
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHyperelatosides A–E, biphenyl ether glycosides from Hypericum elatoides, with neurotrophic activity†
Xi-Tao Yan a,
Zhen Ana,
Dan Tanga,
Guang-Rui Penga,
Chen-Yu Caoa,
Yuan-Zhen Xua,
Chun-Huan Lia,
Pei-Liang Liub,
Zai-Min Jiangc and
Jin-Ming Gao*a
a,
Zhen Ana,
Dan Tanga,
Guang-Rui Penga,
Chen-Yu Caoa,
Yuan-Zhen Xua,
Chun-Huan Lia,
Pei-Liang Liub,
Zai-Min Jiangc and
Jin-Ming Gao*a
aShaanxi Key Laboratory of Natural Products & Chemical Biology, College of Chemistry & Pharmacy, Northwest A&F University, Yangling 712100, China. E-mail: jinminggao@nwsuaf.edu.cn
bKey Laboratory of Resource Biology and Biotechnology in Western China, College of Life Sciences, Northwest University, Xi'an 710069, China
cCollege of Life Sciences, Northwest A&F University, Yangling 712100, China
First published on 25th July 2018
Abstract
Five new biphenyl ether glycosides, hyperelatosides A–E (1–5), one new benzoate glycoside, hyperelatoside F (6), along with nine known phenolic compounds (7–15), were isolated from the aerial parts of Hypericum elatoides. Their structures were elucidated by 1D and 2D NMR spectroscopy and HRESIMS, as well as chemical derivatization. This is the first report of the identification of biphenyl ether glycosides as plant metabolites and their possible biosynthetic pathway is proposed. Except for 3, the new phenolic metabolites exhibited significant neurotrophic activities to enhance nerve growth factor-induced neurite outgrowth in PC12 cells. In addition, the anti-neuroinflammatory and antioxidant activities of compounds 1–15 were preliminarily evaluated in vitro.
Introduction
Plants belonging to the genus Hypericum (Hypericaceae) are well known for their therapeutic uses in neurological disorders.1 In particular, the extract of H. perforatum (St. John's wort), the most known of the Hypericum species, has long been used for the treatment of mild to moderate depression.2 Due to the continuous discovery of structurally novel natural products and their significant biological activities, plants of the genus Hypericum have attracted much attention from researchers in both chemistry and pharmacology. Previous chemical investigations on this genus have resulted in the isolation of phloroglucinol derivatives, xanthones, benzophenones, naphthodianthrones, flavonoids, and coumarins as major constituents.3–6 These secondary metabolites possess extensive biological activities, such as antidepressant, neuroprotective, anti-neurodegenerative, antioxidant, memory-enhancing, anti-inflammatory, anticancer, anti-HIV, and antimicrobial activities.7–13Hypericum elatoides R. Keller is an herbaceous plant distributed in northwest China, especially in the western region of Shaanxi province.14 To date, there have been no phytochemical and biological studies on this plant. In the course of our ongoing search for natural neurotrophic compounds with novel structures for potential neurodegenerative disease therapies,15 15 phenolic compounds including five new uncommon biphenyl ether glycosides named hyperelatosides A–E (1–5), one new benzoate glycoside named hyperelatoside F (6), and nine known, structurally related phenolic derivatives (7–15) were isolated from the MeOH extract of the aerial parts of H. elatoides (Fig. 1). Herein, we described the isolation and structural elucidation of these new phenolic metabolites, as well as the biological evaluation of their promoting effects on nerve growth factor (NGF)-induced neurite outgrowth in PC12 cells, inhibitory effects on lipopolysaccharide (LPS)-induced nitric oxide (NO) production in BV-2 cells, and 1,1-diphenyl-2-picrylhydrazyl (DPPH) free radical-scavenging capacities.
 | ||
| Fig. 1 Chemical structures of compounds 1–6. | ||
Results and discussion
The MeOH extract of H. elatoides was subjected to liquid–liquid fractionation to afford a hexane-soluble fraction, an EtOAc-soluble fraction, and a BuOH-soluble fraction. The active EtOAc fraction was separated by semipreparative reversed phase (RP) HPLC and column chromatography (CC) using silica gel, RP-C18, and Sephadex LH-20 to yield six new compounds (1–6) and nine known phenolic metabolites (7–15).Compound 1 was isolated as a brown amorphous solid. Its molecular formula was determined to be C22H26O13 by HRESIMS (m/z 521.1247 [M + Na]+, calcd for C22H26NaO13, 521.1271). The IR spectrum showed absorption bands at 3418 and 1701 cm−1 for hydroxyl and ester carbonyl groups, respectively. The low-field region of the 1H NMR spectrum (measured in CD3OD) exhibited two para-positional aromatic protons [δH 7.44 (1H, s, H-3) and 6.38 (1H, s, H-6)] and two meta-coupled aromatic protons [δH 6.67 (1H, d, J = 2.9 Hz, H-4′) and 6.10 (1H, d, J = 2.9 Hz, H-6′)] (Table 1), indicating the presence of a 1,2,4,5-tetrasubstituted (A-ring) and a 1,2,3,5-tetrasubstituted (B-ring) benzene rings, respectively. Further, the aliphatic region of the 1H NMR spectrum showed signals of three methoxy groups [δH 3.87 (3H, s), 3.78 (3H, s), and 3.65 (3H, s)] and a set of sugar protons (δH 3.41–4.84). The 13C NMR spectrum (Table 2) supported by DEPT experiments displayed signals of three methoxy carbons (δC 57.1, 56.3, and 52.5), six characteristic glucopyranosyl carbons (δC 104.2, 78.6, 77.7, 75.0, 71.5, and 62.6), an ester carbonyl carbon (δC 167.9), and 12 aromatic carbons (δC 100.5–154.3), including eight quaternary carbons and four methine carbons, corroborating the presence of two tetrasubstituted aromatic rings detected in the 1H NMR spectrum. Furthermore, the 1H NMR spectrum of 1, measured in DMSO-d6, displayed the downfield signals of two hydroxyl groups at δH 10.03 and 8.03 (Table 1), which were attached to C-5 position of A-ring and C-2′ position of B-ring based on the HMBC correlations from the hydroxyl proton at δH 10.03 to C-4, C-5, and C-6 and from the hydroxyl proton at δH 8.03 to C-1′, C-2′, and C-3′. In the HMBC spectrum, the correlations from the methoxy protons at δH 3.78 to the carbonyl carbon at δC 167.9 (C-7), from H-3 (δH 7.44) to C-1 (δC 153.9), C-5 (δC 153.7), and C-7 (δC 167.9), from H-6 (δH 6.38) to C-2 (δC 112.8) and C-4 (δC 145.0), and from the methoxy protons at δH 3.87 to C-4 (δC 145.0) (Fig. 2) suggested the presence of a methoxycarbonyl group and a methoxy group located at the C-2 and C-4 positions of A-ring, respectively. In addition, the locations of a methoxy group at C-5′ and a sugar moiety at C-3′ of B-ring were established according to the HMBC correlations observed from the methoxy protons at δH 3.65 to C-5′ (δC 154.3) and from the anomeric proton at δH 4.84 (1H, d, J = 7.4 Hz, H-1′′) to C-3′ (δC 148.4), respectively. These two aromatic rings were bonded via an oxygen atom based on analysis of the chemical shifts of C-1 (δC 153.9) and C-1′ (δC 147.0) together with the molecular formula. Acid hydrolysis followed by HPLC analysis after arylthiocarbamoyl-thiazolidine derivatization confirmed the characterization of a β-D-glucopyranosyl unit. Thus, the structure of 1 was elucidated as 2-(3-O-β-D-glucopyranosyl-2-hydroxy-5-methoxyphenoxy)-4-hydroxy-5-methoxy-methylbenzoate and was named hyperelatoside A.
