
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfur(VI) fluoride exchange as a key reaction for synthesizing biaryl sulfate core derivatives as potent hepatitis C virus NS5A inhibitors and their structure–activity relationship studies†
Youngsu Youa,
Hee Sun Kimb,
Jung Woo Parkc,
Gyochang Keumd,
Sung Key Jange and
B. Moon Kim *a
*a
aDepartment of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, South Korea. E-mail: kimbm@snu.ac.kr
bDivision of Integrative Biosciences and Biotechnology, Pohang University of Science and Technology, Pohang 37673, South Korea
cSupercomputing Modeling & Simulation Center, Division of Data Analysis, Korea Institute of Science and Technology Information (KISTI), 245 Daehak-ro, Yuseong-gu, Daejeon, 34141, South Korea
dCenter for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology (KIST), Hwarangno 14-gil 5, Seongbuk-gu, Seoul 02455, South Korea
eDepartment of Life Sciences, Pohang University of Science and Technology, Pohang 37673, South Korea
First published on 12th September 2018
Abstract
Extremely potent, new hepatitis C virus (HCV) nonstructural 5A (NS5A) featuring substituted biaryl sulfate core structures was designed and synthesized. Based on the previously reported novel HCV NS5A inhibitors featuring biaryl sulfate core structures which exhibit two-digit picomolar half-maximal effective concentration (EC50) values against HCV genotype 1b and 2a, the new inhibitors equipped with the sulfate core structures containing diversely substituted aryl groups were explored. In this study, highly efficient, chemoselective coupling reactions between an arylsulfonyl fluoride and an aryl silyl ether, known as the sulfur(VI) fluoride exchange (SuFEx) reaction, were utilized. Among the inhibitors prepared based on the SuFEx chemistry, compounds 14, 15 and 29 exhibited two-digit picomolar EC50 values against GT-1b and single digit or sub nanomolar activities against the HCV GT-2a strain. Nonsymmetrical inhibitors containing an imidazole and amide moieties on each side of the sulfate core structures were also synthesized. In addition, a biotinylated probe targeting NS5A protein was prepared for labeling using the same synthetic methodology.
Introduction
Hepatitis C virus (HCV) is known to cause serious liver infection, which may result in liver cirrhosis and eventually hepatocellular carcinoma (HCC).1–7 Currently, more than 170 million people are infected with HCV worldwide, and approximately 3 to 4 million people are newly infected by this insidious disease yearly.8–16In recent years, many pharmaceutical companies and laboratories worldwide have focused their studies on understanding the HCV life cycle and developing direct-acting agents (DAAs) as next-generation therapies.17–22 One of the most notable HCV target proteins is the nonstructural 5A (NS5A) protein, which is believed to be involved in viral replication and assembly of new virions.23–29
Currently, six NS5A inhibitors are on the market for use with ribavirin, NS3/NS4A protease inhibitors, or NS5B inhibitors.7,30–32 In 2014, the US Food and Drug Administration (FDA) approved ledipasvir (GS-5885) with the NS5B inhibitor, sofosbuvir.33,34 Ombitasvir (ABT-267) was also approved by the FDA in combination with paritaprevir (NS3/4A protease inhibitor), ritonavir (protease inhibitor), and dasabuvir (NS5B inhibitor) for HCV genotype 1 infections.35–37 Daclatasvir (BMS-790052) in combination with sofosbuvir (2015), elbasvir (MK-8742) in combination with grazoprevir (NS3/4A protease inhibitor), and velpatasvir (GS-5816) along with sofosbuvir in 2016 are among other FDA-approved HCV DAAs.38–41 Recently, the FDA approved Vosevi® in 2017, which is a combination of sofosbuvir, velpatasvir, and voxilaprevir (NS3/4A inhibitor); a new pan-genotype drug, Mavyret® (glecaprevir/pibrentasvir), was also approved in the same year.32,42,43
Daclatasvir, which was first reported in 2010, exhibits extremely high antiviral activities against a few genetic variants, and against genotype 1b with EC50 value of 9 pM.44–48 Therefore, numerous DAAs have been introduced based on the daclatasvir structure, which has a symmetrical phenylimidazole core with proline–valine–carbamate motif.49–53 Novel, excessively potent NS5A inhibitors have been introduced by many pharmaceutical companies.54–58 However, treatment with daclatasvir quickly resulted in the development of drug-resistance, such as mutation on L31 and Y93 residues in NS5A protein and the antiviral activities were reduced by up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times.59–63
000 times.59–63
We recently reported the design, synthesis, and structure–activity-relationship (SAR) studies of novel NS5A inhibitors based on a biaryl sulfate core structure (Fig. 1).64 These compounds showed high antiviral activities and good additive effects as a combination treatment with an NS5B inhibitor, sofosbuvir, without showing any cytotoxicity.64
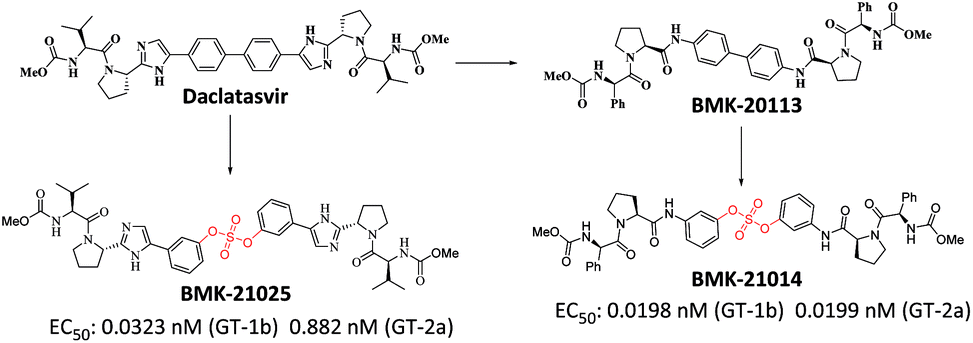 | ||
| Fig. 1 Discovery of biaryl sulfate derivatives as potent HCV NS5A inhibitors. | ||
Encouraged by the initial results, we optimized the biaryl sulfate core part and carried further SAR studies focused on the sulfate core structures. During this investigation, we discovered that some substituted biaryl sulfate core structures were not readily accessible using traditional coupling reactions with sulfonyl diimidazole (SDI). For example, in the cross-coupling reactions of o-fluorophenol derivatives with SDI, only a monomeric aryl sulfonyl imidazole was obtained. Likewise, the sulfonyl diimidazole coupling strategy would not be appropriate for selective construction of nonsymmetrical biaryl sulfate core structures (Fig. 2).
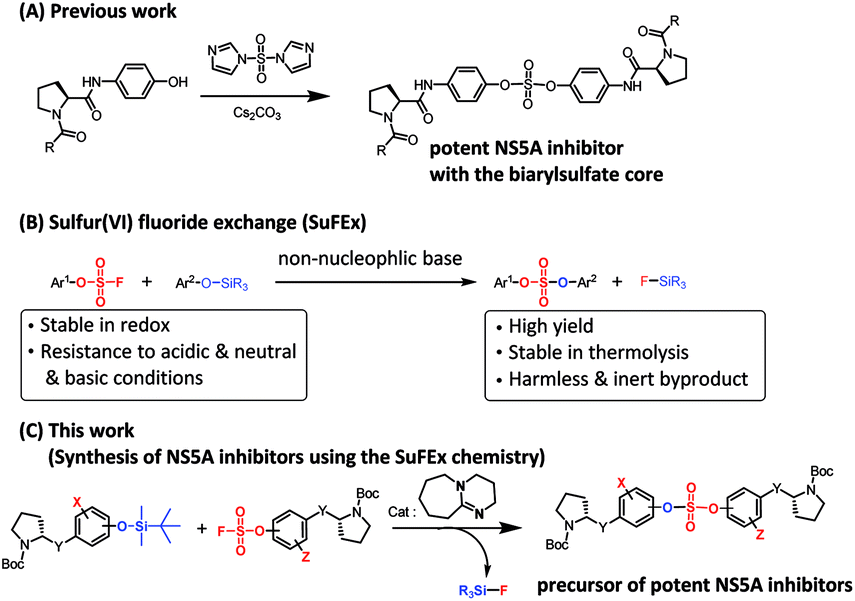 | ||
| Fig. 2 Biaryl sulfate core based nonstructural 5A (NS5A) inhibitors and modification using sulfur(VI) fluoride exchange (SuFEx) chemistry. | ||
Although the first-generation DAA HCV drugs have symmetrical structures similar to daclatasvir, many of the next-generation DAAs developed to treat multi-genotype strains tend to possess nonsymmetric structures.65–69 Since there have been reports of NS5A inhibitors with a high degree of variation at the center core, we envisioned that nonsymmetrical biaryl sulfate core structures with substituted aryl groups would be worth investigating and, thus, were included in our SAR studies.69–73 For the synthesis of the nonsymmetrical biaryl sulfate core structures, SuFEx chemistry appeared to be an extremely attractive strategy.74 Here, we report the fruitful exploitation of SuFEx chemistry for the construction of symmetrical and nonsymmetrical biaryl sulfate-based HCV NS5A inhibitors, which exhibit extremely high inhibitory activities.
In 2014, Sharpless and coworkers introduced the SuFEx click reaction for chemoselectively linking two different phenol derivatives.74,75 The sulfate coupling reaction was accomplished through the reaction of an aryl fluorosulfate and an aryl silyl ether in the presence of a catalytic amount of 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU).76–78 The synthesis of sulfate compounds from the SVI–F group proceeded with high yields and at a fast rate because of the driving force that formed the strong Si–F and S–O bond.74 Furthermore, the resulting compounds exhibited considerably high stabilities against hydrolysis, nucleophilic substitution, thermolysis, and reduction.74,77–79 Moreover, this useful coupling reaction produces only inert silyl fluorides as by-products.75,78,80,81 We envisioned that this chemoselective SuFEx reaction could be used for the synthesis of novel HCV NS5A inhibitors containing biaryl sulfates possessing various substitution at the o-position of the phenol derivatives or a nonsymmetric biaryl sulfate core structures.
