DOI:
10.1039/C8RA05684F
(Paper)
RSC Adv., 2018,
8, 30894-30901
Retracted Article: TRIM22 functions as an oncogene in gliomas through regulating the Wnt/β-catenin signaling pathway
Received
3rd July 2018
, Accepted 20th August 2018
First published on 3rd September 2018
Abstract
The tripartite motif-containing (TRIM) family is a group of proteins that are implicated in a plethora of pathological conditions. TRIM22 has been found to be involved in various cancers; however, the role of TRIM22 in gliomas has not been reported. The present study aimed to evaluate the expression pattern of TRIM22 and its function in gliomas. TRIM22 expressions in glioma tissues and cell lines were measured by RT-PCR and western blot analysis. To knockdown TRIM22 by small hairpin RNAs (shTRIM22), the U118 cells were transfected with pLKO.1-shTRIM22 plasmid or pLKO.1 plasmid. Cell proliferation was measured using CCK-8 assay. Transwell assays were performed to evaluate the migration and invasion. The epithelial–mesenchymal transition (EMT) was assessed by detecting the expressions of E-cadherin, N-cadherin and vimentin with western blot analysis. A xenograft mouse model was established to evaluate the effect of TRIM22 silencing on tumor growth in vivo. The expressions of β-catenin, cyclin D1, and c-Myc were analyzed by western blot analysis. TRIM22 was significantly overexpressed in glioma tissues and cell lines. In vitro studies demonstrated that TRIM22 knockdown inhibited cell proliferation, migration, and invasion. Additionally, TRIM22 silencing increased the expressions of E-cadherin, and decreased the expressions of N-cadherin and vimentin. Nude mouse xenograft assay showed that TRIM22 silencing inhibited tumor growth in vivo. Furthermore, silencing of TRIM22 inhibited the activation of the Wnt/β-catenin pathway. Treatment with LiCl, an activator of the Wnt/β-catenin pathway, attenuated the effects of shTRIM22 on U118 cells. Silencing of TRIM22 inhibited proliferation, migration and invasion, as well as repressing the EMT process in glioma cells. The Wnt/β-catenin pathway was involved in the effect of TRIM22.
1. Introduction
Glioma is a type of tumor originating in the glial cells in the brain.1 It accounts for about 33 percent of all brain tumors, and 80 percent of all malignant brain tumors.2 It represents a relatively serious health burden.1 On the basis of their histopathology, gliomas can be subdivided into grades I–IV according to the World Health Organisation (WHO) classification of tumors in the central nervous system.3 The treatment of gliomas is a combined therapy with surgery, radiation therapy and chemotherapy, which depends on the location and grade of malignancy.4 However, the conventional treatment has an alarmingly dismal prognosis. Gliomas generally recur even after optimal initial treatment. Therefore, novel approaches in the treatment of this disease are still needed.
The tripartite motif-containing (TRIM) family is a group of proteins that are characterized by a conserved structure with a RING domain, one or two B-box domains, a predicted coiled coil, and usually a C-terminal domain.5 Several lines of evidence suggest that TRIM proteins have a broad range of functions in regulation of many cellular processes such as cell differentiation, proliferation/cycle, and apoptosis.6 Consistently, the alterations of these proteins are implicated in a plethora of pathological conditions.6,7 Some members are thought to be important regulators of carcinogenesis.8,9 Among these members, TRIM22 has been proved to be involved in various cancers, such as endometrial cancer,10 leukemia,11 non-small cell lung cancer,12 and breast cancer.13 However, the role of TRIM22 in glioma has not been investigated. The present study aimed to evaluate expression pattern of TRIM22 and its function in glioma.
2. Materials and methods
2.1 Patient samples
The clinical samples used in this study are consisted of 21 glioma tissues and matched normal brain tissues were collected from patients with a mean age of 44 ± 5 years. The patients underwent surgical treatment at Huaihe Hospital of Henan University between May 2014 and September 2017. This study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of Huaihe Hospital of Henan University (Kaifeng, China). All patients were informed and have signed the informed consent.
2.2 Cell culture
Normal microglia cell line HEB, and glioma cell lines including A172, LN229, U251, and U87 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were grown in Dulbecco's modified Eagle's medium (DMEM; Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (FBS; Gibco Laboratories, Grand Island, NY, USA) and 1% penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA) at 37 °C in an atmosphere with 5% CO2.
2.3 Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
Total RNA was extracted from the tissues and cells by Trizol reagent (Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA). Then the obtained RNA was used to synthesize cDNA using a cDNA Reverse Transcription Kit (Fermentas-Thermo Fisher Scientific, USA) following the manufacturer's protocol. The mRNA levels of TRIM22 were quantified using the SYBR® Green PCR Master Mix (Thermo) on an ABI-7300 sequence detection system (Applied Biosystems, Foster, CA, USA). The primers used are as follows: TRIM22, forward 5′-GCAC GCTC ATCT CAGA TCTC C-3′ and reverse 5′-TTTT GGCT TTTC AATG TCCA G-3′; GAPDH, forward 5′-AATC CCAT CACC ATCT TC-3′ and reverse 5′-AGGC TGTT GTCA TACT TC-3′. GAPDH was used as a reference gene. The relative mRNA levels of TRIM22 to GAPDH were calculated by the 2−ΔΔCt method.
2.4 Western blot analysis
Total proteins of tissues and cells were prepared using RIPA buffer (Invitrogen). Protein concentrations were determined using the BCA protein assay kit (Thermo). The proteins (30 μg) were separated by 10% SDS-PAGE and then transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA) using semi-dry method. Subsequently, the membranes were blocked for 1 h with 5% non-fat milk at room temperature. After that, the membranes were subjected to incubation with primary antibodies against TRIM22 (Sigma; 1: 1000); E-cadherin, N-cadherin, vimentin (Thermo; 1:1000); β-catenin, cyclin D1, c-Myc or β-actin (Abcam, Cambridge, MA, USA; 1: 800) overnight at 4 °C. The membranes were then incubated with respective horseradish peroxidase (HPR)-conjugated secondary antibodies (Sigma; 1: 3000) at 37 °C for 1 h. Finally, the signals were detected using enhanced chemiluminescence (ECL) reagents (Thermo). The intensity of bands was analyzed using Quantity One Software (Bio-Rad Laboratories, Hercules, CA, USA).
