
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLivin participates in resistance to trastuzumab therapy for breast cancer through ERK1/2 and AKT pathways and promotes EMT-like phenotype
Fan Li†
 *abc,
Lu Zhang†abc,
Fan Fengabc,
Ke Zhengabc,
YuJing Liabc,
TieLin Wangabc and
GuoSheng Renabc
*abc,
Lu Zhang†abc,
Fan Fengabc,
Ke Zhengabc,
YuJing Liabc,
TieLin Wangabc and
GuoSheng Renabc
aDepartment of Endocrine and Breast Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China. E-mail: LF628@163.com; Fax: +86-23-89012558; Tel: +86-23-89012558
bBreast Cancer Center of Chongqing, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China
cMolecular Oncology and Epigenetics Laboratory, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China
First published on 10th August 2018
Abstract
Trastuzumab resistance has emerged as a major issue in anti-human epidermal growth factor receptor-2 (HER2) therapy for breast cancers. The cell lines maintain overexpression of HER2. Upon treatment with trastuzumab, R-SKBR3 and R-BT474 cell lines displayed an increased growth rate and invasiveness, accompanied by activation of the ERK1/2 and AKT signaling pathways, and also a parental EMT-like transition (epithelial–mesenchymal transition) was promoted, with increases in N-cadherin, vimentin, and fibronectin and a decrease in E-cadherin. A further investigation found that livin played a key role in the development of trastuzumab resistance. Knockdown of the expression of livin by livin-shRNA3 in R-SKBR3 and R-BT474 cells decreased ERK1/2 and AKT, resensitized the resistant cells to the therapeutic activities of trastuzumab by inducing growth arrest, inhibition of proliferation, and G1-S cell cycle checking in the presence of the antibody, and they also exhibited an EMT-like transition (epithelial–mesenchymal transition), with a decrease in N-cadherin and an increase in E-cadherin, and the cell invasiveness was inhibited in response to the downregulation of livin. Conversely, SKBR3 and BT474 cells that had been stably transfected with pcDNA3.1-livin underwent promotion of an EMT-like transition and displayed a significant decrease in E-cadherin and increases in N-cadherin, vimentin, and fibronectin, and ectopic expression of livin in HER2-overexpressing breast cancer cells conferred resistance to trastuzumab. In vivo, the administration of livin AS (antisense oligonucleotides) restored sensitivity to trastuzumab in resistant breast cancer xenografts via the ERK1/2 and AKT signaling pathways. Patients with livin-overexpressing breast cancers exhibited significantly poorer responses to trastuzumab-based therapy than those with normal livin levels. In summary, our data suggest that the upregulation of livin activates the ERK1/2 and AKT signaling pathways and promotes an EMT-like transition. This could be an important mechanism that leads to trastuzumab resistance in HER2-overexpressing breast cancer cells.
1. Introduction
HER2 is aberrantly upregulated in approximately 30% of breast cancer patients, which is correlated with a poor clinical prognosis and chemoresistance, which makes HER2 an important therapeutic target. Amplification of HER2 is linked to aggressive tumor behavior and poor clinical outcomes with shorter disease-free intervals and overall survival in patients with early and advanced breast cancers.1 Tremendous efforts have been made to develop HER2-targeting cancer therapies, and a highly successful strategy involves the recombinant humanized anti-HER2 monoclonal antibody trastuzumab (Herceptin), which specifically binds to the extracellular domain of HER2 and blocks its function. The clinical use of trastuzumab in adjuvant and metastatic settings has been shown to prolong the survival of patients with HER2-overexpressing breast cancers.2 However, the rate of response to trastuzumab monotherapy is less than 35%, whereas 60% of patients with HER2-overexpressing cancers on regimens that combine trastuzumab with microtubule-stabilizing drugs do not respond to treatment.3 Moreover, most patients who achieve an initial response develop resistance to trastuzumab within 1 year.4 Therefore, the identification of the mechanisms responsible for trastuzumab resistance is important for the development of new therapeutic strategies. A number of mechanisms have been suggested for trastuzumab resistance, including dysregulation of downstream signaling pathways and compensated signaling by other members of the EGF family or via alternative pathways.5 Some studies have suggested an increase in homo- and heterodimerization of HER2 with other receptors of the ErbB family, such as epidermal growth factor receptor (EGFR), HER3, and HER4.6,7 These interactions activate intracellular signaling via the mitogen-activated protein kinase (MAPK) or phosphatidylinositol 3-kinase (PI3K) pathways.8–10 However, how these pathways are activated in trastuzumab resistance remains elusive.Livin, which is a novel member of the IAP family, is undetectable in most normal differentiated tissues but displays a high level of expression in a wide variety of human malignancies. Elevated livin activity is associated with a poor prognosis in cancer patients. In our previous research it was demonstrated that livin promotes the progression of breast cancer via the induction of the epithelial–mesenchymal transition and activation of AKT signaling.11 It was also demonstrated that the overexpression of livin affects in vitro drug resistance and the clinical response of patients.12 However, whether livin is involved in trastuzumab resistance has not yet been clarified.
The goal of this study was to understand the mechanisms that lead to trastuzumab resistance in HER2-overexpressing breast tumors and which pathway-specific genes may contribute to resistance.
2. Materials and methods
2.1. Cell lines and cell cultures
The human breast cancer cell lines SKBR3 and BT474 were obtained from the American Type Culture Collection.11 Unless otherwise stated, monolayer cultures of SKBR3 and BT474 cells were maintained in DMEM/F12 medium with 10% fetal bovine serum. The cell lines overexpressed the product of the HER2/c-erb-2 (HER2) gene. The trastuzumab-resistant clones R-SKBR3 and R-BT474 were generated from SKBR3 and BT474 cells, respectively. In order to select trastuzumab-resistant clones, SKBR3 and BT474 cells were plated in 24-well plates at a low density and maintained in growth media containing 1 μg mL−1, 5 μg mL−1 and 10 μg mL−1 of trastuzumab. The R-SKBR3 and R-BT474 clones were maintained in a growth medium containing 5 μg mL−1 of trastuzumab for over 1 year. Both R-SKBR3 and R-BT474 cells were repeatedly confirmed to be insensitive to trastuzumab.2.2. Real-time reverse-transcription (RT)-PCR and semiquantitative RT-PCR
The following PCR methods were followed as previously described.11 Total RNA was extracted from parent and resistant cells using TRIzol reagent (Takara, Dalian, China) according to the manufacturer's instructions. cDNA was generated from total RNA using a PrimeScript RT reagent kit (Takara). To confirm the integrity and primer specificity of cDNA, livin and β-actin were amplified by conventional PCR in an automatic thermal cycler using livin-specific sense primers (forward, 5′-CACCATGACTTCCAAGCTGGC-3′; reverse, 5′-TTATGAATTCTCAGCCCTCTTC-3′) and a β-actin-specific sense primer (5′-TGACGTGGACATCCGCAAAG-3′) and an antisense primer (5′-CTGGAAGGTGGACAGCGAGG-3′). The PCR-amplified products were separated by electrophoresis on 1.5% agarose gels to visualize the products. Quantitative real-time PCR was conducted with a Bio-Rad Miniopticom real-time PCR system using an SYBR® Premix Ex Taq™ II kit (Takara, Dalian, China). All samples were amplified by real-time PCR twice and normalized against β-actin. Data were analyzed by comparison with a series of serial dilutions of cell cDNA.2.3. Measurements of cell viability, proliferation, and cell cycle
