DOI:
10.1039/C8RA05195J
(Paper)
RSC Adv., 2018,
8, 33753-33774
East Indian sandalwood (Santalum album L.) oil confers neuroprotection and geroprotection in Caenorhabditis elegans via activating SKN-1/Nrf2 signaling pathway†
Received
17th June 2018
, Accepted 18th September 2018
First published on 2nd October 2018
Abstract
East Indian Sandalwood Oil (EISO) has diverse beneficial effects and has been used for thousands of years in traditional folk-medicine for treatment of different human ailments. However, there has been no in-depth scientific investigation to decipher the neuroprotective and geroprotective mechanism of EISO and its principle components, α- and β-santalol. Hence the current study was undertaken to assess the protective effects of EISO, and α- and β-santalol against neurotoxic (6-OHDA/6-hydroxydopamine) and proteotoxic (α-synuclein) stresses in a Caenorhabditis elegans model. Initially, we found that EISO and its principle components exerted an excellent antioxidant and antiapoptotic activity as it was able to extend the lifespan, and inhibit the ROS generation, and germline cell apoptosis in 6-OHDA-intoxicated C. elegans. Further, we showed that supplementation of EISO, and α- and β-santalol reduced the 6-OHDA and α-synuclein-induced Parkinson's disease associated pathologies and improved the physiological functions. The genetic and reporter gene expression analysis revealed that an EISO, or α- and β-santalol-mediated protective effect does not appear to rely on DAF-2/DAF-16, but selectively regulates SKN-1 and its downstream targets involved in antioxidant defense and geroprotective processes. Together, our findings indicated that EISO and its principle components are worth exploring further as a candidate redox-based neuroprotectant for the prevention and management of age-related neurological disorders.
1. Introduction
East Indian Sandalwood oil (EISO) is the volatile essential oil usually obtained from the heartwood of Santalum album L. (Santalaceae) trees. S. album is considered as one of the highly valuable industrial and medicinal plants and it has a long history in traditional medicinal systems across the world. The United states Food and Drug Administration (FDA) approved EISO as a natural flavoring agent and its use in combination with flavors for human consumption. EISO has been extensively used in aromatherapy as an antidepressant, to alter cogitation and psychological or physical wellness and is most likely used in fragrance material, incense, cosmetics, personal care products, meditative and spiritual practices.1,2 Some preclinical and clinical research using an integrated approach has demonstrated that EISO and its active principles showed a broad spectrum of pharmacological activities, including chemopreventive,3–5 antihyperglycemic, antioxidant,6 antineoplastic,7,8 antiviral9 and anti-inflammatory activities.10,11 However, the potential benefits of EISO treatment in preserving neural health and the underlying protective mechanisms from intracellular destructive stimuli and extracellular physiological changes are yet to be elucidated.
Neurodegeneration can occur as a consequence of misfolding and dysfunctional trafficking of proteins leading to inflammatory damage and cell death in central or peripheral nervous systems. In addition, such debilitating factors as oxidative stress, mitochondrial dysfunction, environmental factors and/or exposure to neurotoxin intensify with aging also disturb the homeostatic plasticity of neurons. These factors drive the heterogeneous group of neurodegenerative diseases including Alzheimer's disease, amyotrophic lateral sclerosis and Parkinson's disease.12 Parkinson's disease (PD, MIM 168600) is a common devastating neurological condition that can be seen in elderly people from all geographical regions. PD is predominantly characterized as a movement disorder. The neuronal loss in pars compacta of the substantia nigra, which causes a deficiency of catecholamine neurotransmitter in the striatum, and a typical aggregation of intracytoplasmic inclusions (Lewy bodies) inside the brain tissues known as α-synuclein are the two pathological hallmarks of neurodegeneration in PD.13 The loss of dopaminergic (DA) neurotransmission induce the primary motor symptoms (muscle rigidity, tremors, bradykinesia and postural instability) and eventually personality, behavioral and cognitive symptoms (dementia, depression, anxiety and sleep disruption) which are observed in individuals with PD.14,15
The biochemical and molecular neuropathogenesis of PD is quite ambiguous and the exact mechanisms that cause neuronal loss remain to be defined fully. Candidate gene identification approaches and genome wide association studies suggested that the first genetic determinant to be linked with α-synucleinopathies (abnormal accumulation of α-synuclein) was SNCA gene (PARK1), which encodes a 14.5 kDa (140 amino acids) natively unfolded cytoplasmic protein designated as α-synuclein. The SNCA was most likely to be expressed in central nervous system and several missense mutations have been identified so far in SNCA. Interestingly, mutations in the SNCA gene and extension of wild-type α-synuclein were also involved in the etiopathogenesis of PD.16,17 The catecholaminergic neurotoxin 6-OHDA has been widely used for many years as an inducer in in vitro and in vivo experimental PD models. It is a hydroxylated dopamine analogue enters DA neurons via dopamine transporters (DAT) and disrupts the neuronal functions. 6-OHDA is also found in human brain samples and urine of patients with PD.18,19
Mounting clinical evidence suggest that oxidative stress is also one of the major cause of nigral DA neuronal death in the PD brains.20 Additionally, oxidative metabolism of dopamine itself produces several endogenous neurotoxins such as aminochrome, dopamine o-quinone, 6-indolequinone and hydrogen peroxide which are implicated in the pathological events in selective DA neurodegeneration.21 In order to maintain the redox balance, cells have several defense mechanisms to counterbalance the production of oxidants and its damages. The antioxidant response element (ARE) pathway confers oxidative stress resistance and is considered as an essential factor for neuronal survival.22 The nuclear factor-erythroid 2-related factor 2 (Nrf2/NFE2L2) is largely known ARE associated transcription factor that belong to member of cap ‘n’ collar (CNC) subfamily activated under oxidative stress condition and regulates the expression of several genes that promote cellular defense and species longevity. SKiNhead-1 (SKN-1) is the only known functional ortholog of Nrf2 in Caenorhabditis elegans found to be associated in maintaining the cellular redox homeostasis.23,24 Under homeostatic conditions, the activity of SKN-1 was suppressed by binding and tethering of an ubiquitin ligase adaptor protein called WDR-23 (a WD40 repeat-containing protein). Once activated under stress conditions, SKN-1 is uncoupled from WDR-23 and accumulates in nuclei to transactivate the cellular antioxidant and detoxifying genes.25 Genetic analysis showed that loss of SKN-1 can shorten the lifespan26 and activation or over expression of SKN-1 extends lifespan, protects the DA neurodegeneration and improving the age-related proteostasis functions in C. elegans.27–30 Overall, these findings indicate the direct role of SKN-1/Nrf2 signaling in aging and age-related diseases.
Currently, there is no effective therapy available for the treatment of age-associated neuronal dysfunction. Hence, suitable drugs from traditional origin that can delay or halt the onset and progression of neurodegeneration are sorely needed. In this context, we report the first demonstration of the potential neuroprotective mechanisms of EISO and its major active principles, α- and β-santalol by using the powerful genetics of C. elegans.
2. Materials and methods
2.1. Sandalwood oil preparation and isolation of principle components
2.1.1. Preparation of sandalwood oil. The sandalwood oil was extracted by traditional steam distillation from the heartwood of plantation-grown Santalum album L. (Santalaceae) trees under Good Manufacturing Practice (GMP)/Good Laboratory Practice (GLP) regulatory guidelines was obtained from Santalis Pharmaceuticals Inc., (San Antonio, TX, USA). A second distillation under vacuum (rectification) was conducted to remove low-boiling santalenes that are typically present at low levels in EISO.
2.1.2. Isolation of santalol isomers. The santalol isomers were purified from EISO by sequential column chromatography on silica gel31 followed by supercritical fluid chromatography on a chiral support (Averica Discovery Services, Worcester MA, USA). The purity of isolated santalol isomers were determined by various analytical techniques, including gas chromatography-flame ionization detector (GC-FID), fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (1H, 13C and 2D NMR), liquid chromatography-mass spectrometry (LC-MS) and elemental analysis (ESI †).
2.2. DPPH radical-scavenging assay
The antioxidant activities of EISO and santalol isomers were measured by 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical quenching experiment as described in our previous study.32 Briefly, various concentrations of test compounds were added to 0.1 mM DPPH in methanol and the solution was vortexed vigorously. The tubes were then incubated for 20 min in the dark, and the absorbance was taken at 517 nm using UV-Vis spectrophotometer (UV-1800, Shimadzu, Japan). The percentage inhibition of DPPH radicals (IC50-inhibition concentration) by EISO and santalol isomers were calculated as illustrated,
| DPPH scavenging effect (%) = (control OD517 nm − test OD517 nm/control OD517 nm) × 100 |
2.3. C. elegans: strains, genetics and synchronization
The following C. elegans variants were used in this study: Bristol N2 (wild), TK22 (mev-1 [kn1]), MQ887 (isp-1 [qm150]), CB4876 (clk-1 [e2519]), CB1370 (daf-2 [e1370]), RB759 (akt-1 [ok525]), GR1307 (daf-16 [mgDf50]), WU51 (lin-45 [n2520]), MT8666 (mek-2 [n1989]), MH37 (mpk-1 [ku1]), EU1 (skn-1 [zu67]), FX01817 (wdr-23 [tm1817]), MT8735 (egl-1 [n1084n3082]), MT4770 (ced-9 [n1950]), MT2547 (ced-4 [n1162]), MT1522 (ced-3 [n717]), EG1285 (oxIs12 [unc-47p::GFP + lin-15]), LD1 (ldIs7 [skn-1::GFP]), CL2166 (dvIs19 [gst-4::GFP]), LD1171 (ldIs3 [gcs-1::GFP]), GA800 (wuIs151 [ctl-1,2,3::GFP]), MD701 (bcIs39 [lim-7p::ced-1::GFP + lin-15]), NL5901 (pkIs2386 [unc-54p::alpha-synuclein::YFP + unc-119]), and BZ555 (egIs1 [dat-1p::GFP]). All strains of C. elegans were maintained and propagated onto nematode growth medium (NGM) agar plates carrying a lawn of Escherichia coli OP50 (uracil auxotroph) as the food source at 20 °C according to the standardized protocols.33 To obtain synchronized cultures, gravid hermaphrodites were lysed in 5 M sodium hydroxide +5% household bleach and the gathered eggs were incubated overnight at 20 °C in M9 buffer to favor hatching.34 Genotypic and phenotypic descriptions of all C. elegans strains used in this study are given in ESI Table 1.†
2.4. Test compounds, toxicity, reproduction and growth
The oil or santalol isomers were dissolved in dimethyl sulfoxide (DMSO), and the final concentration of DMSO was maintained <0.3% at all experiments. To check the toxicity, age sorted N2 worms (n = 20–25 per replicate) were exposed to 1.0 mL aliquot of test solution containing different pharmacological doses of EISO (0, 3, 6, 12, 24 and 48 μg mL−1), α- and β- santalol (0, 2, 4, 8, 16, 32 and 64 μM) and 6 mg mL−1 heat-killed E. coli OP50. Exposures to test compounds were performed on L4-stage worms for 24 h (acute exposure), or from L1-stage to adult (chronic exposure) in a sterile tissue culture plate at 20 °C. Following exposure, the worms were observed for inactivity under stereo zoom microscope. Nematodes were considered as dead if they fail to respond gentle physical prodding using a metal loop. The toxicity was assessed by the endpoint of the percentage of survived animals with three independent experiments, each performed in triplicates.35 Reproduction was assessed by counting the eggs and offsprings produced by each worm from treated and untreated groups. Growth of the worms was assessed by body size, about 30 randomly-selected day-5 worms per treatment were subjected to analysis for body length measurement using Optika IsView image analyzing system (Optika, Italy).
2.5. Chemotaxis behavior assay
Chemotaxis assay was performed according to Bargmann et al.36 Briefly, the age-synchronized worms (n = 150–200 worms per treatment) were treated with various pharmacological doses of EISO, α- and β-santalol. On day-6 , the worms were transferred to chemotaxis plates divided into four equal quadrant (A, B, C, D) carrying 10 μL attractant (1 M sodium acetate) on one side (A, C) and 10 μL distilled water on the other side (B, D). 25 mM sodium azide (NaN3) was spotted on each side in order to paralyze the attracted worms towards the region. The worms were released at the center of plates and incubated at 20 °C for 90 min and the chemotaxis index (CI) was calculated using the following formula; CI = (A + C) − (B + D)/N where, A and C represents worms in the attractant region, B and D represent worms in the control region and N represent the total number of worms. Three independent trails were performed with appropriate replicates.
2.6. 6-OHDA exposure and EISO, α- and β-santalol treatment
Selective degeneration of DA neurons was carried out by treating the C. elegans with 6-OHDA (Sigma, St. Louis, USA) according to the method described previously.37 In brief, synchronized L3 worms were incubated with 25 mM 6-OHDA and 10 mM ascorbic acid in S-medium blended with OP50 (6 mg mL−1) and 0.1% DMSO (as solvent control), EISO, α- or β-santalol at 20 °C. After 1 h treatment, the worms were washed with M9 buffer and then incubated with 0.1% DMSO, EISO, α- or β-santalol at indicated concentrations for 24 h. Then, 50 mM of 5-fluoro-2′-deoxyuridine (FUdR, Sigma) was added to inhibit the production of progeny. Animals were incubated for 48 h and scored for various assays. Ascorbic acid was used to enhance the toxicity and selectivity of 6-OHDA.38
2.7. Assay for food-sensing behavior
To evaluate the functions of DA neurons-mediated slowing response to food, the food-sensing behavior assay was carried out.39 Assay plates were prepared in a 9 cm diameter Petri dish by spreading E. coli OP50 overnight in a ring with an inner diameter of ∼1 cm and an outer diameter of ∼8 cm on NGM agar. After treatment with 6-OHDA and 6-OHDA/EISO, α- or β-santalol the nematodes (n = 30–40 per treatment) were washed with M9 buffer and released to center of the NGM agar plate spotted with or without E. coli OP50 lawn. After five minutes, the body bends of each nematode was measured for 1 min in the presence or absence of food and the slowing response was calculated using the following formula:
| Slowing rate = (Nwithout food − Nwith food)/Nwithout food |
where, N represents total number of body bends in the presence or absence of bacterial food source.
2.8. Reporter gene expression assay
Age-synchronized L3 stage worms of transgenic strains stably expressing skn-1b/c::GFP (LD-1), gst-4p::GFP (CL2166), gcs-1p::GFP (LD1171) and ctl-1,2,3p::GFP (GA800) were treated with respective pharmacological doses of EISO, α- and β- santalol at 20 °C in the presence or absence of 6-OHAD. After 72 h of treatment, the worms were washed thrice with M9 buffer to remove the adhering bacteria, anesthetized with 25 mM NaN3, mounted onto 3% agarose padded microscopic slides and capped with a cover slip. Photomicrographs of immobilized live worms were taken under upright fluorescence microscope (BX41, Olympus, Japan) which was equipped with a digital camera (E330, Olympus, Japan). A total of 25–30 randomly selected worms from each experiment were measured for GFP signal using ImageJ software (NIH, Bethesda, MD) by determining the mean pixel intensity and expressed as relative fluorescent unit (RFU).32
2.9. Dopaminergic neurodegeneration assay
To explore the neuroprotective effects of EISO, α- and β-santalol against 6-OHDA-induced selective death of DA neurons, the BZ555 worms were treated with 6-OHDA and 6-OHDA/EISO, α- or β-santalol as said above. At the end of described treatments, the GFP images were photographed and the fluorescence intensity in head region was measured using imageJ software as mentioned in clause reporter gene expression assay.
2.10. Longevity assay
Synchronized animals (30–40 individuals per experiment) treated with 6-OHDA or 6-OHDA/EISO, α- or β-santalol as described previously, were transferred to fresh NGM plates containing an appropriate test compound for every two/three day intervals. A total of 50 mM FUdR was added to each plate to prevent the development of offspring. The number of dead and live worms was counted each day during the course of experiment. Worms were considered dead when they failed to respond to repeated mild contact with a metal loop, loss of pharyngeal pumping and lysed body structure. Worms exhibiting internal hatching, desiccation due to crawling off the agar or mechanical death were marked as censored. This experiment was terminated when the last worm became marked as dead or censored. Three independent biological trials were performed for each concentration under the similar conditions.
2.11. Assay for α-synuclein protein aggregation
The α-synuclein protein aggregation was assessed in untreated and EISO, α- or β-santalol treated transgenic NL5901 worms. Age-sorted worms were raised on NGM plates containing 50 mM FUdR and appropriate concentration of EISO, α- or β-santalol from L1 stage. On 2, 4 and 8 days of adulthood, the worms were monitored for α-synuclein aggregation under the fluorescence microscope and quantified using imageJ software.40
2.12. Quantification of lipid deposits
2.12.1. Nile Red staining. Lipid deposition in NL5901 C. elegans was assessed by Nile Red staining as per our previously described method.41 Before staining, the stock solution of Nile Red was prepared by dissolving 0.5 mg in 1 mL of acetone. Treated and untreated worms (n = 25–30 individuals per experiment) were washed and resuspended in M9 buffer, then fixed with 0.2 mL of 40% isopropanol solution for 3 min. Afterwards, the worms were stained with 150 μL Nile Red/isopropanol solution (prepared as follows: 6 μL Nile Red from stock solution were dissolved in 1 mL of 40% isopropanol) and incubated at room temperature for 30 min in the dark followed by imaging with fluorescence microscope.
2.12.2. Oil Red O staining. Oil Red O staining method was performed as described previously.42,43 The adult nematodes (n = 25–30 individuals per experiment) were washed with 1× PBS and resuspended in 400 μL of PBS to which 500 μL freshly prepared 2× MRWB buffer (160 mM KCl, 40 mM NaCl, 14 mM Na2EGTA, mM spermidine-HCl, 0.4 mM spermine, 30 mM NaPIPES (pH 7.4) and 0.2% beta-mercaptoethanol) and 100 μL 20% paraformaldehyde was added. The worms were subjected to three freeze–thaw cycles between dry-ice/ethanol and warm running tap water, followed by spinning at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, washed once in PBS to remove paraformaldehyde. The worms were then resuspended and dehydrated in 60% isopropanol for 20 min. After dehydration, the control and treated worms were stained with 60% Oil Red O staining solution for 12 h at room temperature, avoiding light. Stock solution of Oil Red O was prepared by dissolving 0.5 g of Oil Red O powder in 100 mL isopropanol solution and equilibrated for 3 days with gentle stirring. In order to get 60% working solution, stock solution was freshly diluted with 40% water and filtered through 0.2 μm membrane filter. After staining, the worms were washed twice with 1× PBS to remove the dyes, and the stained worms were mounted on agarose padded glass slide and imaged using an inverted microscope (XDS-2, Optika, Italy). The captured images were separated into RGB channel components and green channel was used to measure the lipid deposition.43
000 rpm, washed once in PBS to remove paraformaldehyde. The worms were then resuspended and dehydrated in 60% isopropanol for 20 min. After dehydration, the control and treated worms were stained with 60% Oil Red O staining solution for 12 h at room temperature, avoiding light. Stock solution of Oil Red O was prepared by dissolving 0.5 g of Oil Red O powder in 100 mL isopropanol solution and equilibrated for 3 days with gentle stirring. In order to get 60% working solution, stock solution was freshly diluted with 40% water and filtered through 0.2 μm membrane filter. After staining, the worms were washed twice with 1× PBS to remove the dyes, and the stained worms were mounted on agarose padded glass slide and imaged using an inverted microscope (XDS-2, Optika, Italy). The captured images were separated into RGB channel components and green channel was used to measure the lipid deposition.43
2.12.3. Sudan Black B staining. Sudan Black B staining for lipid deposition was performed as described previously.44 The worms from control and treatment groups (n = 25–30 individuals per experiment) were fixed as following the same fixation method. After fixation, the worms were then subjected to dehydration through washes in 25%, 50% and 70% ethanol. The fixed worms were stained with 50% saturated solution of Sudan Black B in 70% ethanol for 12 h at room temperature, protecting from light. The images were acquired using an inverted microscope (XDS-2, Optika, Italy).
2.13. Analysis of intracellular ROS levels and CED-1 expression
Control and EISO, α- or β-santalol fed worms (n = 30–40 per experiment) were incubated with 2 μM of H2DCF-DA for 30 min at 20 °C in the dark. After incubation, photographic images were examined for ROS levels by quantifying the fluorescence intensity of DCF.32 To analyze the CED-1 expression, transgenic strain MD701 has been used. Synchronized L3 stage worms were grown on the NGM plates in the presence or absence EISO, α- or β-santalol for 72 h at 20 °C with and without 6-OHDA treatment. After treatment, worms were imaged under a fluorescence microscope and germline apoptosis was analyzed by quantifying the fluorescent pattern of GFP “halos” on the engulfing cells and nascent phagosomes.