2.5 Sh-TRIM22 transfection
Small hairpin RNAs (shRNA) targeting TRIM22 (sh-TRIM22) and its negative control (sh-control) were purchased from Shanghai Genechem Co., Ltd. (Shanghai, China). The shTRIM22 was constructed into pLKO.1 plasmids (Sigma). The pLKO.1-sh-TRIM22 plasmid or pLKO.1-sh-control was transfected into U87 cells. Then the transfected cells were subjected to 14 days of selection in DMEM medium containing 1.0 μg mL−1 puromycin (Sigma). Finally, the individual puromycin-resistant cells were isolated and collected for further experiments as stable expressing cells.
2.6 Cell proliferation assay
Cell counting-kit-8 (CCK-8) assay was used for the determination of cell proliferation. U87 cells were seeded in 96-well plates at a density of 1 × 104 cells per well. Then 10 μl CCK-8 solution (Dojindo, Kumamoto, Japan) was added to each well at 0 h, 24 h, 48 h, and 72 h, and incubated for 1 h at 37 °C. Subsequently, the absorbance values were determined at 450 nm using a microplate reader (Bio-Rad).
2.7 Cell migration and invasion assays
Cell invasive and migratory abilities were assayed using transwell assays with 8 μm transwell plates (Corning Inc., Corning, NY, USA). Cells (100 μl) suspended in DMEM at a density of 1 × 105 cells per ml were added to the upper chambers coated with matrigel (for invasion assay) or without matrigel (for migration assay). And DMEM containing 10% FBS (600 μl) was added to the lower chambers. After incubation for 24 h, the inserts were removed and fixed with 4% methanol for 15 min, followed by staining with 0.1% crystal violet for 15 min. Images of five random fields were captured under a microscope (Olympus Corporation, Tokyo, Japan). The numbers of the cells were counted, and the average numbers were calculated.
2.8 Nude mouse xenograft model
