DOI:
10.1039/C8RA05760E
(Paper)
RSC Adv., 2018,
8, 36150-36160
New photoluminescent iodoargentates with bisimidazole derivatives as countercations†
Received
6th July 2018
, Accepted 12th October 2018
First published on 23rd October 2018
Abstract
In this article, three bisimidazole derivatives (1,4-bis(2-ethylimidazol-1-yl)butane, L1; 4,4′-di(1H-imidazol-1-yl)-1,1′-biphenyl, L2′; and 1,3-bis(2-ethylimidazol-1-yl)propane, L3) were employed to solvothermally react with AgI in an acidic environment, creating three new 1-D chained iodoargentates [H(L1)][Ag5I6]·DMF (DMF = N,N′-dimethylformamide) 1, [L2][Ag3I5] (L22+ = 4,4′-di(1H-imidazol-1-ium)-1,1′-biphenyl) 2, and [H2(L3)][Ag2I4] 3. L22+ in 2 originated from the in situ N-alkylation of L2′ with the CH3OH solvent. X-ray single-crystal diffraction analysis reveals that (i) in 1, Ag+ and I− aggregate to form a 1-D tube-like iodoargentate, which exhibits the same topology as the carbon tube; (ii) the chain structure of the iodoargentate in 2 is based on a kind of trinuclear Ag–I cluster, which can be viewed as a segment of the classical cubic M4I4 cluster; (iii) the chain structure of the iodoargentate in 3 is simple, which can be described as a linear arrangement of the AgI4 tetrahedra by sharing edges. The photoluminescence analysis reveals that at 77 K, (i) 1 and 2 emit strong yellow light with ms-grade photoluminescence lifetimes (5.460 ms for 1, 6.931 ms for 2); (ii) 3 possesses photochromic luminescence properties. Upon excitation at 254 nm, it emits blue-green light, whereas upon excitation at 365 nm, it emits yellow light.
Introduction
Much attention has been paid to the design and synthesis of novel haloargentates with various substances as the countercations due to their structural diversity1 and their potential applications in photoluminescence,2 thermo(photo)chromism,3 dye degradation4 and non-linear optics.5 For example, Chen et al. constructed a series of novel haloargentates by employing pre-synthesized organic base derivatives as the countercations, and found that some of them possess better photoluminescence and non-linear optical properties.6 Fu et al. constructed a series of novel iodoargentates by employing pyridine derivatives as the countercations, and found that some of them have thermo(photo)chromism properties.7 Yue et al. constructed a series of novel iodo(bromo)argentates by employing in situ generated transition-metal complexes as the countercations, and found that some of them can effectively degrade organic dye pollutants.8 In the construction of this type of material, the counteraction plays a key role. It not only directly controls the structures of the haloargentate anion and the final supramolecular network, but also directly or indirectly affects the functional property of the material. For instance, the superoxide ion ·O2− has been confirmed to be the dominant reactive species for the degradation of the organic dyes. Just due to a hybrid process of the inorganic and organic moieties, the electronic structure and the band gap for the material are changed, which makes the photogenerated electrons more easily transfer. Then the electrons are further to be trapped by O2 to form ·O2−.8 The pre-synthesized organic base derivatives (through the N-alkylation of organic bases with organic halides),6 in situ generated complexes,8 and the in situ protonated organic bases1 can all be employed to serve as the countercations. Recently, the in situ N-alkylation of organic bases with alcohol molecules has also been employed to create the new countercations.9 So far, the in situ N-alkylation of pyridine derivatives with alcohol molecules have been extensively employed in the construction of new organic–inorganic hybrid materials, but the in situ N-alkylation of imidazole derivatives with alcohol molecules is seldom considered.10 In addition, based on our investigations, the in situ protonation of imidazole derivatives has seldom employed in the construction of the iodoargentate.
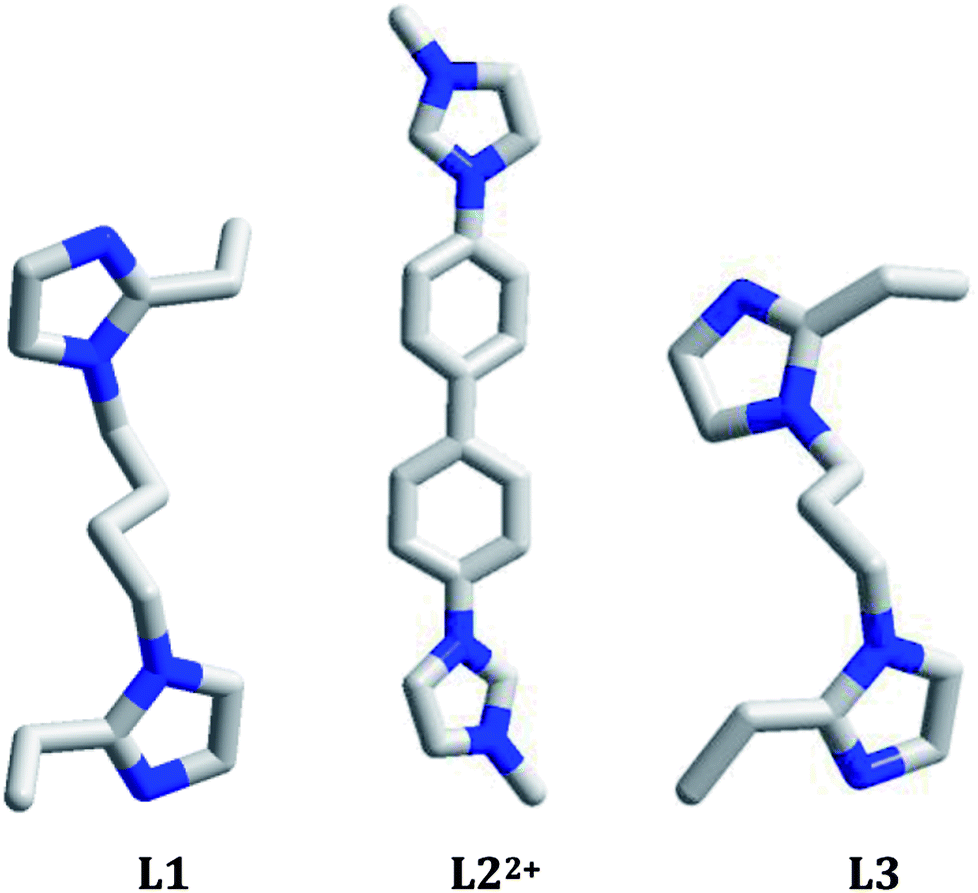
At recent, the tunable photoluminescence materials have gained the considerable interest owing to their potential technological applications in light emitting diodes, bioimaging and memories.11 Although the degradation on organic dyes as well as the thermo(photo)chromism for the hybrid haloargentates have been well developed,3,4,7,8 the investigations on the photoluminescence behaviors for this type of material are still limited. For the reported hybrid haloargentates, only the room-temperature photoluminescence behaviors were characterized, whereas the photoluminescence behaviors with the change of temperature and excitation wavelength (namely the so-called luminescence thermo(photo)chromic properties) as well as the photoluminescence lifetime are rarely investigated.2,6 Many CuI–X cluster-based compounds have been proved to be the excellent photoluminescence thermochromic materials.12 This is mainly due to the following two factors: (i) considerable flexibility for CuI–X cluster. The CuI–I cluster easily distorts upon external stimulus; (ii) considerable sensibility of photoluminescence behavior for CuI–I cluster. A minor distinction in structure can produce a big variation of the photoluminescence behavior.13 As an element of the same group as Cu, do the Ag–I cluster-based hybrids also possess the photoluminescence thermochromic properties? The photoluminescence photochromic property is generally associated with the existence of multiple emissions in the molecule.13 So far, only extremely limited hybrids have been found to possess this property, such as [bmim]2[SbCl5] (bmim+ = 1-butyl-3-methylimidazolium),14 [spy][CdCl2(H2O)2] (spy = N-succinopyridine),15 and [dmdabco]2[Cu4Br5(CN)3] (dmdabco2+ = N,N′-dimethyl-1,4-diazabicyclo[2,2,2]octanium).13 Do the Ag–I cluster-based hybrids have the photoluminescence photochromic properties? What about the photoluminescence lifetimes? Based on these questions, in this article we constructed three new 1-D chained iodoargentates by employing three bisimidazole derivatives (1,4-bis(2-ethylimidazol-1-yl)butane, L1; 4,4′-di(1H-imidazol-1-yl)-1,1′-biphenyl, L2′; 1,3-bis(2-ethylimidazol-1-yl)propane, L3) as the countercations: [H(L1)][Ag5I6]·DMF (DMF = N,N′-dimethylformamide) 1, [L2][Ag3I5] (L22+ = 4,4′-di(1H-imidazol-1-ium)-1,1′-biphenyl) 2, and [H2(L3)][Ag2I4] 3. Of those, L22+ originated from the in situ N-alkylation of 4,4′-di(1H-imidazol-1-yl)-1,1′-biphenyl (L2′) with CH3OH. The photoluminescence behaviors of these three hybrids at 298 K, 177 K, and 77 K were measured. Their photoluminescence behaviors under the different excitation wavelengths were investigated. Their photoluminescence lifetimes were also measured (Scheme 1).
 |
| | Scheme 1 Molecular structures of organic bases in 1–3. | |
Experimental
Materials and physical measurement
All chemicals are of reagent grade quality, obtained from commercial sources without further purification. Elemental analysis (C, H and N) was performed on a Perkin-Elmer 2400LS II elemental analyzer. Infrared (IR) spectrum was recorded on a Perkin Elmer Spectrum 1 spectrophotometer in 4000–400 cm−1 region using a powdered sample on a KBr plate. Powder X-ray diffraction (XRD) data were collected on a Rigaku/max-2550 diffractometer with Cu-Kα radiation (λ = 1.5418 Å). Thermogravimetric (TG) behavior was investigated on a Perkin-Elmer TGA-7 instrument with a heating rate of 10 °C min−1 in air. Ultraviolet-visible (UV-Vis) spectrum was obtained on a Rigaku-UV-3100 spectrophotometer. Fluorescence spectra were recorded on a LS 55 florescence/phosphorescence spectrophotometer at room temperature. Fluorescence lifetime was measured on an Edinburgh Instrument FLS920 steady-state transient fluorescence spectrometer.