Results and discussion
We prepared various aminophenol derivatives (3a–d) as substrates for the SuFEx reaction (Scheme 1). The reduction of nitro groups to amines was carried out with commercially available nitrophenols 1a–b using 10 wt% palladium on activated charcoal (Pd/C).82 The amide linked monomers 3a–d were prepared from amino-phenol derivatives 2a–d and N-Boc-L-proline using general amide coupling methods in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI).83,84 The imidazole-linked monomers 7a–c were prepared from the acetophenone derivatives following an established procedure.64,85 α-Bromination of acetophenone 4 was carried out in the presence of copper(II) bromide to provide α-bromoacetophenone 5 in an 81% yield.85,86 Then, the resulting product 5 was treated with N-Boc-L-proline in the presence of DIPEA to yield ester 6a.85 Likewise, commercially available α-bromoacetophenone derivatives 5b and 5c were converted to the corresponding esters 6b and 6c, respectively. Synthesis of the imidazole derivatives 7a–c was accomplished by the reaction of 6a–c with ammonium acetate.85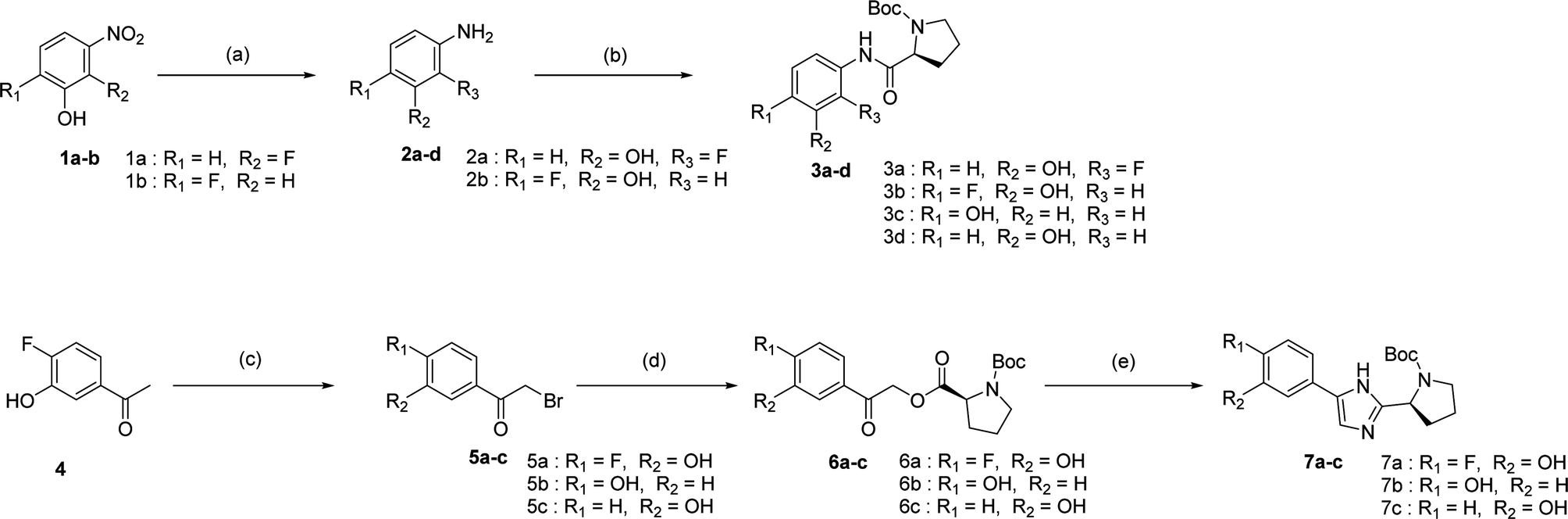 | ||
| Scheme 1 Synthesis of intermediates; reagents and conditions: (a) Pd/C, H2, MeOH, room temperature (rt), 24 h, 99%; (b) EDCI, N-Boc-L-proline, CH2Cl2, rt, 4 h, 59–96%; (c) CuBr2, EA/CHCl3, reflux, 8 h, 81%; (d) N-Boc-L-proline, N,N-diisopropylethylamine (DIPEA), acetonitrile, rt, 5 h, 72–91%; (e) NH4OAc, toluene, 95 °C, 20 h, 45–52%. | ||
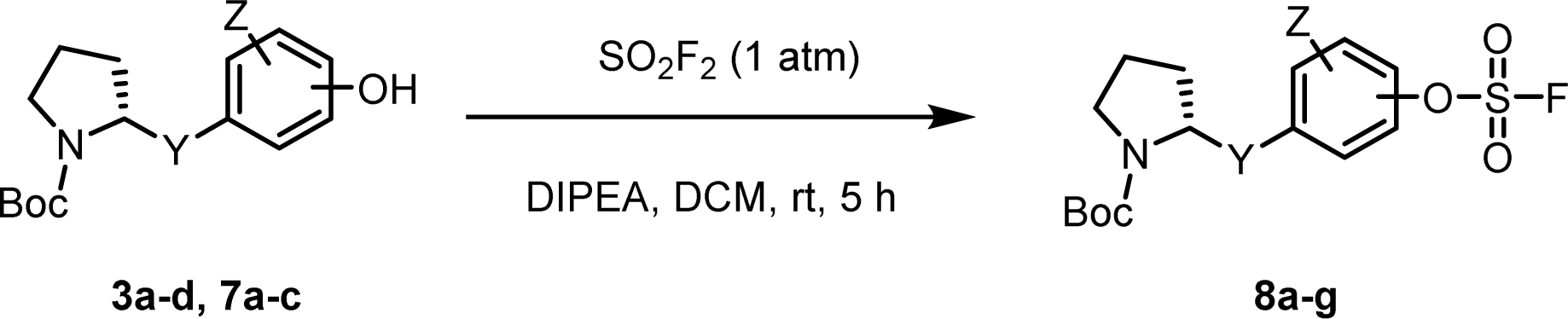
The introduction of a fluorosulfonyl (fosyl) group was achieved by the reaction of the corresponding phenol derivatives with sulfuryl fluoride (SO2F2) and 1.7 equiv. DIPEA in dichloromethane (Table 1).74 Since SO2F2 is a gas, the reaction mixture was stirred rapidly (>1000 rpm) to enhance the liquid–gas contact.76,78 The reaction was complete within 5 h, monitored using thin layer chromatography (TLC). The structures of the fosylated products were confirmed using proton (1H), 13C, and 19F nuclear magnetic resonance (NMR) spectroscopy; especially, 19F NMR chemical shifts of fluorine at fosyl group ranged from ca. 31 to 40 ppm.76,87 These fluorosulfate derivatives exhibited good stability against hydrolysis and could be stored on the shelf for months.77,78,80,88 Compounds 8a–g were obtained in good yields (>80%) as shown in Table 1.
|
|---|
| a Yields of isolated products. |
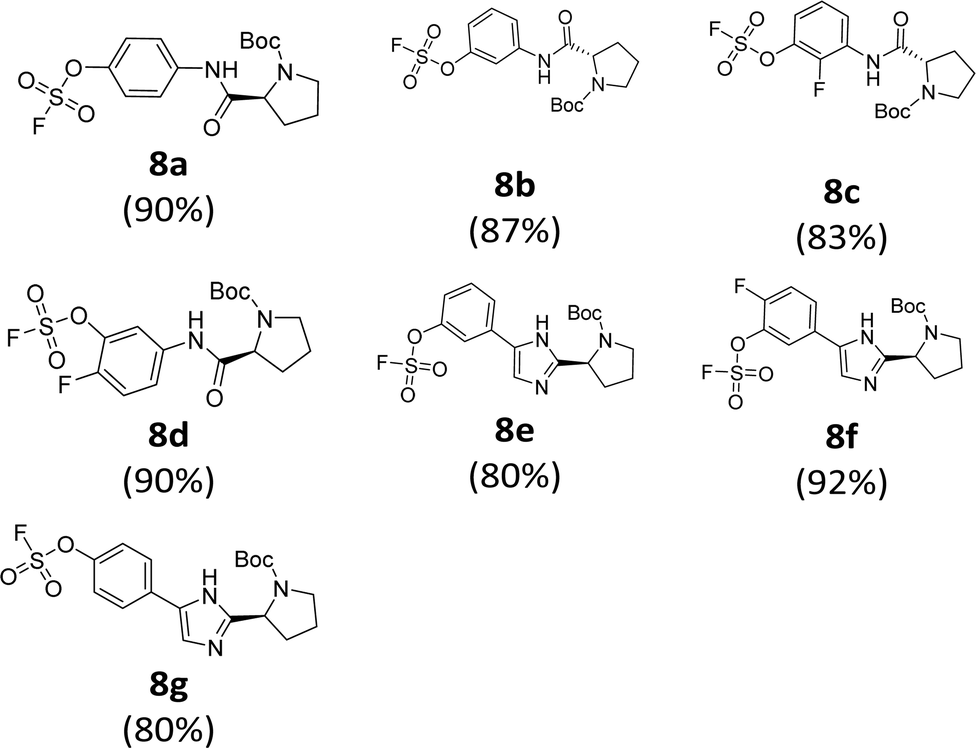 |
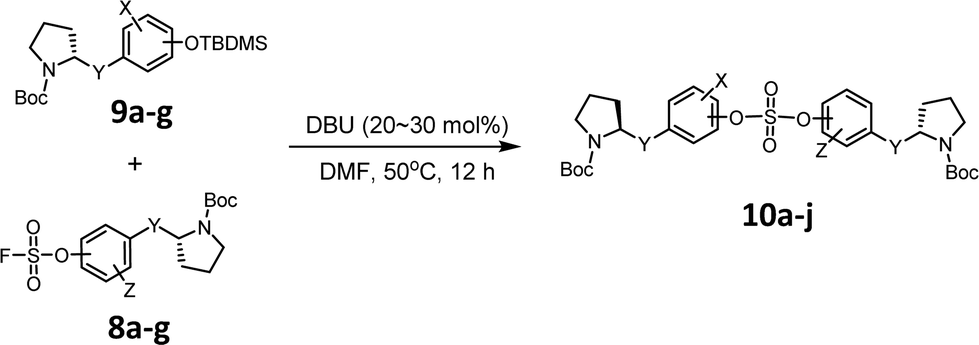
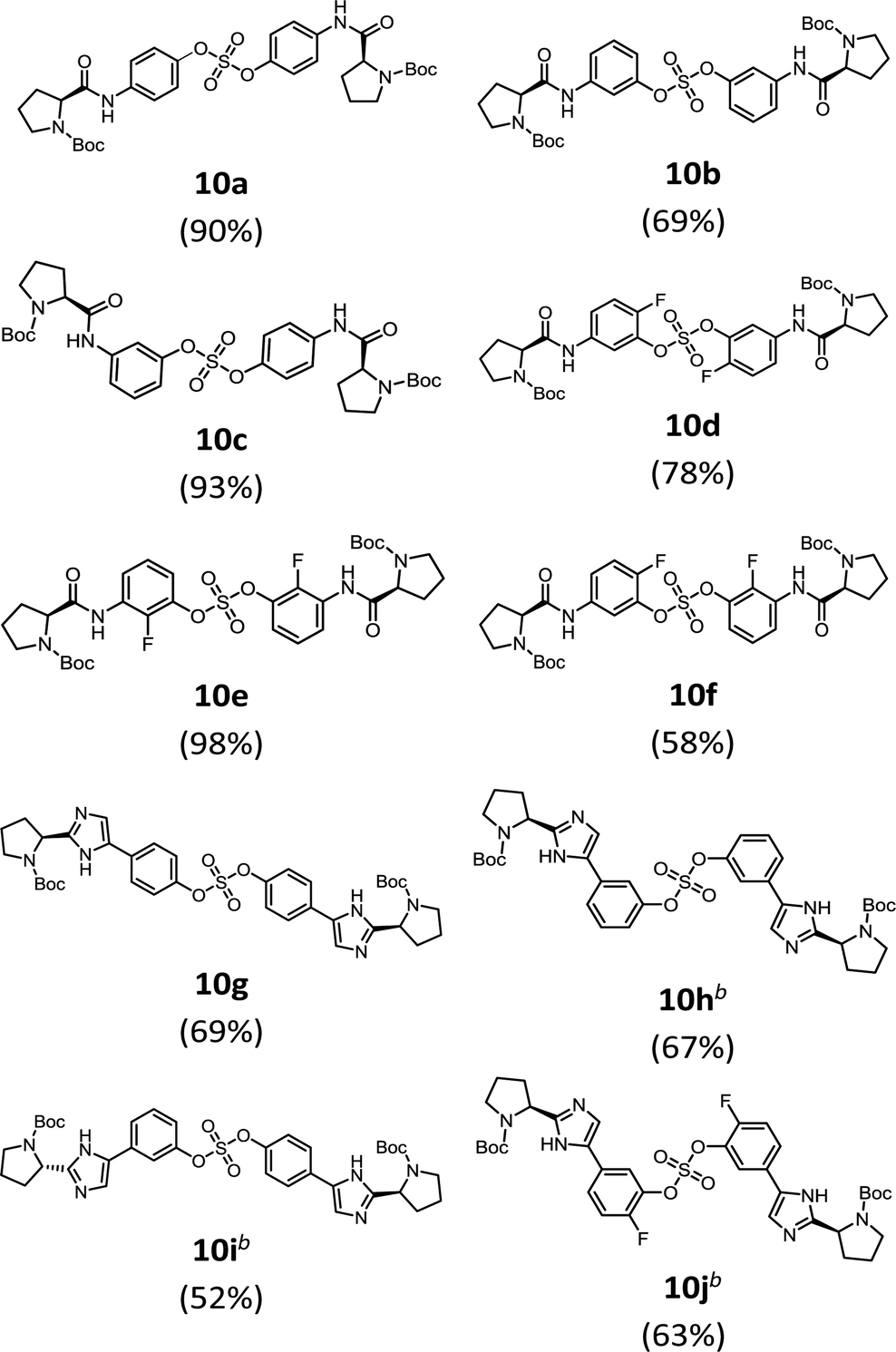
We then constructed the sulfate core structures using aryl silyl ether counterparts through SuFEx chemistry (Table 2).79 Silylation of phenol groups was achieved using the standard method with tert-butyldimethylsilyl chloride (TBDMSCl) and imidazole.89 The coupling reaction between an aryl fosylate and aryl silyl ether was carried out in the presence of DBU in dimethyl formamide (DMF) according to Sharpless' protocol.74,75 Then, the mixture was stirred at 50 °C for 12 h. The biaryl sulfate products (compounds 10a–10f) were formed in good to excellent yields in the presence of catalytic amounts of DBU (20–30 mol%).74 However, in case of the coupling reactions involving the silyl ether or fluorosulfate monomer containing an imidazole moiety (compounds 10g–10j), 2.2–2.3 equiv. DBU was used for the SuFEx coupling products.