Cell viability was measured using an MTT assay. In brief, cells were plated at a density of 5 × 103 cells per well in 96-well plates, incubated for 2 days and then treated with trastuzumab at concentrations of 0, 1 μg mL−1, 2 μg mL−1, 3 μg mL−1, 4 μg mL−1 and 5 μg mL−1 for 3 days. Then, the cells were incubated with 20 μL MTT (10 mg mL−1) for 4 h at 37 °C, and 100 μL DMSO (Sigma Chemical Co.) was pipetted to solubilize the crystalline product for 10 min at room temperature. The absorbance (A) of each well was measured with a microplate reader (Bio-Rad) at a wavelength of 490 nm. This experiment was repeated in triplicate. For a colony formation assay, cells were harvested and resuspended in a medium supplemented with agar with a final concentration of 0.35%. Subsequently, 1 mL of the resulting cell suspension was plated in 12-well plates covered with a medium supplemented with 0.6% agar and cultured at 37 °C with 5% CO2. The percentage of cells that formed a spherical clone was calculated. For cell cycle analysis, cells were trypsinized, pelleted, and then resuspended in a propidium iodide solution (Sigma) that contained 50 g mL−1 of propidium iodide, 0.1 mg mL−1 of RNase A, and 0.05% Triton X-100 for incubation for 40 min at 37 °C, and were analyzed by flow cytometry using an FACSCalibur instrument with CellQuest software (BD Biosciences). In addition, cells in the S-phase were detected via the incorporation of 5-bromo-2-deoxyuridine (BrdU). In brief, BrdU (Upstate) was added to the medium at a concentration of 10 μM for incubation for 8 or 24 h. The incorporation of BrdU into cancer cells was assessed by immunostaining with a phycoerythrin-conjugated anti-BrdU antibody (Upstate, Temecula, CA) and analyzed by flow cytometry.132.4. Western blot analysis
A western blot analysis was performed as described previously.11 The antibodies that were used included anti-livin (SC-30161, 1:1000). Antibodies against ERK1/2 (sc-292838, 1:1000) and phospho-ERK1/2 (sc-23759-R, 1:2000) were purchased from Santa Cruz Biotechnology (USA). Antibodies against HER2 (ab8054, 1:500) were purchased from Abcam. Anti-phospho-AKT (SC-33437, 1:2000), anti-AKT (SC-1618, 1:1000), and mouse anti-fibronectin (SC18827, 1:1000) antibodies were purchased from Santa Cruz Biotechnology. Anti-vimentin (BS1776, 1:1000), anti-E-cadherin (BS1098, 1:1000), and anti-N-cadherin (BS222, 1:1000) antibodies were purchased from Bioworld Technology, and anti-rabbit peroxidase-conjugated secondary antibodies were purchased from Promega. An inhibitor of ERK1/2 (PD98059) was obtained from Santa Cruz Biotechnology (USA) and an inhibitor of AKT (LY294002) was obtained from Sigma-Aldrich (St. Louis, MO, USA).2.5. Indirect immunofluorescence
Cells grown on coverslips were stained by indirect immunofluorescence as described previously.14 In brief, the cells were incubated with primary antibodies against HER2 and then incubated with an FITC-conjugated (Dako, Denmark) secondary antibody against mouse or rabbit IgG. The cells were then counterstained with DAPI and imaged with a fluorescence microscope (Leica DM IRB).2.6. Immunohistochemistry
Immunohistochemical staining was performed as described previously.18 The expression of livin was examined by immunohistochemistry in paraffin-embedded tissue sections. In brief, an anti-livin polyclonal antibody was used as a primary antibody for overnight incubation at 4 °C. The sections were subsequently treated with a goat anti-rabbit secondary antibody, followed by further incubation with a streptavidin-horseradish peroxidase complex. Diaminobenzidine (Dako, Carpinteria, CA) was used as a chromogen, and the sections were lightly counterstained with hematoxylin. The percentage of positively stained tumor cells was calculated per field of view, and at least 20 fields of view per section were examined at a magnification of 400×.112.7. Migration and invasion assays
Boyden chamber–Matrigel cell invasion assays were performed according to the manufacturer's protocol (Chemicon, Billerica, MA, USA). In brief, 3 × 105 cells were plated into cell culture inserts with 8 μm microporous filters (BD, Franklin Lakes, NJ, USA) coated with (invasion) or without (migration) Matrigel and incubated for 48 h. Cells in the upper filters (inside the inserts) were removed, and migrated or invaded cells in the lower filters (outside the inserts) were fixed in methanol, stained with crystal violet, and counted under a microscope. After incubation for 48 h, the invaded cells at the bottom of the membrane were stained and counted under a light microscope at a magnification of 400×. Experiments were performed in triplicate.2.8. In vivo tumor growth
All experimental procedures were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. In animal experiments, athymic nude female mice with ages of 3–4 weeks were subcutaneously implanted with 1.5 mg 60-day release pellets of 17-estradiol (Innovative Research, Sarasota, FL). Trastuzumab-resistant SKBR3 and BT474 cells in 100 μL of PBS per 100 μL of growth factor-reduced Matrigel (BD Biosciences) were injected into the mammary fat pads of the mice. When the xenografts reached a volume of 150 mm3, livin AS or control MIS oligonucleotides (30 g per week) were administered by multiple intratumoral injections 72 h prior to intravenous injection of trastuzumab or IgG as a control (10 mg kg−1 twice a week). An unpaired Student's t-test was used to assess statistical significance.2.9. Patients and tissue samples
Primary ductal carcinomas of the breast with amplification of the HER2 gene were obtained from 24 female patients with breast cancer before and after preoperative neoadjuvant therapy with Herceptin in the breast tumor center at the First Affiliated Hospital of Chongqing Medical University between January 2006 and December 2009. All patients underwent preoperative neoadjuvant therapy with 3–4 cycles of a tri-weekly TCH regimen (Taxotere, 75 mg m−2; carboplatin, AUC 6 mg mL−1 min−1; and Herceptin, loading dose of 8 mg kg−1 followed by 6 mg kg−1 every 3 weeks) according to NCCN guidelines. Breast tumor samples were obtained via core-needle biopsy prior to therapy with trastuzumab and via surgery after treatment. The collected tumor tissues were snap-frozen in liquid nitrogen for PCR and western blotting. In addition, the remaining tissues were embedded for histology and immunohistochemistry studies. All patients who were involved in this study consented to participate in the study and to publication of its results. The experiments were approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University and were conducted in compliance with the Helsinki Declaration.2.10. Statistical analysis
Data were expressed as the mean ± standard deviation (SD). All statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS) 10.0 software package for Windows (SPSS, Inc., Chicago, IL, USA). Student's t-test followed by the least significant difference t-test (LSD-t) was used for group comparisons. Values of p < 0.05 were considered to be statistically significant.3. Results
3.1. Characterization of trastuzumab-resistant cell lines (R-SKBR3 and R-BT474)
