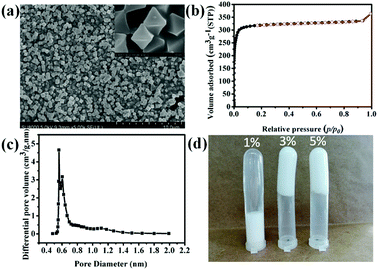 |
| | Fig. 2 SEM image of UiO-66 (a), N2 sorption isothermal curve of UiO-66 (b), pore size distribution of UiO-66 (c) cyclohexane-in-water HIPEs stabilized by different UiO-66 contents. | |
The SEM image of UiO-66 shown in Fig. 2a indicates that the average particle size is approximately in the range of 300–500 nm and almost without aggregation. Furthermore, the pore property of the as synthesized UiO-66 was investigated. The N2 sorption isothermal curve of UiO-66 shown in Fig. 2b belongs to type I isotherm and is indicative of microporous structure. The BET surface area of UiO-66 reached 1312 ± 15 m2 g−1 with octahedral pore diameter of 5.61 Å, which was calculated according to the HK micropore distribution method shown in Fig. 2c.
3.2 FT-IR spectra
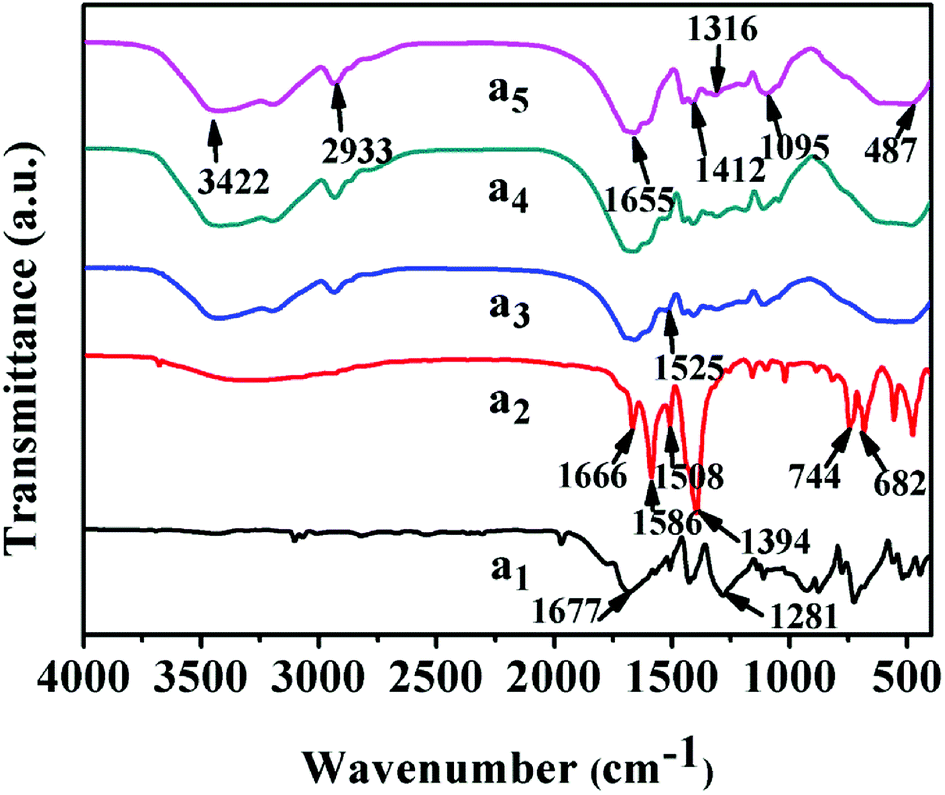
The infrared spectra of UiO-66 and UiO-66/PAM composites are shown in Fig. 3. The characteristic peak of carbon oxygen double bond in UiO-66 (curve a2) is around 1666 cm−1. The adsorption peak at 1586 and 1394 cm−1 were caused by the expansion vibration of COO−, which confirmed the coordination of carboxylic acid in UiO-66. And the vibration peak at 744 cm−1 is consistent with the Zr–(μ3)–O bond, indicating that the existence of zirconium ion in UiO-66, and the octahedral secondary structure unit of UiO-66 is formed through the coordination of μ3-O and terephthalic acid. In curve a5, the characteristic absorption peak at 3422 cm−1, 3195 cm−1 and 2933 cm−1 correspond to the free –NH2, associative –NH2 and the methylene stretching vibration, respectively. The absorption peak at 1655 cm−1 is characteristic absorption peak of carbonyl, which correspond to the amide I band (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration), and that at 1412 cm−1 is the characteristic absorption peak of methylene base deformation. Comparing curves a2, a3 and a4 with curve a5, the peak width at 1655 cm−1 is wider due to the introduction of PAM, which was caused by the overlap of CO in the PAM and H3BTC, confirming the recombination of PAM and UiO-66.
O stretching vibration), and that at 1412 cm−1 is the characteristic absorption peak of methylene base deformation. Comparing curves a2, a3 and a4 with curve a5, the peak width at 1655 cm−1 is wider due to the introduction of PAM, which was caused by the overlap of CO in the PAM and H3BTC, confirming the recombination of PAM and UiO-66.
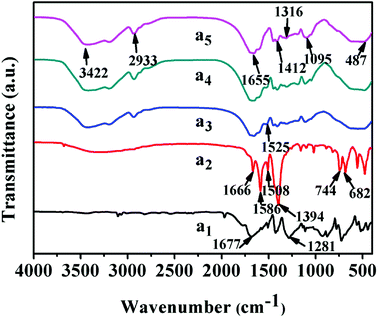 |
| | Fig. 3 FT-IR spectra: (a1) H2BDC, (a2) UiO-66, (a3–a5) UiO-66/PAM composites with 9 wt%, 2 wt% and 0 wt% UiO-66, respectively. | |
3.3 XRD analysis
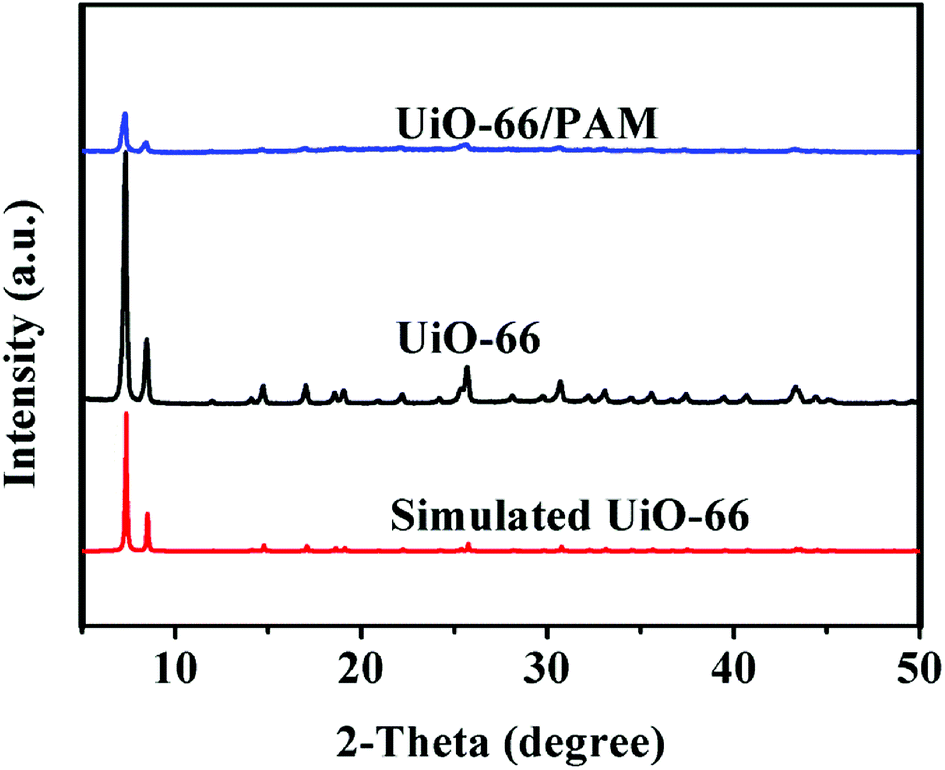
Fig. 4 shows the PXRD of the simulated UiO-66, synthetic UiO-66, and UiO-66/PAM. The PXRD of the as-synthesized UiO-66 (curve b) coincides well with the simulated XRD pattern of UiO-66 (curve c), which proved the formation of crystalline UiO-66. The appearance of the peaks at 2θ = 7.36, 8.48°, 12.04°, 14.15°, 17.08°, 22.25°, 25.68°, and 33.12° in the (111), (002), (022), (113), (004), (115), (224), and (137) reflections of reo nanoregions, respectively. Notably, the diffraction peak of synthetic UiO-66 is higher and sharper than the simulated one, indicating higher crystal phase and bigger particle size. From the three XRD curves, the crystal diffraction peak positions of UiO-66 and UiO-66/PAM correspond to the simulated UiO-66 approximately. The intensity of crystal diffraction peak decreased in UiO-66/PAM, which indicates that UiO-66 is well combined into PAM network.
