
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of multifunctional CuFe2O4–reduced graphene oxide nanocomposite: an efficient magnetically separable catalyst as well as high performance supercapacitor and first-principles calculations of its electronic structures†
Madhurya Chandel,
Debabrata Moitra,
Priyanka Makkar,
Harshit Sinha,
Harshdeep Singh Hora and
Narendra Nath Ghosh *
*
Nano-materials Lab, Department of Chemistry, Birla Institute of Technology and Science, Pilani K K Birla Goa Campus, Goa-403726, India. E-mail: naren70@yahoo.com; Fax: +91 832 2557033; Tel: +91 832 2580318
First published on 3rd August 2018
Abstract
Here, we report an ‘in situ’ co-precipitation reduction based synthetic methodology to prepare CuFe2O4 nanoparticle–reduced graphene oxide (CuFe2O4–RGO) nanocomposites. First principles calculations based on Density Functional Theory (DFT) were performed to obtain the electronic structures and properties of CuFe2O4, graphene and CuFe2O4–graphene composites, and to understand the interfacial interaction between CuFe2O4 and graphene in the composite. The synergistic effect, which resulted from the combination of the unique properties of RGO and CuFe2O4 nanoparticles, was exploited to design a magnetically separable catalyst and high performance supercapacitor. It has been demonstrated that the incorporation of RGO in the composite enhanced its catalytic properties as well as supercapacitance performance compared with pure CuFe2O4. The nanocomposite with 96 wt% CuFe2O4 and 4 wt% RGO (96CuFe2O4–4RGO) exhibited high catalytic efficiency towards (i) reduction of 4-nitrophenol to 4-aminophenol, and (ii) epoxidation of styrene to styrene oxide. For both of these reactions, the catalytic efficiency of 96CuFe2O4–4RGO was significantly higher than that of pure CuFe2O4. The easy magnetic separation of 96CuFe2O4–4RGO from the reaction mixture and good reusability of the recovered catalyst also showed here. 96CuFe2O4–4RGO also demonstrated better supercapacitance performance than pure CuFe2O4. 96CuFe2O4–4RGO showed specific capacitance of 797 F g−1 at a current density of 2 A g−1, along with ∼92% retention for up to 2000 cycles. To the best of our knowledge, this is the first investigation on the catalytic properties of CuFe2O4–RGO towards the reduction of 4-nitrophenol and the epoxidation reaction, and DFT calculations on the CuFe2O4–graphene composite have been reported.
1. Introduction
For the last few years, graphene-based materials have attracted immense attention from scientists and technologists due to their plethora of applications in myriad fields.1–3 The interesting properties of graphene, such as mechanical flexibility, low density, high surface area, 2-dimensional honeycomb structure with sp2 hybridized C-atoms, excellent electrical conductivity, etc., have been exploited in several applications.1–4 It has been well demonstrated that graphene can act as a support for various nanoparticles.1,3,5 The interfacial interactions between the graphene and nanoparticles affect the electronic structure of both the graphene and the nanoparticles, which are immobilized on its surface.6–8 Hence, there is a scope to design tailor-made graphene-based composite materials with superior properties by exploring this synergistic effect.In this paper, we are reporting the synthesis of nanocomposites composed of CuFe2O4 nanoparticles and reduced graphene oxide (RGO). The synthesized nanocomposites displayed their applications in two different but important fields, (i) as a magnetically separable catalyst for two reactions, and (ii) as an active material to fabricate high performing supercapacitor. To evaluate the catalytic activities of CuFe2O4–RGO nanocomposites, we have chosen two reactions (i) reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in presence of excess NaBH4 in an aqueous medium, and (ii) epoxidation of styrene to styrene oxide in presence of tert-butyl hydroperoxide (TBHP). Reduction of 4-NP to 4-AP has been chosen as one of the catalysis reactions because this reaction has been used as a model reaction to study the catalytic property of varieties of nanostructured catalysts by several researchers.9–11 It has been well proven that this reaction is not only simple to perform but also convincing and trustworthy for this purpose. Moreover, 4-NP and its derivatives are the byproducts produced during the manufacture of many synthetic dyes, pesticides, and herbicides.10,12 The industrial effluents containing 4-NP cause severe water pollution because it is harmful to the liver, kidney, central nervous system, etc.10 Therefore, removal or destruction of 4-NP from the polluted aquatic system is an important task. On the other hand, 4-AP, which is produced due to the reduction of 4-NP, is an essential chemical in many pharmaceutical industries for the production of various antipyretic and analgesic drugs. 4-AP is also used in anti-corrosion lubricant, photographic developer, etc.10 The other catalysis reaction we have investigated here is the epoxidation of styrene to styrene oxide because styrene oxide is used in varieties of applications, such as synthesis of fine chemicals, medicines, many commodities, commercial resin, etc.13,14 However, selective epoxidation of styrene is a challenging issue. The conventional processes for styrene oxide production (e.g. non-catalytic process using chlorine, peracids based catalytic process, etc.) suffer from several limitations, including the production of large amount of waste products, chlorine-laden sewage, difficulty in handling corrosive peracids, difficulty in separation of homogeneous catalysts at the end of the reactions, etc.13,15 Hence, there is a need to develop an efficient catalyst which not only will show high selectivity towards styrene oxide formation but also will not produce any pollutant as a waste product, and will be easily separable from the reaction mixture.
We have also explored the possibility to develop supercapacitor using CuFe2O4–RGO nanocomposites because in recent years the development of electrochemical systems for energy storage and energy conversion has intensified many folds to address the issues related to the increasing cost of energy, exhaustion of fossil fuel reserves, and environmental pollution.16,17 Hence, supercapacitors, which are intermediate systems between the battery and a conventional capacitor, are appearing as an attractive alternative due to their excellent property to deliver high power density along with long life cycle.18–21 Generally, charging and discharging times of supercapacitor is in seconds. Though their energy density (∼5 W h kg−1) is lower than the conventional batteries, they generally deliver higher power density (∼10 kW kg−1) for a shorter time.16,20,22,23 Therefore, supercapacitors can find applications in advanced energy storage systems (e.g., uninterrupted power supplies, load leveling, etc.) as either replacement or complement to the batteries.16,24 Electrochemical double layer electrodes (EDL) and pseudocapacitor electrodes are two prominent class of supercapacitor.25–27 Use of EDL electrode in the AIRBUS A380 is one of the examples of the commercial applications of the supercapacitor.28 However, most of the EDL electrodes suffer from lower energy density than batteries, which often limits their wide application.16 Till date several metal oxides (e.g., RuO2, MnO2, Fe3O4), conducting polymers, and their composites are used to construct pseudocapacitive electrodes.16,29–32 Amongst the transition metal oxide, RuO2 and MnO2 have been extensively investigated.30–32 Though RuO2 exhibits high specific capacitance (>600 F g−1) in aqueous medium but RuO2 based electrodes are very expensive for the large-scale common application.16 The usages of MnO2 is limited because it does not possess any oxidation state below 0 V.16 Recently spinel ferrites (MFe2O4 where M = Mn, Co, Ni, Cu, Zn) are utilized as active material for pseudocapacitive electrode, because of the synergistic effect of Fe and M ions results in richer redox reactions to achieve higher specific capacitance.18,25,33,34 Though ferrites have been regarded as promising active materials to construct supercapacitors, their inherent poor electrical conductivity detrimentally affects their supercapacitance performance.25
In this study, we have designed the nanocomposites consist of CuFe2O4 and RGO. In these nanocomposites, CuFe2O4 has been chosen as one of the components due to its magnetic nature, ability to act as a catalyst for various reactions,35–39 and high theoretical capacity (895 mA h g−1).18 To overcome the limitation associated with its low electrical conductivity, CuFe2O4 nanoparticles have been immobilized on the surface of highly conducting RGO sheets. To understand the influence of RGO on the electronic structure of CuFe2O4, first principles quantum mechanical calculations based on Density Functional Theory (DFT) has been performed.
Though CuFe2O4–RGO nanocomposite is a very promising material till date, it has not yet been well explored. Only limited number of publications are available in the literature, where the synthesis of CuFe2O4–RGO by hydrothermal/solvothermal, and one pot co-precipitation method has been reported.5,18,40–44 Researchers have investigated the application of this nanocomposite in the fields of catalysis reactions (e.g. phenol hydroxylation, photocatalysis degradation of methylene blue, oxidative coupling of amine, and chemoselective reduction of nitroarenes), glucose sensing, and energy storage (supercapacitor, anode material for lithium-ion batteries).5,18,40–44
To the best of our knowledge, this is the first time first-principle calculations based on DFT theory has been reported for CuFe2O4–graphene composite to obtain its electronic structure, which helps us to understand how the presence of RGO influences the electronic property of CuFe2O4 in the nanocomposite, and to interpret the enhanced catalytic property as well as supercapacitor property of CuFe2O4–RGO nanocomposites. Here, we firstly reported the synthesis of CuFe2O4–RGO nanocomposites by an ‘in situ’ co-precipitation reduction technique and their structural characterization by several techniques. The electronic structure of the nanocomposite was determined by performing first principle calculations based on DFT. Quantum ESPRESSO package45 was used for DFT calculations because this package was used by several researchers to calculate a variety of semiconductors, ferrites, graphene, and graphene-based composites.46–52 The catalytic performance of the as-synthesized nanocomposites was tested for (i) reduction of 4-NP in presence of NaBH4, and (ii) epoxidation of styrene. Finally, the supercapacitance performance of CuFe2O4–RGO as electrode material was tested first by constructing a three electrode system, and then a two-electrode system.
2. Experimental
2.1. Materials
Copper nitrate trihydrate (Cu(NO3)2·3H2O), iron nitrate nonahydrate (Fe(NO3)3·9H2O), sodium nitrate (NaNO3), 30% H2O2 solution and potassium permanganate (KMnO4) were purchased from Merck, India. Sulphuric acid (H2SO4), sodium hydroxide (NaOH), and 4-nitrophenol (4-NP) were purchased from Fisher Scientific. Styrene, styrene oxide, tert-butyl hydroperoxide (TBHP in 5–6 M decane), acetonitrile, sodium borohydride (NaBH4), polyvinylidene difluoride (PVDF), acetylene black (SA 75 m2 g−1, bulk density 170–230 g L−1), N-methyl-2-pyrrolidinone (NMP) and graphite powder (mean particle size of <20 μm) were bought from Sigma Aldrich. All the chemicals were used without further purification. Distilled water was used throughout the experiment.2.2. Synthesis of CuFe2O4–RGO nanocomposite
Here, we have synthesized CuFe2O4–RGO nanocomposites, having a varying amount of RGO content, by employing an ‘in situ’ co-precipitation reduction technique. The preparation of the nanocomposite was performed in two steps. In the first step graphene oxide (GO) was prepared by employing the modified Hummer's method.53 The detailed preparation procedure of GO has been provided in ESI.† In the second step, in a round-bottomed flask stoichiometric amount of Cu(NO3)2·3H2O, Fe(NO3)3·9H2O and a dispersion of GO in water were taken. This mixture was stirred for 2 h using a magnetic stirrer after addition of a calculated amount of water. Then to this mixture, 2 M aqueous solution of NaOH was added dropwise until the pH of the mixture was reached ∼11. This reaction mixture was then refluxed at 160 °C for 16 h in an oil bath. After completion of the reaction, the reaction mixture was allowed to cool to room temperature and then the reddish black precipitate thus formed was separated from the mixture by applying a magnet externally. The collected precipitate was washed with water several times until the pH of the washing became ∼7. Then it was dried for 10 h at 60 °C. Using this procedure several compositions of CuFe2O4–RGO nanocomposites were prepared, such as 98CuFe2O4–2RGO, 96CuFe2O4–4RGO, 94CuFe2O4–6RGO and 92CuFe2O4–8RGO where 2, 4, 6 and 8 wt% of RGO was present, respectively. Pure CuFe2O4 nanoparticles were also synthesized using the same procedure, but here GO was not added to the reaction mixture.2.3. Catalytic activity tests
| dCt/dt = −kapp Ct | (1) |
| ln(Ct/C0) = ln(At/A0) = −kappt | (2) |
The value of kapp was calculated from the slope of the ln(At/A0) vs. time plot.