Data from the MTT assay showed that the growth of SKBR3 and BT474 parent cells was significantly inhibited by fresh treatment with 5 μg mL−1 and 10 μg mL−1 trastuzumab, whereas the growth of R-SKBR3 and R-BT474 cells that were maintained in media containing 10 μg mL−1 trastuzumab was not inhibited after 5 days (Fig. 1A). Both R-SKBR3 and R-BT474 cells displayed significantly enhanced invasion capacities. The quantitative cellular motility was also determined for both cell lines using the Boyden chamber assay. The resistant breast cancer cells displayed significantly (p < 0.01) increased motility relative to that of the SKBR3 and BT474 parent cells. To investigate the invasion capacity of the transfected breast cancer cells, an in vitro invasion assay was performed. The R-SKBR3 and R-BT474 cells exhibited a significant (p < 0.01) increase in the number of invading cells in comparison with the parent cells (Fig. 1B). The number of cells that overexpressed HER2 was reduced in SKBR3 and BT474 cells upon fresh treatment with trastuzumab (10 μg mL−1) for 72 h. However, R-SKBR3 and R-BT474 cells that were maintained in growth media containing 10 μg mL−1 trastuzumab for a long period of time did not exhibit any significant difference in the overall number of cells that overexpressed HER2 in comparison with the untreated parent cells (Fig. 1C).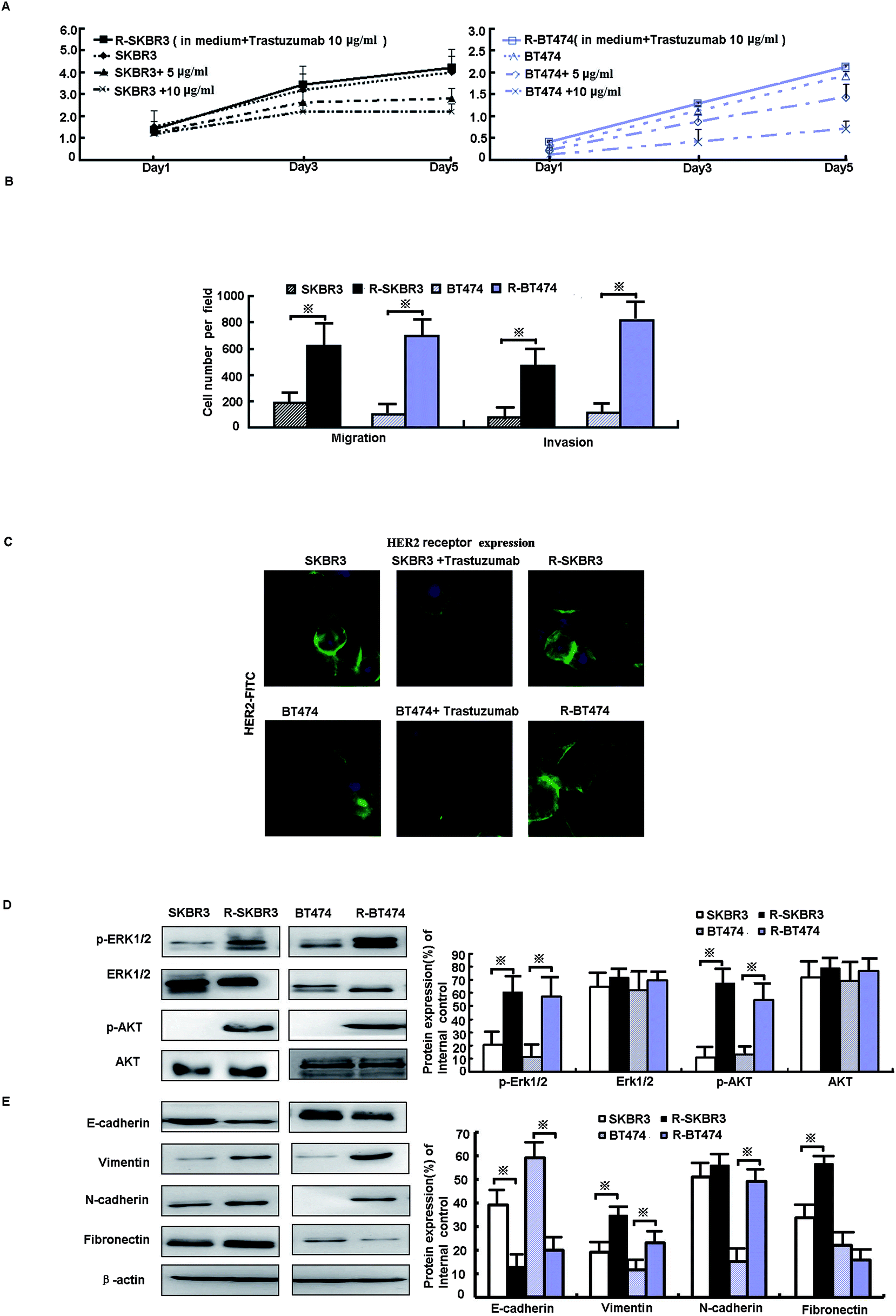 | ||
| Fig. 1 Characterization of trastuzumab-resistant clones. (A) R-SKBR3 and R-BT474 cells were maintained in a growth medium containing 10 μg mL−1 trastuzumab. SKBR3 and BT474 cells were treated with trastuzumab at concentrations of 0, 5 μg mL−1 and 10 μg mL−1, and an MTT assay was performed on the indicated days. (B) The invasiveness of cells in the indicated cell lines was measured by the Boyden chamber invasion assay as described in the Materials and methods section. Invasive cells (mean ± SD for 5 different areas) were counted. *p < 0.05 in comparison with untreated cells. (C) SKBR3 and BT474 cells were treated with or without trastuzumab (10 μg mL−1) for 3 days. HER2 levels were determined by immunofluorescence analysis with an FITC-labeled anti-HER2 antibody (green), and the cell nuclei were labeled with propidium iodide (blue). (D) Protein levels of the indicated genes in SKBR3, R-SKBR3, BT474, and R-BT474 cells were analyzed by western blotting, and β-actin was used as a vehicle control. (E) Immunoblotting of EMT was performed with the indicated antibodies on whole-cell lysates. | ||
3.2. Activation of ERK1/2 and AKT signaling pathways promotes EMT phenotype in trastuzumab-resistant cell lines
As shown in our previous research, activation of EGFR downstream elements, such as mitogen-activated protein kinases (MAPKs), is an important mechanism of drug resistance.15 In addition, extracellular signal-regulated protein kinase-1 and -2 (ERK1/2) are part of a major MAPK pathway cascade. Some studies have suggested that an increase in HER2 activates the MAPK signaling pathway in trastuzumab-resistant cells. To study the response of breast cancer cells to trastuzumab after the development of trastuzumab resistance, levels of ERK1/2 phosphorylation were measured. Treatment with trastuzumab can induce ERK1/2 phosphorylation. The levels of ERK1/2 phosphorylation in trastuzumab-resistant SKBR3 and BT474 cells were higher by factors of 6.1 (p < 0.001) and 2.5 (p < 0.05), respectively, than in the parent cells, whereas the expression of ERK1/2 did not differ in either line (p > 0.05, Fig. 1D). A further mechanistic study showed that HER2-overexpressing cancer cells lead to trastuzumab resistance by enhancing downstream PI3K/AKT phosphorylation and thus preventing trastuzumab-mediated growth arrest.13 p-AKT was upregulated by a factor of ∼3.5 in both R-SKBR3 and R-BT474 clones (p < 0.05, Fig. 1D). The EMT is a crucial step in cancer cell metastasis. Fig. 1E shows that the protein level of E-cadherin decreased and the expression of vimentin, N-cadherin, and fibronectin increased in R-SKBR3 cell lines. A decrease in E-cadherin and increases in vimentin and N-cadherin were generally seen across most R-BT474 cells in comparison with BT474 cells (Fig. 1E). These results confirmed an increase in EMT markers in the trastuzumab-resistant cell lines, as shown in Fig. 1D. The results implied that activation of ERK1/2 and AKT signaling might promote the EMT or a partial EMT-like transition in trastuzumab-resistant cells.3.3. Blockade of livin recovers sensitivity to trastuzumab
As mentioned in our research,11 livin is involved in the development of breast cancer progression, as both R-SKBR3 and R-BT474 cells displayed increases in livin levels in comparison with the parent cell lines at the RNA and protein levels. To confirm that livin played a role in the development of acquired resistance to trastuzumab, transfection of the trastuzumab-resistant cell lines with livin-shRNA3 downregulated the expression of livin by a factor of 4–9 (p < 0.001). In contrast, transfection with pcDNA3.1-livin, instead of pcDNA3.1-N1, increased the expression of livin to a level comparable to that in the trastuzumab-resistant cell lines (p < 0.001). These data suggest that the upregulation of livin is induced in HER2-overexpressing breast cancer cells in the development of acquired trastuzumab resistance (Fig. 2). Next, we examined if a decrease in the expression of livin could sensitize cells in response to trastuzumab. R-SKBR3 and R-BT474 cells were treated with livin-shRNA3, N1-shRNA and mock transfection for 48 h, followed by fresh treatment with 1 μg mL−1 to 5 μg mL−1 trastuzumab for 72 h. The data in Fig. 3A show that 1 μg mL−1 to 5 μg mL−1 trastuzumab inhibited 36% to 44% of cell growth in R-SKBR3 cells transfected with livin-shRNA3, but only ∼8% in R-SKBR3 cells treated with N1-shRNA and 7% in cells treated by mock transfection. The cell growth inhibition rate was 23% to 37% in R-BT474 cells transfected with livin-shRNA3 and treated with 1 μg mL−1 to 5 μg mL−1 trastuzumab, but a maximum of 13% in R-BT474 cells transfected with N1-shRNA and a maximum of 7% in cells treated by mock transfection (Fig. 3A). However, without treatment with trastuzumab, transfection with livin-shRNA3 alone only slightly inhibited cell growth in the resistant cell lines in comparison with N1-shRNA and mock transfection (p < 0.05). We next tested whether livin-shRNA3 may inhibit anchorage-independent growth of resistant breast tumor cells in the presence of trastuzumab. The cells were treated with 10 μg mL−1 trastuzumab for 3 days. As shown by the quantification via a soft-agar assay, transfection with livin-shRNA3 resulted in reductions by factors of 8.2 (p < 0.001) and 6.0 (p < 0.001) in the number of colonies formed by the resistant SKBR3 and BT474 cell lines, respectively (Fig. 3B). However, without trastuzumab, livin-shRNA3 alone reduced colony formation by only 3.18 ± 1.8% (p < 0.05, Fig. 3B). Therefore, downregulation of livin enhances sensitivity to trastuzumab in HER2-overexpressing breast cancer cells with acquired resistance to the drug. During treatment with trastuzumab, the proliferation of breast tumor cells was measured in terms of their ability to incorporate BrdU, and cell cycle profiles were examined by flow cytometric analysis via staining with propidium iodide. In the presence of trastuzumab, the resistant SKBR3 and BT474 breast cancer cells transfected with livin-shRNA3, instead of N1-shRNA, or treated by mock transfection exhibited a substantial (28%) reduction in BrdU staining (p < 0.001, Fig. 3C). However, the absence of trastuzumab resulted in a slight decrease in the incorporation of BrdU into the resistant cell lines (p < 0.05, Fig. 3C). Consistently, transfection of the resistant cell lines with livin-shRNA3 markedly increased the percentage population of cells in the G1 phase in the presence of trastuzumab (p < 0.001) but slightly affected the magnitude of G1-S arrest in the absence of trastuzumab (p < 0.05, Fig. 3D). These data suggested that a decrease in the expression of livin resensitized the resistant breast cancer cells to the antiproliferative and cell cycle-blocking effect of trastuzumab.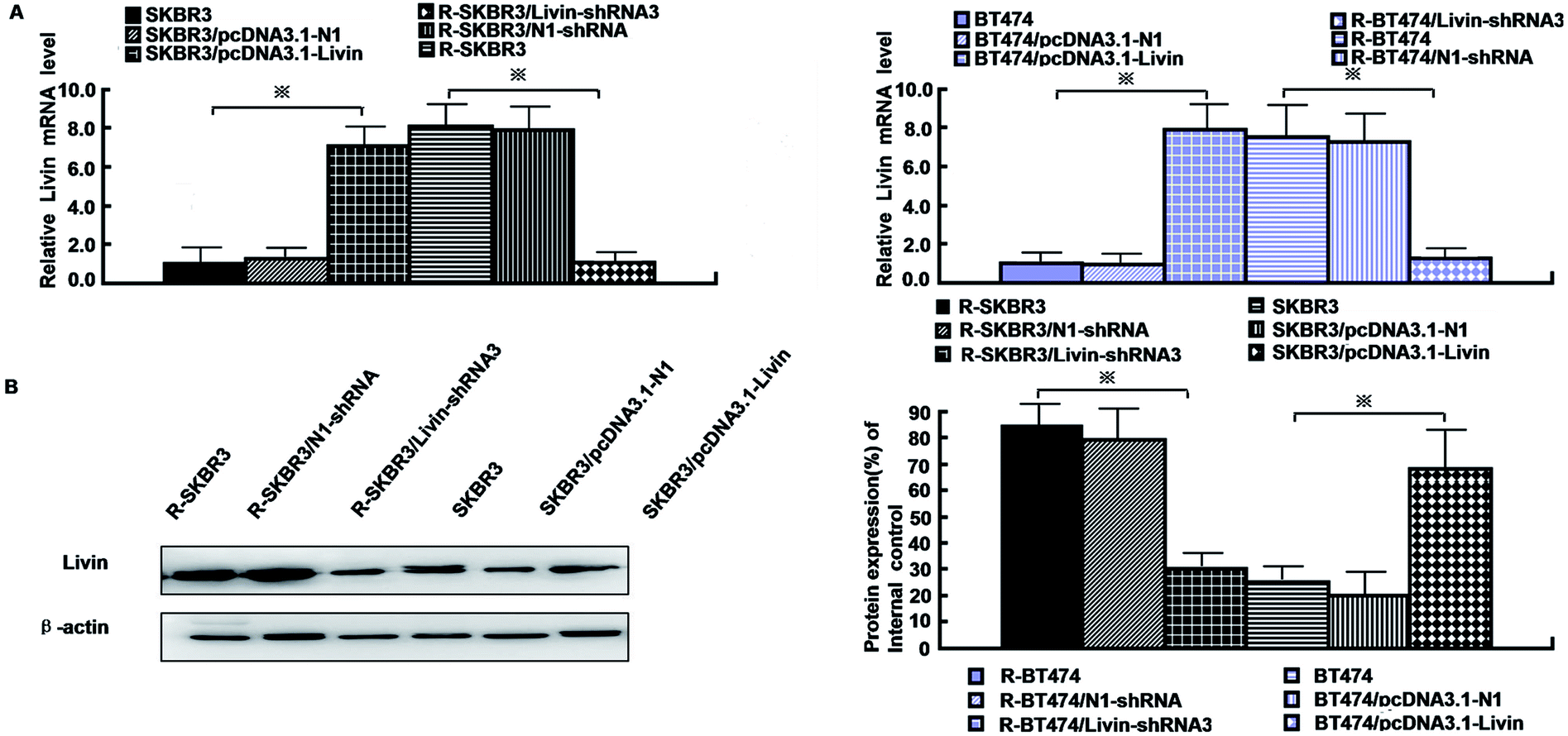 | ||
