
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of polytetrafluoroethylene nanofibrous membranes for guided bone regeneration
Jin-Young Parka,
Jung-Hee Leea,
Chun-Ho Kimb and
Young-Jin Kim *a
*a
aDepartment of Biomedical Engineering, Daegu Catholic University, Gyeongsan 38430, Republic of Korea. E-mail: yjkim@cu.ac.kr; Fax: +82-53-359-6752; Tel: +82-53-850-2512
bLaboratory of Tissue Engineering, Korea Institute of Radiological and Medical Sciences, Seoul 01812, Republic of Korea
First published on 8th October 2018
Abstract
In this study, we first prepared the precursor polytetrafluoroethylene (PTFE)/poly(ethylene oxide) (PEO) nanofibrous membranes by electrospinning with different PTFE/PEO weight ratios. These membranes exhibited three-dimensional interconnected pore structures. The average diameter of the precursor nanofibres decreased with increased PTFE contents from 633 ± 34 nm (PTFE/PEO weight ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to 555 ± 63 nm (PTFE/PEO weight ratio of 7:1) because of the decrease in solution viscosity. Then, the precursor membranes were sintered with different temperatures to obtain the PTFE nanofibrous membranes, resulting in the average diameter of the nanofibres increasing from 633 ± 34 nm to 947 ± 78 nm with the increase in sintering temperature; consequently, the membrane became more compact. This compaction caused a decrease in porosity from 76.5 ± 2.9% to 69.1 ± 2.6% and an increase in water contact angle from 94.1 ± 4.2° to 143.3 ± 3.5°. In addition, the mechanical properties of the PTFE nanofibrous membranes increased with increasing sintering temperature. Cytocompatibility test results revealed that the PTFE350 membrane, which was sintered at 350 °C, promoted the proliferation and differentiation of MC3T3-E1 cells more rapidly than other membrane types. These results suggested that the PTFE nanofibrous membranes could be ideal biomaterials in tissue engineering for bone regeneration.
:1) to 555 ± 63 nm (PTFE/PEO weight ratio of 7:1) because of the decrease in solution viscosity. Then, the precursor membranes were sintered with different temperatures to obtain the PTFE nanofibrous membranes, resulting in the average diameter of the nanofibres increasing from 633 ± 34 nm to 947 ± 78 nm with the increase in sintering temperature; consequently, the membrane became more compact. This compaction caused a decrease in porosity from 76.5 ± 2.9% to 69.1 ± 2.6% and an increase in water contact angle from 94.1 ± 4.2° to 143.3 ± 3.5°. In addition, the mechanical properties of the PTFE nanofibrous membranes increased with increasing sintering temperature. Cytocompatibility test results revealed that the PTFE350 membrane, which was sintered at 350 °C, promoted the proliferation and differentiation of MC3T3-E1 cells more rapidly than other membrane types. These results suggested that the PTFE nanofibrous membranes could be ideal biomaterials in tissue engineering for bone regeneration.
1 Introduction
Guided bone regeneration (GBR) is a common therapy used for the treatment of lesions in the alveolar bone or the mandible caused by infections or trauma. The principle underlying the GBR treatment is the use of a membrane to create a secluded space in the defect site and prevent faster-growing connective tissue from migration into the defect site which favours the proliferation of bone-forming cells, resulting in new bone formation. Therefore, this membrane is expected to have a three-dimensional (3D) structure which can be structurally and mechanically equivalent to the neighbouring tissue.1 It should also have suitable biocompatibility to support cell growth and tissue reconstruction.2 Moreover, the membrane should have an interconnecting porous microarchitecture, which can act as a physical flow path to enable efficient transport of nutrients, oxygen and metabolic waste in the whole region of the membrane.1To fabricate biomedical membranes with desirable properties, various methods such as phase separation, solid freeform fabrication and electrospinning have been used.1,3,4 Among those methods, electrospinning is an attractive method owing to its simplicity, versatility, and ability to produce a variety of polymer nanofibres. Electrospun polymer nanofibres have unique advantages in biomedical fields by providing porous structures which mimic the features of extracellular matrix; thus, they have been widely used as drug carriers and tissue engineering scaffolds.3,5,6
Polytetrafluoroethylene (PTFE) has many remarkable characteristics that include outstanding thermal and chemical stability, high fracture toughness, low surface friction and biocompatibility.7 These features are due to the low surface energy and the strong carbon–carbon and carbon–fluorine bonds as well as the high degree of crystallinity, which in turn make PTFE an excellent candidate in medical applications such as a vascular graft and GBR membrane.8–10 Particularly, the porous PTFE membranes have shown good biocompatibility and excellent support of the structural integrity of the defect sites during healing, leading to positive results in experimental studies and clinical GBR procedures.8,9
The porous PTFE membranes have been prepared through many procedures including blending, jet-blowing and thermo-mechanical stretching.7,11,12 The extent of PTFE membrane processing was limited due to the lack of common solvents and high melt viscosity. Nonetheless, PTFE membranes are usually manufactured by thermo-mechanical stretching.7 However, this method includes processing steps of PTFE mixing with much lubricant, billet formation and extrusion, resulting in considerable environmental pollution by lubricant.7,13 Therefore, in this study, we offered an alternative route that would be more environmentally friendly. PTFE nanofibrous membranes were fabricated by sintering the previously electrospun PTFE/poly(ethylene oxide) (PEO) nanofibrous membranes. We systematically examined the effects of PTFE/PEO weight ratios and the sintering temperatures on the structure and physicochemical properties of the PTFE nanofibrous membranes obtained. In addition, the cytocompatibility of the resulting PTFE membranes was evaluated through assays of the cell proliferation and differentiation and the bone mineralisation.
2 Materials and methods
2.1 Materials
Polytetrafluoroethylene (PTFE, 60 wt% dispersion in water), poly(ethylene oxide) (PEO, Mv = 600000 g mol−1), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), alizarin red S (ARS), cetylpyridinium chloride and lipopolysaccharide (LPS) were purchased from Sigma-Aldrich Co. and were used without further purification. The mouse calvaria preosteoblast cell line (MC3T3-E1) was obtained from the American Type Culture Collection (ATCC, USA). Alpha minimum essential medium (α-MEM), Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin and Dulbecco's phosphate-buffered saline (DPBS, pH 7.4) were obtained from Gibco BRL (USA). The QuantiChrom™ alkaline phosphatase (ALP) assay kit (DALP-250) was purchased from BioAssay Systems (USA). Mouse osteocalcin enzyme-linked immunosorbent assay (ELISA) kits were purchased from MyBioSource (USA). The other reagents and solvents were obtained from commercial sources and were used as received.
2.2 Fabrication of PTFE nanofibrous membranes
The electrospinning setup utilised in this study consisted of a syringe and needle (ID = 0.41 mm), a ground electrode, and a high voltage supply (Chungpa EMT Co., Korea). The needle was connected to the high voltage supply, which could generate positive DC voltages up to 40 kV. Before electrospinning PTFE/PEO precursor membranes, a mixture of aqueous PTFE dispersion and PEO was dissolved in deionised water to obtain a concentration of 20 wt% solution, in which PEO was used as a binder for improving electrospinnability. We varied the weight ratios of PTFE and PEO as 5:1, 6:1 and 7:1. This solution was then electrospun under the conditions of a 1.5 mL h−1 feed rate, 17 kV voltage and 12 cm working distance. All experiments were carried out at room temperature and below 60% RH. After electrospinning, the precursor membranes were dried at 40 °C for 12 h and subsequently sintered at 200 °C or 350 °C for 30 min at a heating rate of 2 °C min−1 to obtain the different membranes. The sintering process was conducted to remove PEO for fabricating pure PTFE nanofibrous membranes.