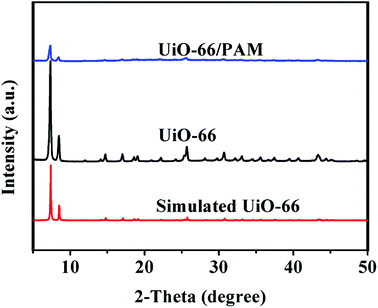 |
| | Fig. 4 The PXRD patterns of UiO-66/PAM (a), UiO-66 (b), Simulated UiO-66 (c). | |
3.4 UiO-66/PAM morphology
Fig. 5 shows the FE-SEM images of UiO-66/PAM with different contents of UiO-66. A great difference in the void size and pore interconnectivity was found among these PolyHIPE structures. The composite with lower contents of UiO-66 are typical closed-cell structure (such as 2 wt% and 3 wt%, recipes are shown in Table 1). The thicker wall covers the closed-cell between neighboring pores in the composite of 2 wt% UiO-66. Fig. 5(a) show lesser voids compared with Fig. 5(b), indicating that the phase separation may have occurred at a lower UiO-66 content (2 wt%). Thus, the interface of CO2–water cannot be well stabilized. Notably, the void becomes larger and appears opened-cell structure with increasing UiO-66 mass ratio, this was most likely due to the lower interfacial tension. Similar results have been reported by Yang S et al.36
 |
| | Fig. 5 FE-SEM images of the UiO-66/PAM for samples with different contents of UiO-66: the mass ratio of UiO-66 refer to water is 2 wt% (a), 3 wt% (b), 5 wt% (c), 7 wt% (d), 9 wt% (e). | |
As showed in the Fig. 6b shows a more homogeneous and smaller void diameter compared to image a. The void wall gradually becomes thinner with the increasing density of CO2. When density of CO2 reached a certain value, UiO-66 and the co-stabilizer (PVA) will not prevent flocculation. From the above result, various pore sizes and porosity of porous PolyHIPE composites could be readily adjusted by varying density of CO2. The similar conclusion was also obtained in Wang J et al.'s work.37
 |
| | Fig. 6 SEM images of under different density of CO2: 12 MPa (a), 17 MPa (b). | |
Table 1 shows the effect of reaction condition on the true density of PolyHIPEs. As for the different UiO-66 contents in PolyHIPEs, the void diameter increased with the increase in UiO-66 content to some extent, and the thickness of the void wall becomes thin at first and then thickened (Fig. 5). The results led to the increase in the density of UiO-66/PAM (2–9 wt% of UiO-66). On the other hand, the PolyHIPEs with lower crosslinking degree have larger void diameter and lower density, whereas higher crosslinking degree PolyHIPEs has smaller void diameter and higher density. Third, PolyHIPEs were obtained