| No. | 1a | 1b | 2a | 3a | 4a | 4b | 5a |
|---|---|---|---|---|---|---|---|
| a Date were recorded in CD3OD.b Date were recorded in DMSO-d6. | |||||||
| 3 | 7.44, s | 7.32, s | 7.35, s | 7.34, s | 6.85, d (8.8) | 6.84, d (8.8) | 6.84, d (8.8) |
| 4 | 6.59, dd (8.8, 2.9) | 6.58, dd (8.8, 3.0) | 6.56, dd (8.8, 2.9) | ||||
| 6 | 6.38, s | 6.26, s | 6.40, s | 6.72, s | 6.46, d (2.9) | 6.45, d (3.0) | 6.39, d (2.9) |
| 4′ | 6.67, d (2.9) | 6.64, d (2.9) | 6.68, d (2.8) | 6.72, d (2.9) | 6.24, d (2.9) | 6.19, d (2.9) | 6.67, d (2.9) |
| 6′ | 6.10, d (2.9) | 6.14, d (2.9) | 6.33, d (2.8) | 6.54, d (2.9) | 5.91, d (2.9) | 5.74, d (2.9) | 6.17, d (2.9) |
| 1′′ | 4.84, d (7.4) | 4.72, d (7.4) | 4.83, d (7.4) | 4.94, d (7.7) | 4.73, d (7.6) | 4.60, d (7.5) | 4.80, d (7.6) |
| 2′′ | 3.52, m | 3.31, m | 3.49, m | 3.52, m | 3.46, m | 3.25, m | 3.52, m |
| 3′′ | 3.50, m | 3.30, m | 3.48, m | 3.47, m | 3.41, m | 3.27, m | 3.49, m |
| 4′′ | 3.41, m | 3.16, m | 3.39, m | 3.38, m | 3.42, m | 3.17, m | 3.41, m |
| 5′′ | 3.45, m | 3.37, m | 3.44, m | 3.45, m | 3.25, m | 3.34, m | 3.44, m |
| 6′′ | 3.71, dd (12.1, 5.8) | 3.47, dd (11.8, 5.9) | 3.70, dd (12.0, 5.9) | 3.67, dd (12.1, 5.9) | 3.69, dd (12.0, 4.7) | 3.45, dd (11.7, 5.8) | 3.71, dd (12.1, 5.7) |
| 3.92, dd (12.1, 2.1) | 3.75, m | 3.91, dd (12.0, 2.2) | 3.86, dd (12.1, 2.2) | 3.77, dd (12.0, 2.4) | 3.62, m | 3.91, dd (12.1, 2.0) | |
| 4-OMe | 3.87, s | 3.76, s | 3.85, s | 3.87, s | |||
| 5-OMe | 3.68, s | 3.62, s | 3.66, s | ||||
| 7-OMe | 3.78, s | 3.73, s | |||||
| 5′-OMe | 3.65, s | 3.62, s | 3.70, s | 3.77, s | 3.65, s | 3.59, s | 3.66, s |
| 2-OH | 8.74, s | ||||||
| 5-OH | 10.03, s | ||||||
| 2′-OH | 8.03, s | ||||||
| 3′-OH | 9.08, br s | ||||||
| 2′′-OH | 5.44, d (2.8) | 6.05, br s | |||||
| 3′′-OH | 5.08, br s | 5.12, d (4.4) | |||||
| 4′′-OH | 5.05, d (5.3) | 4.99, d (4.6) | |||||
| 6′′-OH | 4.64, t (5.5) | 4.31, t (5.7) | |||||
| No. | 1a | 1b | 2a | 3a | 4a | 4b | 5a |
|---|---|---|---|---|---|---|---|
| a Date were recorded in CD3OD.b Date were recorded in DMSO-d6. | |||||||
| 1 | 153.9 | 152.5 | 152.9 | 158.0 | 146.0 | 143.9 | 146.4 |
| 2 | 112.8 | 110.0 | 117.6 | 111.2 | 143.3 | 142.2 | 143.1 |
| 3 | 115.3 | 114.4 | 115.0 | 115.6 | 118.4 | 117.2 | 118.2 |
| 4 | 145.0 | 142.8 | 144.9 | 147.1 | 110.6 | 109.5 | 110.1 |
| 5 | 153.7 | 151.9 | 151.8 | 155.8 | 155.0 | 152.4 | 155.0 |
| 6 | 108.0 | 105.2 | 107.2 | 108.4 | 107.4 | 106.7 | 106.6 |
| 7 | 167.9 | 165.0 | 172.8 | 166.4 | |||
| 1′ | 147.0 | 143.9 | 146.8 | 154.0 | 152.1 | 150.9 | 146.8 |
| 2′ | 133.7 | 132.5 | 134.7 | 130.0 | 131.4 | 129.7 | 133.7 |
| 3′ | 148.4 | 147.0 | 148.6 | 150.8 | 153.1 | 151.4 | 148.5 |
| 4′ | 100.5 | 99.1 | 101.2 | 101.8 | 98.2 | 96.3 | 100.4 |
| 5′ | 154.3 | 151.8 | 154.0 | 159.2 | 158.8 | 156.2 | 154.4 |
| 6′ | 101.1 | 100.2 | 102.7 | 101.0 | 97.3 | 95.4 | 101.2 |
| 1′′ | 104.2 | 102.2 | 103.8 | 103.1 | 107.4 | 105.8 | 104.3 |
| 2′′ | 75.0 | 73.3 | 75.0 | 74.9 | 75.5 | 73.8 | 75.0 |
| 3′′ | 77.7 | 75.8 | 77.7 | 77.8 | 77.9 | 76.2 | 77.8 |
| 4′′ | 71.5 | 70.0 | 71.6 | 71.5 | 71.0 | 69.6 | 71.6 |
| 5′′ | 78.6 | 77.3 | 78.5 | 78.6 | 78.5 | 77.1 | 78.6 |
| 6′′ | 62.6 | 60.8 | 62.6 | 62.6 | 62.2 | 60.8 | 62.6 |
| 4-OMe | 57.1 | 56.1 | 57.0 | 57.0 | |||
| 5-OMe | 56.4 | 55.2 | 56.4 | ||||
| 7-OMe | 52.5 | 51.5 | |||||
| 5′-OMe | 56.3 | 55.3 | 56.4 | 56.5 | 56.1 | 55.4 | 56.3 |
 | ||
| Fig. 2 Key HMBC (blue arrows) correlations of compounds 1–6. | ||
Compound 2 was obtained as a brown amorphous solid. Its molecular formula was deduced to be C21H24O13 by HRESIMS ion peak at m/z 483.1150 [M − H]− (calcd for C21H23O13, 483.1139). The 1H and 13C NMR data of 2 (Tables 1 and 2) showed similarity to those of 1, except for the lack of one methoxy signals as well as the chemical shift of C-7 (δC 172.8 in 2 and δC 167.9 in 1). These observations indicated a carboxyl group in 2 instead of the methoxycarbonyl group in 1 at C-2 position, which was supported by the molecular formula. In addition, the HMBC correlations of 2 (Fig. 2) confirmed the same connectivity as in 1. Therefore, the structure of 2 was determined as 2-(3-O-β-D-glucopyranosyl-2-hydroxy-5-methoxyphenoxy)-4-hydroxy-5-methoxy benzoic acid and was given the name hyperelatoside B.
Compound 3 was obtained as a brown amorphous solid. Its molecular formula was established as C21H22O12 by HRESIMS (m/z 489.0998 [M + Na]+, calcd for C21H22NaO12, 489.1009), accounting for 11 degrees of unsaturation. The UV, IR, and NMR data of 3 were similar to those of 1 and 2 (Table 1), indicating that the structure of 3 is a biphenyl ether glycoside. The 1H NMR spectrum of 3 revealed the presence of a 1,2,4,5-tetrasubstituted benzene ring, a 1,2,3,5-tetrasubstituted benzene ring, two methoxy groups, as well as a sugar moiety, which were consistent with the constituent units of 2. Similar HMBC correlations (Fig. 2) established that a sugar moiety, a carbonyl group, and two methoxy groups were located at the same positions as 2. Compared with the chemical shifts of C-7 of 1 and 2 (Table 2), the appearance of an ester carbonyl carbon at δC 166.4 in 3 suggested that an ester group presented between these two aromatic rings via the linkage of C-7–O–C-2′, consistent with the 11 degrees of unsaturation required by the molecular formula. Acid hydrolysis result confirmed the sugar to be a β-D-glucopyranose. Hence, the structure of 3 was characterized as 2′,5-dihydroxy-4,5′-dimethoxy-3′-O-β-D-glucopyranosyl-2-carboxy-diphenyl ether 2,2′-lactone and was named hyperelatoside C.