2.3 Characterisation of PTFE nanofibrous membranes
Field-emission scanning electronic microscopy (FE-SEM, JSM-6335F, JEOL, Japan) images were recorded at an acceleration voltage of 5 kV to examine the morphologies of the PTFE nanofibrous membranes. Before SEM observation, all samples were coated with gold by low-vacuum sputter coating for 2 min. The average diameters of the nanofibres were determined by analysing the SEM images with Image-Pro Plus (Media Cybernetics Inc., USA). An ALPHA spectrometer (Bruker Optics, USA) was used to record the attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectra at wavenumbers ranging from 400 cm−1 to 4000 cm−1 with a spectral resolution of 4 cm−1. X-ray diffraction (XRD) patterns were recorded using a Panalytical X'Pert Pro X-ray diffractometer (The Netherlands) equipped with Cu Kα radiation operated at 40 kV and 30 mA to examine the crystalline phases of the PTFE nanofibrous membranes. The samples were scanned at a 2θ range of 10° to 50° at a rate of 2° min−1. The surface chemical compositions of the samples were analysed by X-ray photoelectron spectroscopy (XPS) measurements acquired with a Quantera SXM (ULVAC-PHI Inc., Japan). The incident X-ray used was monochromated Al Kα radiation (1486 eV). Survey scans were recorded at a take-off angle of 45° relative to the sample surface. A contact angle meter (DSA 100, KRÜSS, Germany) was used to measure the contact angle between the water and the external surface of the membrane so as to evaluate the membrane hydrophobicity. To minimise experimental error, the contact angles of each sample were measured five times and then averaged. A mercury porosimeter (AutoPore IV 9520, Micrometrics Instrument Corp., USA) was used to measure the porosity of the membranes. The porosity was defined as the volume of the pores divided by the total volume of the porous membrane. The thermal stability of the membrane was evaluated by thermogravimetric analysis (TGA, Q500, TA Instruments, USA). After drying all the samples under vacuum at 40 °C for 24 h, TGA measurement was carried out under a nitrogen atmosphere at a heating rate of 10 °C min−1 from 30 °C to 800 °C. The mechanical strengths of the membranes were assessed with a universal testing system (TO-101, Testone Co., Republic of Korea) using a force load cell of 10 kN capacity. Strip-shaped specimens (5 cm × 1 cm) were tested 5 times for each sample at a loading speed of 1.0 mm min−1.2.4 Degradation of PTFE nanofibrous membranes
The PTFE nanofibrous membranes were cut into a size of 3 cm × 3 cm and immersed into DPBS at 37 °C for 3 days to examine their degradation behaviour. This experimental condition was selected to simulate the membrane in a real physiological condition for a biomedical application such as GBR. After 3 days, the samples were washed and subsequently dried in a vacuum oven at room temperature for 24 h. The morphological changes of the membranes were observed using SEM.2.5 Cell proliferation assay
A cell viability assay of the MC3T3-E1 cells was employed to examine the in vitro cytotoxicity of the PTFE nanofibrous membranes. All the cultures were prepared in α-MEM supplemented with 10% of FBS and 5% of penicillin–streptomycin. The cells were incubated at 37 °C under humidified 5% CO2. When the cells reached 80% confluence, they were harvested using 0.25% trypsin–EDTA and seeded into a new tissue culture plate for subculture.Cell proliferation on the PTFE nanofibrous membranes was investigated using the MTT assay. Prior to cell seeding, the membranes were sterilised with 70% ethanol and UV radiation for 3 h and then rinsed each membrane 5 times with DPBS and culture medium. Further, the MC3T3-E1 cells (5 × 104 cells per well) were seeded onto the sterilized membranes in a 24 well tissue culture plate and cultured for 1, 3, 5 and 7 days at 37 °C. To evaluate the cell proliferation, 0.2 mL of the MTT solution (5 mg mL−1 in DPBS) was added to the cultured cells, followed by further incubation at 37 °C for 4 h. After removing the remaining medium, 1 mL of DMSO was added into each well to solubilise the precipitate. Next, 0.2 mL of the obtained supernatant was transferred into a 96 well microplate, and the optical density, which is proportional to the number of viable cells, was measured at 570 nm using a microplate reader (OPSYS-MR, Dynex Technology Inc., USA). To confirm the cell viability on the PTFE nanofibrous membranes, the interactions between the MC3T3-E1 cells and the membranes were observed using SEM. The cultured cells for 1, 3, 5 and 7 days were gently rinsed with DPBS and fixed in 4% glutaraldehyde for 1 h at room temperature. After washing with DPBS, the cells were dehydrated using a graded series of ethanol (25%, 50%, 70% and 100%). The samples were subjected to critical desiccation, followed by coating with gold for SEM observations.
2.6 Cell differentiation and bone mineralisation assay
As an early indicator of osteoblastic differentiation of the MC3T3-E1 cells during culturing on the PTFE nanofibrous membranes, ALP activity was determined using ALP assay kits. After culturing for 3, 5, 7 and 14 days in α-MEM supplemented with 10% of FBS, the cell layer was harvested and treated with 0.2% Triton X-100 cell lysis medium. The cell lysates were then centrifuged at 5000 rpm for 10 min at 4 °C. The supernatant was checked for ALP activity using p-nitrophenyl phosphate as the substrate. The p-nitrophenol produced in the presence of ALP was quantified from its absorbance at 405 nm. Osteocalcin, a non-collagenous protein produced by mature osteoblasts during late-stage osteoblastic differentiation, was determined in the media using osteocalcin ELISA kits on days 3, 5, 7 and 14. The absorbance related to the osteocalcin produced at 450 nm was measured using a microplate reader.Calcium mineralisation was determined by ARS staining of the MC3T3-E1 cells in a 24 well tissue culture plate. ARS is a dye that selectively binds to the calcium salts and thus usually used for quantitative and qualitative detection of calcium mineral deposition. The cells were cultured for 3, 5, 7 and 14 days and then washed thrice with DPBS, before fixing them in 70% cold ethanol for 1 h. The ethanol-fixed specimens were stained with 40 mM ARS (pH 4.2) for 30 min and washed thrice with deionised water. For the quantitative assessment, the stain was eluted with 10% cetylpyridinium chloride for 1 h. The absorbance of the solution was recorded at 540 nm using a microplate reader.