Synthesis of 1–3
[H(L1)][Ag5I6]·DMF 1. The yellow block crystals of 1 were obtained from a simple solvothermal self-assembly of AgI (12 mg, 0.05 mmol) and L1 (13 mg, 0.05 mmol) in a mixed solvent of 2 mL C2H5OH and 3 mL DMF (pH = 5) at 100 °C for 3 days. Yield: ca. 20% based on Ag(I). Anal. Calcd C17H30N5OAg5I6 1: C 12.59, H 1.87, N 4.32. Found: C 12.29, H 2.05, N 4.53%. IR (cm−1): 1672 s, 1442 w, 1387 m, 1255 m, 1092 s, 1058 w, 1046 w, 920 w, 794 m, 745 s, 723 s, 662 m, 625 w.
[L2][Ag3I5] 2. The yellow block crystals of 2 were obtained from a simple solvothermal self-assembly of AgI (23 mg, 0.1 mmol), L2 (29 mg, 0.1 mmol) and HI (1 mL 45%) in a mixed solvent of 4 mL CH3OH and 1 mL CH3CN (pH = 2) at 120 °C for 3 days. Yield: ca. 35% based on Ag(I). Anal. Calcd C20H20N4Ag3I5 2: C 18.85, H 1.58, N 4.40. Found: C 18.21, H 1.42, N 4.72%. IR (cm−1): 3092 w, 3016 s, 1628 w, 1589 w, 1494 m, 1460 w, 1419 w, 1347 w, 1289 w, 1260 w, 1227 s, 1074 m, 952 w, 851 m, 826 s, 763 m, 724 w, 703 w, 614 m, 517 m.
[H2(L3)][Ag2I4] 3. The yellow block crystals of 3 were obtained from a simple solvothermal self-assembly of AgI (23 mg, 0.1 mmol), L3 (18 mg, 0.1 mmol) and HI (0.25 mL 45%) in 7 mL C2H5OH (pH = 2) at 120 °C for 3 days. Yield: ca. 25% based on Ag(I). Anal. Calcd C13H22N4Ag2I4 3: C 16.30, H 2.30, N 5.85. Found: C 16.20, H 2.47, N 5.96%. IR (cm−1): 1593 s, 1450 m, 1419 m, 1393 m, 1299 s, 1109 s, 1064 w, 1030 w, 1004 w, 980 w, 923 m, 870 w, 854 w, 806 w, 794 w, 770 m, 742 s.
X-ray crystallography
The data were collected with Mo-Kα radiation (λ = 0.71073 Å) on a Rigaku R-AXIS RAPID IP diffractometer for 1 and 2, and on a Siemens SMART CCD diffractometer for 3. With SHELXTL program, the structures of 1 and 2 were solved using direct methods, whereas the structure of 3 was solved using heavy-atom methods.16 The non-hydrogen atoms were assigned anisotropic displacement parameters in the refinement. The hydrogen atoms were treated using a riding model. The structures were then refined on F2 using SHELXL-2018.16 The CCDC numbers are 1818391 for 1, 1818388 for 2 and 1818390 for 3, respectively (Table 1).
Table 1 Crystal data of 1–3
| |
1 |
2 |
3 |
| Formula |
C17H30N5OAg5I6 |
C20H20N4Ag3I5 |
C13H22N4Ag2I4 |
| M |
1621.21 |
1274.51 |
957.69 |
| T (K) |
293(2) |
293(2) |
299(2) |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
Cm |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
21.078(4) |
18.655(4) |
7.2734(2) |
| b (Å) |
21.543(4) |
13.100(3) |
9.8577(2) |
| c (Å) |
7.9092(16) |
12.832(3) |
16.4819(4) |
| α (o) |
90 |
90 |
81.4680(10) |
| β (o) |
107.59(3) |
110.08(3) |
87.2330(10) |
| γ (o) |
90 |
90 |
85.0890(10) |
| V (Å3) |
3423.5(11) |
2945.3(10) |
1163.61(5) |
| Z |
4 |
4 |
2 |
| Dc (g cm−3) |
3.145 |
2.874 |
2.733 |
| μ (mm−1) |
8.240 |
7.232 |
6.992 |
| Reflections collected |
13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 589 589 |
25104 |
25513 |
| Unique reflections |
6062 |
6547 |
5759 |
| Rint |
0.0279 |
0.0413 |
0.0239 |
| Gof |
1.043 |
1.034 |
1.038 |
| R1, I > 2σ(I) |
0.0298 |
0.0392 |
0.0248 |
| wR2, all data |
0.0644 |
0.0869 |
0.0530 |
Results and discussion
Synthetic analysis
All of the reactions were carried out under the solvothermal conditions. In general, such a reaction of AgI, organic bisimidazole molecule and HI will be investigated, as exemplified by the reactions of preparing 2 and 3. Here HI possesses the following several roles: (i) providing a strong acid source. This is helpful to the thorough protonation of the organic bisimidazole molecule. On the other hand, the in situ N-alkylation of organic bisimidazole molecules with alcohol molecules also needs a strongly acidic environment; (ii) providing the extra I− source; (iii) improving the solubility of the reactive precursors in the solvent. Obviously, the reaction of preparing 1 shows an exceptional situation. In the presence of HI, no any crystalline solid was obtained. But without HI, the title compound 1 was luckily obtained. Without HI, the acidic degree of reactive system is not high, so the bisimidazole molecule is just to be monoprotonated. The formation of the iodoargentate in 1 ([Ag5I6]−) should be associated with the +1 oxidation state of the bisimidazole molecule. The reactions were generally performed in an alcohol solvent. The aim is to hope that the in situ N-alkylation of organic bisimidazole molecule with alcohol molecule could occur, creating a new countercation. Regretfully, only in the reaction of synthesizing 2, the in situ N-alkylation occurred. In the reactions of synthesizing 1 and 3, the in situ alkylation was not observed. This might be due to the existence of the substituted –C2H5 group on the imidazole moiety of L1 and L3. As exemplified by the reactions of preparing 1 and 2, another kind of strong polar solvent was sometimes used to aid the alcohol molecule to dissolve the reactive precursors (DMF for 1, CH3CN for 2). Interestingly, the solvent DMF was introduced in the final backbone of 1. We are sure that DMF also plays a crucial role in the formation of the inorganic anionic framework for 1, because when DMA (N,N-dimethylacetamide) instead of DMF was used, a different iodoargentate was obtained. Regretfully, the crystal data of this compound do not pass the cif-checking examination. In addition, 1,4-di(1H-imidazol-1-yl)benzene (L4) was also employed to react with AgI, creating the compound [H2(L4)][Ag2I4] 4. Regretfully, due to the severe disorder of the imidazole rings, the crystal data of 4 do not pass the cif-checking examination, either. A bistriazole molecule (1,3-di(1H-1,2,4-triazol-1-yl)propane, L5′) was also employed to react with AgI in C2H5OH, producing the compound [L5][Ag2I4] (L52+ = 2-ethyl-1-(3-(4-ethyl-1H-1,2,4λ4-triazol-1-yl)propyl)-1H-1,2λ4,4-triazolium) 5. Note that the in situ N-alylation of bistriazole with C2H5OH was observed for the first time. Regretfully, the pure 5 was not obtained so far.
Structural description
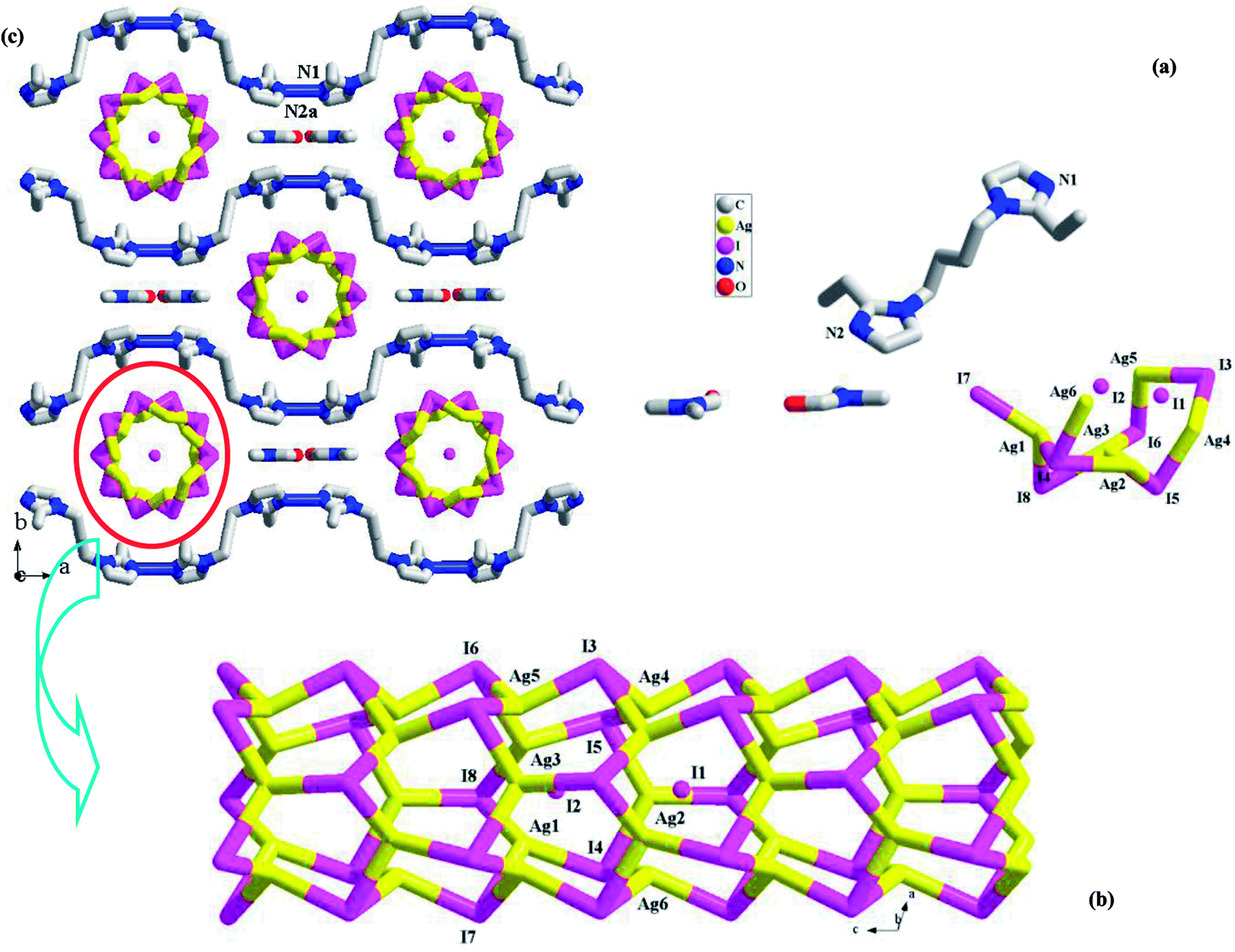
[H(L1)][Ag5I6]·DMF 1. X-ray single-crystal diffraction analysis reveals that 1 is a 1-D tubed iodoargentate with H(L1)+ as the countercation. The solvent molecule DMF also appears in the final framework of 1. It crystallizes in a non-centric space group Cm, and the asymmetric unit (see Fig. 1a) is found to be composed of six Ag+ ions (occupancy ration: 1 for Ag1, Ag2, Ag3, Ag4; 0.5 for Ag5, Ag6; 5 for all), eight I− ions (occupancy ratio: 1 for I4, I5, I6, I8; 0.5 for I1, I2, I4, I7; 6 for all), one L1 molecule, and two DMF molecules (occupancy ratio: 0.5 for each atom). L1 should be monoprotonated in order to balance the anionic charge ([Ag5I6]−). Fig. 1b illustrates the structure of the inorganic anion [Ag5I6]−. All of the Ag+ ions together with six I− ions (I3–I8) construct a 1-D endless tube, running down the c-axial direction. The other two I− ions (I1, I2) are distributed on the centric axis of the tube. Actually, all of the Ag+ ions form the covalent bonds to I1 or I2. For clarity and better understanding its structure, these bonds are omitted. All of the Ag+ ions in 1 are tetrahedrally coordinated by four I− ions. The Ag–I1/I2 bonds (2.9320(19)–3.0042(11) Å) are obviously longer than the others (2.8151(11)–2.8766(13) Å). Eight I− ions adopt two types of geometric configurations. The I− ions on the tube all adopt a trigonal pyrimidal configuration, whereas I1 and I2 adopt a pentagonal pyrimidal configuration (see Fig. S1†). According to the topological method, Ag+ and I− on the tube are all viewed as a 3-connected node, so this tube has the same topology as the carbon tube. Fig. 1c is the projection plot of 1 in the (001) direction. Via the N1imidazole–H⋯N2aimidazole interactions (N1⋯N2a = 2.707(9) Å), the adjacent H(L1)+ molecules self-assemble into a 1-D castellated supramolecular chain, extending along the a-axial direction. Along the c-axial direction, the neighboring supramolecular chains array in a parallel way, while along the b-axial direction, the adjacent supramolecular chains array in the form of mirror symmetry. Thus, two types of spaces appear. The anionic tube occupies the larger space, whereas the DMF molecules occupy the smaller space. The shortest Ag⋯Ag contact distance in 1 is Ag1⋯Ag3 = 3.1832(14) Å.