Compound 4 was obtained as a brown amorphous solid with a molecular formula of C20H24O11 based on HRESIMS (m/z 463.1205 [M + Na]+, calcd for C20H24NaO11, 463.1216). The 1H NMR data of 4 (measured in CD3OD, Table 1) exhibited a set of characteristic ABX aromatic protons at δH 6.85 (1H, d, J = 8.8 Hz, H-3), 6.59 (1H, dd, J = 8.8, 2.9 Hz, H-4), and 6.46 (1H, d, J = 2.9 Hz, H-6) and two meta-coupled aromatic protons at δH 6.24 (1H, d, J = 2.9 Hz, H-4′) and 5.91 (1H, d, J = 2.9 Hz, H-6′), indicating the presence of a 1,2,4-trisubstituted (A-ring) and a 1,2,3,5-tetrasubstituted (B-ring) benzene rings, respectively. It also showed aliphatic signals for two methoxy groups [δH 3.68 (3H, s) and 3.65 (3H, s)] and a set of sugar protons (δH 3.25–4.73), including an anomeric proton at δH 4.73 (1H, d, J = 7.6 Hz, H-1′′). The 13C NMR spectrum displayed signals for 12 aromatic carbons (δC 97.3–158.8), two methoxy carbons (δC 56.4 and 56.1), and a characteristic glucopyranosyl moiety (δC 107.4, 78.5, 77.9, 75.5, 71.0, and 62.2) (Table 2). Acid hydrolysis result of 4 confirmed the sugar to be a β-D-glucopyranose. The assignment of signals corresponding to A-ring was based on the HMBC correlations from H-3 (δH 6.85) to C-1 (δC 146.0) and C-5 (δC 155.0), from H-4 (δH 6.59) to C-2 (δC 143.3) and C-6 (δC 107.4), and from H-6 (δH 6.46) to C-2 (δC 143.3) and C-4 (δC 110.6) (Fig. 2). The methoxy protons at δH 3.68 exhibited a HMBC correlation to C-5 (δC 155.0), which indicated the connection of a methoxy group at C-5 position of A-ring. Furthermore, the 1H NMR spectrum of 4, measured in DMSO-d6, displayed a hydroxyl proton signal at δH 8.74 (1H, s, 2-OH), which showed HMBC correlations to C-1, C-2, and C-3, supporting that a hydroxyl group was attached to C-2 position of A-ring. Complete assignment of the 1H and 13C NMR data of B-ring was achieved by the HMBC correlations from H-4′ (δH 6.24) to C-2′ (δC 131.4), C-3′ (δC 153.1), C-5′ (δC 158.8), and C-6′ (δC 97.3) and from H-6′ (δH 5.91) to C-1′ (δC 152.1), C-2′ (δC 131.4), C-4′ (δC 98.2), and C-5′ (δC 158.8) (Fig. 2). The HMBC correlations from the methoxy protons at δH 3.65 to C-5′ and from the anomeric proton at δH 4.73 to C-2′ suggested the locations of a methoxy group at C-5′ and a β-D-glucopyranosyl moiety at C-2′, respectively. An ether linkage between C-1 (δC 146.0) of A-ring and C-1′ (δC 152.1) of B-ring was then established due to their downfield chemical shifts and the molecular formula. Accordingly, compound 4 was identified as 2,3′-dihydroxy-5,5′-dimethoxy-2′-O-β-D-glucopyranosyl-diphenyl ether and was named hyperelatoside D.
Compound 5 was obtained as a brown amorphous solid. Its molecular formula was determined to be C20H24O11 based on a deprotonated ion peak at m/z 439.1247 [M − H]− (calcd for C20H23O11, 439.1240) in HRESIMS, identical to that found for compound 4. Moreover, the UV and IR spectra of 5 showed similar absorption bands to those of 4, indicating their similar structures. The 1H and 13C NMR data (Tables 1 and 2) of A-ring of 5 were almost superimposable to those of 4, but they differed in their B-ring, suggesting that the locations of the substituent groups on B-ring were different between 4 and 5. The glycosylation site at C-3′ position of B-ring of 5 was inferred from an upfield shift of δC 4.6 ppm for C-3′ (ipso-C) and downfield shifts of δC 2.3 ppm for C-2′ (ortho-C), 2.2 ppm for C-4′ (ortho-C), and 3.9 ppm for C-6′ (para-C) (Table 2). This was further supported by a HMBC cross-peak between the anomeric proton at δH 4.80 (H-1′′) and the corresponding aglycone carbon at δC 148.5 (C-3′) (Fig. 2). The β-D-glucopyranosyl moiety was determined by the same method as described for 1. Therefore, the structure of 5 was defined as 2,2′-dihydroxy-5,5′-dimethoxy-3′-O-β-D-glucopyranosyl-diphenyl ether and was named hyperelatoside E.
Compound 6 was purified as white needles with a melting point of 220–222 °C. The molecular formula, C15H20O10, was deduced from HRESIMS (m/z 383.0946 [M + Na]+, calcd for C15H20NaO10, 383.0954). The IR spectrum showed absorption bands corresponding to a hydroxyl group (3439 cm−1) and a conjugated carbonyl group (1666 cm−1). The 1H NMR spectrum exhibited signals attributable to one hydroxyl group [δH 10.46 (1H, s, 2-OH)], one 1,2,4,5-tetrasubstituted benzene ring [δH 7.22 (1H, s, H-6) and 6.71 (1H, s, H-3)], one anomeric proton at δH 5.04 (1H, d, J = 7.2 Hz, H-1′), and two methoxy groups [δH 3.88 (3H, s, H-8) and 3.73 (3H, s, H-9)] (Table 3). The 13C NMR spectrum displayed 15 carbon resonances, which were classified as an ester carbonyl carbon (δC 169.3), six aromatic carbons (δC 103.5–156.6), a characteristic glucopyranosyl moiety (δC 99.4, 77.1, 76.7, 73.0, 69.5, and 60.6), and two methoxy carbons (δC 56.1 and 52.3) (Table 3). The β-D-glucopyranosyl unit was verified by the same method as described for 1. In the HMBC spectrum of 6, the methoxy protons at δH 3.88 (H-8) exhibited a correlation with the carbonyl carbon at δC 169.3 (C-7), which suggested that 6 is a derivative of methyl benzoate. The key HMBC correlations from H-6 (δH 7.22) to C-1 (δC 104.1), C-2 (δC 156.6), C-4 (δC 153.3), C-5 (δC 142.1), and C-7 (δC 169.3), from the hydroxyl proton at δH 10.46 to C-1 (δC 104.1), C-2 (δC 156.6), and C-3 (δC 103.5), and from the methoxy protons at δH 3.73 (H-9) to C-5 (δC 142.1) (Fig. 2), indicated the locations of a hydroxyl group and a methoxy group at C-2 and C-5, respectively. In addition, the anomeric proton at δH 5.04 (H-1′) exhibited a HMBC correlation with C-4 (δC 153.3), indicating that the glucopyranosyl moiety was attached to C-4 position. Thus, compound 6 was assigned as 4-O-β-D-glucopyranosyl-2-hydroxy-5-methoxy-methylbenzoate and was named hyperelatoside F.
| No. | δH (J in Hz) | δC |
|---|---|---|
| 1 | 104.1 | |
| 2 | 156.6 | |
| 3 | 6.71, s | 103.5 |
| 4 | 153.3 | |
| 5 | 142.1 | |
| 6 | 7.22, s | 111.5 |
| 7 | 169.3 | |
| 8 | 3.88, s | 52.3 |
| 9 | 3.73, s | 56.1 |
| 4a | ||
| 4b | ||
| 8a | ||
| 9a | ||
| 1′ | 5.04, d (7.2) | 99.4 |
| 2′ | 3.26, m | 73.0 |
| 3′ | 3.28, m | 76.7 |
| 4′ | 3.16, m | 69.5 |
| 5′ | 3.39, m | 77.1 |
| 6′ | 3.45, dd (11.7, 5.2) | 60.6 |
| 3.66, dd (11.7, 6.0) | ||
| 1-OH | ||
| 2-OH | 10.46, s | |
| 7-OH | ||
| 2′-OH | 5.34, d (5.1) | |
| 3′-OH | 5.13, d (4.6) | |
| 4′-OH | 5.06, d (5.3) | |
| 6′-OH | 4.58, t (5.6) |
The remaining compounds were elucidated on the basis of NMR and ESIMS data analysis as well as by comparison with literature data. They were identified as 3,5-dihydroxy-4-methoxyxanthone (7),16 1,5,6-trihydroxy-7-methoxyxanthone (8),17 1,3,7-trihydroxy-2-(2-hydroxy-3-methyl-3-butenyl)-xanthone (9),18 2,4-dihydroxy-3-methyl-6-O-β-D-glucopyranosyl benzophenone (10),19 garcimangosone D (11),20 1-(2-methylbutyryl)-phloroglucinol-glucopyranoside (12),21 quercetin 3-O-α-L-rhamnopyranoside (13),22 quercetin 3-O-β-D-galactopyranoside (14),23 and kaempferol 3-O-α-L-rhamnopyranoside (15)24 (Fig. S54, ESI†). Among them, compounds 7, 10, and 12 were isolated from the genus Hypericum for the first time.
Naturally occurring biphenyl ethers have been reported to be in some fungi in recent years, especially Aspergillus species,25 but are rarely found in plants.26 In fungi, these kinds of compounds are formed biosynthetically from the anthraquinone emodin, via sulochrin, and the grisandienes such as geodin was previously reported.27 However, these intermediates of desmethylsulochrin, sulochrin, dihydrogeodin, and geodin are rarely as the plant metabolites. Given the isolation of benzoic acid derivatives (6 and 12), benzophenones (10 and 11), and xanthones (7–9) from H. elatoides, the possible biosynthetic pathway of hyperelatosides A–E (1–5) is proposed in Scheme 1. The key step of this pathway is the xanthone ring cleavage reaction to form biphenyl ether skeleton, the immediate precursors of which may be 1,3,6,7- and 1,3,5,8-tetrahydroxyxanthones. Although the formation of biphenyl ethers from xanthones catalyzed by an enzyme system involving an oxygenase, has not yet detected in plants, the similar reaction has been observed in the biosynthetic studies of biphenyl ethers from anthraquinones in fungi. An anthraquinone ring cleavage enzyme, named questin oxygenase, was found in the cell-free extract of Aspergillus terreus and the reaction mechanism underlying the ring cleavage of anthraquinone is likely to be a chemical Baeyer–Villiger oxidation and hydrolysis of the formed lactone intermediate.27,28 This suggested that the same type of enzymes as questin oxygenase may be involved in the formation of biphenyl ethers from xanthones in H. elatoides. Xanthones and benzophenones are two major constituents of the plants of the genus Hypericum.1eThe biosynthesis of xanthone products has been studied in H. androsaemum, H. calycinum, and H. perforatum.29 2,3′,4,6-Tetrahydroxybenzophenone, which is formed from benzoic acid by the condensation of one benzoyl CoA and three malonyl CoA units catalyzed by benzophenone synthase, is cyclized regioselectively to yield either 1,3,7- or 1,3,5-trihydroxyxanthone, which is then regioselectively hydroxylated to 1,3,6,7- or 1,3,5,8-tetrahydroxyxanthone.30 Subsequent xanthone ring cleavage reaction gives the biphenyl ether skeleton, which may generate the structures of 1–5 via enzymatic reactions of decarboxylation, O-methylation using S-adenosylmethionine (SAM), and glycosylation.