from the different internal phase densities, the higher internal phase density resulted in a slightly higher density of composite. In reality, the increase of internal phase density increases led to the thinner void wall of composites, causing the occurrence of flocculation that produced lower true density of composites. This inconsistency needs further study.
3.5 EDS mapping image
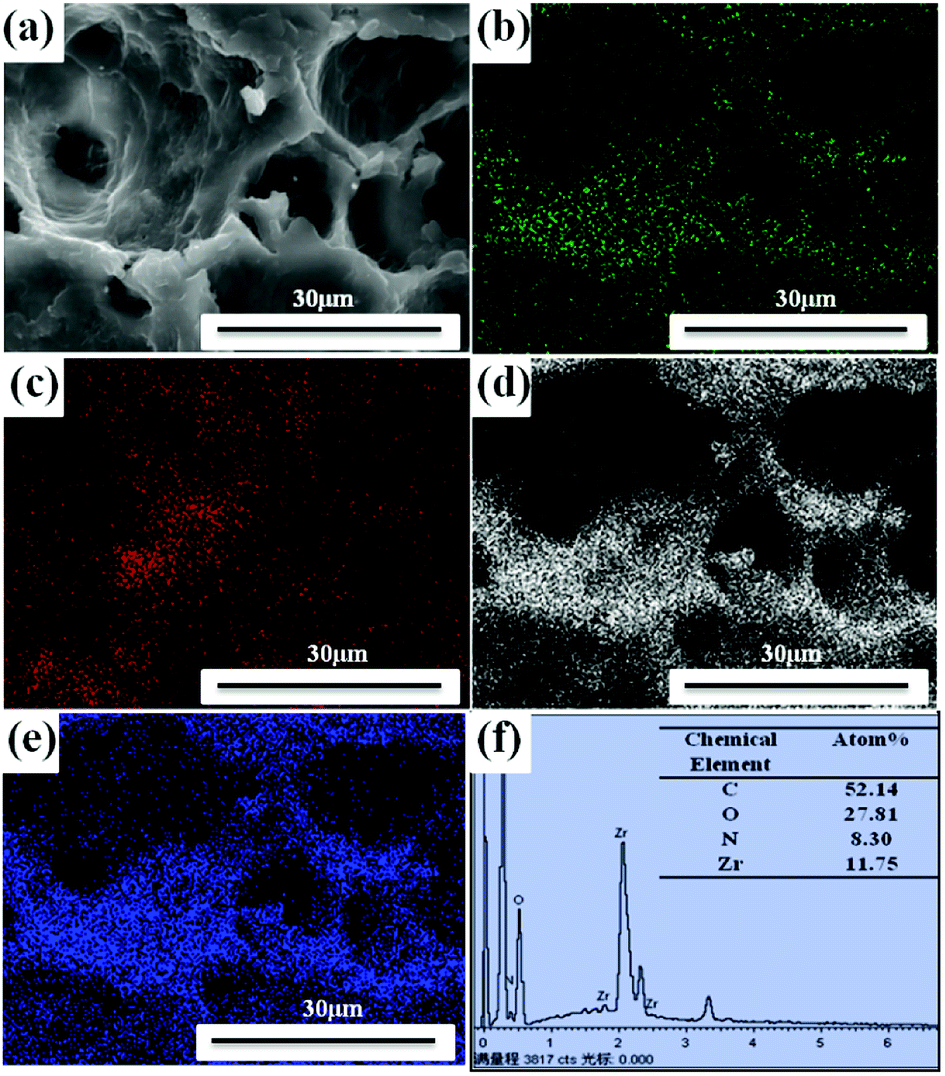
The elements analysis of UiO-66/PAM composite can be detected by the energy dispersive spectrometer analysis. The result in Fig. 7 indicates that the UiO-66 has successfully incorporated with PAM PolyHIPE. Further elemental analysis of the spatial distribution of UiO-66 in the UiO-66/PAM composite was performed through elemental mapping. In Fig. 7(b–e), elemental mapping images exhibit a uniform distribution of N, Zr, C and O elements in the UiO-66/PAM composites. Thus, the EDS and elemental mapping analysis testified that UiO-66 nanoparticles are distributed uniformly in the PolyHIPE
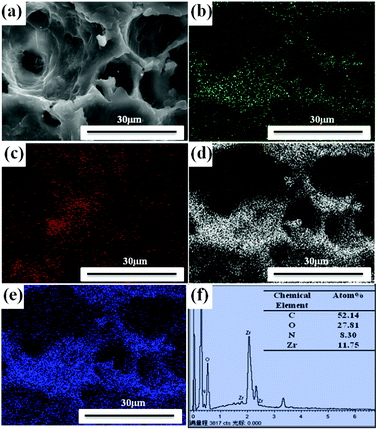 |
| | Fig. 7 SEM images of UiO-66/PAM (a) with the corresponding elemental mapping (b, c, d and e) of N, Zr, C, O, respectively, and EDS analysis (f). | |
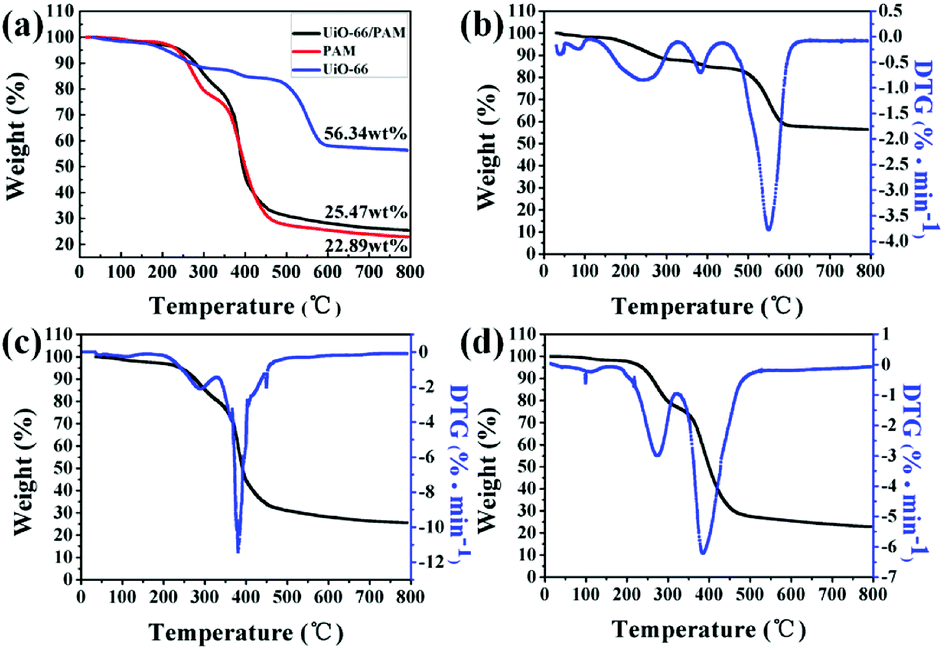
3.6 TG analysis
The TGA–DTG curves are shown in Fig. 8(a–d). The curves in Fig. 8(b) and (c) reflect that the weights were standardized with respect to residual Zr6O6(BDC)6, which was oxidized to six ZrO2 after heating to 800 °C. The curves b, c, and d presented in Fig. 8 indicated that the three samples exhibit three steps of weight loss. The first stage of weight loss (approximately 4%) ascribed to the physical desorption of surface adsorbed water and methyl alcohol in 40–200 °C. The second stage weight loss of UiO-66 occurred in the temperature range of 200–350 °C, resulting from the desorption of solvent molecules adsorbed in the PolyHIPEs and MOF pores. The weight loss of UiO-66/PAM in the 200–500 °C arose from the decomposition of polymer organics and some residual monomers. The gross weight losses were 43.66%, 74.53%, and 77.11% for the UiO-66, UiO-66/PAM, and PAM, respectively. As shown in Fig. 8b, the weight loss corresponds to the structural decomposition of Zr6O6(BDC)6 to 6 ZrO2 and volatile compounds over 400 °C. The thermal stability of PolyHIPEs has improved with the addition of UiO-66.