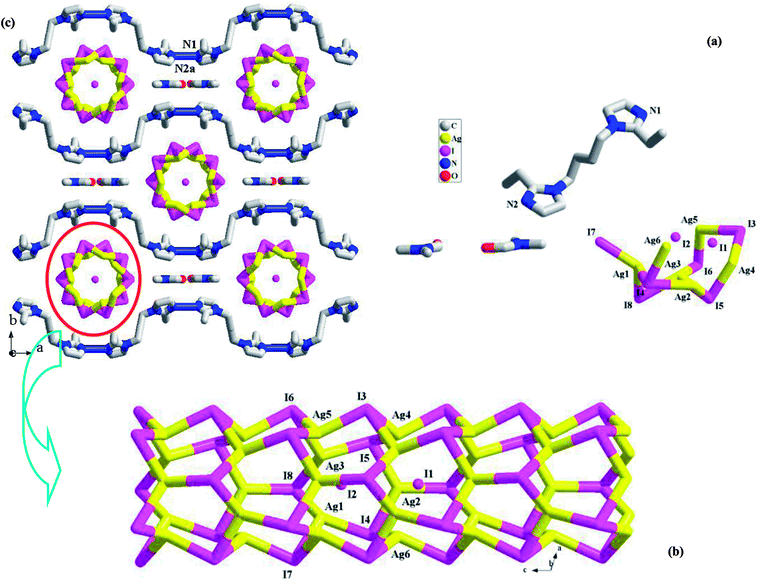 |
| | Fig. 1 Asymmetric unit (a), 1-D chain (b), and projection plot in (001) direction (c) in 1 (a: x + 1/2, −y + 1/2, z). | |
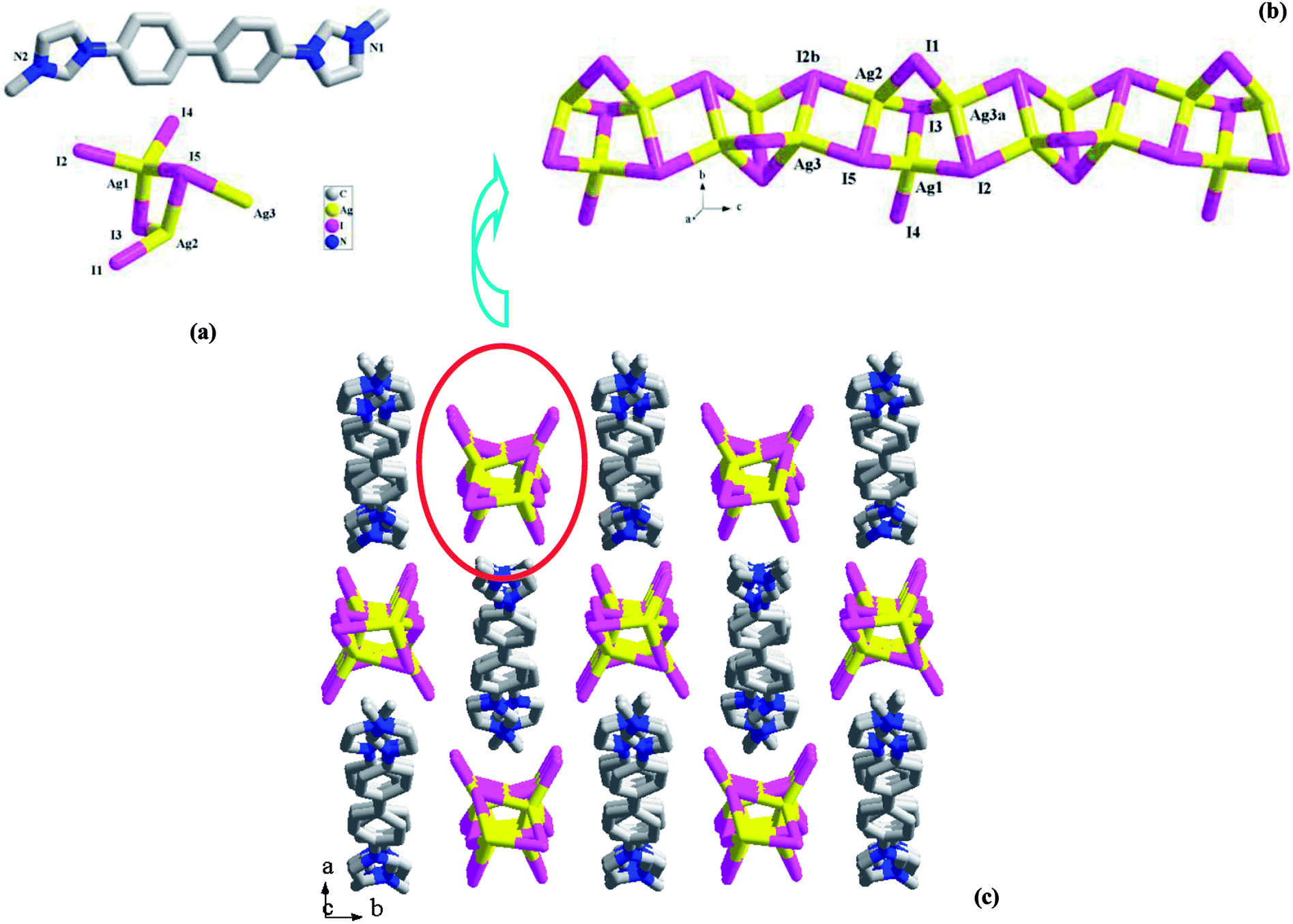
[L2][Ag3I5] 2. 2 is a L22+-templated 1-D chained iodoargentate. Obviously, the in situ N-alkylation of L2′ with CH3OH really occurred, producing L22+. Compound 2 crystallizes in the space group P21/c, and the asymmetric unit is found to be composed of three Ag+ ions (Ag1, Ag2, Ag3), five I− ions (I1, I2, I3, I4, I5), and one L22+ molecule (see Fig. 2a). Fig. 2b illustrates the chain structure of the inorganic anion [Ag3I5]2−. The chain is based on a kind of trinuclear Ag–I cluster. This trinuclear cluster can be described as a moiety of the classical cubic M4I4 cluster (M+ = Cu+, Ag+).17 The removal of one M+ ion for cubic M4I4 cluster forms this trinuclear cluster. Then via the intercluster Ag–I interactions (Ag3–I5 = 2.8467(9) Å, Ag2–I2b = 2.8559(9) Å), the neighboring trinuclear clusters are linked together to form the title 1-D chain of 2, which extends along the c-axial direction. In 2, three Ag+ ions are all in a tetrahedral site, and coordinated by four I− ion. The Ag–I bond length range is 2.7360(9)–2.9517(14) Å. Five I− ions are involved in three types of coordination modes: a terminal mode for I4; a double bridged mode for I1; a triple bridged mode with a trigonal pyrimidal configuration for I2, I3 and I5. The shortest Ag⋯Ag contact distance in 2 is Ag1⋯Ag2 = 3.0325(11) Å. Fig. 2c is the projection plot of 2 in the (001) direction. Via the π⋯π interactions between the L22+ molecules, the adjacent L22+ molecules self-assemble into a supramolecular chain, running down the c-axial direction. Each four supramolecular chains enclose a square channel. The inorganic anion [Ag3I5]2− occupies the channel space. Between the L22+ molecule and the inorganic anion, there exist the C–H⋯I hydrogen-bonded interactions, stabilizing the crystal structure of 2. For clarity, these C–H⋯I interactions are omitted.