2.7 Quantification of inflammatory cytokine
The effect of the PTFE nanofibrous membranes on the amount released of the pro-inflammatory factor interleukin-6 (IL-6) was determined using the murine macrophage cell line RAW 264.7 which was obtained from the Korean cell line bank (KCLB, Korea). The RAW 264.7 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS and antibiotic/antimycotic in a humidified atmosphere under 5% CO2 at 37 °C. For the cytokine immunoassay, the cells (2 × 104 cells per well) were seeded on the nanofibrous membranes in a 24 well tissue culture plate and cultured for 24 h at 37 °C. After that, 1 μg mL−1 of LPS was added to the cell culture plate to activate the cells for 24 h. The concentration of IL-6 was determined using a commercial mouse enzyme-linked immunosorbent assay kit (R&D Systems, USA). The kit was used according to the manufacture's specifications.2.8 Statistical analysis
All the data were expressed as means ± standard deviation. Statistical analyses were performed using one-way ANOVA with Turkey test. Comparison of the different groups and significant difference were determined using SigmaPlot 13.0 (Systat Software, CA) where p* < 0.05, p** < 0.01.3 Results and discussion
3.1 Fabrication of PTFE nanofibrous membranes
PTFE nanofibrous membranes have been proven to be convenient and effective in providing mechanical support and osteoconductivity to the growing cells in bone regeneration.8,9 In this study, we first prepared the precursor PTFE/PEO nanofibrous membranes by electrospinning with the PTFE/PEO weight ratios of 5:1 (PTEO51), 6:1 (PTEO61) and 7:1 (PTEO71). In the electrospinning process, PEO is usually added because of its good electrospinnability performance and enables the formation of continuous fibres.14 Moreover, PEO allows for the processing using water as solvent, which may be also suitable for dispersing PTFE. The SEM observations revealed that the precursor membranes formed thoroughly interconnected pore structures (Fig. 1). The average diameter of the nanofibres decreased with increasing PTFE contents, which was 633 ± 34 nm for PTEO51, 620 ± 75 nm for PTEO61 and 555 ± 63 nm for PTEO71. The use of aqueous PTFE dispersion having low viscosity reduced the solution viscosity from 4.15 Pa s to 1.54 Pa s. The solution viscosity influenced the morphological structure and average diameter of the nanofibres, whereby a higher viscosity increased the nanofibre's diameter.15 In addition, PTEO51 exhibited a more uniform nanofibre formation than the other prepared membranes.
 | ||
| Fig. 1 SEM micrographs of (a) PTEO51, (b) PTEO61 and (c) PTEO71 nanofibrous membranes. (d) Average fibre diameters of nanofibrous membranes determined by analyzing the SEM images using image analysis method (n = 4). | ||
For the preparation of PTFE nanofibrous membranes, the PTEO51 precursor membrane was used to examine the heating rate during the sintering treatment. When the membranes were sintered at 350 °C for 30 min at heating rates of 10 °C min−1 and 20 °C min−1, the precursor membranes were significantly shrunk due to the PTFE melting (Fig. 2). The nanofibres were enormously fused and stuck together under these conditions, resulting in the disappearing of the membrane pores. However, as the heating rate decreased, the melted PTFE could easily fill the cavities which were vacated by the decomposition of PEO.16 Therefore, during the sintering treatment at a heating rate of 2 °C min−1, the nanofibre shape and the membrane shape were almost maintained, although the membranes were a little shrunk. Accordingly, this heating rate was used in the following experiment.
 | ||
| Fig. 2 (a) Digital and (b) SEM images of the PTFE nanofibrous membranes before and after sintering at 350 °C for 30 min with different heating rates. | ||