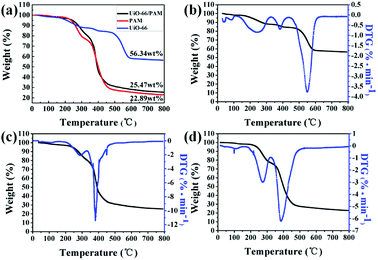 |
| | Fig. 8 The thermogravimetric analysis curves (TGA) of samples (a), the TGA–DTG curves the UiO-66 (b), UiO-66 (5 wt%)/PAM (c) and PAM (d). | |
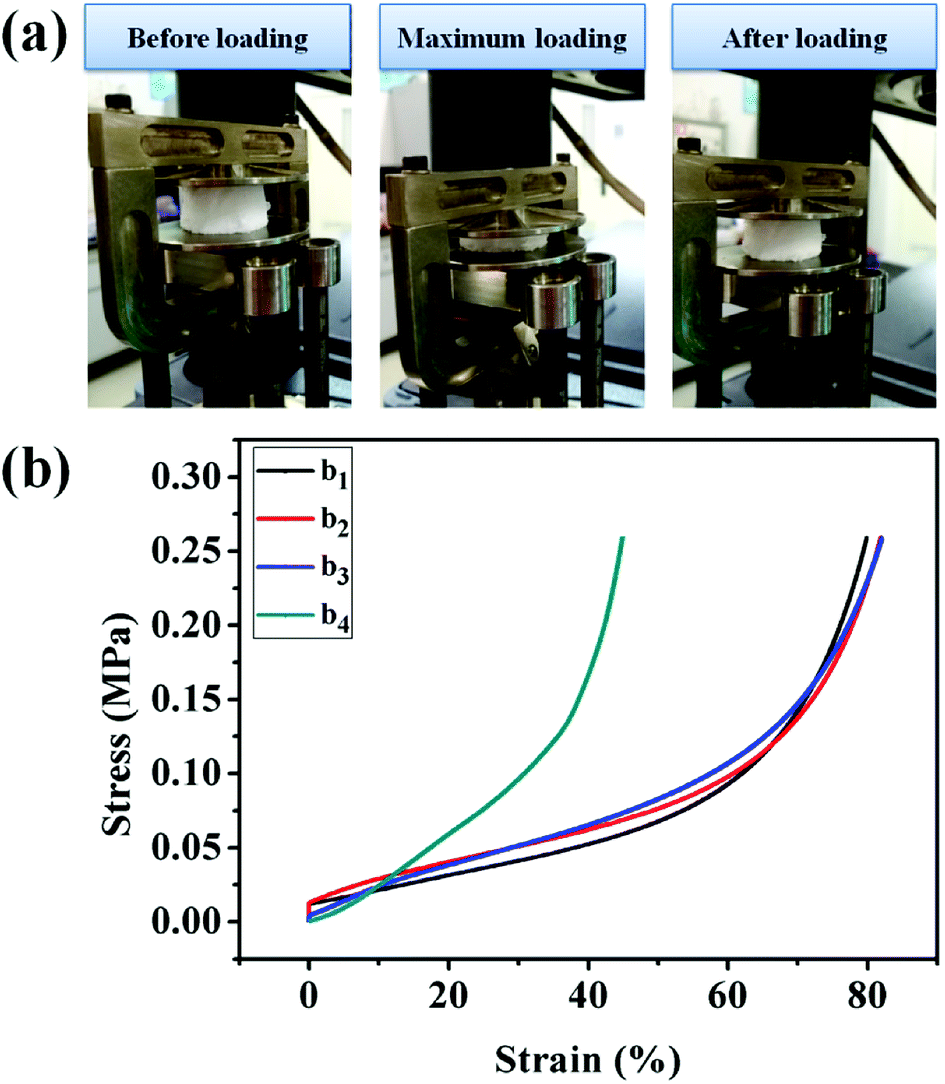
3.7 Mechanical property analysis
The UiO-66/PAM composites with typical scaffold appearance in the three processes of the compressive test are presented in Fig. 9. The UiO-66/PAM composites exhibit good compressive resistance and can restore the original appearance rapidly after removing the compressive force. The compression modulus of UiO-66/PAM HIPE porous composites is closed to 260 kPa, which means that the structure can provide sufficient mechanical support resist external forces and protect external voids. The mechanical properties of UiO-66/PAM materials are similar to that of PAM foams reported by Tan H et al.38 Compared with the compression modulus, UiO-66/PAM material was superior to graphene–PAM hydrogels.39 UiO-66/PAM (0–9 wt%) composites are well deformable under compressive strains of 79.91%, 81.88%, 82.02% and 44.90%, respectively. It indicates that the UiO-66/PAM have a better elasticity and compressive strain capacity than pure PAM. Meanwhile, the compression modulus of UiO-66/PAM have increased with the introduction of UiO-66, and strain obviously weakened in the case of 9.0 wt% UiO-66, which was caused by the increased interaction effect between UiO-66 and the PAM skeleton. UiO-66 plays the role of physical cross-linking point in composite, which enhanced the stiffness modulus of UiO-66/PAM.39–41
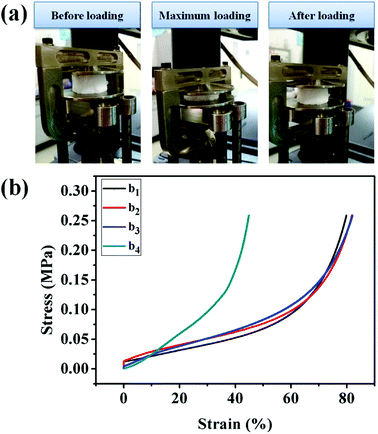 |
| | Fig. 9 The three progress images of compression (a), the data of compression tests: 0 wt% (b1), 3 wt% (b2), 5 wt% (b3), 9 wt% (b4). | |
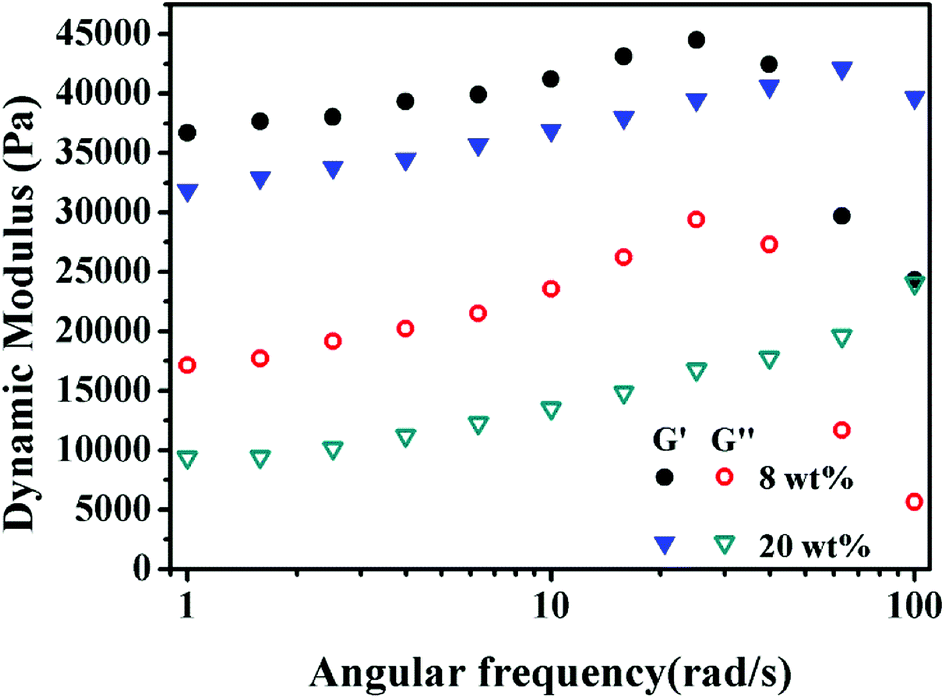
3.8 Dynamic rheological property
As shown in Fig. 10, the storage modulus (G′) is higher than the loss modulus (G′′) in the entire frequency range, indicating that the elastic modulus is the dominant characteristic of these PolyHIPEs. The maximum G′ of 8 wt% and 20 wt% MBAM PolyHIPE were 44.5 kPa and 42.1 kPa, respectively. The G′ of 8 wt% MBAM PolyHIPE is higher than that of 20 wt% MBAM PolyHIPE at low angular frequency. The result is opposite to that at high shear stress. In reality, although the monoliths have excellent elasticity under low angular frequency, the original state unable to restore and the internal structure was damaged when reaching beyond the linear viscoelastic plateau. Because the chain arrangement of PolyHIPEs is mainly branched, the UiO-66/PAM has high flexibility and poor elasticity in the lower cross-linking degree. With the increase of the cross-linking degree, the mechanical stability and the characteristic of resistance to external forces of the PolyHIPEs can be enhanced.