2.14. Visualization of germ cell apoptosis
Worms at the indicated time points were stained with 25 μg mL−1 acridine orange (AO) in M9 buffer containing heat-killed E. coli OP50 (6 mg mL−1) for 1 h at 20 °C in the dark and then transferred to fresh NGM plates for a recovery time of 1 h. Afterwards, animals were mounted on agarose pads and the apoptotic cells were visualized under fluorescence microscope. The gonad arm in the posterior part of control and treated C. elegans were photographed, because lipofuscin accumulation in the intestine shaded the gonad arm in the anterior part of the body. The apoptotic cells appeared yellow or yellow-orange after stained with AO representing increased DNA fragmentation, while intact cells appeared green in color.
2.15. Determination of acetylcholinesterase activity
L3 stage wild-type worms (n = 2000–2500) were treated with 6-OHDA and 6-OHDA/EISO, α- or β-santalol for 48 h at 20 °C. After the exposure period, the worms were washed twice with M9 buffer and homogenized in potassium phosphate buffer (0.1 M, pH 7.8) on ice followed by centrifugation at 12000 rpm for 10 min at 4 °C. The lysate supernatant was used for AChE activity determination according to the method of Ellman et al.,45 and the absorbance was measured at 414 nm. Bradford protein assay was performed to determine the total protein content46 and bovine serum albumin was used as the standard.
2.16. Molecular docking and ADME predication
To investigate the possible binding mode of α- and β-santalol to AChE, Keap1 and WDR-23, a molecular docking analysis was performed. The crystal structure of AChE (4M0E) and Kelch domain of Keap1 (4L7B) was retrieved from RCSB Protein Data Bank (http://www.rcsb.org). The amino acid sequence of WDR-23 was retrieved from the WormBase (http://www.wormbase.org) and the model was generated based on sequence homology by means of the Schrödinger program Prime. The structure of α- and β-santalol was downloaded from PubChem database (CID: 5281531 and 6857681, respectively). The binding pose of the ligands was scored and ranked by extra-precision glide docking (Glide XP), which docks ligand flexibility, was performed by using Schrödinger molecular modeling suite 9.3 (Schrödinger, LLC, New York, USA). The ADME (absorption, distribution, metabolism, and excretion) properties were predicted for santalol isomers using QikProp.
2.17. Statistical analysis
Regular statistical analysis and bar diagrams were generated using Excel, Microsoft office 2010 (Microsoft Corporation, Redmond, WA, USA). Survival of C. elegans were estimated using Kaplan–Meier survival method and analyzed by log-rank test using MedCalc 14.8.1 (MedCalc, Ostend, Belgium). Data was subjected to one-way analysis of variance (ANOVA; 95% confidence interval), followed by Bonferroni's post-hoc test for the determination of significance level using IBM SPSS 17.0 (IBM Corporation, Armonk, NY, USA). Data are presented as mean ± standard error of the mean (SEM) and probability levels of p < 0.05 were considered as statistically significant.
3. Results and discussion
3.1. Chemical composition of EISO
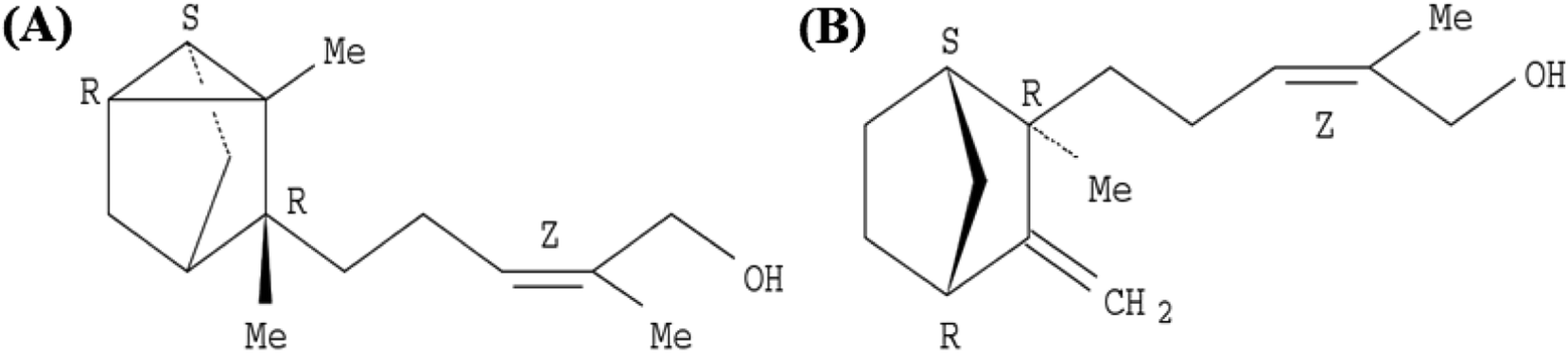
EISO contains a mixture of terpenoid compounds, primarily sesquiterpenoids. The predominant components in the mixture are α- and β-santalol, which together comprise approximately 60–80% of the mixture which are responsible for pleasant fragrance of EISO.2 The principle components of EISO used in this study were shown in the Table 1. The important sesquiterpenols like α-, β-, and epi-β-santalol and α-bergamotol alone make up ∼90% of the EISO. According to the GC-FID analysis, the EISO used in these studies contains 46.8% α-santalol and 18.7% β-santalol. The α- and β-santalol isomers were purified from EISO by sequential column chromatography on silica gel31 followed by supercritical fluid chromatography on a chiral support. Purified α- and β-santalols had 98.2% and 93.2% purity respectively, and the structures of two santalol isomers are shown in Fig. 1. The purified (93.2%) β-santalol also contains probably ∼2.5% α-santalol, a small amount of (Z)-α-trans-bergamotol, epi β-santalol and solvents (mostly hexane) and ∼0.23% water.
Table 1 Chemical composition of plantation-grown East Indian sandalwood (S. album L.) oil
| Sesquiterpenes |
% concentration |
| (Z)-α-Santalol |
46.8 |
| (Z)-β-Santalol |
18.7 |
| (Z)-α-trans-Bergamotol |
5.7 |
| epi-β-Santalol |
3.7 |
| cis Nuciferol |
2.7 |
| (Z)-Lanceol |
2.4 |
| (Z)-β-Curcumen-12-ol |
0.9 |
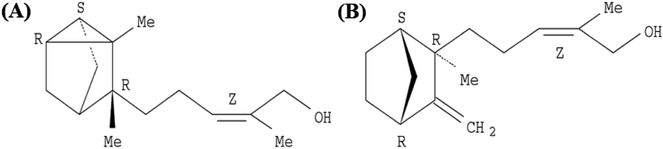 |
| | Fig. 1 Structure of (A) (+)-(Z)-α-santalol and (B) (−)-(Z)-β-santalol. | |
3.2. Safety evaluation of EISO on C. elegans
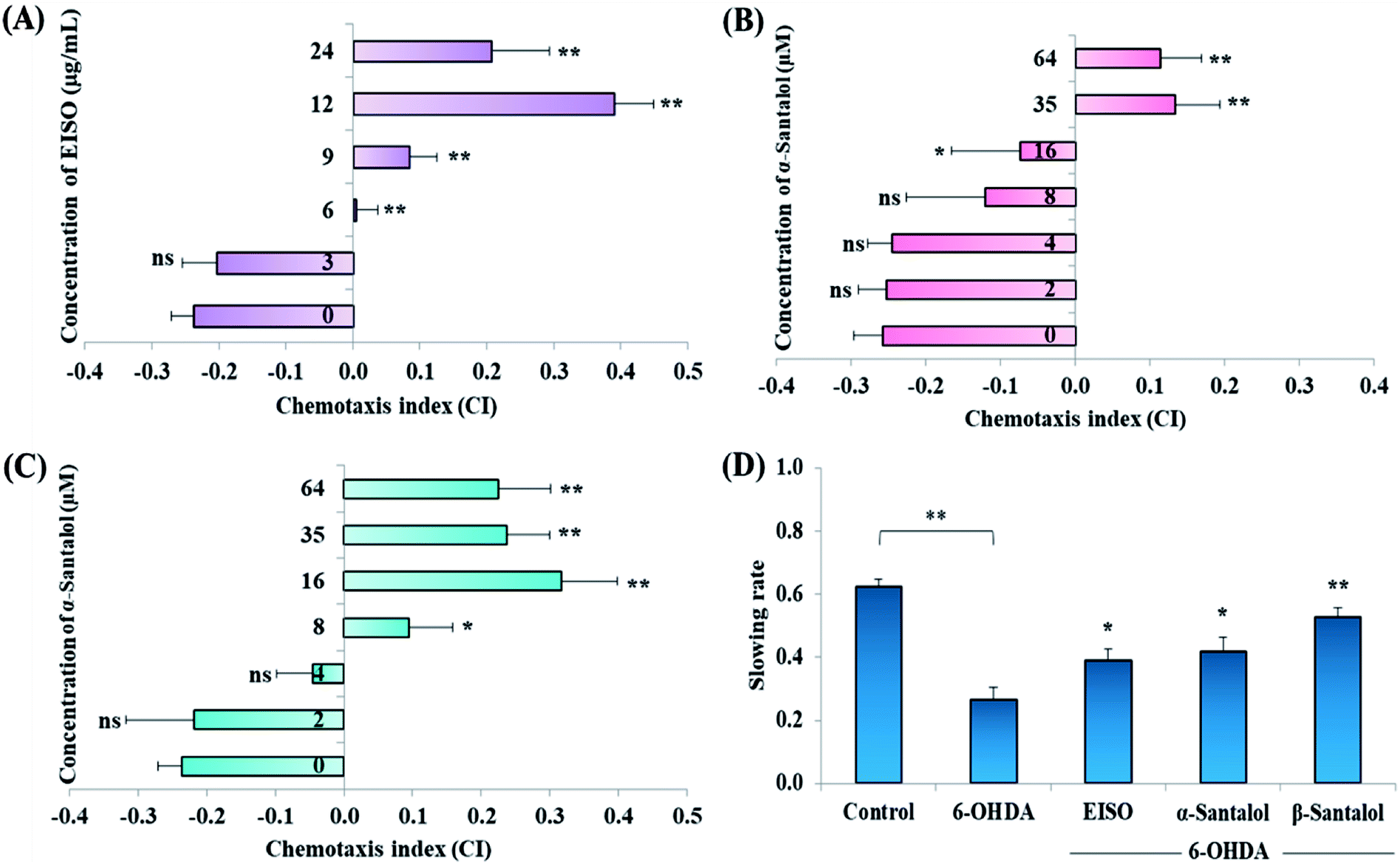
Acute and prolonged exposure of the studied pharmacological doses of EISO (0, 3, 6, 12, 24 and 48 μg mL−1), α- and β-santalol (0, 2, 4, 8, 16, 32 and 64 μM) did not adversely affect the survival, fecundity rate and development of C. elegans (ESI Tables 2 and 3†). To further investigate the effect of EISO, α- and β-santalol treatment on response to chemical stimuli, we examined the chemotaxis behavior of day-6 adult worms. We observed that wild-type worms pre-conditioned with EISO, α- and β-santalol showed an enhanced chemotaxis behavior than control worms, it indicates the healthy status of chemosensory neurons (Fig. 2A–C). This chemosensory behavior of C. elegans was reported to decay with age,47 that would be a reason for the reduction of chemotaxis behavior noticed in control worms. In addition, EISO and santalol isomers did not obliviously alter the morphology of D-type GABAergic motor neurons (ESI Fig. 1†). It is well-known that the reproductive organs and neurons are important secondary target of toxicants and drug compounds in C. elegans.48 Interestingly, the optimal doses of EISO (24 μg mL−1), α-santalol (32 μM) and β-santalol (16 μM) significantly reversed the natural reproduction and extended the mean lifespan of NL5901 C. elegans (PD model) under standard laboratory conditions; perhaps due to the modulation of intracellular stress levels (ESI Fig. 2 and ESI Table 4†). Also, the optimal dose of EISO, α- and β-santalol attenuated the 6-OHDA-induced food searching deficits in wild-type C. elegans (Fig. 2D). It's known that dopamine-mediated neural networks modulate the basal slowing response to food in C. elegans.49
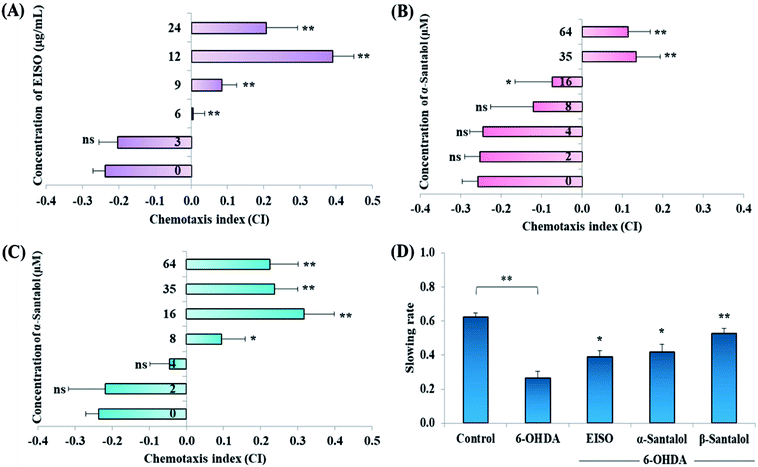 |
| | Fig. 2 Effect of EISO, α- and β-santalol on chemotaxis behavior and slowing response to food of wild-type C. elegans. Wild-type worms treated with (A) EISO, (B) α-santalol and (C) β-santalol exhibited an enhanced chemotaxis behavior at tested concentrations. (D) Effect of EISO, α- and β-santalol on food searching behavior in C. elegans. Data are presented as mean ± SEM, probability levels of p < 0.05 were considered as statistically significant, *p < 0.05, **p < 0.01, ns, not significant. | |
3.3. EISO extends the mean lifespan of wild-type C. elegans
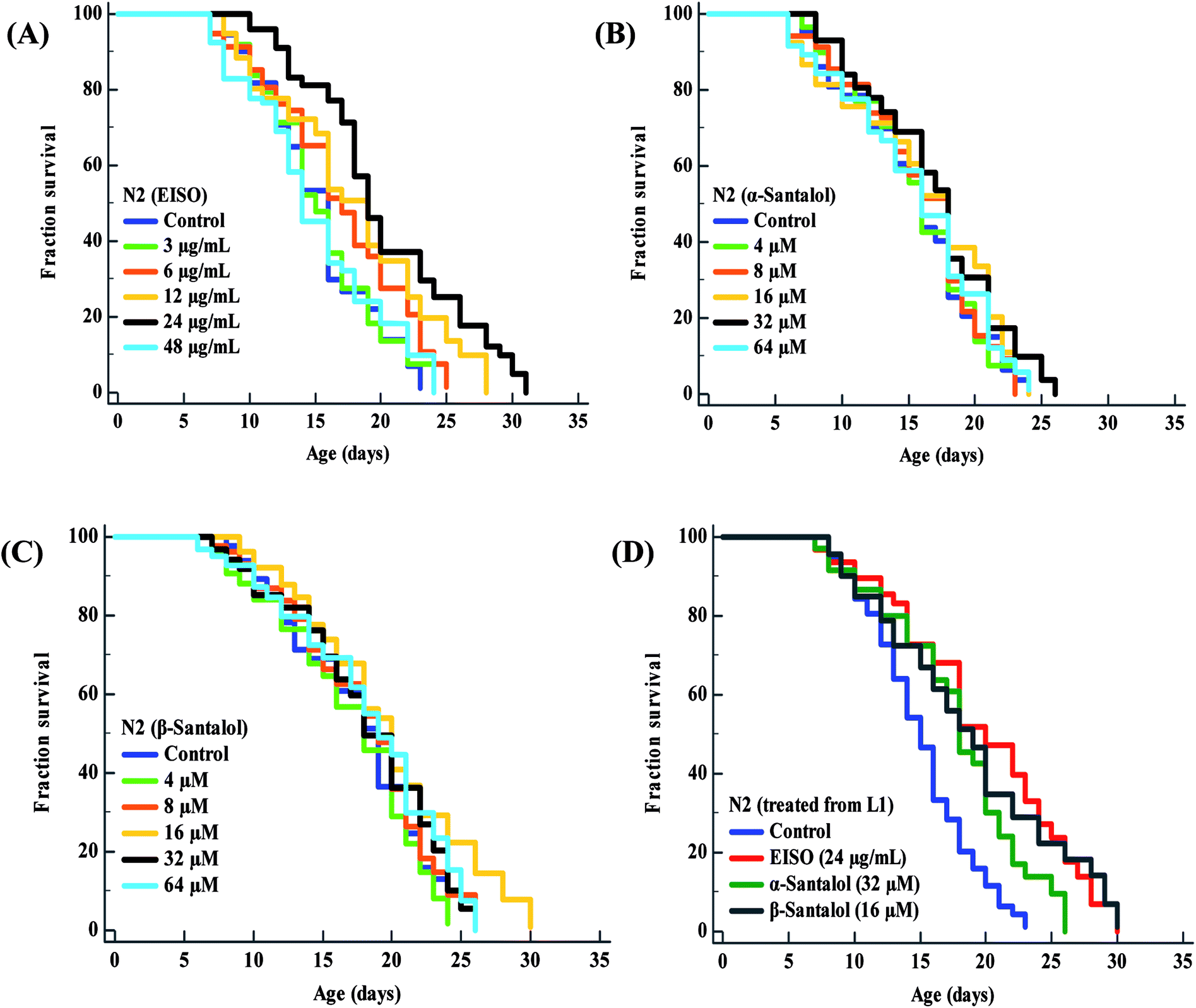
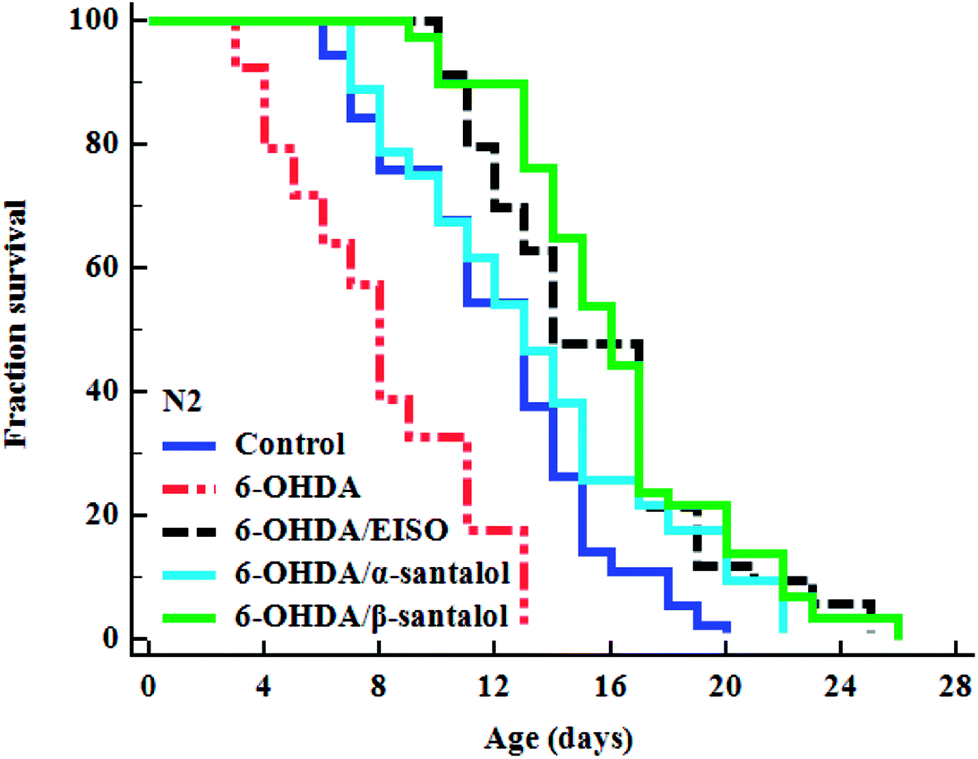
To examine whether EISO and its active components has an effect on the lifespan of C. elegans, age-sorted wild-type populations were cultured under monoxenic conditions with different pharmacological doses (as mentioned in materials and methods) on NGM plates at 20 °C starting from young adult stage. Under normal culture conditions, wild-type control worms lived an average of 15.31 ± 0.42 days and maximum of 23 days. We have noticed that different pharmacological doses of EISO and santalol isomers exhibited a concentration-dependent biphasic effect on the lifespan of C. elegans. It was found that 24 μg mL−1 of EISO (Fig. 3A), 32 μM of α-santalol (Fig. 3B) and 16 μM of β-santalol (Fig. 3C) being the most effective concentration and it could extend the mean lifespan of wild-type worms by up to 26.36%, 10.31% and 12.56% respectively (ESI Table 5†). In addition, studies suggest that life stages of worms influenced the beneficial effects of longevity-promoting interventions.50,51 Therefore, we treated worms from embryo to till death with respective concentration of EISO and santalol isomers at 20 °C. Our results showed that the mean lifespan of wild-type worms treated with EISO, α- and β-santalol was further extended by up to 31.32% (p < 0.0001), 18.04% (p < 0.0001) and 25.38% (p < 0.0001), respectively (Fig. 3D; ESI Table 5†). Previous studies have clearly demonstrated that supplementation of essential oils and their active constituents, mostly sesquiterpene, appear to prolong the lifespan of C. elegans,51–53 and our results are well supported by these reports. Based on the results obtained, the optimal concentrations of EISO (24 μg mL−1), α- (32 μM) and β-santalol (16 μM) were considered as relatively safe and beneficial in C. elegans and used for most subsequent experiments.