The animal experiments were approved by the Institutional Animal Care and Use Committee of Huaihe Hospital of Henan University (China). The experiments were performed according to the National Institute of Health guidelines. Twelve female 6 week-old athymic nude mice (HFKBio, Peking, China) were randomly separated into two groups: control group (n = 6) and TRIM22 silencing group (n = 6). The pLKO.1-shTRIM22 plasmid or pLKO.1 plasmid transfected cells (5 × 106 cells per mouse) were subcutaneously inoculated into left right of the dorsal midline. The xenografts were allowed to grow for 4 weeks. The tumor sizes (mm) were measured every seven days using digital caliper. The tumor volumes (v, mm3) were calculated using the following formula: length (mm) × width (mm) × width (mm)/2. After 28 days, all mice were sacrificed with general anesthesia, and the tumors were removed and weighted.
2.9 Statistical analysis
All data were expressed as the means ± SD, and analyzed by SPSS 21.0 (SPSS Inc., Chicago, IL, USA). The differences among groups were analyzed by ANOVA or unpaired Student's t-test. Differences were considered statistically significant when p value < 0.05.
3. Results
3.1 TRIM22 was highly expressed in glioma tissues and cell lines
Compared with the TRIM22 expression levels observed in normal brain tissues, the protein and mRNA levels of TRIM22 were found to be significant abundant in the glioma tissues (Fig. 1A and B). Furthermore, we detected the TRIM22 expression levels in normal microglia cell line HEB, and glioma cell lines including A172, LN229, U251, and U87. As shown in Fig. 1C and D, the protein and mRNA levels of TRIM22 were markedly higher in glioma cell lines than that of in HEB cells. Because the highest expression level of TRIM22 was detected in U87 cell line, thus, the U87 cells were applied in the following studies.
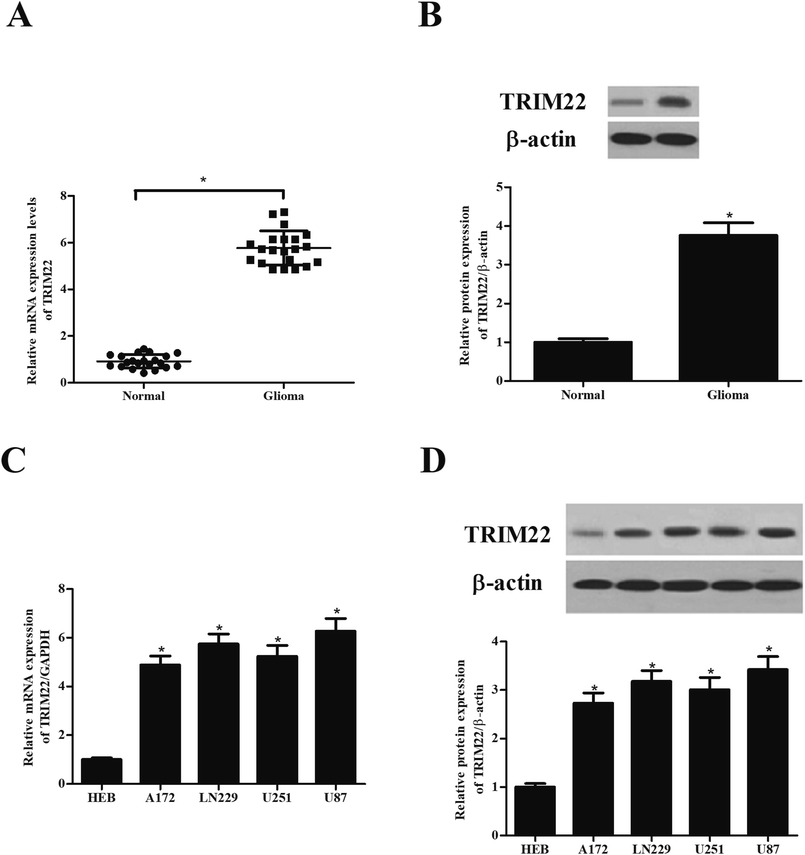 |