We synthesized nonsymmetric or o-fluoro substituted biaryl sulfate based NS5A inhibitors using appropriate precursors (Tables 3 and 4). The tert-butyloxycarbonyl (Boc)-protecting group for the proline moiety was deprotected with 50% (v/v) TFA in CH2Cl2.64,90,91 Volatiles were removed from the reaction mixture under reduced pressure and the residue was directly coupled with a capping group such as N-methyloxycarbonyl-protected valine (val) or phenylglycine (phg) in the presence of EDCI and HOBt.64 Finally, the biaryl sulfate based HCV NS5A inhibitors were obtained after purification of the crude products using silica gel chromatography.64
|
|||||
|---|---|---|---|---|---|
| Entry | Compounds | Biaryl sulfate group | Capping group (R) | Replicon EC50b (GT-1b, nM) | HCVcc EC50b (GT-2a, nM) |
| a EC50, half-maximal (50%) effective concentration; GT-1b, genotype-1b; GT-2a, genotype-2a.b All experiments were performed three times except for compounds that exhibited >1 nM EC50 for GT-1b.c Experiment was performed once.d Not determined. | |||||
| 1 | BMK-21007 | 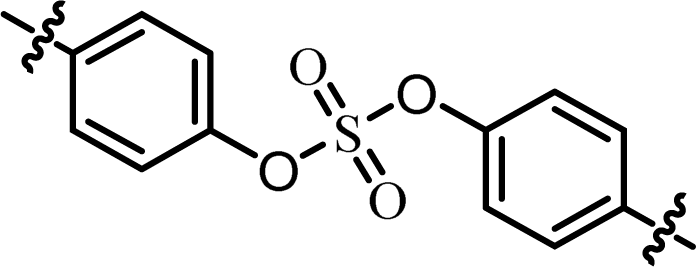 |
(R)-Phenyl | 0.0100 | 0.128 |
| 2 | BMK-21014 | 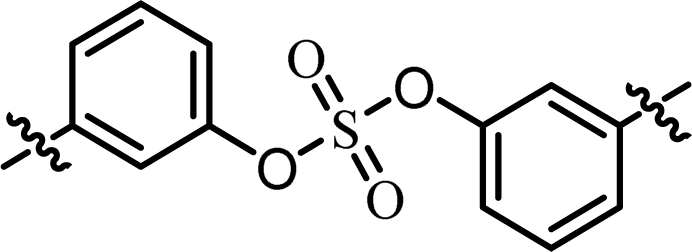 |
(R)-Phenyl | 0.0198 | 0.0199 |
| 3 | BMK-21028 | 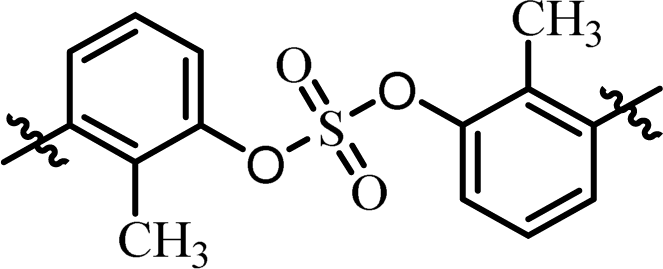 |
(R)-Phenyl | 23.0 | —d |
| 4 | BMK-21020 | 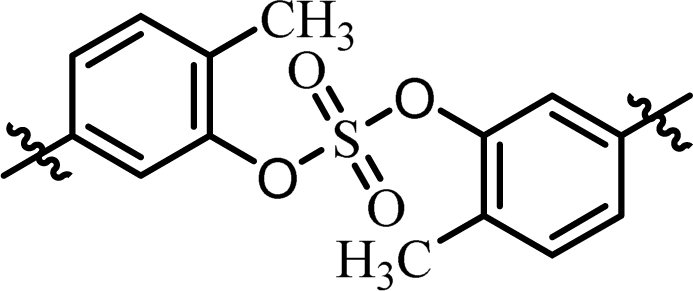 |
(R)-Phenyl | 0.110 | 2.23 |
| 5 | 11 | 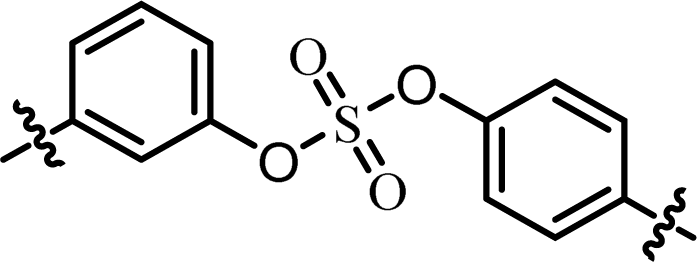 |
(R)-Phenyl | 0.361 | 0.129 |
| 6 | 12 | 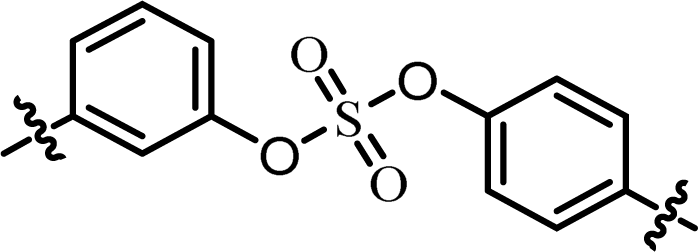 |
(R)-Isopropyl | >100c | —d |
| 7 | 13 | 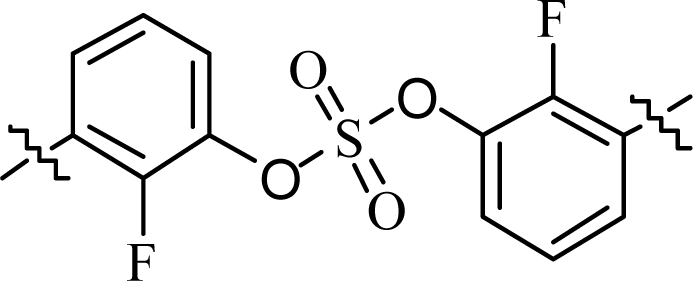 |
(R)-Phenyl | 0.0204 | >10.0c |
| 8 | 14 | 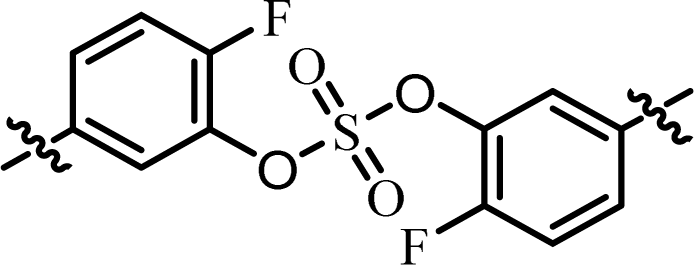 |
(R)-Phenyl | 0.0224 | 1.22 |
| 9 | 15 |  |
(R)-Phenyl | 0.0241 | 0.627 |
| 10 | 16 | 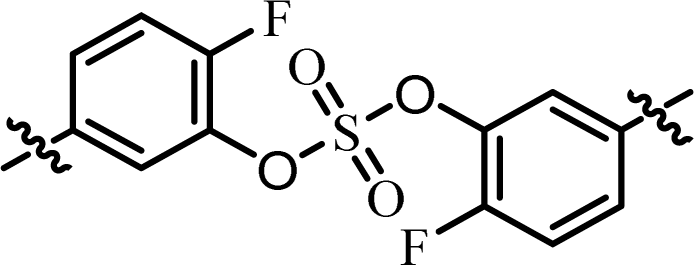 |
(R)-Isopropyl | >100c | —d |
| 11 | 17 | 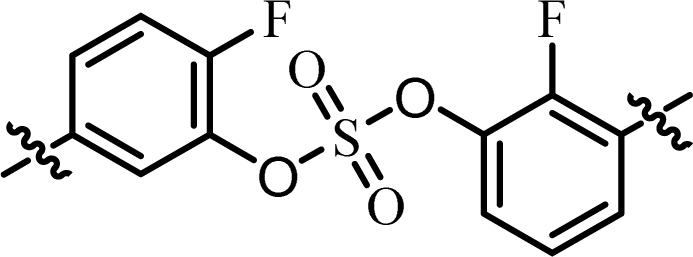 |
(R)-Isopropyl | >100c | —d |
| 12 | 18 | 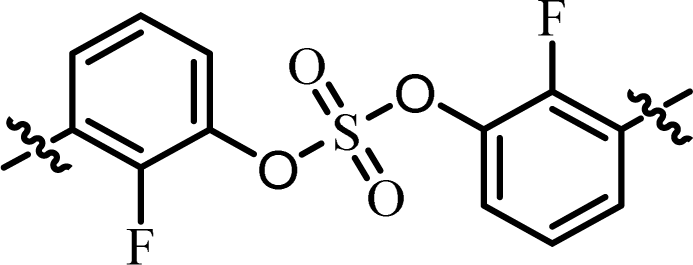 |
(R)-Isopropyl | >1.00c | —d |
|
|||||
|---|---|---|---|---|---|
| Entry | Compounds | Biaryl sulfate group | Capping group (R) | Replicon EC50 (GT-1b)b (nM) | HCVcc EC50 (GT-2a)b (nM) |
| a EC50, half-maximal (50%) effective concentration; GT-1b, genotype-1b; GT-2a, genotype-2a.b All experiments were performed three times except for compounds that exhibited >1 nM EC50 for GT-1b.c Experiment was performed once.d Not determined. | |||||
| 1 | BMK-21025 | 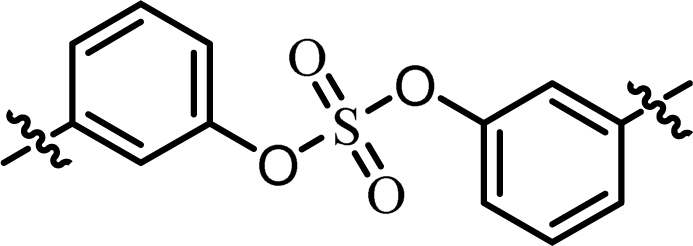 |
(S)-Isopropyl | 0.0323 | 0.882 |
| 2 | 19 | 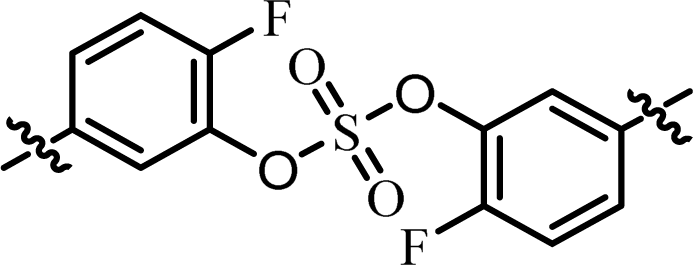 |
(R)-Isopropyl | >100c | —d |
| 3 | 20 | 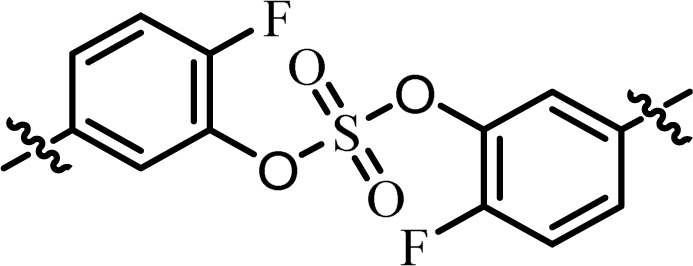 |
(S)-Isopropyl | >10.0c | —d |
| 4 | 21 | 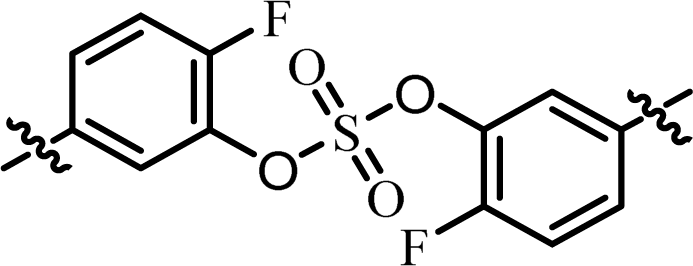 |
(R)-Phenyl | 0.584 | 9.70 |
| 5 | 22 | 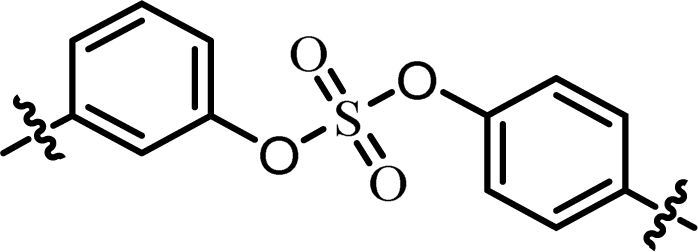 |
(R)-Phenyl | 0.139 | 3.59 |
| 6 | 23 | 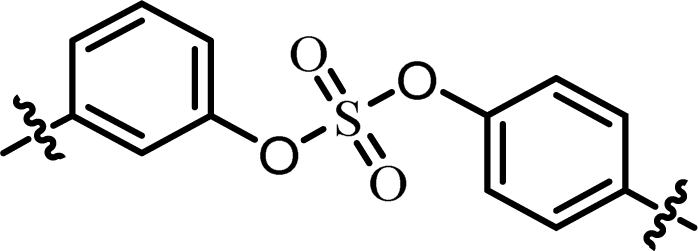 |
(R)-Isopropyl | >100c | —d |
| 7 | 24 | 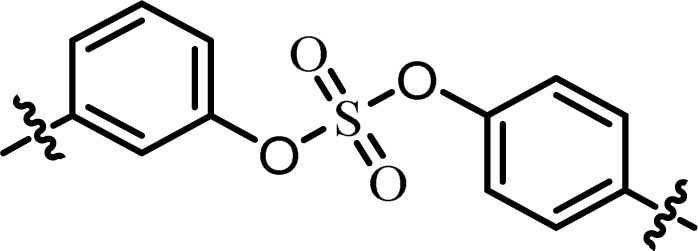 |
(S)-Isopropyl | 0.260 | >10.0c |
To determine the antiviral activities of each compound, we measured the EC50 of inhibitors for genotype-1b (GT-1b) and 2a (GT-2a) using the HCV replicon and HCV cell culture (HCVcc) systems, respectively.91 We used the human hepatoma Huh 7.5.1 cell line to investigate the EC50 of the compounds in the HCV replicon systems for GT-1b, which encodes the bicistronic NK5.1 gene and Gaussia luciferase (Gluc) reporter gene.9,91–93
On the other hand, the inhibitory activities of GT-2a were measured using the HCVcc system with JFH 5a-Rluc-ad34, which is a derivative of JFH1 containing a Renilla luciferase reporter and cell culture adaptive mutations.90,94,95
The antiviral activities (EC50) of the tested compounds containing amide groups on both side of the sulfate core against replicon (GT-1b) and HCVcc (GT-2a) are listed in Table 3. We first selected the compounds that exhibited <1 nM EC50 for GT-1b, and then measured the HCVcc EC50 for GT-2a. In the amide linker series, the D-valine or D-phenylglycine derivatives were chosen as only capping groups because these groups showed the highest levels of antiviral activities in our previous study.64
We first carried out the SAR studies of inhibitors containing a non-symmetric sulfate core. The inhibitor 11 (Table 3, entry 5) containing 3-aminophenyl (4-aminophenyl) sulfate derivative equipped with a D-phenylglycine capping moiety showed 36-fold lower potency against the GT-1b than the symmetric p,p′-substituted inhibitor BMK-21007 did, while the antiviral activities on GT-2a were reduced by >6-fold compared to those of the symmetrically m,m′-substituted biaryl sulfate inhibitor BMK-21014 (Table 3, entries 1 and 2, respectively).64 The replacement of the D-phenylglycine with D-valine moieties as shown in compound 12 induced no apparent inhibitory activity against GT-1b at 100 nM concentration (Table 3, entry 6).