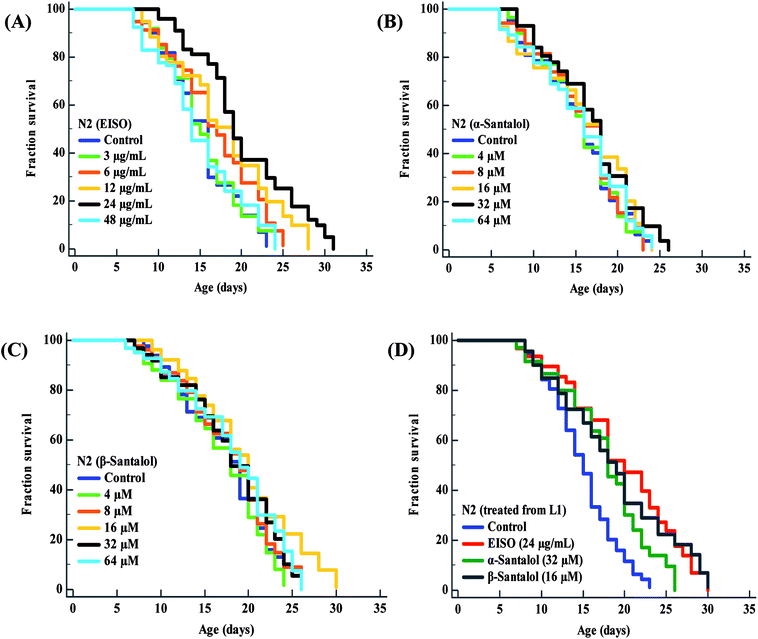 |
| | Fig. 3 Effect of EISO, α- and β-santalol on C. elegans lifespan. Survival rate of wild-type C. elegans cultured on NGM plates (from late L4 stage) containing different pharmacological doses of (A) EISO, (B) α-santalol and (C) β-santalol under normal culture conditions. (D) Survival curve of wild-type worms treated from embryo and raised at normal culture conditions on NGM plates carrying either 0.1% DMSO (solvent control) or EISO (24 μg mL−1), α-santalol (32 μM), and β-santalol (16 μM). The survival curves were plotted using Kaplan–Meier survival analysis and significance levels were estimated by long-rank test in Medcalc statistical tool. The representative Kaplan–Meier survival curves from three independent runs were presented. Statistical details of these experiments are summarized in ESI Table 5.† | |
3.4. EISO exerted an excellent antioxidant activity and reduced the 6-OHDA-induced elevation in intracellular ROS levels
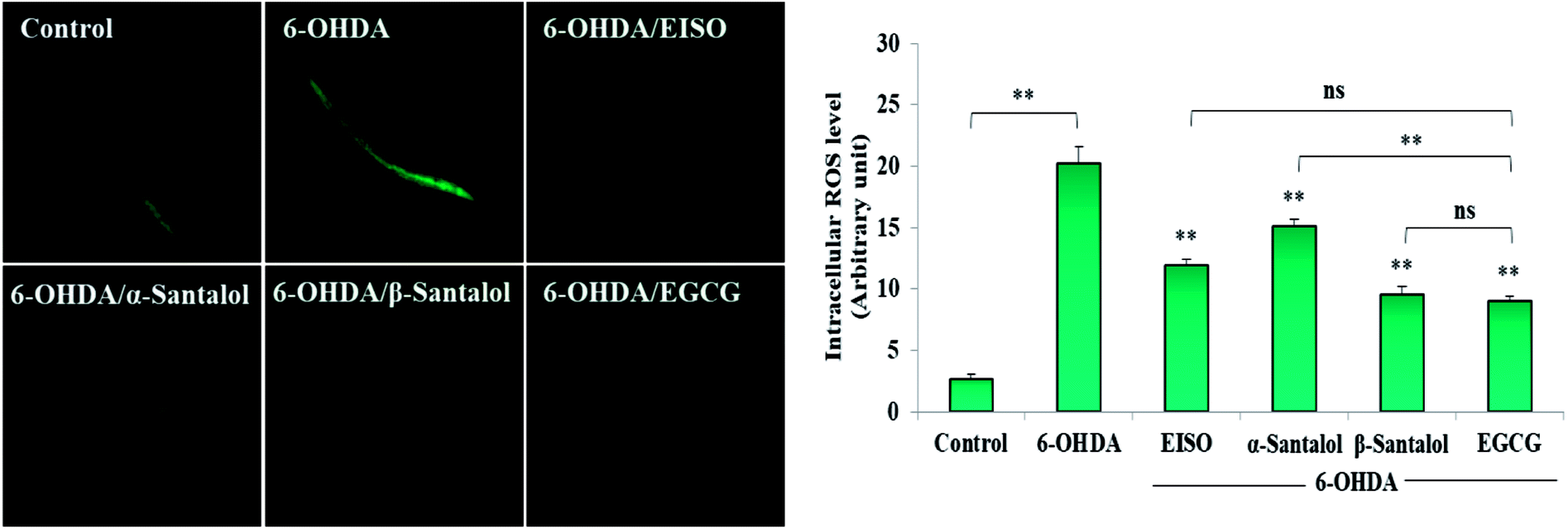
Oxidative stress produced by the excessive free radicals seems to play an important role in long list of human aliments including aging and degenerative diseases.54,55 At present, experiments were carried out to discover the in vitro and in vivo antioxidant activity of EISO and its active components. Initially, the antioxidant activity of EISO, santalol isomers and reference antioxidant compound (vitamin c) was tested in a cell free system using DPPH. The magnitude of DPPH reduction (IC50 DPPH) by EISO, α- and β-santalol was observed at a concentration of 65.11 ± 0.41 μg mL−1, 42.34 ± 0.71 μM and 34.87 ± 0.97 μM, respectively. The free radical scavenging activity of compounds were tested against vitamin c (IC50 = 3.26 μg mL−1 or 18.51 μM) (Table 2). β-Santalol exhibit reactively higher antioxidant activity than α-santalol it may due to the presence of unsaturated active alkene group in the second position over the eight member ring. The results of DPPH assay confirmed that EISO, α- and β-santalol possess noticeable antioxidant activity as compared to vitamin c in vitro. According to Harman's free-radical theory of aging (FRTA), overproduction of ROS causes damage to vital macromolecules which accelerates the normal aging process and progression of several life-limiting diseases.56 Application of antioxidants suppresses the oxidative stress via scavenging the excessively produced ROS. Therefore, we measured the effect of EISO, α- and β-santalol on the intracellular ROS levels. As shown in Fig. 4, exposure of wild-type C. elegans to 6-OHDA led to an increase in the level of intracellular ROS. The increase in intracellular ROS level was significantly decreased (p < 0.01) in the worms exposed to EISO, α- and β-santalol. Moreover, EISO and β-santalol have far similar ROS inhibiting activity when compared to the positive control EGCG (Fig. 4). These results suggest that the antioxidant activity of EISO and its constituents protect the worms from 6-OHDA toxicity probably via modulating the intracellular redox status.
Table 2 In vitro free radical scavenging activities of EISO and its active components. Data are presented as mean ± SEM, vitamins c was used as positive control
| S. no. |
Particulars |
No. of trails |
IC50 DPPH mean ± SD |
| 1 |
EISO |
4 |
65.11 ± 0.41 μg mL−1 |
| 2 |
α-Santalol |
4 |
42.34 ± 0.71 μM |
| 3 |
β-Santalol |
4 |
34.87 ± 0.97 μM |
| 4 |
Vitamin C |
3 |
18.51 μM/3.26 μg mL−1 |
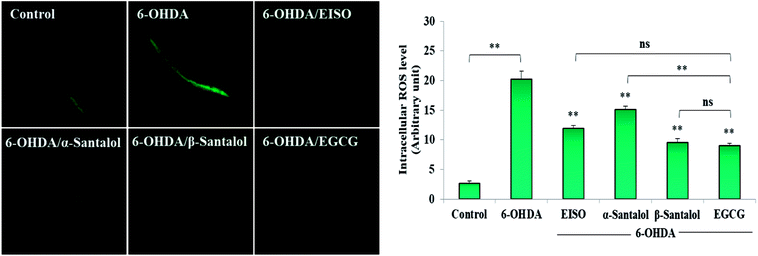 |
| | Fig. 4 Effect of EISO, α- and β-santalol on the inhibition of ROS generation induced by 6-OHDA in C. elegans (n = 30–40 worms per experiment). The relative formation of ROS in control and treated C. elegans was quantified using fluorescence probe H2DCF-DA. Epigallocatechin gallate (EGCG, 50 μM) was used as positive control. Data were pooled from biological triplicates in experimental duplicates and presented as mean ± SEM. Probability levels of p < 0.05 were considered as statistically significant. *p < 0.05, **p < 0.01, ns, not significant. | |
3.5. EISO suppresses the apoptotic features induced by 6-OHDA in C. elegans
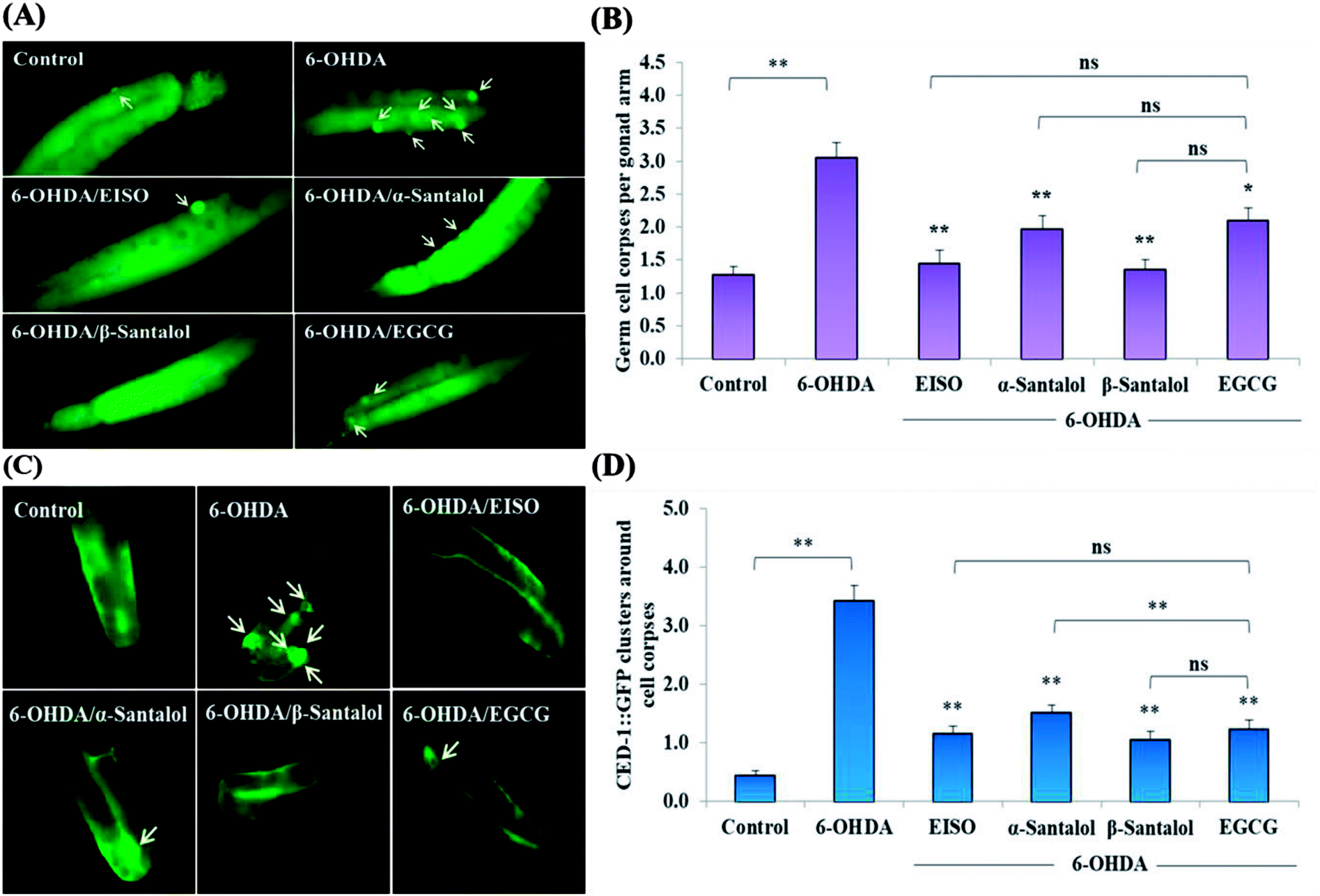
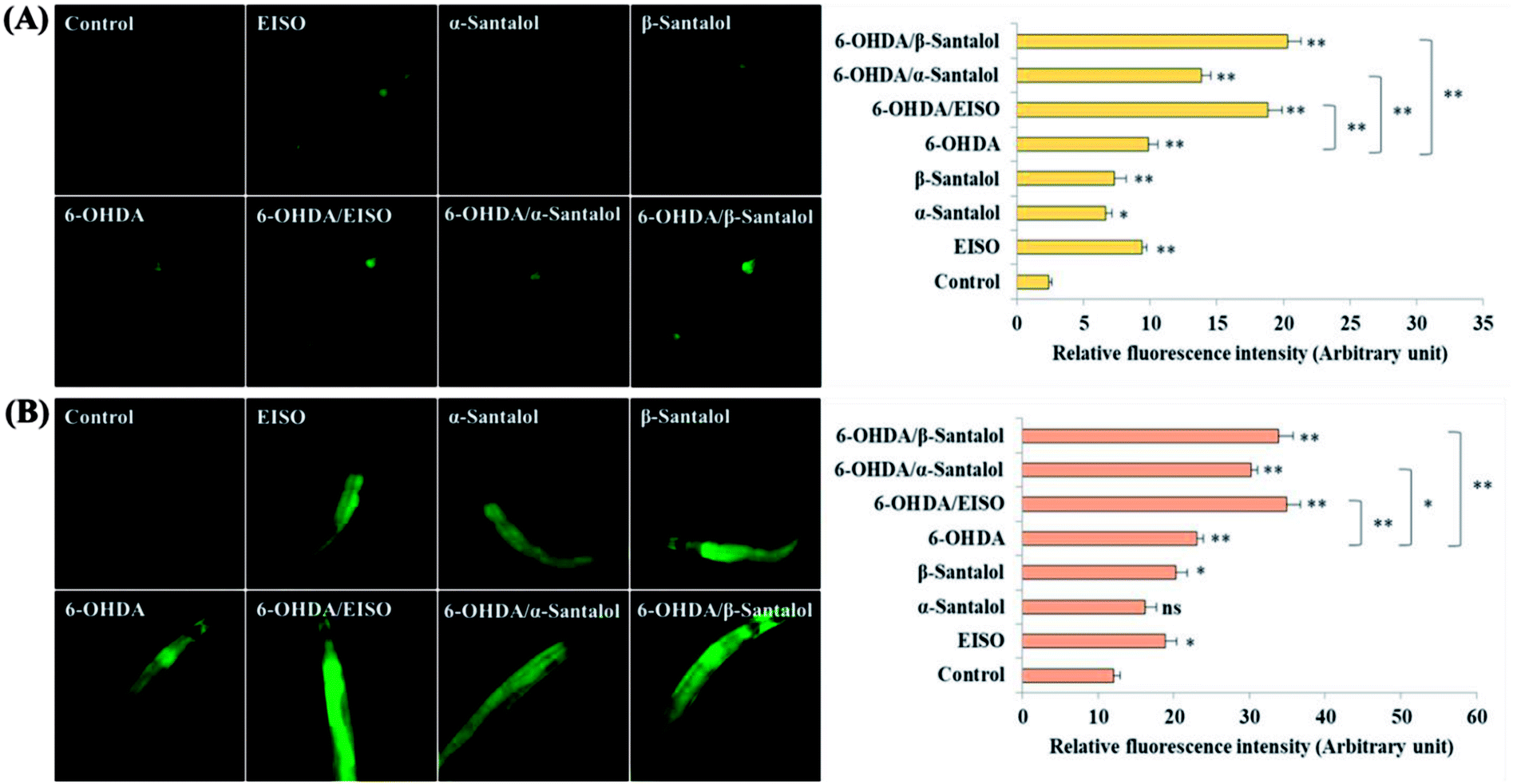
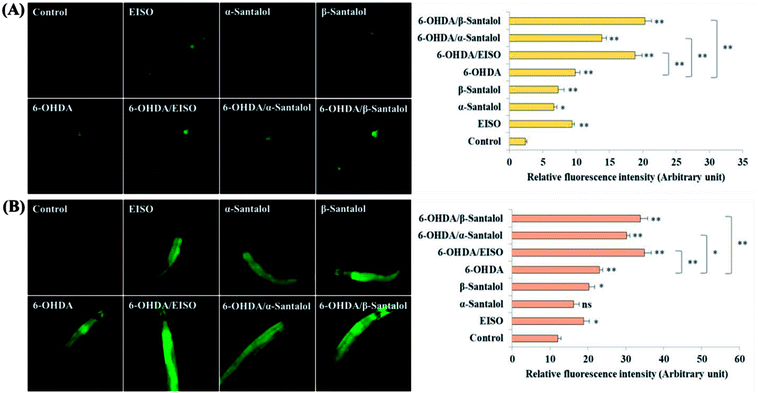
We first used AO staining techniques to investigate the protective effect of EISO and its active components against 6-OHDA exposure induced germline cell apoptosis in pachytene cells of gonad in wild-type C. elegans. Based on the AO staining results, exposure to 6-OHDA caused the production of more germ cell corpses in posterior part of gonadal arm; however, the worms conditioned with EISO, α- and β-santalol significantly reduced the germ cell corpses by about 52.46%, 35.25% and 55.74%, respectively. Furthermore, the subcellular localization and expression pattern of ced-1::GFP was analyzed to identify cells which had undergone apoptosis. In C. elegans, ced-1 gene encodes a single-pass transmembrane protein homologous to human low density lipoprotein receptor-related protein 1 (CD91/LRP1). CED-1 is an important component of apoptotic pathway and it initiate a signaling pathway in phagocytic cells to promote cell corpse engulfment, phagosome maturation and apoptotic cell degradation.57 Exposure of C. elegans transgenic strain carrying a fusion protein for CED-1::GFP to 6-OHDA led to an enhanced expression of CED-1::GFP clusters around cell corpses compared with control group worms indicating that 6-OHDA induced apoptosis in germline cells (Fig. 5A). EISO, α- and β-santalol treatment significantly decreased such increase in expression around cell corpses by about 66.08%, 55.56% and 69.01% respectively (Fig. 5B), while EISO α- and β-santalols alone had no obvious toxic effects on CED-1::GFP expression (ESI Table 6†).