| | Fig. 1 TRIM22 was highly expressed in glioma tissues and cell lines. (A and B) The protein and mRNA levels of TRIM22 in glioma tissues and normal brain tissues. *p < 0.05 vs. normal brain tissues. (C and D) The protein and mRNA levels of TRIM22 in normal microglia cell line (HEB) and glioma cell lines (A172, LN229, U118, U251, and U87). n = 3. *p < 0.05 vs. HEB cells. | |
3.2 Silencing of TRIM22 inhibited the proliferation of glioma cells
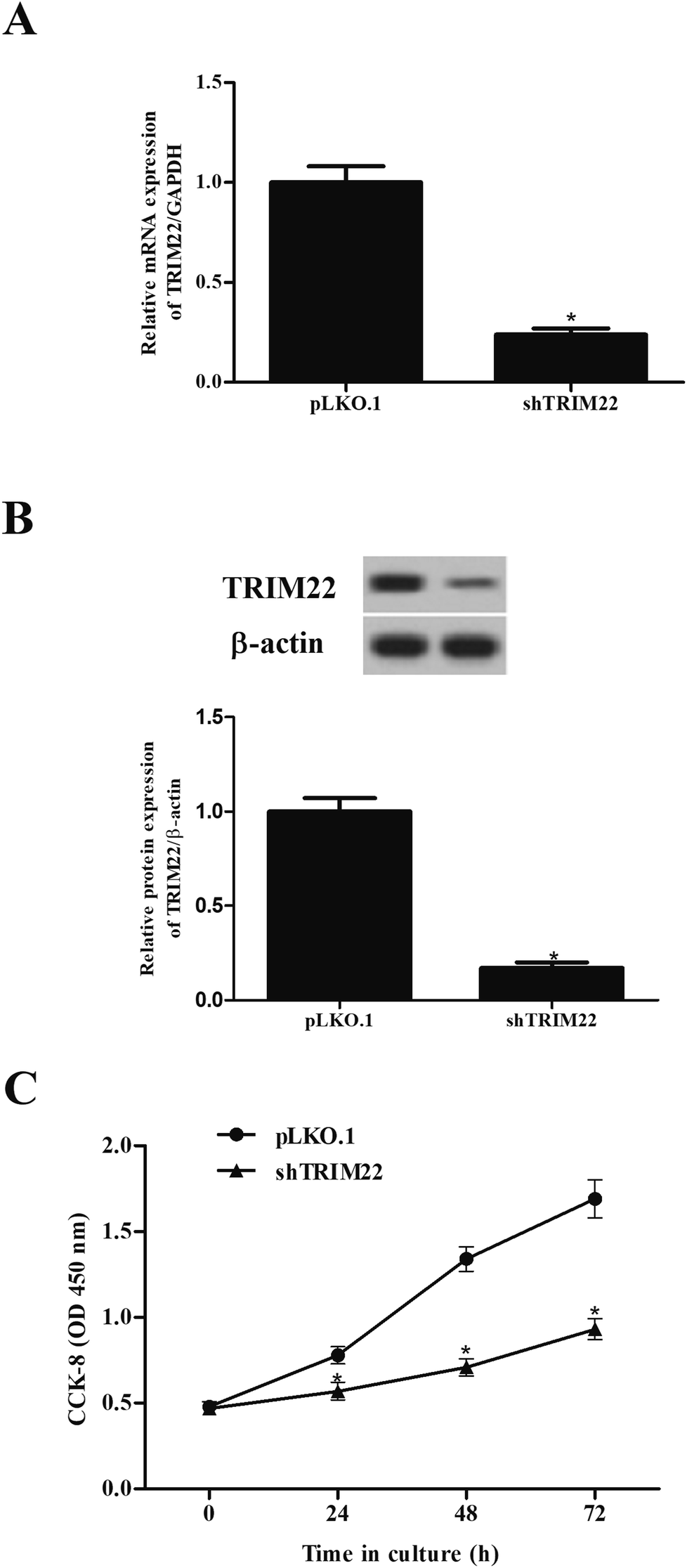
The expressions of TRIM22 in the transfected cells were assessed. The RT-PCR and western blotting showed that TRIM22 expression was significantly reduced in the cells transfected with pLKO.1-shTRIM22 plasmid in both mRNA and protein levels, as compared to the cells transfected with pLKO.1 plasmid (Fig. 2A and B). Next, we examined the cell proliferation using CCK-8 assay. A trend towards a significant decrease in cell proliferation was detected in cells transfected with pLKO.1-shTRIM22 plasmid when compared with the cells transfected with pLKO.1 plasmid (Fig. 2C).
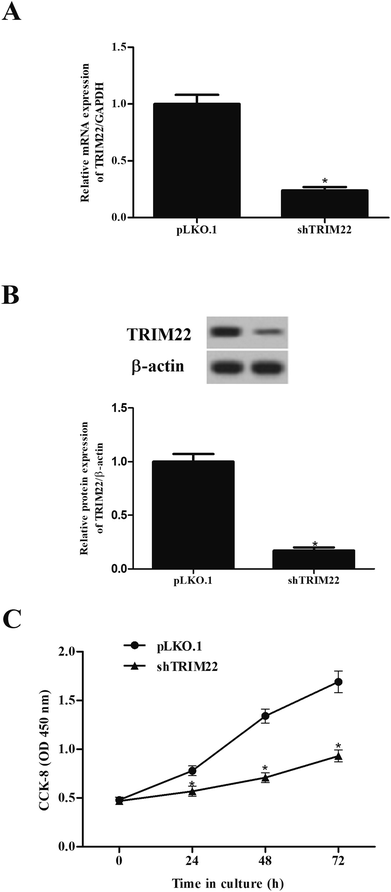 |
| | Fig. 2 Silencing of TRIM22 inhibited the cell proliferation of U87 cells. U118 cells were transfected with pLKO.1-shTRIM22 plasmid or pLKO.1 plasmid. The expressions of TRIM22 in the transfected cells were assessed by RT-PCR (A) and western blotting (B). (C) The cell proliferation was measured using CCK-8 assay. n = 3. *p < 0.05 vs. pLKO.1 plasmid transfected cells. | |
3.3 Silencing of TRIM22 inhibited the migration and invasion of glioma cells
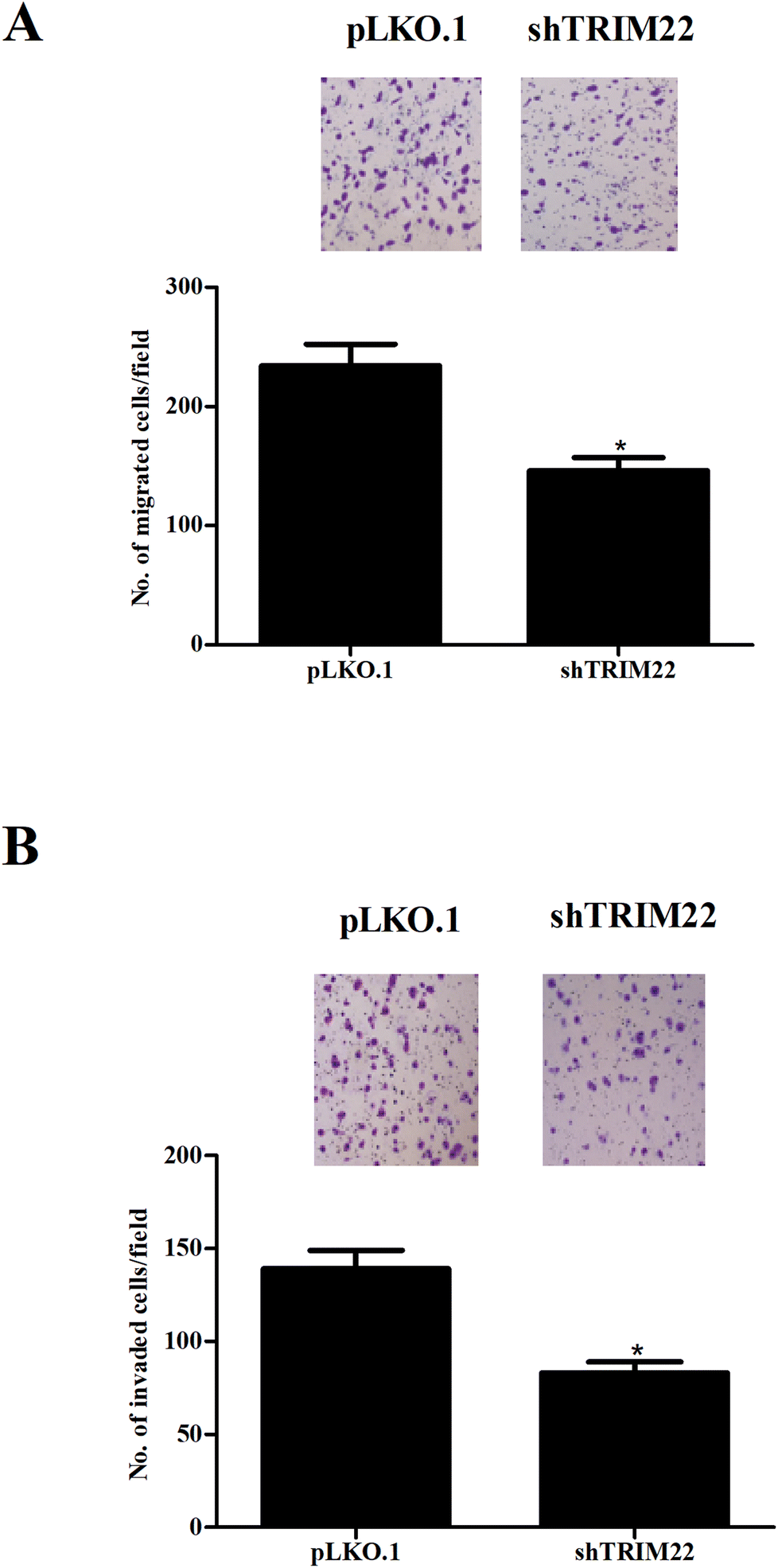
Transwell assays were performed to evaluate the migration and invasion of glioma cells. Fig. 3A showed that the migration was markedly decreased after pLKO.1-shTRIM22 transfection. Fig. 3B showed that pLKO.1-shTRIM22 transfection also led to a reduction in invasion ability of U87 cells. The results indicated that silencing of TRIM22 dramatically inhibited migration and invasion of U87 cells.