| Fig. 2 Upregulation of livin is induced in HER2-overexpressing breast cancer cells in the development of acquired trastuzumab resistance. (A) Real-time PCR showed the mRNA expression of livin by livin-shRNA3 in resistant cells and pcDNA3.1-livin in parent cells. (B) Western blot analyses revealed that the protein expression of livin was significantly inhibited in the livin-shRNA3 transfectants and elevated by pcDNA3.1-livin. | ||
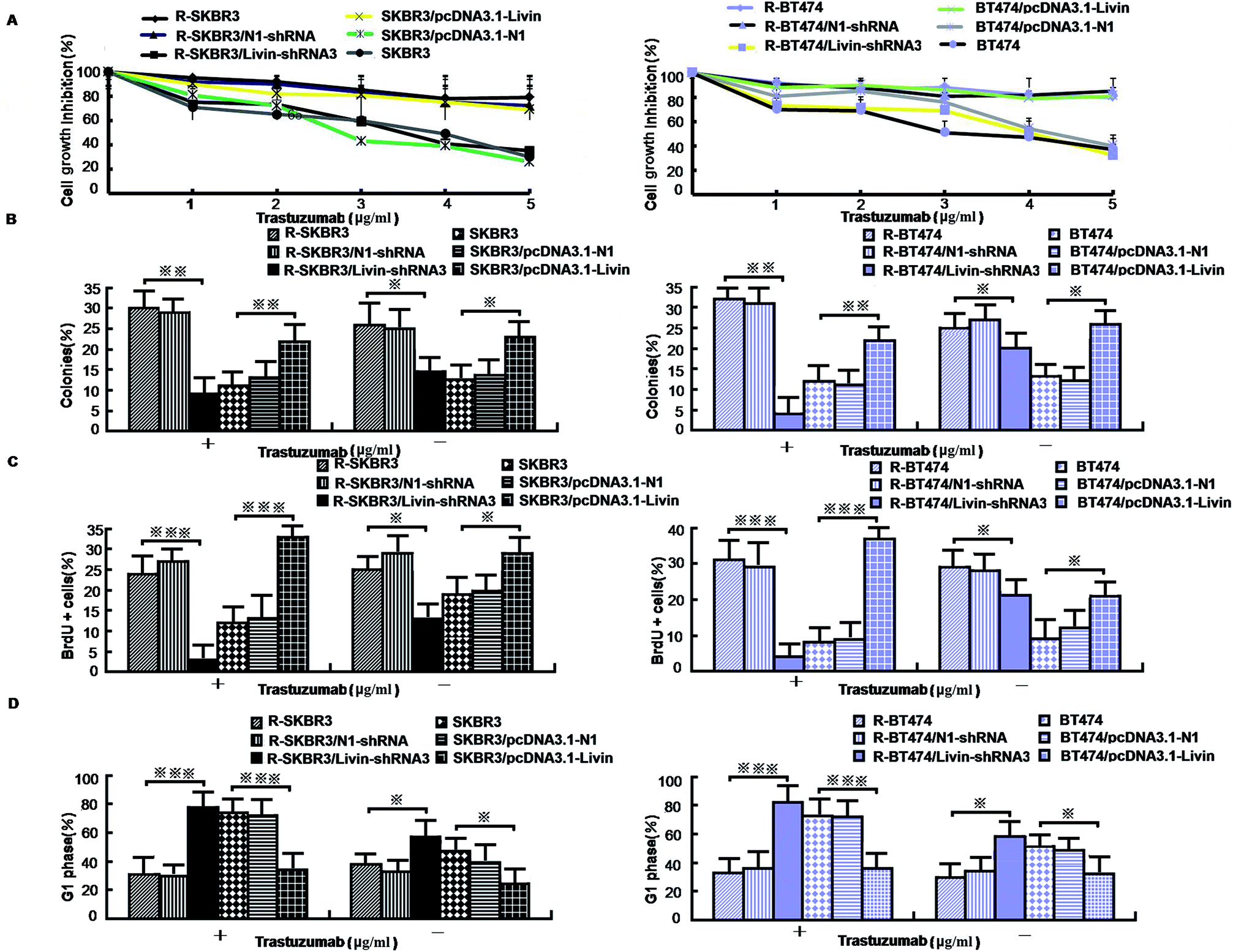 | ||
| Fig. 3 Effect of expression of livin on the proliferation and apoptosis of trastuzumab-resistant breast cancer cells. (A) [3H] thymidine incorporation assays on trastuzumab-resistant SKBR3 and BT474 cells that were transfected with livin-shRNA3 and SKBR3 and BT474 cells that were treated with pcDNA3.1-livin. (B) Percentage formation of anchorage-independent colonies determined by soft-agar assays in trastuzumab-resistant and parent cells treated as in A (*p < 0.05, **p < 0.01 and ***p < 0.001 versus cells treated by mock transfection). (C) Percentage of cells that incorporated BrdU determined by flow cytometric analysis of BrdU immunostaining and representative flow cytometric histograms of BrdU immunostaining in cells treated as in A (*p < 0.05, **p < 0.01 and ***p < 0.001 versus cells treated by mock transfection). (D) Percentage of cells in the G1 phase determined by flow cytometric analysis of cell cycles and representative flow cytometric histograms of the cell cycle for cells treated as in A (*p < 0.05, **p < 0.01 and ***p < 0.001 versus cells treated by mock transfection). | ||
3.4. Ectopic expression of livin confers trastuzumab resistance on breast cancer cells
Because our data showed that a decrease in the expression of livin in trastuzumab-resistant breast cancer lines recovers their sensitivity, we next investigated whether enforced expression of livin in sensitive HER2-overexpressing cancer cells may lead to trastuzumab resistance. In contrast to the decrease in the expression of livin in trastuzumab-resistant cells that overexpressed HER2, transfection of the parent SKBR3 and BT474 cells with pcDNA3.1-livin, but neither transfection with irrelevant pcDNA3.1-N1 nor mock transfection, significantly reduced their sensitivity to the growth-inhibiting effect of trastuzumab. Fig. 3A shows that after treatment with trastuzumab (1 μg mL−1 to 5 μg mL−1) for 3 days, cell growth was inhibited by 38% to 44% in the cell line transfected with pcDNA3.1-N1 and 31% to 45% in the cell line treated by mock transfection. However, cells transfected with pcDNA3.1-livin displayed significantly decreased sensitivity to trastuzumab. The inhibition of cell growth by 1 μg mL−1 to 5 μg mL−1 trastuzumab was less than 10% in SKBR3 cells transfected with pcDNA3.1-livin. However, in the absence of trastuzumab, pcDNA3.1-livin increased inhibition by nearly 5% (p < 0.05). We next hypothesized whether overexpression of livin may reverse the insensitivity of tumor cells to the antiproliferative effect of trastuzumab. A soft-agar assay demonstrated that colony formation by cells transfected with pcDNA3.1-livin was 1.8–2.3 times higher than that by control transfected cells (p < 0.001, Fig. 3B). Moreover, treatment with trastuzumab did not result in any changes in colony formation by cells with enforced expression of livin (p < 0.05). However, in the absence of trastuzumab, pcDNA3.1-livin increased colony formation by 9% (p < 0.01) in the parent cell lines. These data provided evidence that an increase in the expression of livin conferred trastuzumab resistance on HER2-overexpressing breast cancer cells. Furthermore, the incorporation of BrdU in the presence of 10 μg mL−1 trastuzumab for 3 days remained unchanged in the parent SKBR3 and BT474 cell lines that were transfected with pcDNA3.1-livin (p < 0.05, Fig. 3C) but was substantially reduced in control cells that were treated by mock transfection or transfected with pcDNA3.1-N1 (p < 0.01, Fig. 3C). Accordingly, during treatment with trastuzumab, the parent cell lines transfected with pcDNA3.1-livin did not exhibit any significant enrichment of the population of cells in the G1 phase (p < 0.05), as determined by staining with propidium iodide followed by flow cytometric analysis,13 whereas a notable increase in the number of cells treated by mock transfection in the G1 phase was observed (p < 0.01, Fig. 3D). Taken together, these results indicate that the ectopic expression of livin overcomes the G1-S arrest and inhibition of proliferation mediated by trastuzumab treatment and thus induces drug resistance.3.5. Relations between livin and ERK1/2 and AKT pathways, EMT-like transition, motility and invasiveness
Livin is highly expressed in highly invasive breast cancer cells and breast cancer tissues, which is clinically correlated with breast tumors. Because activated livin played a role in trastuzumab resistance (as described above), to investigate the invasion capacity of the transfected breast cancer cells, an in vitro invasion assay was performed. Motility was also significantly inhibited in R-SKBR3 and R-BT474 cells treated with livin-shRNA3 in comparison with cells treated with N1-shRNA. The SKBR3 and BT474 cells transfected with livin exhibited significantly (p < 0.01) increased motility with respect to that of the SKBR3 and BT474 parent cells.The SKBR3 and BT474 cells transfected with livin exhibited a significant (p < 0.01) increase in the number of invading cells in comparison with the cells transfected with the vector. In addition, fewer invading cells were observed in the trastuzumab-resistant cell groups transfected with livin-shRNA3 (p < 0.05, Fig. 4A). Western blotting showed that knockdown of livin by shRNA3 increased E-cadherin and decreased N-cadherin, vimentin, and fibronectin in both R-SKBR3 and R-BT474 cells in comparison with SKBR3 and BT474 cells. pcDNA3.1-livin decreased E-cadherin and increased the expression of vimentin, N-cadherin and fibronectin in both R-SKBR3 and R-BT474 cells in comparison with SKBR3 and BT474 cells. The protein level of N-cadherin, however, was downregulated significantly in R-SKBR3 cells treated with pcDNA3.1-livin. Conversely, the protein level of vimentin was not changed in R-SKBR3 cells after transfection with livin. The protein expression of E-cadherin was upregulated in R-SKBR3 cells after livin was downregulated, which was consistent with its mRNA level (Fig. 4B). To investigate whether the livin-induced MAPK signaling pathway is responsible for trastuzumab resistance in breast cancer cells, transfection of the trastuzumab-resistant breast cancer lines with livin-shRNA3 substantially decreased p-ERK1/2 protein, whereas pcDNA3.1-livin significantly increased the protein expression of p-ERK1/2 in the parent cell lines, although levels of ERK1/2 remained unchanged in trastuzumab-resistant SKBR3 and BT474 cells in comparison with the parent cell lines. As shown in Fig. 4B, in trastuzumab-resistant cells livin-shRNA3 could lower the levels of p-AKT in the presence of trastuzumab.