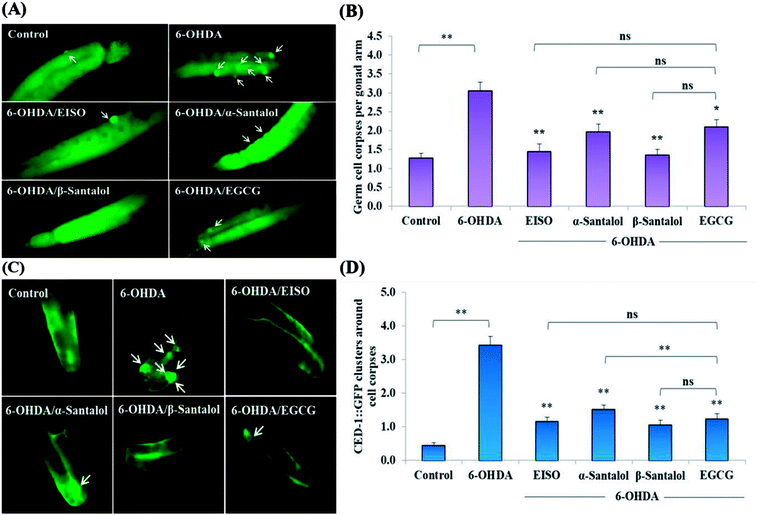 |
| | Fig. 5 Effect of EISO, α- and β-santalol on the inhibition of apoptosis induced by 6-OHDA in C. elegans (n = 30–40 worms per experiment). (A) Fluorescence micrographs depicting the 6-OHDA exposure-induced germline cell death/apoptosis in wild-type N2 C. elegans. Arrow marks indicating bright distinct circles represent germ cell corpses. (B) Bar graph depicts the quantified germ cell corpses in posterior part of gonad arm of each animal across various treatments. (C) Fluorescence micrographs illustrating the protective effects of EISO against 6-OHDA-induced apoptosis and its influences on CED-1::GFP expression in engulfing cells/phagosomal surface around the cell corpses in posterior gonadal arm of MD701 C. elegans. (D) Quantified CED-1::GFP halos on the engulfing cells and nascent phagosomes. For all the experiments, 50 μM epigallocatechin gallate (EGCG) was used as positive control. Data were pooled from biological triplicates in experimental duplicates and presented as mean ± SEM. Probability levels of p < 0.05 were considered as statistically significant. *p < 0.05, **p < 0.01, ns, not significant. | |
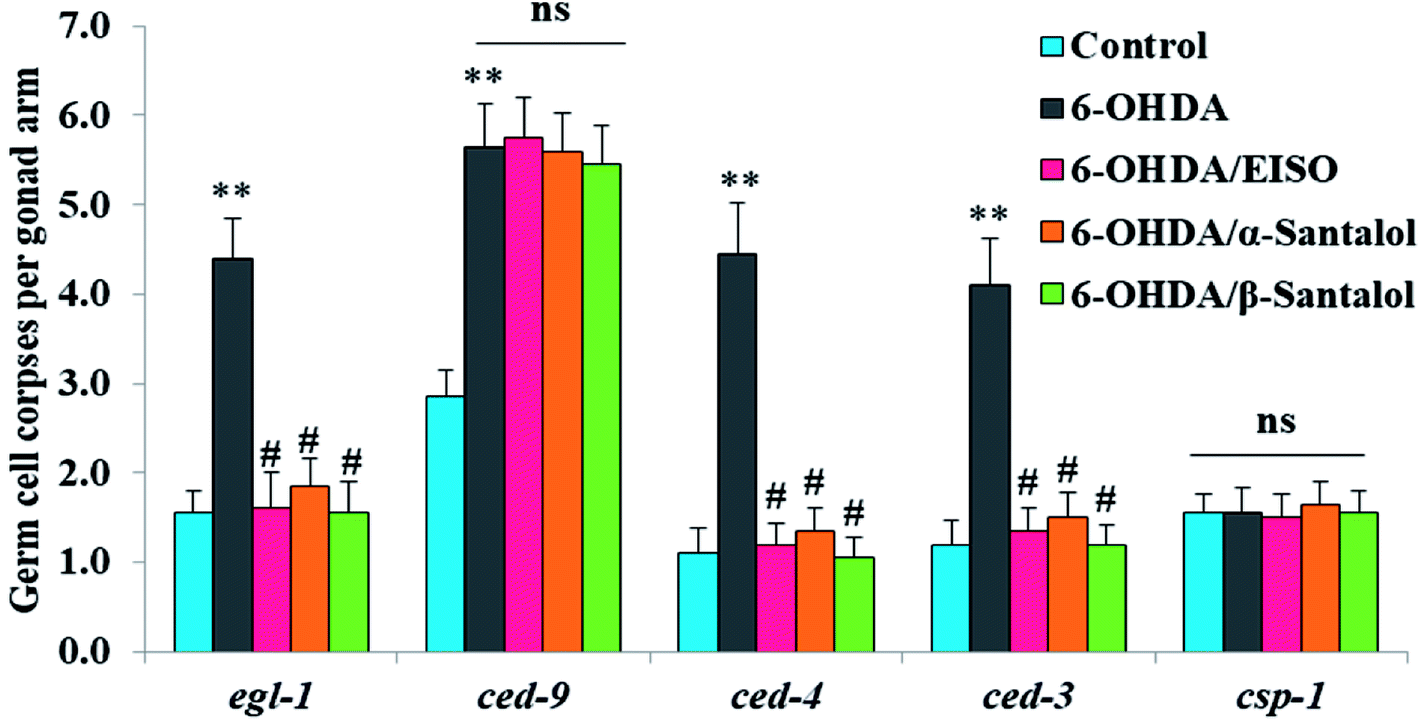
In order to identify the potential mechanism underlying the EISO-mediated protection against 6-OHDA-induced apoptosis, the worms deficient in core components of apoptotic machinery, egl-1(n1084n3082), ced-9(n1950), ced-4(n1162) and ced-3(n717), were used. In C. elegans, EGL-1, a BH3-containing protein, positively regulates the cell death by antagonizing the functions of CED-9, a homolog of the mammalian cell-death inhibitor Bcl-2.58 CED-9 negatively regulates Apaf-1 like protein CED-4, which promotes apoptotic cell death by activating CED-3 caspase.59 As shown in Fig. 6, after exposed to 6-OHDA, the egl-1, ced-9, ced-4 and ced-3 mutants showed a much higher induction of apoptotic germ cells as seen in wild-type worms. These results suggest that the apoptosis induced by 6-OHDA was not regulated by the canonical apoptosis pathway in C. elegans, indicating that other mechanism(s) may be involved in this process. Whereas EISO, α- and β-santalol treatment significantly suppressed the apoptotic phenotype observed in egl-1, ced-4 and ced-3 mutants but not in ced-9 mutants when compared to that of the 6-OHDA treated worms, signifying the essential role of ced-9 in EISO-meditated antiapoptotic ability. Previous studies have also shown that 6-OHDA-mediated apoptosis does not influences the transcript levels of egl-1, ced-9, ced-4 and ced-3 in wild-type C. elegans,37,39,60 suggested that the canonical cell-death execution pathway was not contributed to germ cell apoptosis in 6-OHDA treated animals. It is interesting to note that an another parkinsonism-inducing neurotoxin, 1-methyl-4-phenylpyridinium (MPP+), have been implicated with DA neurodegeneration independent of CED-4 pathway in C. elegans.61 However, substantial amount of data accumulated over the years specified that 6-OHDA triggered the caspase-dependent, mitochondrial-mediated apoptosis in human neural cells.62,63 This inconsistency may be due to the difference in the experimental models. Genetic studies have evidenced that apoptosis can occur in a programmed manner but independent of CED-3/caspase activation by CED-4 in C. elegans.64,65 The C. elegans genome contains four caspase genes viz., csp-1, csp-2, csp-3 and ced-3, of which csp-2 and csp-3 negatively regulates the CED-3 function in germline and soma, respectively. Whereas, CSP-1 found to promotes cell death in a caspase-independent manner (parallel with CED-4) and is not regulated by CED-9 or CED-4.64 Therefore, we next investigated the possibility of whether 6-OHDA exposure could trigger the apoptosis in a caspase-independent pathway, we quantified the germ cell corpses in csp-1 mutant worms exposed to 6-OHDA. It was found that 6-OHDA exposure did not obviously alter the germline cell apoptosis in csp-1 worms when compare to its corresponding control. On the other hand, 6-OHDA/EISO, α- or β-santalol treatment exhibited negligible effect on the germline apoptosis. The present results imply that EISO and its active components may impede the function of csp-1, thus protect the cells from 6-OHDA-induced apoptosis. Taken collectively, it was clear that EISO hampers the apoptosis induced by 6-OHDA through regulating the CSP-1 and Bcl-2 family protein CED-9 that negatively regulates the CED-3 caspase activation via inhibition of Apaf-1 homolog CED-4.
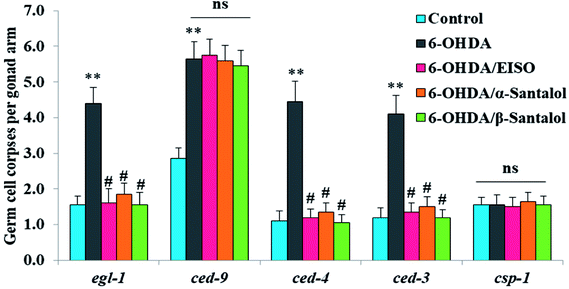 |
| | Fig. 6 Role of apoptotic signaling pathways in 6-OHDA-induced germline apoptosis and the protective effect of EISO and santalol isomers. Data pooled from three independent biological experiments with appropriate replicates. Probability levels of p < 0.05 were considered as statistically significant. An asterisk (**) indicates significant difference between control and 6-OHDA treated groups. A hash (#) mark indicates significant difference between 6-OHDA treated and 6-OHDA/EISO, α- or β-santalol treated groups (** and #p < 0.01, ns, not significant). | |
3.6. EISO confers the PD related pathologies in NL5901 worms
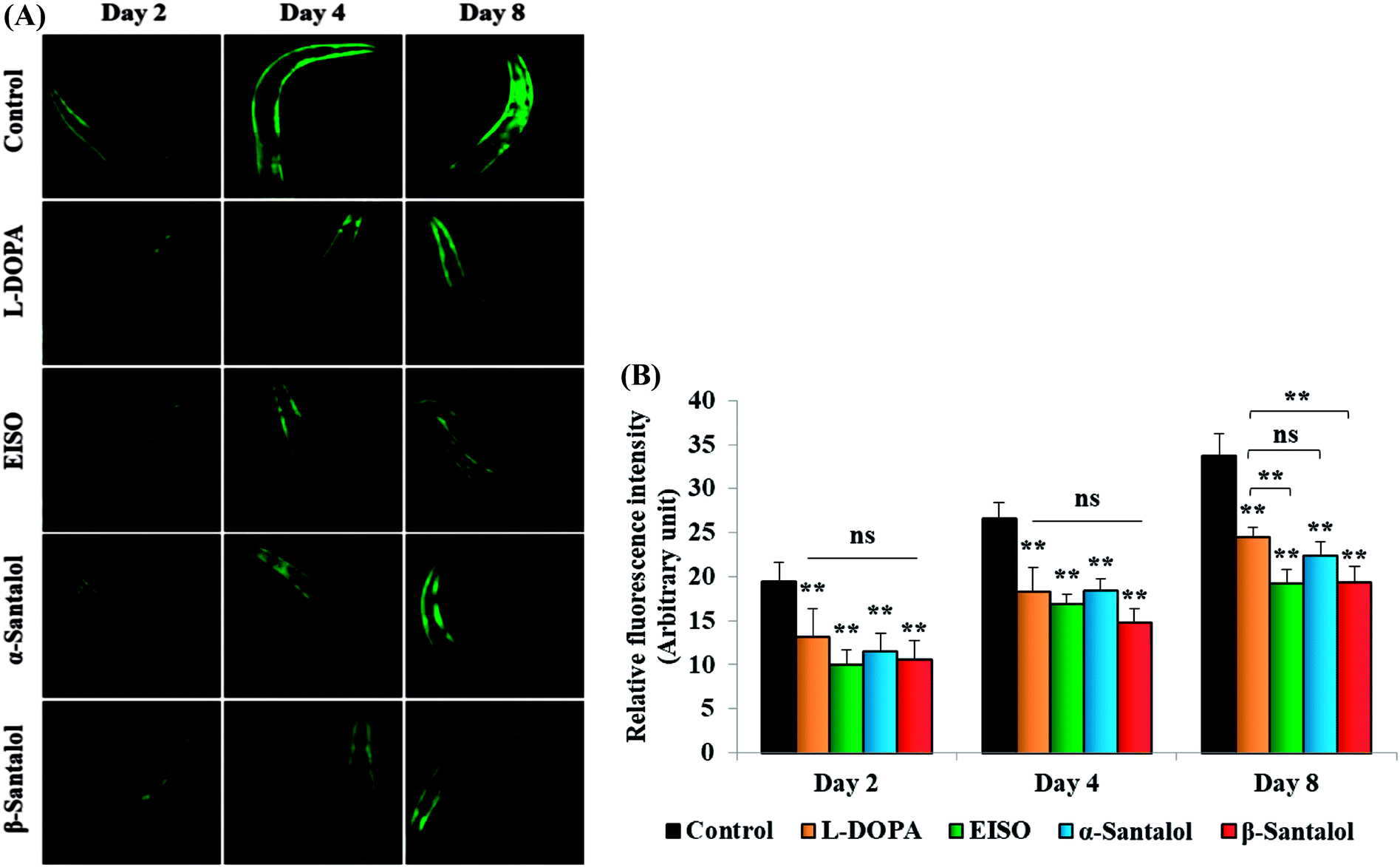
C. elegans does not carry the human ortholog gene for α-syncline. However, this nematode comprises genetic plasticity to allow transgenic expression of human gene for α-syncline fused with yellow fluorescent protein (YFP) in body muscle cells (strain NL5901) which is governed by the unc-54 promoter offers a perfect platform to examine the quantitative aggregation of α-syncline.66 NL5901 worms of EISO, α- and β-santalol treated and untreated groups were examined for their α-syncline protein aggregation at various adulthood stages (Fig. 7). It was observed that solvent control does not influence the α-syncline aggregation, while EISO, α- and β-santalol significantly reduced (p < 0.05/p < 0.01) the fluorescence intensities of α-syncline::YFP expression in NL5901 worms at different time points. The effect of EISO on α-syncline aggregation was comparable to that of L-DOPA, a mainstay prodrug for PD treatment.67 No significant changes were observed in α-syncline aggregation between L-DOPA and EISO, α- or β-santalol treatments.
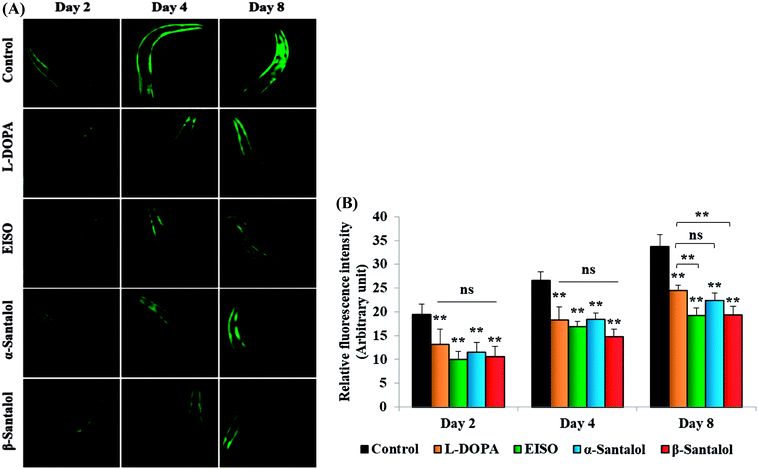 |
| | Fig. 7 Influence of EISO, α- and β-santalol on α-synuclein aggregation in C. elegans. (A) Photographic representation of α-synuclein aggregation in NL5901 worms (anterior region) (n = 30–40 individuals per experiment) treated with EISO, α- and β-santalol. (B) Graphical representation of estimated α-synuclein levels in C. elegans across different time points. Data are presented as mean ± SEM of three independent runs, and processed by one way analysis of variance (ANOVA) followed by Bonferroni post-hoc test. Probability levels of p < 0.05 were considered as statistically significant, *p < 0.05, **p < 0.01, ns, not significant. | |
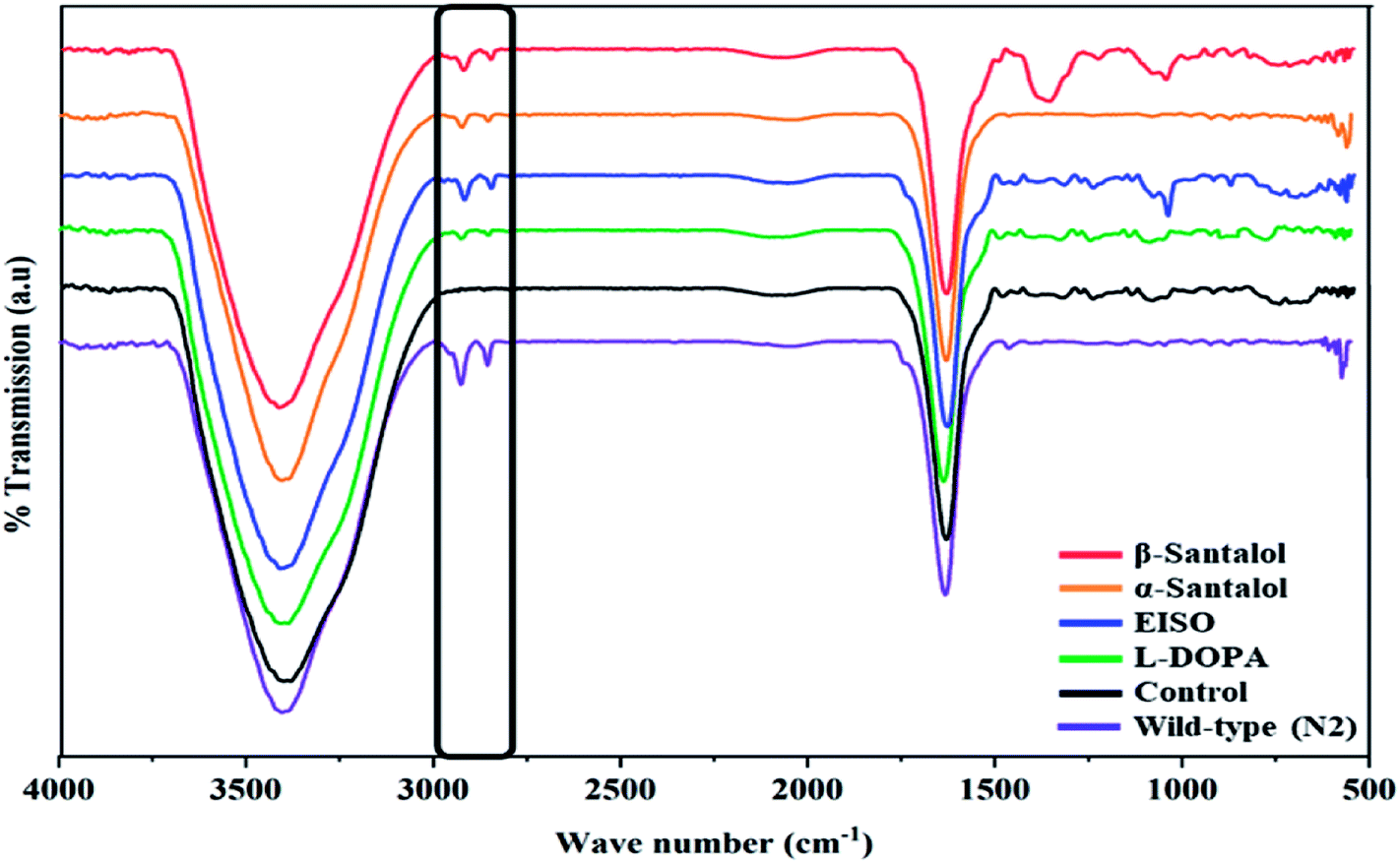
Pathogenesis of PD is associated with fluctuated levels of lipid and fatty acid content due to the expression of α-synuclein. Hitherto, there are several studies reported that α-synuclein amalgamates with free polyunsaturated fatty acids (PUFAs) to form a soluble oligomer which then transforms and aggregate into a high molecular weight insoluble inclusions. Such combinatorial associations of α-synuclein with oxidized lipid metabolites triggered the mitochondrial dysfunction result in DA neuronal death. Evidences indicated that an enhanced lipid peroxidation was found in autopsy brain of PD victims.68–70 With this understanding, we investigated the lipid content in the NL5901 worms exposed to EISO, α- and β-santalol by labeling the lipid droplets using lipid specific molecular probes like Nile Red (Fig. 8A), Oil Red O (Fig. 8B) and Sudan Black B (Fig. 8C). The wild-type worms showed an optimal level of lipid content. The worms expressing α-synuclein (NL5901) exhibited significantly reduced lipid content as compared to wild-type worms. At the same time, the worms treated with L-DOPA, EISO, α- and β-santalol restored the lipid level throughout the body in NL5901 worms. Furthermore, Fourier transform infrared (FTIR) spectroscopy was used as a powerful tool to authenticate the lipid levels in treated C. elegans. FTIR is a non-invasive and label-free technique that can be used to study the biomolecular complexes.71 The results of FTIR analysis showed that the C–H stretching band area between 3000–2800 cm−1 corresponding to fatty acids71 was observed in untreated wild-type worms and NL5901 worms treated with L-DOPA, EISO, α- and β-santalol, however, the peak was not observed in untreated NL5901 worms (Fig. 9). Generally in cells, the large number of C–H bonds are in long-chain fatty acids, thus, the signature peaks at 3000–2800 cm−1 region is closely connected to the amount of lipids.71,72 Meanwhile, the spectral ranges between 3000-2800 cm−1 corresponding to fatty acids was not observed in the untreated worms expressing α-synuclein, it undisputedly confirmed the ability of α-synuclein to disrupt the lipid homeostasis. The FTIR data strongly confirmed that EISO, α- and β-santalol prevents the disturbed lipid levels in NL5901 worms by reducing the aggregation and toxicity of α-synuclein. The enormous number of researches has been reported that the central and peripheral nervous systems are highly enriched in lipids that are essential for maintaining the structural integrity and signal transduction.