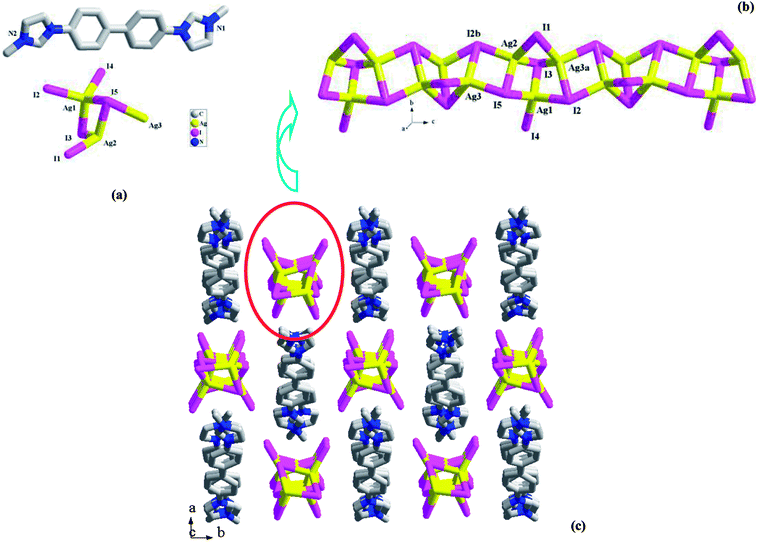 |
| | Fig. 2 Asymmetric unit (a), 1-D chain (b), and projection plot in (001) direction (c) in 2 (a: x, −y + 3/2, z + 1/2; b: x, −y + 3/2, z − 1/2). | |
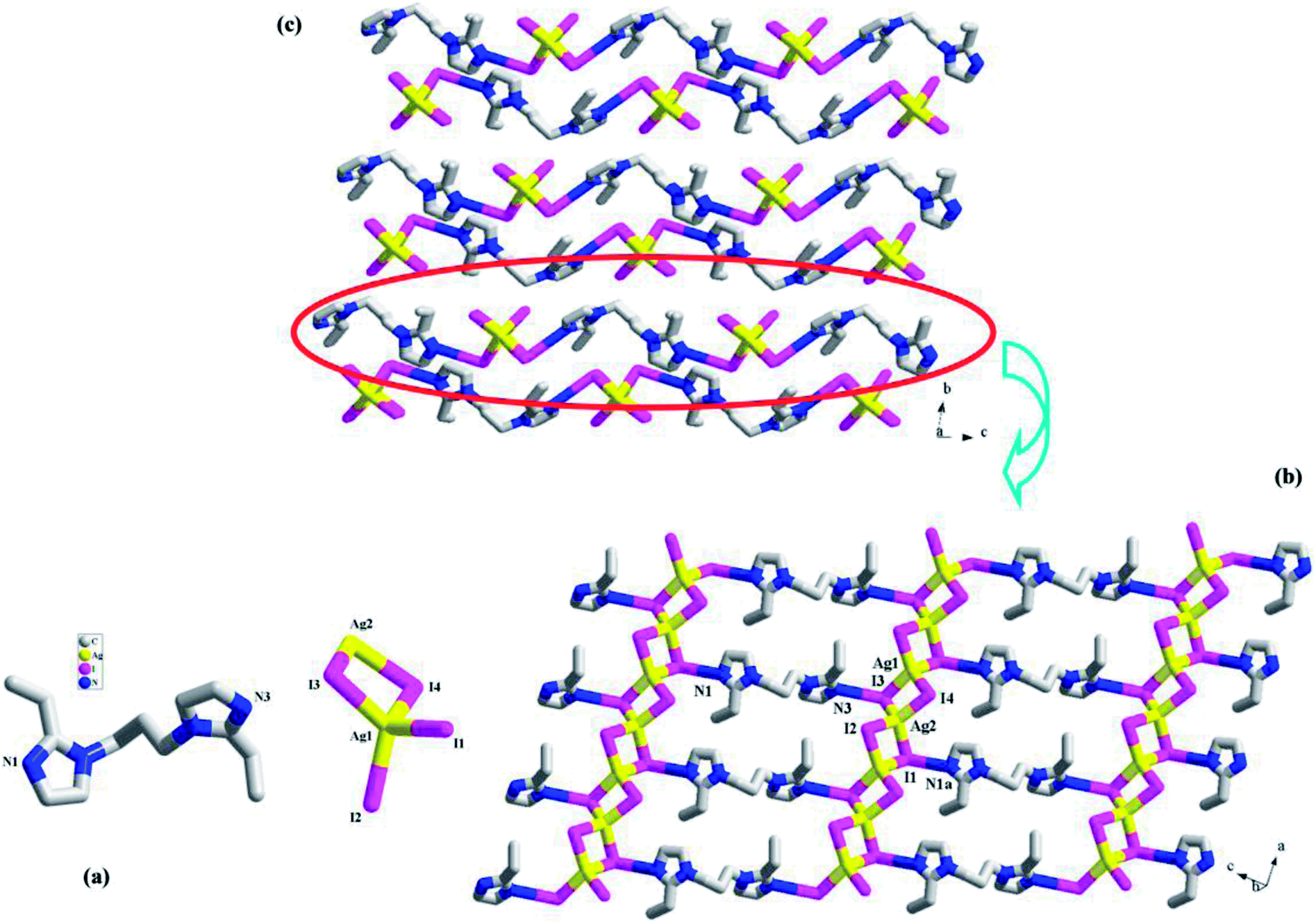
[H2(L3)][Ag2I4] 3. 3 is a H2(L3)2+-templated 1-D chained iodoargentate. It crystallizes in the space group P, and the asymmetric unit is found to be composed of two Ag+ ions (Ag1, Ag2), four I− ions (I1, I2, I3, I4), and one H2(L3)2+ molecule (see Fig. 3a). Fig. 3b illustrates a 2-D supramolecular layer network in 3, which is constructed up from the Ag–I chains by the H2(L3)2+ molecules. In the Ag–I chain, two crystallographically independent Ag+ ions (Ag1, Ag2) are in a tetrahedral site, and surrounded by four I− ions. The Ag–I bond length range is 2.8300(4)–2.8926(4) Å. Four I− ions all adopt a μ2-bridging mode. The I− ions doublely bridge the tetrahedral Ag+ ions to form this 1-D Ag–I chain of 3, which can also be described as a linear arrangement of the AgI4 tetrahedra by sharing the edges. The shortest Ag⋯Ag contact distance in the chain is Ag1⋯Ag2 = 3.645 Å. I1 and I3 form the hydrogen bonds to the imidazole N atoms (N1a, I1⋯N1a = 3.489(3) Å; N3, I3⋯N3 = 3.500(3) Å). Via these N–H⋯I interactions, the H2(L3)2+ molecules link the [Ag2I4]2− chains into a 2-D supramolecular layer network. If Ag+, I2 and I4 are ignored, this 2-D supramolecular layer network can be simplified into a 63 net. Fig. 3c is the projection plot of 3 in the (100) direction. Between the supramolecular layers, there exists the weak C–H⋯I interactions. For clarity, they are omitted.
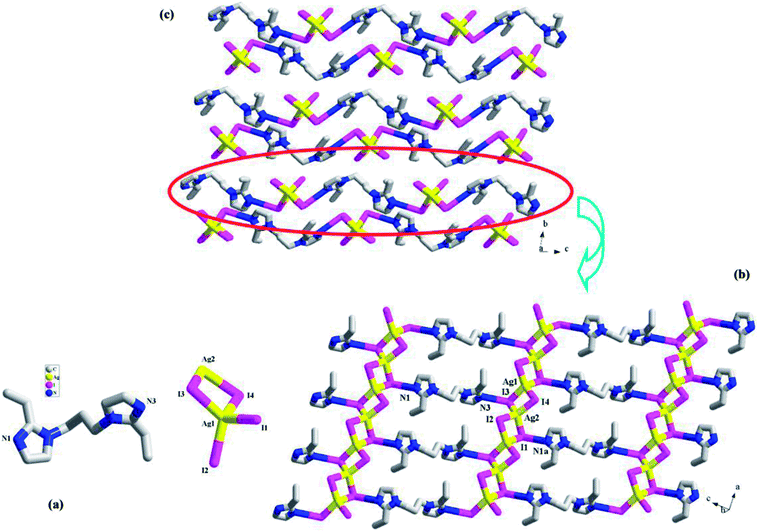 |
| | Fig. 3 Asymmetric unit (a), 2-D supramolecular layer (b), and projection plot in (100) direction (c) in 3 (a: x + 1, y, z − 1). | |
Structural discussion
All of the title compounds are new, and have never been reported. In 1–3, the different organic bisimidazole molecules are employed, producing three different iodoargentates: the tubed iodoargentate in 1; the cluster-based chained iodoargentate in 2; the Ag–I single chain in 3. The formation of the iodoargentate should be related to the organic bisimidazole molecule. Through the charge, size, form, and the various hydrogen-bonded interactions with the iodoargentate, the bisimidazole molecule directly controls the backbone structure of the iodoargentate. For example, (i) in 1, the L1 molecule is just to be monoprotonated, and has a +1 charge, so an iodoargentate with a −1 charge is formed; (ii) the cubic M4I4 cluster is a classical structure. The aim of the appearance for the terminal I− ion as well as the removal of an apical Ag+ ion in 2 should be to balance the +2 charge of the counteraction (L22+); (iii) in 1, due to the better flexibility of L1, they self-assemble into a 1-D castellated supramolecular chain, which leads to the appearance of the larger space. Such a space should be associated with the formation of the iodoargentate tube in 1; (iv) in the packing structures of 1–3, many N–H⋯I and C–H⋯I hydrogen-bonded interactions are found (see Table S1†). Their roles should not be ignored. The formation of the iodoargentate should also be related to the solvent molecule, as exemplified by 1. The Ag–I single chain in 3 has been frequently observed in the reported iodoargentates.1a,d,2e However, only in the limited examples,2b,4c,7h the tubed iodoargentate in 1 and the cluster-based iodoargentate in 2 have ever been found. Even though the same iodoargentates are found in the different compounds, the Ag–I and Ag⋯Ag interactions are generally different. This will directly influence some functional property of the material. Meanwhile, the different packing structures will be formed. For instance, the same Ag–I singe chains are found in 3 and 4, but in 3, the 2-D supramolecular layer is found, whereas in 4, the pore yet appears (ca. 500 Å3).