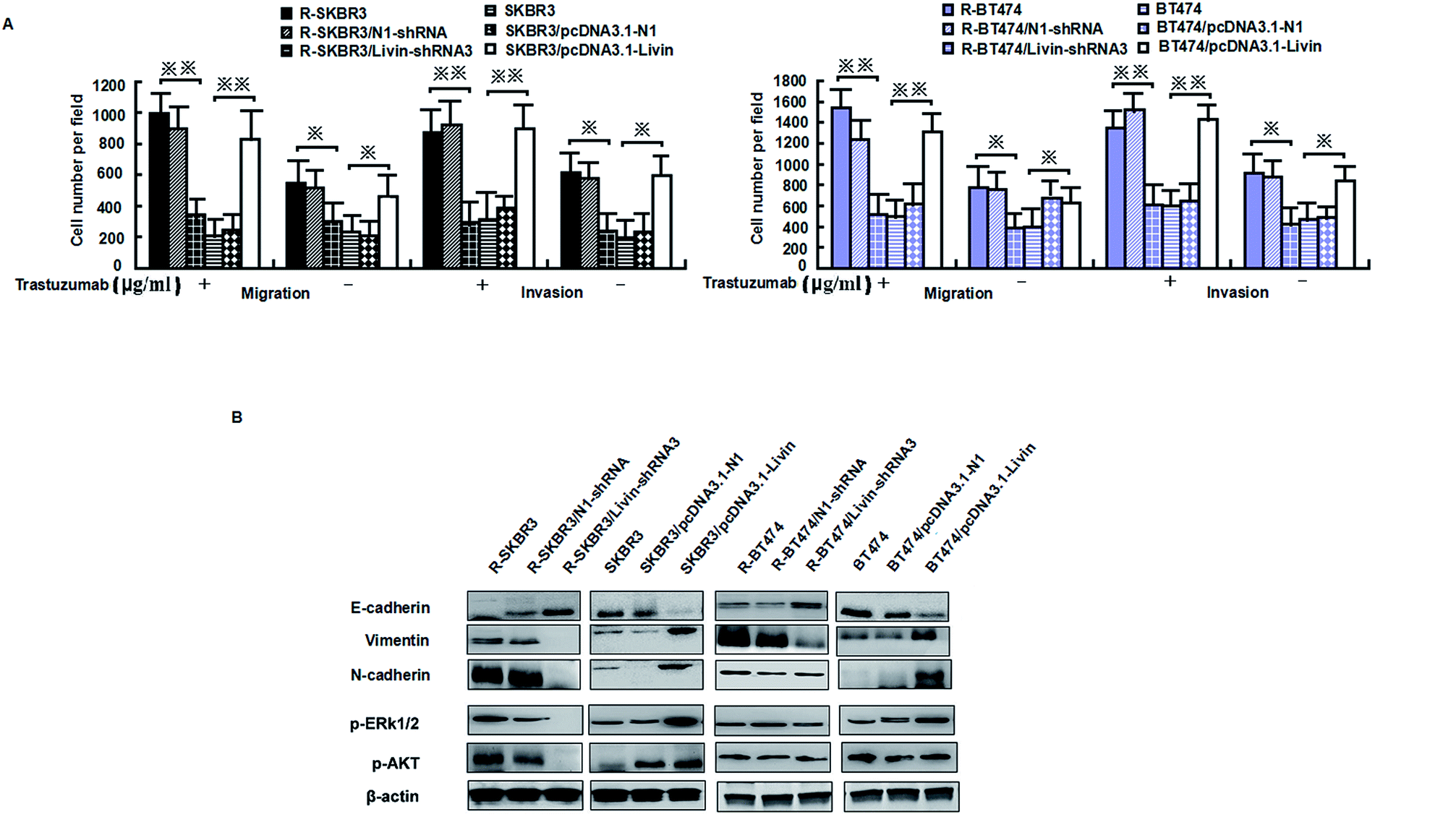 | ||
| Fig. 4 Livin promotes invasiveness, the epithelial–mesenchymal transition and ERK1/2 and AKT signaling in trastuzumab-resistant cell lines. (A) Migratory and invasive abilities of cells expressing livin or the control vector as assessed by a transwell assay. *p < 0.05, **p < 0.01 relative to the vector used as a control (Student's t-test). Bars correspond to mean ± standard deviation (SD) of 3 independent experiments. (B) Immunoblotting was performed with the indicated antibodies on whole-cell lysates. The β-actin was used as loading control. | ||
We further confirmed that the ERK1/2 and PI3K/AKT pathways were involved in the livin-mediated proliferation and metastasis of SKBR3 cells. Inhibitors of ERK1/2 (PD98059) and PI3K/AKT (LY294002) were used to pretreat SKBR3 cells for 30 min, which were then transfected with pcDNA3.1-livin, followed by treatment with 10 μg mL−1 trastuzumab. The results showed that the phosphorylation of ERK1/2 and AKT induced by pcDNA3.1-livin was reversed by PD98059 (p < 0.05) and LY294002 (p < 0.05) independently (Fig. 5A). Then, proliferation and metastasis were investigated after the addition of PD98059 or LY294002. The results showed that the proliferation and metastasis of SKBR3 cells after transfection with pcDNA3.1-livin were reversed by PD98059 and LY294002 (Fig. 5B and C). These results suggest that the ERK1/2 and PI3K/AKT pathways were involved in the livin-mediated proliferation and metastasis of SKBR3 cells in trastuzumab resistance. Taken together, the data suggested that livin acted as a key mediator in the localization of ERK1/2 and AKT and controlled the EMT-like transition, which resulted in increases in cell invasion and proliferation. These events contributed to the increase in resistance to trastuzumab in HER2-overexpressing breast cancer cells.
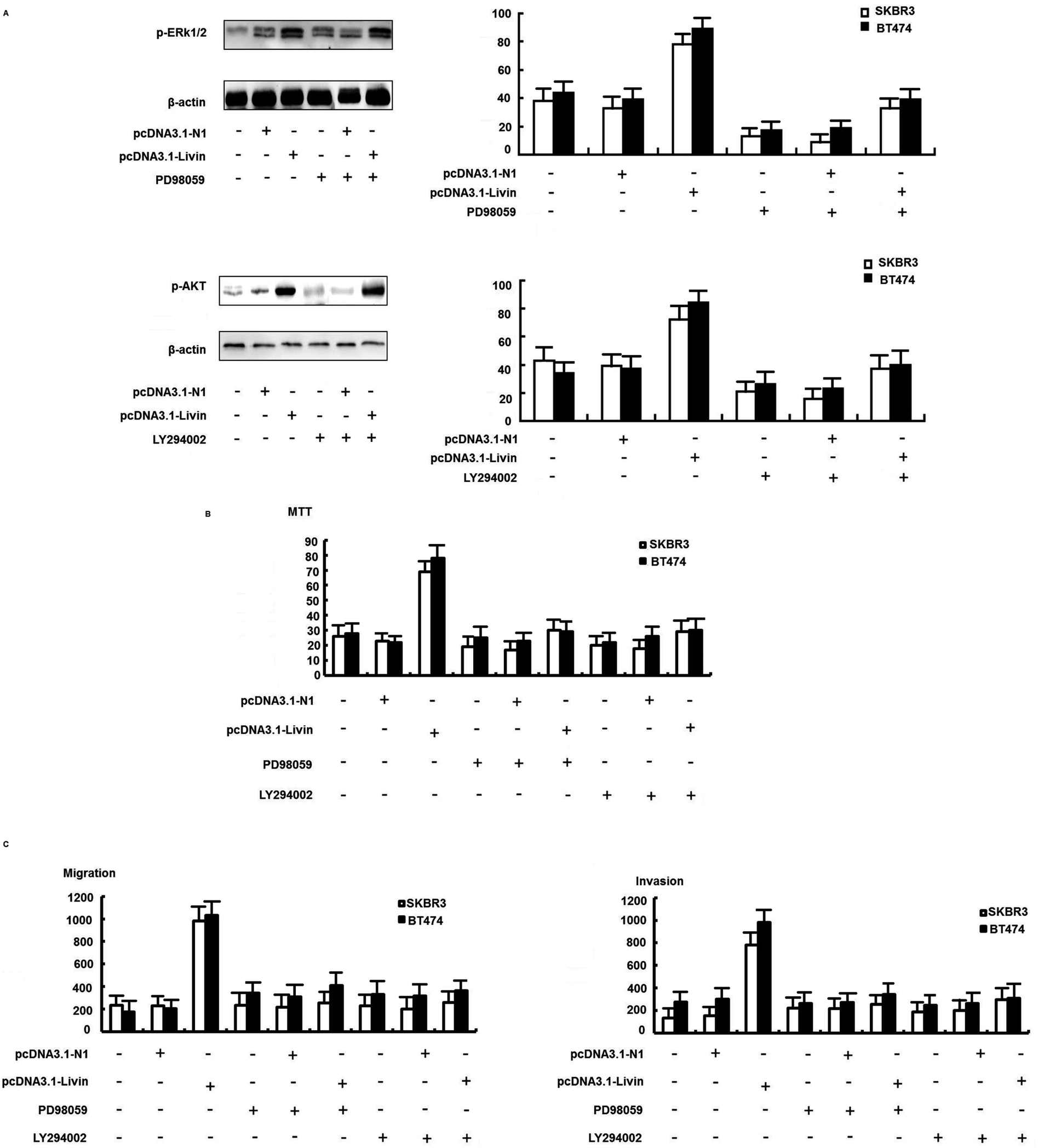 | ||
| Fig. 5 The ERK1/2 and PI3K/AKT signaling pathways were involved in livin-mediated trastuzumab resistance. (A) Effect of an ERK1/2 inhibitor (PD98059) on the expression of ERK1/2 and phospho-ERK1/2. Effect of a PI3K/AKT inhibitor (LY294002) on the expression of AKT and phospho-AKT. SKBR3 cells were transfected with pcDNA3.1-livin in the presence of PD98059 or LY294002 for 72 h. (B) An MTT assay showed that proliferation induced by pcDNA3.1-livin was reversed by PD98059 and LY294002. (C) A transwell assay showed that the invasion induced by pcDNA3.1-livin was reversed by PD98059 and LY294002. All experiments were performed in triplicate. The bars represent the SD. | ||
3.6. Livin confers trastuzumab resistance on breast tumor xenografts
To achieve this, we transfected R-SKBR3 breast cancer cells with livin antisense (AS) oligonucleotides, which effectively reduced the endogenous expression of livin in comparison with cells treated by mock transfection or cells transfected with control mismatched (MIS) oligonucleotides (Fig. 6A). In comparison with cells treated with the MIS control, which exhibited rapid dephosphorylation of ERK1/2 by trastuzumab, dephosphorylation of ERK1/2 and AKT was attenuated in cells treated with livin AS. This indicates that livin is required for the rapid phosphorylation of ERK1/2 and AKT by trastuzumab. Cells treated with livin AS that had reduced livin levels underwent significantly less EMT in the presence of trastuzumab than R-SKBR3 cells transfected with the MIS control that had normal expression of livin (Fig. 6B). Cells treated with livin AS that had reduced livin levels experienced significantly more inhibition of growth by trastuzumab than R-SKBR3 cells transfected with the MIS control that had normal expression of livin. Without trastuzumab treatment, the reduction of livin levels by livin AS alone had no significant effect on cell proliferation, which was consistent with the fact that levels of ERK1/2 phosphorylation in cells treated with livin AS alone were similar to those in cells treated with the MIS control (Fig. 6C). The fact that blockade of livin only reduced the antiproliferative effect of trastuzumab but had no effect on cells without trastuzumab treatment confirmed that the activity of livin is important for the function of trastuzumab, which closely corresponds to our finding that livin is activated by trastuzumab. Hence, livin led to resistance to the antiproliferative effect of trastuzumab in these cells.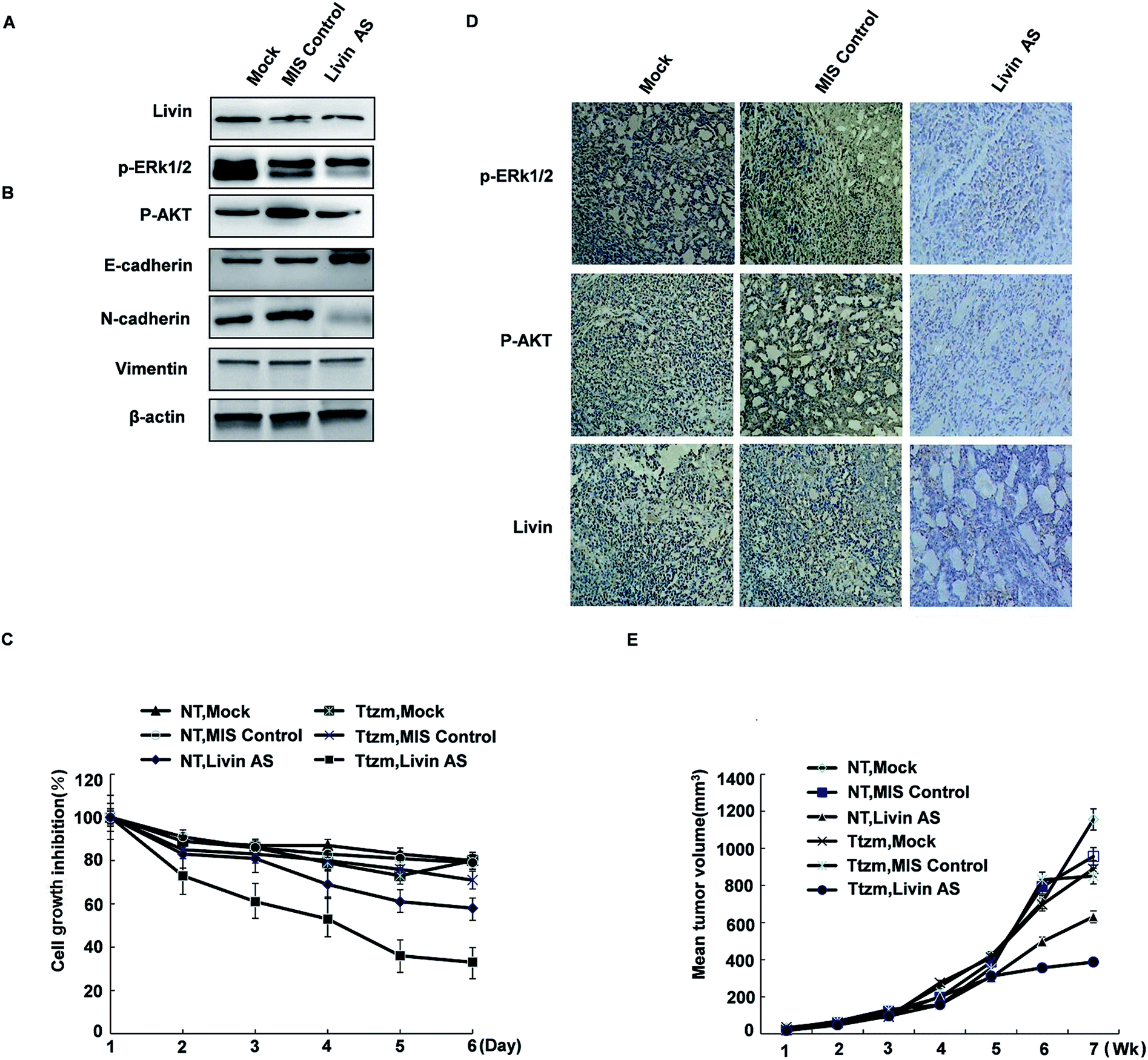 | ||
| Fig. 6 The reduction of livin levels by livin AS oligonucleotides reverses resistance to the antiproliferative effects of trastuzumab in vitro and in vivo. (A) Protein levels of livin are reduced in R-SKBR3 cells transfected with livin AS (25 nM) in comparison with cells treated by mock transfection or transfected with mismatched (MIS) oligonucleotides. (B) Trastuzumab-mediated dephosphorylation of AKT was attenuated in R-SKBR3 cells transfected with livin AS. R-SKBR3 cells transfected with MIS or livin AS (as in A) were treated with trastuzumab (2 μg mL−1) for the indicated times, and cell lysates were immunoblotted. (C) Trastuzumab inhibited cell proliferation to a greater extent in R-SKBR3 cells treated with livin AS in comparison with cells transfected with MIS. R-SKBR3 cells transfected with MIS or livin AS (as in A) were treated with trastuzumab (2 μg mL−1) for 3 and 5 days. Cell masses were determined by an MTT assay. The percentage cell numbers represent the percentage cell counts for each treatment with respect to untreated cells. (D) The expression of livin was reduced in R-SKBR3 xenografts injected with livin AS. R-SKBR3 cells were inoculated into the mfp of female nude mice. After the tumor volume reached 150 mm3, MIS or livin AS was injected intratumorally (15 μg per injection, twice per week) for 1 week. Removed tumors were stained with antibodies to livin, p-ERK1/2 and p-AKT. (E) Livin-deficient R-SKBR3 xenografts were less resistant to trastuzumab than R-SKBR3 xenografts. After treatment with livin AS or MIS for 1 week, mice were treated with trastuzumab (10 mg kg−1) or vehicle twice per week. The arrows indicate the starting days of treatment with AS/MIS (AS) and treatment with trastuzumab/vehicle (Ttzm). The results shown are the mean tumor volumes ± SE; *p < 0.05. | ||
To investigate whether livin may confer resistance to trastuzumab on breast cancer cells in vivo, we injected a tumorigenic subline of R-SKBR3 cells into the mammary fat pad (mfp) of athymic nude mice. When tumor xenografts reached a volume of ≈150 mm3, we injected livin AS or MIS oligonucleotides into the tumor xenografts.