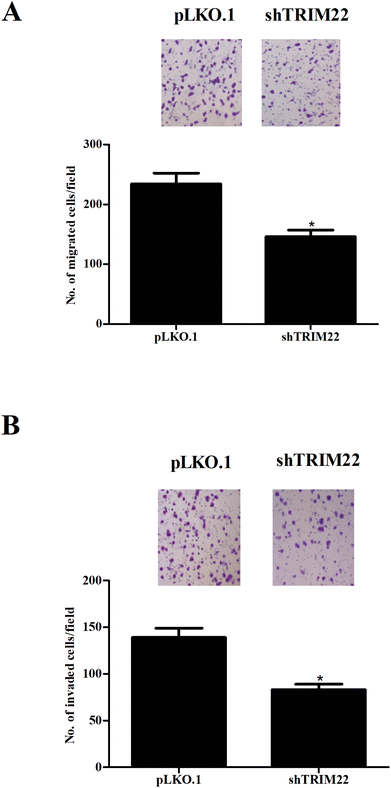 |
| | Fig. 3 Silencing of TRIM22 inhibited the migration and invasion of U87 cells. Transwell assays were performed to evaluate the migration (A) and invasion (B) of U118 cells with 8 μm transwell plates. The migration and invasion were reflected by the numbers of transferred cells. n = 3. *p < 0.05 vs. pLKO.1 plasmid transfected cells. | |
3.4 Silencing of TRIM22 inhibited the epithelial–mesenchymal transition (EMT) in glioma cells
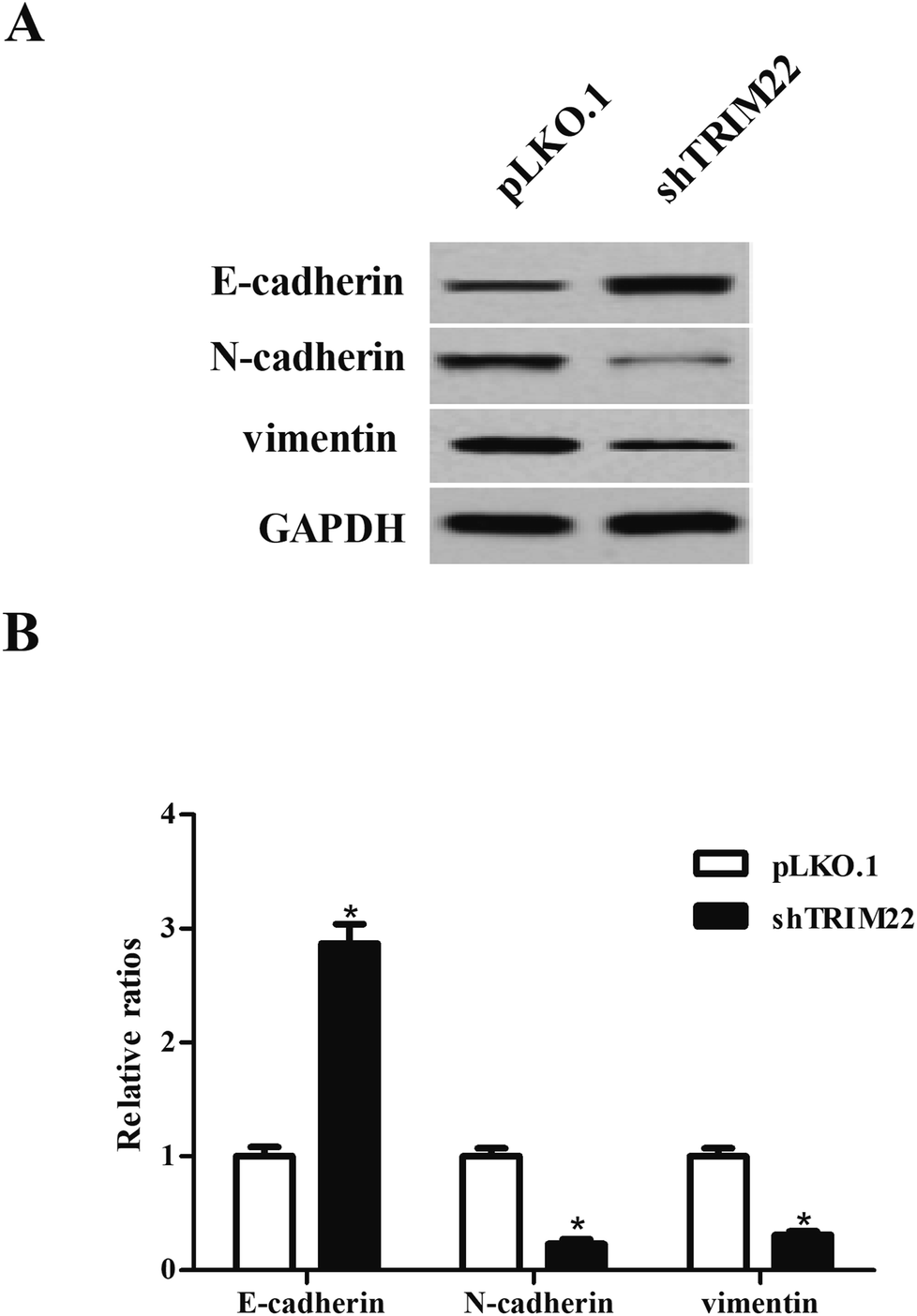
EMT is a critical process that occurs in the initiation of metastasis and progression of malignant tumors.14 The biomarkers of EMT including E-cadherin, N-cadherin and vimentin were measured by western blot. As shown in Fig. 4, there were obvious higher expressions of E-cadherin as well as lower expressions of N-cadherin and vimentin in pLKO.1-shTRIM22 transfected cells than those in pLKO.1 transfected cells.
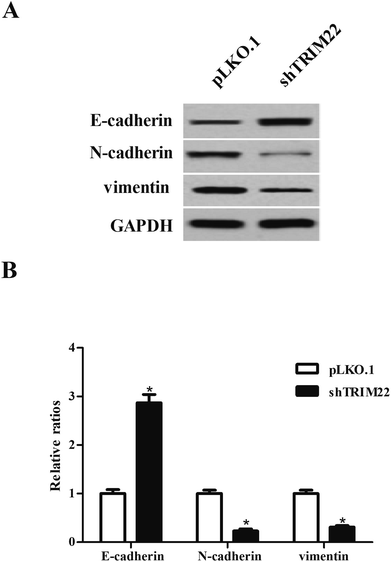 |
| | Fig. 4 Silencing of TRIM22 inhibited the EMT process in U87 cells. (A) The biomarkers of EMT including E-cadherin, N-cadherin, vimentin were measured by western blot to evaluate the process of EMT. (B) Quantification analysis was performed using Gel-Pro Analyzer version 4.0 software. n = 3. *p < 0.05 vs. pLKO.1 plasmid transfected cells. | |
3.5 Silencing of TRIM22 attenuated xenograft tumor growth in nude mice
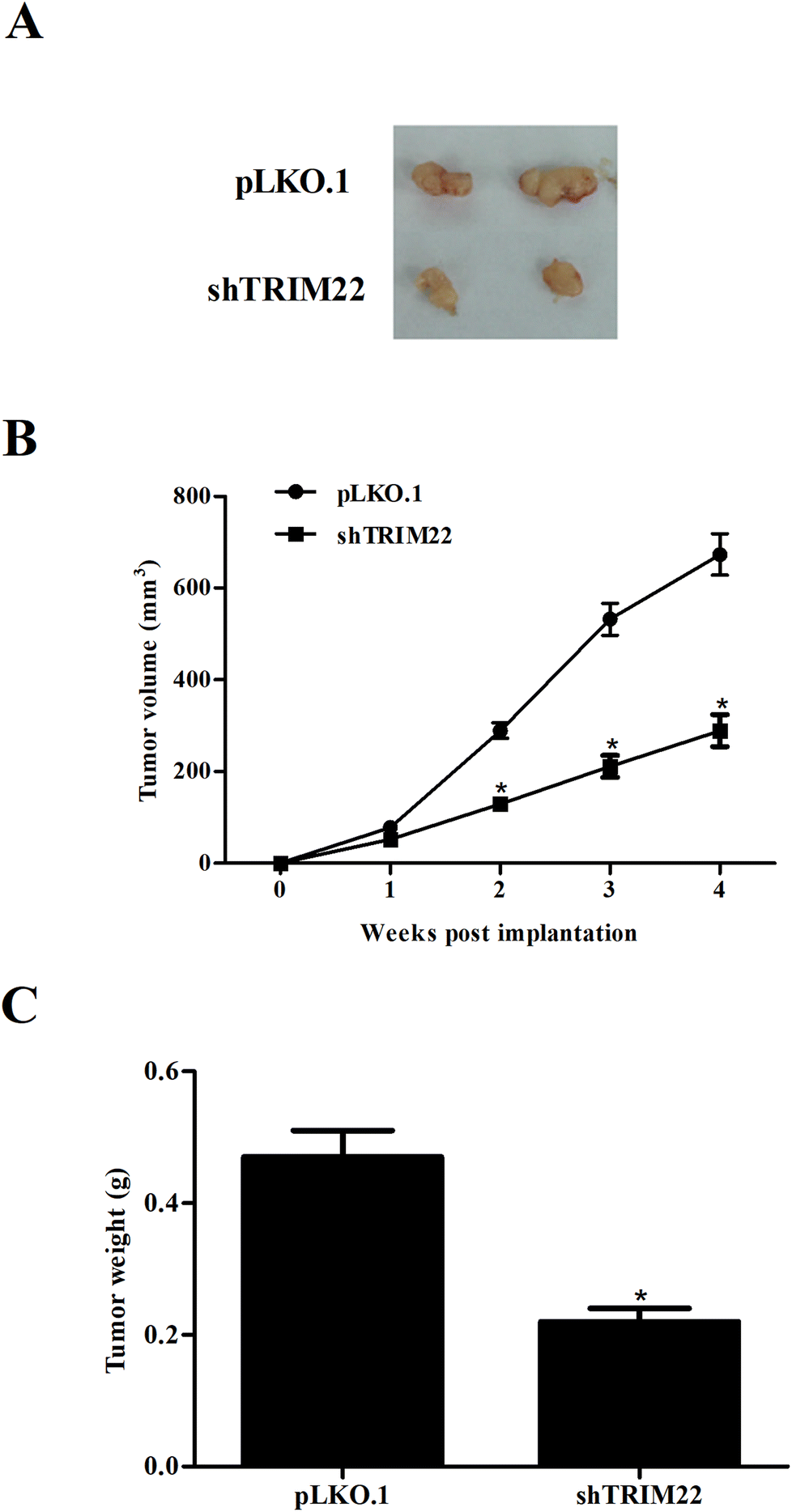
Next, we established xenograft model to evaluate the effect of TRIM22 silencing on tumor growth in vivo. As illustrated in Fig. 5A, the subcutaneous tumors in control group were dramatically larger than that in TRIM22 silencing group. Fig. 5B revealed that the growth rate of xenograft tumors was obviously reduced in the TRIM22 silencing group compared with control group. Furthermore, compared with control group, a decrease in tumor weight was observed in TRIM22 silencing group (Fig. 5C).