We next investigated the inhibitors containing o-fluoro substituted biaryl groups, expecting increased potency based on the restricted rotational conformation around the sulfate core. In a previous study, compound BMK-21028 (Table 3, entry 3), which was substituted with methyl at 2- and 2′-positions, showed activity at two-digit nanomolar levels in GT-1b.64 However, replacement of the methyl groups of BMK-21028 with fluorine (compound 13) improved the inhibitory activity against GT-1b strain with 0.0204 nM EC50 (Table 3, entry 7). This result, surprisingly, was a 1127-fold higher inhibitory activity than that of BMK-21028 was, suggesting that the o-position in the biaryl groups is extremely important for increasing the potency.
The C2 symmetric 6,6′-difluoro-substituted phenol derivatives containing the D-phenylglycine moieties 14 (Table 3, entry 8) also showed increased inhibitory activities against both GT-1b and GT-2a, compared to BMK-21020, which has o-methyl groups at 6- and 6′-positions of the biaryl groups (Table 3, entry 4).64 Although the inhibitory activities against GT-1b were increased, compound 14 showed low GT-2a inhibitory activity (single nanomolar level EC50) and compound 13 showed no inhibitory activities at 10 nM.
Nonsymmetrically substituted difluorobiarylsulfate 15 (Table 3, entry 9) with 2- and 2′-fluoro-substitution exhibited 5-fold weaker activity for GT-1b and 2-fold higher potency for GT-2a compared to symmetric compound 13. The replacement of the D-phenylglycine with D-valine moiety (compounds 16 and 17) showed lower inhibitory activities except for compound 18, which had GT-1b potency to single nanomolar range (Table 3, entries 10, 11, and 12, respectively). The fact that the SAR results were highly dependent on the substitution and the sulfate linking the position of the biaryl groups indicates that the inhibitory activity of various HCV genotype is highly sensitive to the structural features of the core moiety. These results indicate that ortho-substitution in a phenyl group with fluorine blocks the free rotation, which lowers the antiviral activities against GT-1b. Nevertheless, replacing it with hydrogen, which lacks steric hindrance relatively, enhanced the inhibitory activities against the GT-2a gene.
As shown in Table 2, we designed the NS5A inhibitor series consisting of imidazole linkers with the aim of enhancing the antiviral activities and solubility.96–98 In our previous report, the imidazole linked biaryl sulfate inhibitors containing an L-valine capping group (BMK-21025, Table 4, entry 1) showed good activities against GT-1b and GT-2a.64 Thus, we introduced nonsymmetric or substituted sulfate core structures in imidazole-linked inhibitors to determine if it would influence the inhibitory activities and the results are summarized in Table 4. Compound 19, containing o-fluoro substituted biaryl sulfate with D-valine derivative, showed no inhibitory activity at 100 nM against replicon GT-1b (Table 4, entry 2). Compound 20 with an N-methoxycarbonyl-L-valine capping group resulted in two-digit nM EC50 on GT-1b (Table 4, entry 3). The replacement of the L-valine moiety with D-phenylglycine moiety as in compound 21 dramatically enhanced the antiviral activity against GT-1b (Table 4, entry 4). However, these results showed 25-fold lower activity than that of inhibitors linked with amides such as compound 15. Introduction of m-, p′-disubstituted biaryl sulfate core as in compound 22 demonstrated that the potencies for both GT-1b and GT-2a were better than that of compound 21 with an o-fluoro substituent (Table 4, entry 5). While the D-valine derivative 23 showed no inhibition at 100 nM, the opposite stereoisomeric L-valine derivative 24 showed improved potency for GT-1b (Table 4, entries 6 and 7, respectively). However, it showed no marked antiviral activity against GT-2a.
Based on the SAR study, it can be concluded that compounds with asymmetric biaryl sulfate core and D-phenylglycine capping derivatives (compounds 11 and 22) showed the best activities for both genotypes. We proceeded to synthesize nonsymmetric compounds, exploiting the chemoselectivity of SuFex chemistry. In our previous study, we reported that m,m′-biaryl sulfate derivatives exhibit high inhibitory activities against NS5A.64 Especially, C2-symmetric biaryl sulfate comprising amide (BMK-21014) and imidazole (BMK-21025) linkers showed extremely high antiviral activities against two genotypes. Based on these results, we synthesized compound 29 having a hybridized structure of the two compounds. Due to the orthogonality of SuFEx, we were able to synthesize the biaryl sulfate compound containing imidazole-proline-L-valine carbamate derivative and amide-proline-D-phenylglycine carbamate derivative in moderate yields as shown in Scheme 2.99 The nonsymmetric compound 29 exhibited excellent inhibitory activity for GT-1b. However, it exhibited nanomolar levels of antiviral activity against GT-2a, which indicated no dramatic change in activity.