To obtain the PTFE nanofibrous membranes with good biocompatibility, the sintering temperature of the PTEO51 precursor membrane was also assessed at 200 °C (PTFE200) and 350 °C (PTFE350). All the samples prepared at different sintering temperatures exhibited an interconnected nanofibrous network (Fig. 3). After sintering at 200 °C, the shrinkage of membrane was hardly observed whereas unmelted PTFE particles were still observed in the PTFE200 membrane. However, increasing the sintering temperature to 350 °C induced further melting of these PTFE particles which was accompanied by the decomposition of PEO, leading to an increase in the average diameter of nanofibres from 633 ± 34 nm to 947 ± 78 nm; hence, the membrane more closely packed. This compaction caused the decrease in PTFE350 porosity and the increase in water contact angle (Table 1).
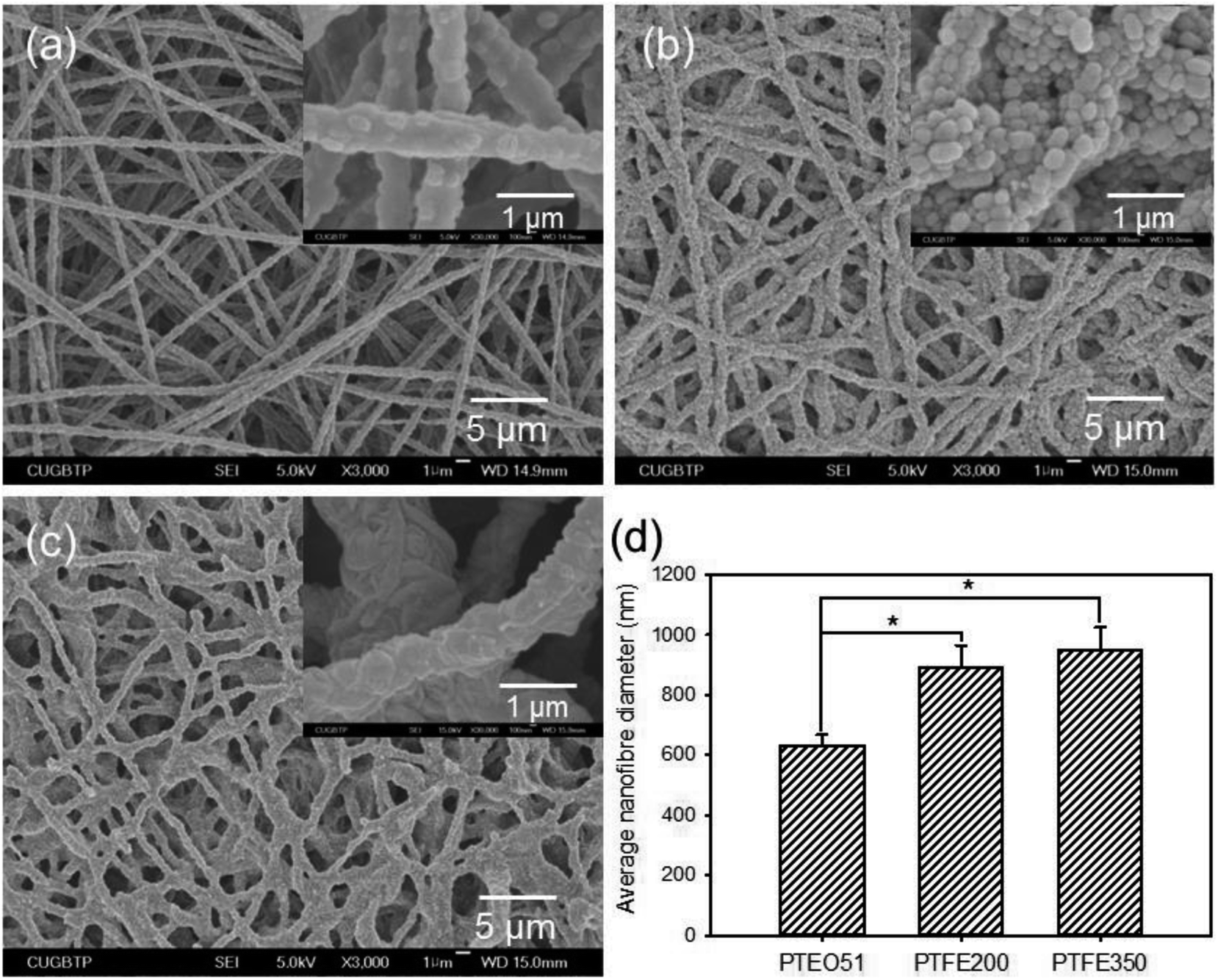 | ||
| Fig. 3 SEM micrographs of (a) PTEO51, (b) PTFE200 and (c) PTFE350 nanofibrous membranes. (d) Average fibre diameters of nanofibrous membranes determined by analyzing the SEM images using image analysis method (n = 4, p* < 0.05). | ||
| Sample | Porosity (%) | Contact angle (°) |
|---|---|---|
| PTEO51 | 76.5 ± 2.9 | 94.1 ± 4.2 |
| PTFE200 | 75.5 ± 3.1 | 133.9 ± 3.9 |
| PTFE350 | 69.1 ± 2.6 | 143.3 ± 3.5 |
3.2 Physicochemical properties of PTFE nanofibrous membranes
ATR-FTIR analysis was carried out to identify the functional groups of the PTFE nanofibrous membranes which in turn provided information about the constitution and phase composition of the products. Absorption bands were observed at 2878 cm−1, 1464 cm−1 and 1345 cm−1 in the spectrum of PTEO51corresponding to the stretching, bending and wagging vibration modes of the CH2 groups present in PEO, respectively.17 The two bands observed at 1097 cm−1 and 958 cm−1 corresponded to the C–O–C stretching and the CH2 rocking modes in PEO (Fig. 4a). PTEO51 also exhibited absorption peaks at 1195 cm−1, 1144 cm−1 and 635 cm−1, corresponding to the stretching and wagging vibrations of the CF bonds in PTFE, respectively.16 The characteristic peaks at 552 cm−1 and 504 cm−1 corresponded to the bending and rocking modes of the CF bonds. By increasing the sintering temperature (i.e. 350 °C) that consequently decreased the PEO content, the intensity of the PTFE peaks became stronger while the PEO peaks disappeared in the PTFE350 spectrum.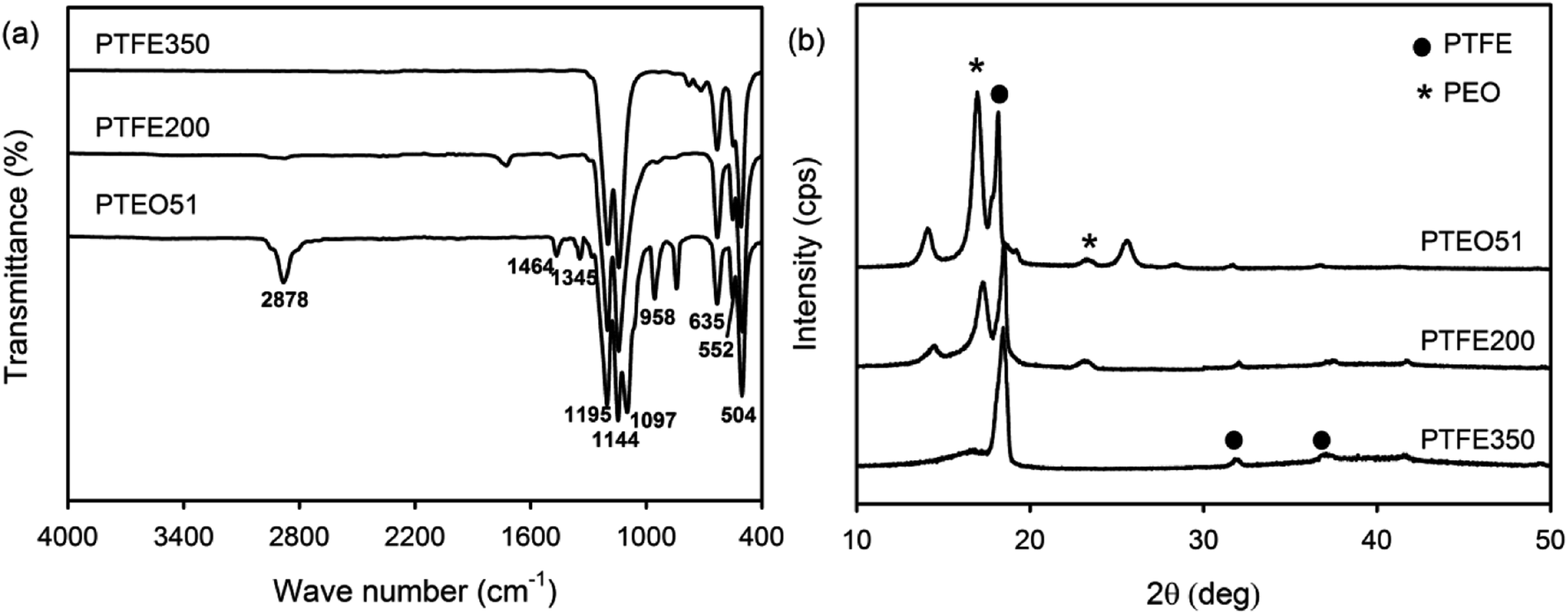 | ||
| Fig. 4 (a) ATR-FTIR and (b) XRD spectra of PTEO51, PTFE200 and PTFE350 nanofibrous membranes. | ||
XRD patterns were recorded to examine the crystalline phases of the PTFE nanofibrous membranes (Fig. 4b). The XRD pattern of PTEO51 precursor membrane showed peaks attributed to the PEO crystalline phase at 16.8° and 25.5°, which were indexed to the (120) and (112) planes, respectively.17 In addition, diffraction peaks ascribed to the PTFE crystalline phase were observed at 18.3°, 31.8° and 37.1°; these peaks corresponded to the (100), (110) and (107) planes, respectively.18 The PTFE200 membrane sintered at 200 °C still exhibited diffraction peaks due to the PEO and PTFE crystalline phases. However, the PTFE350 membrane sintered at 350 °C exhibited peaks ascribed to the PTFE crystalline phase only.
The chemical composition of the PTFE nanofibrous membranes was determined by XPS. The XPS survey scan spectrum of PTEO51 comprised five separated peaks assigned to PEO and PTFE: F 1s (692 eV), O 1s (534 eV), C 1s (292 eV), F 2s (36 eV) and F 2p (12 eV) (Fig. 5a). Meanwhile, O 1s peak attributed to PEO completely disappeared after sintering at 350 °C which was in agreement with the ATR-FTIR results. Additionally, the high-resolution XPS C 1s spectra were deconvoluted using a curve fitting technique to obtain insights into the chemical bonds present on the surface of the nanofibrous membranes. Fig. 5b shows that the C 1s peaks of PTEO51 were decomposed into five components, and the fractions of the different carbon functional groups are given in Table 2, i.e. a peak at 284.6 eV corresponded to the C–C bond, a peak at 286.1 eV corresponded to the C–O–C bond, a peak at 286.6 eV corresponded to the C–CF bond, a peak at 288.8 eV corresponded to the CF bond, and a peak at 291.7 eV corresponded to the CF2 bond.19 The areas of the C 1s peaks for the C–C and C–O–C bonds decreased with an increasing sintering temperature (attributed to the decomposition of the PEO residues), whereas those values assigned to the C–CF, CF, CF2 and CF3 bonds increased with the sintering treatment (Table 2).