Characterization
Fig. S2† presents the experimental and simulated powder XRD patterns of 1–3. The experimental powder XRD pattern for each compound is in accord with the simulated one generated on the basis of structural data, confirming that the as-synthesized product is pure phase. The TG behaviors of the title compounds in the temperature range of 30–800 °C were investigated. Based on their TG carves (Fig. S3†), we can know that (i) in the temperature range of 30–800 °C, the weigh loss for all do not end; (ii) 2 and 3 possess the better thermobility, and can be thermal stable up to ca. 340 °C; (iii) the initial minor weight loss for 1 (ca. 4.2%) should be attributed to the sublimation of DMF (calcd: 4.5%). Ca. 19% weight loss for the second step for 1 should be assigned to the removal of L1 (calcd: 18.8%). At this time, the intermediate should be H[Ag5I6]; (iv) the initial weight loss for 2 should correspond to the departure of [L2]I2, even though the platform is not obvious; (v) the initial two steps of weight loss for 3 should be assigned to the removal of [H2(L3)]I2 (calcd: 51%; found: 50%). The solid-state UV-Vis spectra of the title compounds were investigated. As shown in Fig. S4,† the adsorption edges at 416 nm for 1, 395 nm for 2 and 354 nm for 3, suggest that the energy gaps for three hybrids are 2.95 eV for 1, 3.11 eV for 2 and 3.47 eV for 3, respectively. Compared with the energy gap of AgI (2.81 eV),18 the energy gaps for the title three hybrids all become wide, which should be due to the hybrid. Fig. S5† illustrates the IR spectra of 1–3.
Photoluminescence property
In order to preliminarily know the photoluminescence properties of the title three compounds, we first recorded their photoluminescence behaviors under the UV lamp. At the room temperature, the emissions for all are not obvious. However, after the samples were soaked in the liquid nitrogen for a period of time, the different situations appeared. At the low temperature, they all emit light. For 1 and 2, they exhibit the strong yellow-light emissions (Fig. 4a and b). The emissions of 3 are somewhat weak, compared with the emissions of 1 and 2. But interestingly, it shows the different emissions upon excitation at the different wavelengths: a blue-green-light emission when excited at 254 nm (Fig. 4c); a yellow-light emission when excited at 365 nm (Fig. 4d). This suggests that 3 might be a potential photochromic luminescence material.
 |
| | Fig. 4 Photoluminescence behaviors of 1 (a), 2 (b) and 3 (c and d) under UV lamp at low temperature. | |
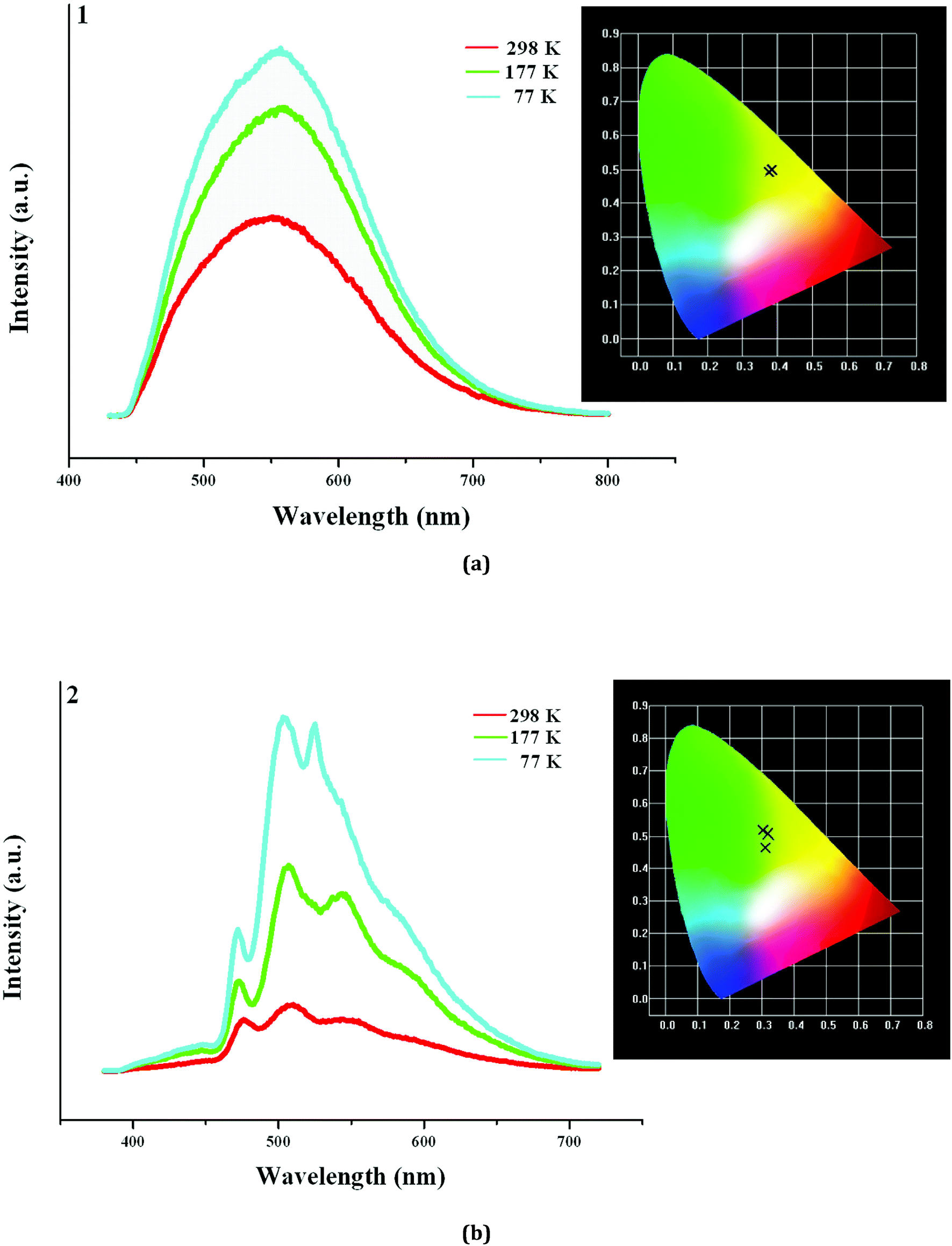
In order to further understand the photoluminescence properties of the title three compounds, we measured their emission spectra at the different temperature. As shown in Fig. 5a, 1 actually emits light at 298 K, but the intensity is not strong. Upon excitation at 398 nm, 1 shows a broad emission, and the maximum appears at 553 nm (τ1 = 13.84 μs, τ2 = 598 μs). The corresponding CIE chromaticity coordinates of (0.382, 0.4971) suggests that the broad emission for 1 is a yellow-light one. With the decrease of the temperature, the peak positions hardly change, but the emission intensities are obviously strengthened (τ1 = 94.67 μs, τ2 = 803 μs, τ3 = 5.460 ms at 77 K). Fig. 5b displays the emission spectra of 2 at the different temperature. Indeed, 2 also emits light upon excitation (λex = 358 nm) at 298 K, but the emission is extremely weak. Three peaks are found in the emission spectrum of 2, which appear at 475 nm, 508 nm, and 553 nm, respectively (τ1 = 2.323 μs, τ2 = 28.73 μs, τ3 = 104.9 μs). At 177 K, the emission intensity for 2 is obviously improved. The peak positions at 475 nm and 508 nm have no change, but the peak at 553 nm slightly blue-shifts to 543 nm. When the temperature is declined to 77 K, the emission intensity for 2 is further improved. The peak positions at 475 nm and 508 nm still have no change, but the peak at 553 nm further blue-shifts to 523 nm (τ1 = 436.9 μs, τ2 = 1.663 ms, τ3 = 6.931 ms at 77 K). Although a large blue shift (30 nm) for the peak at 553 nm occurs, this does not alter the emission of 2. From 298 K to 77 K, 2 always emits the yellow light, based on the corresponding CIE chromaticity coordinates ((0.3091, 0.4642) at 298 K; (0.321, 0.507) at 177 K; (0.3021, 0.5199) at 77 K).