The effects of combination therapy with livin AS and trastuzumab on trastuzumab-resistant xenografts were studied. One week later, we treated the mice with trastuzumab (10 mg kg−1) or vehicle twice per week. Trastuzumab-resistant tumors were visible after 35 to 42 days in ovariectomized female athymic nude mice. In these experiments, the mean volume of tumors treated with ethanol (control group) increased by a factor of 3.2 over 56 days, whereas the mean volume of tumors in the group treated with MIS oligonucleotides did not significantly differ from that in the control group. However, combination treatment remarkably inhibited cell growth in trastuzumab-resistant xenografts during the intervention. At the end of treatment, the group that received combination treatment had an approximately twofold reduction in tumor volume in comparison with the control group (Fig. 6E). The extent of tumor inhibition was significantly greater in animals that received trastuzumab than in those not treated with the antibody (p < 0.01). Moreover, this inhibition resulted in no obvious toxicity, as body weights did not change greatly. Trastuzumab only inhibited tumors treated with livin AS that expressed decreased levels of livin, but not tumors treated with the MIS control with normal livin levels. Without trastuzumab treatment, tumors treated with livin AS and MIS had similar growth rates. The immunoreactivity and expression of p-ERK1/2 and phospho-AKT decreased in tumors injected with livin AS in comparison with mice injected with the control vector (Fig. 6D). These results imply that livin is a stimulatory factor in the growth of trastuzumab-resistant xenografts, and the inhibition of the activation of livin by targeted therapy could restore the curative effect of endocrine treatment of trastuzumab-resistant breast cancer (Fig. 6D and E).
3.7. Breast cancer patients with livin overexpression have a poor clinical response to trastuzumab
To further assess the clinical significance of the overexpression of livin in trastuzumab resistance, we examined the expression of livin in primary breast cancers from 24 patients who were receiving neoadjuvant therapy with trastuzumab and correlated it with the response of the tumor to the drug, as determined using the Response Evaluation Criteria in Solid Tumors (RECIST). This was further confirmed by qRT-PCR, which showed that livin levels were 2.7 times higher in trastuzumab-resistant tumors than in sensitive tumors (p < 0.01). Moreover, the expression of livin was further upregulated after trastuzumab therapy in both resistant and sensitive tumors, but the difference did not reach statistical significance (Fig. 7A–C), which implies that, given a prolonged period of treatment with trastuzumab, acquired trastuzumab resistance due to the induction of livin may occur. In accordance with the upregulation of livin, the protein expression of p-ERK1/2 and p-AKT was significantly higher in trastuzumab-resistant tumors in comparison with sensitive tumors, as determined by immunohistochemistry before and after treatment with trastuzumab (Fig. 7D–G). Moreover, the expression of livin in breast cancer cells was positively correlated with the expression of p-ERK1/2 (Fig. 7H), and the correlation was stronger for tumors after trastuzumab therapy (Fig. 7F, p < 0.01) than before therapy (Fig. 7F, p < 0.05). The expression of livin in breast cancer cells was also positively correlated with the expression of p-AKT (Fig. 7I), and the correlation was stronger in tumors after trastuzumab therapy (Fig. 7F, p < 0.01) than before therapy (Fig. 7F, p < 0.05). These data suggest that the upregulation of livin is associated with the MAPK signaling pathway and AKT in trastuzumab resistance in breast cancers.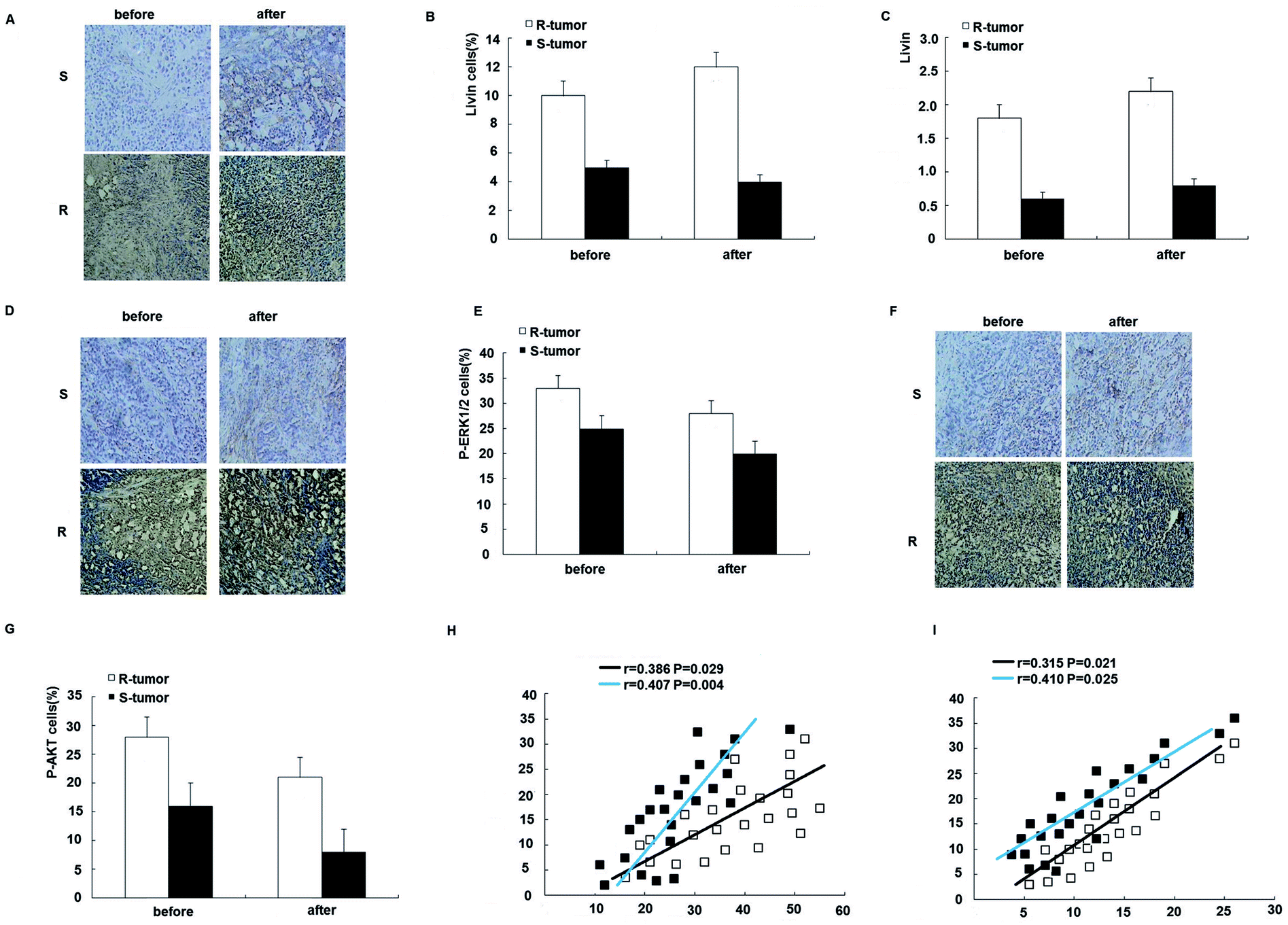 | ||
| Fig. 7 Overexpression of Livin is associated with trastuzumab resistance in breast cancer patients. (A) Microscopic images (200×) of in situ hybridization of livin in trastuzumab-resistant (R) and -sensitive tumors (S) before and after neoadjuvant therapy with trastuzumab. (B) Percentage of livin-positive (livin+) cells in trastuzumab-resistant and -sensitive tumors before and after neoadjuvant therapy with trastuzumab. p < 0.01 for R-tumors versus S-tumors. (C) Quantitative RT-PCR for the expression of livin in trastuzumab-resistant and -sensitive tumors before and after neoadjuvant therapy with trastuzumab. p < 0.01 for R-tumors versus S-tumors. (D) Microscopic images (200×) of immunohistochemical staining for the expression of p-ERK1/2 in trastuzumab-resistant and -sensitive tumors before and after trastuzumab therapy. (E) Percentage of p-ERK1/2-positive (p-ERK1/2+) cells in trastuzumab-resistant and -sensitive tumors before and after neoadjuvant therapy with trastuzumab. p < 0.01 for R-tumors versus S-tumors. (F) Microscopic images (200×) of immunohistochemical staining for the expression of p-AKT in trastuzumab-resistant and -sensitive tumors before and after trastuzumab therapy. (G) Percentage of p-AKT-positive (p-AKT+) cells in trastuzumab-resistant and -sensitive tumors before and after neoadjuvant therapy with trastuzumab. p < 0.01 for R-tumors versus S-tumors. (H) Expression of livin and p-ERK1/2 in each individual case for 24 breast cancer patients before (E) and after (F) neoadjuvant therapy with trastuzumab illustrated in a scatter diagram. (I) Expression of livin and p-AKT in each individual case for 24 breast cancer patients before (E) and after (F) neoadjuvant therapy with trastuzumab illustrated in a scatter diagram. Correlation coefficients were determined, and p < 0.01 for R-tumors versus S-tumors. | ||
4. Discussion
Trastuzumab is an example of highly specific targeted therapy for HER2-overexpressing breast cancers, but tumors may be initially resistant to the antibody or acquire the trait of resistance in the course of treatment.16 The underlying mechanisms are still unclear. In this study, we investigated the role of livin in the development of trastuzumab resistance in hormone-dependent breast cancer. Livin promotes trastuzumab resistance in patients with breast cancer during endocrine treatment. Responsiveness to trastuzumab depends not only on the downregulation of HER2 and the inhibition of downstream events related to HER2, but also on the status of livin. Indeed, our data for cultured breast cancer cell lines, mouse tumor xenografts, and, most importantly, clinical samples from breast cancer patients consistently demonstrated that livin is a molecular factor that conferred resistance to trastuzumab-based therapy on HER2-overexpressing breast cancers and that the upregulation of livin was associated with trastuzumab resistance in patients.HER2 is overexpressed in 25–30% of human breast cancers, and this overexpression is associated with a poor prognosis and poor response to chemotherapy.17,18 Trastuzumab is currently approved as a front-line therapy in patients with HER2-positive breast cancer and has also shown efficacy in combination with conventional chemotherapeutic agents.19–21 However, for HER2-overexpressing patients treated with trastuzumab alone, the overall response rates are only 23–26% for patients without prior chemotherapy22,23 and 12–15% for patients with prior chemotherapy.24,25 In addition, a significant percentage of women who initially respond to trastuzumab may develop acquired resistance to therapy after prolonged treatment. Better predictive markers of response to trastuzumab are therefore needed.