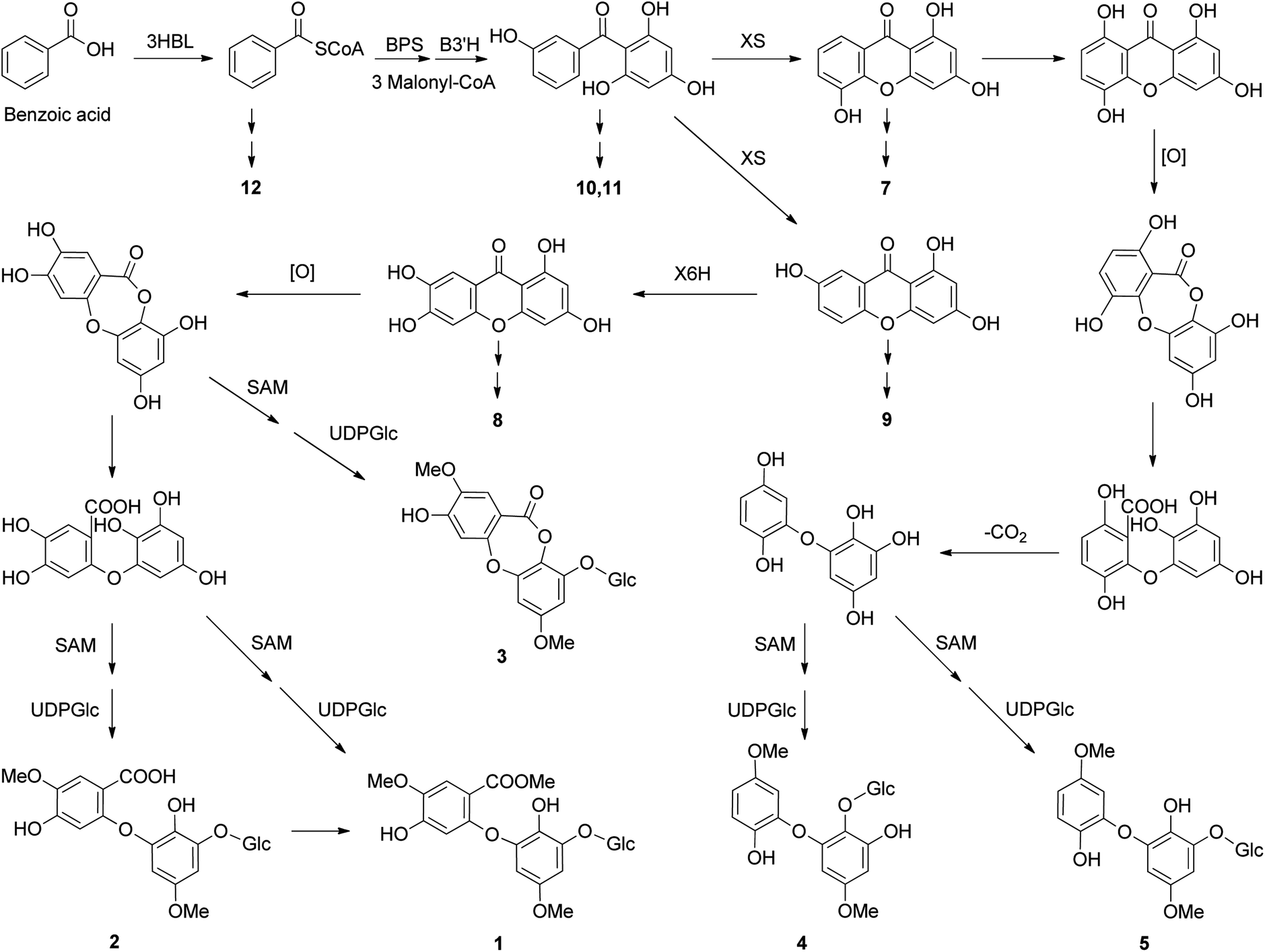 | ||
| Scheme 1 Proposed biosynthetic pathway for biphenyl ether glycosides identified from H. elatoides (3HBL, 3-hydroxybenzoate:CoA ligase; BPS, benzophenone synthase; B3′H, benzophenone 3′-hydroxylase; XS, xanthone synthase; X6H, xanthone 6-hydroxylase; SAM, S-adenosylmethionine; UDPGlc, uridine diphosphate glucose; Glc, glucopyranosyl unit). | ||
The neurotrophic activities of new compounds 1–6 were investigated in terms of their ability to enhance NGF-induced neurite outgrowth using rat pheochromocytoma (PC12) cells as a model system of neuronal differentiation. Fig. 3 showed that, relative to the NGF control (100%), compounds 1, 2, and 4–6 exerted a significant increase in neurite-bearing cells at 1 μM. In particular, the percentages of neurite-bearing cells for cells treated with hyperelatosides B (2) and D (4) in the presence of NGF (20 ng mL−1) reached up to 192.78 ± 17.71% and 228.70 ± 12.51% relative to the NGF control, respectively. Interestingly, among the biphenyl ether glycosides (1–5), only 3 had no effect on NGF-medicated neurite outgrowth in PC12 cells at 1 μM, suggesting that the presence of a carboxyl or hydroxyl group at C-2 position of A-ring may be responsible for the neurotrophic activity. Compound 2 showed much stronger activity than 1, indicating that attachment of an active hydrogen atom to the O-7 position of the carboxyl group could significantly improve this activity than attachment of a methyl group to O-7 position.