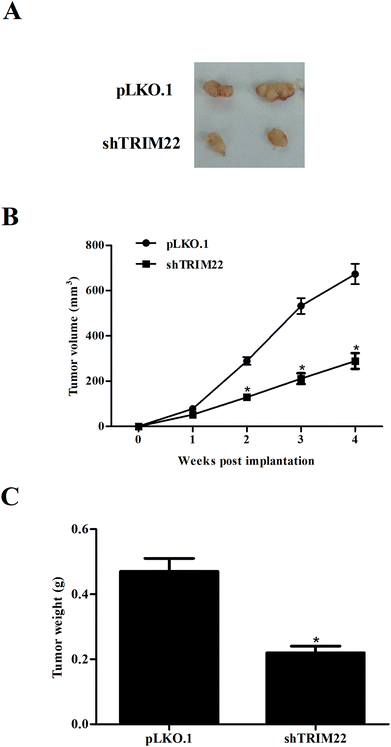 |
| | Fig. 5 Silencing of TRIM22 attenuated xenograft tumor growth in nude mice. Xenograft model was established in nude mice to evaluate the effect of TRIM22 silencing on tumor growth in vivo. The tumor sizes (mm) were measured every 7 days from the tenth day post injection, and the tumor volumes (v, mm3) were calculated using the following formula: length (mm) × width (mm) × width (mm)/2. The tumors were removed and weighted at the last period of the experiments. (A) Tumor volume in each group. (B) Growth rate of xenograft tumors in each group. (C) Tumor weight in each group. n = 6. *p < 0.05 vs. control group (mice injected with pLKO.1 plasmid transfected cells). | |
3.6 Silencing of TRIM22 inhibited the activation of Wnt/β-catenin pathway in glioma cells
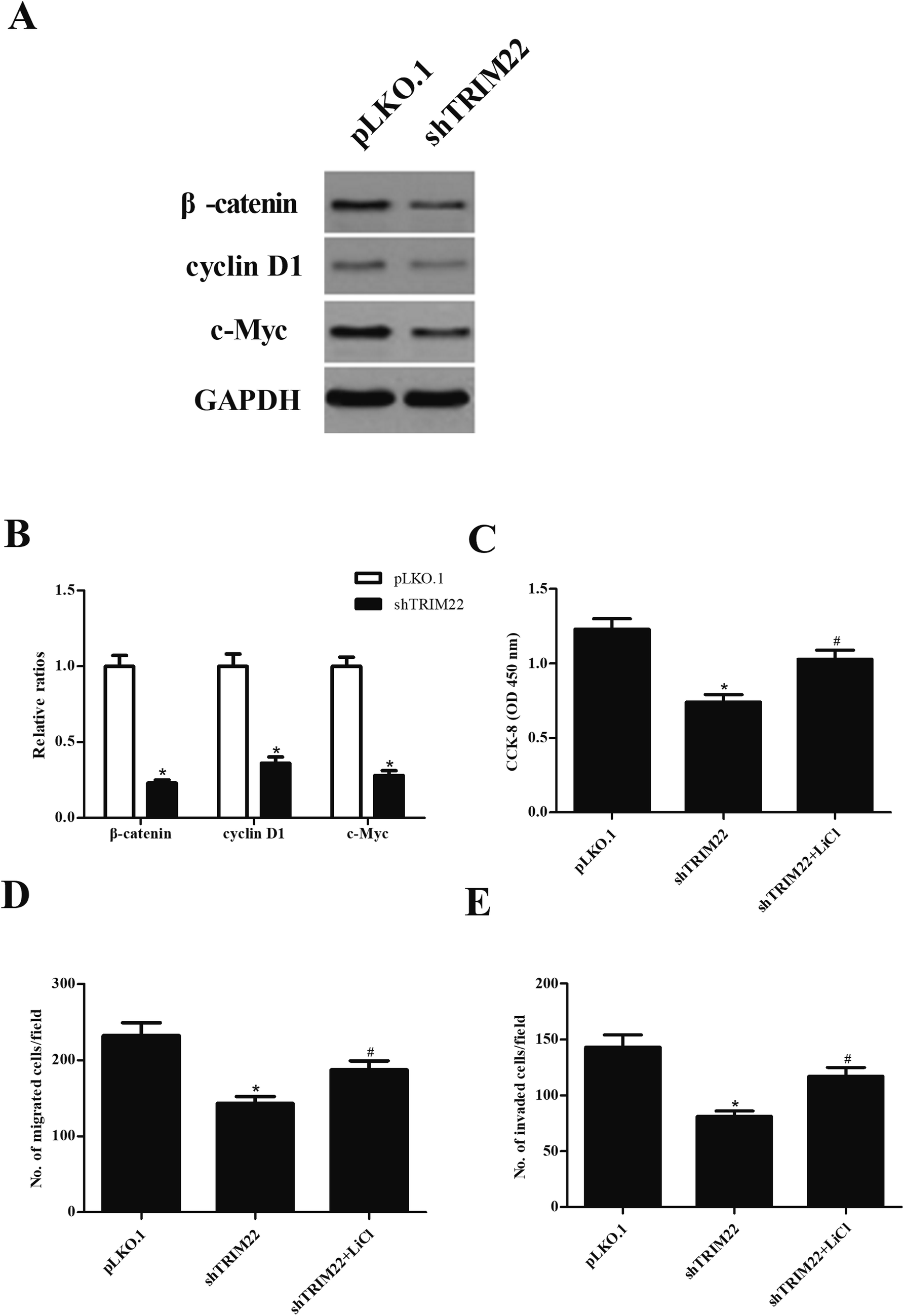
Wnt/β-catenin pathway has been demonstrated to play an important role in caner development.15 To investigate whether the Wnt/β-catenin pathway was involved in the effect of TRIM22 in glioma cells, the expressions of β-catenin, cyclin D1, and c-Myc were analyzed by western blot. As indicated in Fig. 6A, shTRIM22 resulted in significant decrease in expressions of β-catenin, cyclin D1, and c-Myc in U87 cells. Additionally, we applied the LiCl for the activation of Wnt/β-catenin pathway. Fig. 6C–E showed that LiCl attenuated the inhibitory effects of shTRIM22 on cell proliferation, migration, and invasion in U87 cells.