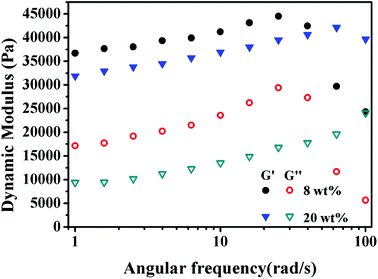 |
| | Fig. 10 Frequency-dependent storage modulus (G′) and loss modulus (G′′) of the PolyHIPEs slices with 8 wt% and 20 wt% MBAM content. | |
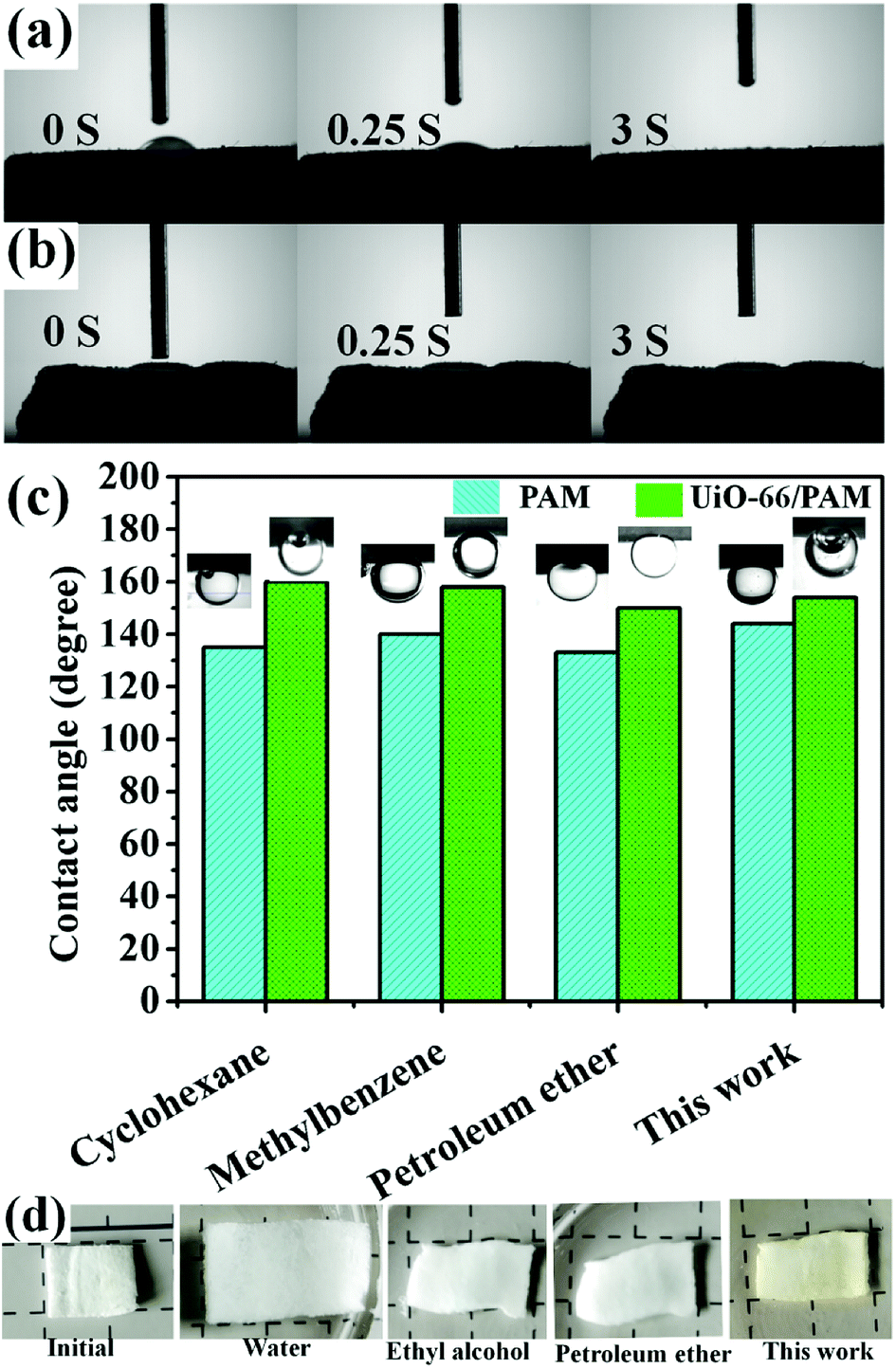
3.9 Oil–water separation tests
The measured water contact angles of UiO-66/PAM and PAM slices are 30° and 0°, respectively. It shows that the wettability of PAM is better than UiO-66/PAM. The oil contact angles of the above sample are shown in the Fig. 11(c). UiO-66/PAM and PAM exhibit superhydrophobicity for different oils. For all four cases (Fig. 11c), the oil contact angle of UiO-66/PAM are more than 150° (θow > 150°) and PAM are less than 150° (θow < 150°), hence the UiO-66/PAM exhibits an enhanced superoleophobicity underwater. The result also indicated that the UiO-66/PAM had low oil-adhesion and feasibility of oil/water separation. In order to explain the separation mechanism, the separated membrane states, which soaked in different oil products was shown in the Fig. 11(d). The slice can shrink and harden rapidly when it encounters different oil products, which makes it impossible to pass through. The principle can be concluded that the hydrophilic end accumulates in a segment and increase the hydrogen bonding interactions.
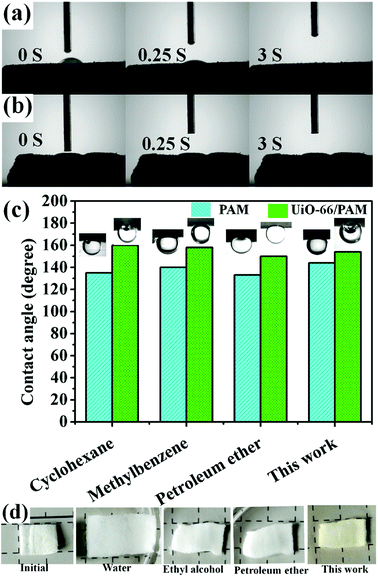 |
| | Fig. 11 The contact angle of UiO-66 (6 wt%)/PAM (a) and PAM (b) slices for water/air. The oil contact angles of UiO-66/PAM and PAM slices for oil/water (c) and the separated slices states (d). | |
In the Fig. 12(e and f), the average void pore diameter of the sample 6 is larger than that of the sample 7. In the process of oil–water separation (obtain the edible oil), the PolyHIPE slices with 8 and 20 wt% MBAM content were installed in the device, and the separation procedure was completed after 120 and 150 min, respectively. In Fig. 12b, after the completion of the separation procedure, the device mounted sample 6 slices and the beaker contained few oil droplets on the water surface. In Fig. 12(c), the sample 7 slices were used, and the beaker only contained water. However, UiO-66/PAM with lower cross-linking degrees exhibit faster separation speed. Besides, as shown in the Fig. 12(d), although the PAM slices exhibit faster separation speed and performed within 30 min, the separation efficiency is poor.