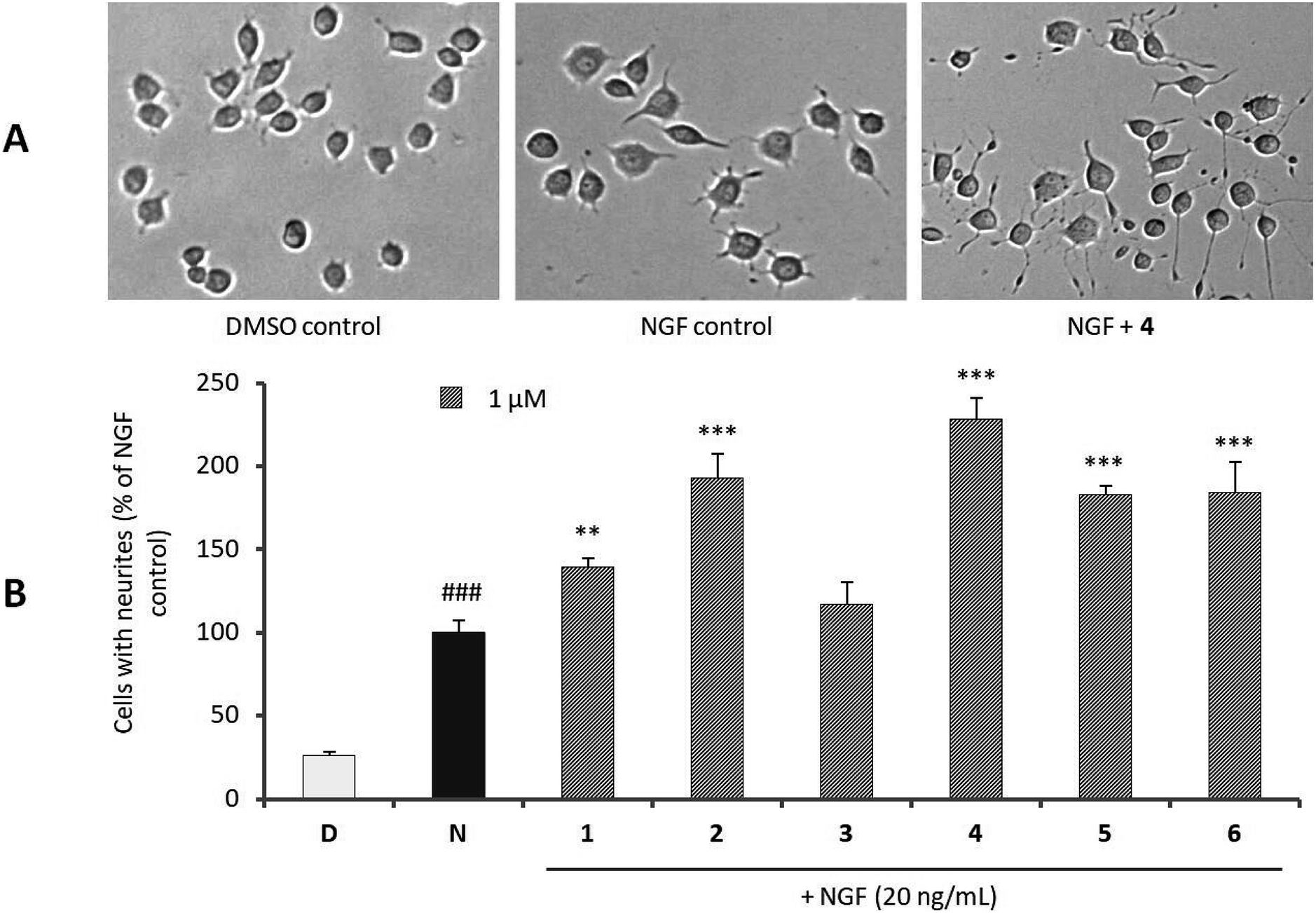 | ||
| Fig. 3 Effects of compounds 1–6 on neurite outgrowth in PC12 cells. (A) Morphological changes of PC12 cells treated with 0.1% DMSO, NGF (20 ng mL−1), and NGF (20 ng mL−1) + 4 (1.0 μM) were visualized under light microphotography. The cells treated with 4 served as a representative example to demonstrate the enhancement of neurite outgrowth in the presence of NGF. (B) Neurite-bearing cells were quantitatively analyzed as described in measurement of neurite outgrowth. Data are expressed as percentages of the value of NGF-treated cells (means ± standard deviations, n = 3) (D, 0.1% DMSO, control; N, 20 ng mL−1 NGF, positive control; ###p < 0.001 vs. control; *p < 0.05, **p < 0.01, ***p < 0.001 vs. NGF control). | ||
Neuroinflammation has been typically involved in the pathology of neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease. Therefore, the inhibitory effects of compounds 1–15 on LPS-stimulated NO production in BV-2 microglial cells were investigated to establish their anti-neuroinflammatory activities. As shown in Fig. 4, compounds 5, 8, and 14 exhibited inhibitory effects against NO production in BV-2 cells at 25 μM. In order to investigate whether the inhibitory activities of the active compounds were due to their cytotoxicity towards BV-2 cells, the effects of compounds 5, 8, and 14 on cell proliferation/viability were measured by 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Of these, compounds 5 and 8 showed no cytotoxicity with LPS treatment for 24 h.
 | ||
| Fig. 4 Effects of compounds 1–15 on LPS-induced NO production in BV-2 microglial cells. Data are expressed as percentages of the value obtained with LPS-treated cells. (C, untreated control, 0.1% DMSO; L, LPS-treated control; Q, quercetin, 5 μM, positive control; ###p < 0.001 compared with control group, **p < 0.01, ***p < 0.001 compared with LPS group). | ||
In addition, severe oxidative stress has been also implicated in the pathogenesis of neurodegenerative diseases. The antioxidant activities of compounds 1–15 were preliminarily evaluated for their DPPH free radical scavenging properties at 1, 5, 10, 25, 50, and 100 μM using ascorbic acid as the positive control (IC50 = 12.45 ± 0.51 μM). The result showed that compound 14 exhibited the most significant DPPH radical scavenging activity with IC50 value of 11.22 ± 0.38 μM (Table 4), which showed stronger antioxidant activity than the positive control. Its potent activity may be due to the presence of two hydroxyl groups at C-3 and C-4 positions of the aromatic ring, which agrees with previous studies.31 Moreover, compounds 2, 4, 5, 8, and 13 exhibited moderate antioxidant activities. The remaining compounds were proved to have weak or no DPPH radical scavenging activities at concentrations of 1–100 μM.
Experimental section
General experimental procedures
Optical rotations were measured with an Auton Paar MCP300 automatic polarimeter (Anton Paar, Graz, Austria). The UV spectra were obtained on a Thermo Scientific Evolution-300 UV-visible spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with KBr pellets (Bruker, Billerica, MA, USA). The NMR spectra were obtained on a Bruker Avance III 500 spectrometer (Bruker, Billerica, MA, USA). HRESIMS were recorded on an AB Triple TOF® 4600 mass spectrometer (AB SCIEX, Redwood City, CA, USA). Semipreparative HPLC was performed with a system consisting of LC-20AP pumps (Shimadzu, Kyoto, Japan) and a SPD-20A UV/vis detector (Shimadzu, Kyoto, Japan) equipped with YMC Packed C18 columns (5 μm, 250 × 10.0 mm and 150 × 4.6 mm, YMC Co., Ltd., Kyoto, Japan). Column chromatography was performed on silica gel (100–200 and 200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, China), RP-C18 resins (YMC Gel ODS-A-HG, 50 μm particle size, YMC Co., Ltd., Kyoto, Japan), and Sephadex LH-20 (Pharmacia Fine Chemicals, Uppsala, Sweden). Thin-layer chromatography (TLC) was performed on silica gel 60 F254 and RP-18 F254S plates (Merck KGaA, Darmstadt, Germany). Fractions were monitored by TLC, and spots were visualized by UV light (254 and 365 nm) and spraying with 10% H2SO4 in ethanol, followed by heating. The purity of obtained compounds were determined by analysis of 1H NMR spectra and HPLC. Horse serum (HS), fetal bovine serum (FBS), and Dulbecco's modified Eagle medium (DMEM) were purchased from Gibco (Thermo Fisher Scientific, Bremen, Germany). NGF, authentic sugars, ascorbic acid, quercetin, dimethylsulfoxide (DMSO), DPPH, the growth substrate poly-L-lysine, and nutrient mixture F-12 (Ham) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).Plant material
The aerial parts of H. elatoides were collected in Dongjue Mountain, Qishan County, Shaanxi Province, China, in September 2016. The plant was identified by Professor Zai-Min Jiang, College of Life Sciences, Northwest A&F University. A voucher specimen (Jiang 1043) was deposited in the herbarium of Northwest A&F University (WUK), Yangling, China.Extraction and isolation
The aerial parts (1.3 kg) of H. elatoides were dried in shade, cut into small pieces, and extracted three times (each for 8 h) with 12 L of MeOH under reflux conditions (55 °C). After filtration and evaporation in vacuo, the obtained extract (334.8 g) was suspended in distilled water (3 L) and partitioned successively with n-hexane (3 L × 3), EtOAc (3 L × 3), and n-BuOH (3 L × 3) to yield a hexane-soluble fraction (55.6 g), an EtOAc-soluble fraction (49.2 g), and a BuOH-soluble fraction (71.0 g), respectively. The EtOAc fraction was subjected to silica gel CC and eluted with a gradient of CH2Cl2/MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:1) to yield fifteen fractions (E1–E15). Fr. E3 was further subjected to RP-C18 CC (acetone–H2O, 1:6) to give ten subfractions (E3.1–E3.10). Fr. E3.5 was subjected to silica gel CC (CHCl3–MeOH, 100:1) to afford compound 7 (63.1 mg). Fr. E6 was subjected to silica gel CC (CH2Cl2–MeOH, 30:1 to 20:1) to yield eight subfractions (E6.1–E6.8). Fr. E6.3 was purified on a RP-C18 column (MeOH–H2O, 3:1) to yield compound 8 (26.5 mg). Fr. E6.4 was purified on a RP-C18 column (acetone–MeOH, 1:5) to yield compound 9 (24.6 mg). Fr. E9 was subjected to silica gel CC and eluted with a gradient of CHCl3–MeOH–H2O (15:1:0.05 to 4:1:0.1) to yield eight fractions (E9.1–E9.8). Fr. E9.4 was further separated by RP-C18 CC (MeOH–H2O, 1:2 to 1:1) to yield seven subfractions (E9.4.1–E9.4.7). Compound 6 (5.3 mg) was obtained by the separation of Fr. E9.4.3 on a Sephadex LH-20 column (MeOH–H2O, 1:2). Compounds 1 (34.2 mg), 2 (21.5 mg), 3 (19.4 mg), and 4 (10.0 mg) were obtained by the separation of Fr. E9.4.5 on a RP-C18 column (acetone–H2O, 1:4 to 1:2.5). Fr. E9.4.6 was subjected to RP-C18 CC (acetone–H2O, 1:2.5) to yield compound 10 (35.9 mg). Fr. E9.5 was separated by RP-C18 CC (acetone–H2O, 1:6 to 1:4) to give seven subfractions (E9.5.1–E9.5.7). Fr. E9.5.2 was subjected to RP-C18 CC (MeOH–H2O, 1:3) to yield compound 11 (3.4 mg). Fr. E9.5.6 was subjected to RP-C18 CC (MeOH–H2O, 1:1.4) followed by RP-C18 CC (acetone–H2O, 1:2) to provide compound 12 (4.2 mg). Fr. E9.5.7 was purified on a RP-C18 column (MeOH–H2O, 1:1.5) to yield compound 15 (7.6 mg). Fr. E10 was separated by silica gel CC (CHCl3–MeOH–H2O, 15:1:0.05 to 3:1:0.1) to afford seven subfractions (E10.1–E10.7). Fr. E10.3 was subjected to RP-C18 CC (acetone–H2O, 1:5) to yield four fractions (E10.3.1–E10.3.4). Fr. E10.3.2 was subjected to RP-C18 CC (MeOH–H2O, 1:2.5 to 1:1.5) to afford eight fractions (E10.3.2.1–E10.3.2.8). Compound 5 (7.6 mg) was obtained by the separation of Fr. E10.3.2.2 on a RP-C18 column (MeOH–H2O, 1:2). Fr. E10.6 was separated on a silica gel column (CHCl3–MeOH–H2O, 4:1:0.1) to yield compound 13 (960.0 mg). Fr. E11 was subjected to silica gel CC and eluted with a gradient of CHCl3–MeOH–H2O (6:1:0.1 to 2:1:0.1) to afford six subfractions (E11.1–E11.6). Fr. E11.4 was further subjected to RP-C18 CC (acetone–H2O, 1:3 to 1:2) to give compound 14 (161.3 mg).