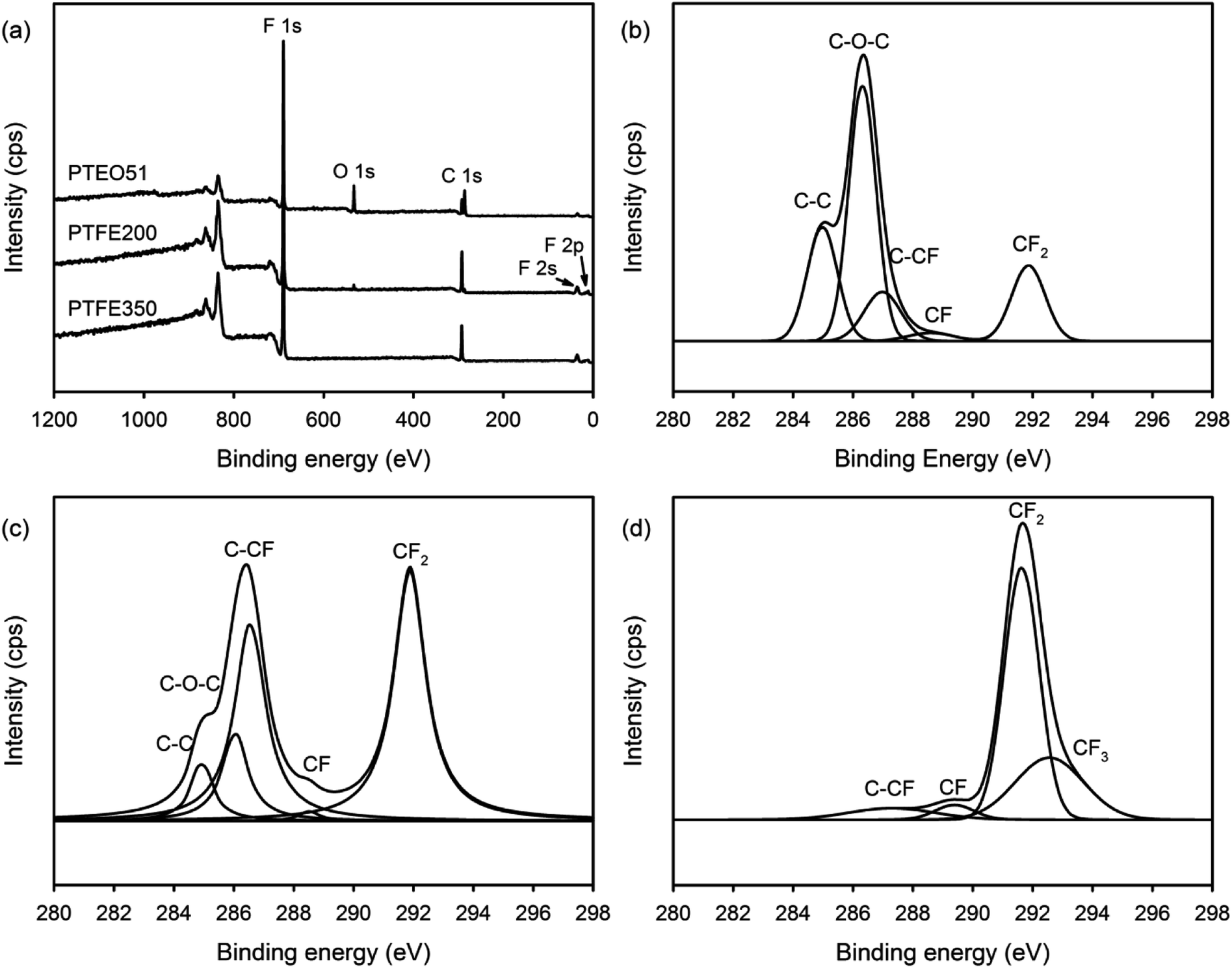 | ||
| Fig. 5 (a) XPS survey scan spectra of the nanofibrous membranes and high-resolution XPS C 1s spectra with peak deconvolutions for (b) PTEO51, (c) PTFE200 and (d) PTFE350 nanofibrous membranes. | ||
| Sample | C–C (%) (284.6 eV) | C–O–C (%) (286.1 eV) | C–CF (%) (286.6 eV) | CF (%) (288.8 eV) | CF2 (%) (291.7 eV) | CF3 (%) (292.5 eV) |
|---|---|---|---|---|---|---|
| PTEO51 | 23.0 | 43.3 | 14.4 | 2.8 | 16.7 | — |
| PTFE200 | 7.3 | 12.7 | 34.3 | 1.1 | 44.5 | 0.1 |
| PTFE350 | — | — | 6.7 | 3.9 | 60.5 | 28.9 |
The thermal properties of the PTFE nanofibrous membranes were investigated by TGA under a nitrogen atmosphere at a heating rate of 10 °C min−1. Fig. 6 shows the typical TGA curve where the amount of weight loss was plotted against the temperature. PTEO51 precursor membrane exhibited a pattern of weight loss that consisted of two main stages. The first stage of weight loss was observed over the temperature ranging from 160 °C to 430 °C. This stage mainly was attributed to the decomposition of PEO.20 Then, the second stage of weight loss in the temperature range of 480–620 °C was possibly due to the decomposition of PTFE. However, PTFE350 did not exhibit weight loss in the first stage due to the decomposition of PEO residues, whereas PTFE200 exhibited a slight weight loss in the first stage because of some undecomposed PEO residues. This result was also in good agreement with the results of ATR-FTIR and XPS analyses.
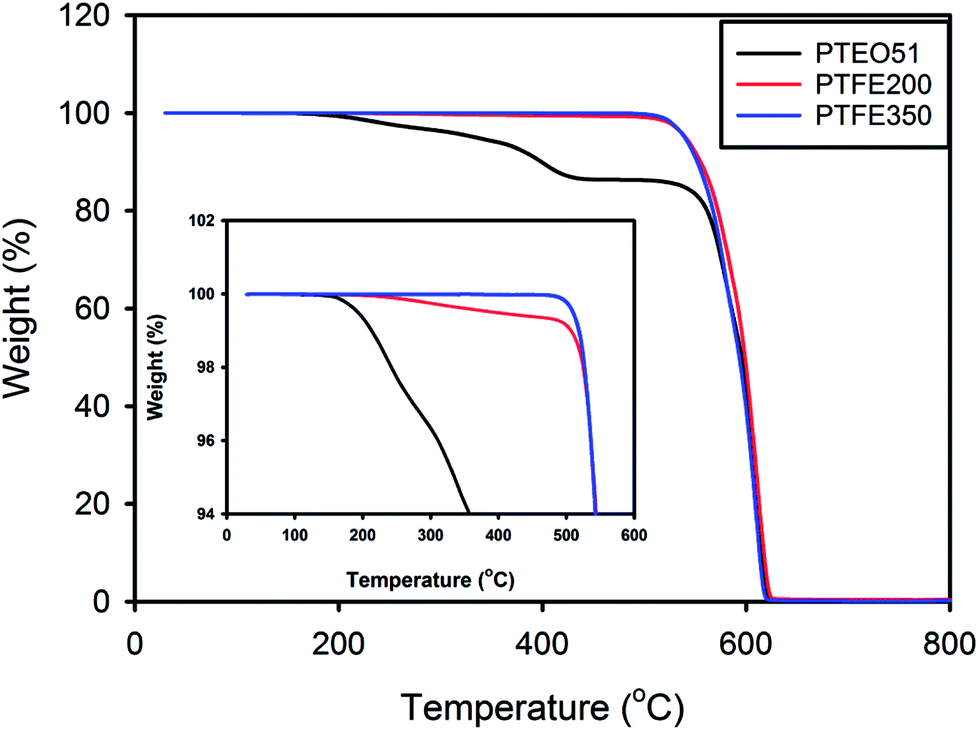 | ||
| Fig. 6 TGA curves of the PTFE nanofibrous membranes. | ||
The mechanical property of GBR membrane is very important, as it needs to provide a biomechanical profile for the cells before new tissue is formed. Therefore, the mechanical properties of the nanofibrous membranes were measured. Based on the strain–stress measurements of these membranes, tensile strength, Young's modulus and elongation at break were summarised in Table 3. The results revealed a correlation with the sintering temperature, meaning that both the tensile strength and Young's modulus were enhanced with increasing the sintering temperature. PTFE350 exhibited significantly enhanced mechanical strength compared with that of PTEO51 precursor membrane but PTFE200 exhibited very low mechanical strength. This is attributable to the compaction of membrane by melting of PTFE particles after sintering at 350 °C as previously described.