 |
| | Fig. 12 Self-made equipment (a), oil/water separation effect diagram: UiO-66/PAM with different cross-linking agent content 8 wt% (b) and 20 wt% slices (c), PAM slices (d). SEM image of the different cross-linking agent content of UiO-66/PAM: 8 wt%, sample 6 (e) and 20 wt%, sample 7 (f). | |
3.10 Adsorption experiment
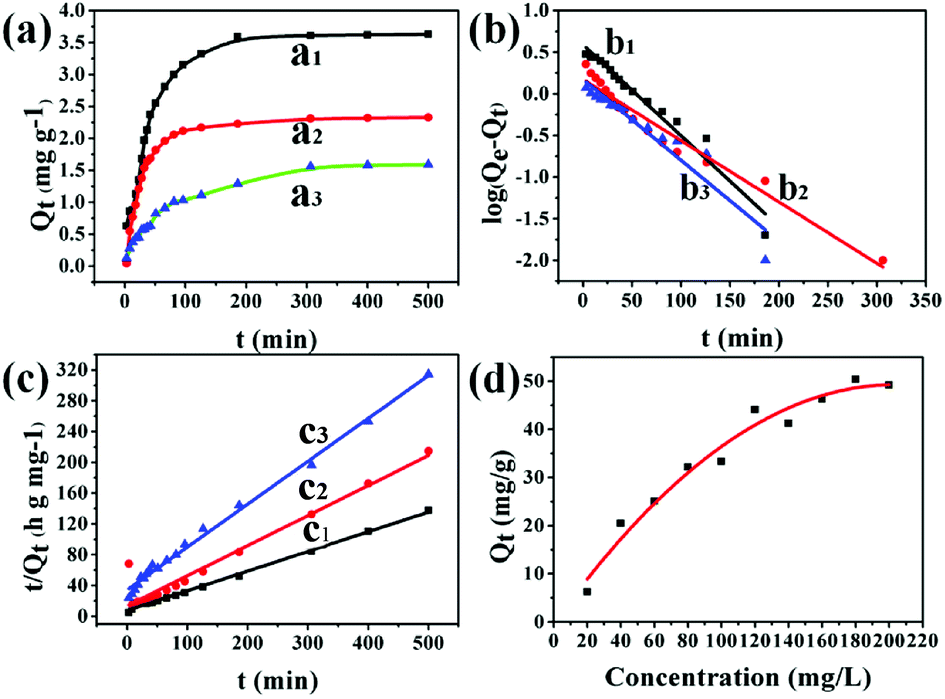
The adsorption capacity of UiO-66/PAM was studied by using different concentration MB solutions. Fig. 13(a–c) exhibits the adsorption kinetic behavior. The composites with different MBAM contents (1.5 wt%, 8 wt% and 20 wt%) were soaked in MB solution (pH = 8), and the adsorption reached saturated adsorption point (3.61, 2.32, and 1.30 mg g−1) after 4 h. As shown in Fig. 13(c), the time-dependence of the MB adsorption was better-fitted to a pseudo-second-order kinetic model, in which the rate constant k2 is the expected diffusion-limited behavior of UiO-66. The results indicated that the PVA layers do not prevent the accessibility of the UiO-66 pores. Different concentrations of MB solution were used to study the maximum saturation adsorption. Fig. 13(d) shows the PolyHIPEs with the maximum saturation adsorption of 50 mg g−1. Moreover, the maximum adsorption capacity of UiO-66/PAM for MB was higher than that of some reported materials, as shown in Table 3. Thus, it could be concluded that UiO-66/PAM HIPE monoliths have a considerable potential to removal of MB from waste water.
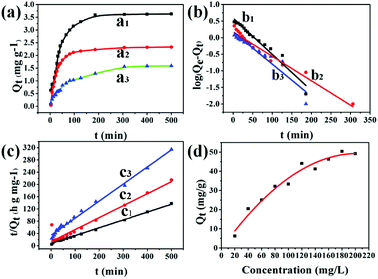 |
| | Fig. 13 Adsorption kinetics of MB by various crosslinking agent content of PolyHIPE: 1.5 wt% (a1), 8 wt% (a2), 20 wt% (a3) (T = 303 K, m = 50 mg, V = 50 mL, C0 = 10 mg L−1). Pseudo-first-order kinetic model: 1.5 wt% (b1), 8 wt% (b2), 20 wt% (b3) (T = 303 K, m = 50 mg, V = 50 mL). Pseudo-second-order kinetic model: 1.5 wt% (c1), 8 wt% (c2), 20 wt% (c3) (T = 303 K, m = 50 mg, V = 50 mL). The variation of adsorption capacity with various initial MB concentrations (d) (1.5 wt% of crosslinking agent, T = 303 K, m = 50 mg, V = 50 mL). | |
However, the two models cannot define and explain the mechanism of adsorption. Thus, the intra-particle diffusion model was implemented to detect adsorption mechanisms, which is shown in the following equations:
| |
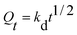 | (4) |
where
Qt (mg g
−1) is the adsorption capacity and
kd (mg g
−1 min
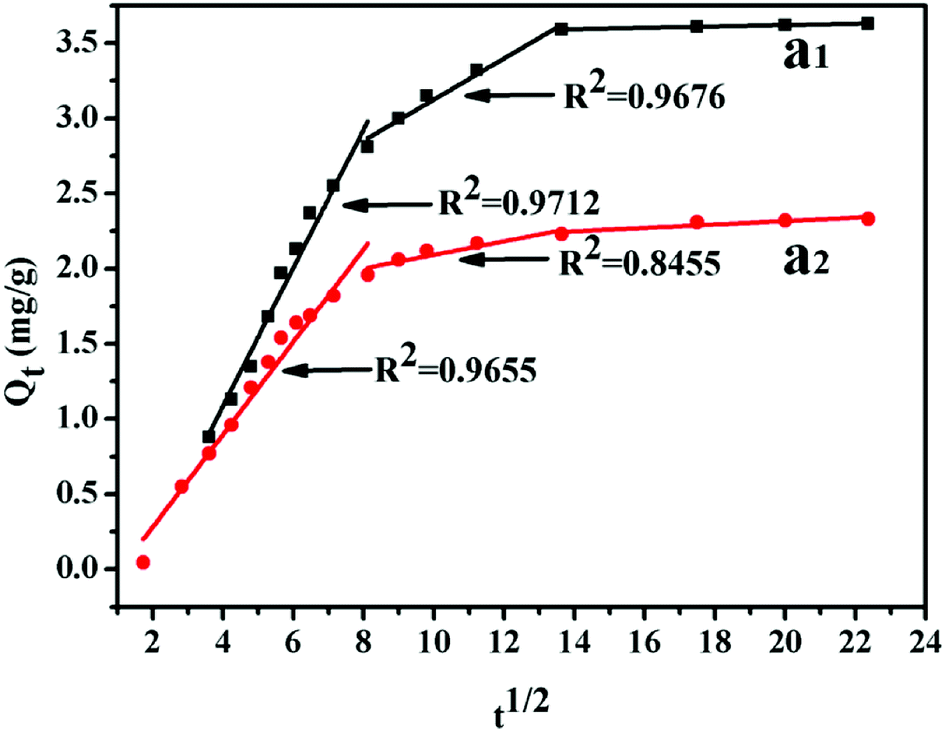
−1/2) is the intra-particle diffusion model constant. There are three stages of adsorption process for MB. Mainly external surface adsorption or layer diffusion in the first stage and the intra-particle of MB diffusion process in the second stage.