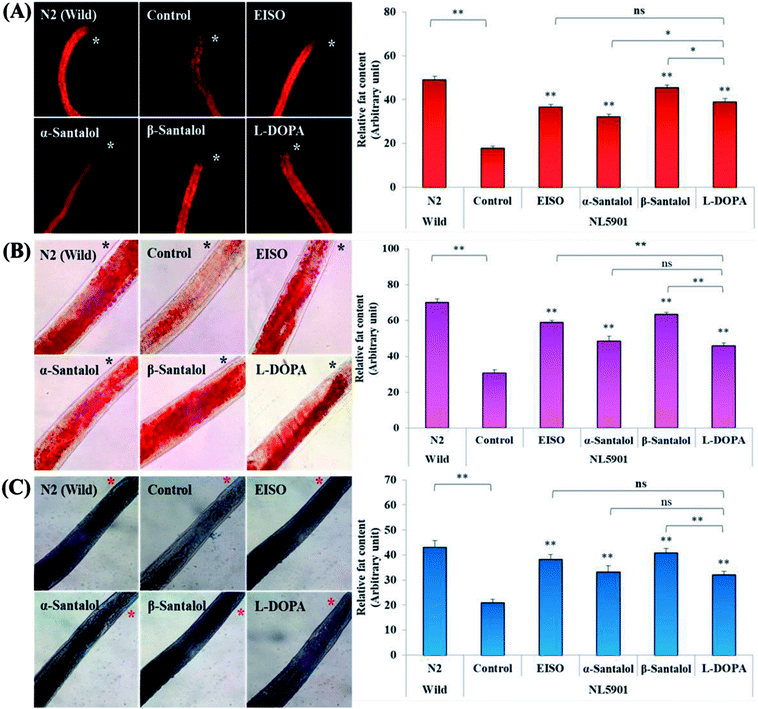 |
| | Fig. 8 EISO, α- and β-santalol treatment alleviates the altered lipid content in worms expressing α-synuclein. Representative images of lipid after treatment with EISO, α- and β-santalol in NL5901 C. elegans by (A) Nile Red, (B), Oil Red O, and (C) Sudan Black B staining. L-DOPA (Levodopa) was used as positive control. Asterisk (*) indicated in the photo micrographs represent the anterior region of the worm. Data are presented as mean ± SEM of three independent runs, and processed by one way analysis of variance (ANOVA) followed by Bonferroni post-hoc test. Probability levels of p < 0.05 were considered as statistically significant. *p < 0.05, **p < 0.01, ns, not significant. | |
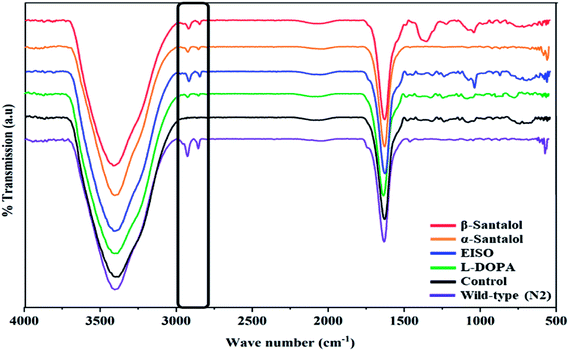 |
| | Fig. 9 Fourier transform infrared (FTIR) spectroscopy analysis of worms treated with EISO, α- and β-santalol. Untreated and treated worms of NL5901 strain (n = 80–100 worms per treatment) were ground with 100 mg of potassium bromide pellets and dried under vacuum evaporator for 48 h. The infrared spectra were scanned from 400 to 4000 cm−1 using Bruker Tensor 27 series spectrophotometer. | |
3.7. The 6-OHDA induced DA neurodegeneration was attenuated with EISO
To explore the neuroprotective effects of EISO, α- and β-santalol on 6-OHDA induced neuronal injury, neuronal viability was analyzed by quantifying the expression of GFP transgene construct tagged in all DA neurons of BZ555 worms. The C. elegans hermaphrodite contains four pairs of DA neurons, including two pairs of cephalic (CEP) neuron and one pair of anterior deirids (ADE) neurons within the head region and one pair of posterior deirids (PDE) neurons in a posterior lateral position.73 As shown in Fig. 10, the GFP intensity of 6-OHDA exposed BZ555 worms were decreased by about 52.83% in comparison with that of unexposed worms (p < 0.01), indicating the capacity of 6-OHDA to damage DA neurons. When the worms were treated with EISO, α- and β-santalol, the neurons displayed an enhanced expression of GFP in all DA neurons to 48.17%, 31.97% and 71.11% respectively. Epigallocatechin gallate (EGCG) has been used as an internal positive control74 which protects the DA neurons by about 79.20% (p < 0.01). Effect of β-santalol ability on protecting the DA neurons was on par with EGCG treatment. Moreover, 6-OHDA treatment significantly reduced the mean lifespan to 8.05 ± 0.35 (33.80%, p < 0.0001) days as compared to unexposed worms (12.16 ± 0.36 days), suggesting that 6-OHDA triggered the aging process in C. elegans.37 We observed that, EISO (24 μg mL−1), α-santalol (32 μM) and β-santalol (16 μM) treatments enhanced the lifespan of 6-OHDA intoxicated worms to 15.44 ± 0.41 (27.04%), 13.39 ± 0.46 (10.13%) and 16.18 ± 0.37 (33.13%) days respectively (Fig. 11; Table 3). From these observations, it was apparent that antioxidant and antiapoptotic activity of EISO and its principle components might drive the neuroprotective and geroprotective functions in C. elegans.
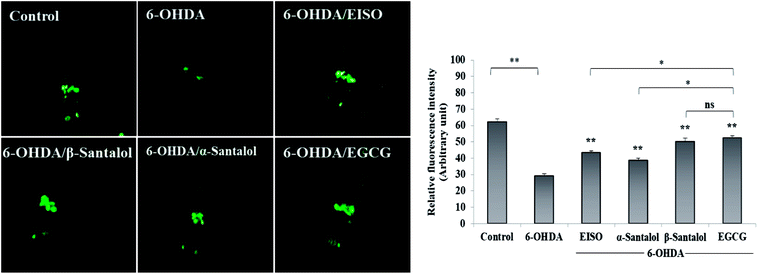 |
| | Fig. 10 Rescuing effect of EISO, α- and β-santalol on 6-OHDA induced DA neurodegeneration in C. elegans. Representative fluorescence micrograph depicting the GFP expression pattern (anterior region) of 6-OHDA treated BZ555 (dat-1::gfp) C. elegans. Graphical representation of estimated GFP intensity in all eight DA neurons. 50 μM epigallocatechin gallate (EGCG) was used as positive control. Data are presented as mean ± SEM of three independent runs, and processed by one way analysis of variance (ANOVA) followed by Bonferroni post-hoc test. Probability levels of p < 0.05 were considered as statistically significant. *p < 0.05, **p < 0.01, ns, not significant. | |
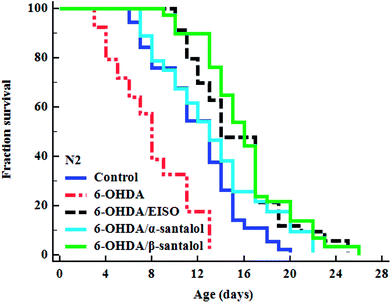 |
| | Fig. 11 The cumulative survival graph depicting the effect of EISO and its major active principles on the lifespan of 6-OHDA intoxicated N2 C. elegans. The 6-OHDA induced reduction in longevity was significantly attenuated with EISO, α- and β-santalol treatments (shown as turquoise blue, black and green survival curves, respectively). The data were processed using the Kaplan–Meir survival analysis and significance levels were estimated by long-rank test. | |
Table 3 Lifespan analysis of N2 worms. EISO, α- and β-santalol treatment reduced the senescence induced by 6-OHDA in C. elegans. The mean lifespan was calculated as the average number of days animal survived in tested pharmacological dose(s) and analyzed using log-rank test. For the analysis of statistical significance, 6-OHDA alone treated group was compared with untreated control, and 6-OHDA/EISO, α- or β-santalol treated groups were compared with 6-OHDA alone treated group
| Genotype |
Treatment |
Mean survival (mean ± SEM) |
Maximum lifespan (days) |
Sample size (N) |
% change |
p value |
| N2 |
Control |
12.156 ± 0.357 |
20 |
109 |
|
|
| 6-OHDA |
8.047 ± 0.347 |
13 |
92 |
(−) 33.80 |
<0.0001 |
| 6-OHDA/EISO |
15.443 ± 0.411 |
25 |
103 |
(+) 27.04 |
<0.0001 |
| 6-OHDA/α-santalol |
13.388 ± 0.456 |
22 |
108 |
(+) 10.13 |
<0.0001 |
| 6-OHDA/β-santalol |
16.183 ± 0.375 |
26 |
110 |
(+) 33.13 |
<0.0001 |
3.8. EISO and santalol isomers modulates the cholinergic denervation
Neuropathological studies have demonstrated that cognitive impairment of PD has also been driven by the degeneration of cholinergic system functions. Studies stated that a significant loss of nucleus basalis of Meynert (nbM) cholinergic neurons has been seen in PD patients, especially in patients with parkinsonian dementia and lewy body dementia. An interesting fact is that cholinergic denervation is a key neuropathological hallmark of Alzheimer's disease (AD) and is often less severe than in PD.75 This suggests that cholinergic dysfunction play a role, may be at least in part in PD pathogenesis as in AD. For this reason, present therapies of neurodegenerative diseases mainly targets cholinergic modulation. In C. elegans, acetylcholine (ACh) plays an important role in the regulation of several biological functions including locomotion, egg lying, pharyngeal pumping, defecation cycle and mating, whereas acetylcholinesterase (AChE) able to terminate the action of ACh by rapid enzymatic hydrolysis at neuronal junction.76 Hence, it is believed that modulation of cholinesterase may be beneficial for the treatment of patients with parkinsonian dementia and lewy body dementia.77
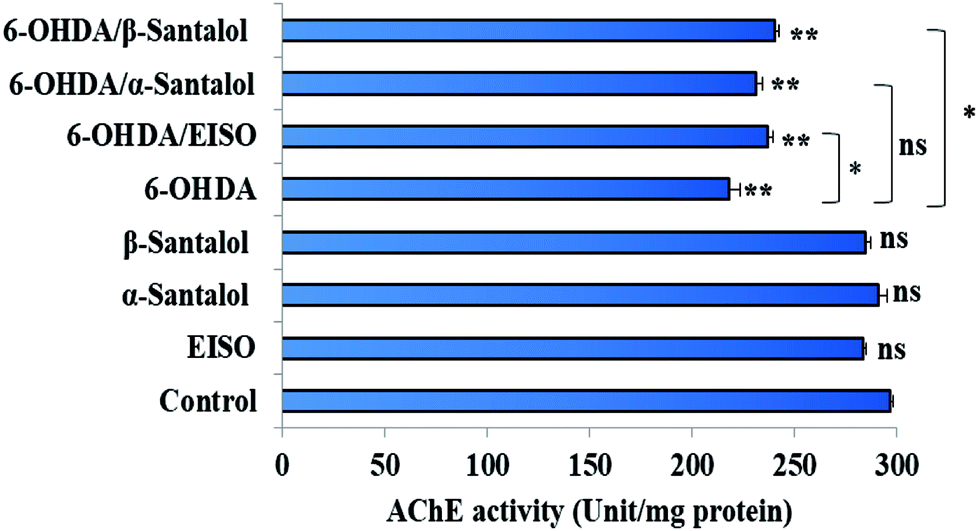
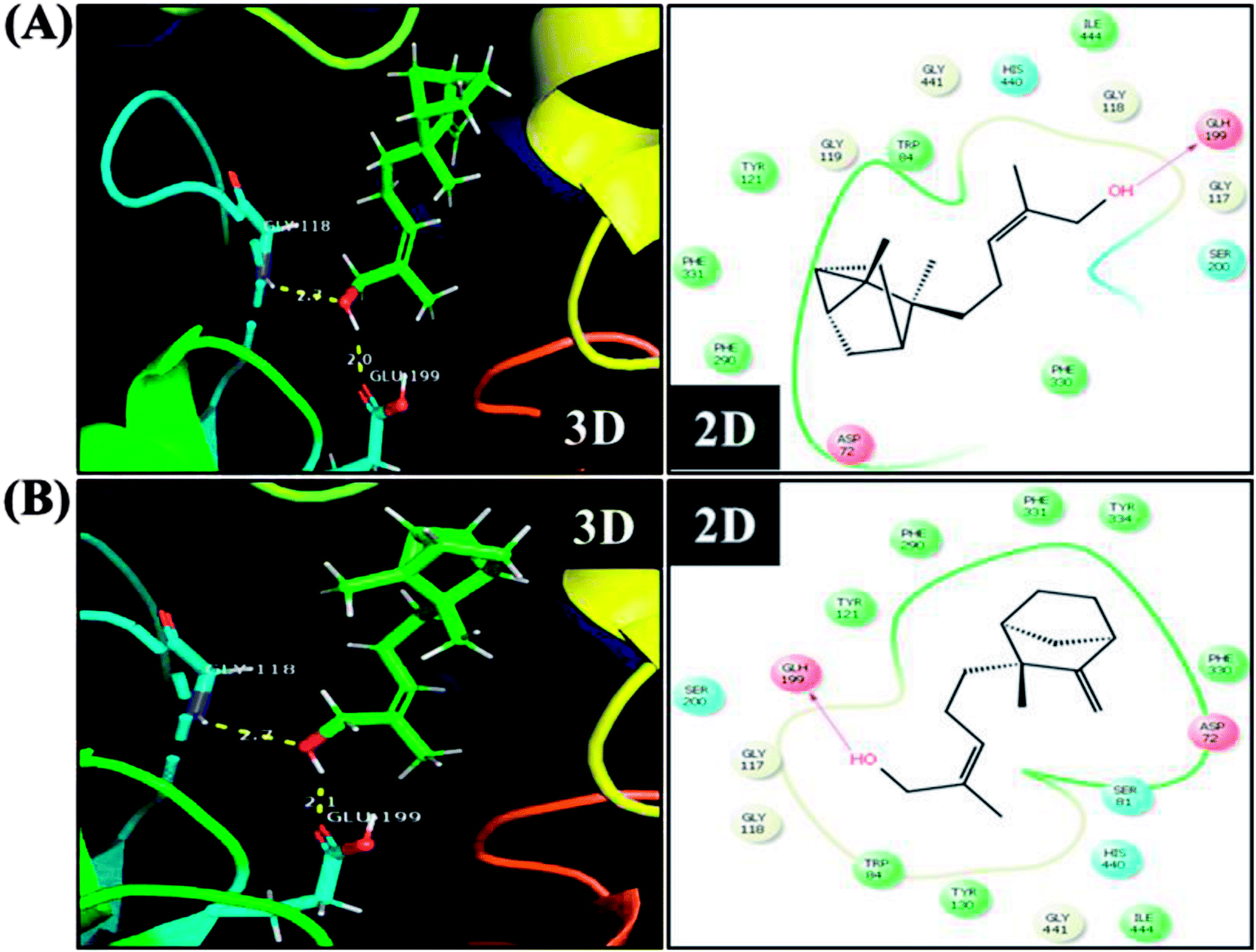
In order to detect the effect of EISO, α- and β-santalol on cholinergic functions, AChE activity assay was conducted. In our experiments, the worms exposed with 6-OHDA exhibited a significant reduction (26.46%, p < 0.01) in AChE activity compared to that in control worms. However, worms pre-conditioned with EISO, α- and β-santalol alone showed a slight reduction in AChE activity (4.48%, 2.79% and 4.04%, respectively), but the reduction was insignificant (p > 0.05) (Fig. 12). Further, the molecular docking analysis was performed to get insight into the inhibition of AChE by santalol isomers. The docking models for binding interaction are shown in Fig. 13, which confirms that α- and β-santalol fit snugly within the active site pocket of AChE through hydrogen bonds with a glide score of −7.13 and −7.49 kcal mol−1 respectively; it clearly indicates that santalol isomers decreasing the breakdown rate of ACh possibly by inhibiting the AChE activity at a moderate level. When compared to in silico results the in vivo AChE inhibitory potential of EISO and santalol isomers are rather poor, it could be influenced by bioavailability, amount of uptake, and metabolism in whole organism. Surprisingly, when the worms were exposed to EISO, α- and β-santalol along with 6-OHDA, only a marginal decrease (20.18%, 21.97% and 18.83%, respectively) in AChE activity was observed (Fig. 12). Results evidenced that EISO, α- and β-santalol treatments offered only a marginal protection against 6-OHDA induced AChE inhibition. In other words, EISO and santalol isomers significantly (p < 0.05) restored the AChE levels in worms exposed to 6-OHDA. We speculate that EISO may limit the persistence of ACh in neuronal junction leading to repetitive stimulation of acetylcholine receptors in target tissues of worms. Our results showed that EISO and its active components regulating the cholinergic dysfunction possibly by preserving the AChE levels. These results are in line with previous report of Nidheesh et al.,78 who studied the neuroprotective and antioxidant potential of chitooligomers on monocrotophos induced neurodegeneration in C. elegans. They found that monocrotophos exposure caused significant reduction in AChE level, whereas chitooligomers treatment marginally protect the monocrotophos induced lesion in AChE activity even though it has AChE inhibitory potential. Further studies are, however, required to identify the molecular mechanism(s) involved in the partial restoration of AChE activity by EISO and its active components. Several clinical trials evidenced that commercially available cholinesterase inhibitors effectively improved the cognitive declines in PD patients.77 In addition, earlier report showed that inhibition or deficiency of AChE can protects the DA neurons via decreasing the apoptotic events in PD models.79 Hence, it is believed that optimal AChE modulating efficacy of EISO and santalol isomers might involve or at least in part in reduction of germ cell corpses and DA neuronal preservation in 6-OHDA treated worms. Together, the results indicated that cholinergic modulating potential of EISO and santalol isomers may be useful for the treatment of patients with parkinsonian dementia and lewy body dementia.