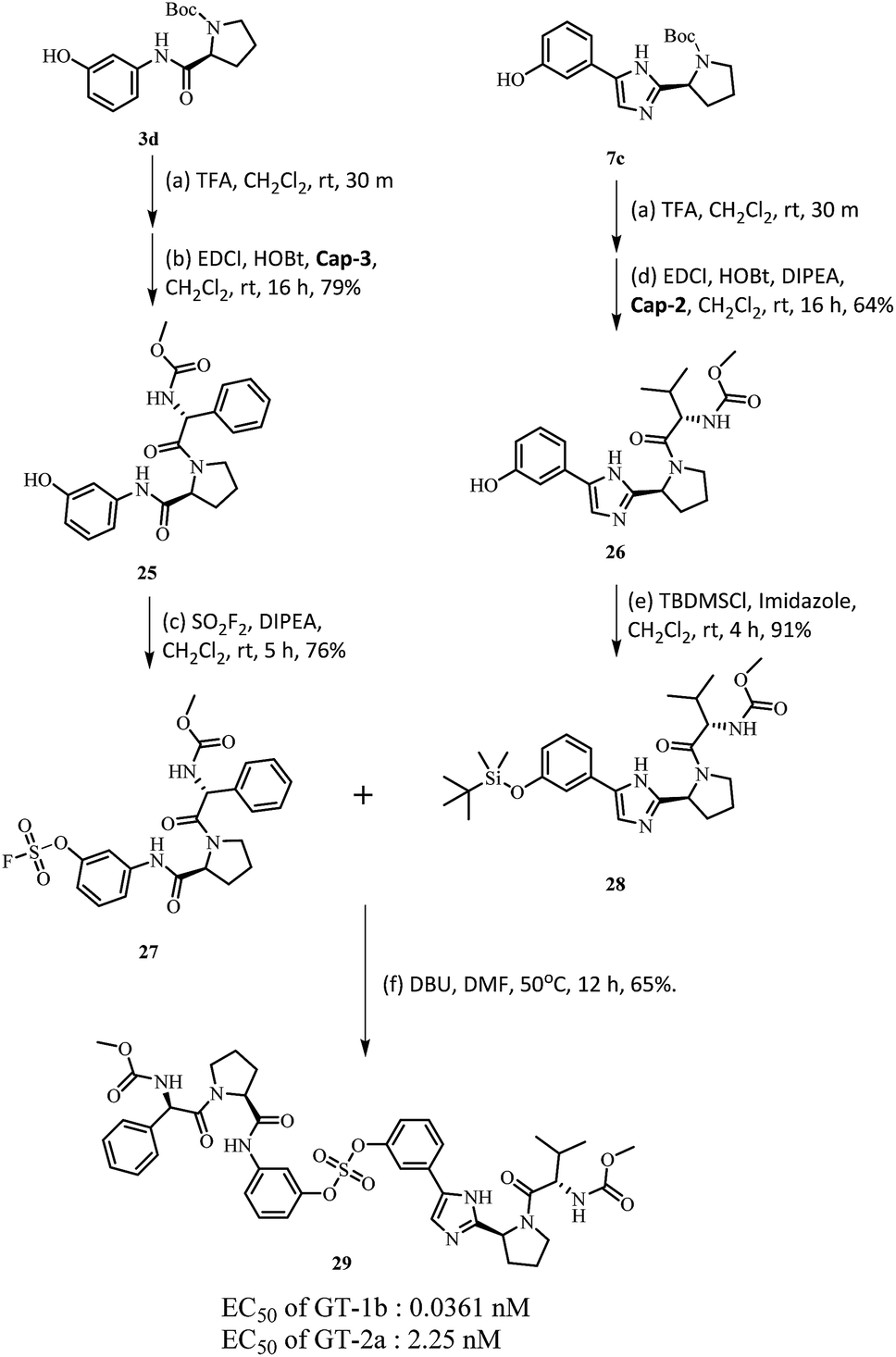 | ||
| Scheme 2 Synthesis of 3-(2-((S)-1-((methoxycarbonyl)-L-valyl)pyrrolidin-2-yl)-1H-imidazol-5-yl)phenyl(3-((S)-1-((R)-2-((methoxycarbonyl)amino)-2-phenylacetyl)pyrrolidine-2-carboxamido)phenyl) sulfate. | ||
We also used SuFEx chemistry to prepare the biotin-tagged, biaryl sulfate core based NS5A inhibitor as shown in Scheme 3. Biotin binds specific proteins such as streptavidin and avidin with extremely high affinity.44 Because of this specificity, biotinylation of NS5A inhibitors has been reported.100,101 The introduction of biotin into one proline was readily carried out using SuFex chemistry. Although the NS5A inhibitor containing the biaryl sulfate with biotin-tag 32 showed a slightly decreased EC50 against genotype-1b (9.59 nM), it was 10 times higher than the previously reported biotinylated tag molecules based on BMS compound (BMS-671 and BMS-690).63,102
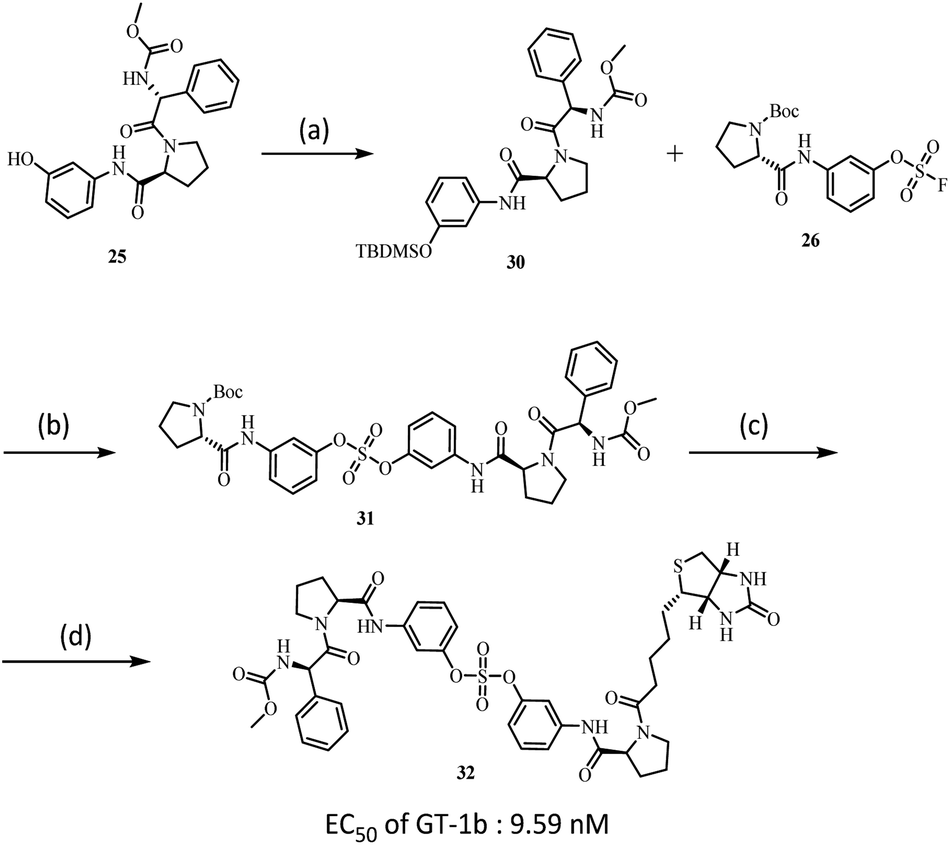 | ||
| Scheme 3 (a) TBDMSCl, Imidazole, CH2Cl2, rt, 4 h, 81%; (b) DBU, DMF, 50 °C, 12 h, 75%; (c) TFA, CH2Cl2, rt, 30 m; (d) EDCI, HOBt, DIPEA, Biotin, CH2Cl2, rt, 16 h, 96%. | ||
To understand the observed antiviral activities (EC50) of inhibitors from a 3D structural perspective, three representative inhibitors, BMK-21007, BMK-21014, and 11, were docked in an NS5A protein dimer model. The model was constructed using the NS5A domain 1 crystal structure (3FQQ)103 and amino terminal alpha helical NMR structure (1R7G).104 Fig. 3 illustrates the 3D-interaction models of BMK-21007, BMK-21014, and 11 with the NS5A dimer. The docking model demonstrates that inhibitors bound across the dimer interface with Thr95A, which forms a hydrogen bond with the nonsymmetric biaryl sulfate core. The interactions of inhibitors and the residues at the binding site are detailed in two dimensional (2D) docking representation (ESI, Fig. S4†). In the case of BMK-21007, the biaryl sulfate moiety can engage in hydrophobic interaction with Gln54A and Gln54B, whereas the methyl groups of the carbamate groups make π-alkyl interaction with Pro58 and Tyr93 of the NS5A protein dimer (Fig. 3A and S4A†). The amide linkers of BMK-21014 make hydrogen bond with Gln54, Thr56, and Gln62, whereas both methyl groups of the carbamates involve π-alkyl interaction with Pro58 and Tyr93 (Fig. 3B and S4B†). In compound 11 (Fig. 3C), the sulfate moiety appears to interact with Thr95 by hydrogen bonding. Interestingly, Tyr93A has π-alkyl interaction with the pyrrolidine ring and the methyl moiety of methyl carbamate, whereas Tyr93B shows π-donor hydrogen bond interaction with the phenyl group of D-phenylglycine. There are no interactions with the residues of Pro58 (ESI, Fig. S4C†).
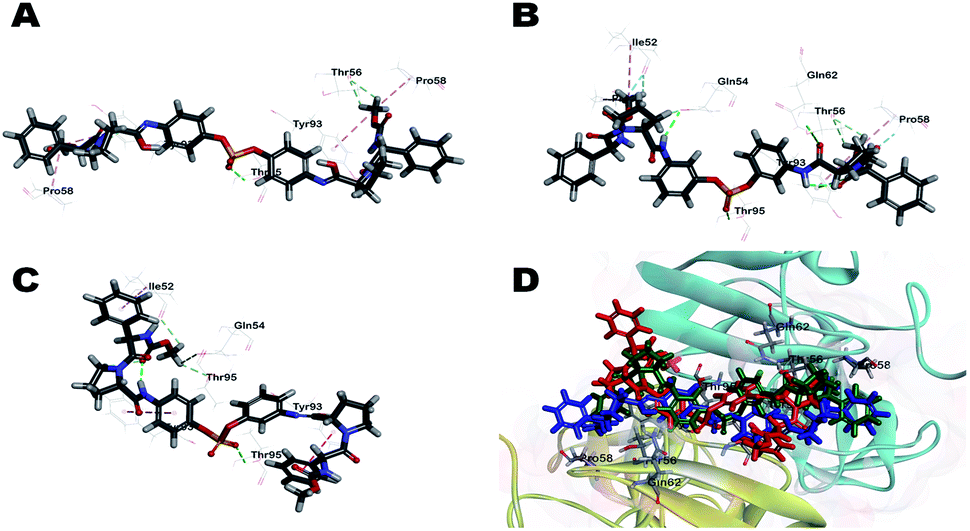 | ||
| Fig. 3 Docking of (A) BMK-21007, (B) BMK-21014, and (C) 11 to an NS5A (3FQQ) dimer model. For clarity purpose, only residues having interactions were shown. BMK-21007, BMK-21014, and 11 are shown in stick and surrounding residues were shown in line. Various interactions are shown in dashes. (For more clarity see the ESI, Fig. S4†). (D) Superposed model of inhibitors inside 3FQQ dimer model. BMK-21007, BMK-21014, and 11 are shown blue, green and red, respectively. | ||
As shown in a superposed model (Fig. 3D, S4A and S4B†), the symmetric inhibitors BMK-21007 and BMK-21014 pose reduced steric hindrance of D-phenylglycine moiety compared to nonsymmetrical inhibitor 11, where only one methyl group of the carbamates is involved in π-alkyl interaction with Tyr93A. Also, CDocker interaction energies of BMK-21007, BMK-21014, and 11 are −58.41, −57.20, and −46.19 kcal mol−1, respectively, which suggests that BMK-21007 and BMK-21014 might be better placed in the binding mode compared to 11. In the case of inhibitor 22, hydrogen bond interactions are found not only between the sulfate and Gly54A but also between the imidazole and Thr56 (ESI, Fig. S4D†). Both ends of the carbamate groups appear to show π-alkyl interaction with Tyr93A and Tyr93B, and especially Tyr93A is engaged in hydrogen bond interaction with the carbonyl moiety of one carbamate. Interestingly, one methyl group of the carbamate makes hydrophobic interaction with Pro58B, whereas the other side correlates with Pro97B. Therefore, the D-phenylglycine of inhibitor 22 poses reduced steric hindrance compared to inhibitor 11 due to the hydrogen bonding of the carbamate group (ESI, Fig. S6†). CDocker interaction energy of inhibitor 22 is −54.76 kcal mol−1, which bodes well with its EC50 activity.