Altered expression and dysfunction of livin have been well documented in nearly all types of human malignancies and are involved in the formation and progression of tumors by regulating the expression and action of many oncogenes and tumor suppressor genes. Livin is consistently upregulated in a variety of cancers, including melanoma, leukemia, bladder cancer, cervical cancer, nasopharyngeal cancer, and lung cancer.11,26 Interestingly, the expression of livin is barely detectable, or it is present at substantially lower levels, in normal tissues.11,27 As shown in our previous research, livin is significantly upregulated in a subgroup of breast cancers, and high expression of livin has been associated with unfavorable pathological and molecular features of the disease, including advanced tumor stage, high tumor grade, lymph node metastasis, hormone receptor status, and poor patient survival. In addition, livin has been shown to promote oncogenesis and progression of breast cancers in vitro and in vivo.11 Likewise, overexpression of livin also contributes to resistance of malignant tumors, including breast cancers, to chemotherapeutic agents.
Here, our study shows that high expression of livin is sufficient to induce trastuzumab resistance in HER2-expressing breast cancers in vitro and in vivo, whereas downregulation of livin alone is capable of restoring sensitivity to trastuzumab in breast cancer cells with acquired resistance in vitro and in vivo. Therefore, livin is not only crucial in the oncogenesis and development of breast cancer, but is also a key factor in the development of resistance to anticancer therapies. In our present study, the expression of livin was dramatically increased when HER2-overexpressing breast cancer cells were induced to acquire resistance to trastuzumab by long-term exposure to the antibody in culture. Upon phosphorylation of its tyrosine kinase domain, HER2 promotes proliferation by activating the RAS-MAPK pathway and survival via the PI3K-AKT pathway.28 Among these, activation of the MAPK pathway is due to increased homodimerization of EGFR or heterodimerization of EGFR/HER3.29 The present model shows variable activation of the EGFR downstream cascade. The release of HB-EGF allows it to activate the EGFR signaling pathway, which results in the induction of ERK1/2 phosphorylation with consequent stimulation of cell growth.30 Levels of phosphorylated ERK1/2 increased transiently in our trastuzumab-resistant cells and in models of long-term tamoxifen treatment reported by other researchers.31,32 On examining the downstream MAPK/ERK1/2 signaling pathway, total levels of ERK1/2 were all sustained in breast cancer subclones in the absence of trastuzumab, but phosphorylated ERK1/2 was significantly upregulated in resistant cells exposed to trastuzumab. Our experiments, which demonstrated sustained levels of ERK1/2 and AKT and phosphorylation during continuous exposure to trastuzumab, suggested that resistant cells might also retain sensitivity to kinase inhibitors in the presence of trastuzumab. In the in vitro isolation and characterization of trastuzumab-resistant cells, trastuzumab also rendered trastuzumab-resistant subclones equally sensitive to the ERK1/2 kinase inhibitor PD98059 and the AKT kinase inhibitor LY294002, even though resistant subclones were relatively resistant in the absence of trastuzumab. Livin-shRNA3 significantly inhibited phosphorylation of ERK1/2 and AKT, as did the inhibitors PD98059 and LY294002. Inhibition of the expression of ERK1/2 and AKT in trastuzumab-resistant cells restores their sensitivity to the antibody, whereas ectopic expression of livin in previously sensitive cells confers resistance to trastuzumab. This suggests that resistant subclones acquired a trastuzumab-resistant mechanism of MAPK/ERK1/2 and AKT signaling. The data suggest that combination therapy with trastuzumab and an inhibitor of ERK1/2 or AKT signaling might be a viable therapeutic strategy for trastuzumab-resistant human breast carcinomas. In these scenarios, livin-mediated trastuzumab resistance acts in concert with other mechanisms of resistance and may form positive feed-forward loops with other pathways to exert the resistance effect.
The implications of the above findings extend beyond acquired trastuzumab resistance, as overexpression of livin occurred in breast cancers that responded poorly to neoadjuvant therapy with trastuzumab. Moreover, the enforced expression of livin in sensitive breast cancer cells via transfection confers resistance to trastuzumab on these cells. Therefore, the upregulation of livin is not only crucial to acquired trastuzumab resistance but may also play a critical role in inherent primary resistance. Here, we show that inhibition of the expression of livin in trastuzumab-resistant cell lines with livin-shRNA3 or livin-ASO ultimately leads to the inhibition of p-ERK1/2 and p-AKT and induction of growth arrest. Moreover, treatment with PD98059 and LY294002 also removes trastuzumab resistance induced by pcDNA3.1-livin. In light of this, blocking the action of livin with ASO provides tremendous therapeutic value in combination with trastuzumab. Because the enforced expression of livin or direct inhibition of the MAPK/ERK1/2 and AKT signaling pathways is not only technically challenging but may also interfere with normal cellular functions, specifically targeting upregulated oncogenic livin in cancer cells may prove to be a more feasible therapeutic modality. However, this will require the determination of how to systemically deliver ASO into HER2-expressing cancer cells in vivo, and the selection of delivery vectors with a targeting moiety against HER2, such as a fragment of an anti-HER2 antibody fused to protamine, may facilitate the in vivo delivery of livin-ASO for therapeutic purposes. Our results provide experimental evidence of the important role of livin in the development of trastuzumab resistance and establish a new therapeutic target for delaying drug resistance or improving the response to endocrine treatment in cases in which trastuzumab resistance develops. Clinically, the upregulation of livin accompanied by increased p-ERK1/2 and p-AKT is associated with a poor response to trastuzumab in breast cancers. The role of livin in acquired trastuzumab resistance can be reflected by our findings that blocking the action of livin alone with ASO is sufficient to restore the inhibition of ERK1/2 and AKT by trastuzumab in resistant cells and is capable of sensitizing resistant cells to trastuzumab in vitro and in vivo.