After each reaction cycle, the catalyst was separated from the reaction mixture by applying an external magnet (N35 grade NdFeB magnet having energy product BHmax = 33–36 MGO) and then washed with distilled water and ethanol. It was observed that no unreacted 4-NP molecule was remained adsorbed in the catalyst. After washing, the catalyst was dried for the next reaction cycle.
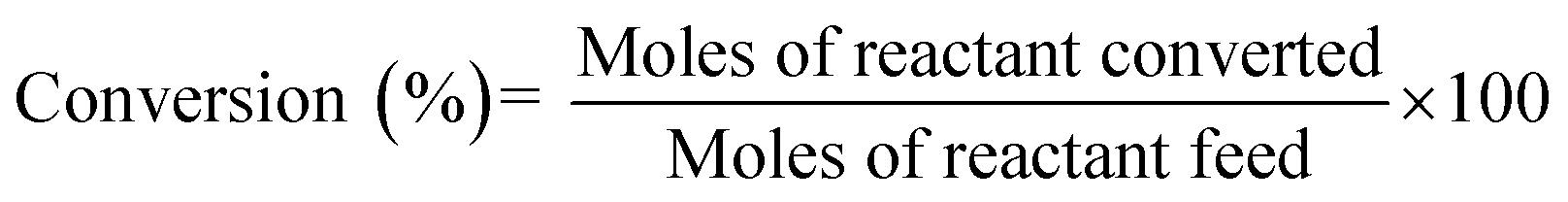 | (3) |
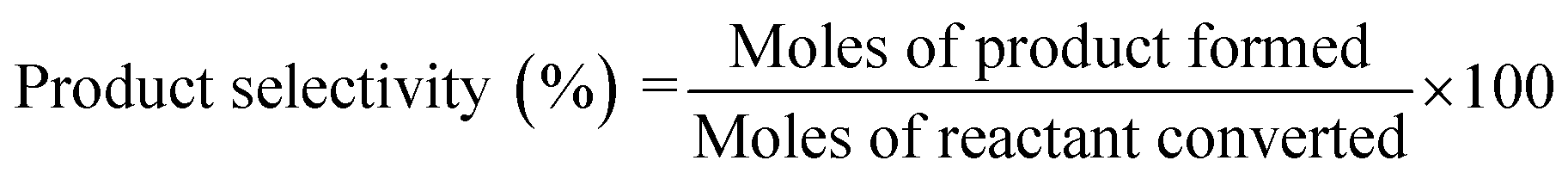 | (4) |
2.4. Electrochemical testing
In the present study, to determine the electrochemical properties of the synthesized materials, the three-electrode cell was constructed for electrochemical measurements. Here, to fabricate working electrode pure CuFe2O4, RGO and CuFe2O4–RGO nanocomposites were used as active materials. First, a viscous paste was made by mixing 80 wt% of active material, 10 wt% acetylene black, and 10 wt% polyvinylidene difluoride in N-methyl-2-pyrrolidinone. This viscous paste was cast on nickel foam (1.5 cm × 1.5 cm × 0.2 mm). Then this electrode was dried at 80 °C for 12 h in a vacuum oven. In this working electrode, the amount of active materials load on Ni Foam was ∼3 mg. In the three electrode system apart from this working electrode, an Ag/AgCl electrode was used as the reference electrode and a Pt wire as the counter electrode. Electrochemical measurements were performed using two electrolytes (i) 3 M KOH aqueous solution, and (ii) an aqueous mixture of 3 M KOH and 0.1 M K4[Fe(CN)6].The cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were performed using a galvanostat–potentiostat. All the CV measurements were conducted in the working potential window between 0 to 0.6 V, and using various scan rates between 10 to 100 mV s−1. GCD measurements were carried out at different current densities ranging from 2 to 10 A g−1.
GCD measurements were employed to determine the specific capacitance (Cs) of pure CuFe2O4 and 96CuFe2O4–4RGO nanocomposite and the following equation was used:18,24
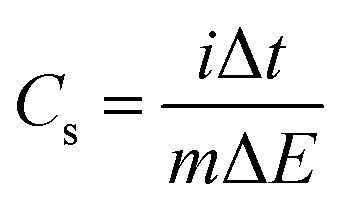 | (5) |
The energy density (E), and power density (P) of the electrodes were calculated from the GCD by using the following equations:18,24
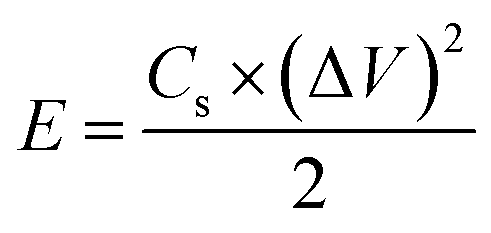 | (6) |
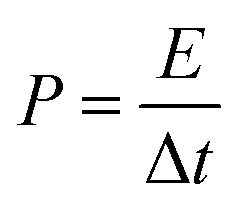 | (7) |
Electrochemical Impedance Spectroscopy (EIS) measurements were conducted in the frequency range of 0.01–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz at open circuit potential with an alternating current amplitude of 0.01 V.
000 Hz at open circuit potential with an alternating current amplitude of 0.01 V.
2.5. Characterization and instrumentation
Room temperature powder X-ray diffraction (XRD) patterns of the synthesized materials were recorded using a powder X-ray diffractometer (Mini Flex II, Rigaku, Japan) with Cu Kα (λ = 0.15405 nm) radiation at a scanning speed of 3° min−1. Fourier Transform Infrared spectra (FT-IR) were recorded in KBr by using spectrophotometer (IR Affinity-1, Shimadzu, Japan). Thermogravimetric analysis (TGA) was carried out using DTA-60 (Shimadzu, Japan). Multiple point BET (Brunauer–Emmett–Teller) surface areas were measured with a surface area and porosity analyzer (Micromeritics Tristar 3000, USA). Samples were degassed at 100 °C for ∼6 h under the nitrogen atmosphere before the analysis. Field Emission Scanning Electron Microscope (FESEM) images of samples were obtained using Quanta 250 FEG (FEI). Energy dispersive X-ray spectra of the synthesized material were recorded using an EDAX ELEMENT electron microscope. High-Resolution Transmission Electron Microscope (HRTEM) images of the samples were obtained using a JEOL JEM 1400, Japan. Raman spectra was recorded on a Renishaw In Via Raman microscope with a 633 nm laser excitation. Room temperature magnetization with respect to an external magnetic field was measured for the synthesized catalysts using a Vibrating Sample Magnetometer (VSM) (EV5, ADE Technology, USA). UV-Vis diffuse reflectance spectra (DRS) was recorded using JASCO V-770 spectrophotometer and energy band gap was calculated from the plot of Kubelka–Munk function versus photon energy. IVIUMSTAT (10 V/5 A/8 MHz) workstation was used to perform the electrochemical studies.2.6. First-principles calculations
In the present work, the first principles quantum mechanical calculations based on DFT were performed to obtain the electronic structures of CuFe2O4, graphene, and CuFe2O4–graphene nanocomposites. The ground state structures, binding energy, and Density of States (DOS) have been calculated with the Quantum ESPRESSO computational package45 using a plane wave set and pseudopotentials. DFT was used with Generalized Gradient Approximation (GGA)54 and parameterized by Perdew, Burke, and Ernzerhof (PBE).55 Kohn–Sham orbitals were expanded in a plane-wave basis set up to the kinetic energy cutoff 30 Ry (408 eV). The convergence criteria for the self-consistent calculation was 10−6 Ry, and the energy tolerance was 1 × 10−4 Ry (1.36 × 10−3 eV), while the force tolerance was set to 1 × 10−3 Ry per Bohr (0.0257 eV Å−1). It is a well-known fact that the GGA approach underestimates the band gap for semiconductors and insulators,46 and the weak interactions are not well described by standard PBE function.56–58 Therefore, for calculating the final electronic properties of the structures, an empirical dispersion- corrected density functional theory (DFT-D2) approach, which has proposed by Grimme57,59,60 was adopted. The Grimme-D2 (ref. 57–59) correction was used to account for the weak intermolecular interactions and van der Waals (vdW) interactions. All calculations were performed considering spin polarization.Here three systems were calculated: (i) superlattice of CuFe2O4 with face-centered cubic structure (space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m),61,62 (ii) graphene, and (iii) CuFe2O4–graphene composite. Ultrasoft pseudopotential for these systems were constructed by using 17, 14, 6, and 4 electrons for Cu (3p63d104s1), Fe (3p63d64s2), O (2s22p4), and C (2s22p2), respectively. The pseudopotentials for Cu, Fe, O, and C were chosen from the Quantum ESPRESSO website.63 The Monkhorst–Pack approach64,65 was used to select the k-point mesh (the details of k points for each system are provided as computational details in ESI†). The Brillouin zone integration was carried out with Methfessel–Paxton smearing technique66 for CuFe2O4 unit cell and Marzari–Vanderbilt smearing technique67 for CuFe2O4 (111) slab, graphene, and CuFe2O4 (111)-graphene superlattice and the smearing parameter was 0.005 Ry. The binding energy was calculated from the difference between the total energy of the CuFe2O4–graphene and the sum of each system (i.e. CuFe2O4 and graphene) alone. The sizes of the unit cells of the simulated systems are listed in Table S1 (ESI†) Details of the sample input files for geometric optimization of CuFe2O4, graphene, and CuFe2O4–graphene superlattice are provided in the ESI.†