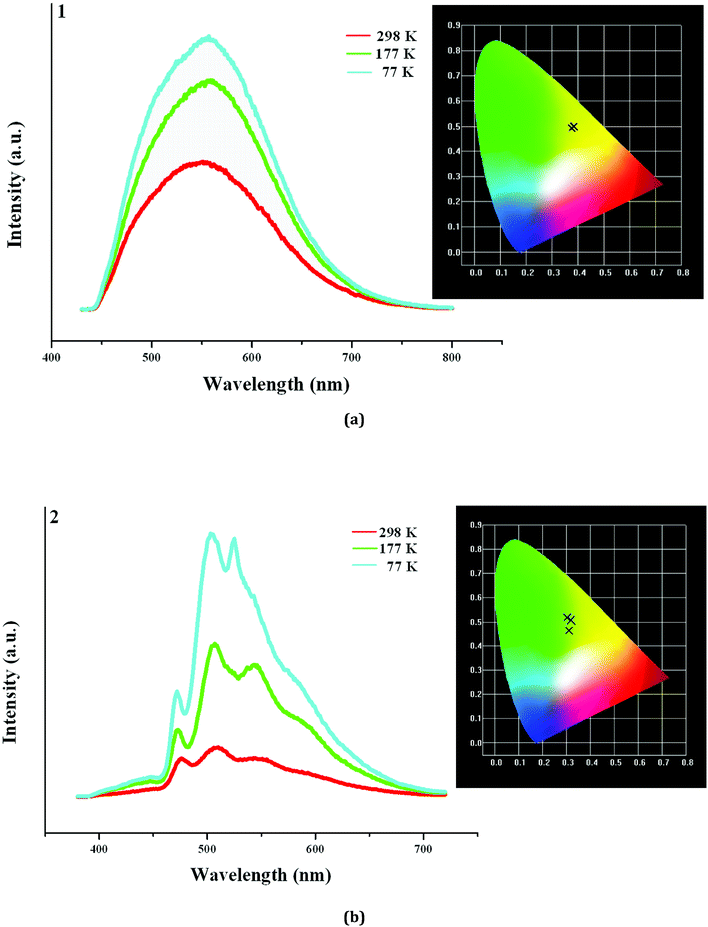 |
| | Fig. 5 Emission spectra of 1 (a) and 2 (b) at 298 K, 177 K and 77 K (inserted are corresponding CIE chromaticity diagrams). | |
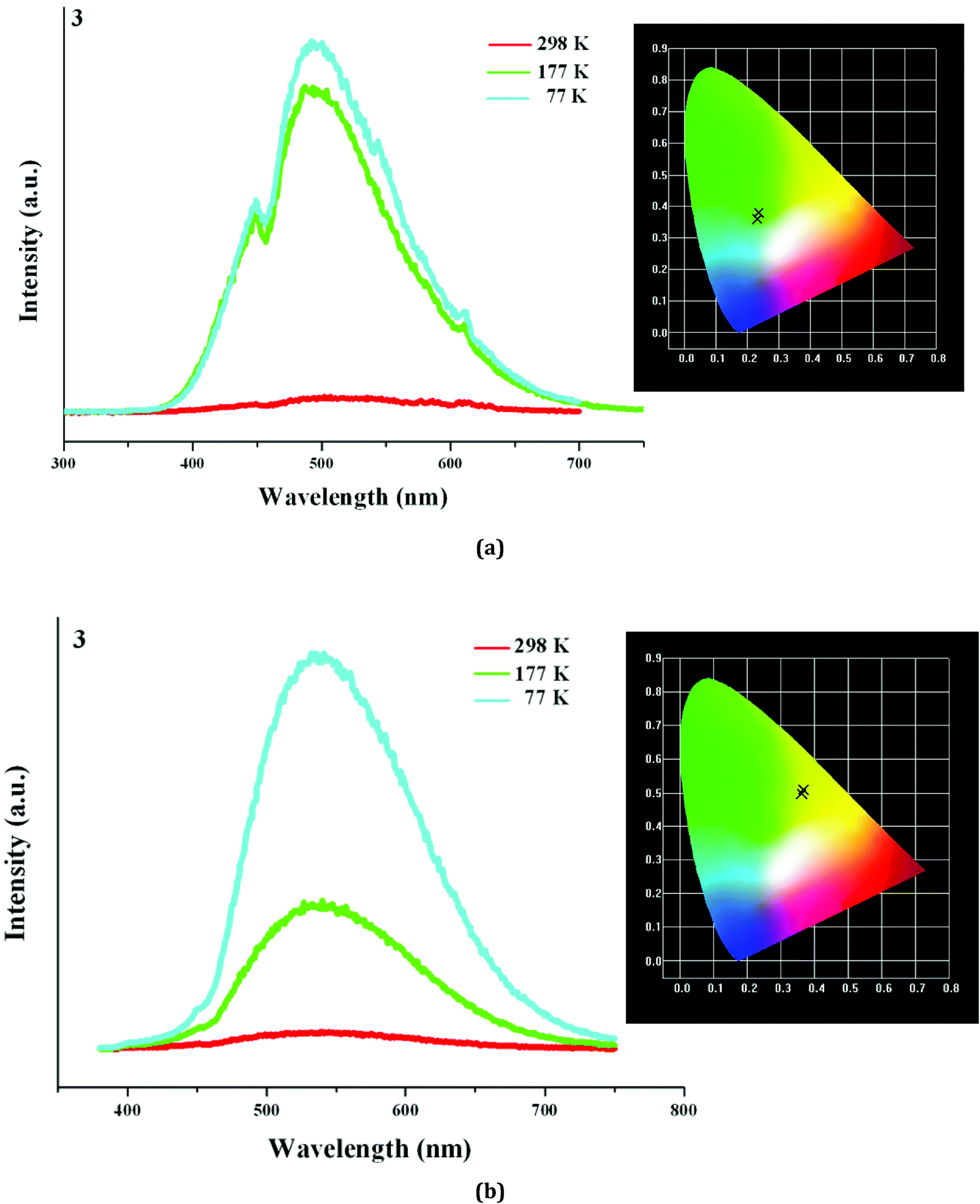
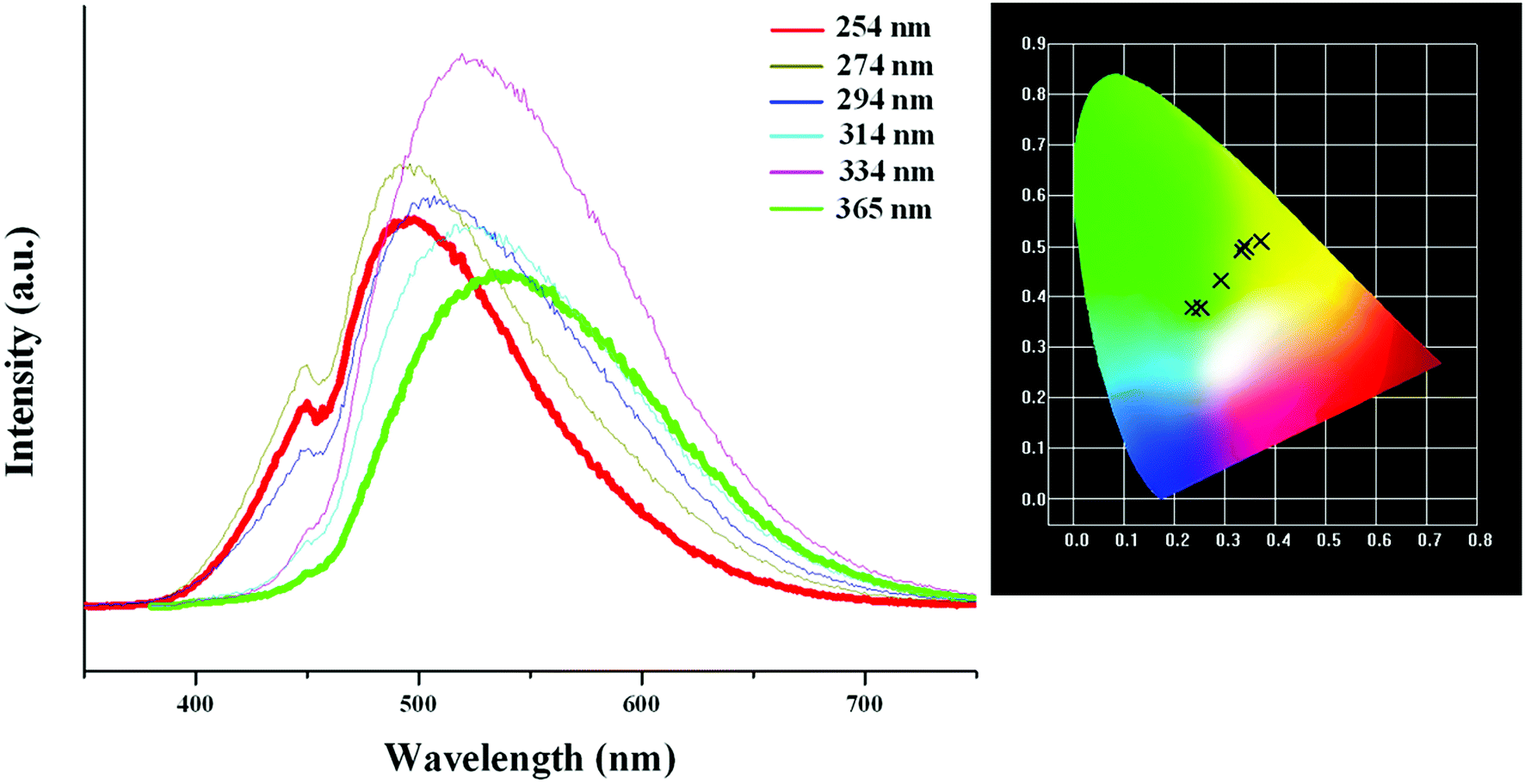
Based on the photoluminescence behavior of 3 under the UV lamp, we first measured the emission spectra of 3 at the different temperature when excited at 254 nm. As shown in Fig. 6(a), at 298 K, 3 really does not emit light upon excitation at 254 nm. At 177 K, 3 exhibits a strong emission with the maximum at 494 nm. The corresponding CIE chromaticity coordinates of (0.2288, 0.359) suggests a blue-green-light emission. At 77 K, the emission intensity of 3 is slightly strengthened, but the peak position does not change. Then we measured the emission spectra of 3 at the different temperature when excited at 365 nm. At 298 K, 3 still does not emit light. At 177 K, 3 exhibits a weak emission with the maximum at 540 nm, which is defined as a yellow-light emission based on the corresponding the CIE chromaticity coordinates of (0.3622, 0.4966). At 77 K, 3 still emit the yellow light (CIE chromaticity coordinates: (0.3684, 0.508)), but the intensity is largely improved (see Fig. 6b). The statements above mean that at the low temperature, 3 emits the different light upon excited at the different wavelengths: a blue-green-light upon excitation at 254; a yellow-light upon excitation at 365 nm. That is to say, 3 possesses the luminescence photochromic property, and it is a potential photochromic luminescence material. The results from the emission spectra of 3 are completely in agreement with the photoluminescence behaviors of 3 under the UV lamp. In order to deeply understand the luminescence photochromic property, we measured the emission spectra of 3 at 77 K when excited at the different wavelengths from 254 nm to 365 nm. As shown in Fig. 7, with the change of excitation wavelength from 254 nm to 365 nm, the corresponding CIE chromaticity coordinates of the emission spectra gradually transfer from the blue green to the final yellow.