| Sample | Young's modulus (MPa) | Tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|
| PTEO51 | 3.11 ± 0.27 | 0.83 ± 0.14 | 380.5 ± 40.9 |
| PTFE200 | 0.10 ± 0.05 | 0.03 ± 0.01 | 18.1 ± 3.4 |
| PTFE350 | 8.15 ± 0.61 | 2.38 ± 0.44 | 135.3 ± 16.2 |
3.3 Cell proliferation
The in vitro cytotoxicity of the PTFE nanofibrous membranes was evaluated for assessing their potential as biomedical materials. The proliferation of the mouse calvaria pre-osteoblasts (MC3T3-E1) on the PTFE nanofibrous membranes were assessed using the MTT assay. The time-dependent changes in the cellular behaviour were observed for all the investigated samples (Fig. 7). Differences in the chemical composition of the membrane surface affected the cellular activities. The MC3T3-E1 cells proliferated more rapidly on PTFE350 than on PTFE200 at all tested time points. These results were related to the degradation of the nanofibrous membranes in the latter membrane, indicating that the cells were probably detached from the surface of PTFE200.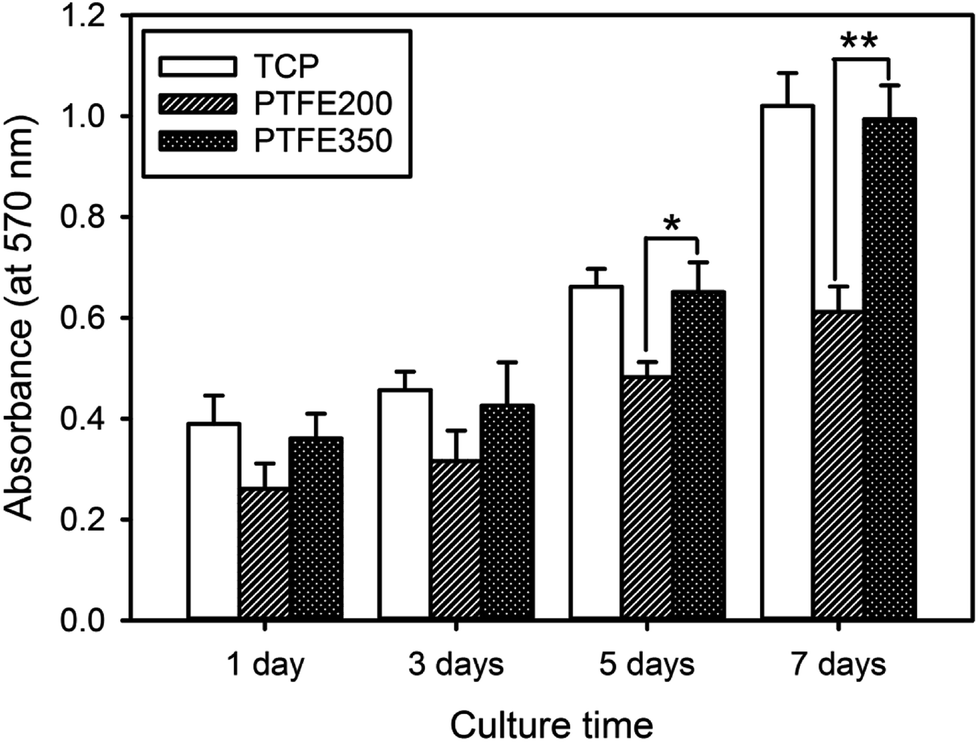 | ||
| Fig. 7 Cell proliferation as a function of culture time for the MC3T3-E1 cells incubated on the nanofibrous membranes (n = 5, p* < 0.05, p** < 0.01). | ||
The degradation rate of the polymer membranes could influence the response of the biological system toward them. To assess the degradation behaviour of the PTFE nanofibrous membranes, they were incubated in DPBS at 37 °C for 3 days. The morphological changes of the nanofibrous membranes were observed by SEM. As expected, the morphological change in PTFE350 was not observed, whereas PTFE200 exhibited a significant morphological change (Fig. 8). PEO was easily dissolved in water, and thus, the degradability of PTFE200 was increased by the amount of undecomposed PEO residues. As a result, the proliferation of MC3T3-E1 cells was affected by the degradation rate of the nanofibrous membranes.
 | ||
| Fig. 8 Morphological changes of (a) PTFE200 and (b) PTFE350 nanofibrous membranes after immersion in DPBS for 3 days. | ||
SEM was employed to observe the interactions between the cells and the membranes to confirm the cell viability on the PTFE nanofibrous membranes (Fig. 9). The cell proliferation increased in a time-dependent manner for all the samples, indicating good biocompatibility of the substrates. However, after culturing for 7 days, the highest number cells were found to spread over most of the PTFE350 surface. This result was consistent with that obtained from the MTT assay.
 | ||
| Fig. 9 SEM micrographs of the MC3T3-E1 cells grown on (a) PTFE200 and (b) PTFE350 nanofibrous membranes for different durations. | ||
3.4 Cell differentiation and bone mineralisation
ALP is a key component of the bone matrix vesicles that catalyses the cleavage of organic phosphate esters. ALP plays a significant role in the formation of bone mineral, and is an early indicator of immature osteoblast activity.1,6 The ALP activity is also a marker of early osteoblastic differentiation. Thus, the ALP activity was evaluated to examine the ability of the PTFE nanofibrous membranes to promote osteoblastic differentiation. The value of PTFE350 was normalised to the value of blank tissue culture plate (TCP) after culturing for 3 days by setting TCP as a control (100%). Results revealed that the cells seeded on PTFE350 expressed higher ALP activity than that seeded on PTFE200 at all time points (Fig. 10a). However, because the ALP activity, which represents an early-stage marker of osteoblastic differentiation, is nonspecific to osteoblastic differentiation, a late-stage indicator such as osteocalcin expression should be evaluated to confirm the actual cell phenotype. Fig. 10b shows the osteocalcin secretion detected in cell culture media for each sample, as measured by ELISA, and normalised to the value of blank TCP after culturing for 3 days by setting TCP as a control (100%). Results revealed that osteocalcin secretion increases in a time-dependent manner in all samples. PTFE350 exhibited higher osteocalcin expression than PTFE200 at all time points.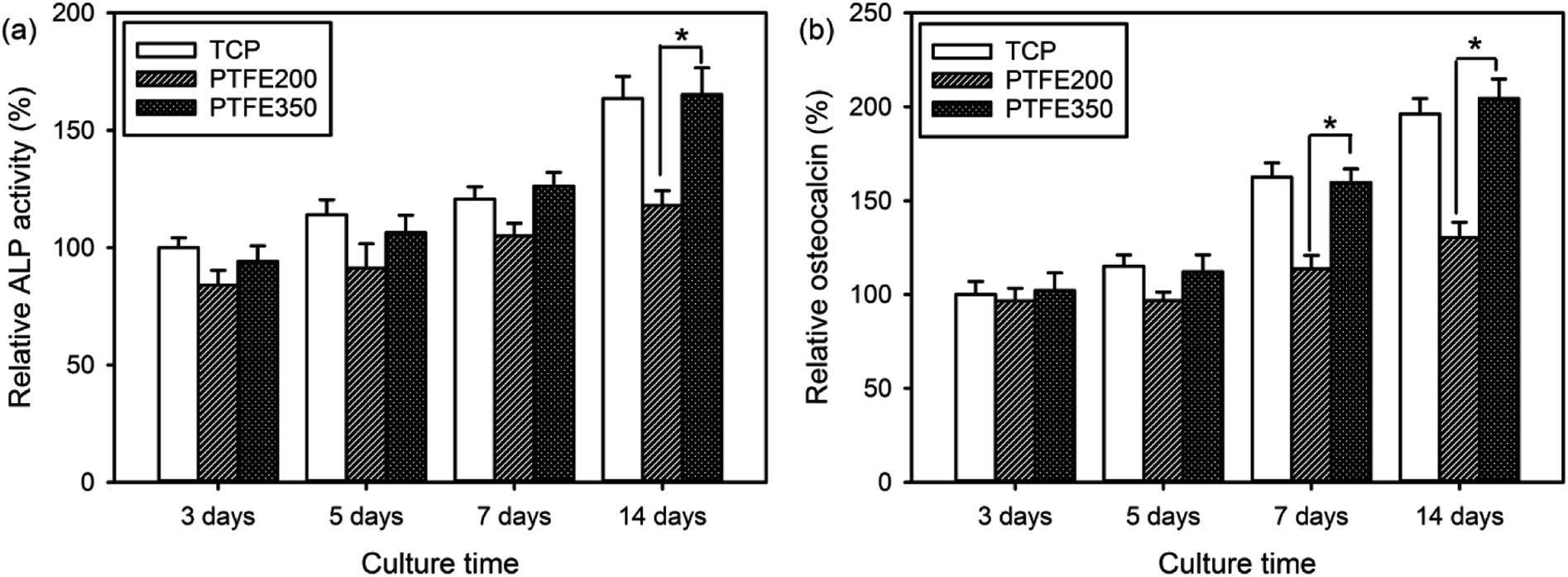 | ||