m),61,62 (ii) graphene, and (iii) CuFe2O4–graphene composite. Ultrasoft pseudopotential for these systems were constructed by using 17, 14, 6, and 4 electrons for Cu (3p63d104s1), Fe (3p63d64s2), O (2s22p4), and C (2s22p2), respectively. The pseudopotentials for Cu, Fe, O, and C were chosen from the Quantum ESPRESSO website.63 The Monkhorst–Pack approach64,65 was used to select the k-point mesh (the details of k points for each system are provided as computational details in ESI†). The Brillouin zone integration was carried out with Methfessel–Paxton smearing technique66 for CuFe2O4 unit cell and Marzari–Vanderbilt smearing technique67 for CuFe2O4 (111) slab, graphene, and CuFe2O4 (111)-graphene superlattice and the smearing parameter was 0.005 Ry. The binding energy was calculated from the difference between the total energy of the CuFe2O4–graphene and the sum of each system (i.e. CuFe2O4 and graphene) alone. The sizes of the unit cells of the simulated systems are listed in Table S1 (ESI†) Details of the sample input files for geometric optimization of CuFe2O4, graphene, and CuFe2O4–graphene superlattice are provided in the ESI.†
3. Results and discussion
3.1. Structure and morphology of the synthesized CuFe2O4, RGO, and CuFe2O4–RGO nanocomposites
The structural characterizations of the synthesized pure CuFe2O4, RGO, and CuFe2O4–RGO were performed by using room temperature powder XRD, FT-IR, Raman spectroscopy, TGA, FESEM, and TEM. Fig. 1 shows the XRD patterns of GO, CuFe2O4, and CuFe2O4–RGO nanocomposites. For GO sample, the presence of an intense diffraction peak at 2θ = 9.76°, and a small peak at 42.14° corresponding to (001) and (101) planes of GO were observed.3,46,68,69 In the XRD patterns of CuFe2O4, and CuFe2O4–RGO diffraction peaks at 2θ = 18.16°, 30.11°, 35.62°, 37.36°, 39.01°, 43.27°, 57.46°, and 62.72° were present which correspond to (111), (220), (311), (202), (222), (400), (511), and (440) planes of cubic of CuFe2O4 (JCPDS card no. 77-0010). This fact indicated the presence of CuFe2O4 nanoparticles in CuFe2O4–RGO nanocomposites. The crystallite size of CuFe2O4 nanoparticle was determined by Scherrer's equation using (311) plane of CuFe2O4 and found to be ∼14 nm. It was also observed that for CuFe2O4–RGO samples no diffraction peaks of GO were present. Therefore, it was concluded that during the synthesis of CuFe2O4–RGO via ‘in situ’ co-precipitation method GO was converted to RGO along with the formation of CuFe2O4 nanoparticles. Moreover, no impurity phases were detected in the XRD patterns of CuFe2O4–RGO. The conversion of GO to RGO was also indicated by FT-IR, Raman spectroscopy, and TG analysis.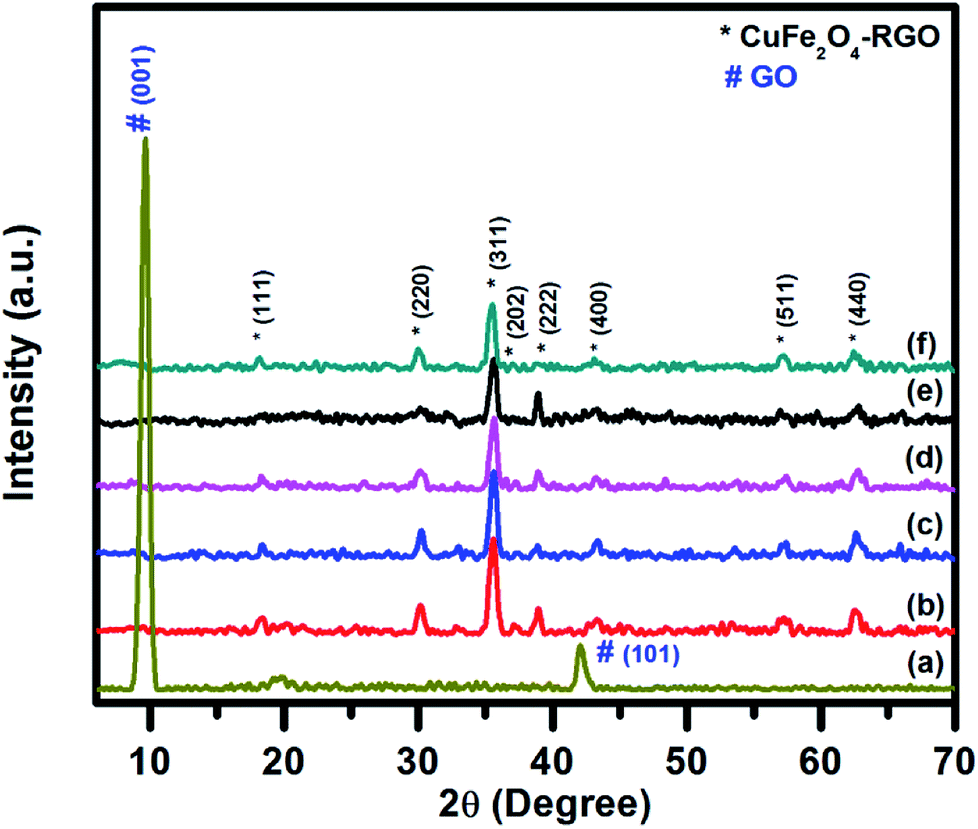 | ||
| Fig. 1 Room temperature wide angle powder XRD pattern of (a) GO, (b) pure CuFe2O4, (c) 98CuFe2O4–2RGO, (d) 96CuFe2O4–4RGO, (e) 94CuFe2O4–6RGO, and (f) 92CuFe2O4–8RGO. | ||
Fig. 2 displays the TEM micrographs of pure CuFe2O4 nanoparticles, and CuFe2O4–RGO nanocomposite. These micrographs revealed that the average particle size of the CuFe2O4 nanoparticles are 15–20 nm and these CuFe2O4 nanoparticles are immobilized on the surface of (the nanometer thin) RGO sheets. SAED patterns (Fig. 2b) show the Debye–Scherrer diffraction rings for pure CuFe2O4. In HRTEM micrographs (Fig. 2c) well-resolved lattice fringes corresponding to (311) plane of CuFe2O4 were observed. EDS analysis of these nanocomposites also confirmed their compositions. As a representative EDS of 96CuFe2O4–4RGO is shown in Fig. S1 (ESI†).
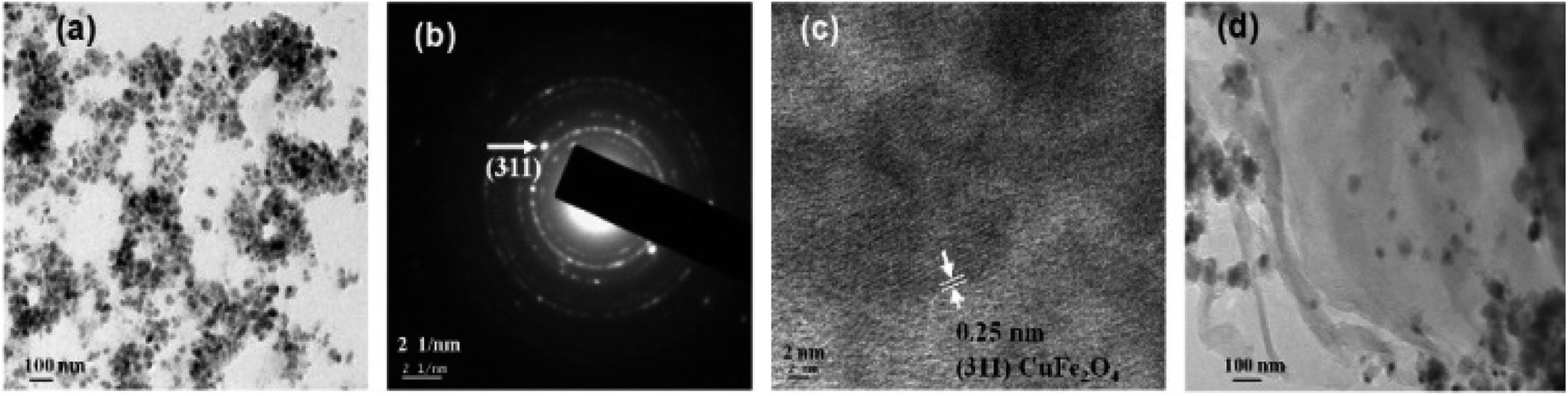 | ||
| Fig. 2 TEM micrographs of (a) pure CuFe2O4, (b) SAED pattern of CuFe2O4, (c) HRTEM micrograph of a typical portion of a corresponding CuFe2O4 and (d) CuFe2O4–RGO. | ||
The FT-IR spectra of GO (Fig. S2, ESI†) showed the presence of peaks at (i) 1232 cm−1 corresponding to the C–O stretching vibration of epoxy groups, (ii) 1728 cm−1 for the carbonyl groups, (iii) 1382 cm−1 corresponding to the –C–O stretching vibration of carboxylic groups, and (iv) 1056 cm−1 for the C–O stretching vibration. Therefore, the FT-IR spectra clearly indicated the presence of carbonyl group, carboxylic group, and epoxy groups on the surface of GO. Moreover, the characteristic peak of the skeletal vibration of the graphitic domains at 1621 cm−1 was also observed.68,69 For CuFe2O4–RGO samples, the disappearance of the peaks at 1728 cm−1, 1231 cm−1 and the decrease of the intensity of the peak at 1382 cm−1 indicated that during formation of CuFe2O4–RGO nanocomposites the oxygen-containing groups (e.g. epoxy, carbonyl, carboxyl) of GO have been reduced significantly and GO has converted to RGO. In the spectra of CuFe2O4–RGO, the presence of a band at 594 cm−1 was also observed, which can be attributed to the lattice absorption of M − O (M = Fe3+, Cu2+), indicating the formation of CuFe2O4.
Raman spectra of GO, RGO, and 96CuFe2O4–4RGO composite are shown in Fig. S3 (ESI†). In Raman spectra of pure GO, the peaks were observed at 1345 and 1587 cm−1, which are the characteristic peaks of D and G band, respectively. In case of 96CuFe2O4–4RGO these peaks appeared at 1332 and 1572 cm−1 whereas, for RGO, characteristic peaks for D and G band were found at 1328 and 1580 cm−1, respectively. It has been reported that when GO is reduced to RGO, the values which correspond to the D and G bands of GO move to the lower values.40,69 For GO sample, the value of ID/IG was ∼0.9 whereas for RGO and 96CuFe2O4–4RGO this ratio was ∼1.04 and ∼1.02, respectively. This increase of ID/IG value for RGO and 96CuFe2O4–4RGO can be attributed to the decrease in the average size of sp2 domains upon reduction of GO during formation of 96CuFe2O4–4RGO composite.5,44
TGA thermograms of pure CuFe2O4, 96CuFe2O4–4RGO, 92CuFe2O4–8RGO and GO are shown in Fig. S4 (ESI†). TGA thermogram of GO showed that (i) ∼16% weight loss in the temperature range of 30–100 °C, which might be due to the loss of surface adsorbed water molecules, and (ii) ∼5% weight loss in 100–200 °C range, and ∼26 wt% loss in 200–275 °C range, which were due to the loss of oxygen-containing groups (e.g. carbonyl, carboxyl, epoxy groups, etc.) from the surface of GO sheets,3,69 (iii) in the temperature range of ∼300 to 600 °C, the oxidative decomposition of C atoms of GO occurred. In case of CuFe2O4–RGO nanocomposites, no weight loss in the temperature range of 200–275 °C was observed, which indicated the conversion of GO to RGO during the synthesis of nanocomposites and the functional groups of GO have been reduced in this process to a great extent.3,69 The thermograms of 96CuFe2O4–4RGO, and 92CuFe2O4–8RGO showed 4% and 8% weight loss in the temperature range of 275–500 °C. This weight loss was due to the decomposition of the carbon content of RGO in the composites. Pure CuFe2O4 was found to be quite stable in the temperature range of 30 to 800 °C.
The surface area of the synthesized pure CuFe2O4 and 96CuFe2O4–4RGO were obtained by conducting multipoint BET surface area analysis (Fig. S5, ESI†). The specific surface area of the pure CuFe2O4 and 96CuFe2O4–4RGO was 133 and 141 m2 g−1, respectively. The total pore volume calculated for pure CuFe2O4 and 96CuFe2O4–4RGO were ∼0.02 cm3 g−1 and 0.04 cm3 g−1, respectively.