ε) 226 (3.9), 260 (2.4), 292 (2.0) nm; IR (KBr) νmax 3418, 2952, 1701, 1618, 1515, 1440, 1379, 1273, 1209, 1076, 1028, 630 cm−1; 1H and 13C NMR data (CD3OD and DMSO-d6), see Tables 1 and 2; HRESIMS (positive) m/z 521.1247 [M + Na]+ (calcd for C22H26NaO13, 521.1271).ε) 225 (5.4), 260 (1.0) nm; IR (KBr) νmax 3415, 1624, 1514, 1444, 1274, 1211, 1170, 1027, 630 cm−1; 1H and 13C NMR data (CD3OD), see Tables 1 and 2; HRESIMS (negative) m/z 483.1150 [M − H]− (calcd for C21H23O13, 483.1139).ε) 225 (5.4), 260 (2.4), 292 (1.0) nm; IR (KBr) νmax 3434, 2954, 1640, 1514, 1454, 1019, 661 cm−1; 1H and 13C NMR data (CD3OD), see Tables 1 and 2; HRESIMS (positive) m/z 489.0998 [M + Na]+ (calcd for C21H22NaO12, 489.1009).ε) 290 (2.3) nm; IR (KBr) νmax 3402, 2950, 2837, 1650, 1455, 1410, 1111, 1022, 671 cm−1; 1H and 13C NMR data (CD3OD and DMSO-d6), see Tables 1 and 2; HRESIMS (positive) m/z 463.1205 [M + Na]+ (calcd for C20H24NaO11, 463.1216).ε) 290 (3.0) nm; IR (KBr) νmax 3392, 2950, 2836, 1651, 1457, 1411, 1111, 1023, 671 cm−1; 1H and 13C NMR data (CD3OD), see Tables 1 and 2; HRESIMS (negative) m/z 439.1247 [M − H]− (calcd for C20H23O11, 439.1240).ε) 222 (3.3), 256 (2.9), 319 (3.2) nm; IR (KBr) νmax 3439, 2954, 1666, 1626, 1509, 1445, 1366, 1267, 1226, 1062, 694, 652 cm−1; 1H and 13C NMR data (DMSO-d6), see Table 3; HRESIMS (positive) m/z 383.0946 [M + Na]+ (calcd for C15H20NaO10, 383.0954).
:1 to 2:1) to yield fifteen fractions (E1–E15). Fr. E3 was further subjected to RP-C18 CC (acetone–H2O, 1:6) to give ten subfractions (E3.1–E3.10). Fr. E3.5 was subjected to silica gel CC (CHCl3–MeOH, 100:1) to afford compound 7 (63.1 mg). Fr. E6 was subjected to silica gel CC (CH2Cl2–MeOH, 30:1 to 20:1) to yield eight subfractions (E6.1–E6.8). Fr. E6.3 was purified on a RP-C18 column (MeOH–H2O, 3:1) to yield compound 8 (26.5 mg). Fr. E6.4 was purified on a RP-C18 column (acetone–MeOH, 1:5) to yield compound 9 (24.6 mg). Fr. E9 was subjected to silica gel CC and eluted with a gradient of CHCl3–MeOH–H2O (15:1:0.05 to 4:1:0.1) to yield eight fractions (E9.1–E9.8). Fr. E9.4 was further separated by RP-C18 CC (MeOH–H2O, 1:2 to 1:1) to yield seven subfractions (E9.4.1–E9.4.7). Compound 6 (5.3 mg) was obtained by the separation of Fr. E9.4.3 on a Sephadex LH-20 column (MeOH–H2O, 1:2). Compounds 1 (34.2 mg), 2 (21.5 mg), 3 (19.4 mg), and 4 (10.0 mg) were obtained by the separation of Fr. E9.4.5 on a RP-C18 column (acetone–H2O, 1:4 to 1:2.5). Fr. E9.4.6 was subjected to RP-C18 CC (acetone–H2O, 1:2.5) to yield compound 10 (35.9 mg). Fr. E9.5 was separated by RP-C18 CC (acetone–H2O, 1:6 to 1:4) to give seven subfractions (E9.5.1–E9.5.7). Fr. E9.5.2 was subjected to RP-C18 CC (MeOH–H2O, 1:3) to yield compound 11 (3.4 mg). Fr. E9.5.6 was subjected to RP-C18 CC (MeOH–H2O, 1:1.4) followed by RP-C18 CC (acetone–H2O, 1:2) to provide compound 12 (4.2 mg). Fr. E9.5.7 was purified on a RP-C18 column (MeOH–H2O, 1:1.5) to yield compound 15 (7.6 mg). Fr. E10 was separated by silica gel CC (CHCl3–MeOH–H2O, 15:1:0.05 to 3:1:0.1) to afford seven subfractions (E10.1–E10.7). Fr. E10.3 was subjected to RP-C18 CC (acetone–H2O, 1:5) to yield four fractions (E10.3.1–E10.3.4). Fr. E10.3.2 was subjected to RP-C18 CC (MeOH–H2O, 1:2.5 to 1:1.5) to afford eight fractions (E10.3.2.1–E10.3.2.8). Compound 5 (7.6 mg) was obtained by the separation of Fr. E10.3.2.2 on a RP-C18 column (MeOH–H2O, 1:2). Fr. E10.6 was separated on a silica gel column (CHCl3–MeOH–H2O, 4:1:0.1) to yield compound 13 (960.0 mg). Fr. E11 was subjected to silica gel CC and eluted with a gradient of CHCl3–MeOH–H2O (6:1:0.1 to 2:1:0.1) to afford six subfractions (E11.1–E11.6). Fr. E11.4 was further subjected to RP-C18 CC (acetone–H2O, 1:3 to 1:2) to give compound 14 (161.3 mg).
ε) 226 (3.9), 260 (2.4), 292 (2.0) nm; IR (KBr) νmax 3418, 2952, 1701, 1618, 1515, 1440, 1379, 1273, 1209, 1076, 1028, 630 cm−1; 1H and 13C NMR data (CD3OD and DMSO-d6), see Tables 1 and 2; HRESIMS (positive) m/z 521.1247 [M + Na]+ (calcd for C22H26NaO13, 521.1271).ε) 225 (5.4), 260 (1.0) nm; IR (KBr) νmax 3415, 1624, 1514, 1444, 1274, 1211, 1170, 1027, 630 cm−1; 1H and 13C NMR data (CD3OD), see Tables 1 and 2; HRESIMS (negative) m/z 483.1150 [M − H]− (calcd for C21H23O13, 483.1139).ε) 225 (5.4), 260 (2.4), 292 (1.0) nm; IR (KBr) νmax 3434, 2954, 1640, 1514, 1454, 1019, 661 cm−1; 1H and 13C NMR data (CD3OD), see Tables 1 and 2; HRESIMS (positive) m/z 489.0998 [M + Na]+ (calcd for C21H22NaO12, 489.1009).ε) 290 (2.3) nm; IR (KBr) νmax 3402, 2950, 2837, 1650, 1455, 1410, 1111, 1022, 671 cm−1; 1H and 13C NMR data (CD3OD and DMSO-d6), see Tables 1 and 2; HRESIMS (positive) m/z 463.1205 [M + Na]+ (calcd for C20H24NaO11, 463.1216).ε) 290 (3.0) nm; IR (KBr) νmax 3392, 2950, 2836, 1651, 1457, 1411, 1111, 1023, 671 cm−1; 1H and 13C NMR data (CD3OD), see Tables 1 and 2; HRESIMS (negative) m/z 439.1247 [M − H]− (calcd for C20H23O11, 439.1240).ε) 222 (3.3), 256 (2.9), 319 (3.2) nm; IR (KBr) νmax 3439, 2954, 1666, 1626, 1509, 1445, 1366, 1267, 1226, 1062, 694, 652 cm−1; 1H and 13C NMR data (DMSO-d6), see Table 3; HRESIMS (positive) m/z 383.0946 [M + Na]+ (calcd for C15H20NaO10, 383.0954).Acid hydrolysis
The absolute configurations of the sugar moieties of 1–6 were determined by the acid hydrolysis method.32 Each compound (approximately 1.0 mg) was separately dissolved in 1 N HCl (0.5 mL), and then heated at 90 °C in a water bath for 2 h. After extraction with EtOAc two times, the H2O-soluble fraction was evaporated to dryness under reduced pressure. The residue was dissolved in pyridine (0.1 mL) containing L-cysteine methyl ester hydrochloride (0.5 mg) and heated at 60 °C for 1 h. A 100 μL solution of o-tolylisothiocyanate (0.5 mg) in pyridine was added and the reaction mixture was heated at 60 °C for 1 h. Then each reaction mixture was analyzed by the Waters 1525 HPLC system using a Waters 2489 UV/vis detector (at 250 nm). Analytical HPLC was performed on the YMC Packed C18 column (5 μm, 150 × 4.6 mm) with a linear gradient elution (CH3CN–H2O, 20:80 to 40:60) for 30 min. The derivative of D-glucose was identified in 1–6 by a comparison of the retention time with authentic D-glucose (tR 16.3 min), which was subjected to the same derivatization procedure.