42 Table 2 shows that the parameters were obtained by fitting the kinetic adsorption process. In the case of different cross-linking agent contents, the correlation coefficient of pseudo-second-order model was higher than that of the pseudo-first-order model in general. Meanwhile, the rate-limiting step might be the second phase and chemical adsorption during the removal of methylene blue. The results show that the adsorption kinetics type is more correlated with pseudo-second-order model. This finding demonstrates that the interaction between dye cationic ion and UiO-66/PAM is mainly due to the electrostatic interaction (
Fig. 14).
Table 2 List of the pseudo-first-order and the pseudo-second-order kinetic rate and intra-particle diffusion constant (k1, k2) and correlation coefficient R2 of samples (5–7)
| Samples |
Pseudo-first-order |
Pseudo-second-order |
| k1 (min−1) |
R2 |
k2 (min−1) |
R2 |
| 5 |
0.0088 |
0.9711 |
0.2555 |
0.9961 |
| 6 |
0.0074 |
0.9642 |
0.3907 |
0.9990 |
| 7 |
0.0054 |
0.9386 |
0.5572 |
0.9934 |
| Intra-particle diffusion |
| |
Kd1(min−1) |
R2 |
Kd2 (min−1) |
R2 |
| 5 |
0.4593 |
0.9712 |
0.13687 |
0.9676 |
| 6 |
0.3079 |
0.9655 |
0.04518 |
0.8455 |
Table 3 Others adsorption materials and adsorption capacity
| Materials types |
Example |
Adsorption capacity (mg g−1) |
Ref. |
| Synthetic materials |
Mn-25@BCF |
46.3 |
43 |
| UiO-66 |
24.5 |
44 |
| Adsorbent-ultrasonic spray |
289.85 |
45 |
| UiO-66 |
67.5 |
46 |
| Acid-promoted UiO-66 |
13.2 |
46 |
| CMt nanocomposites |
138.1 |
47 |
| Natural inorganic materials |
Zeolite |
10.82 |
48 |
| Amorphous silica |
22.66 |
48 |
| Kaolinite |
35.91 |
49 |
| Bioadsorbents |
Green alga Ulva lactuca |
40.2 |
50 |
| Rice straw |
40.02 |
51 |
| In this work |
UiO-66/PAM HIPE |
50 |
|
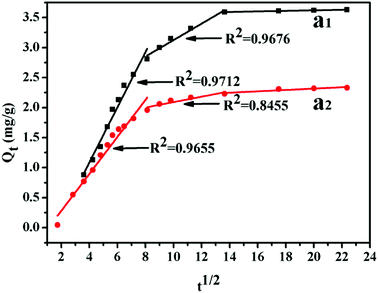 |
| | Fig. 14 Intra-particle diffusion plots of adsorption MB: 1.5 wt% (a1), 8 wt% (a2). | |
The isothermal adsorption data are usually explained by Langmuir and Freundlich, which are shown in the following equations, respectively:
| |
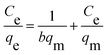 | (5) |
| |
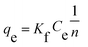 | (6) |
where
Ce is equilibrium concentration of MB solution (mg L
−1). The value of
qe (mg g
−1) and
qm (mg g
−1) represent the equilibrium adsorption capacity and the maximum adsorption capacity, respectively.
b (L mg
−1) and
qm are Langmuir constants.
Kf (mg g
−1) and
n are Freundlich constants.
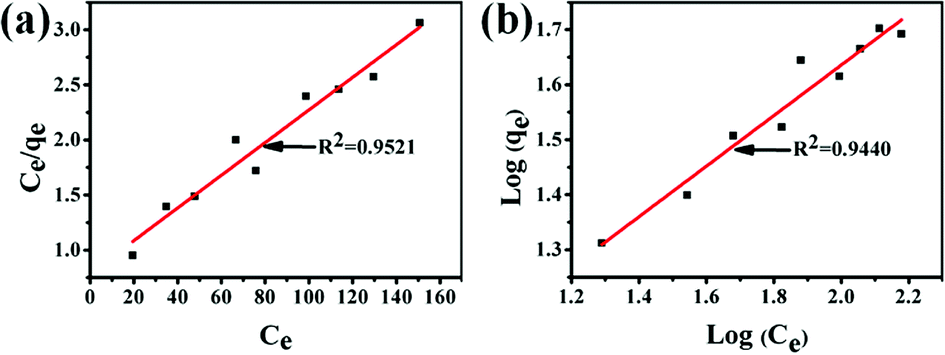
The Langmuir and Freundlich adsorption equilibrium curves of UiO-66/PAM for MB are shown in the Fig. 15. The constants value of Langmuir and Freundlich model for MB adsorption are qm = 67.57 mg g−1, b = 0.0187 L mg−1 (TC: 0.0045 L mg−1), Kf = 5.2199 mg g−1 and n = 2.1744, respectively. Compared with the qm value of Langmuir, the adsorption value in the actual experiment is basically the same (50 mg g−1 at 30 °C). Meanwhile, UiO-66/PAM adsorption of MB, which the correlation coefficients (R2) of Langmuir curve and Freundlich curve are 0.9521 and 0.9440, respectively. Thus, the concentration-dependence isotherm process of the MB adsorption were better-fitted to Langmuir model, which has a more reasonable description of the adsorption process of the UiO-66/PAM material. This means that the adsorbate (MB) is single-layer coverage on the surface of UiO-66/PAM.
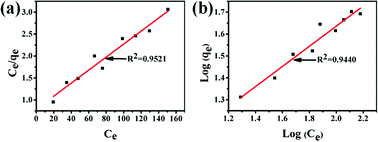 |
| | Fig. 15 Langmuir adsorption equilibrium curve (a) and Freundlich adsorption equilibrium curve (b). | |
4 Conclusion
UiO-66/PAM composite monoliths with interconnected pores were successfully synthesized using the C/W HIPE templated method. Through the different dosages of cross-linking agent, UiO-66, and CO2 internal phase amount, the void pore diameter increased with the increase of the UiO-66 dosage in the composite. The mechanical performances of UiO-66/PAM monolithic composites were also improved compared with the poly(AM)HIPE, resulting in a strain of more than 80%. The UiO-66/PAM containing 20 wt% MBAM exhibits high storage modulus of 42.1 kPa at high angular frequency. For the oil–water separation experiment, the UiO-66/PAM has superoleophobicity that can separate the edible oil and water with 1/3 volume after 2 h. The adsorption kinetics curves showed that the saturated adsorption point can be determined after 4 h, and the maximum saturation adsorption is 50 mg g−1 in soaking 200 mg L−1 initial MB solution. These porous composite monoliths exhibit potential applications in biocatalysis, adsorption-separation, or using as the scaffold. Future work will focus on the demonstration of UiO-66/PAM biocompatibility.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the research grant provided by National Natural Science Foundation of China (No. 51763020).
References
- Z. Wang, J. Liu, H. K. Arslan, S. Grosjean, T. Hagendorn, H. Gliemann, S. Brase and C. Woll, Langmuir, 2013, 29, 15958 CrossRef PubMed.
- U. S. F. Arrozi, H. W. Wijaya, A. Patah and Y. Permana, Appl. Catal., A, 2015, 506, 77 CrossRef.
- D. Saha, D. K. Hazra, T. Maity and S. Koner, Inorg. Chem., 2016, 55, 5729 CrossRef PubMed.
- T. Yang, H. Cui, C. Zhang, L. Zhang and C. Y. Su, Inorg. Chem., 2013, 52, 9053 CrossRef PubMed.