The magnetic property of the synthesized materials was measured by VSM. As representative, the M–H hysteresis loops of CuFe2O4 and 96CuFe2O4–4RGO are presented in Fig. S6, ESI† and saturation magnetization values were 13, and 12 emu g−1, respectively.
3.2. First-principles calculations of electronic structure
In order to understand the inner mechanism of the interaction between CuFe2O4 and graphene in the CuFe2O4–graphene nanocomposite, the first-principles calculations based on DFT were performed and the interfacial interactions were investigated. The superlattice structures of CuFe2O4, graphene, and CuFe2O4–graphene nanocomposites before and after full relaxation are shown in Fig. S7 and S8 (ESI†). The structural parameters obtained after optimization of structures of the graphene (Table S2, ESI†) and CuFe2O4 unit cell match well with the reported values obtained by several researchers from their theoretical calculations (Table S3, ESI†), and experiments (Table S4, ESI†). In CuFe2O4–graphene superlattice, lattice distortion of the CuFe2O4 slab was observed after relaxation and ∼4.3% expansion was found in the z-direction. The binding energy between CuFe2O4 and graphene in CuFe2O4–graphene superlattice was −4.38 eV. These facts indicated the existence of a strong interaction between CuFe2O4 and graphene. The equilibrium interlayer distance between CuFe2O4 and graphene was 2.85 Å. The charge density distribution of CuFe2O4–graphene composite shows the clear interaction between C–O, C–Cu, and C–Fe atoms (Fig. 3). Band structures (Fig. S9, ESI†) and Projected Density Of States (PDOS) (Fig. S10, ESI†) were calculated to understand the details of the interaction between CuFe2O4 and graphene. First, the electronic properties of graphene and CuFe2O4 superlattice were studied. Fig. S9a (ESI†) shows the band structures of graphene superlattice, which showed the conduction bands and the valence bands are either separated by a gap or overlapped with each other, which intersect in two inequivalent points (Dirac points in the first Brillouin zone).7,8,70 The band structure plot of graphene also showed the band gap of graphene is 0 eV. From the band structure of the CuFe2O4 unit cell (Fig. S9b and c, ESI†), we have obtained the value of the majority and minority spin band gap of 1.62 and 1.43 eV, respectively. These band gap values of CuFe2O4 closely match with the experimentally determined optical band gap value of CuFe2O4 (1.50 eV) (measured by Diffusive Reflection Spectra Fig. S11, ESI†), as well as the reported values in the literature Table S3 (ESI†). In the band structure of CuFe2O4–graphene (Fig. S9d and e, ESI†) the appearance of new bands near the Fermi level compared with the CuFe2O4 indicated the effect of graphene on the electronic properties of CuFe2O4–graphene superlattice and these new bands are generated from the C 2p state of graphene. Projected density of states of CuFe2O4–graphene superlattice (Fig. S10c, ESI†) also indicated the strong hybridization between C 2p states of graphene and O 2s, O 2p, Cu 3d, and Fe 3d orbitals of CuFe2O4 at both valance and conduction band. This hybridization can enhance the conductivity of CuFe2O4–graphene nanocomposites. The Electrochemical Impedance Spectroscopy (EIS) measurement also showed this enhancement of electrical conductivity of CuFe2O4–RGO, which has been discussed in Section 3.4. Based on these finding, we can predict that this enhancement of conductivity will positively affect the catalytic properties of CuFe2O4–RGO for the reactions, where the charge transfers between the catalyst and reactant molecules are critical. This factor will also influence the supercapacitance property of the nanocomposite. These predictions agree well with our experimental results which are discussed is in Sections 3.3 and 3.4.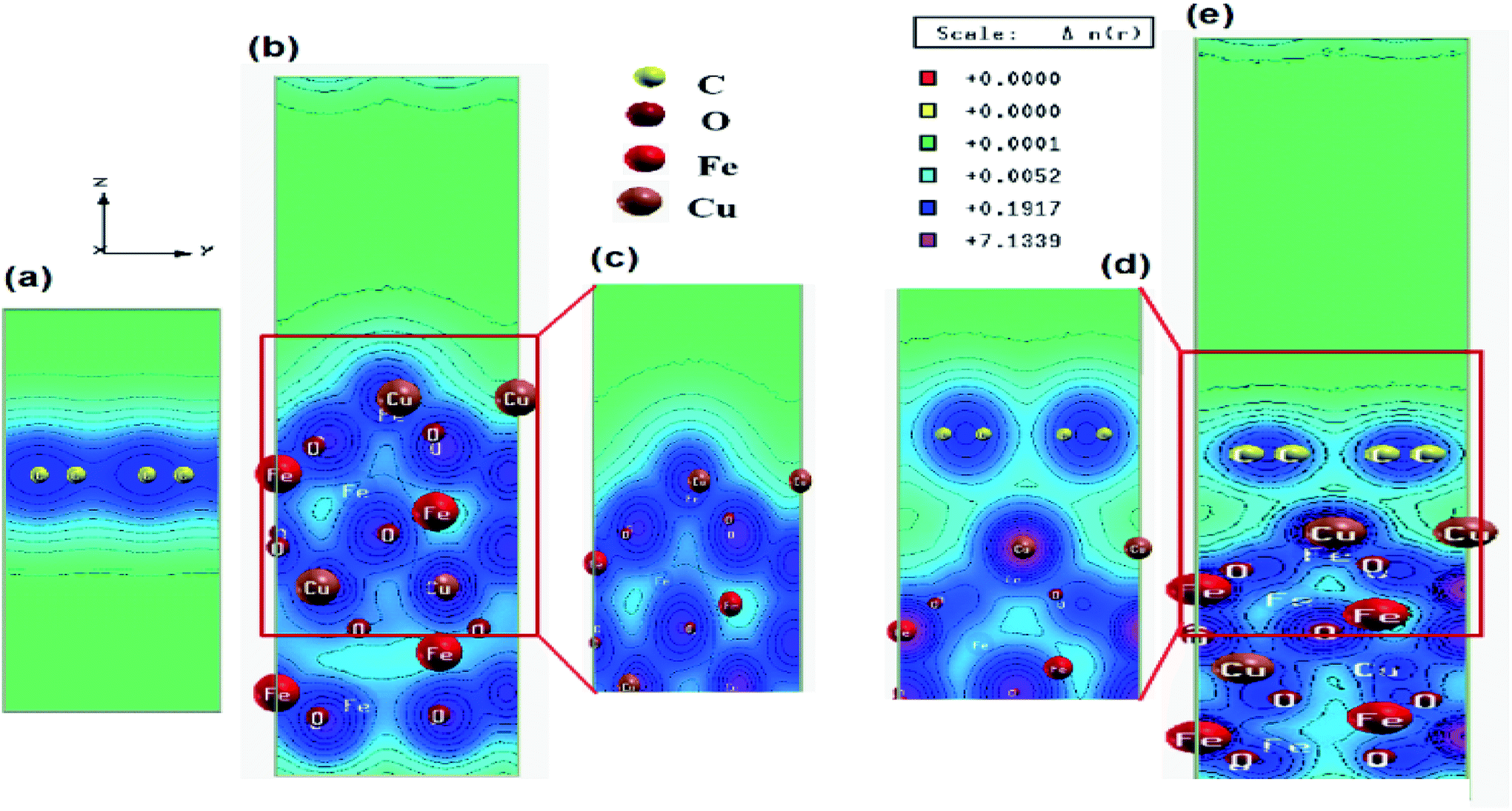 | ||
| Fig. 3 Electronic total charge density plot of (a) graphene, (b) CuFe2O4 slab, (c) enlarged selected area of CuFe2O4 slab, (d) enlarged selected area of CuFe2O4–graphene superlattice and (e) CuFe2O4–graphene superlattice. | ||
3.3. Catalytic activity of CuFe2O4–RGO nanocomposite
To investigate the catalytic activity of synthesized CuFe2O4–RGO, we have chosen two reactions: (i) aqueous phase reduction of 4-nitrophenol in the presence of NaBH4, and (ii) epoxidation of styrene in presence of TBHP.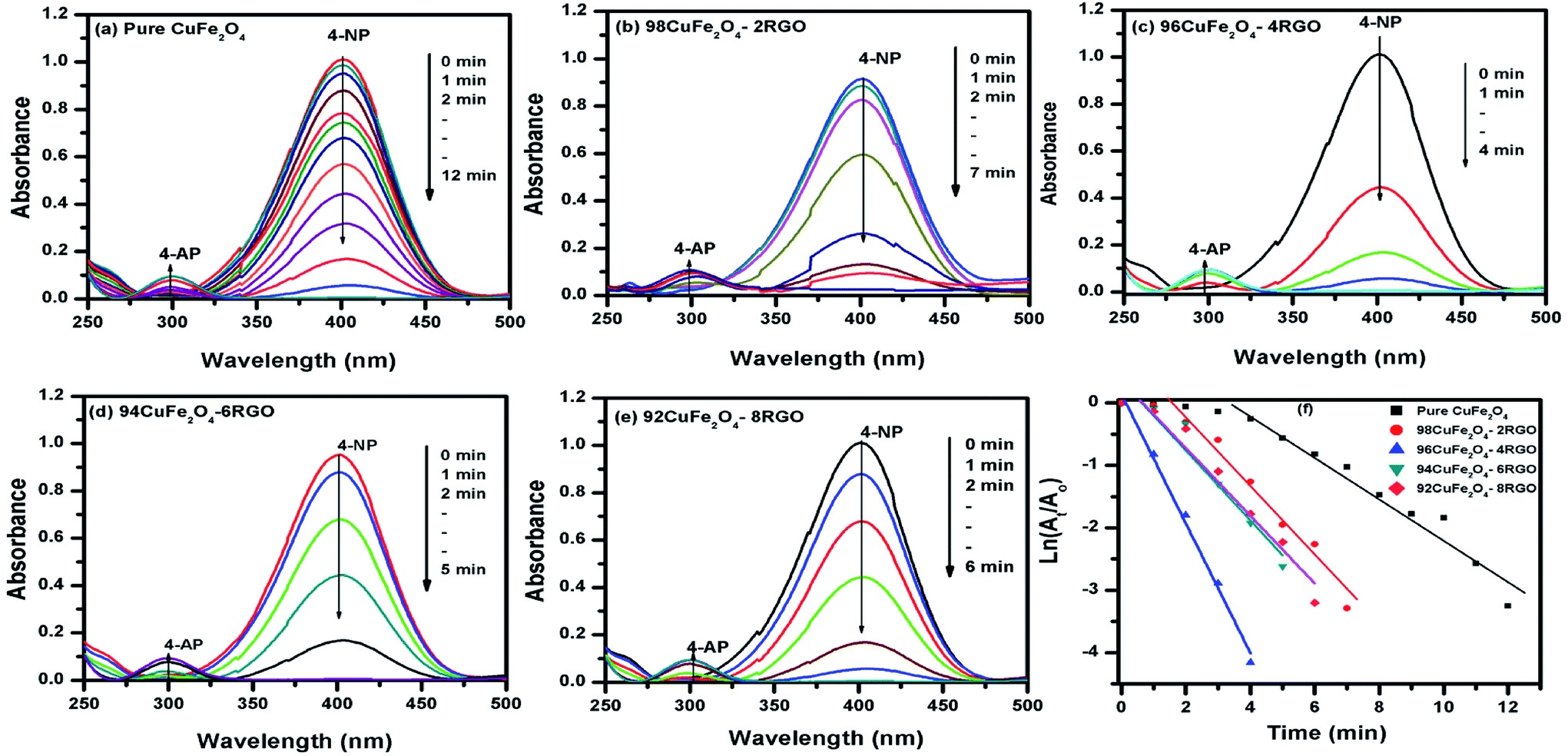 | ||
| Fig. 4 Time dependent UV-Vis spectral changes of the reaction mixture of 4-NP catalyzed by (a) pure CuFe2O4, (b) 98CuFe2O4–2RGO, (c) 96CuFe2O4–4RGO, (d) 94CuFe2O4–6RGO, (e) 92CuFe2O4–8RGO, and (f) pseudo first order kinetic plot of 4-NP reduction reaction with pure CuFe2O4, 98CuFe2O4–2RGO, 96CuFe2O4–4RGO, 94CuFe2O4–6RGO, and 92CuFe2O4–8RGO. | ||
The reduction of 4-nitrophenol to 4-aminophenol with NaBH4 is a six electron transfer reduction reaction. In the aqueous medium BH4− ions were first adsorbed on the surface of the catalyst. Then the H-atom, which was generated from BH4−, after electron transfer to the catalytically active site (CuFe2O4), attacked 4-NP molecule to reduce it to 4-AP. This electron transfer induced hydrogenation occurred spontaneously.10 As the transfer of electron plays a critical role in this reduction reaction, the presence of RGO in the catalyst became beneficial for the enhancement of its catalytic performance. CuFe2O4–RGO exhibited superior catalytic activity over pure CuFe2O4 because of the following reasons (i) according to Rout et al. RGO possesses high adsorption capacity of 4-NP due to π–π stacking interactions.11 Due to the electron withdrawing nature of –NO2 group, 4-NP molecules are adsorbed efficiently on the electron-rich sites of heterogeneous catalysts (e.g. metal nanoparticles, ferrites, etc) as well as on RGO, where its surface functional group act as strong Lewis acids. Using DFT study Rout et al. have shown that, due to this strong adsorption of 4-NP on RGO, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of –NO2 group becomes elongated (from 1.23 Å to 1.28 Å), which helps to activate the –NO2 group of 4-NP.4,11 This activation facilitates the reduction reaction of 4-NP. (ii) In this present study, DFT calculation showed the existence of a strong interaction in the electron density level in the interface between CuFe2O4 and graphene in the CuFe2O4–graphene composite. Due to this electronic interaction, CuFe2O4–RGO showed superior electrical conductivity over pure CuFe2O4 (EIS measurement results also support this and has been discussed in Section 3.4). This enhancement of conductivity in CuFe2O4–RGO catalyst accelerates the electron transfer process during the reduction of 4-NP to 4-AP. Therefore, 96CuFe2O4–4RGO exhibited significantly higher catalytic performance than that of pure CuFe2O4. The catalytic activity of 96CuFe2O4–4RGO towards the reduction of 4-NP was found to be comparable and in some cases superior to several reported catalysts. (Table S6, ESI†).