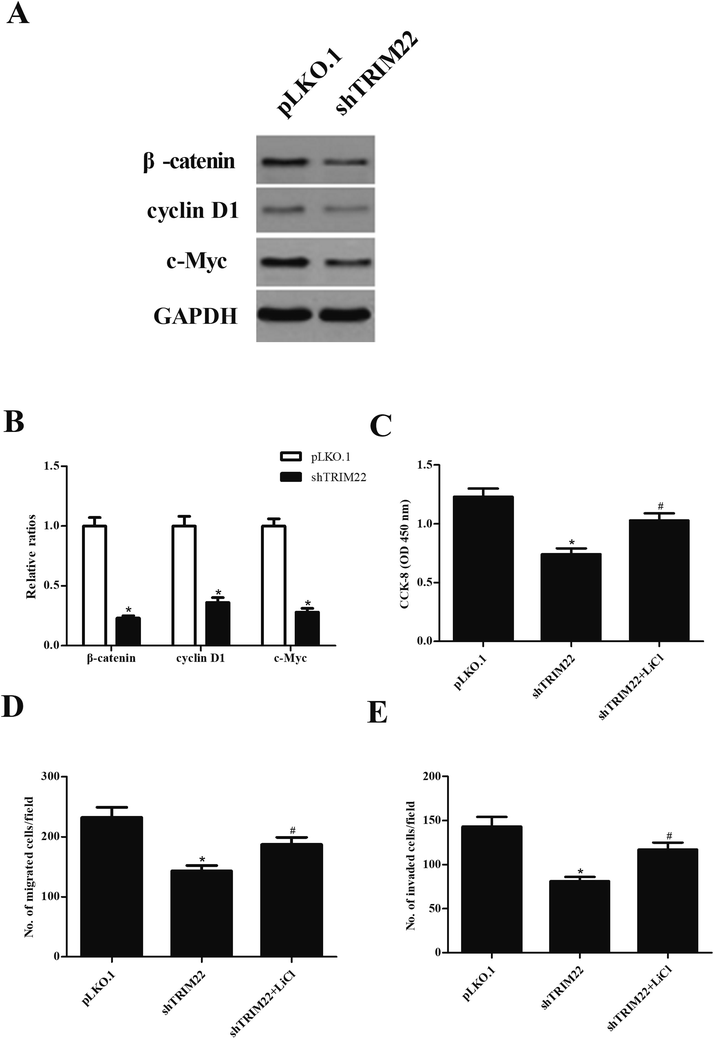 |
| | Fig. 6 Silencing of TRIM22 inhibited the activation of Wnt/β-catenin pathway in U87 cells. (A) The expressions of β-catenin, cyclin D1, and c-Myc were analyzed by western blot. To further investigate the role of Wnt/β-catenin pathway in the effect of TRIM22, the cells were treated with LiCl, which is an activator of Wnt/β-catenin pathway. (B) Quantification analysis of β-catenin, cyclin D1 and c-Myc. (C) Effect of LiCl on cell proliferation. (D) Effect of LiCl on cell migration. (E) Effect of LiCl on cell invasion. n = 3. *p < 0.05 vs. cells transfected with pLKO.1, #p < 0.05 vs. cells transfected with pLKO.1-shTRIM22. | |
4. Discussion
Despite significant advances are achieved in diagnostics and therapeutics of glioma over the past decades, the therapies for patients with high-grade glioma are inadequate. In recent decades, targeted therapy, a form of molecular medicine, has attracted many attentions for the treatment of cancers with more effective and less harmful properties.16 TRIM proteins are a large family that consists of more than 80 members, and most of the members have E3 ubiquitin ligase activity.17 Since the ubiquitin system is responsible for degradation of target protein and plays crucial roles in multiple cellular processes, therefore, TRIM proteins have various functions.18 Accumulating studies have shown that TRIM proteins have unique and important roles in several dysregulation diseases such as immunological diseases, developmental disorders, and cancers.19 Recently, pathological analyses prove that aberrant expressions of TRIM proteins are intensely correlated with the malignancy of cancers and prognosis.19 To date, many TRIM proteins have been shown to play critical roles in EMT, transformation, and metastasis, which are considered as the most important factors in the cancers development.6,9
TRIM22, also known as Staf50, has been identified as an interferon (IFN)-inducible protein.20 It was reported that TRIM22 could suppress hepatitis C virus replication in Con1b and Huh7.5.1 cells.21 TRIM22 has been found to be a direct target gene p53, which is a tumor suppressor gene that mediates cell cycle arrest and apoptosis.22,23 TRIM22 is up-regulated in response to p53, and inhibits clonogenic growth of leukemic U-937 cells.22 TRIM22 has been proven to be associated with proliferation and differentiation of leukemic U-937 cells, indicating that TRIM22 may function as a tumor suppressor.22 However, Li et al.11 reported that TRIM22 knockdown inhibits cell proliferation, and induces cell cycle arrest and cell apoptosis of chronic myeloid leukemia through regulation of PI3K/Akt/mTOR pathway. In addition, TRIM22 was found to be up-regulated in 55.6% cases of non-small cell lung cancer (NSCLC) tissues.12 TRIM22 overexpression is correlated with advanced TNM stage, positive nodal metastasis and poor prognosis in patients with NSCLC, and induces proliferation, colony formation, invasion and EMT in A549 cells.12 These findings suggest that TRIM22 serves as an oncogene in NSCLC. Consequently, the role of TRIM22 in cancer is controversial. These dual roles of TRIM22 could attribute to organ-specific actions and different cellular contexts. Consistent with the data of TRIM22 in NSCLC, the present study showed that TRIM22 was up-regulated in glioma tissues and cell lines. TRIM22 knockdown inhibited cell proliferation, migration, invasion, and EMT in U118 cells, as well as attenuated tumor growth in vivo. Our results suggested that TRIM22 might function as an oncogene in glioma. Therefore, it is important to developing inhibitory agents of TRIM22 for their use as therapeutic tools for glioma treatment.