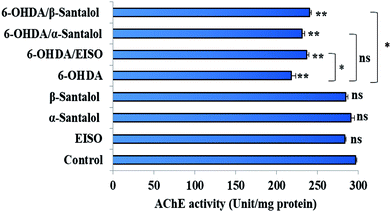 |
| | Fig. 12 Effect of EISO, α- and β-santalol on AChE activity in wild-type C. elegans. Graph depicts the AChE activity levels in worms treated with 6-OHDA, EISO and santalol isomers. Results shown here are mean ± SEM, the probability levels of p < 0.05 were considered as statistically significant. *p < 0.05, **p < 0.01, ns, not significant. | |
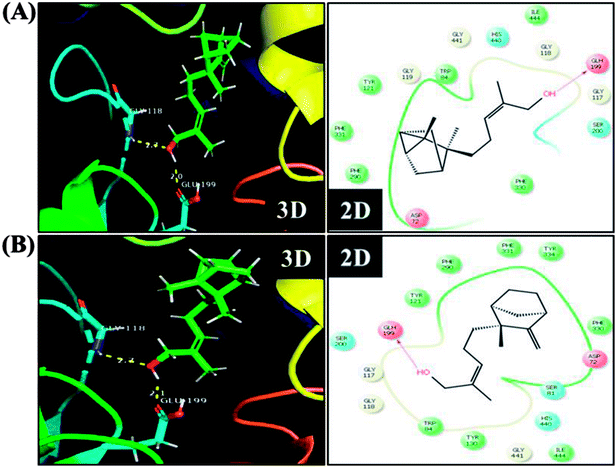 |
| | Fig. 13 The representative three-dimensional and two-dimensional interaction map shows the binding mode of (A) α-santalol and (B) β-santalol within the active site pocket of AChE. Santalol isomers form a strong hydrogen bonding (yellow dashed lines) with amino acid residue (GLH199) of AChE. | |
3.9. Protective effect of EISO involves mitochondrial electron transport chain and ERK-MAPK pathway but not via DAF-2/DAF-16
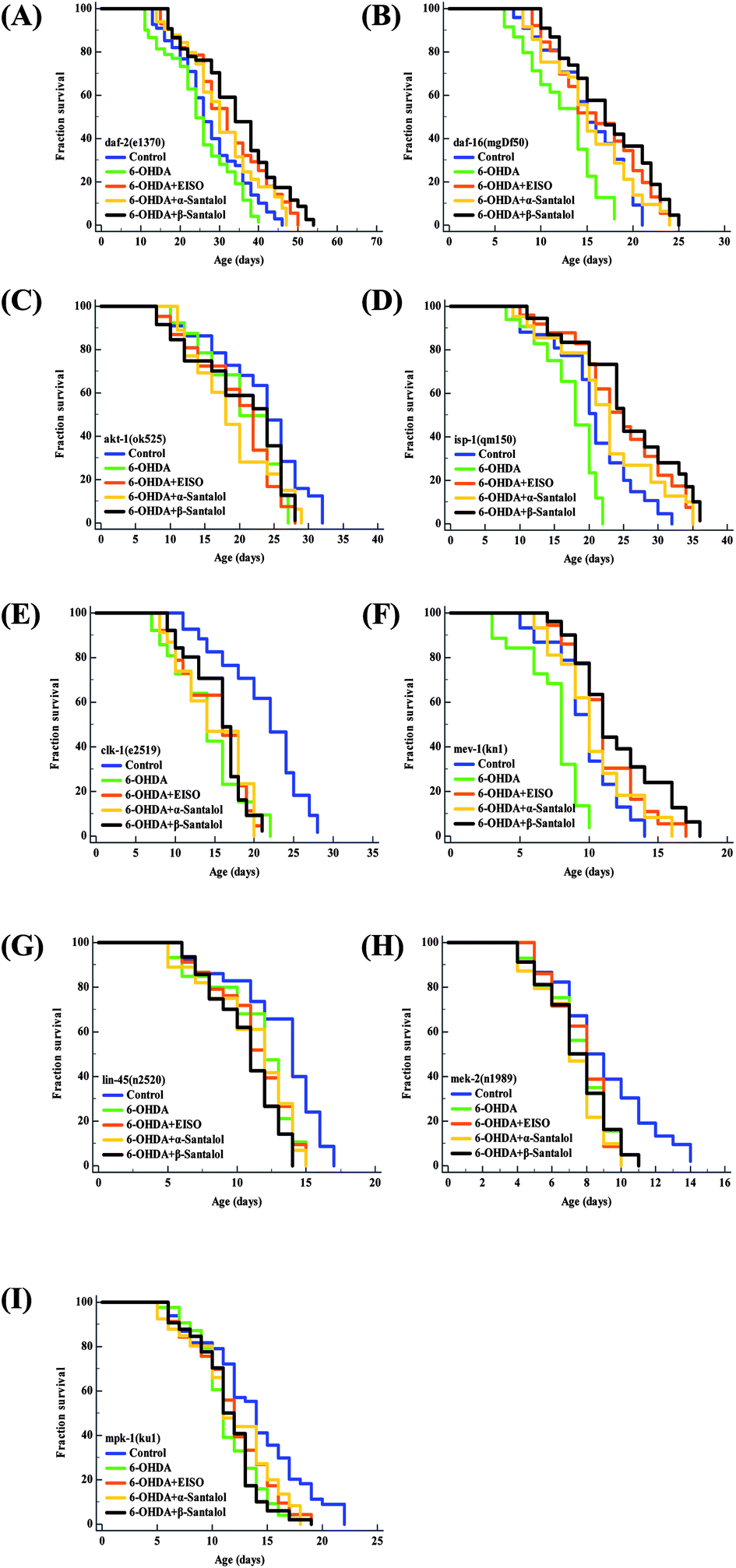
Aging is the major risk factor for the development of late-onset neurodegenerative disease. The progression of neurodegeneration and alterations in the molecular mechanism of aging are usually associated with the reduction in life expectancy.80 Therefore, to elucidate the molecular mechanism of neuroprotection and geroprotection mediated by EISO and its active principles, specific mutant C. elegans strains deficient in the insulin/insulin like growth factor (IIS: daf-2(e1370), akt-1(ok525), and daf-16(mgDf50)), extracellular-signal-regulated kinase-MAPK (ERK-MAPK: lin-45(n2520), mek-2(n1989) and mpk-1(ku1)) and mitochondrial electron transport chain (mitochondrial ETC: mev-1(kn1), isp-1(qm150) and clk-1(e2519)) signaling pathways were used. Our mechanistic study showed that EISO, α- and β-santalol treatment significantly extended (p < 0.0001) the lifespan of daf-2(e1370) (Fig. 14A) and daf-16(mgDf50) (Fig. 14B) mutants, as seen in wild-type worms under 6-OHDA. However, no corresponding lifespan extension was observed in the akt-1(ok525) worms (Fig. 14C). These results indicated that EISO, α- and β-santalol might acted on DAF-2 and DAF-16 in an independent and AKT-1 dependent manner. DAF-2 and DAF-16 are the key components of IIS pathways and mediates several key biological functions.81 We next wanted to investigate whether mitochondrial function was the underlying mechanism of neuroprotection in C. elegans. Hence, we studied the effect of EISO, α- and β-santalol on lifespan of mev-1(kn-1), isp-1(qm150) and clk-1(e2519) mutants, which encodes succinate dehydrogenase cytochrome b560 subunit of mitochondrial complex II, subunit of the mitochondrial complex III and demethoxyubiquinone hydroxylase, respectively.82,83 As shown in Fig. 14D and E, EISO, α- and β-santalol significantly prolongs the lifespan of isp-1(qm150) mutants, while no significant lifespan extension was observed in clk-1(e2519) mutants. A study stated that mutation in isp-1 and clk-1 showed an optimal elevation of ROS production which triggers hormesis-mediated longevity extension in C. elegans via enhancing the activity of hypoxia-inducible factor-1 (HIF-1).84 We noted that EISO, α- and β-santalol significantly reduced the ROS accumulation to optimal level in isp-1 worms, but they fails to alter the ROS production in clk-1 worms under 6-OHDA (ESI Fig. 3A†). These data strongly indicated that EISO, α- and β-santalol required clk-1 but not isp-1 for longevity extension under 6-OHDA intoxicated condition. Furthermore, we found that mev-1(kn-1) mutants are hypersensitive to 6-OHDA, whereas EISO, α- and β-santalol significantly prolongs their lifespan under 6-OHDA (p < 0.0001) (Fig. 14F). Interestingly, EISO, α- and β-santalol fails to reduce the internal ROS generation in mev-1(kn1) mutant worms (ESI Fig. 3A†) which has shortened lifespan resulted in the overproduction of superoxide anion.82 This result implicated that the protective effect of EISO, α- and β-santalol acted independently of MEV-1 and their direct antioxidant activity does not contributed to the lifespan extension in mev-1(kn1) mutant worms. A study indicated that supernumerary apoptosis also involved in life shortening phenotype in mev-1(kn1) mutant C. elegans.85 Therefore, it was expected that EISO, α- and β-santalol might prolongs the lifespan of mev-1(kn1) mutant worms by reducing the apoptosis. To test the idea, we examined the effect of EISO, α- and β-santalol on the germ cell apoptosis by means of AO staining. Our results showed the spontaneous physiological germ cell corpses were comparatively higher in mev-1(kn1) mutants. Following exposure to 6-OHDA, the germ cell apoptosis in mev-1(kn1) worms was further increased when compared to that of in wild-type worms. However, treatment with EISO, α- and β-santalol resulted in a significantly reduced germ cell apoptosis in mev-1(kn1) worms (ESI Fig. 3B†). These results provides clear evidence that EISO, α- and β-santalol might prolongs the lifespan of mev-1(kn1) mutant worms by reducing the apoptosis instead of rescuing the worms from lethal oxidative stress. We also clarify the possibility of whether EISO, α- and β-santalol required MAPK pathway to elicit neuroprotection, the lifespan of ERK-MAPK pathway loss-of-function mutants were measured. The ERK-MAPK signaling module suggested to controlling diverse cellular processes in C. elegans,86 but their role in neuroprotection is poorly characterized. In this study we identified that, EISO, α- and β-santalol treatment fails to augment the survival of lin-45(n2520) (Fig. 14G), mek-2(n1989) (Fig. 14H) and mpk-1(ku1) (Fig. 14I) loss-of-function mutant worms under 6-OHDA exposed condition. To summarize, the data indicated that mitochondrial ETC, and ERK-MAPK pathways regulates the EISO-mediated neuroprotection and geroprotection in C. elegans.
 |
| | Fig. 14 Lifespan of C. elegans mutants treated with EISO, α- and β-santalol. Survival curves of (A) daf-2(e1370) (B) daf-16(mgDf50) (C) akt-1(ok525) (D) isp-1(qm150) (E) clk-1(e2519) (F) mev-1(kn1) (G) lin-45(n2520) (H) mek-2(n1989) and (I) mpk-1(ku1) mutant strains treated with 6-OHDA and 6-OHDA/EISO, α- or β-santalol till their end of life. The survival curves were plotted using Kaplan–Meier survival analysis and significance levels were estimated by long-rank test in Medcalc statistical software v.14. The representative Kaplan–Meier survival curves from three independent runs were presented. Statistical details of these experiments are summarized in ESI Table 7.† | |
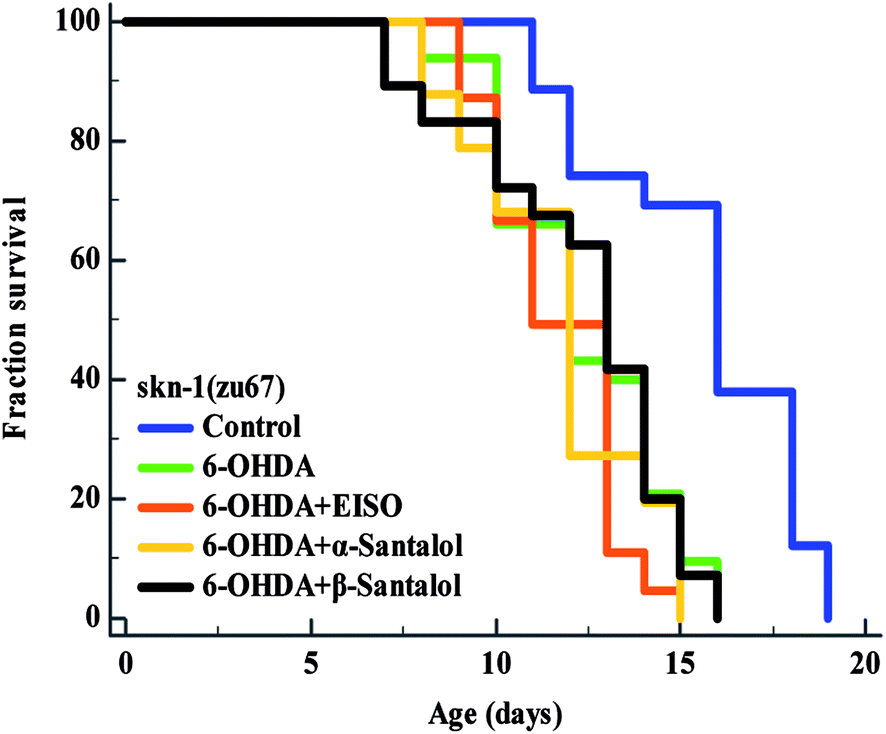
3.10. EISO triggers the ERK-MAPK mediated SKN-1 dependent pathway to exert the protective effect
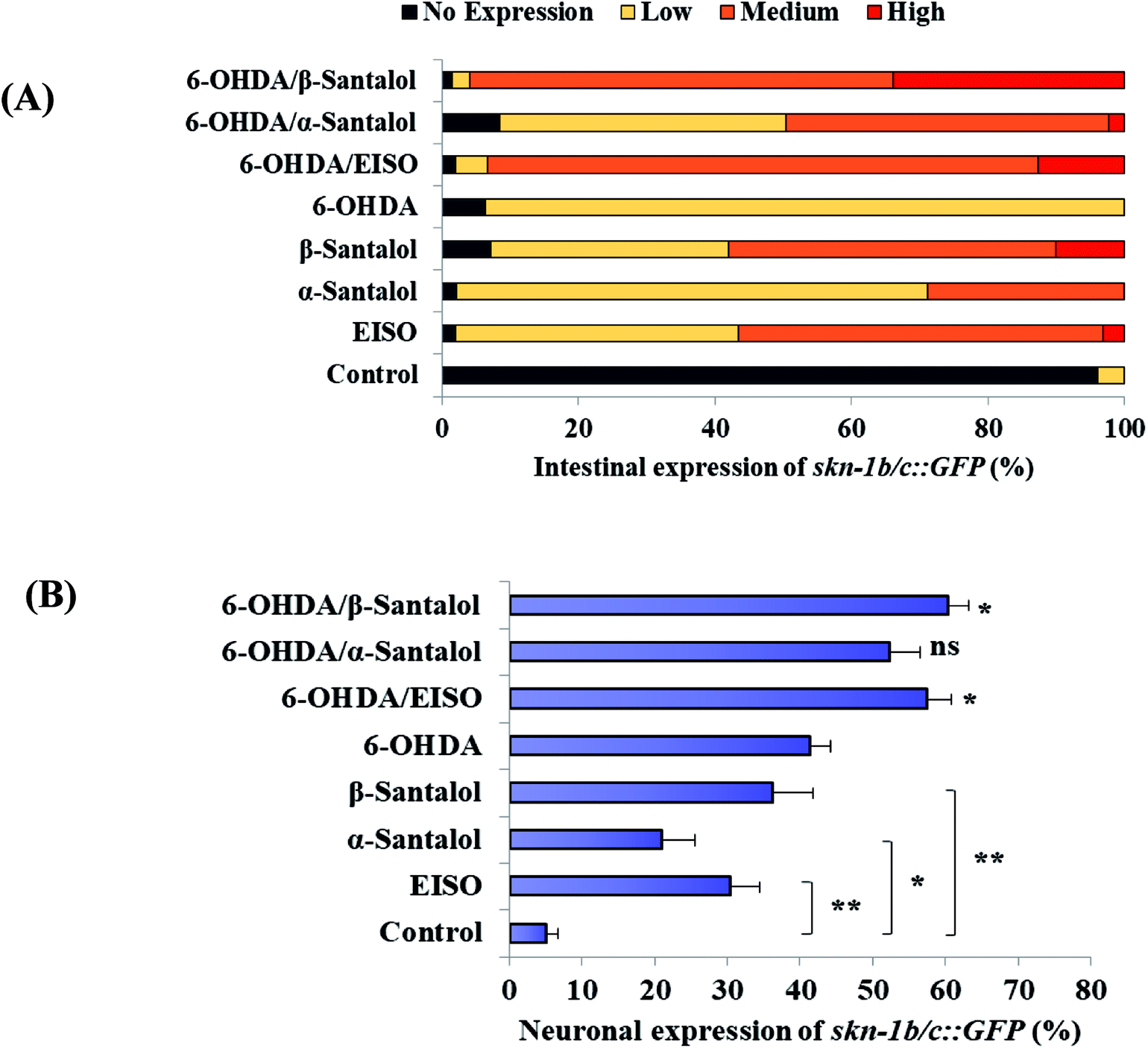
The data presented above indicates that the ERK-MAPK pathway play an indispensable role in EISO-mediated stress protection in C. elegans. Hence, further studies were carried out to understand the influences of ERK-MAPK on SKN-1 activity. The SKN-1, a CNC family transcription factor, regulates the expression of plethora of genes that promotes survival of an organism under stress.24,27 In order to determine the role of SKN-1 in EISO-mediated protection, the magnitude of the lifespan extension in EISO, α- and β-santalol pretreated C. elegans deficient in skn-1(zu67) exposed to 6-OHDA was measured. The results showed that worms loss-of-function in skn-1(zu67) was susceptible to 6-OHDA treatment (p < 0.0001). It is also interesting to note that EISO, α- and β-santalol treatment would not further extends the lifespan of skn-1(zu67) mutants under 6-OHDA exposure (Fig. 15), suggesting that EISO, α- and β-santalol might act on the SKN-1 dependent manner. In C. elegans, the transcriptional activation of SKN-1 resulted in transactivation of specific antioxidants and phase II class detoxification genes including gamma-glutamine cysteine synthetase-1 (gcs-1) and glutathione S-transferase 4 (gst-4) which offers a conserved protection against oxidative stress and promotes longevity in parallel.24 In this study, we found that treatment with 6-OHDA alone triggered the constitutive localization of skn-1 b/c::GFP into nuclei of intestinal cells, whereas EISO, α- and β-santalol additionally enhanced the SKN-1 in intestinal cells as well in ASI neurons (Fig. 16; ESI Fig. 4†). Further, we examined the effect of EISO, α- and β-santalol on expression of gcs-1 and gst-4 genes, the transgenic strains carrying reporter transgenes for gcs-1p::GFP and gst-4p::GFP were treated with respective doses of test compounds. As a result, the expression levels of gcs-1p::GFP and gst-4p::GFP, the direct readouts of SKN-1, were shown to increase after exposure with 6-OHDA and addition of either EISO, α- or β-santalol further up-regulate the gcs-1p::GFP (Fig. 17) and gst-4p::GFP (Fig. 17). Additionally the GFP signal was no longer observed in the skn-1 b/c::GFP, gcs-1p::GFP and gst-4p::GFP worms exposed to EISO, α- and β-santalol for 96 h under 6-OHDA. This indicate that an enhanced activation of SKN-1 and downstream targets by EISO, α- and β-santalol treatment might suppressed the 6-OHDA-induced oxidative stress. Surprisingly, EISO, α- and β-santalol treatments alone substantially influenced skn-1 b/c::GFP, gcs-1p::GFP and gst-4p::GFP expression in C. elegans (Fig. 16 and 17). Moreover, similar effect has also been observed in worms carrying reporter transgene for ctl-1,2,3p::GFP (ESI Fig. 5†). The ctl genes which encode antioxidant enzyme catalases protects C. elegans from ROS induced damages.