O bond of –NO2 group becomes elongated (from 1.23 Å to 1.28 Å), which helps to activate the –NO2 group of 4-NP.4,11 This activation facilitates the reduction reaction of 4-NP. (ii) In this present study, DFT calculation showed the existence of a strong interaction in the electron density level in the interface between CuFe2O4 and graphene in the CuFe2O4–graphene composite. Due to this electronic interaction, CuFe2O4–RGO showed superior electrical conductivity over pure CuFe2O4 (EIS measurement results also support this and has been discussed in Section 3.4). This enhancement of conductivity in CuFe2O4–RGO catalyst accelerates the electron transfer process during the reduction of 4-NP to 4-AP. Therefore, 96CuFe2O4–4RGO exhibited significantly higher catalytic performance than that of pure CuFe2O4. The catalytic activity of 96CuFe2O4–4RGO towards the reduction of 4-NP was found to be comparable and in some cases superior to several reported catalysts. (Table S6, ESI†).
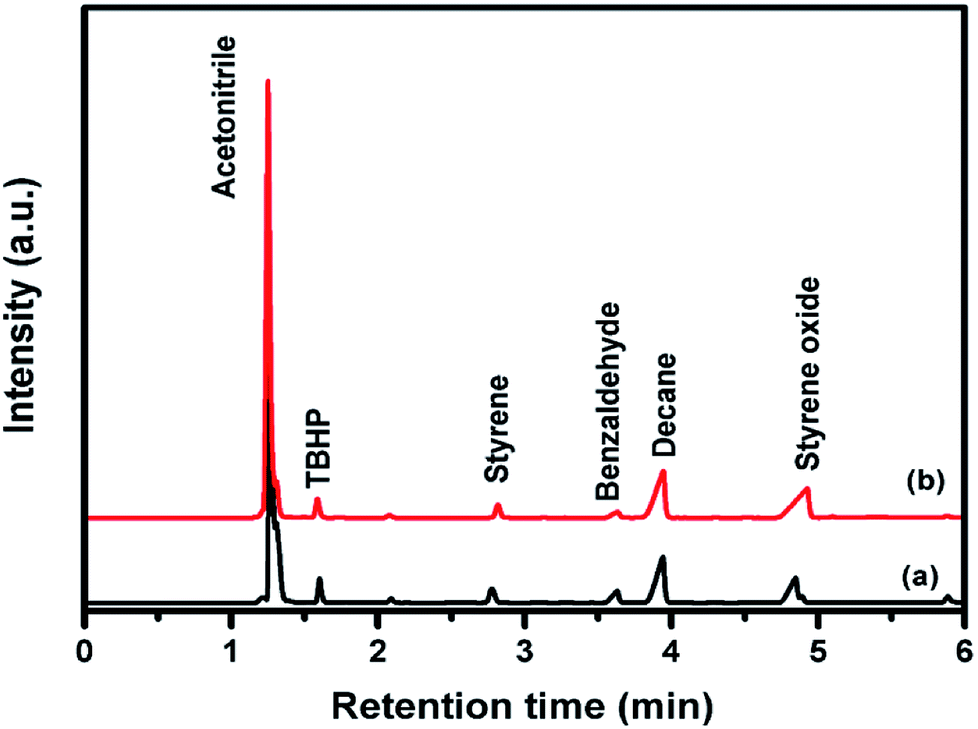 | ||
| Fig. 5 Gas chromatogram analysis of styrene epoxidation reaction with (a) pure CuFe2O4 (b) 96CuFe2O4–4RGO nanocomposite. | ||
The reaction mechanism for epoxidation of styrene to styrene oxide in presence of TBHP and catalyst has been reported by many researchers.6,14,15 The reaction proceeds via formation of a metal–alkylperoxy species when TBHP interacts with the active site of the catalyst. This metal–alkylperoxy species undergoes one electron transfer reduction. Addition of oxygen to the terminal double bond of styrene leads to the formation of two types of intermediates which are the isomers of oxametallacyclic species.15 Quiller et al. reported that DFT study indicated both isomers (branched and linear) are relatively stable.71 Conversion of products from this intermediate is the rate-determining step. From the oxametallacyclic intermediate epoxide forms via sharpless mechanism, where the transfer of oxygen occurs to the olefinic bond.69 On the other hand, the breakage of the C–C bond of the intermediate leads to the formation of benzaldehyde.15 In the present study, it has been observed that the presence of RGO in the catalyst (96CuFe2O4–4RGO) resulted in better styrene conversion with higher styrene oxide formation selectivity compared to pure CuFe2O4. This can be explained by the presence of RGO in the catalyst helps to adsorb styrene better on the surface of the catalyst than the catalyst without RGO (i.e. pure CuFe2O4). This might be due to the π–π interaction between the π system of RGO and π system of styrene.6,69 This interaction accelerates the electron transfer process between sp2 hybridized C-atoms of RGO and benzene ring of styrene and facilitates the combination of O– atom of TBHP and C– atoms of vinyl group of styrene.69,72 As a result, the conversion of styrene and selectivity of styrene oxide formation were improved when 96CuFe2O4–4RGO was present as a catalyst. The catalytic activity of 96CuFe2O4–4RGO towards epoxidation of styrene was found to be comparable and in some cases superior to many reported catalysts (Table S7, ESI†).
As CuFe2O4–RGO nanocomposites are magnetic in nature, we have explored the feasibility of the separation of the catalyst from the reaction mixture by the magnetic separation process. After completion of the reactions, the catalysts were removed from the reaction mixture by applying a permanent magnet externally. The magnetic separation of the catalysts is shown in Fig. S13a and S14a (ESI†). After separation, the catalyst was thoroughly washed and dried. The recovered catalyst was then used for the next cycle of the reaction. The efficiency of the catalyst was almost remained the same up to 5th cycle (Fig. S13b and S14b, ESI†). From XRD and TEM analysis of the recovered catalysts showed no significant changes in their crystal structures and morphology (Fig. S15, ESI†) and indicated the robustness of the structure of the catalyst.
As in these catalysis reactions we have observed that presence of 4 wt% RGO in the nanocomposite (96CuFe2O4–4RGO) significantly enhanced the catalytic activity compared to pure CuFe2O4, we have then investigated the electrochemical properties of pure CuFe2O4, and 96CuFe2O4–4RGO to construct high performing supercapacitor.