Measurement of neurite outgrowth
PC12 cells were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). PC12 cells were maintained in nutrient mixture F-12 medium supplemented with 10% inactivated HS, 5% inactivated FBS, penicillin G (100 U mL−1), streptomycin (100 μg mL−1), and sodium bicarbonate (2.5 g L−1) at 37 °C in humidified atmosphere of 5% CO2. Morphological analysis and quantification of neurite-bearing cells were performed using a phase-contrast microscope as previously described.15 Briefly, PC12 cells were seeded in poly-L-lysine-coated 24-well plates at a density of 2 × 104 cells per mL with normal serum medium for 24 h. The F-12 medium containing low serum (2% HS and 1% FBS) was replaced prior to exposure to vehicle (0.1% DMSO) or indicated reagents. The cells were treated with tested compounds in the presence of NGF (20 ng mL−1). Cells without treatment served as a negative control. Cells treated with 20 ng mL−1 of NGF served as a positive control. One concentration experiment was repeated in three wells. After an additional 48 h of incubation, neurite outgrowth of PC12 cells was observed under an inverted microscope using phase-contrast objectives and photographed by the digital camera. Eight images were selected randomly under a microscope for each well. At least 100 cells in each of eight randomly separated fields were scored. The cells with neurites greater than or equal to the length of one cell body were positive for neurite outgrowth and expressed as a percentage of the total cell number in eight fields. Experiments were repeated at least three times, and data are expressed as means ± standard deviations.Determination of NO production
BV-2 microglial cells were purchased from Peking Union Medical College Cell Bank (Beijing, China) and cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, penicillin G (100 U mL−1), and streptomycin (100 μg mL−1) at 37 °C in a humidified incubator with 5% CO2. The NO concentration was detected by the Griess reagent.33 Quercetin was used as a positive control. BV-2 cells were seeded at the density of 1.5 × 105 cells per mL in 96-well culture plate and treated with each compound and LPS (1.0 μg mL−1) for 24 h. After that, 50 μL of cell-free supernatant was allowed to react with an equal volume of Griess reagent (Beyotime Institute of Biotechnology, Shanghai, China) for 15 min at room temperature in the dark. Then, the absorbance was measured at 540 nm using a microplate reader. The cell viability of the cultured cells was detected by MTT-based colorimetric method.Antioxidant activity
The DPPH radical scavenging assay was performed according to a previously described method.34 Ascorbic acid was used as a positive control. The absorbance of the resulting solution of each compound was measured at 517 nm with a spectrophotometer. Results were expressed as IC50 values.Conclusions
In summary, this study is the first report to describe the chemical constituents and their biological activities of H. elatoides. Chemical investigation of the aerial parts of H. elatoides led to the isolation of six new phenolic compounds including five unusual biphenyl ether glycosides, hyperelatosides A–E (1–5), and one benzoate glycoside, hyperelatoside F (6). To the best of our knowledge, this represents the first report of the isolation of biphenyl ether glycosides from plants and their putative biosynthetic pathway is proposed. Furthermore, except for 3, all the new isolates possessed potent ability to potentiate the activity of NGF to stimulate neurite outgrowth in PC12 cells. Compounds 5 and 8 exhibited moderate anti-neuroinflammatory activities and 14 showed significant antioxidant activity in vitro. Our findings show that the aerial parts of H. elatoides may be an excellent source of neurotrophic phytochemicals for the prevention and treatment of neurodegenerative diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Scientific Startup Foundation for Doctors of Northwest A&F University (No. 2452015356) and the Fundamental Research Funds for the Central Universities (No. 2452017173) and the Natural Science Foundation of China (No. 21572182).Notes and references
- (a) C. C. Sánchez-Mateo, B. Prado and R. M. Rabanal, J. Ethnopharmacol., 2002, 79, 119 CrossRef; (b) M. F. Rieli, R. Mattei and C. E. L. de Araujo, Fitoterapia, 2002, 73, 462 CrossRef; (c) A. Viana, J. C. do Rego, G. von Poser, A. Ferraz, A. P. Heckler, J. Costentin and S. M. K. Rates, Neuropharmacology, 2005, 49, 1042 CrossRef PubMed; (d) O. Grundmann, Y. Lv, O. Kelber and V. Butterweck, Neuropharmacology, 2010, 58, 767 CrossRef PubMed; (e) J. Zhao, W. Liu and J. C. Wang, Chem. Biodiversity, 2015, 12, 309 CrossRef PubMed.
- (a) S. S. Chatterjee, S. K. Bhattacharya, M. Wonnemann, A. Singer and W. E. Müller, Life Sci., 1998, 63, 499 CrossRef PubMed; (b) J. Sarris, Phytother. Res., 2007, 21, 703 CrossRef PubMed; (c) S. Kasper, F. Caraci, B. Forti, F. Drago and E. Aguglia, Eur. Neuropsychopharmacol., 2010, 20, 747 CrossRef PubMed.
- (a) R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963 CrossRef PubMed; (b) I. P. Singh and S. B. Bharate, Nat. Prod. Rep., 2006, 23, 558 RSC; (c) J. J. Zhang, J. Yang, Y. Liao, X. W. Yang, J. Z. Ma, Q. L. Xiao, L. X. Yang and G. Xu, Org. Lett., 2014, 16, 4912 CrossRef PubMed; (d) X. W. Yang, R. B. Grossman and G. Xu, Chem. Rev., 2018, 118, 3508 CrossRef PubMed.
- (a) Z. Q. Yin, Y. Wang, W. C. Ye and S. X. Zhao, Biochem. Syst. Ecol., 2004, 32, 521 CrossRef; (b) N. Tanaka, Y. Kashiwada, S. Y. Kim, M. Sekiya, Y. Ikeshiro and Y. Takaishi, Phytochemistry, 2009, 70, 1456 CrossRef PubMed; (c) W. J. Xu, R. J. Li, O. Quasie, M. H. Yang, L. Y. Kong and J. Luo, J. Nat. Prod., 2016, 79, 1971 CrossRef PubMed.
- (a) A. Hecka, B. Maunit, F. Aubriet and J. F. Muller, Rapid Commun. Mass Spectrom., 2009, 23, 885 CrossRef PubMed; (b) M. A. Farag and L. A. Wessjohann, Planta Med., 2012, 78, 488 CrossRef PubMed; (c) A. Porzel, M. A. Farag, J. Mülbradt and L. A. Wessjohann, Metabolomics, 2014, 10, 574 CrossRef.
- (a) C. A. Ang'edu, J. Schmidt, A. Porzel, P. Gitu, J. O. Midiwo and G. Adam, Pharmazie, 1999, 54, 235 Search PubMed; (b) S. A. F. Tanemossu, K. Franke, N. Arnold, J. Schmidt, H. K. Wabo, P. Tane and L. A. Wessjohann, Phytochemistry, 2014, 105, 171 CrossRef PubMed.
- (a) L. R. Jat, Int. J. Pharm. Pharm. Sci., 2013, 5, 9 Search PubMed; (b) Z. B. Zhou, Z. R. Li, X. B. Wang, J. G. Luo and L. Y. Kong, J. Nat. Prod., 2016, 79, 1231 CrossRef PubMed.