- J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277 CrossRef PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef PubMed.
- M. Zhang, G. Feng, Z. Song, Y. P. Zhou, H. Y. Chao, D. Yuan, T. T. Tan, Z. Guo, Z. Hu and B. Z. Tang, J. Am. Chem. Soc., 2014, 136, 7241 CrossRef PubMed.
- J. Zhuang, C. H. Kuo, L. Y. Chou, D. Y. Liu, E. Weerapana and C. K. Tsung, ACS Nano, 2014, 8, 2812 CrossRef PubMed.
- J. D. Rocca, D. Liu and W. Lin, J. Shanghai Norm. Univ., Nat. Sci., 2012, 44, 957 Search PubMed.
- S. Ma and H. C. Zhou, Chem. Commun., 2010, 46, 44 RSC.
- J. Goldsmith, A. G. Wongfoy, M. J. Cafarella and D. J. Siegel, Chem. Mater., 2013, 25, 3373 CrossRef.
- L. Arnold, G. Averlant, S. Marx, M. Weickert, U. Müller, J. Mertel, C. Horch, M. Peksa and F. Stallmach, Chem. Ing. Tech., 2013, 85, 1726 CrossRef.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. Mcdonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef PubMed.
- B. Zhang, J. Zhang, C. Liu, L. Peng, X. Sang, B. Han, X. Ma, T. Luo, X. Tan and G. Yang, Sci. Rep., 2016, 6, 21401 CrossRef PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- A. D. Vos, K. Hendrickx, P. V. D. Voort, V. V. Speybroeck and K. Lejaeghere, Chem. Mater., 2017, 29, 3008 Search PubMed.
- X. W. Chen, J. M. Wang, J. Guo, Z. L. Wan, S. W. Yin and X. Q. Yang, Food Funct., 2017, 8, 823 RSC.
- A. Patel, Y. Rodriguez, A. Lesaffer and K. Dewettinck, RSC Adv., 2014, 4, 18136 RSC.
- J. M. Hughes, P. M. Budd, K. Tiede and J. Lewis, J. Appl. Polym. Sci., 2014, 132, 41229 Search PubMed.
- S. Yu, H. Tan, W. Jin, L. Xin and K. Zhou, ACS Appl. Mater. Interfaces, 2015, 7, 6745 CrossRef PubMed.
- T. Zhang, Y. Wu, Z. Xu and Q. Guo, Chem. Commun., 2014, 50, 13821 RSC.
- Y. Ma, H. D. Asfaw and K. Edström, Chem. Mater., 2015, 27, 3957 CrossRef.
- V. O. Ikem, A. Menner and A. Bismarck, Angew. Chem., 2009, 48, 8277 CrossRef PubMed.
- I. Capron and B. Cathala, Biomacromolecules, 2013, 14, 291 CrossRef PubMed.
- H. Yu, Z. Qin, C. Yan and J. Yao, ACS Sustainable Chem. Eng., 2014, 2, 875 CrossRef.
- E. Passaslagos and F. Schuth, Langmuir, 2015, 31, 7749 CrossRef PubMed.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed. Engl., 2016, 128, 6771 CrossRef PubMed.
- A. I. Cooper, J. Mater. Chem., 2000, 10, 207 RSC.
- M. Hou, L. Cao, J. Wang, J. Lin, M. Zhao and G. Wang, Polym. Adv. Technol., 2014, 25, 693 CrossRef.
- H. Chi, L. Cao and J. Wang, RSC Adv., 2016, 6, 4434 RSC.
- C. C. Hwang, J. J. Tour, C. Kittrell, L. Espinal, L. B. Alemany and J. M. Tour, Nat. Commun., 2014, 5, 3961 CrossRef PubMed.
- R. Butler, C. M. Davies and A. I. Cooper, Adv. Mater., 2001, 13, 1459 CrossRef.
- H. Zhu, Q. Zhang and S. Zhu, Chem.–Eur. J., 2016, 22, 8751 CrossRef PubMed.
- C. Liu, J. Zhang, L. Zheng, J. Zhang, X. Sang, X. Kang, B. Zhang, T. Luo, X. Tan and B. Han, Angew. Chem., 2016, 55, 11372 CrossRef PubMed.
- M. R. Armstrong, K. Y. Y. Arredondo, C. Y. Liu, J. E. Stevens, A. Mayhob, B. Shan, S. Senthilnathan, C. J. Balzer and B. Mu, Ind. Eng. Chem. Res., 2015, 54, 3957 CrossRef.
- S. Yang, Y. Wang, Y. Jia, X. Sun, P. Sun, Y. Qin, R. Li, H. Liu and C. Nie, Colloid Polym. Sci., 2018, 296, 1005 CrossRef.
- J. Wang, Y. Li, Y. Gao, Z. Xie, M. Zhou, Y. He, H. Wu, W. Zhou, X. Dong and Z. Yang, Ind. Crops Prod., 2018, 112, 281 CrossRef.
- H. Tan, J. Wei, G. Sun, C. Mu, W. Lin and T. Ngai, Soft Matter, 2017, 13, 3871 RSC.
- S. Das, F. Irin, L. Ma, S. K. Bhattacharia, R. C. Hedden and M. J. Green, ACS Appl. Mater. Interfaces, 2013, 5, 8633 CrossRef PubMed.
- Z. Zhao, Y. Liu, K. Zhang, S. Zhuo, R. Fang, J. Zhang, L. Jiang and M. Liu, Angew. Chem., 2017, 129, 13464 CrossRef PubMed.
- F. Gao, Y. Zhang, Y. Li, B. Xu, Z. Cao and W. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 8956 CrossRef PubMed.
- B. Hui and L. Ye, J. Ind. Eng. Chem., 2016, 35, 309 CrossRef.
- J. Wang, L. Kou, Z. Huang and L. Zhao, RSC Adv., 2018, 8, 21577 RSC.
- J. Yang, J. Colloid Interface Sci., 2017, 505, 178 CrossRef PubMed.
- S. Cheng, L. Zhang, H. Xia, J. Peng, J. Shu, C. Li, X. Jiang and Q. Zhang, RSC Adv., 2017, 7, 27331 RSC.
- J. Qiu, Y. Feng, X. Zhang, M. Jia and J. Yao, J. Colloid Interface Sci., 2017, 499, 151 CrossRef PubMed.
- D. S. Tong, C. W. Wu, M. O. Adebajo, G. C. Jin, W. H. Yu, S. F. Ji and C. H. Zhou, Appl. Clay Sci., 2018, 161, 256 CrossRef.
- C. D. Woolard, J. Strong and C. R. Erasmus, Appl. Geochem., 2003, 18, 1279 CrossRef.
- W. Gao, S. Zhao, H. Wu, W. Deligeer and S. Asuha, Appl. Clay Sci., 2016, 126, 98 CrossRef.
- A. E. Sikaily, A. Khaled, A. E. Nemr and O. Abdelwahab, Chem. Ecol., 2006, 22, 149 CrossRef.
- S. Zhang, Z. Wang, Y. Zhang, H. Pan and L. Tao, Procedia Environ. Sci., 2016, 31, 3 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Jing Lib,
Yongxia Yanga and
Jide Wang
*a,
Jing Lib,
Yongxia Yanga and
Jide Wang