3.4. Electrochemical studies
To evaluate the supercapacitance nature of CuFe2O4, and 96CuFe2O4–4RGO nanocomposite, their electrochemical performances were investigated with CV, GCD, and EIS measurements. The CV measurements have been conducted at different scan rates in the fixed potential window of 0.0 V to 0.6 V (versus Ag/AgCl electrode) in 3 M KOH electrolyte. The CV curves of pure CuFe2O4 and 96CuFe2O4–4RGO electrodes are shown in Fig. 6. The presence of a pair of cathodic and anodic peaks (0.35/0.48 V) in the CV curve of pure CuFe2O4 electrode indicated the fast redox reaction of CuFe2O4 in KOH solution. This redox reaction can be presented as eqn (8):34
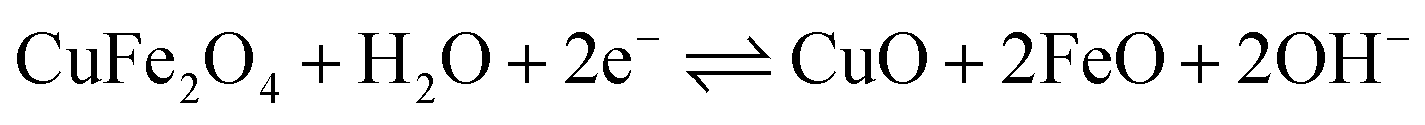 | (8) |
 | ||
| Fig. 6 Cyclic voltammetry curves of (a) pure CuFe2O4, (b) 96CuFe2O4–4RGO electrodes in 3 M KOH electrolyte, and (c) 96CuFe2O4–4RGO electrodes in 3 M KOH + 0.1 M K4[Fe(CN)6] electrolyte at scan rate 10 mV s−1. | ||
In the CV curve of 96CuFe2O4–4RGO electrode a pair of redox peak was also observed at 0.29 V and 0.43 V (Fig. 6), which indicated the fast redox reaction of CuFe2O4 in the composite. Hence, we have considered that the capacitance of CuFe2O4 and 96CuFe2O4–4RGO were mainly originated from the pseudocapacitance. The excellent conductivity of RGO might cause the negative shifting of redox peaks of 96CuFe2O4–4RGO electrode. Moreover, compared to pure CuFe2O4, the higher response of the peak current, and larger area under the CV curve of 96CuFe2O4–4RGO indicated the higher specific capacitance of 96CuFe2O4–4RGO (Fig. 6). This might be due to the synergistic effect of CuFe2O4 and RGO in the 96CuFe2O4–4RGO nanocomposites arising from the high redox activity of CuFe2O4 nanoparticles, combined with the highly conductive backbone provided by RGO, which not only facilitates the faster electron transport but also offers mechanical support that maintains the integrity of the electrode during the electrochemical process.18,73–75
To evaluate the supercapacitance performance at various scan rate, CV measurements were also conducted with different scan rate (10–100 mV s−1) in 3 M KOH for both CuFe2O4 and 96CuFe2O4–4RGO electrodes, Fig. S16a and b (ESI†). It was observed that the change in scan rate affected both the peak potential and peak current response. The increase of the peak current with increasing scan rate for both CuFe2O4 and 96CuFe2O4–4RGO electrodes indicated their excellent rate capability. With the progressive increase of scan rate, a well linear increase of peak current was observed for both the electrodes in their Randles–Sevcik plot (Fig. S16c, ESI†). This suggested that the electrochemical reaction, which is occurring on the electrode surfaces (pure CuFe2O4, and 96CuFe2O4–4RGO), is a diffusion-controlled redox process. Moreover, it was also observed that the current response was increased with increasing scan rate. This is due to the fact that, the scan rate affects the migration of electrolytic ions, and their diffusion into the electrode. When the scan rate is relatively low, the growth of a thick diffusion layer on the surface of the electrode limits the flux of electrolytic ions towards the electrode and results in the lower current. But at a higher scan rate, this diffusion layer cannot grow on the electrode surface. Hence, in this condition, the electrolyte flux towards the electrode enhances, which leads to the increase of current at the higher scan rate.76 In the CV curves (Fig. S16, ESI†), with increasing scan rate the shifting of both the upper and lower peaks towards positive and negative direction respectively, was also observed. This can be due to the development of overpotential which limits the faradic reaction.76
GCD behavior of the pure CuFe2O4 with varying current densities from 2 to 10 A g−1 in 3 M KOH shown in Fig. S17a (ESI†). The GCD curves suggest that pure CuFe2O4 electrode possess good pseudo-electrochemical character and typical battery like behavior. It was noted that the largest capacitance value for pure CuFe2O4 could be reached up to 83 F g−1 when the current density is 2 A g−1. The value of specific capacitance reported here is higher than that of some of the reported CuFe2O4 electrodes. For example, CuFe2O4 hollow fibres, which were prepared by electrospinning and direct annealing by Zhao et al. exhibited a specific capacitance of 28 F g−1 at 0.5 A g−1.77 Spray pyrolyzed thin films of CuFe2O4 showed specific capacitance of 5.7 F g−1 at 0.3 μA cm−2.78 Hydrothermally synthesized CuFe2O4 nanosphere showed capacitance of ∼81.5 F g−1 at 1 A g−1.18 In the present case the CuFe2O4 nanoparticles, which were synthesized by co-precipitation method showed that with increasing current density (2–10 A g−1) its capacitance was decreased from 83–9 F g−1. This can be explained by the decrease of the diffusion rate of electrolyte anions (OH−) into the electrode with the increase of current density.34
The change in specific capacitance with increasing current density (ranging from 2–10 A g−1) of the 96CuFe2O4–4RGO electrode (which contains 4 wt% RGO) is shown in Fig. S17b.† The discharge time of 96CuFe2O4–4RGO was found to be higher than that of pure CuFe2O4, which is a suggestive of the higher charge capacity of 96CuFe2O4–4RGO than that of CuFe2O4, which was consistent with the CV results. The largest specific capacitance of 96CuFe2O4–4RGO could be reached up to 313 F g−1 when current density was 2 A g−1. For 96CuFe2O4–4RGO electrode also the specific capacitance was decreased from 313 to 174 F g−1 with increasing charge density from 2 to 10 A g−1 like CuFe2O4 electrode. The retention of the initial capacitance was increased from 10% (for pure CuFe2O4) to 56% (96CuFe2O4–4RGO) when CuFe2O4 nanoparticles were immobilized on RGO sheets.
As it was observed that the presence of 4 wt% RGO in CuFe2O4–RGO nanocomposite significantly enhanced the supercapacitance of CuFe2O4, the GCD behavior of pure RGO electrode was also studied. The highest specific capacitance of RGO was 152 F g−1 at the charge density of 2 A g−1, and it was decreased from 152 to 72 F g−1 when current density was increased from 2–10 A g−1 (Fig. S18, ESI†). Though this specific capacitance value was lower than that of the theoretical value of a single layer RGO (550 F g−1), this present value is comparable with the values reported by others.18 The agglomeration of RGO sheets due to their π–π interaction might be the main cause of the lower specific capacitance of synthesized RGO. FESEM micrograph of synthesized RGO reveals the agglomeration of RGO sheets (Fig. S19, ESI†). However, when CuFe2O4–RGO nanocomposites were synthesized by immobilizing CuFe2O4 nanoparticles on the surface of RGO sheets these nanoparticles acted as a spacer between the sheets and prevented the agglomeration of RGO sheets to a great extent (Fig. 2d). Therefore, 96CuFe2O4–4RGO nanocomposite exhibited much higher supercapacitance than that of pure RGO.
EIS is an important tool to investigate the fundamental behavior of electrode materials for supercapacitors. The Nyquist plot is generally generated from the data obtained from EIS measurements. In this plot, the presence of a semicircle in the high-frequency region indicates the charge transfer resistance at the electrode–electrolyte interface. In the high-frequency region, the intercept of the semicircle on the X-axis (Z′) indicates the internal resistance (Rs) of the cell. The ionic resistance of the electrolyte, the intrinsic resistance of the electrode materials, and the contact resistance between the electrode and the current collector are the three major factors, which influence Rs. From the diameter of the semicircle, the charge transfer resistance (Rct) of the electrode–electrolyte interface can be estimated. The straight line at the low-frequency region signifies a diffusion process, which is known as Warburg diffusion. Fig. 7 shows the Nyquist plots of CuFe2O4, RGO, and 96CuFe2O4–4RGO electrodes. The smaller radius of the semicircle of 96CuFe2O4–4RGO than that of pure CuFe2O4 indicated the Rct of 96CuFe2O4–4RGO is lower than that of pure CuFe2O4. The estimated Rct values of pure CuFe2O4 and 96CuFe2O4–4RGO were ∼26 and ∼5 Ω respectively. This relatively lower value of Rct of 96CuFe2O4–4RGO indicated its better charge transferability than pure CuFe2O4. The origin of the smaller value of Rct of 96CuFe2O4–4RGO might be due to the well dispersion of CuFe2O4 nanoparticles which are in intimate contact with the highly conductive RGO. This causes strong interfacial interaction between CuFe2O4 and RGO in 96CuFe2O4–4RGO nanocomposite. The orbital level interaction between RGO and CuFe2O4 as observed from DFT calculation in their interface causes to enhance the electrical conductivity of CuFe2O4–RGO nanocomposite.
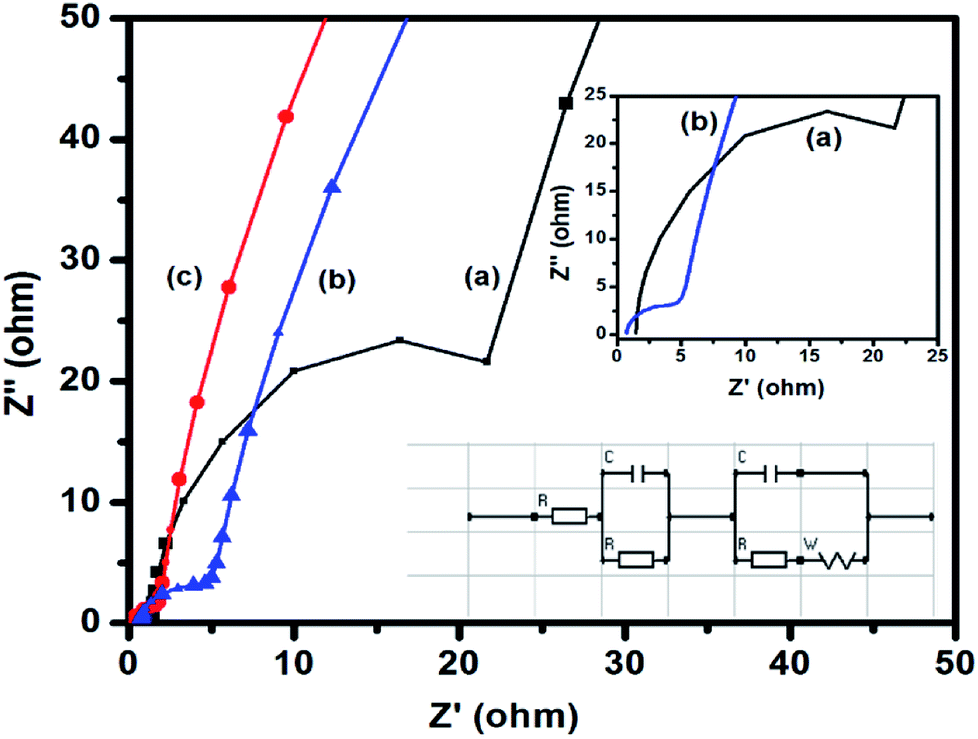 | ||
| Fig. 7 Electrochemical impedance spectra of (a) pure CuFe2O4, (b) 96CuFe2O4–4RGO, and (c) RGO, electrodes in 3 M KOH electrolyte. Inset shows the high-frequency region of the impedance spectra and equivalent circuit used for fitting the Nyquist plots. | ||
To evaluate the effect of electrolyte on the supercapacitance performance of the 96CuFe2O4–4RGO electrode, its electrochemical measurements were performed using a mixture of 3 M KOH and 0.1 M K4[Fe(CN)6] as the electrolyte. The CV curves of 96CuFe2O4–4RGO electrodes in 3 M KOH and 3 M KOH + 0.1 M K4[Fe(CN)6] are shown in Fig. 6. In the CV curve of 96CuFe2O4–4RGO electrode, the presence of a distinct pair of redox peaks at 0.32/0.40 V was observed, which also indicated the fast faradic redox reaction. It was noted that due to the addition of 0.1 M K4[Fe(CN)6] in 3 M KOH electrolyte system the peak area under the CV curve and peak current were increased significantly. During CV measurements of 96CuFe2O4–4RGO electrode at different scan rate (10–100 mV s−1), the increasing trend of peak current with increasing scan rate was observed which is shown in Fig. S20 (ESI†). This indicates the good rate capability of the 96CuFe2O4–4RGO electrode in this modified electrolyte system.