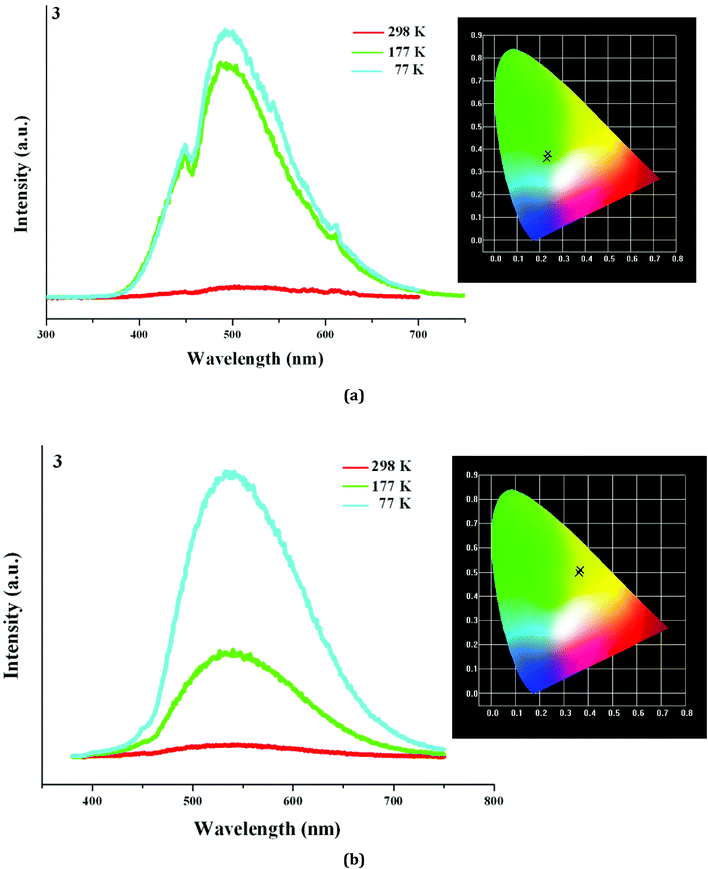 |
| | Fig. 6 Emission spectra of 3 at 298 K, 177 K and 77 K when excited at 254 nm (a) and 365 nm (b). | |
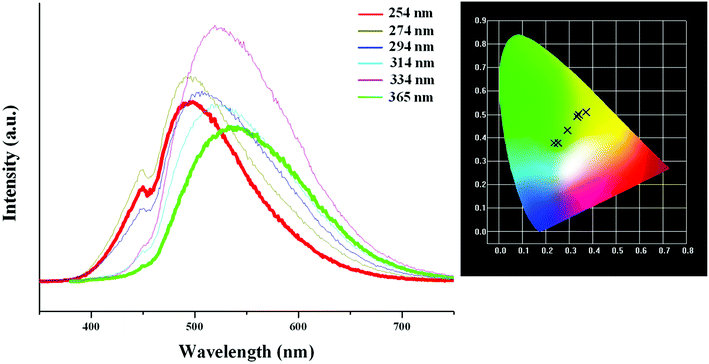 |
| | Fig. 7 CIE chromaticity coordinates of 3, showing color change from blue-green to yellow upon varying excitation wavelength from 254 nm to 365 nm. | |
With the decrease of the temperature, even though the emission intensities for all are improved, they still emit the same light. This suggests that the title three compounds are not the luminescence thermochromic materials. We speculate that a slight distortion for the Ag–I clusters in 1–3 should occur at the low temperature. However, maybe the photoluminescence behavior for the Ag–I cluster is not sensitive, so this minor change in molecular structure does not lead to a large variation of the photoluminescence property. The yellow-light emissions at 553 nm for 1 and 508 nm for 2 should be attributed to a mixture of the I-to-Ag charge transfer and the AgI d10–d9s1 transition based on the short Ag⋯Ag interactions, because some reported hybrid iodoargentates exhibit a similar emission, and they have been assigned to this attribution.2,6 With the decrease of the temperature, the photoluminescence lifetimes for 1 and 2 are longer. This should be related to the change of molecular thermal vibration. At 298 K, the molecular thermal vibration is active, and partial energy is consumed in the form of nonradiative transition. While at 77 K, the molecular thermal vibration is not active, and more energy participates in the radiative transition. Interestingly, with the change of excitation wavelength, the emission of 3 exhibits a color change from the blue green to the yellow. This suggests that 3 is a potential luminescence photochromic material. 3 is just a simple hybrid, in which there only exist two potential charge transferring paths: the ligand-centered electronic excitations; a mixture of the I-to-Ag charge transfer and the AgI d10–d9s1 transition. Therefore, the emission centered at 494 nm in 3 should be originated from the former (L3 exhibits a similar emission with the maximum at 460 nm (Fig. S6†)), whereas the emission centered at 540 nm should be derived from the latter. The existence of these two emissions should be responsible for the photoluminescence photochromic property of 3. For 1 and 2, the ligand-centered electronic excitations do not occur. This is to say, the electron in the excited-state energy level does not decays nonradiatively to the π* orbitals of the organic molecule,19 so 1 and 2 do not exhibit the photoluminescence photochromic behaviors. Fig. S7† gives the decay curves of 1 and 2 at 298 K and 77 K.
Conclusion
In summary, we reported the synthesis, structural characterization, and the photoluminescence properties of three new bisimidazole-based iodoargentates. Synthetically, pH plays a key role in the reactions. It not only affects the protonation of bisimidazole molecules, but also affects the N-alylation of bisimidazole molecules with alcohol solvents. Structurally, three different chained iodoargentates are observed in 1–3: a tube-like chain in 1; a cluster-based chain in 2; a simple single chain in 3. The bisimidazole molecule as the countercation plays a key role. By the size, charge, flexibility/rigidity and the hydrogen-bonded interactions, it controls the formation of the inorganic iodoargentates. Meanwhile, it also determines the structure of the overall supramolecular network for the as-synthesized hybrid. Only in the limited examples, the iodoargentates in 1 and 2 are found. Even though the iodoargentate in 3 is simple, the photoluminescence analysis reveals that at the low temperature, it possesses the photoluminescence photochromic property: a blue-green-light emission upon excitation at 254 nm; a yellow-light emission upon excitation at 365 nm. The photoluminescence analysis also indicates that at 77 K, 1 and 2 emit the strong yellow light with the ms-grade photoluminescence lifetimes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the financial aid from the National Natural Science Foundation of China (Grant No. 21771076, 21271083).
References
-
(a) T. L. Yu, G. X. Wu, Z. G. Wang and Y. L. Fu, J. Cluster Sci., 2018, 29, 443–449 CrossRef CAS;
(b) L. S. Song, H. M. Wang, Y. Y. Niu, H. W. Hou and Y. Zhu, CrystEngComm, 2012, 14, 4927–4938 RSC;
(c) Q. Q. Wu, P. Huang, L. T. Fan, M. Li, Z. R. Chen, Y. Li and H. H. Li, Chin. J. Struct. Chem., 2016, 35, 1637–1644 CAS;
(d) H. B. Duan, S. S. Yu and Z. Hong, Mater. Res. Bull., 2015, 65, 137–141 CrossRef CAS;
(e) G. Saha, K. K. Sarker, C. J. Chen, J. Cheng, T. H. Lu, G. Mostafa and C. Sinha, Polyhedron, 2009, 28, 3586–3592 CrossRef CAS;
(f) X. Chen, Z. Y. Yao, C. Xue, Z. X. Yang, J. L. Liu and X. M. Ren, CrystEngComm, 2018, 20, 356–361 RSC;
(g) S. S. Yu, S. X. Liu, Y. X. Zhu, H. B. Duan and H. Zhang, RSC Adv., 2017, 7, 56153–56156 RSC;
(h) S. Mishra, E. Jeanneau, S. Daniele and G. Ledoux, Dalton Trans., 2008, 6296–6304 RSC;
(i) J. M. Yue, Y. Y. Niu, B. Zhang, S. W. Ng and H. W. Hou, CrystEngComm, 2011, 13, 2571–2577 RSC;
(j) G. N. Liu, L. L. Liu, Y. N. Chu, Y. Q. Sun, Z. W. Zhang and C. C. Li, Eur. J. Inorg. Chem., 2015, 478–487 CrossRef CAS;
(k) S. Mishra, E. Jeanneau, G. Ledoux and S. Daniele, Inorg. Chem., 2014, 53, 11721–11731 CrossRef CAS PubMed.
-
(a) Q. Y. Chen, X. Cheng, T. Wang, Z. H. Yu, C. Zhang, S. K. Liu, H. H. Li and Z. R. Chen, Z. Anorg. Allg. Chem., 2014, 640, 439–443 CrossRef CAS;
(b) Y. L. Shen, J. L. Lu, C. Y. Tang, W. Fang, Y. Zhang and D. X. Jia, RSC Adv., 2014, 4, 39596–39605 RSC;
(c) T. L. Yu, J. J. Shen, Y. B. Fu and Y. L. Fu, CrystEngComm, 2014, 16, 5280–5289 RSC;
(d) T. L. Yu, H. H. Li, P. F. Hao, J. J. Shen and Y. L. Fu, Eur. J. Inorg. Chem., 2016, 4878–4884 CrossRef CAS;
(e) Y. Z. Qiao, W. Z. Fu, J. M. Yue, X. C. Liu and H. W. Hou, CrystEngComm, 2012, 14, 3241–3249 RSC.
- D. H. Wang, L. M. Zhao, X. Y. Lin, Y. K. Wang, W. T. Zhang, K. Y. Song, H. H. Li and Z. R. Chen, Inorg. Chem. Front., 2018, 5, 1162–1173 RSC.
-
(a) M. Liu, Y. Liang, C. H. Wang, C. J. Ma and Y. Y. Niu, J. Cluster Sci., 2015, 26, 1723–1733 CrossRef CAS;
(b) C. Y. Yue, B. Hu, X. W. Lei, R. Q. Li, F. Q. Mi, H. Gao, Y. Li, F. Wu, C. L. Wang and N. Lin, Inorg. Chem., 2017, 56, 10962–10970 CrossRef CAS PubMed;
(c) X. W. Lei, C. Y. Yue, L. J. Feng, Y. F. Han, R. R. Meng, J. T. Yang, H. Ding, C. S. Gao and C. Y. Wang, CrystEngComm, 2016, 18, 427–436 RSC;
(d) X. W. Lei, C. Y. Yue, S. Wang, H. Gao, W. Wang, N. Wang and Y. D. Yin, Dalton Trans., 2017, 46, 4209–4217 RSC.
-
(a) G. C. Xiao, J. Cluster Sci., 2006, 17, 457–466 CrossRef CAS;
(b) Y. Y. Niu, Y. L. Song, H. W. Hou and Y. Zhu, Inorg. Chem., 2005, 44, 2553–2559 CrossRef CAS PubMed;
(c) H. H. Li, Z. R. Chen, J. Q. Li, C. C. Huang, Y. F. Zhang and G. X. Jia, Eur. J. Inorg. Chem., 2006, 2447–2453 CrossRef CAS.
-
(a) H. H. Li, Z. R. Chen, J. Q. Li, J. Q. Li, C. C. Huang, X. L. Hu, B. Zhao and Z. X. Ni, J. Cluster Sci., 2005, 16, 537–545 CrossRef CAS;
(b) H. H. Li, S. Y. Chen, H. J. Dong, Y. L. Wu and Z. R. Chen, J. Chem. Crystallogr., 2011, 41, 858–863 CrossRef CAS;
(c) H. H. Li, Z. R. Chen, J. Q. Li, C. C. Huang, Y. F. Zhang and G. X. Jia, Eur. J. Inorg. Chem., 2006, 2447–2453 CrossRef CAS;
(d) H. H. Li, Z. R. Chen, J. Q. Li, C. C. Huang, Y. F. Zhang and G. X. Jia, Cryst. Growth Des., 2006, 6, 1813–1820 CrossRef CAS;
(e) H. H. Li, Y. Y. Xing, Z. X. Lian, A. W. Gong, H. Y. Wu, Y. Li and Z. R. Chen, CrystEngComm, 2013, 15, 1721–1728 RSC;
(f) H. H. Li, J. X. Wu, H. J. Dong, Y. L. Wu and Z. R. Chen, J. Mol. Struct., 2011, 987, 180–185 CrossRef CAS;
(g) H. H. Li, Z. R. Chen, L. C. Cheng, G. M. Feng, H. D. Zheng and J. Q. Li, Dalton Trans., 2009, 4888–4895 RSC.