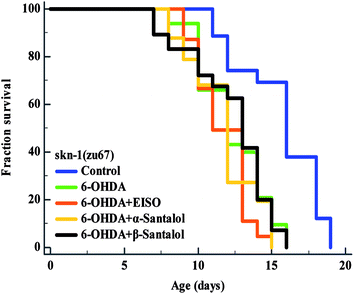 |
| | Fig. 15 Survival curves of skn-1(zu67) loss-of-function C. elegans raised on culture plates carrying 6-OHDA and 6-OHDA/EISO, α- or β-santalol at 20 °C. The curves were plotted using Kaplan–Meier survival analysis and significance levels were estimated by long-rank test in Medcalc statistical software v.14. The representative Kaplan–Meier survival curves from three independent runs were presented. Statistical details of this experiment are summarized in ESI Table 7.† | |
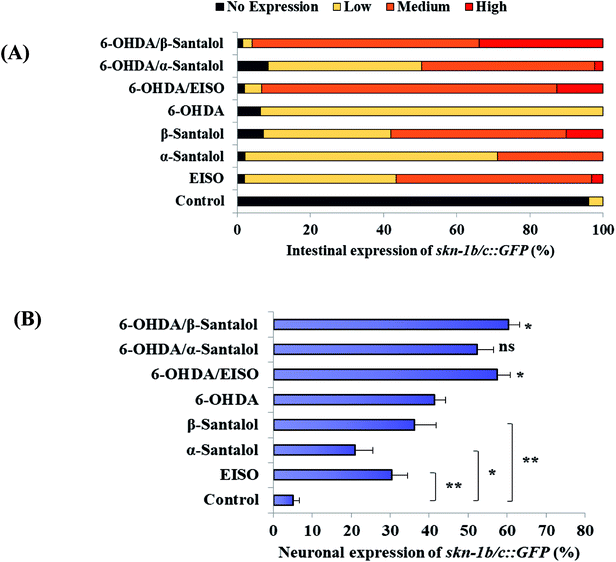 |
| | Fig. 16 EISO, α- and β-santalol modulate SKN-1 expression in C. elegans. Graphical representation illustrating the level of (A) intestinal expression and (B) neuronal expression of skn-1b/c::GFP transgene in LD1 C. elegans treated with and without 6-OHDA, EISO, α-and β-santalol. Data were acquired from three independent biological experiments and presented as mean ± SEM and classified into no, low, medium and high expression. | |
 |
| | Fig. 17 Influence of EISO, α- and β-santalol on the expression of antioxidant defense genes in C. elegans (n = 25–30 per experiment). Representative fluorescence images of (A) gcs-1p::GFP transgene in LD1171, and (B) gst-4p::GFP transgene in CL2166 C. elegans. Graphs depict the quantified expression rate of gcs-1p::GFP and gst-4p::GFP. Data shown here are mean ± SEM, the probability levels of p < 0.05 were considered as statistically significant. *p < 0.05, **p < 0.01 and ns, not significant. | |
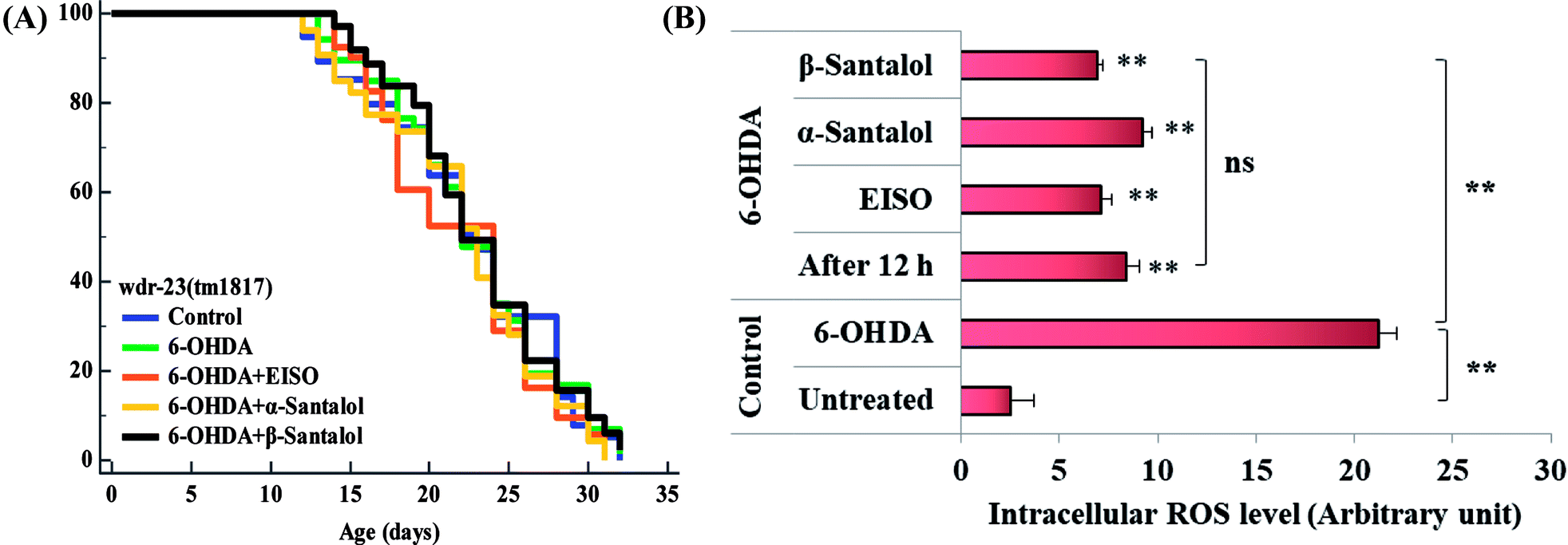
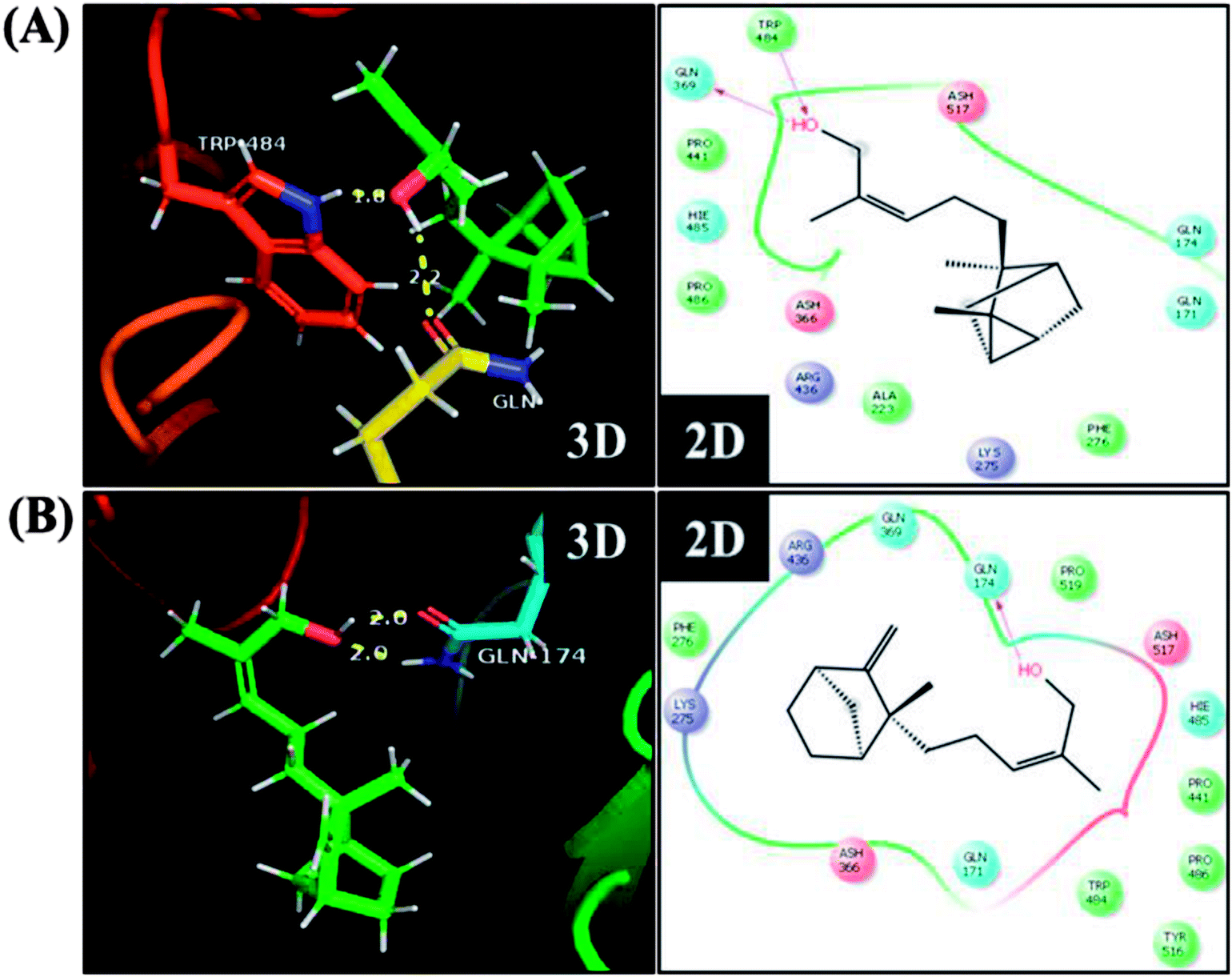
To further understand the possibility that EISO and santalol isomers could modulates the SKN-1 activity, we examined the effect of EISO, α- and β-santalol on the lifespan of wdr-23(tm1817) loss-of-function mutants. It is well-known that the activity of SKN-1 was suppressed by the direct physical interaction of WDR-23 and mutation/loss-of-function of wdr-23 causes nuclear accumulation of SKN-1 thus leading to activation of downstream genes.25 We found that the mean lifespan of wdr-23(tm1817) mutant exposed to 6-OHDA was neither increased nor decreased compared with that of untreated control group (Fig. 18A). Moreover, significant ROS production could be detected in wdr-23 worms exposed to 6-OHDA, however, this increase in ROS level was decreased remarkably after 12 h exposure (Fig. 18B). This signified that 6-OHDA-induced oxidative stress was further detoxified by the transcriptional activation of SKN-1 target genes gcs-1 and gst-4 which may attribute to the constant lifespan. A study documented that loss of wdr-23 increased resistance to stress in SKN-1 dependent manner.25 At the same time, the mean lifespan of wdr-23(tm1817) mutant worms fed with EISO, α- and β-santalol did not increased significantly when compared to 6-OHDA exposed worms. Meanwhile, 6-OHDA/EISO, α- and β-santalol treatment did not obviously altered the intracellular ROS level (Fig. 18B). This result suggested that EISO, α- and β-santalol modulates the SKN-1 activity via regulating the WDR-23 functions. To further strengthen this result, we therefore performed molecular docking analysis to predict the interaction of santalol isomers with WDR-23. The results revealed that both α- and β-santalol form a connection between amino acid residues like GLN369/TRP484 and GLN174 of WDR-23 through strong hydrogen bonding with a glide score of −4.15 and −4.33 kcal mol−1, respectively (Fig. 19). This direct interaction might perturb the protein–protein interaction of WDR-23 with SKN-1 thereby hinders its degradation by the ubiquitin proteasome system thus leading to activation and nuclear accumulation of SKN-1, which consistent with the results of in vivo experiments. The current result was also supported by previous work done by Ogawa and his co-workers.28 Currently, we have been working to validate the biophysical interaction of α- and β-santalol with WDR-23 in C. elegans. Additionally, we have also evaluated whether santalol isomers has similar effect on vertebrate CNC family transcription factor suppressor Kelch-like erythroid cell-derived protein with CNC homology (ECH) associated protein 1 (Keap1) of Nrf2 through molecular docking. The proposed docked model presented in ESI Fig. 6† suggest that santalol isomers binds to the residues of Kelch domain of Keap1 with a binding affinity of −5.17 and −5.56 kcal mol−1. This indicates EISO and santalol isomers may exert similar effect in vertebrates, which is an important issue to be validated in future studies. C. elegans lacks a Keap1 homology, whereas the function of WDR-23 is far similar to Keap1.25 In summary, the results confirmed that EISO and santalol isomers require SKN-1 to exert its beneficial effects.
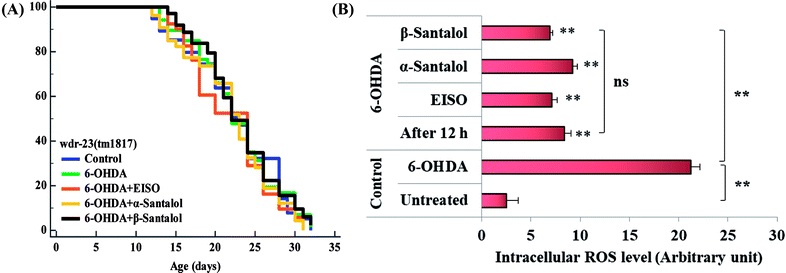 |
| | Fig. 18 Lifespan analysis and ROS accumulation in wdr-23(tm1817) loss-of-function worms exposed to 6-OHDA and 6-OHDA/EISO, α- and β-santalol. (A) Lifespan of wdr-23(tm1817). Survival curves were plotted using Kaplan–Meier survival assay and p value was calculated by log-rank rest. Three biological replicates were performed. Statistical details of this experiment are summarized in ESI Table 7.† (B) Graph depicts the intracellular generation of ROS in wdr-23(tm1817) mutant after treated with 6-OHDA or 6-OHDA/EISO, α- and β-santalol. Data were acquired from three independent biological experiments and presented as mean ± SEM, the probability levels of p < 0.05 were considered as statistically significant. **p < 0.01 and ns, not significant. | |
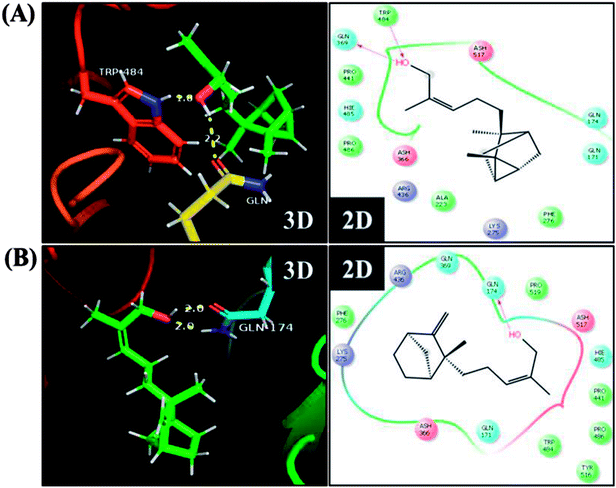 |
| | Fig. 19 Proposed interaction of (A) α-santalol and (B) β-santalol on the binding cleft of WDR-23. The representative three-dimensional (3D) and two-dimensional (2D) interaction map shows the binding mode of α- and β-santalol (stick style in a green color) through hydrogen bonds (yellow dashed lines) with WDR-23 (shown in ribbon style and colored shape). The pink arrow in two-dimensional image indicates potential interactions between amino acid residues and α- or β-santalol. | |
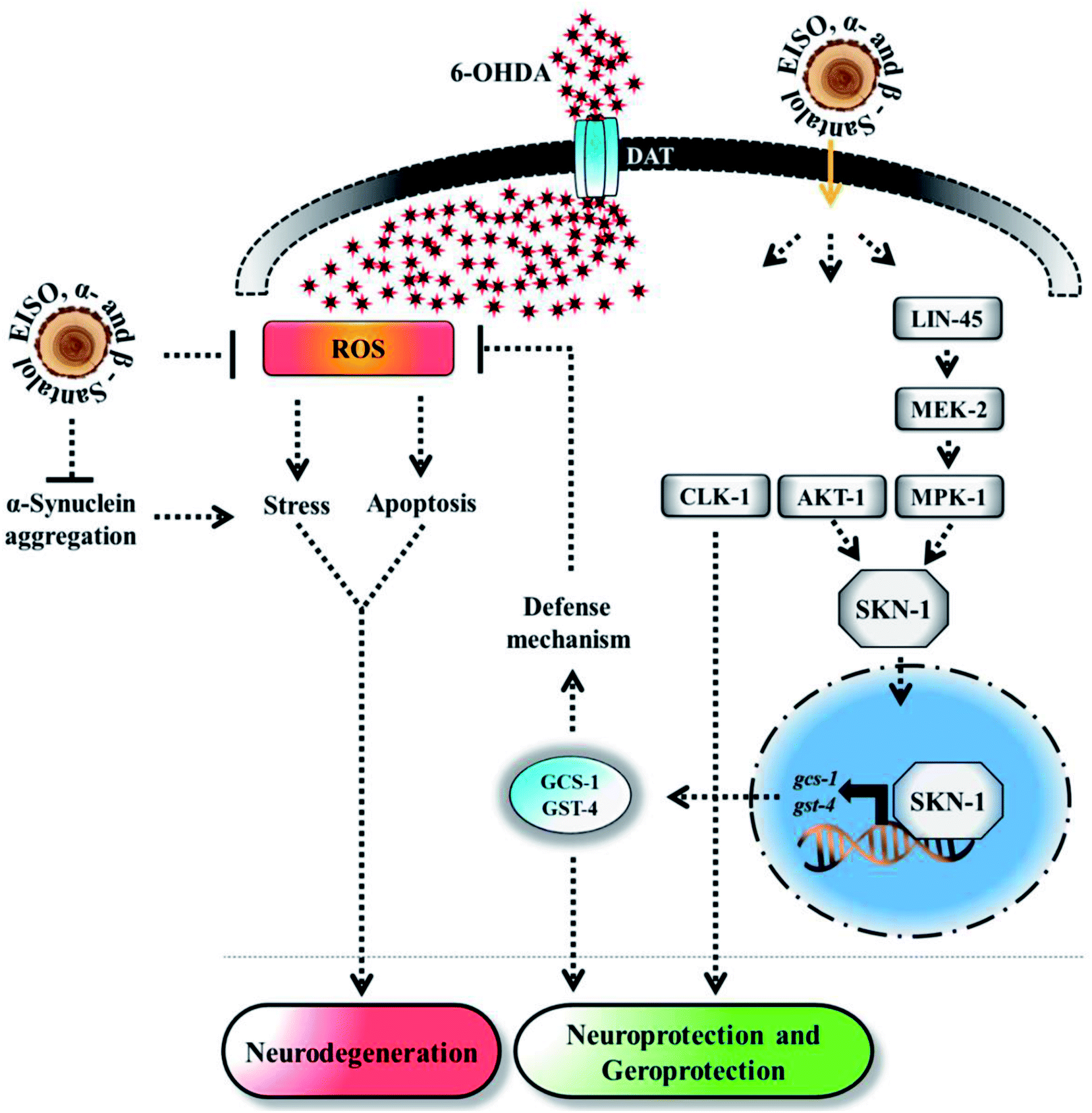
Experimental evidences showed that upregulation of SKN-1 maintains the cellular redox homeostasis and extend lifespan by promoting the transcription of cytoprotection genes, while its disruption accelerates the normal aging process in C. elegans.27,87 Thus, like mammalian Nrf2, SKN-1 is implicated in a wide range of detoxification processes and biological functions. Certain cellular signaling pathways directly regulate the SKN-1 transcriptional activity in C. elegans. In fact, ERK-MAPK plays a dynamic function in the regulation of dopamine signaling88 and it associated with α-synuclein, signifying its potential role in PD.89 In one study, it was clearly elucidated that activation of ERK in the presence of 6-OHDA promotes the survival of DA neurons.90 It has been demonstrated that valproic acid conferred the DA neurodegeneration via the ERK pathway in C. elegans.91 Signaling through the ERK and AKT-1 phosphorylates SKN-1 on the key site leading to its accumulation in nucleus that promotes lifespan and healthspan.86 A study revealed the novel role of SKN-1 in the regulation of synaptic transmission at neuromuscular junctions. SKN-1 has been shown to perform a wide range of protective functions and its expression in DA neurons protects the C. elegans from methylmercury-induced toxicity.92 A recent study showed that mulberry anthocyanin extract ameliorates the paraquat toxicity through enhanced activation of SKN-1.93 It has also been confirmed that hyperactivation of SKN-1 improves the defense against 6-OHDA-induced DA neurodegeneration.94 Furthermore, the genes that are switched on by SKN-1 or itself directly regulate the lipid metabolism in C. elegans.95 This suggests that SKN-1 might play an important role in the restoration of lipid content in the worms expressing α-synuclein. Cooper and his coworkers recently proved that slowing down the aging process alleviated the PD pathogenesis in C. elegans.96 In accordance to these observations, our results indicated that EISO and its principle components activate AKT-1 and ERK-MAPK mediated SKN-1 dependent pathway in C. elegans to resist against proteotoxic and neurotoxic stresses (Fig. 20). Finally, our in silico ADME prediction study showed that santalol isomers exhibited promising drug-likeness properties with high oral bioavailability (ESI Table 8†). Therefore, results concluded that α- and β-santalol from EISO might consider as lead compounds for the development of potent neuroprotectant to limit the neuronal death.