Conclusions
In conclusion, using SuFEx chemistry we achieved chemoselective coupling of aryl fluorosulfates and aryl silyl ethers in the presence of a catalytic amount of a base. Furthermore, this method was used to synthesize new NS5A inhibitors containing various substituted sulfate core structures with high yields. These compounds exhibited high inhibitory activities against HCV viral proliferation by binding NS5A protein. We easily synthesized inhibitors containing nonsymmetric or o-fluoro substituted biaryl sulfate core structures using the SuFEx reaction in a “click chemistry” manner. Furthermore, the monomeric halves of BMK-21014 and BMK-21025, which showed excellent activity in our previous report, were easily coupled using SuFEx chemistry to form nonsymmetric biarylsulfate core-based inhibitor 29. Compounds 14, 15, and 29 had two-digit pM EC50 values against GT-1b and single digit or sub nanomolar values for GT-2a. In addition, a sulfate core-containing compound conjugated with biotin 32 was synthesized in a straightforward manner and showed high activity compared to that of reference biotinylated compounds. Based on these results, SuFEx chemistry proved to be highly useful in constructing symmetric and nonsymmetric sulfate core-containing NS5A inhibitors, and could potentially be applied to the synthesis of a wider range of compounds in medicinal chemistry.Experimental
General methods and material
The 1H, 13C and 19F NMR-spectra were measured with a Varian/Oxford As-500 (500 MHz), an Agilent 400-MR DD2 Magnetic Resonance System (400 MHz), or Bruker/Avance DPX-300 (300 MHz) spectrophotometer. Chemical shifts were measured as part per million (d values) from Tetramethylsilane (TMS) as an internal standard at probe temperature in CD3OD or CDCl3 or DMSO-D6 for neutral compounds. Coupling constants are provided in Hz, with the following spectral pattern designations: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; br, broad; app, apparent. Reactions were conducted, purified, and analyzed according to methods widely practiced in the field, while taking necessary precautions in the exclusion of moisture and/or oxygen where appropriate. Evaporation of solvents was performed at reduced pressure using a rotary evaporator. TLC was performed using silica gel 60F254 coated on an aluminium sheet (E. Merck, Art.5554). High resolution mass spectra (HRMS) were recorded on a ThermoFinnigan LCQ™ Classic, Quadrupole Ion-Trap Mass Spectrometer. HPLC analyses were carried out on an Agilent HP1100 system (Santa Clara, CA, USA), composed of an auto sampler, quaternary pump, photodiode array detector (DAD), and HP Chemstation software. The separation was carried out on a poroshell 120 EC-C18 column 50 × 4.6 mm, 2.7 mm with acetonitrile (A), 0.1% TFA in water (B), as a mobile phase at a flow rate of 1 mL min−1 at 20 °C. Method: 5% A and 95% B (0 min), 50% A and 50% B (15 min), 95% A and 5% B (24 min), 95% A and 5% B (25 min), 5% A and 95% B (26 min), 5% A and 95% B (27 min). All materials were purchased from a commercial supplier and used without further purification unless otherwise noted. The 1H NMR-spectra of intermediates and final products exhibit a mixture of rotamers at ambient temperature in DMSO-d6.General procedure for synthesis of capping groups64,90,91
Na2CO3 (2.6 mmol) was added to aq. NaOH (5 mL of 1 M/H2O, 5 mmol) solution of an amino acid (5.00 mmol) and the resulting solution was cooled with ice-water bath. Methyl chloroformate (5.40 mmol) was added dropwise, the cooling bath was removed and the reaction mixture was stirred at ambient temperature for 3.25 h. The reaction mixture was washed with ether (3 × 9 mL), and the aqueous phase was cooled with ice-water bath and acidified with concentrated HCl to a pH region of 1–2, and extracted with CH2Cl2 (3 × 9 mL). The organic phase was dried (MgSO4), filtered, and concentrated in vacuo to afford capping groups as a white solid.General procedure for synthesis of aminophenol
A stirred mixture of nitro phenol (12.7 mmol) in MeOH (50 mL) at room temperature was treated with 10 wt% palladium on charcoal (100 mg). Hydrogen gas was passed through the mixture for 24 h, and then the palladium on charcoal was removed by filtration through Celite. The filtrate was concentrated in vacuo, and was obtained as solid product.General procedure for synthesis of amide linked intermediate
A mixture of amino phenol (12.6 mmol), N-Boc-L-proline (3.25 g, 15.1 mmol), and EDCI (2.11 g, 16.4 mmol) in CH2Cl2 (30 mL) was stirred at room temperature for 4 h. The mixture was then poured in 1 N aq. HCl solution and Extracted with CH2Cl2. The organic layer dried over magnesium sulfate, and then was purified by the column chromatography (n-hexane/ethyl acetate). The residue was obtained as solid product.General procedure for synthesis of keto ester derivatives
2-Bromo-acetophenone (0.416 mmol) and N-Boc-L-proline (0.499 mmol) in acetonitrile (10 mL) put into the flask. Diisopropylethylamine (0.541 mmol) was added dropwise to the mixture and was stirred at room temperature for 5 h. The mixture was then poured in 1 N aq. HCl solution and extracted with CH2Cl2. The organic layer dried over magnesium sulfate, and then was purified by the column chromatography (n-hexane/ethyl acetate). The residue was obtained as white solid.General procedure for synthesis of imidazole linked intermediate
A mixture of keto ester (0.554 mmol) and ammonium acetate (8.30 mmol) were suspended in toluene (10 mL). The reaction mixture was heated to 90 °C and stirred for 20 h. The mixture was then poured in H2O and extracted with ethyl acetate. The organic layer dried over magnesium sulfate and evaporated in vacuo. After evaporation, the residue was purified by the column chromatography (n-hexane/ethyl acetate). Then product was obtained as yellow solid.General procedure for synthesis of fosylated intermediate
A mixture of phenol derivative (0.800 mmol) and N,N-diisopropylethylamine (1.20 mmol) in CH2Cl2 (5 mL) was stirred under sulfuryl fluoride at room temperature for 5 h. The mixture was then poured in 1 N aq. HCl solution and extracted with CH2Cl2. The organic layer dried over magnesium sulfate, filtered, and concentrated in vacuo. Without any purification, the residue was obtained as an oil.General procedure for silyl protection
A mixture of phenol derivative (2.29 mmol), tert-butyldimethylsilyl chloride (3.44 mmol), imidazole (6.87 mmol) in THF (10 mL) was stirred at room temperature for 4 h. The mixture was then poured in H2O and extracted with CH2Cl2. The organic layer dried over magnesium sulfate, and then was purified by the column chromatography (n-hexane/ethyl acetate). The residue was obtained as sticky oil.General procedure for SuFEx reaction
A mixture of silylated monomer (0.408 mmol), fosylated monomer (0.272 mmol) and DBU (0.05 mmol) in DMF (10 mL) was stirred at 50 °C for 12 h. The mixture was then poured in 1 N aq. HCl solution and extracted with CH2Cl2. The organic layer dried over magnesium sulfate, and then was purified by the column chromatography (n-hexane/ethyl acetate). The residue was obtained as solid.General procedure for synthesis of inhibitors
A mixture of precursor (0.151 mmol) in CF3CO2H (3 mL)/CH2Cl2 (3 mL) was stirred at room temperature for 30 m. The volatile component was removed in vacuo, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (0.392 mmol), hydroxy-benzotriazole hydrate (0.392 mmol), capping group (0.362 mmol) were added in batches over 4 min to a solution of N,N-diisopropylethylamine (0.754 mmol) in CH2Cl2 (10 mL) and the reaction mixture stirred at room temperature for overnight. The mixture was then poured in 1 N aq. HCl solution and extracted with CH2Cl2. The organic layer dried over magnesium sulfate, and then was purified by the column chromatography (CH2Cl2/MeOH). The residue was obtained as solid.Measurement of the anti-HCV activities of compounds using HCVcc
Cell line and cell culture. Huh 7.5.1 cells were grown in Dulbecco's Modified Eagle's medium (DMEM; Gibco) containing antibiotics (100 U mL−1 penicillin, 10 mg mL−1 streptomycin) and 10% heat-inactivated fetal bovine serum (DFBS; Hyclone) at 37 °C in a humidified 6.0% CO2 incubator.Virus production
In vitro transcribed RNA of JFH5a-Rluc-ad34, which is a derivative of JFH1 with adaptive mutations in the E2 and p7 regions, was transfected into Huh7.5.1 cells by electroporation. JFH5a-Rluc-ad34 virus contains a reporter Renilla luciferase (Rluc) for convenient virus proliferation assay.92 HCV-containing cell culture media were collected 3–5 days after electroporation and filtered through a 0.45 mM pore size filter.Antiviral activity test with infectious HCV particles
Huh 7.5.1 cells were inoculated with a JFH5a-Rluc-ad34 virus stock and cultivated for 3 h. At 3 h after the virus inoculation, HCV infected Huh7.5.1 cells were cultured with media containing serially diluted compounds. At 3 days after the chemical treatment, the cells were harvested and their luciferase activities were measured using a Renilla luciferase assay system (Promega) according to the manufacturer's direction. Finally, the luciferase activities were normalized to those obtained from mock-treated cells.Cell line and cell culture
Huh 7.5.1 cells containing a bicistronic HCV replicon NK5.1-Gluc cell line (genotype 1b) were employed for the interrogation of the anti-HCV activities of the compounds.64 The first open reading frame (ORF) of the replicon contains the Gaussia luciferase gene fused with foot-and-mouth disease virus (FMDV) 2a gene and the neomycin phosphotransferase gene, and the second ORF contains HCV nonstructural genes NS3-5. The replicon-containing cells were cultivated under the same conditions as described above in presence of an additional antibiotic G418 (0.5 mg mL−1, Calbiochem).Antiviral activity assay test with HCV replicon
Huh7.5.1 cells containing HCV replicon (NK5.1-Gluc) were planted on a 12-well plate. At 16 h after cultivation, the replicon containing NK/R2AN-containing cells were incubated with media containing serially diluted compounds for 3 days. After the chemical treatment, the cell culture media were collected and luciferase activities were measured through the use of the Renilla luciferase assay system (Promega) according to manufacturer's direction, and then normalized to those obtained from mock-treated cells.Molecular docking
The X-ray structure of 3FQQ and NMR structure of 1R7G were retrieved from the protein data bank (PDB) and prepared via homology modeling with Discovery Studio Client. The default ligand settings were used to prepare all tautomers, and further energy minimized. The inhibitors were then docked into each of the NS5A dimer models using the CDOCKER.Conflicts of interest
There are no conflicts of interest to declare regarding this study.Acknowledgements
We gratefully acknowledge the financial support of the Bio R&D Program (No. 2012M3A9A9054974 and No. 2017M3A9F6029755) through the National Research Foundation funded by the MSIT, Republic of Korea. This research also was supported by the Korea Institute of Science and Technology Information (KISTI) Institutional Program.Notes and references
- A. M. Di Bisceglie, Lancet, 1998, 351, 351–355 CrossRef.
- A. M. Di Bisceglie, Hepatology, 2000, 31, 1014–1018 CrossRef PubMed.
- W. R. Kim, Microbes Infect., 2002, 4, 1219–1225 CrossRef.
- N. Horscroft, V. C. H. Lai, W. Cheney, N. Yao, J. Z. Wu, Z. Hong and W. Zhong, Antiviral Chem. Chemother., 2005, 16, 1–12 CrossRef PubMed.
- S. A. Qureshi, Med. Res. Rev., 2007, 27, 353–373 CrossRef PubMed.
- T. O. Moore, M. Paradowski and S. E. Ward, Org. Biomol. Chem., 2016, 14, 3307–3313 RSC.
- L. Tong, W. Yu, L. Chen, O. Selyutin, M. P. Dwyer, A. G. Nair, R. Mazzola, J. H. Kim, D. Sha, J. Yin, R. T. Ruck, I. W. Davies, B. Hu, B. Zhong, J. Hao, T. Ji, S. Zan, R. Liu, S. Agrawal, E. Xia, S. Curry, P. McMonagle, K. Bystol, F. Lahser, D. Carr, L. Rokosz, P. Ingravallo, S. Chen, K. I. Feng, M. Cartwright, E. Asante-Appiah and J. A. Kozlowski, J. Med. Chem., 2017, 60, 290–306 CrossRef PubMed.
- K. J. Blight, A. A. Kolykhalov and C. M. Rice, Science, 2000, 290, 1972–1974 CrossRef PubMed.
- C. Liang, E. Rieder, B. Hahm, S. K. Jang, A. Paul and E. Wimmer, Virology, 2005, 333, 41–53 CrossRef PubMed.
- T. W. Bell, ChemMedChem, 2010, 5, 1663–1665 CrossRef PubMed.