The EMT-like phenotype has been observed in the development of drug resistance33,34 and could promote tumor invasion and metastases, as well as mediating drug resistance.35,36 To confirm the specific role of livin in the development of trastuzumab resistance, livin levels in R-SKBR3 and R-BT474 cells were decreased by livin-shRNA3. These cells exhibited upregulation of E-cadherin and downregulation of EMT markers such as N-cadherin. Furthermore, the cell invasiveness was reduced and the growth-inhibiting effects of trastuzumab were restored. The clones stably transfected with livin tended to undergo an EMT-like transition with downregulation of E-cadherin and upregulation of N-cadherin. The livin transfectants were less responsive to trastuzumab-induced growth inhibition. This could be due to redundant expression of livin. Data from our current study suggest that specifically targeting livin could overcome trastuzumab resistance and benefit breast cancer patients with HER2-overexpressing tumors.
We have shown in the current study that the development of trastuzumab resistance in HER2-overexpressing breast cancer cells is accompanied by a partial EMT-like transition. Remarkably, our finding that livin confers resistance to trastuzumab is strongly supported by clinical data that patients with livin-overexpressing tumors did not respond to trastuzumab as well as those with normal livin, and the difference was statistically highly significant (p < 0.01), even in the small cohort. Hence, the livin expression status of HER2-overexpressing breast tumors may be used as a predictive factor for patients and can also serve as an indication that the patient needs combination therapy with inhibitors of ERK1/2 and AKT to overcome livin-mediated trastuzumab resistance and thus receive the maximum therapeutic benefit of trastuzumab. Large-scale clinical investigations will be performed to further validate these ideas. The activation by livin of the MAPK and AKT signaling pathways is one of the underlying mechanisms of the development of resistance to trastuzumab.
5. Conclusion
It will be important to determine if the features of trastuzumab resistance seen in our study are present in trastuzumab-resistant breast cancers. If so, the retention of livin in trastuzumab-resistant cells suggests that livin should remain a possible target for radioimmunotherapy in trastuzumab-resistant cancers using radiolabeled trastuzumab or another anti-Her2 conjugate. The sustained activity of ERK1/2 kinase in trastuzumab-resistant cells suggests that PD98059 and LY294002 may be effective in some cases and might be particularly useful in targeting trastuzumab-resistant breast cancers.Funding
This study was supported by the Chongqing Health Bureau grant (No. 30872520) and the National Natural Science Foundation of China (No. 81202089).Conflicts of interest
The authors declare no conflict of interest.Notes and references
- T. Cooke, J. Reeves, A. Lanigan and P. Stanton, Ann. Oncol., 2001, 12(suppl 1), S23–S28 CrossRef PubMed.
- G. Mariani, A. Fasolo, E. De Benedictis and L. Gianni, Nat. Clin. Pract. Oncol., 2009, 6(2), 93–104 CrossRef PubMed.
- A. Prat, C. Fan, A. Fernández, K. A. Hoadley, R. Martinello, M. Vidal, M. Viladot, E. Pineda, A. Arance, M. Muñoz, L. Paré, M. C. Cheang, B. Adamo and C. M. Perou, BMC Med., 2015, 13(1), 303 CrossRef PubMed.
- F. J. Esteva, V. Valero, D. Booser, L. T. Guerra, J. L. Murray, L. Pusztai, M. Cristofanilli, B. Arun, B. Esmaeli, H. A. Fritsche, N. Sneige, T. L. Smith and G. N. Hortobagyi, J. Clin. Oncol., 2002, 20(7), 1800–1808 CrossRef PubMed.
- P. R. Pohlmann, I. A. Mayer and R. Mernaugh, Clin. Cancer Res., 2009, 15(24), 7479–7491 CrossRef PubMed.
- H. S. Cho, K. Mason, K. X. Ramyar, A. M. Stanley, S. B. Gabelli, D. W. Denney Jr and D. J. Leahy, Nature, 2003, 421(6924), 756–760 CrossRef PubMed.
- R. Nahta, M. C. Hung and F. J. Esteva, Cancer Res., 2004, 64(7), 2343–2346 CrossRef PubMed.
- C. T. Chan, M. Z. Metz and S. E. Kane, Breast Cancer Res. Treat., 2005, 91(2), 187–201 CrossRef PubMed.
- B. Weigelt, A. T. Lo, C. C. Park, J. W. Gray and M. J. Bissell, Breast Cancer Res. Treat., 2010, 122(1), 35–43 CrossRef PubMed.
- Y. Wu, H. Mohamed, R. Chillar, I. Ali, S. Clayton, D. Slamon and J. V. Vadgama, Breast Cancer Res., 2008, 10(1), R3 CrossRef PubMed.
- F. Li, X. Yin, X. Luo, H. Y. Li, X. Su, X. Y. Wang, L. Chen, K. Zheng and G. S. Ren, Cell. Signalling, 2013, 25(6), 1413–1422 CrossRef PubMed.
- B. Nachmias, Y. Ashhab, V. Bucholtz, O. Drize, L. Kadouri, M. Lotem, T. Peretz, O. Mandelboim and D. Ben-Yehuda, Cancer Res., 2003, 63(19), 6340–6349 Search PubMed.
- C. Gong, Y. Yao, Y. Wang, B. Liu, W. Wu, J. Chen, F. Su, H. Yao and E. Song, J. Biol. Chem., 2011, 286(21), 19127–19137 CrossRef PubMed.
- T. Xiang, L. Li, X. Yin, C. Yuan, C. Tan, X. Su, L. Xiong, T. C. Putti, M. Oberst, K. Kelly, G. Ren and Q. Tao, PLoS One, 2012, 7(1), e29783 CrossRef PubMed.
- Mo, M. Liu, F. Yang, H. Luo, Z. Li, G. Tu and G. Yang, Breast Cancer Res., 2013, 15, R114 CrossRef PubMed.
- H. Lyu, X. H. Yang, S. M. Edgerton, A. D. Thor, X. Wu, Z. He and B. Liu, Oncotarget, 2016, 7(3), 2921–2935 CrossRef PubMed.
- D. J. Slamon, W. Godolphin, L. A. Jones, J. A. Holt, S. G. Wong, D. E. Keith, W. J. Levin, S. G. Stuart, J. Udove and A. Ullrich, et al., Science, 1989, 244(4905), 707–712 CrossRef PubMed.
- L. Ning, Z. Guo-Chun, A. Sheng-Li, L. Xue-Rui, W. Kun, Z. Jian, R. Chong-Yang, W. Ling-Zhu and L. Hai-Tong, Tumor Biol., 2016, 37(4), 5445–5454 CrossRef PubMed.
- V. Diéras, P. Beuzeboc, V. Laurence, J. Y. Pierga and P. Pouillart, Oncology, 2001, 61(suppl 2), 43–49 CrossRef PubMed.
- D. J. Slamon, B. Leyland-Jones, S. Shak, H. Fuchs, V. Paton, A. Bajamonde, T. Fleming, W. Eiermann, J. Wolter, M. Pegram, J. Baselga and L. Norton, N. Engl. J. Med., 2001, 344(11), 783–792 CrossRef PubMed.
- M. D. Pegram and D. J. Slamon, Semin. Oncol., 1999, 26(4 suppl 12), 89–95 Search PubMed.
- C. L. Vogel, M. A. Cobleigh, D. Tripathy, J. C. Gutheil, L. N. Harris, L. Fehrenbacher, D. J. Slamon, M. Murphy, W. F. Novotny, M. Burchmore, S. Shak and S. J. Stewart, Oncology, 2001, 61(suppl 2), 37–42 CrossRef PubMed.
- C. Vogel, M. A. Cobleigh, D. Tripathy, J. C. Gutheil, L. N. Harris, L. Fehrenbacher, D. J. Slamon, M. Murphy, W. F. Novotny, M. Burchmore, S. Shak and S. J. Stewart, Eur. J. Cancer, 2001, 37(suppl 1), S25–S29 CrossRef PubMed.
- J. Baselga, Oncology, 2001, 61(suppl 2), 14–21 CrossRef PubMed.
- M. A. Cobleigh, C. L. Vogel, D. Tripathy, N. J. Robert, S. Scholl, L. Fehrenbacher, J. M. Wolter, V. Paton, S. Shak, G. Lieberman and D. J. Slamon, J. Clin. Oncol., 1999, 17(9), 2639–2648 CrossRef PubMed.
- J. Choi, Y. K. Hwang, K. W. Sung, S. H. Lee, K. H. Yoo, H. L. Jung, H. H. Koo, H. J. Kim, H. J. Kang, H. Y. Shin and H. S. Ahn, Blood, 2007, 109, 471–477 CrossRef PubMed.
- J. Gong, N. Chen, Q. Zhou, B. Yang, Y. Wang and X. Wang, J. Clin. Pathol., 2005, 58, 1081–1085 CrossRef PubMed.
- S. Massarweh, C. K. Osborne, C. J. Creighton, L. Qin, A. Tsimelzon, S. Huang, H. Weiss, M. Rimawi and R. Schiff, Cancer Res., 2008, 68(3), 826–833 CrossRef PubMed.
- F. Talotta, A. Cimmino, M. R. Matarazzo, L. Casalino, G. De Vita, M. D'Esposito, R. Di Lauro and P. Verde, Oncogene, 2009, 28(1), 73–84 CrossRef PubMed.
- E. J. Filardo, J. A. Quinn, K. I. Bland and A. R. Frackelton Jr, Mol. Endocrinol., 2000, 14(10), 1649–1660 CrossRef PubMed.
- J. A. McCubrey, L. S. Steelman, S. L. Abrams, J. T. Lee, F. Chang, F. E. Bertrand, P. M. Navolanic, D. M. Terrian, R. A. Franklin, A. B. D'Assoro, J. L. Salisbury, M. C. Mazzarino, F. Stivala and M. Libra, Adv. Enzyme Regul., 2006, 46, 249–279 CrossRef PubMed.
- J. Shou, S. Massarweh, C. K. Osborne, A. E. Wakeling, S. Ali, H. Weiss and R. Schiff, J. Natl. Cancer Inst., 2004, 96(12), 926–935 CrossRef PubMed.
- S. Burdette-Radoux and H. B. Muss, Clin. Breast Cancer, 2009, 9(suppl 1), S37–S41 CrossRef PubMed.
- Z. H. Xu, J. B. Hang, J. A. Hu and B. L. Gao, Int. J. Clin. Exp. Pathol., 2013, 6(8), 1493–1504 Search PubMed.
- S. Gupta, K. Iljin, H. Sara, J. P. Mpindi, T. Mirtti, P. Vainio, J. Rantala, K. Alanen, M. Nees and O. Kallioniemi, Cancer Res., 2010, 70(17), 6735–6745 CrossRef PubMed.
- M. K. Asiedu, J. N. Ingle, M. D. Behrens, D. C. Radisky and K. L. Knutson, Cancer Res., 2011, 71(13), 4707–4719 CrossRef PubMed.
Footnote |
| † First author. |
| This journal is © The Royal Society of Chemistry 2018 |