From GCD analysis, the specific capacitance of 96CuFe2O4–4RGO electrode was also measured in 3 M KOH + 0.1 M K4[Fe(CN)6] electrolyte and shown in Fig. S21 (ESI†). In this electrolyte system, the specific capacitance value was significantly increased (up to 797 F g−1 at 2 A g−1) compared to 3 M KOH electrolyte (313 F g−1 at 2 A g−1). The presence of 0.1 M K4[Fe(CN)6] in this electrolyte system was found to be beneficial because it provides an additional complementary redox couple, [Fe(CN)6]4−/[Fe(CN)6]3− with matching potential (0.20/0.37 V) to the electrolyte system, which can act as an electron buffer source in the electrochemical reaction at 96CuFe2O4–4RGO electrode/electrolyte interface.24,79,80 Moreover, as the Born radius of [Fe(CN)6]4−/[Fe(CN)6]3− is ∼0.4 nm (ref. 24) and the total pore volume of 96CuFe2O4–4RGO was ∼0.04 cm3 g−1 (obtained from N2 adsorption–desorption analysis Fig. S5 ESI†), a large number of redox additive anions can be accommodated within the pore of the 96CuFe2O4–4RGO electrode. The decrease of ionic diffusion resistance of the electrolytic ions due to the addition of K4[Fe(CN)6] is one of the major factors which enhances the redox kinetics and hence the supercapacitance of the electrodes. The positive influence of K4[Fe(CN)6] redox additive on the supercapacitance of the electrode is shown in Fig. 8a. However, the value of specific capacitance in this modified electrolyte system was decreased from 797 to 266 F g−1 when current density increase from 2 to 10 A g−1. In this system 96CuFe2O4–4RGO electrode retained ∼33% of its initial capacitance. This significant decrease of supercapacitance value with increasing current density (particularly at comparatively higher current density) was observed which may be due to the sluggish redox reaction kinetics, and low penetration of electrolyte ions in and out of the electrode at higher current density.
 | ||
| Fig. 8 (a) Change of specific capacitance of pure CuFe2O4 and 96CuFe2O4–4RGO electrodes with changing current density from 2 to 10 A g−1, (b) Ragone plots of CuFe2O4 and 96CuFe2O4–4RGO electrodes. | ||
As a reasonable energy density with high power density required for a high performing supercapacitor, we have calculated the energy density of CuFe2O4 and 96CuFe2O4–4RGO electrodes at different power densities. Fig. 8b shows the Ragone plots of these electrodes when 3 M KOH and 3 M KOH + 0.1 M K4[Fe(CN)6] were used as the electrolyte. In 3 M KOH electrolyte 96CuFe2O4–4RGO delivered an energy density of 11 W h kg−1 at a power density of 543 W kg−1, which was significantly higher than that of CuFe2O4 (energy density 2.4 W h kg−1 at a power density of 460 W kg−1). When 3 M KOH + 0.1 M K4[Fe(CN)6] was used as the electrolyte, 96CuFe2O4–4RGO exhibited larger values of the energy density of 16 W h kg−1 at a power density of 380 W kg−1. The energy density was decreased from 16 to 5 W h kg−1 with increasing power density from 380 to 1900 W kg−1. These values are comparable with commercial supercapacitor (3–9 W h kg−1 at 3000 to 10000 W kg−1).24,74 The supercapacitor performance of 96CuFe2O4–4RGO electrode was found to be comparable and in some cases better than the previously reported ferrite and ferrite-RGO based electrodes (Table S8, ESI†).
Sustainability of high electro-capacitance value at high current density is an important factor to determine the performance of the electrode. The high specific capacitance value at high current density demonstrates the better performance of 96CuFe2O4–4RGO electrode than the work reported by others. Moreover, the symmetric nature of GCD profile even at high current density also indicated high rate performance of the 96CuFe2O4–4RGO electrode.
As 96CuFe2O4–4RGO electrode exhibited better supercapacitance behavior than pure CuFe2O4, we have investigated its cycling behavior to understand its stability. Cycling behavior of 96CuFe2O4–4RGO electrode has been investigated at a constant current density of 6 A g−1 for more than 2000 cycles using both the electrolytes (3 M KOH, and 3 M KOH + 0.1 M K4[Fe(CN)6]), and shown in Fig. 9. The equilibrium was reached after the first few cycles and then the steady capacitance was reached for the subsequent cycles. The 96CuFe2O4–4RGO electrode exhibited a higher specific capacitance (343 F g−1 at 6 A g−1) when the mixture of 3 M KOH and 0.1 M K4[Fe(CN)6] was used as electrolyte than when 3 M KOH was the electrolyte (specific capacitance = 172 F g−1). After 2000 cycle 96CuFe2O4–4RGO electrode exhibited ∼85% retention of its specific capacitance when KOH was used as the electrolyte. However, this retention value was increased up to ∼92% in 3 M KOH + 0.1 M K4[Fe(CN)6] electrolyte, which demonstrated its high cycling stability.
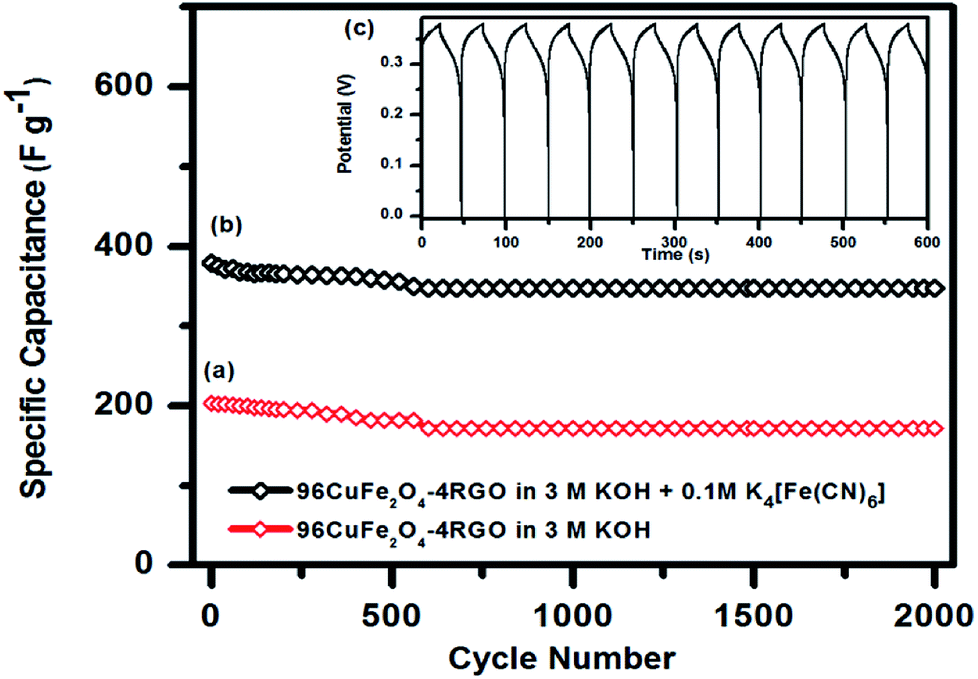 | ||
| Fig. 9 Cyclic stability of the 96CuFe2O4–4RGO electrode in (a) 3 M KOH and (b) 3 M KOH + 0.1 M K4[Fe(CN)6] showing the capacitance retention after 2000 cycles using a charge/discharge at constant current density 6 Ag−1. (c) The inset shows the charge–discharge curves of the 96CuFe2O4–4RGO electrode in 3 M KOH + 0.1 M K4[Fe(CN)6]. | ||
The electrochemical property measurements of the electrode by using a three-electrode system generally provide the insight of the materials electrochemical behavior for its application as a supercapacitor. But as the two electrode test cell measurement system mimics the configuration of the commercial supercapacitor, the results it provides are more significant for the practical purposes.18,81 Hence, we have constructed a two electrode test cell using 96CuFe2O4–4RGO nanocomposite and measured its specific capacitance with 3 M KOH + 0.1 M K4[Fe(CN)6] electrolyte. Fig. S22 (ESI†) shows the CV curves of 96CuFe2O4–4RGO electrode at different scan rates and GCD curves with varying current densities which were measured in two electrode system. The value of specific capacitance in this condition obtained was 160 F g−1 at a current density of 2 A g−1. Though this value is lower than the specific capacitance value, which was obtained from three-electrode measurement system (797 F g−1 at 2 A g−1), this trend is quite normal and consistent with the observations reported by different researchers for various systems.18,74,81
As a demonstration for the real application of the 96CuFe2O4–4RGO electrode as a supercapacitor device, two symmetric cells were fabricated using 96CuFe2O4–4RGO as an active electrode material and they were connected in series and charged for 10 min using a 9 V battery. After charging the cell, a yellow light-emitting diode (LED) (1.8 V) was connected with it. The connected LED was light-up for 6 min. The light-up of LED after connecting with 96CuFe2O4–4RGO electrode is shown in Fig. 10.
 | ||
| Fig. 10 A yellow light-emitting diode (LED) (1.8 V) powered by two 96CuFe2O4–4RGO capacitors connected in series. Glowing LED at a different time interval. | ||
4. Conclusion
Here, the preparation of CuFe2O4 nanoparticle-RGO nanocomposites by using an ‘in situ’ co-precipitation reduction technique, and their catalytic properties, as well as electrochemical properties, has been described. In the synthesized nanocomposites, CuFe2O4 nanoparticles were immobilized on the surface of nanometer thin RGO sheets. In these nanocomposites, RGO provided a highly conducting support due to which the electrical conductivity of the nanocomposites was increased. First-principles based DFT calculations showed the presence of strong interfacial interaction between CuFe2O4 and graphene via hybridization of atomic orbitals of C, Cu, Fe, and O. This interfacial interaction influenced the electronic structure of CuFe2O4 in CuFe2O4–graphene composite and enhanced the conductivity of the nanocomposite, which in turn positively affected the catalytic and supercapacitance property of the composites.Here, it has been demonstrated that the presence of RGO caused significant enhancement of the catalytic activities of the CuFe2O4–RGO nanocomposite compared to pure CuFe2O4 for two reactions (i) reduction of 4-nitrophenol, and (ii) epoxidation of styrene. The nanocomposite, which was composed of 96 wt% of CuFe2O4 and 4 wt% RGO (96CuFe2O4–4RGO), exhibited a high catalytic activity towards the reduction of 4-nitrophenol to 4-aminophenol with kapp = 17.2 × 10−3 s−1, and the epoxidation of styrene with ∼90% styrene conversion and ∼65% selectivity of styrene oxide formation. Whereas pure CuFe2O4 showed kapp of 5.7 × 10−3 s−1 for reduction of 4-nitrophenol, and ∼85% styrene conversion with ∼37% selectivity of styrene oxide formation in epoxidation reaction. The magnetic nature of the nanocomposite was exploited for its easy magnetic separation from the reaction mixtures after the completion of the reactions. The recovered catalyst exhibited a very good reusability.
Due to the presence of RGO, 96CuFe2O4–4RGO also exhibited higher supercapacitance performance and cyclic performance compare to pure CuFe2O4. When 3 M KOH was used as the electrolyte, the 96CuFe2O4–4RGO electrode showed a specific capacitance of 313 F g−1, whereas pure CuFe2O4 electrode showed 83 F g−1 at 2 A g−1 current density. Addition of 0.1 M K4[Fe(CN)6] in 3 M KOH solution was found to be beneficial as the electrolyte. In this electrolyte at 2 A g−1 current density 96CuFe2O4–4RGO showed a maximum specific capacitance of 797 F g−1 and retention of ∼92% specific capacitance up to 2000 cycle. It also showed a high energy density of 16 W h kg−1 at a power density of 380 W kg−1.