Wnt signaling is an important intracellular signal transduction pathway that is implicated in various cellular functions.24 Wnt signaling is classified as canonical (β-catenin dependent) and the non-canonical (β-catenin independent) pathway.24 In canonical pathway, Wnt signaling can be activated by binding of Wnt proteins to surface receptors, which leads to the inhibition of phosphorylation and degradation of β-catenin. The stabilized β-catenin then translocates into the nucleus and regulates the transcription of target genes such as c-Myc and cyclin D1.24 It has recently been proven that Wnt/β-catenin pathway exerts oncogenic activities in regulating proliferation, apoptosis, invasion and EMT in glioma cells.25 Therefore, inhibition of the Wnt/β-catenin pathway activation might be a new therapeutic approach for glioma. In the present study, our results showed that silencing of TRIM22 inhibited the activation of Wnt/β-catenin pathway. In addition, LiCl treatment attenuated the effects of TRIM22 on U87 cells. These findings suggested that TRIM22 executed its effects, at least in part, through regulating Wnt/β-catenin pathway. Several other pathways are known to contribute to glioma cell invasiveness and motility, such as PI3K/AKT, JAK/STAT3 and NF-κB signaling pathway,26–28 thus, more studies are needed to clarify the role of TRIM22 in these pathways.
5. Conclusion
In summary, the present study demonstrated that TRIM22 was significantly up-regulated in glioma tissues and cell lines. In vitro studies demonstrated that TRIM22 knockdown inhibited cell proliferation, migration, invasion, and EMT in U118 cells. Nude mouse xenograft assay denoted that TRIM22 silencing inhibited tumor growth in vivo. Furthermore, TRIM22 executed its effects through regulating the Wnt/β-catenin pathway. Our results suggested that TRIM22 might function as an oncogene in glioma.
Conflicts of interest
The authors have no competing interests to disclose.
Acknowledgements
None.
References
- J. S. Rao, Nat. Rev. Cancer, 2003, 3, 489–501 CrossRef PubMed.
- M. L. Goodenberger and R. B. Jenkins, Cancer Genet., 2012, 205, 613–621 CrossRef PubMed.
- T. W. Rogers, G. Toor, K. Drummond, C. Love, K. Field, R. Asher, A. Tsui, M. Buckland and M. Gonzales, J. Neuro-Oncol., 2018, 137, 181–189 CrossRef PubMed.
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer, R. O. Mirimanoff, European Organisation for T. Treatment of Cancer Brain Radiotherapy and National Cancer Institute of Canada Clinical Trials, N. Engl. J. Med., 2005, 352, 987–996 CrossRef PubMed.
- S. Nisole, J. P. Stoye and A. Saib, Nat. Rev. Microbiol., 2005, 3, 799–808 CrossRef PubMed.
- S. Hatakeyama, Trends Biochem. Sci., 2017, 42, 297–311 CrossRef PubMed.
- G. Meroni, Adv. Exp. Med. Biol., 2012, 770, 1–9 Search PubMed.
- V. Cambiaghi, V. Giuliani, S. Lombardi, C. Marinelli, F. Toffalorio and P. G. Pelicci, Adv. Exp. Med. Biol., 2012, 770, 77–91 CrossRef PubMed.
- S. Hatakeyama, Nat. Rev. Cancer, 2011, 11, 792–804 CrossRef PubMed.
- M. Saito-Kanatani, T. Urano, H. Hiroi, M. Momoeda, M. Ito, T. Fujii and S. Inoue, J. Steroid Biochem. Mol. Biol., 2015, 154, 217–225 CrossRef PubMed.
- L. Li, Y. Qi, X. Ma, G. Xiong, L. Wang and C. Bao, Cell Biol. Int., 2018, 42, 1192–1199 CrossRef PubMed.
- L. Liu, X. M. Zhou, F. F. Yang, Y. Miao, Y. Yin, X. J. Hu, G. Hou, Q. Y. Wang and J. Kang, Oncotarget, 2017, 8, 62069–62080 Search PubMed.
- Y. Sun, G. H. Ho, H. N. Koong, G. Sivaramakrishnan, W. T. Ang, Q. M. Koh and V. C. Lin, Biochem. Biophys. Res. Commun., 2013, 441, 600–606 CrossRef PubMed.
- S. Lamouille, J. Xu and R. Derynck, Nat. Rev. Mol. Cell Biol., 2014, 15, 178–196 CrossRef PubMed.
- R. C. Arend, A. I. Londono-Joshi, J. M. Straughn Jr and D. J. Buchsbaum, Gynecol. Oncol., 2013, 131, 772–779 CrossRef PubMed.
- A. M. Tsimberidou, Cancer Chemother. Pharmacol., 2015, 76, 1113–1132 CrossRef PubMed.
- J. M. Doyle, J. Gao, J. Wang, M. Yang and P. R. Potts, Mol. Cell, 2010, 39, 963–974 CrossRef PubMed.
- A. Ciechanover, Nat. Rev. Mol. Cell Biol., 2015, 16, 322–324 CrossRef PubMed.
- M. Watanabe and S. Hatakeyama, J. Biochem., 2017, 161, 135–144 Search PubMed.
- R. Singh, G. Gaiha, L. Werner, K. McKim, K. Mlisana, J. Luban, B. D. Walker, S. S. Karim, A. L. Brass, T. Ndung'u and C.A.I.S. Team, J. Virol., 2011, 85, 208–216 CrossRef PubMed.
- H. Tian and Z. K. He, Yonsei Med. J., 2018, 59, 511–518 CrossRef PubMed.
- S. Obad, H. Brunnstrom, J. Vallon-Christersson, A. Borg, K. Drott and U. Gullberg, Oncogene, 2004, 23, 4050–4059 CrossRef PubMed.
- C. F. Labuschagne, F. Zani and K. H. Vousden, Biochim. Biophys. Acta, 2018 Search PubMed , S0304-419X(17)30222-30226.
- N. Krishnamurthy and R. Kurzrock, Cancer Treat. Rev., 2018, 62, 50–60 CrossRef PubMed.
- K. Zhang, J. Zhang, L. Han, P. Pu and C. Kang, J. Neuroimmune Pharmacol., 2012, 7, 740–749 CrossRef PubMed.
- F. Zhang, Y. Huang, B. Wang, C. Zhong, X. Liu and S. Ding, NeuroReport, 2018, 29, 718–722 CrossRef PubMed.
- C. Song, B. Fan and Z. Xiao, Biomed. Pharmacother., 2018, 98, 440–445 CrossRef PubMed.
- X. Xu, X. Wan and X. Wei, Mol. Med. Rep., 2017, 15, 963–968 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a