- W. Cun, J. Jiang and G. Luo, J. Virol., 2010, 84, 11532–11541 CrossRef PubMed.
- J. L. Romine, D. R. St. Laurent, J. E. Leet, S. W. Martin, M. H. Serrano-Wu, F. Yang, M. Gao, D. R. O'Boyle, J. A. Lemm, J.-H. Sun, P. T. Nower, X. Huang, M. S. Deshpande, N. A. Meanwell and L. B. Snyder, ACS Med. Chem. Lett., 2011, 2, 224–229 CrossRef PubMed.
- D. A. DeGoey, J. T. Randolph, D. Liu, J. Pratt, C. Hutchins, P. Donner, A. C. Krueger, M. Matulenko, S. Patel, C. E. Motter, L. Nelson, R. Keddy, M. Tufano, D. D. Caspi, P. Krishnan, N. Mistry, G. Koev, T. J. Reisch, R. Mondal, T. Pilot-Matias, Y. Gao, D. W. Beno, C. J. Maring, A. Molla, E. Dumas, A. Campbell, L. Williams, C. Collins, R. Wagner and W. M. Kati, J. Med. Chem., 2014, 57, 2047–2057 CrossRef PubMed.
- M. S. Sulkowski, D. F. Gardiner, M. Rodriguez-Torres, K. R. Reddy, T. Hassanein, I. Jacobson, E. Lawitz, A. S. Lok, F. Hinestrosa, P. J. Thuluvath, H. Schwartz, D. R. Nelson, G. T. Everson, T. Eley, M. Wind-Rotolo, S.-P. Huang, M. Gao, D. Hernandez, F. McPhee, D. Sherman, R. Hindes, W. Symonds, C. Pasquinelli and D. M. Grasela, N. Engl. J. Med., 2014, 370, 211–221 CrossRef PubMed.
- T. I. Ng, P. Krishnan, T. Pilot-Matias, W. Kati, G. Schnell, J. Beyer, T. Reisch, L. Lu, T. Dekhtyar, M. Irvin, R. Tripathi, C. Maring, J. T. Randolph, R. Wagner and C. Collins, Antimicrob. Agents Chemother., 2017, 61, e02558-16 CrossRef PubMed.
- B. Liu, K. Gai, H. Qin, X. Liu, Y. Cao, Q. Lu, D. Lu, D. Chen, H. Shen, W. Song, Y. Zhang, X. Wang, H. Xu and Y. Zhang, Eur. J. Med. Chem., 2018, 148, 95–105 CrossRef PubMed.
- S. L. Tan, A. Pause, Y. Shi and N. Sonenberg, Nat. Rev. Drug Discovery, 2002, 1, 867–881 CrossRef PubMed.
- R. De Francesco and G. Migliaccio, Nature, 2005, 436, 953–960 CrossRef PubMed.
- K. Garber, Nat. Biotechnol., 2011, 29, 963–966 CrossRef PubMed.
- P. J. Pockros, Drugs, 2012, 72, 1825–1831 CrossRef PubMed.
- J. J. Kiser and C. Flexner, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 427–449 CrossRef PubMed.
- M. Zhong, E. Peng, N. Huang, Q. Huang, A. Huq, M. Lau, R. Colonno and L. Li, Bioorg. Med. Chem. Lett., 2014, 24, 5738–5742 CrossRef PubMed.
- S. L. Tan and M. G. Katze, Virology, 2001, 284, 1–12 CrossRef PubMed.
- T. L. Tellinghuisen, M. J. Evans, T. von Hahn, S. You and C. M. Rice, J. Virol., 2007, 81, 8853–8867 CrossRef PubMed.
- R. Bartenschlager, F. Penin, V. Lohmann and P. Andre, Trends Microbiol., 2011, 19, 95–103 CrossRef PubMed.
- J. Schlutter, Nature, 2011, 474, S5–S7 CrossRef.
- M. Belema, V. N. Nguyen, D. R. St. Laurent, O. D. Lopez, Y. Qiu, A. C. Good, P. T. Nower, L. Valera, D. R. O'Boyle, J.-H. Sun, M. Liu, R. A. Fridell, J. A. Lemm, M. Gao, J. O. Knipe, N. A. Meanwell and L. B. Snyder, Bioorg. Med. Chem. Lett., 2013, 23, 4428–4435 CrossRef PubMed.
- M. Issur and M. Götte, Viruses, 2014, 6, 4227–4241 CrossRef PubMed.
- J. H. Nettles, R. A. Stanton, J. Broyde, F. Amblard, H. Zhang, L. Zhou, J. Shi, T. R. McBrayer, T. Whitaker, S. J. Coats, J. J. Kohler and R. F. Schinazi, J. Med. Chem., 2014, 57, 10031–10043 CrossRef PubMed.
- D. Lavanchy, Liver Int., 2009, 29(suppl. 1), 74–81 CrossRef PubMed.
- E. R. Feeney and R. T. Chung, BMJ, 2014, 348, g3308 CrossRef PubMed.
- R. Wagner, J. T. Randolph, S. V. Patel, L. Nelson, M. A. Matulenko, R. Keddy, J. K. Pratt, D. Liu, A. C. Krueger, P. L. Donner, D. K. Hutchinson, C. Flentge, D. Betebenner, T. Rockway, C. J. Maring, T. I. Ng, P. Krishnan, T. Pilot-Matias, C. Collins, N. Panchal, T. Reisch, T. Dekhtyar, R. Mondal, D. F. Stolarik, Y. Gao, W. Gao, D. A. Beno and W. M. Kati, J. Med. Chem., 2018, 61, 4052–4066 CrossRef PubMed.
- N. A. Terrault, S. Zeuzem, A. M. Di Bisceglie, J. K. Lim, P. J. Pockros, L. M. Frazier, A. Kuo, A. S. Lok, M. L. Shiffman, Z. Ben Ari, L. Akushevich, M. Vainorius, M. S. Sulkowski, M. W. Fried, D. R. Nelson and H.-T. S. Group, Gastroenterology, 2016, 151, 1131–1140 CrossRef PubMed.
- A. Geddawy, Y. F. Ibrahim, N. M. Elbahie and M. A. Ibrahim, J. Transl. Int. Med., 2017, 5, 8–17 Search PubMed.
- D. J. Cada, J. Leonard, T. L. Levien and D. E. Baker, Hosp. Pharm., 2015, 50, 396–412 CrossRef PubMed.
- A. Tamori, M. Enomoto and N. Kawada, Mediators Inflammation, 2016, 2016, 6841628 Search PubMed.
- J. R. King and R. M. Menon, Clin. Pharmacol. Drug Dev., 2017, 6, 201–205 CrossRef PubMed.
- P. McCormack, Drugs, 2015, 75, 515–524 CrossRef PubMed.
- A. M. Bell, J. L. Wagner, K. E. Barber and K. R. Stover, Int. J. Hepatol., 2016, 2016, 3852126 Search PubMed.
- S. M. McConachie, S. M. Wilhelm and P. B. Kale-Pradhan, Expert Rev. Clin. Pharmacol., 2016, 9, 287–302 CrossRef PubMed.
- J. Yu, Z. Zhou, K. H. Owens, T. K. Ritchie and I. Ragueneau-Majlessi, Drug Metab. Dispos., 2017, 45, 86–108 CrossRef PubMed.
- E. Mogalian, P. German, B. P. Kearney, C. Y. Yang, D. Brainard, J. Link, J. McNally, L. Han, J. Ling and A. Mathias, Antimicrob. Agents Chemother., 2017, 61, e02084–16 CrossRef PubMed.
- E. B. Chahine, D. Kelley and L. M. Childs-Kean, Ann. Pharmacother., 2018, 52, 352–363 CrossRef PubMed.
- M. Gao, R. E. Nettles, M. Belema, L. B. Snyder, V. N. Nguyen, R. A. Fridell, M. H. Serrano-Wu, D. R. Langley, J. H. Sun, D. R. O'Boyle 2nd, J. A. Lemm, C. Wang, J. O. Knipe, C. Chien, R. J. Colonno, D. M. Grasela, N. A. Meanwell and L. G. Hamann, Nature, 2010, 465, 96–100 CrossRef PubMed.
- R. E. Nettles, M. Gao, M. Bifano, E. Chung, A. Persson, T. C. Marbury, R. Goldwater, M. P. DeMicco, M. Rodriguez-Torres, A. Vutikullird, E. Fuentes, E. Lawitz, J. C. Lopez-Talavera and D. M. Grasela, Hepatology, 2011, 54, 1956–1965 CrossRef PubMed.
- M. Belema, O. D. Lopez, J. A. Bender, J. L. Romine, D. R. St Laurent, D. R. Langley, J. A. Lemm, D. R. O'Boyle 2nd, J. H. Sun, C. Wang, R. A. Fridell and N. A. Meanwell, J. Med. Chem., 2014, 57, 1643–1672 CrossRef PubMed.
- D. R. St Laurent, M. H. Serrano-Wu, M. Belema, M. Ding, H. Fang, M. Gao, J. T. Goodrich, R. G. Krause, J. A. Lemm, M. Liu, O. D. Lopez, V. N. Nguyen, P. T. Nower, D. R. O'Boyle 2nd, B. C. Pearce, J. L. Romine, L. Valera, J. H. Sun, Y. K. Wang, F. Yang, X. Yang, N. A. Meanwell and L. B. Snyder, J. Med. Chem., 2014, 57, 1976–1994 CrossRef PubMed.
- N. A. Meanwell, J. Med. Chem., 2016, 59, 7311–7351 CrossRef PubMed.
- C. Wang, H. Huang, L. Valera, J.-H. Sun, D. R. O'Boyle II, P. T. Nower, L. Jia, D. Qiu, X. Huang, A. Altaf, M. Gao and R. A. Fridell, Antimicrob. Agents Chemother., 2012, 56, 1350–1358 CrossRef PubMed.
- A. Aghemo and R. De Francesco, Hepatology, 2013, 58, 428–438 CrossRef PubMed.
- E. De Clercq, Biochem. Pharmacol., 2014, 89, 441–452 CrossRef PubMed.
- S. Pol and M. Corouge, Med. Mal. Infect., 2014, 44, 449–454 CrossRef PubMed.
- A. Hill, B. Simmons, D. Gotham and J. Fortunak, J. Virus Erad., 2016, 2, 28–31 Search PubMed.
- P. J. Pockros, Expert Opin. Biol. Ther., 2011, 11, 1611–1622 CrossRef PubMed.
- C. Sheridan, Nat. Biotechnol., 2011, 29, 553–554 CrossRef PubMed.
- N. M. Dabbouseh and D. M. Jensen, Nat. Rev. Gastroenterol. Hepatol., 2013, 10, 268–276 CrossRef PubMed.
- J. A. Henderson, D. Bilimoria, M. Bubenik, C. Cadilhac, K. M. Cottrell, F. Denis, E. Dietrich, N. Ewing, G. Falardeau, S. Giroux, L. L'Heureux, B. Liu, N. Mani, M. Morris, O. Nicolas, O. Z. Pereira, C. Poisson, T. J. Reddy, S. Selliah, R. S. Shawgo, L. Vaillancourt, J. Wang, J. Xu, N. Chauret, F. Berlioz-Seux, L. C. Chan, S. K. Das, A. L. Grillot, Y. L. Bennani and J. P. Maxwell, Bioorg. Med. Chem. Lett., 2015, 25, 948–951 CrossRef PubMed.
- I. J. Kang, S. J. Hsu, H. Y. Yang, T. K. Yeh, C. C. Lee, Y. C. Lee, Y. W. Tian, J. S. Song, T. A. Hsu, Y. S. Chao, A. Yueh and J. H. Chern, J. Med. Chem., 2017, 60, 228–247 CrossRef PubMed.
- R. A. Fridell, C. Wang, J. H. Sun, D. R. O'Boyle 2nd, P. Nower, L. Valera, D. Qiu, S. Roberts, X. Huang, B. Kienzle, M. Bifano, R. E. Nettles and M. Gao, Hepatology, 2011, 54, 1924–1935 CrossRef PubMed.