-
(a) Y. R. Qiao, P. F. Hao and Y. L. Fu, Inorg. Chem., 2015, 54, 8705–8710 CrossRef CAS PubMed;
(b) J. J. Shen, F. Wang, X. X. Li, T. T. Yu, P. F. Hao and Y. L. Fu, RSC Adv., 2016, 6, 98916–98920 RSC;
(c) Y. C. Zhu, T. L. Yu, P. F. Hao, J. J. Shen and Y. L. Fu, J. Cluster Sci., 2016, 27, 1283–1291 CrossRef CAS;
(d) P. F. Hao, Y. R. Qiao, T. L. Yu, J. J. Shen, D. T. Dai and Y. L. Fu, RSC Adv., 2016, 6, 87628–87636 RSC;
(e) T. L. Yu, P. F. Hao, J. J. Shen, H. H. Li and Y. L. Fu, Dalton Trans., 2016, 45, 16505–16510 RSC;
(f) T. L. Yu, J. J. Shen, Y. L. Wang and Y. L. Fu, Eur. J. Inorg. Chem., 2015, 1989–1996 CrossRef CAS;
(g) P. F. Hao, L. F. Zhang, J. J. Shen and Y. L. Fu, Dyes Pigm., 2018, 153, 284–290 CrossRef CAS;
(h) C. F. Zhang, J. J. Shen, Q. Guan, T. L. Yu and Y. L. Fu, Solid State Sci., 2015, 46, 14–18 CrossRef CAS;
(i) J. J. Shen, C. F. Zhang, T. L. Yu, L. An and Y. L. Fu, Cryst. Growth Des., 2014, 14, 6337–6342 CrossRef CAS;
(j) T. L. Yu, L. An, L. Zhang, J. J. Shen, Y. B. Fu and Y. L. Fu, Cryst. Growth Des., 2014, 14, 3875–3879 CrossRef CAS.
-
(a) X. W. Lei, C. Y. Yue, F. Wu, X. Y. Jiang and L. N. Chen, Inorg. Chem. Commun., 2017, 77, 64–67 CrossRef CAS;
(b) X. W. Lei, C. Y. Yue, J. Q. Zhao, Y. F. Han, J. T. Yang, R. R. Meng, C. S. Gao, H. Ding, C. Y. Wang and W. D. Chen, Cryst. Growth Des., 2015, 15, 5416–5426 CrossRef CAS;
(c) X. W. Lei, C. Y. Yue, J. Q. Zhao, Y. F. Han, J. T. Yang, R. R. Meng, C. S. Gao, H. Ding, C. Y. Wang, W. D. Chen and M. C. Hong, Inorg. Chem., 2015, 54, 10593–10603 CrossRef CAS PubMed;
(d) C. Y. Yue, X. W. Lei, Y. F. Han, X. X. Lu, Y. W. Tian, J. Xu, X. F. Liu and X. Xu, Inorg. Chem., 2016, 55, 12193–12203 CrossRef CAS PubMed;
(e) X. W. Lei, C. Y. Yue, J. C. Wei, R. Q. Li, Y. Lia and F. Q. Mi, Dalton Trans., 2016, 45, 19389–19398 RSC;
(f) C. Y. Yue, X. W. Lei, X. X. Lu, Y. Li, J. C. Wei, W. Wang, Y. D. Yin and N. Wang, Dalton Trans., 2017, 46, 9235–9244 RSC;
(g) X. W. Lei, C. Y. Yue, J. Q. Zhao, Y. F. Han, Z. R. Ba, C. Wang, X. Y. Liu, Y. P. Gong and X. Y. Liu, Eur. J. Inorg. Chem., 2015, 4412–4419 CrossRef CAS;
(h) X. W. Lei, C. Y. Yue, J. C. Wei, R. Q. Li, F. Q. Mi, Y. Li, L. Gao and Q. X. Liu, Chem.–Eur. J., 2017, 23, 14547–14553 CrossRef CAS PubMed.
-
(a) G. N. Liu, X. M. Jiang, Q. S. Fan, M. B. Hussain, K. Li, H. Sun, X. Y. Li, W. Q. Liu and C. Li, Inorg. Chem., 2017, 56, 1906–1918 CrossRef CAS PubMed;
(b) J. J. Hou, S. L. Li, C. R. Li and X. M. Zhang, Dalton Trans., 2010, 39, 2701–2707 RSC;
(c) H. Chan, Y. Chen, M. Dai, C. N. Lü, H. F. Wang, Z. G. Ren, Z. J. Huang, C. Y. Ni and J. P. Lang, CrystEngComm, 2012, 14, 466–473 RSC;
(d) Y. Chen, Z. Yang, C. X. Guo, C. Y. Ni, Z. G. Ren, H. X. Li and J. P. Lang, Eur. J. Inorg. Chem., 2010, 5326–5333 CrossRef CAS;
(e) G. M. Wang, J. Q. Jiao, X. Zhang, X. M. Zhao, X. Yin, Z. H. Wang, Y. X. Wang and J. H. Lin, Inorg. Chem. Commun., 2014, 39, 94–98 CrossRef CAS;
(f) G. M. Wang, J. H. Lin, J. Q. Jiao, X. Zhang, X. M. Zhao, X. Yin, J. S. Huang, Y. X. Wang and J. H. Lin, Inorg. Chem. Commun., 2014, 43, 105–109 CrossRef CAS.
- R. Y. Wang, X. Zhang, Q. S. Huo, J. H. Yu and J. Q. Xu, RSC Adv., 2017, 7, 19073 RSC.
-
(a) Z. Deng, L. Tong, M. Flores, S. Lin, J. X. Cheng, H. Yan and Y. Liu, J. Am. Chem. Soc., 2011, 133, 5389–5396 CrossRef CAS PubMed;
(b) K. Neinhaus, H. Nar, R. Heiker, J. Wiedenmann and G. U. Nienhaus, J. Am. Chem. Soc., 2008, 130, 12578–12579 CrossRef PubMed;
(c) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 12, 1126–1162 CrossRef PubMed.
-
(a) Z. X. Fu, J. Lin, L. Wang, C. Li, W. B. Yan and T. Wu, Cryst. Growth Des., 2016, 16, 2322–2327 CrossRef CAS;
(b) T. Hayashi, A. Kobayashi, H. Ohara, M. Yoshida, T. Matsumoto, H. Chang and M. Kato, Inorg. Chem., 2015, 54, 8905–8913 CrossRef CAS PubMed;
(c) K. Kirakci, K. Fejfarová, J. Martinčí, M. Nikl and K. Lang, Inorg. Chem., 2017, 56, 4609–4614 CrossRef PubMed;
(d) S. S. Zhao, L. Wang, Y. J. Liu, L. Chen and Z. G. Xie, Inorg. Chem., 2017, 56, 13975–13981 CrossRef CAS PubMed;
(e) S. Z. Zhan, M. Li, J. Zheng, Q. J. Wang, S. W. Ng and D. Li, Inorg. Chem., 2017, 56, 13446–13455 CrossRef CAS PubMed;
(f) S. Q. Bai, D. Kai, K. L. Ke, M. Lin, L. Jiang, Y. Jiang, D. J. Young, X. J. Loh, X. Li and T. S. A. Hor, ChemPlusChem, 2015, 80, 1235–1240 CrossRef CAS;
(g) D. Sun, S. Yuan, H. Wang, H. F. Lu, S. Y. Feng and D. F. Sun, Chem. Commun., 2013, 49, 6152–6154 RSC;
(h) S. Yuan, H. Wang, D. X. Wang, H. F. Lu, S. Y. Feng and D. Sun, CrystEngComm, 2013, 15, 7792–7802 RSC;
(i) X. C. Shan, F. L. Jiang, D. Q. Yuan, H. B. Zhang, M. Y. Wu, L. Chen, J. Wei, S. Q. Zhang, J. Pan and M. C. Hong, Chem. Sci., 2013, 4, 1484–1489 RSC;
(j) J. H. Wang, M. Li, J. Zheng, X. C. Huang and D. Li, Chem. Commun., 2014, 50, 9115–9118 RSC;
(k) L. Maini, D. Braga, P. P. Mazzeo, L. Maschio, M. Rérat, I. Manete and B. Ventura, Dalton Trans., 2015, 44, 13003–13006 RSC;
(l) G. Zeng, S. H. Xing, X. Han, B. J. Xin, X. R. Wang, G. H. Li, Z. Shi and S. H. Feng, RSC Adv., 2015, 5, 40792–40797 RSC;
(m) F. Farinella, L. Maini, P. P. Mazzeo, V. Fattori, F. Monti and D. Braga, Dalton Trans., 2016, 45, 17939–17947 RSC;
(n) M. S. Deshmukh, A. Yadav, R. Pant and R. Boomishankar, Inorg. Chem., 2015, 54, 1337–1345 CrossRef CAS PubMed;
(o) D. M. Zink, T. Baumann, J. Friedrichs, M. Nieger and S. Bräse, Inorg. Chem., 2013, 52, 13509–13520 CrossRef CAS PubMed.
- S. L. Li, J. Wang, F. Q. Zhang and X. M. Zhang, Cryst. Growth Des., 2017, 17, 746–752 CrossRef CAS.
- Z. P. Wang, J. Y. Wang, J. R. Li, M. L. Feng, G. D. Zou and X. Y. Huang, Chem. Commun., 2015, 51, 3094–3097 RSC.
- L. Z. Cai, M. S. Wang, M. J. Zhang, G. E. Wang, G. C. Guo and J. S. Huang, CrystEngComm, 2012, 14, 6196–6200 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
(a) L. Chen, J. Ma, Q. H. Chen, R. Feng, F. L. Jiang and M. C. Hong, Inorg. Chem. Commun., 2012, 15, 208–211 CrossRef CAS;
(b) H. Park, E. Kwon, H. Chiang, H. Im, K. Y. Lee, J. Kim and T. H. Kim, Inorg. Chem., 2017, 56, 8287–8294 CrossRef CAS PubMed.
- T. Yu, Y. Fu, Y. Wang, P. Hao, J. Shen and Y. Fu, CrystEngComm, 2015, 17, 8752–8761 RSC.
- J. Lu, K. Zhao, Q. R. Fang, J. Q. Xu, J. H. Yu, X. Zhang, H. Y. Bie and T. G. Wang, Cryst. Growth Des., 2005, 5, 1091–1098 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Jie-Hui Yu
*b,
Jie-Hui Yu