To the best our knowledge, this is the first time the ability of CuFe2O4–RGO nanocomposite as a magnetically separable catalyst for the reduction reaction of 4-nitrophenol, and epoxidation reaction of styrene, and the investigations on its electronic structures by DFT calculations have been reported. The easy synthetic methodology, high catalytic activity with very good reusability, and excellent supercapacitance performance make 96CuFe2O4–4RGO nanocomposites as an attractive multifunctional material.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr N. N. Ghosh gratefully acknowledges DRDO Jodhpur for VSM analysis. Dr Ghosh is thankful to the Central Sophisticated Instrumentation Facility (CSIF) of BITS Pilani K K Birla Goa campus for providing the FESEM facility. Dr Ghosh is also thankful to Mahatma Gandhi University, Kottayam for providing TEM facility.Notes and references
- M. Hu, Z. Yao and X. Wang, Ind. Eng. Chem. Res., 2017, 56, 3477 CrossRef.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711 CrossRef PubMed.
- D. Moitra, M. Chandel, B. K. Ghosh, R. K. Jani, M. K. Patra, S. R. Vadera and N. N. Ghosh, RSC Adv., 2016, 6, 76759 RSC.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef PubMed.
- Y. Zhao, G. He, W. Dai and H. Chen, Ind. Eng. Chem. Res., 2014, 53, 12566 CrossRef.
- W. Li, Y. Gao, W. Chen, P. Tang, W. Li, Z. Shi, D. Su, J. Wang and D. Ma, ACS Catal., 2014, 4, 1261 CrossRef.
- C. Gong, G. Lee, B. Shan, E. M. Vogel, R. M. Wallace and K. Cho, J. Appl. Phys., 2010, 108, 123711 CrossRef.
- P. Khomyakov, G. Giovannetti, P. Rusu, G. v. Brocks, J. Van den Brink and P. J. Kelly, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195425 CrossRef.
- S. Gu, S. Wunder, Y. Lu, M. Ballauff, R. Fenger, K. Rademann, B. Jaquet and A. Zaccone, J. Phys. Chem. C, 2014, 118, 18618 CrossRef.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410 RSC.
- L. Rout, A. Kumar, R. S. Dhaka, G. N. Reddy, S. Giri and P. Dash, Appl. Catal., A, 2017, 538, 107 CrossRef.
- B. Naik, S. Hazra, V. S. Prasad and N. N. Ghosh, Catal. Commun., 2011, 12, 1104 CrossRef.
- V. Choudhary, D. Dumbre, N. Patil, B. Uphade and S. Bhargava, J. Catal., 2013, 300, 217 CrossRef.
- D.-H. Zhang, G.-D. Li, J.-X. Li and J.-S. Chen, Chem. Commun., 2008, 29, 3414 RSC.
- D. Nepak and D. Srinivas, Appl. Catal., A, 2016, 523, 61 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef PubMed.
- S. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401 CrossRef.
- W. Zhang, B. Quan, C. Lee, S.-K. Park, X. Li, E. Choi, G. Diao and Y. Piao, ACS Appl. Mater. Interfaces, 2015, 7, 2404 CrossRef PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef PubMed.
- B. Bashir, W. Shaheen, M. Asghar, M. F. Warsi, M. A. Khan, S. Haider, I. Shakir and M. Shahid, J. Alloys Compd., 2017, 695, 881 CrossRef.
- Z. Wang, X. Zhang, Y. Li, Z. Liu and Z. Hao, J. Mater. Chem. A, 2013, 1, 6393 RSC.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum Publishers, New York, 1st edn, 1999 Search PubMed.
- Y.-Q. Zhao, M. Lu, P.-Y. Tao, Y.-J. Zhang, X.-T. Gong, Z. Yang, G.-Q. Zhang and H.-L. Li, J. Power Sources, 2016, 307, 391 CrossRef.
- S. Maiti, A. Pramanik and S. Mahanty, Chem. Commun., 2014, 50, 11717 RSC.
- L. Li, H. Bi, S. Gai, F. He, P. Gao, Y. Dai, X. Zhang, D. Yang, M. Zhang and P. Yang, Sci. Rep., 2017, 7, 43116 CrossRef PubMed.
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Adv. Mater., 2014, 26, 849 CrossRef PubMed.
- X. Fan, B. D. Phebus, L. Li and S. Chen, Sci. Adv. Mater., 2015, 7, 1916 CrossRef.
- L. L. Zhang and X. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863 CrossRef PubMed.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184 CrossRef.
- Y. Hou, Y. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727 CrossRef PubMed.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595 CrossRef.
- S. Bandgar, M. M. Vadiyar, Y.-C. Ling, J.-Y. Chang, S.-H. Han, A. V. Ghule and S. S. Kolekar, ACS Appl. Energy Mater., 2018, 2, 638 CrossRef.
- M. Zhu, D. Meng, C. Wang and G. Diao, ACS Appl. Mater. Interfaces, 2013, 5, 6030 CrossRef PubMed.
- J. Feng, L. Su, Y. Ma, C. Ren, Q. Guo and X. Chen, Chem. Eng. J., 2013, 221, 16 CrossRef.
- K. Swapna, S. N. Murthy, M. T. Jyothi and Y. V. D. Nageswar, Org. Biomol. Chem., 2011, 9, 5989 RSC.
- D. Yang, X. Zhu, W. Wei, M. Jiang, N. Zhang, D. Ren, J. You and H. Wang, Synlett, 2014, 25, 729 CrossRef.
- X. Zhu, D. Yang, W. Wei, M. Jiang, L. Li, X. Zhu, J. You and H. Wang, RSC Adv., 2014, 4, 64930 RSC.
- A. Bazgir, G. Hosseini and R. Ghahremanzadeh, ACS Comb. Sci., 2013, 15, 530 CrossRef PubMed.
- J. Wang, Q. Deng, M. Li, K. Jiang, J. Zhang, Z. Hu and J. Chu, Sci. Rep., 2017, 7, 8903 CrossRef PubMed.
- H. Zhang, S. Gao, N. Shang, C. Wang and Z. Wang, RSC Adv., 2014, 4, 31328 RSC.
- Z. Shahnavaz, P. M. Woi and Y. Alias, Ceram. Int., 2015, 41, 12710 CrossRef.
- Y. Fu, Q. Chen, M. He, Y. Wan, X. Sun, H. Xia and X. Wang, Ind. Eng. Chem. Res., 2012, 51, 11700 CrossRef.
- R. Dhanda and M. Kidwai, RSC Adv., 2016, 6, 53430 RSC.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- D. Moitra, S. Dhole, B. K. Ghosh, M. Chandel, R. K. Jani, M. K. Patra, S. R. Vadera and N. N. Ghosh, J. Phys. Chem. C, 2017, 121, 21290 CrossRef.
- R. M. del Castillo, L. F. del Castillo, A. G. Calles and C. Vicente, J. Mater. Chem. C, 2018, 6, 7401 RSC.
- X. Shi, S. L. Bernasek and A. Selloni, J. Phys. Chem. C, 2016, 120, 14892 CrossRef.
- H. Asadi and M. Vaezzadeh, Mater. Res. Express, 2017, 4, 075039 CrossRef.
- A. M. Ukpong, Comput. Condens. Matter, 2015, 2, 1 CrossRef.
- A. Ramazani, S. Kazemiabnavi and R. Larson, Acta Mater., 2016, 116, 231 CrossRef.
- B. D. Kong, S. Paul, M. B. Nardelli and K. W. Kim, Phys. Rev. B, 2009, 80, 033406 CrossRef.
- W. Humers and R. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- I. Solovyev, P. Dederichs and V. Anisimov, Phys. Rev. B, 1994, 50, 16861 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef PubMed.
- W. Geng, X. Zhao, H. Liu and X. Yao, J. Phys. Chem. C, 2013, 117, 10536 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef PubMed.
- V. Barone, M. Casarin, D. Forrer, M. Pavone, M. Sambi and A. Vittadini, J. Comput. Chem., 2009, 30, 934 CrossRef PubMed.
- R. Zhang, Q. Yuan, R. Ma, X. Liu, C. Gao, M. Liu, C.-L. Jia and H. Wang, RSC Adv., 2017, 7, 21926 RSC.
- Z. Jiang, W. Zhang, W. Shangguan, X. Wu and Y. Teraoka, J. Phys. Chem. C, 2011, 115, 13035 CrossRef.
- Quantum ESPRESSO, Files: C.pbe-n-rrkjus_psl.1.0.0.UPF, Cu.pbe-spn-rrkjus_psl.1.0.0.UPF, Fe.pbe-spn-rrkjus_psl.1.0.0.UPF, and O.pbe-nl-rrkjus_psl.1.0.0.UPF, https://www.quantum-espresso.org/pseudopotentials (accessed February 2018).
- G. Giovannetti, P. Khomyakov, G. Brocks, V. v. Karpan, J. Van den Brink and P. J. Kelly, Phys. Rev. Lett., 2008, 101, 026803 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- M. Methfessel and A. Paxton, Phys. Rev. B, 1989, 40, 3616 CrossRef.
- N. Marzari, D. Vanderbilt, A. De Vita and M. Payne, Phys. Rev. Lett., 1999, 82, 3296 CrossRef.
- D. Moitra, B. Ghosh, M. Chandel, R. Jani, M. Patra, S. Vadera and N. Ghosh, RSC Adv., 2016, 6, 14090 RSC.
- D. Moitra, B. K. Ghosh, M. Chandel and N. N. Ghosh, RSC Adv., 2016, 6, 97941 RSC.
- B. Luo, X. Wang, E. Tian, H. Gong, Q. Zhao, Z. Shen, Y. Xu, X. Xiao and L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3340 CrossRef PubMed.
- R. Quiller, T. Baker, X. Deng, M. Colling, B. Min and C. Friend, J. Chem. Phys., 2008, 129, 064702 CrossRef PubMed.
- M. Liu, X. Wang, Y. Chen and L. Dai, RSC Adv., 2015, 5, 61481 RSC.
- D. Ghosh, S. Giri and C. K. Das, ACS Sustainable Chem. Eng., 2013, 1, 1135 CrossRef.
- D. Moitra, C. Anand, B. K. Ghosh, M. Chandel and N. N. Ghosh, ACS Appl. Energy Mater., 2018, 1, 464 CrossRef.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132 CrossRef PubMed.
- M. Alam, K. Karmakar, M. Pal and K. Mandal, RSC Adv., 2016, 6, 114722 RSC.
- J. Zhao, Y. Cheng, X. Yan, D. Sun, F. Zhu and Q. Xue, CrystEngComm, 2012, 14, 5879 RSC.
- D. Ham, J. Chang, S. Pathan, W. Kim, R. Mane, B. Pawar, O.-S. Joo, H. Chung, M.-Y. Yoon and S.-H. Han, Curr. Appl. Phys, 2009, 9, S98 CrossRef.
- S. Maiti, A. Pramanik and S. Mahanty, ACS Appl. Mater. Interfaces, 2014, 6, 10754 CrossRef PubMed.
- L.-H. Su, X.-G. Zhang, C.-H. Mi, B. Gao and Y. Liu, Phys. Chem. Chem. Phys., 2009, 11, 2195 RSC.
- R. Rakhi, W. Chen, D. Cha and H. N. Alshareef, J. Mater. Chem., 2011, 21, 16197 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05393f |
| This journal is © The Royal Society of Chemistry 2018 |