- (a) M. A. G. del Rio, M. I. Sanchez-Reus, I. Iglesias, M. A. Pozo, M. Garcia-Arencibia, J. Fernandez-Ruiz, L. Garcia-Garcia, M. Delgado and J. Benedi, CNS Neurol. Disord.: Drug Targets, 2013, 12, 665 CrossRef; (b) D. Zheleva-Dimitrova, P. Ncdialkov and G. Momekov, Pharmacogn. Mag., 2013, 9, 1 CrossRef PubMed.
- D. Ben-Eliezer and E. Yechiam, Sci. Rep., 2016, 6, 35700 CrossRef PubMed.
- N. Huang, L. Rizshsky, C. Hauck, B. J. Nikolau, P. A. Murphy and D. F. Birt, Phytochemistry, 2011, 72, 2015 CrossRef PubMed.
- L. C. Brito, A. L. R. Berenger and M. R. Figueiredo, Food Chem. Toxicol., 2017, 109, 847 CrossRef PubMed.
- H. Zhu, C. Chen, J. Yang, X. N. Li, J. Liu, B. Sun, S. X. Huang, D. Li, G. Yao, Z. Luo, Y. Li, J. Zhang, Y. Xue and Y. Zhang, Org. Lett., 2014, 16, 6322 CrossRef PubMed.
- H. Jayasuriya, A. M. Clark and J. D. McChesney, J. Nat. Prod., 1991, 54, 1314 CrossRef.
- (a) P. L. Liu, C. Du, Z. M. Jiang and J. Cai, Xibei Zhiwu Xuebao, 2011, 31, 1268 Search PubMed; (b) Q. Sun, P. L. Liu, Q. Zhang, L. X. Cao and Z. M. Jiang, Xibei Zhiwu Xuebao, 2012, 32, 276 Search PubMed.
- (a) X. W. Shi, L. Liu, J. M. Gao and A. L. Zhang, Eur. J. Med. Chem., 2011, 46, 3112 CrossRef PubMed; (b) C. C. Zhang, X. Yin, C. Y. Cao, J. Wei, Q. Zhang and J. M. Gao, Bioorg. Med. Chem. Lett., 2015, 25, 5078 CrossRef PubMed; (c) R. Bai, C. C. Zhang, X. Yin, J. Wei and J. M. Gao, J. Nat. Prod., 2015, 78, 783 CrossRef PubMed; (d) C. C. Zhang, C. Y. Cao, M. Kubo, K. Harada, X. T. Yan, Y. Fukuyama and J. M. Gao, Int. J. Mol. Sci., 2017, 18, 1659 CrossRef PubMed.
- Z. Zhang, H. N. ElSohly, M. R. Jacob, D. S. Pasco, L. A. Walker and A. M. Clark, Planta Med., 2002, 68, 49 CrossRef PubMed.
- N. Tanaka and Y. Takaishi, Phytochemistry, 2006, 67, 2146 CrossRef PubMed.
- M. Iinuma, H. Tosa, T. Ito, T. Tanaka and M. Aqil, Phytochemistry, 1995, 40, 267 CrossRef.
- S. E. Ukwueze, P. O. Osadebe and F. B. C. Okoye, Nat. Prod. Res., 2015, 29, 1728 CrossRef PubMed.
- Y. L. Huang, C. C. Chen, Y. J. Chen, R. L. Huang and B. J. Shieh, J. Nat. Prod., 2001, 64, 903 CrossRef.
- G. Bohr, C. Gerhaeuser, J. Knauft, J. Zapp and H. Becker, J. Nat. Prod., 2005, 68, 1545 CrossRef PubMed.
- T. Fukunaga, K. Nishiya, I. Kajikawa, Y. Watanabe, N. Suzuki, K. Takeya and H. Itokawa, Chem. Pharm. Bull., 1988, 36, 1180 CrossRef.
- K. Yasukawa and M. Takido, Phytochemistry, 1987, 26, 1224 CrossRef.
- C. Y. Chen, S. L. Hsieh, M. M. Hsieh, S. F. Hsieh and T. J. Hsieh, Chin. Pharm. J., 2004, 56, 141 Search PubMed.
- (a) R. Liu, W. Zhu, Y. Zhang, T. Zhu, H. Liu, Y. Fang and Q. Gu, J. Antibiot., 2006, 59, 362 CrossRef PubMed; (b) T. Bunyapaiboonsri, S. Yoiprommarat, K. Intereya and K. Kocharin, Chem. Pharm. Bull., 2007, 55, 304 CrossRef PubMed; (c) M. J. Fang, H. Fang, W. J. Li, D. M. Huang, Z. Wu and Y. F. Zhao, Nat. Prod. Res., 2012, 26, 1224 CrossRef PubMed; (d) W. Peng, F. You, X. L. Li, M. Jia, C. J. Zheng, T. Han and L. P. Qin, Zhongguo Tianran Yaowu, 2013, 11, 673 Search PubMed; (e) L. L. Xu, C. C. Zhang, X. Y. Zhu, F. Cao and H. J. Zhu, Nat. Prod. Res., 2017, 31, 1875 CrossRef PubMed; (f) P. Phainuphong, V. Rukachaisirikul, S. Phongpaichit, S. Preedanon and J. Sakayaroj, Tetrahedron, 2017, 73, 5920 CrossRef.
- N. Li, H. S. Lee, N. Zhang, Y. N. Sun, J. L. Li, S. S. Xing, J. G. Chen and L. Cui, Phytochem. Lett., 2015, 13, 286 CrossRef.
- (a) R. F. Curtis, C. H. Hassall, D. W. Jones and T. W. Williams, J. Chem. Soc., 1960, 4838 RSC; (b) Z. G. Chen, I. Fujii, Y. Ebizuka and U. Sankawa, Phytochemistry, 1995, 38, 299 CrossRef; (c) K. Huang, I. Fujii, Y. Ebizuka, K. Gomi and U. Sankawa, J. Biol. Chem., 1995, 270, 21495 CrossRef PubMed; (d) J. Hargreaves, J. Park, E. L. Ghisalberti, K. Sivasithamparam, B. W. Skelton and A. H. White, J. Nat. Prod., 2002, 65, 7 CrossRef PubMed.
- I. Fujii, Y. Ebizuka and U. Sankawa, J. Biochem., 1988, 103, 878 CrossRef PubMed.
- (a) W. Schmidt and L. Beerhues, FEBS Lett., 1997, 420, 143 CrossRef PubMed; (b) M. M. Gaid, D. Sircar, A. Müller, T. Beuerle, B. Liu, L. Ernst, R. Hänsch and L. Beerhues, Plant Physiol., 2012, 160, 1267 CrossRef PubMed; (c) I. El-Awaad, M. Bocola, T. Beuerle, B. Liu and L. Beerhues, Nat. Commun., 2016, 7, 11472 CrossRef PubMed.
- (a) W. Schmidt, S. Peters and L. Beerhues, Phytochemistry, 2000, 53, 427 CrossRef PubMed; (b) H. R. El-Seedi, M. A. El-Barbary, D. M. H. El-Ghorab, L. Bohlin, A. K. Borg-Karlson, U. Göransson and R. Verpoorte, Curr. Med. Chem., 2010, 17, 854 CrossRef PubMed.
- (a) P. V. Kiem, N. X. Cuong, N. X. Nhiem, V. K. Thu, N. K. Ban, C. V. Minh, B. H. Tai, T. N. Hai, S. H. Lee, H. D. Jang and Y. H. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 633 CrossRef PubMed; (b) X. T. Yan, S. H. Lee, W. Li, Y. N. Sun, S. Y. Yang, H. D. Jang and Y. H. Kim, Food Chem., 2014, 156, 408 CrossRef PubMed.
- T. Tanaka, T. Nakashima, T. Ueda, K. Tomii and I. Kouno, Chem. Pharm. Bull., 2007, 55, 899 CrossRef PubMed.
- (a) Z. Huang, Z. X. Zhu, Y. T. Li, D. R. Pang, J. Zheng, Q. Zhang, Y. F. Zhao, D. Ferreira, J. K. Zjawiony, P. F. Tu and J. Li, J. Nat. Prod., 2015, 78, 2276 CrossRef PubMed; (b) J. Li, N. Li, X. Li, G. Chen, C. Wang, B. Lin and Y. Hou, J. Nat. Prod., 2017, 80, 3081 CrossRef PubMed; (c) J. Wei, W. H. Guo, C. Y. Cao, R. W. Kou, Y. Z. Xu, M. Górecki, L. D. Bari, G. Pescitelli and J. M. Gao, Sci. Rep., 2018, 8, 2175 CrossRef PubMed.
- (a) O. P. Sharma and T. K. Bhat, Food Chem., 2009, 113, 1202 CrossRef; (b) C. D. Zheng, G. Li, H. Q. Li, X. J. Xu, J. M. Gao and A. L. Zhang, Nat. Prod. Commun., 2010, 5, 1759 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D NMR, 2D NMR, and HRESIMS of 1–6; chemical structures of 7–15. See DOI: 10.1039/c8ra05322g |
| This journal is © The Royal Society of Chemistry 2018 |