| Fig. 10 (a) ALP activity and (b) osteocalcin as a function of culture time for MC3T3-E1 cells incubated on the PTFE nanofibrous membranes (n = 4). The value of TCP after culturing for 3 days was set as a control (100%) (p* < 0.05). | ||
Upon osteoblastic differentiation, the cells enter into the mineralisation phase to deposit the mineralised extracellular matrix. The capacity of the cells to deposit minerals is a market for osteogenic efficiency and can be monitored by ARS staining of the cells cultured on different membranes.6 The absorbance of ARS extracted from the stained PTFE nanofibrous membranes (i.e. PTFE200 and PTFE350) during the incubation time was measured and normalised to the value of blank TCP after culturing for 3 days by setting TCP as a control (100%). These normalised values were used to assess the calcium deposition on the PTFE nanofibrous membranes. The calcium deposition on PTFE200 and PTFE350 for 5 days was similar to each other (Fig. 11). However, the calcium deposition on PTFE350 for 14 days was significantly higher than that on PTFE200. These results indicated that the chemical composition and surface characteristics of the membranes significantly affected the MC3T3-E1 cells in the osteoblastic differentiation and bone mineralisation.
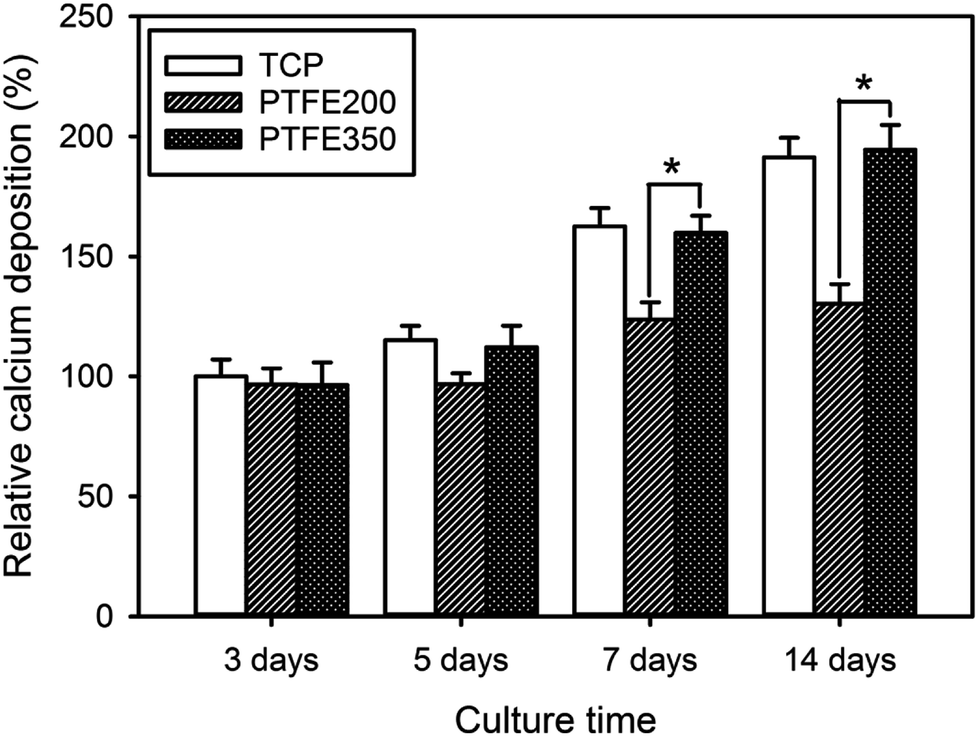 | ||
| Fig. 11 Calcium deposition as a function of culture time for MC3T3-E1 cells incubated on the PTFE nanofibrous membranes (n = 5). The value of TCP after culturing for 3 days was set as a control (100%) (p* < 0.05). | ||
3.5 Quantification of inflammatory cytokine
Cytokines play a major role in the immune response, but have also been reported to be involved in bone metabolism.19 A variety of inflammatory molecules, such as IL-1β, IL-6, and IL-17, promote osteoclastogenesis synergistically with the receptor activator of nuclear factor kappa-B ligand to induce pathological bone resorption in inflammatory settings.21 In addition, IL-6 demonstrates a retroactive mechanism whereby it can induce both phases of bone formation and bone resorption depending on the microenvironment. Most of all, a significant release of IL-6 can reduce cell viability and induce histological damage. Thus, the expression of IL-6 in our cell cultures was analysed to assess the cytocompatibility of the PTFE nanofibrous membranes. PTFE200 stimulated the production of the inflammatory cytokine IL-6 considerably, meaning that it could be contaminated easily by LPS which then could trigger macrophage activation (Fig. 12). Additionally, PTFE200 contained undecomposed PEO residues, and this impurity could also induce the inflammatory response to PTFE200. However, when compared with PTFE200, the IL-6 expression in PTFE350 was remarkably reduced and nearly the same as the one in the control TCP. Therefore, controlling the sintering temperature was a crucial factor in reducing the IL-6 expression on the PTFE nanofibrous membranes.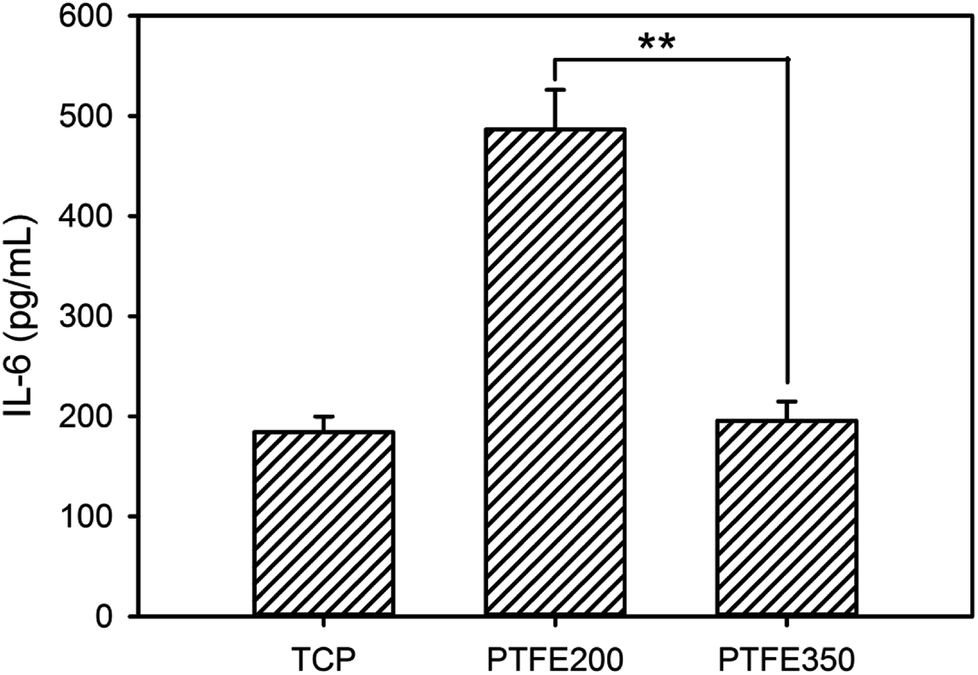 | ||
| Fig. 12 Levels of IL-6 released from the RAW 264.7 cells after incubation for 24 h on the PTFE nanofibrous membranes (n = 5, p** < 0.01). | ||
4 Conclusions