- E. Poveda, D. L. Wyles, Á. Mena, J. D. Pedreira, Á. Castro-Iglesias and E. Cachay, Antiviral Res., 2014, 108, 181–191 CrossRef PubMed.
- C. Wang, L. Jia, D. R. O'Boyle 2nd, J. H. Sun, K. Rigat, L. Valera, P. Nower, X. Huang, B. Kienzle, S. Roberts, M. Gao and R. A. Fridell, Antimicrob. Agents Chemother., 2014, 58, 5155–5163 CrossRef PubMed.
- D. Ross-Thriepland and M. Harris, J. Gen. Virol., 2015, 96, 727–738 CrossRef PubMed.
- J. H. Sun, D. R. O'Boyle 2nd, R. A. Fridell, D. R. Langley, C. Wang, S. B. Roberts, P. Nower, B. M. Johnson, F. Moulin, M. J. Nophsker, Y. K. Wang, M. Liu, K. Rigat, Y. Tu, P. Hewawasam, J. Kadow, N. A. Meanwell, M. Cockett, J. A. Lemm, M. Kramer, M. Belema and M. Gao, Nature, 2015, 527, 245–248 CrossRef PubMed.
- Y. You, H. S. Kim, I. H. Bae, S. G. Lee, M. H. Jee, G. Keum, S. K. Jang and B. M. Kim, Eur. J. Med. Chem., 2017, 125, 87–100 CrossRef PubMed.
- A. V. Ivachtchenko, O. D. Mitkin, P. M. Yamanushkin, I. V. Kuznetsova, E. A. Bulanova, N. A. Shevkun, A. G. Koryakova, R. N. Karapetian, V. V. Bichko, A. S. Trifelenkov, D. V. Kravchenko, N. V. Vostokova, M. S. Veselov, N. V. Chufarova and Y. A. Ivanenkov, J. Med. Chem., 2014, 57, 7716–7730 CrossRef PubMed.
- W. M. Kazmierski, A. Maynard, M. Duan, S. Baskaran, J. Botyanszki, R. Crosby, S. Dickerson, M. Tallant, R. Grimes, R. Hamatake, M. Leivers, C. D. Roberts and J. Walker, J. Med. Chem., 2014, 57, 2058–2073 CrossRef PubMed.
- J. J. Kohler, J. H. Nettles, F. Amblard, S. J. Hurwitz, L. Bassit, R. A. Stanton, M. Ehteshami and R. F. Schinazi, Infect. Drug Resist., 2014, 7, 41–56 Search PubMed.
- S. Giroux, D. Bilimoria, C. Cadilhac, K. M. Cottrell, F. Denis, E. Dietrich, N. Ewing, J. A. Henderson, L. L'Heureux, N. Mani, M. Morris, O. Nicolas, T. J. Reddy, S. Selliah, R. S. Shawgo, J. Xu, N. Chauret, F. Berlioz-Seux, L. C. Chan, S. K. Das, A.-L. Grillot, Y. L. Bennani and J. P. Maxwell, Bioorg. Med. Chem. Lett., 2015, 25, 940–943 CrossRef PubMed.
- J. O. Link, J. G. Taylor, L. Xu, M. Mitchell, H. Guo, H. Liu, D. Kato, T. Kirschberg, J. Sun, N. Squires, J. Parrish, T. Keller, Z.-Y. Yang, C. Yang, M. Matles, Y. Wang, K. Wang, G. Cheng, Y. Tian, E. Mogalian, E. Mondou, M. Cornpropst, J. Perry and M. C. Desai, J. Med. Chem., 2014, 57, 2033–2046 CrossRef PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef PubMed.
- F. Amblard, H. Zhang, L. Zhou, J. Shi, D. R. Bobeck, J. H. Nettles, S. Chavre, T. R. McBrayer, P. Tharnish, T. Whitaker, S. J. Coats and R. F. Schinazi, Bioorg. Med. Chem. Lett., 2013, 23, 2031–2034 CrossRef PubMed.
- S. Giroux, J. Xu, T. J. Reddy, M. Morris, K. M. Cottrell, C. Cadilhac, J. A. Henderson, O. Nicolas, D. Bilimoria, F. Denis, N. Mani, N. Ewing, R. Shawgo, L. L'Heureux, S. Selliah, L. Chan, N. Chauret, F. Berlioz-Seux, M. N. Namchuk, A. L. Grillot, Y. L. Bennani, S. K. Das and J. P. Maxwell, ACS Med. Chem. Lett., 2014, 5, 240–243 CrossRef PubMed.
- D. B. Ascher, J. Wielens, T. L. Nero, L. Doughty, C. J. Morton and M. W. Parker, Sci. Rep., 2014, 4, 4765 CrossRef PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2014, 53, 9430–9448 CrossRef PubMed.
- J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed. Engl., 2014, 53, 9466–9470 CrossRef PubMed.
- S. Li, L. T. Beringer, S. Chen and S. Averick, Polymer, 2015, 78, 37–41 CrossRef.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- J. Yatvin, K. Brooks and J. Locklin, Chem.–Eur. J., 2016, 22, 16348–16354 CrossRef PubMed.
- J. Yatvin, K. Brooks and J. Locklin, Angew. Chem., Int. Ed. Engl., 2015, 54, 13370–13373 CrossRef PubMed.
- A. Dondoni and A. Marra, Org. Biomol. Chem., 2017, 15, 1549–1553 RSC.
- B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083–1088 CrossRef PubMed.
- K. S. Lim, D. W. Kang, Y. S. Kim, M. S. Kim, S. G. Park, S. Choi, L. V. Pearce, P. M. Blumberg and J. Lee, Bioorg. Med. Chem. Lett., 2011, 21, 299–302 CrossRef PubMed.
- J. Shi, L. Zhou, F. Amblard, D. R. Bobeck, H. Zhang, P. Liu, L. Bondada, T. R. McBrayer, P. M. Tharnish, T. Whitaker, S. J. Coats and R. F. Schinazi, Bioorg. Med. Chem. Lett., 2012, 22, 3488–3491 CrossRef PubMed.
- C. A. Coburn, P. T. Meinke, W. Chang, C. M. Fandozzi, D. J. Graham, B. Hu, Q. Huang, S. Kargman, J. Kozlowski, R. Liu, J. A. McCauley, A. A. Nomeir, R. M. Soll, J. P. Vacca, D. Wang, H. Wu, B. Zhong, D. B. Olsen and S. W. Ludmerer, ChemMedChem, 2013, 8, 1930–1940 CrossRef PubMed.
- M. Zhong, E. Peng, N. Huang, Q. Huang, A. Huq, M. Lau, R. Colonno and L. Li, Bioorg. Med. Chem. Lett., 2014, 24, 5731–5737 CrossRef PubMed.
- L. C. King and G. K. Ostrum, J. Org. Chem., 1964, 29, 3459–3461 CrossRef.
- S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef PubMed.
- R. Zelli, S. Tommasone, P. Dumy, A. Marra and A. Dondoni, Eur. J. Org. Chem., 2016, 5102–5116 CrossRef.
- B. Kim, C.-E. Yeom, H. Kim and S. Lee, Synlett, 2007, 1, 0146–0150 CrossRef.
- I. H. Bae, J. K. Choi, C. Chough, S. J. Keum, H. Kim, S. K. Jang and B. M. Kim, ACS Med. Chem. Lett., 2014, 5, 255–258 CrossRef PubMed.
- I. H. Bae, H. S. Kim, Y. You, C. Chough, W. Choe, M. K. Seon, S. G. Lee, G. Keum, S. K. Jang and B. Moon Kim, Eur. J. Med. Chem., 2015, 101, 163–178 CrossRef PubMed.
- C. S. Kim, S. J. Keum and S. K. Jang, PLoS One, 2011, 6, e22808 CrossRef PubMed.
- P. J. Lim, U. Chatterji, D. Cordek, S. D. Sharma, J. A. Garcia-Rivera, C. E. Cameron, K. Lin, P. Targett-Adams and P. A. Gallay, J. Biol. Chem., 2012, 287, 30861–30873 CrossRef PubMed.
- B. D. Lindenbach, M. J. Evans, A. J. Syder, B. Woelk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating and C. M. Rice, Science, 2005, 309, 623–626 CrossRef PubMed.
- G. Randall, M. Panis, J. D. Cooper, T. L. Tellinghuisen, K. E. Sukhodolets, S. Pfeffer, M. Landthaler, P. Landgrafe, S. Kan, B. D. Lindenbach, M. Chien, D. B. Weir, J. J. Russo, J. Ju, M. J. Brownstein, R. Sheridan, C. Sander, M. Zavolan, T. Tuschl and C. M. Rice, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12884–12889 CrossRef PubMed.
- O. D. Lopez, V. N. Nguyen, D. R. St. Laurent, M. Belema, M. H. Serrano-Wu, J. T. Goodrich, F. Yang, Y. Qiu, A. S. Ripka, P. T. Nower, L. Valera, M. Liu, D. R. O'Boyle, J.-H. Sun, R. A. Fridell, J. A. Lemm, M. Gao, A. C. Good, N. A. Meanwell and L. B. Snyder, Bioorg. Med. Chem. Lett., 2013, 23, 779–784 CrossRef PubMed.
- M. Belema, V. N. Nguyen, J. L. Romine, D. R. St. Laurent, O. D. Lopez, J. T. Goodrich, P. T. Nower, D. R. O'Boyle II, J. A. Lemm, R. A. Fridell, M. Gao, H. Fang, R. G. Krause, Y.-K. Wang, A. J. Oliver, A. C. Good, J. O. Knipe, N. A. Meanwell and L. B. Snyder, J. Med. Chem., 2014, 57, 1995–2012 CrossRef PubMed.
- S. Giroux, D. Bilimoria, C. Cadilhac, K. M. Cottrell, F. Denis, E. Dietrich, N. Ewing, J. A. Henderson, L. L'Heureux, N. Mani, M. Morris, O. Nicolas, T. J. Reddy, S. Selliah, R. S. Shawgo, J. Xu, N. Chauret, F. Berlioz-Seux, L. C. Chan, S. K. Das, A. L. Grillot, Y. L. Bennani and J. P. Maxwell, Bioorg. Med. Chem. Lett., 2015, 25, 936–939 CrossRef PubMed.
- S. Li, D. Cohen-Karni, L. T. Beringer, C. Wu, E. Kallick, H. Edington, M. J. Passineau and S. Averick, Polymer, 2016, 99, 7–12 CrossRef.
- P. Targett-Adams, E. J. S. Graham, J. Middleton, A. Palmer, S. M. Shaw, H. Lavender, P. Brain, T. D. Tran, L. H. Jones, F. Wakenhut, B. Stammen, D. Pryde, C. Pickford and M. Westby, J. Virol., 2011, 85, 6353–6368 CrossRef PubMed.
- M. Belema and N. A. Meanwell, J. Med. Chem., 2014, 57, 5057–5071 CrossRef PubMed.
- D. R. O'Boyle Ii, J. H. Sun, P. T. Nower, J. A. Lemm, R. A. Fridell, C. Wang, J. L. Romine, M. Belema, V. N. Nguyen, D. R. Laurent, M. Serrano-Wu, L. B. Snyder, N. A. Meanwell, D. R. Langley and M. Gao, Virology, 2013, 444, 343–354 CrossRef PubMed.
- R. A. Love, O. Brodsky, M. J. Hickey, P. A. Wells and C. N. Cronin, J. Virol., 2009, 83, 4395–4403 CrossRef PubMed.
- F. Penin, V. Brass, N. Appel, S. Ramboarina, R. Montserret, D. Ficheux, H. E. Blum, R. Bartenschlager and D. Moradpour, J. Biol. Chem., 2004, 279, 40835–40843 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05471a |
| This journal is © The Royal Society of Chemistry 2018 |