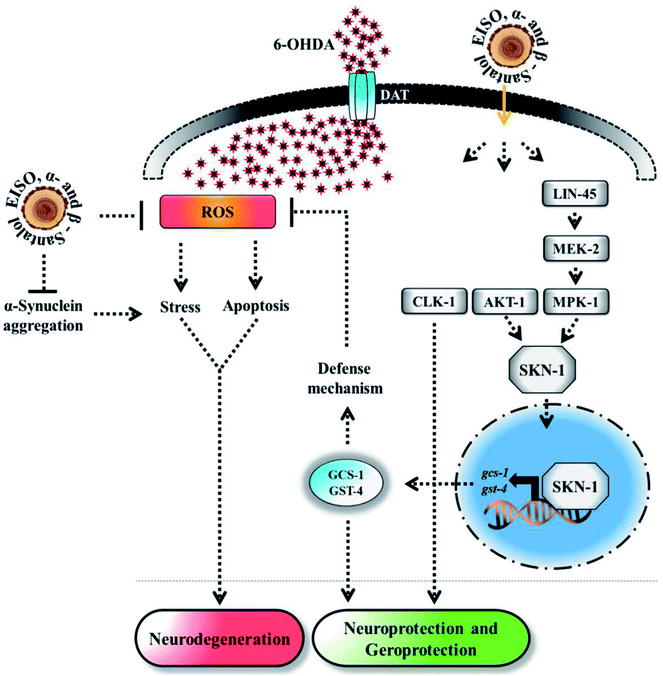 |
| | Fig. 20 The proposed neuroprotective and geroprotective mechanism of EISO and its active components on C. elegans. EISO and santalol isomers likely acted through AKT-1 and ERK-MAPK mediated SKN-1 dependent pathway, which transactivates the stress-responsive genes gcs-1 and gst-4 that enhanced the tolerance to stress and extend the mean lifespan. The direct antioxidant activity, α-synuclein inhibitory potential, antiapoptotic activity, and CLK-1 dependency of EISO, α- and β-santalol can also contribute for the reduction of neurodegeneration and longevity extension in C. elegans. | |
4. Conclusion
In this study, we demonstrated that EISO and its active components isolated from plantation grown S. album L., trees have the capacity to protect C. elegans from 6-OHDA-induced neurodegeneration and other PD associated pathologies without obviously altering the overall physiological functions. We also observed that antioxidant and antiapoptotic activity of EISO, α- and β-santalol which are capable of reducing the intracellular ROS levels and apoptotic features induced by 6-OHDA. The protective activity against neurotoxic and proteotoxic stress in C. elegans offered by EISO and santalol isomers were dependent on multiple cellular signaling mechanisms linked with longevity and stress resistances including mitochondrial ETC, ERK-MAPK and SKN-1/Nrf2 signaling but not via DAF-2/DAF-16 pathway. Our data provided the first insights into the neuroprotective and geroprotective mechanism of EISO and its active components in C. elegans, which will pave a way for developing the possibility of novel pharmaceutical products from EISO to improve the neuronal health.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Santalis Pharmaceuticals Inc., (San Antonio, TX, USA) provided funding to support the studies as well as providing some reagents used in the experiments. Dr Corey Levenson helped to define the scope of the work to be undertaken, reviewed and commented on the data and provided editorial input on the preparation of the manuscript. The authors gratefully acknowledge Caenorhabditis Genetic Centre (CGC, University of Minnesota, MN, USA) which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), for providing C. elegans strains. Special thanks go to Dr Shohei Mitani of the National BioResource Project (NBRP), Women's Medical University, Tokyo, Japan for providing wdr-23(tm1817) worm strain. We acknowledge the Bioinformatics Infrastructure Facility, Bharathiar University, India for providing computational facility. AM thanks Dr D. Nataraj, Dr R. Prabhakaran, Sankar Ganesh, Manikandan, Maharajan, Ruban Durairaj and Logesh for their suggestions and kind assistance during the work.
References
- E. Heuberger, T. Hongratanaworakit and G. Buchbauer, Planta Med., 2006, 72, 792–800 CrossRef CAS PubMed.
- G. A. Burdock and I. G. Carabin, Food Chem. Toxicol., 2008, 46, 421–432 CrossRef CAS PubMed.
- S. E. Dickinson, E. R. Olson, C. Levenson, J. Janda, J. J. Rusche, D. S. Alberts and G. T. Bowden, Arch. Biochem. Biophys., 2014, 558, 143–152 CrossRef CAS PubMed.
- B. Lee, J. Bohmann, T. Reeves, C. Levenson and A. L. Risinger, J. Nat. Prod., 2015, 78, 1357–1362 CrossRef CAS PubMed.
- Y. Matsuo and Y. Mimaki, Phytochemistry, 2012, 77, 304–311 CrossRef CAS PubMed.
- B. B. Misra and S. Dey, Phytomedicine, 2013, 20, 409–416 CrossRef CAS PubMed.
- S. Santha, A. Bommareddy, B. Rule, R. Guillermo, R. S. Kaushik, A. Young and C. Dwivedi, PLoS One, 2013, 8, e56982 CrossRef CAS PubMed.
- A. Bommareddy, S. Brozena, J. Steigerwalt, T. Landis, S. Hughes, E. Mabry, A. Knopp, A. L. VanWert and C. Dwivedi, Nat. Prod. Res., 2017, 1–17 CrossRef PubMed.
- M. Paulpandi, S. Kannan, R. Thangam, K. Kaveri, P. Gunasekaran and C. Rejeeth, Phytomedicine, 2012, 19, 231–235 CrossRef CAS PubMed.
- M. Sharma, C. Levenson, I. Clements, P. Castella, K. Gebauer and M. E. Cox, Front. Pharmacol., 2017, 8, 125 Search PubMed.
- M. Sharma, C. Levenson, J. C. Browning, E. M. Becker, I. Clements, P. Castella and M. E. Cox, Front. Pharmacol., 2018, 9, 1–12 CrossRef PubMed.
- J. Avila, Nat. Med., 2010, 16, 1372 CrossRef CAS PubMed.
- C. Hansen and J. Y. Li, Trends Mol. Med., 2012, 18, 248–255 CrossRef CAS PubMed.
- H. Braak, K. Del Tredici, U. Rüb, R. A. I. De Vos, E. N. H. Jansen Steur and E. Braak, Neurobiol. Aging, 2003, 24, 197–211 CrossRef PubMed.
- M. Coelho and J. J. Ferreira, Nat. Rev. Neurol., 2012, 8, 435–442 CrossRef CAS PubMed.
- U. Dettmer, A. J. Newman, F. Soldner, E. S. Luth, N. C. Kim, V. E. von Saucken, J. B. Sanderson, R. Jaenisch, T. Bartels and D. Selkoe, Nat. Commun., 2015, 6, 7314 CrossRef PubMed.
- B. Singleton, M. Farrer, J. Johnson, A. Singleton, S. Hague, J. Kachergus, M. Hulihan, T. Peuralinna, A. Dutra, R. Nussbaum, S. Lincoln, A. Crawley, M. Hanson, D. Maraganore, C. Adler, M. R. Cookson, M. Muenter, M. Baptista, D. Miller, J. Blancato, J. Hardy and K. Gwinn-Hardy, Science, 2003, 302, 841 CrossRef PubMed.
- H. C. Curtius, M. Wolfensberger, B. Steinmann, U. Redweik and J. Siegfried, J. Chromatogr. A, 1974, 99, 529–540 CrossRef CAS PubMed.
- R. Andrew, D. G. Watson, S. A. Best, J. M. Midgley, H. Wenlong and R. K. H. Petty, Neurochem. Res., 1993, 18, 1175–1177 CrossRef CAS PubMed.
- J. Blesa, I. Trigo-Damas, A. Quiroga-Varela and V. R. Jackson-Lewis, Front. Neuroanat., 2015, 9, 91 Search PubMed.
- J. Segura-Aguilar, I. Paris, P. Muñoz, E. Ferrari, L. Zecca and F. A. Zucca, J. Neurochem., 2014, 129, 898–915 CrossRef CAS PubMed.
- W. Xiong, A. E. M. C. Garfinkel, Y. Li, L. I. Benowitz and C. L. Cepko, J. Clin. Invest., 2015, 125, 1433–1445 CrossRef PubMed.
- A. K. Walker, R. See, C. Batchelder, T. Kophengnavong, J. T. Gronniger, Y. Shi and T. K. Blackwell, J. Biol. Chem., 2000, 275, 22166–22171 CrossRef CAS PubMed.
- T. K. Blackwell, M. J. Steinbaugh, J. M. Hourihan, C. Y. Ewald and M. Isik, Free Radical Biol. Med., 2015, 88, 290–301 CrossRef CAS PubMed.
- L. Tang and K. P. Choe, Mech. Ageing Dev., 2015, 149, 88–98 CrossRef CAS PubMed.
- J. H. An and T. K. Blackwell, Genes Dev., 2003, 17, 1882–1893 CrossRef CAS PubMed.
- J. M. A. Tullet, J. W. Green, C. Au, A. Benedetto, M. A. Thompson, E. Clark, A. F. Gilliat, A. Young, K. Schmeisser and D. Gems, Aging Cell, 2017, 16, 1191–1194 CrossRef CAS PubMed.
- T. Ogawa, Y. Kodera, D. Hirata, T. K. Blackwell and M. Mizunuma, Sci. Rep., 2016, 6, 21611 CrossRef CAS PubMed.
- E. Dehghan, Y. Zhang, B. Saremi, S. Yadavali, A. Hakimi, M. Dehghani, M. Goodarzi, X. Tu, S. Robertson, R. Lin, A. Chudhuri and H. Mirzaei, Nat. Commun., 2017, 8, 2223 CrossRef PubMed.
- S. Alavez, M. C. Vantipalli, D. J. S. Zucker, I. M. Klang and G. J. Lithgow, Nature, 2011, 472, 226–230 CrossRef CAS PubMed.
- P. P. Daramwar, P. L. Srivastava, B. Priyadarshini and H. V. Thulasiram, Analyst, 2012, 137, 4564 RSC.
- G. Devagi, A. Mohankumar, G. Shanmugam, S. Nivitha, F. Dallemer, P. Kalaivani, P. Sundararaj and R. Prabhakaran, Sci. Rep., 2018, 8, 7688 CrossRef CAS PubMed.
- S. Brenner, Genetics, 1974, 77, 71–94 CAS.
- T. J. Fabian and T. E. Johnson, J. Gerontol., 1994, 49, B145–B156 CrossRef CAS PubMed.
- G. Devagi, G. Shanmugam, A. Mohankumar, P. Sundararaj, F. Dallemer, P. Kalaivani and R. Prabhakaran, J. Organomet. Chem., 2017, 838, 12–23 CrossRef CAS.
- C. I. Bargmann, E. Hartwieg and H. R. Horvitz, Cell, 1993, 74, 515–527 CrossRef CAS PubMed.
- R.-H. Fu, Y.-C. Wang, C.-S. Chen, R.-T. Tsai, S.-P. Liu, W.-L. Chang, H.-L. Lin, C.-H. Lu, J.-R. Wei, Z.-W. Wang, W.-C. Shyu and S.-Z. Lin, Neuropharmacology, 2014, 82, 108–120 CrossRef CAS PubMed.
- S. F. Wang, L. F. Liu, M. Y. Wu, C. Z. Cai, H. Su, J. Tan, J. H. Lu and M. Li, Sci. Rep., 2017, 7, 8398 CrossRef PubMed.
- R. H. Fu, H. J. Harn, S. P. Liu, C. S. Chen, W. L. Chang, Y. M. Chen, J. E. Huang, R. J. Li, S. Y. Tsai, H. S. Hung, W. C. Shyu, S. Z. Lin and Y. C. Wang, PLoS One, 2014, 9, e85305 CrossRef PubMed.
- S. Govindan, M. Amirthalingam, K. Duraisamy, T. Govindhan, N. Sundararaj and S. Palanisamy, Biomed. Pharmacother., 2018, 102, 812–822 CrossRef CAS PubMed.
- G. Shanmugam, A. Mohankumar, D. Kalaiselvi, S. Nivitha, E. Murugesh, P. Shanmughavel and P. Sundararaj, Biomed. Pharmacother., 2017, 95, 1693–1703 CrossRef CAS PubMed.
- A. a Soukas, E. a Kane, C. E. Carr, J. a Melo and G. Ruvkun, Genes Dev., 2009, 23, 496–511 CrossRef PubMed.
- K. Yen, T. T. Le, A. Bansal, S. D. Narasimhan, J. X. Cheng and H. A. Tissenbaum, PLoS One, 2010, 5, 1–10 CrossRef PubMed.
- C. Shu, X. Yu, Q. Wu, Z. Zhuang, W. Zhang and D. Wang, RSC Adv., 2015, 5, 8942–8951 RSC.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- C. F. Glenn, D. K. Chow, L. David, C. A. Cooke, M. S. Gami, W. B. Iser, K. B. Hanselman, I. G. Goldberg and C. A. Wolkow, J. Gerontol., Ser. A, 2004, 59, 1251–1260 CrossRef.
- Q. Wu, L. Yin, X. Li, M. Tang, T. Zhang and D. Wang, Nanoscale, 2013, 5, 9934 RSC.
- D. T. Omura, D. A. Clark, A. D. T. Samuel and H. R. Horvitz, PLoS One, 2012, 7, e38649 CrossRef CAS PubMed.
- A. Dillin, A. L. Hsu, N. Arantes-Oliveira, J. Lehrer-Graiwer, H. Hsin, A. G. Fraser, R. S. Kamath, J. Ahringer and C. Kenyon, Science (80- )., 2002, 298, 2398–2401 CrossRef CAS PubMed.
- A. Pant, S. K. Saikia, V. Shukla, J. Asthana, B. A. Akhoon and R. Pandey, Exp. Gerontol., 2014, 57, 81–95 CrossRef CAS PubMed.
- L. Gao, R. Zhang, J. Lan, R. Ning, D. Wu, D. Chen and W. Zhao, J. Nat. Prod., 2016, 79, 3039–3046 CrossRef CAS PubMed.
- C. W. Yu, W. H. Li, F. L. Hsu, P. L. Yen, S. T. Chang and V. H. C. Liao, J. Agric. Food Chem., 2014, 62, 6159–6165 CrossRef CAS PubMed.
- S. Ghavami, S. Shojaei, B. Yeganeh, S. R. Ande, J. R. Jangamreddy, M. Mehrpour, J. Christoffersson, W. Chaabane, A. R. Moghadam, H. H. Kashani, M. Hashemi, A. A. Owji and M. J. Łos, Prog. Neurobiol., 2014, 112, 24–49 CrossRef CAS PubMed.
- K. J. Barnham, C. L. Masters and A. I. Bush, Nat. Rev. Drug Discovery, 2004, 3, 205–214 CrossRef CAS PubMed.
- D. Harman, J. Gerontol., 1956, 11, 298–300 CrossRef CAS PubMed.
- Z. Zhou, E. Hartwieg and H. R. Horvitz, Cell, 2001, 104, 43–56 CrossRef CAS PubMed.
- B. Conradt and H. R. Horvitz, Cell, 1998, 93, 519–529 CrossRef CAS PubMed.
- X. Yang, H. Y. Chang and D. Baltimore, Science (80- ), 1998, 281, 1355–1357 CrossRef CAS.
- H. Li, R. Shi, F. Ding, H. Wang, W. Han, F. Ma, M. Hu, C. W. Ma and Z. Huang, Oxid. Med. Cell. Longevity, 2016, 2016, 1–10 Search PubMed.
- P. Pu and W. Le, Cell Res., 2008, 18, 978–981 CrossRef CAS PubMed.
- M. Gomez-Lazaro, M. F. Galindo, C. G. Concannon, M. F. Segura, F. J. Fernandez-Gomez, N. Llecha, J. X. Comella, J. H. M. Prehn and J. Jordan, J. Neurochem., 2008, 104, 1599–1612 CrossRef CAS PubMed.
- C. Cui, N. Cui, P. Wang, S. Song, H. Liang and A. Ji, Biochem. Biophys. Res. Commun., 2016, 470, 375–383 CrossRef CAS PubMed.
- D. P. Denning, V. Hatch and H. R. Horvitz, PLoS Genet., 2013, 9, e1003341 CrossRef CAS PubMed.
- X. Geng, Q. H. Zhou, E. Kage-Nakadai, Y. Shi, N. Yan, S. Mitani and D. Xue, Cell Death Differ., 2009, 16, 1385–1394 CrossRef CAS PubMed.
- T. J. van Ham, K. L. Thijssen, R. Breitling, R. M. W. Hofstra, R. H. A. Plasterk and E. A. A. Nollen, PLoS Genet., 2008, 4, e1000027 CrossRef PubMed.
- H. T. Li, D. H. Lin, X. Y. Luo, F. Zhang, L. N. Ji, H. N. Du, G. Q. Song, J. Hu, J. W. Zhou and H. Y. Hu, FEBS J., 2005, 272, 3661–3672 CrossRef CAS PubMed.
- V. Ruipérez, F. Darios and B. Davletov, Prog. Lipid Res., 2010, 49, 420–428 CrossRef PubMed.
- R. Sharon, I. Bar-Joseph, M. P. Frosch, D. M. Walsh, J. A. Hamilton and D. J. Selkoe, Neuron, 2003, 37, 583–595 CrossRef CAS PubMed.
- V. V. Shvadchak, D. A. Yushchenko, R. Pievo and T. M. Jovin, FEBS Lett., 2011, 585, 3513–3519 CrossRef CAS PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- D. Helm, H. Labischinski, G. Schallehn and D. Naumann, J. Gen. Microbiol., 1991, 137, 69–79 CrossRef CAS PubMed.
- J. Sulston, M. Dew and S. Brenner, J. Comp. Neurol., 1975, 163, 215–226 CrossRef CAS PubMed.
- D. Chen, A. G. Kanthasamy and M. B. Reddy, Parkinson's Dis., 2015, 2015, 1–10 CrossRef PubMed.
- N. I. Bohnen and R. L. Albin, Behav. Brain Res., 2011, 221, 564–573 CrossRef CAS PubMed.
- J. Rand, WormBook, 2007, 1–21 Search PubMed.
- J. L. Cummings and J. L. Cummings, J. Neurol., Neurosurg. Psychiatry, 2005, 76, 903–904 CrossRef CAS PubMed.
- T. Nidheesh, C. Salim, P. S. Rajini and P. V. Suresh, Carbohydr. Polym., 2016, 135, 138–144 CrossRef CAS PubMed.
- X. Zhang, L. Lu, S. Liu, W. Ye, J. Wu and X. Zhang, Int. J. Biochem. Cell Biol., 2013, 45, 265–272 CrossRef CAS PubMed.
- C. W. Hung, Y. C. Chen, W. L. Hsieh, S. H. Chiou and C. L. Kao, Ageing Res. Rev., 2010, 9 CrossRef CAS PubMed.
- C. J. Kenyon, Nature, 2010, 464, 504–512 CrossRef CAS PubMed.
- N. Ishii, M. Fujii, P. S. Hartman, M. Tsuda, K. Yasuda, N. Senoo-Matsuda, S. Yanase, D. Ayusawa and K. Suzuki, Nature, 1998, 394, 694–697 CrossRef CAS PubMed.
- J. Feng, F. Bussière and S. Hekimi, Dev. Cell, 2001, 1, 633–644 CrossRef CAS PubMed.
- S. J. Lee, A. B. Hwang and C. Kenyon, Curr. Biol., 2010, 20, 2131–2136 CrossRef CAS PubMed.
- N. Senoo-Matsuda, P. S. Hartman, A. Akatsuka, S. Yoshimura and N. Ishii, J. Biol. Chem., 2003, 278, 22031–22036 CrossRef CAS PubMed.
- T. Okuyama, H. Inoue, S. Ookuma, T. Satoh, K. Kano, S. Honjoh, N. Hisamoto, K. Matsumoto and E. Nishida, J. Biol. Chem., 2010, 285, 30274–30281 CrossRef CAS PubMed.
- J. M. A. Tullet, M. Hertweck, J. H. An, J. Baker, J. Y. Hwang, S. Liu, R. P. Oliveira, R. Baumeister and T. K. Blackwell, Cell, 2008, 132, 1025–1038 CrossRef CAS PubMed.
- A. Iwata, M. Maruyama, I. Kanazawa and N. Nukina, J. Biol. Chem., 2001, 276, 45320–45329 CrossRef CAS PubMed.
- N. Ostrerova, L. Petrucelli, M. Farrer, N. Mehta, P. Choi, J. Hardy and B. Wolozin, J. Neurosci., 1999, 19, 5782–5791 CrossRef CAS PubMed.
- E. Lin, J. E. Cavanaugh, R. K. Leak, R. G. Perez and M. J. Zigmond, J. Neurosci. Res., 2008, 86, 108–117 CrossRef CAS PubMed.
- B. B. Kautu, A. Carrasquilla, M. L. Hicks, K. A. Caldwell and G. A. Caldwell, Neurosci. Lett., 2013, 541, 116–119 CrossRef CAS PubMed.
- N. VanDuyn, R. Settivari, G. Wong and R. Nass, Toxicol. Sci., 2010, 118, 613–624 CrossRef CAS PubMed.
- F. Yan, Y. Chen, R. Azat and X. Zheng, Oxid. Med. Cell. Longevity, 2017, 2017, 1–12 Search PubMed.
- S. L. Offenburger, E. Jongsma and A. Gartner, PLoS Genet., 2017, 14, e1007106 CrossRef PubMed.
- S. Pang, D. A. Lynn, J. Y. Lo, J. Paek and S. P. Curran, Nat. Commun., 2014, 5, 5048 CrossRef CAS PubMed.
- J. F. Cooper, D. J. Dues, K. K. Spielbauer, E. Machiela, M. M. Senchuk and J. M. Van Raamsdonk, NPJ Parkinsons Dis., 2015, 1, 15022 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05195j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
G. Shanmugama,
D. Kalaiselvia,
C. Levensonb,
S. Nivithac,
G. Thiruppathia and
P. Sundararaj*a
*a,
G. Shanmugama,
D. Kalaiselvia,
C. Levensonb,
S. Nivithac,
G. Thiruppathia and
P. Sundararaj*a