Porous membranes for bone tissue engineering should possess high porosity to accommodate a large number of cells, as well as fully interconnected pores to facilitate uniform distribution of cells and the diffusion of oxygen and nutrients. In this study, PTFE nanofibrous membranes were fabricated as potential scaffolds for bone regeneration by the combination of electrospinning and sintering. The resulting membranes exhibited a porous morphology formed by the interlay of nanofibres. The sintering temperature significantly affected the chemical composition, surface characteristics and mechanical properties of the membranes which eventually influenced the cytotoxicity of the PTFE nanofibrous membranes. PTFE350 supported the MC3T3-E1 proliferation and osteoblastic differentiation more effectively than PTFE200. The results suggested that our simple method for preparing PTFE nanofibrous membranes could aid in the development of new artificial bone materials.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by the Institute for Enterprise R&BD Program of the Innopolis Foundation grant funded by the Ministry of Science and ICT (MSIT) and by the Technology Innovation Program (10053595, Development of functionalized hydrogel scaffold based on medical grade biomaterials with 30% or less of molecular weight reduction) funded by the Ministry of Trade, Industry & Energy (MOTIE), Republic of Korea.References
- H. Lee, H. Hwang, Y. Kim, H. Jeon and G. Kim, Chem. Eng. J., 2014, 250, 399–408 CrossRef CAS.
- B. Dorj, M. K. Kim, J. E. Won and H. W. Kim, Mater. Lett., 2011, 65, 3359–3562 CrossRef.
- A. G. B. Castro, M. Diba, M. Kersten, J. A. Jansen, J. J. J. P. van den Beucken and F. Yang, Mater. Sci. Eng., C, 2018, 85, 154–161 CrossRef CAS PubMed.
- I. Yoshimoto, J. Sasaki, R. Tsuboi, S. Yamaguchi, H. Kitagawa and S. Imazato, Dent. Mater., 2018, 34, 538–550 CrossRef CAS PubMed.
- H. Zhang, Q. W. Fu, T. W. Sun, F. Chen, C. Qi, J. Wu, Z. Y. Cai, Q. R. Qian and Y. J. Zhu, Colloids Surf., B, 2015, 136, 27–36 CrossRef CAS PubMed.
- C. Dhand, S. T. Ong, N. Dwivedi, S. M. Diaz, J. R. Venugopal, B. Navaneethan, M. H. U. T. Fazil, S. Liu, V. Seitz, E. Wintermantel, R. W. Beuerman, S. Ramakrishna, N. K. Verma and R. Lakshminarayanan, Biomaterials, 2016, 104, 323–338 CrossRef CAS PubMed.
- A. Ranjbarzadeh-Dibazar, J. Barzin and P. Shokrollahi, Polymer, 2017, 121, 75–87 CrossRef CAS.
- L. Canullo and V. A. Malagnino, Int. J. Oral Maxillofac. Implants, 2008, 23, 858–866 Search PubMed.
- P. Gentile, V. Chiono, C. Tonda-Turo, A. M. Ferreira and G. Ciardelli, Biotechnol. J., 2011, 6, 1187–1197 CrossRef CAS PubMed.
- R. A. Hoshi, R. VanLith, M. C. Jen, J. B. Allen, K. A. Lapidos and G. Ameer, Biomaterials, 2013, 34, 30–41 CrossRef CAS PubMed.
- K. M. Ainslie, E. M. Bachelder, S. Borkar, A. S. Zahr, A. Sen, J. V. Badding and M. V. Pishko, Langmuir, 2007, 23, 747–754 CrossRef CAS PubMed.
- Q. L. Huang, C. F. Xiao and X. Y. Hu, J. Mater. Sci., 2010, 45, 6569–6573 CrossRef CAS.
- J. Ju, W. Kang, L. Li, H. He, C. Qiao and B. Cheng, Mater. Lett., 2016, 171, 236–239 CrossRef CAS.
- F. Acevedo, J. Hermosilla, C. Sanhueza, B. Mora-Lagos, I. Fuentes, M. Rubilar, A. Concheiro and C. Alvarez-Lorenzo, Eur. J. Pharm. Sci., 2018, 119, 49–61 CrossRef CAS PubMed.
- A. R. Unnithan, N. A. M. Barakat, P. B. T. Pichiah, G. Gnanasekaran, R. Nirmala, Y. S. Cha, C. H. Jung, M. El-Newehy and H. Y. Kim, Carbohydr. Polym., 2012, 90, 1786–1793 CrossRef CAS PubMed.
- Y. Huang, Q.-L. Huang, H. Lin, C.-X. Zhang, Y.-W. You and N.-N. Li, J. Membr. Sci., 2017, 523, 317–326 CrossRef CAS.
- C. Lu, S. W. Chiang, H. Du, J. Li, L. Gan, X. Zhang, X. Chu, Y. Yao, B. Li and F. Kang, Polymer, 2017, 115, 52–59 CrossRef CAS.
- Y. Liang, J. Ju, N. Deng, X. Zhou, J. Yan, W. Kang and B. Cheng, Appl. Surf. Sci., 2018, 442, 54–64 CrossRef CAS.
- Y. Oishi, Y. Watanabe, S. Shinoda, M. Naka, Y. Ozawa, T. Matsuyama, K. Morozumi and Y. Fuke, Gene, 2012, 504, 75–83 CrossRef CAS PubMed.
- B. Darbasizadeh, H. Motasadizadeh, B. Foroughi-Nia and H. Farhadnejad, J. Pharm. Biomed. Anal., 2018, 153, 63–75 CrossRef CAS PubMed.
- B. Zhao and L. B. Ivashkiv, Arthritis Res. Ther., 2011, 13, 234 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |