
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando structural study of non-aqueous Li–air batteries using synchrotron-based X-ray diffraction†
Chulho Song ab,
Kimihiko Itoa,
Osami Sakataab and
Yoshimi Kubo*a
ab,
Kimihiko Itoa,
Osami Sakataab and
Yoshimi Kubo*a
aGlobal Research Center for Environment and Energy based on Nanomaterials Science (GREEN), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: KUBO.Yoshimi@nims.go.jp; Tel: +81-029-860-4773
bSynchrotron X-ray Station at SPring-8, Research Network and Facility Services Division, National Institute for Materials Science (NIMS), 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan
First published on 23rd July 2018
Abstract
Non-aqueous lithium–air batteries (LABs) attract attention as a candidate technology for next-generation energy storage devices. It is crucial to understand how the discharge product Li2O2 is formed and decomposed by the electrochemical reactions to improve the cycle performance and decrease the charge voltage, which are the most important subjects for LAB development. Here, operando X-ray diffraction with high-brilliant X-rays in a transmission mode was used to observe the intensity and structural changes of crystalline Li2O2 in an operating non-aqueous LAB in real time, and the Li–O2 electrochemical reaction involving Li2O2 formation and decomposition was clearly demonstrated. The electrochemically formed Li2O2, which had an anisotropic domain size of 10 nm in the c-direction and 40–70 nm in the ab-plane, grew due to the increase of the number of domains during the discharge process. No other reaction products with a crystalline phase such as LiOH were found in either the cathode or anode of the LAB, whereas the accelerated decomposition rate of the domains was accompanied with the change of the domain shape and lattice constant of the c-axis in the latter half of the charge process with voltage higher than 4 V.
1 Introduction
Rechargeable non-aqueous lithium–air batteries (LABs) are attractive as next-generation energy storage devices to enable the shift from fossil fuels to renewable energy sources because of their high theoretical specific energy and specific capacity.1–6 However, non-aqueous LAB technology is still in its infancy regarding commercialisation for use as energy storage devices. Current drawbacks such as poor cycle ability7 and a large overpotential in the charge process8 need to be overcome for a commercial application of LABs. To improve the cycle performance and lower the charge voltage of LABs, it is important to elucidate the Li–O2 electrochemical reaction that has the following equation:| 2Li+ + O2 + 2e− ↔ Li2O2 (s), E0 = 2.96 V vs. Li/Li+ | (1) |
Namely, for discharge/charge processes of non-aqueous LABs, the formation and decomposition of Li2O2 as a reaction product need to be thoroughly understood. Understanding of side reactions, particularly LiOH formation, during the operating process of LABs is also necessary.
A general technique used to identify the existence of crystalline Li2O2 in discharged cathodes is conventional9,10/ex situ X-ray diffraction (XRD).4,11,12 Previous studies using conventional/ex situ XRD have involved the removal of the cathode from the LAB cell after full discharge and then collecting XRD data for the cathode using a laboratory-based XRD instrument with a low signal-to-noise (S/N) ratio. Thus, information of the growth process of crystalline Li2O2 could not be obtained especially both in the initial stage of discharge and the final stage of recharge processes, where the amount of discharge product of Li2O2 is small. In addition, there is an unavoidable risk that Li2O2 qualitatively changes by extracting the cathode from a test cell (e.g. formation of LiOH).
In situ or operando XRD is a powerful technique to monitor the structural changes of reaction products with a crystalline phase in LAB cells during operation. To investigate the reaction products of LABs, it is very important to design a LAB cell for measurement purposes that minimises unintended exposure to the ambient environment during an operation. A laboratory-based in situ XRD technique in reflection mode has been used to study the Li–O2 electrochemical reaction in specially designed LAB cells.13,14 However, this in situ XRD technique used an X-ray source of low-brilliance (Cu Kα), so it required a long scan time of 30–70 min to obtain XRD patterns including only three diffraction peaks. This long scan time is inconvenient for studying the Li–O2 electrochemical reaction. A synchrotron-based in situ X-ray technique15–20 has been used to collect high-quality XRD patterns in a short scan time. Storm et al.15 designed a capillary-based Li–O2 battery consisting of an electrolyte-filled capillary with the anode and cathode on opposite ends coated on stainless steel wires. Shui and colleagues16,17 used a microfocused synchrotron XRD technique to investigate the reversibility of anodic lithium, and Li2O2 grain growth and its distribution inside a Swagelok-type cell. They observed the conversion of metallic Li to LiOH on the anode in their investigation of the reversibility of anodic lithium formation.16 This phenomenon is a cause of cycle performance degradation in LABs. The conversion of metallic Li to LiOH may originate from the following two reasons: (1) reaction of Li with water produced from electrolyte decomposition; (2) issues controlling the ambient environment of the Li–O2 battery cell. It is necessary to confirm which of these reasons results in LiOH formation. The information derived from these studies is of value to understand the electrochemical reactions that occur in LABs.
In this study, we aim to clarify the relationship between the reversible electrochemical reaction and structure of crystalline Li2O2 and examine the LiOH formation on both a cathode and an anode in a non-aqueous LAB during an operation. For this purpose, we have to strictly eliminate the influence from an ambient water and detect structural information about reaction products in a LAB cell in a very short time without stopping discharge and charge operation. We made a special airtight cell and employed synchrotron-based XRD in a transmission mode with multi-channel detectors to detect simultaneously diffracted X-rays all over angular range of interest. Those enable us to measure high quality XRD data and carry out Rietveld refinement analysis which finally lead to attain real-time structural evolution of crystalline Li2O2 in an operating LAB cell.
2 Experimental
2.1 Sample preparation
2.2 Synchrotron-based XRD in a transmission mode
Operando synchrotron-based XRD experiments in a transmission mode were performed at beamline BL15XU (SPring-8, Japan) using an X-ray of energy 19 keV (λ = 0.653 Å). The experimental set-up used for operando synchrotron-based XRD measurements is shown in Fig. S2 of the ESI.† Diffraction patterns were collected at angle intervals of 0.01° using six one-dimensional detectors. Each XRD profile was collected in the 2θ region of 1.63–74.37° every 5 min. The exposure time was 22 s. This measurement technique has several advantages for LAB investigation. The high-energy X-rays can penetrate the entire LAB cell without disturbing cell operation; therefore, one can observe phase and structural changes simultaneously as they occur. The exposure time of 22 s is much shorter than the operating duration of the LAB cell, making it feasible to observe the formation/decomposition of crystalline Li2O2 at the cathode region in real time.3 Results and discussion
3.1 Discharge/charge voltage profiles
Fig. 1 shows the discharge/charge profiles of the investigated LAB. The discharge processes were limited at a cut-off voltage of 2.2 V. The discharge processes were carried out at three different current densities (0.8, 0.6, and 0.4 mA cm−2) until a capacity of 3.54 mA h cm−2 was reached. The charge process was carried out at a current density of 0.3 mA cm−2 from 3.54 to 0 mA h cm−2. The charging potential curve is in agreement with that reported in the literature.13,17 As it can be seen in Fig. 1, the voltage steeply decreases just after the beginning of discharge until the amount of the capacity reaches at ∼0.15 mA h cm−2. The current of the region mainly originates from the discharge in an electrical double-layer between a carbon and an electrolyte in a cathode (double-layer capacitance). Its capacity can be roughly estimated to be 25 mF from the slope of a discharge profile. This value can be easily verified from the specifications of the cathode. The specific surface is about 0.8 m2 mg−1 (800 m2 g−1 for CNovel carbon). The capacity of the double layer at the interface between the carbon and the electrolyte is expected to be 28 mF when relative permittivity of electrolyte and the thickness of the double layer is assumed to be 4 and 1 nm, respectively.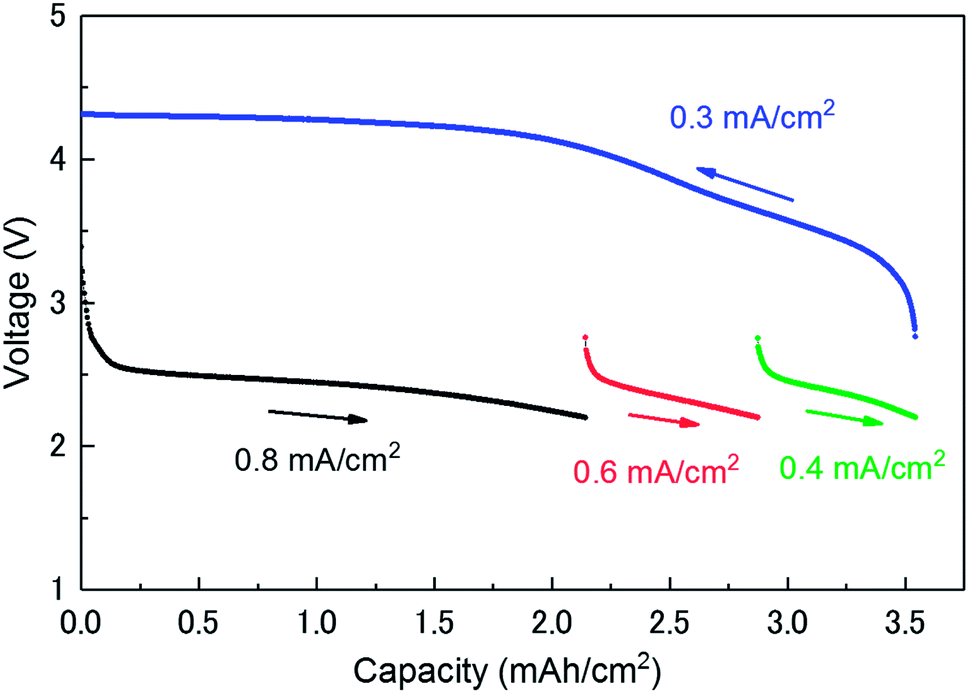 | ||
| Fig. 1 Discharge/charge voltage profiles of the investigated lithium–air battery. | ||
3.2 Operando XRD patterns
Fig. 2 depicts the change of XRD patterns for the reaction products in the LAB cell collected during the discharge/charge processes illustrated in Fig. 1. Fig. 2A and B show time-dependent XRD patterns from 2θ = 13.3 to 31.0° for the discharge and charge process, respectively. Fig. 2C and D are the enlarged XRD patterns of 2θ = 13.3 to 15.1° for Fig. 2A and B, respectively. Miller indices, shown for number of peaks in Fig. 2, indicate the reflections of crystalline Li2O2. The direction of the solid arrows, shown next to the capacity axes in Fig. 2, represents the increase of operation time of the LAB cell. The XRD patterns were obtained by subtracting the XRD profile of the fresh LAB cell from each XRD profile of the discharged LAB cells. Details of XRD pattern collection are presented in Section 2 of the ESI.† Notably, regardless of a very short scanning time of 22 s, the S/N ratio of the obtained XRD patterns were better than that of the previous study14 of a laboratory-based in situ XRD. These high-quality XRD patterns enable us to conduct more accurate structural analysis during the operating process of LABs as precisely described after the next section.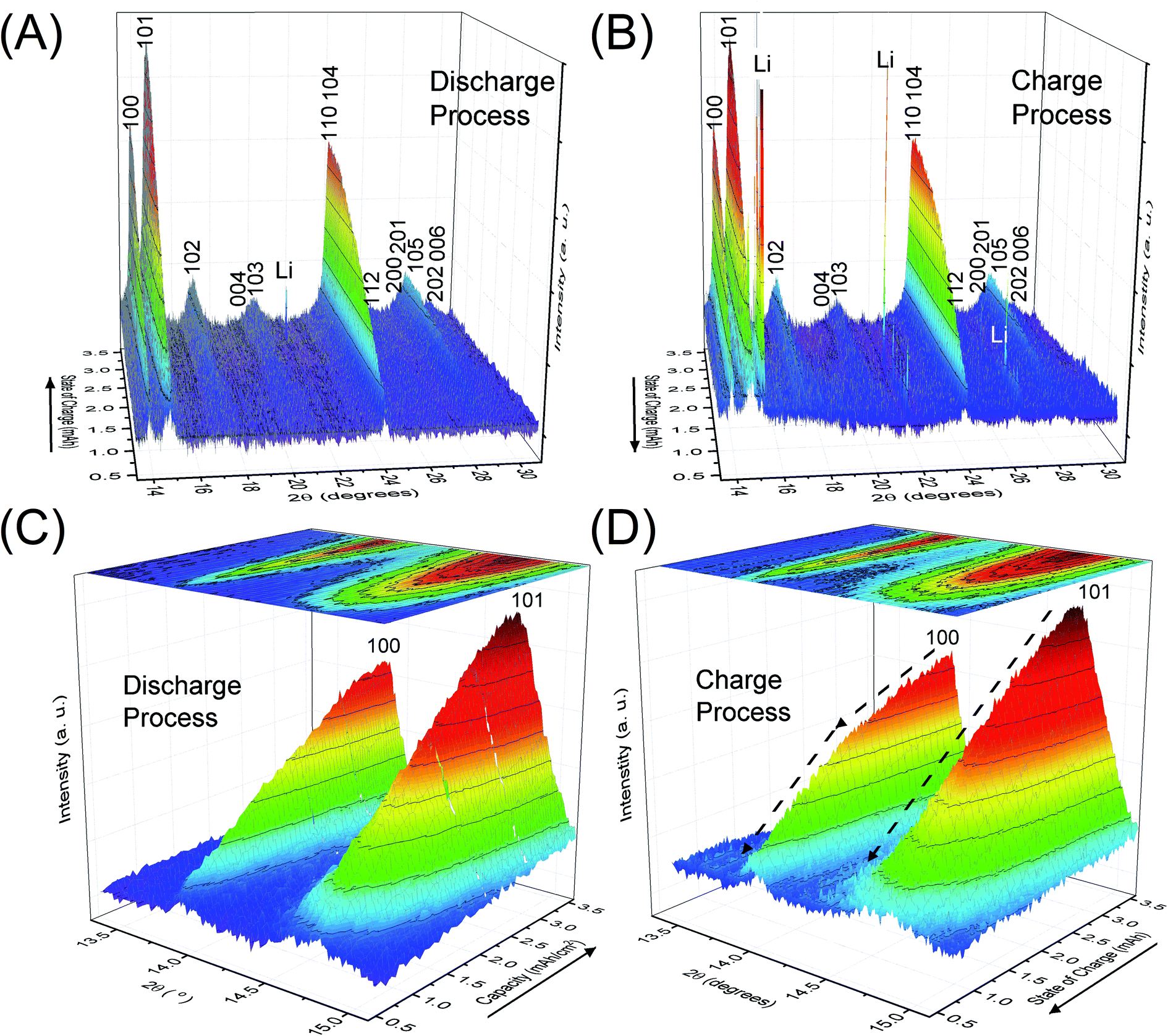 | ||
| Fig. 2 Operando XRD patterns of crystalline Li2O2 in the lithium–air battery cell during (A) and (C) the discharge and (B) and (D) the charge processes. The direction of the solid arrows represents the flow of time. | ||
3.3 Rietveld refinement
The benefit of the XRD data with a high S/N ratio superior to the previous reports,13,14 as shown in Fig. 2, enables us to perform the Rietveld refinement analysis using the pseudo-Voigt function with a reflection asymmetry to precisely elucidate the structural changes of crystalline Li2O2 in an operating LAB. The results of the Rietveld refinement analysis for a discharge product Li2O2 are presented in Fig. S4 of the ESI.† The Bragg R factor (RB), which shows the accuracy of the refinement procedure, is also presented in Fig. S5 of the ESI.† As a typical example, the Rietveld refinement of the Li2O2 XRD pattern after full discharge (3.54 mA h cm−2) of the LAB is presented in Fig. 3, and the obtained RB is kept less than 5%. Such good RBs are obtained for the cathode discharged more than 1 mA h, and even for the case of residual charge of less than 1 mA h, RBs are usually kept less than 20%. The RBs of residual components are small enough to discuss the structural change in the discharge product as shown in Fig. S4 of the ESI.† RBs larger than 20% sometimes elucidated are due to appearance of diffraction line from polycrystalline Li metal stronger than that of Li2O2, which tends to appear near the end region of the charging process. It is noted that peaks corresponding to the Li anode occasionally appear at 15.2°, 21.5°, and 26.5° as indicated by “Li” in Fig. 2, which frequently appear during the charge process than during the discharge process. This result indicates the expected anode reaction in LAB where Li ions are electrochemically deposited on the surface of Li anode in a polycrystalline form with small grain size, thus X-rays diffracted from those are often detected during the charging process. On the other hand, grain size of the original Li foil used in our experiments is so large that its diffraction pattern is spotty similar as a single crystal material. Such diffracted X-rays are rarely detected in our experimental setup at the initial and during the discharge process. It is noted again that: (1) no XRD peaks from other reaction products such as Li2O, LiOH, and Li2CO3 with a crystalline phase were found at the cathode or anode during the operation, although these results do not exclude the possible existence of non-crystalline products due to side reactions in the LAB system during the operation. Shui et al.17 observed the main reaction product in the cathode is Li2O2. While they reported the conversion of metallic Li to LiOH on the anode,16 we hardly observed crystalline LiOH. This discrepancy may be due to differences in the components such as electrolyte, and/or the difference in airtightness of the operando cell. (2) The XRD patterns for Li2O2 could be refined using the P63/mmc space group. In addition to the phase identification, some important structural parameters were calculated from the results of Rietveld analysis, such as formation/decomposition rate, anisotropic domain size, and lattice parameters. The full width at half maximum (FWHM) of XRD peaks in the c-direction is anisotropically broadened, unlike that of commercial bulk Li2O2. Details of the FWHM and relative intensity ratio of the XRD peaks of commercial bulk Li2O2 and discharge product Li2O2 are described in Section 3 of the ESI.† The details of analysis results for the series of XRD patterns are given in the following sections. From the such control of the experimental environments, which leads an accurate Rietveld analysis, we can discuss the truly real-time (operando) appearance and disappearance of peaks corresponding to the reaction product of crystalline Li2O2 in Fig. 2 and confirm that the electrochemical reaction proceeds dominantly according to eqn (1). In more detail in the evolution of Li2O2 as found in Fig. 2C and D, the change of peak intensity corresponding to the 100 reflection for the discharge process was clearly different to that for the charge process; the peak intensity of the 100 reflection increases linearly on discharge (Fig. 2C) whereas it decreases nonlinearly with two steps during charging (Fig. 2D). Similar decrease in peak intensities during the charge process was observed in the previous in situ XRD study,14 which reported that the peak intensities of the 100 and 101 reflections decrease nonlinearly with two steps. As it can be seen in Fig. S7 and Table S2 of the ESI,† the relative intensity ratio for 100 peaks hardly change and the FWHM for 100 peaks decrease during the charge process. The nonlinearity in the charge process implies that the decomposition of crystalline Li2O2 involves two stages and structural changes are greatly influenced by crystallographic direction during the charge process. Here, it should be noted that the 2nd step starts from around 2.28 mA h cm−2 corresponding to the charge voltage of 4 V. | ||
| Fig. 3 Rietveld refinement of a crystalline Li2O2 XRD pattern after full discharge (3.54 mA h cm−2) of the lithium–air battery. The black, red, and blue line represent the measured pattern (Iobs), the calculated pattern (Ical), and the difference (Iobs − Ical), respectively. | ||
3.4 Variation of the integrated and normalised areas of crystalline Li2O2 peaks
Fig. 4 shows the variation of the integrated and normalised areas of crystalline Li2O2 peaks in the 2θ range of 13.3–31° during the discharge/charge processes. Here, the integrated area was calculated by summing all areas of 13 Li2O2 peaks in the results of the Rietveld analysis of each XRD pattern, which were normalised by the integrated area of a fully discharged LAB (3.54 mA h cm−2). The error bars in Fig. 4 were presented by using RB. For a discharge process, the integrated and normalised areas of Li2O2 peaks increase linearly, without a change of slope, irrespective of current density (0.8, 0.6, and 0.4 mA cm−2 in Fig. 1). The extrapolated curves for the discharge process do not cross the origin (capacity = 0.114 mA h cm−2 for integrated and normalised areas = 0) (see Fig. S8 of the ESI†). This amount of charge is almost the same as 0.15 mA h cm−2 and is interpreted by discharge from the double-layer explained previously, thus it is reasonable that Li2O2 does not grow in the range from 0 to 0.114 mA h cm−2 because the electrical current in this region does not have electrochemical contribution (namely, Faraday current). It is also hard to conclude that Li2O2 exists in an amorphous state until 0.114 mA h cm−2, or some electrons from the Li anode in the LAB cell participate in a reaction other than Li2O2 formation. For the charge process, at first glance overall, the integrated and normalised areas of Li2O2 peaks also decrease linearly, which indicates that the reversible Li–O2 electrochemical reaction involving crystalline Li2O2 formation/decomposition occurs. A plateau region at the beginning of the charge process between 3.54 and 3.25 mA h cm−2, showing little change of integrated and normalised areas, can be explained again by charging to the double-layer as mentioned above. In more detail, the decreases in the integrated and normalised areas of Li2O2 peaks seem to accelerate slightly from the latter half of the charge process (below 2.28 mA h cm−2). As seen in the intensity change of 100 reflection shown in Fig. 2, it is confirmed again that the manner of electrochemical reaction switched to a somewhat different one after 2.28 mA h cm−2 during the charging process.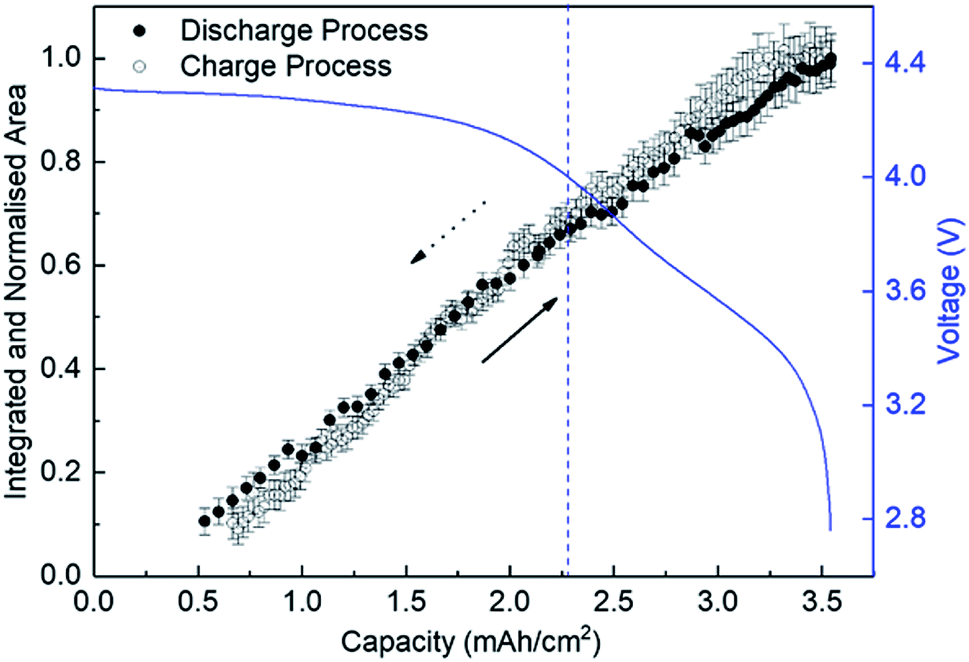 | ||
| Fig. 4 Integrated and normalised areas of crystalline Li2O2 peaks during the discharge and charge processes. The line-arrows and dotted-arrows in a black colour show the directions of discharge and charge processes, respectively. | ||
3.5 Variation of the domain size of crystalline Li2O2
Fig. 5A shows the variation of domain size of crystalline Li2O2 calculated from the FWHM of the 100, 101, and 004 peaks during the discharge/charge processes. The FWHM values are presented in Table S2 of the ESI.† The notable difference of FWHM values of 100, 101, and 004 peaks indicates that the crystalline Li2O2 possesses anisotropic morphology. The domain size was calculated from the Scherrer equation:
D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ
| (2) |
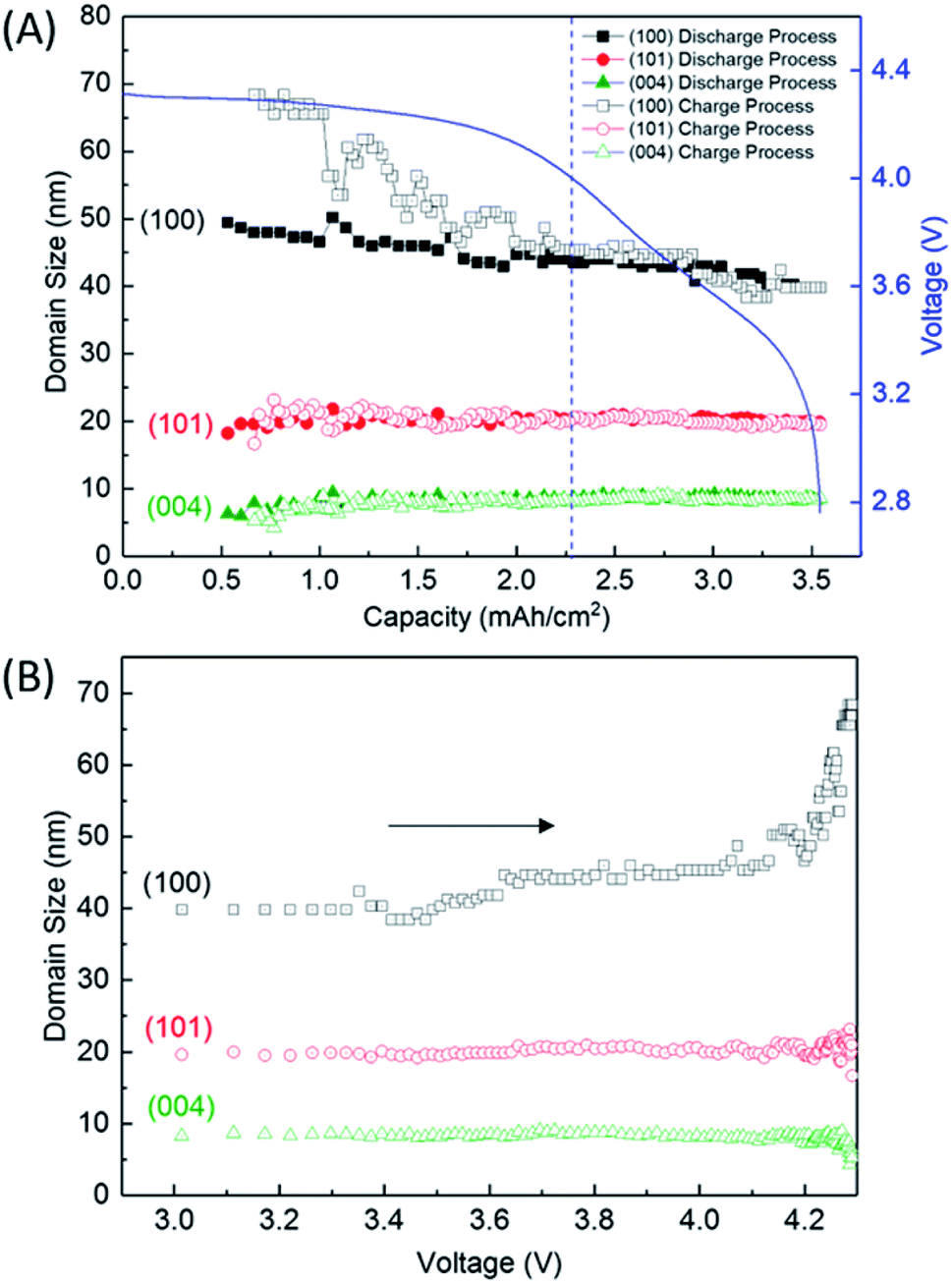 | ||
| Fig. 5 Domain sizes of crystalline Li2O2 (A) as a function of capacity during the discharge and charge processes and (B) as a function of voltage during the charge process, which was replotted by converting amount of charge in (A) to voltage using the charging profile. The direction of the arrows in (B) represents the flow of charge process. | ||
We finally discussed the variation of lattice constants of crystalline Li2O2 during the operation in Section 7 of the ESI.† The lattice constant of the a-axis has no variation during the discharge process. However, the lattice constant of the c-axis looks to increase gradually from 2.28 mA h cm−2 during the charge process. In addition to this result, the increase of domain size for the 100 reflection in Fig. 5 seems to be accompanied with the accelerated electrochemical decomposition of crystalline Li2O2 above 4 V for the charge process, as shown in Fig. 4. It is hard to explain such changes especially in domain size and lattice constant during the charging process under the assumption of the main electrochemical reaction described by eqn (1). It is natural to think that the path of electrochemical reaction involving Li2O2 switches to different one when the voltage exceeds the threshold of the electrochemically stable window of the electrolyte or carbon. In this case, 4 V might be the criterion voltage. As a result, the decomposition rate and the shape of domain of Li2O2 are affected. As a supporting evidence, the drastic evolution of CO2, instead of O2, was actually observed at voltage higher than 4 V during a charge process for a TEGDME-based Li–air battery cell, as shown in Fig. S11 of the ESI.† This result indicates the occurrence of electrochemical decomposition of a solvent or carbon cathode itself. Advanced analysing technique, which enables us to understand microscopically the chemical state of lithium, are needed to elucidate more precisely the detail of this electrochemical reaction.
Finally, it should be emphasized from the technical point of view that such small changes in the structural parameters of crystalline Li2O2 in a LAB cell in operation can be observed in real time using our operando XRD measurement system.
4 Conclusions
To elucidate the structural changes of crystalline Li2O2 and examine the LiOH formation in an operating LAB, operando synchrotron radiation-based XRD experiments in a transmission mode were conducted. The Li–O2 electrochemical reaction involving the formation and decomposition of crystalline Li2O2 was clearly demonstrated. Rietveld refinement analysis revealed that the domain size of anisotropic crystalline Li2O2 platelets, which were 10 nm in the c-direction and 40–70 nm in the ab-plane, was not influenced by the current density, and the growth of disc- and toroid-shaped Li2O2 particles was not accompanied with the growth of crystalline domains during the discharge process. No other reaction products with a crystalline phase as LiOH were observed in either the cathode or anode of LAB during the operation, whereas the accelerated electrochemical decomposition of the crystalline Li2O2 was accompanied with the increase of the lattice constant of the c-axis/the domain size for the 100 reflection and the drastic evolution of CO2 in voltage higher than 4 V of the charge process. This information about the structural changes within a non-aqueous LAB during an operation furthers our understanding of the nature of the Li–O2 electrochemical reaction in non-aqueous LABs, and should aid fabrication of high-performance non-aqueous LABs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Advanced Low Carbon Technology Research and Development program (ALCA) conducted by the Japan Science and Technology Agency (JST) and by the MEXT program for the development of environmental technology using nanotechnology. Synchrotron-based operando XRD measurements were carried out at SPring-8 with the approval of the Japan Synchrotron Radiation Institute under proposal No. 2012A4501, 2012A4502, 2014A4505, 2014B4500, 2015A4501, 2015B4501, 2016A4504 and 2016B4501.References
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1–5 CrossRef.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef PubMed.
- H.-G. Jung, J. Hassoun, J.-B. Park, Y.-K. Sun and B. Scrosati, Nat. Chem., 2012, 4, 579–585 CrossRef PubMed.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef PubMed.
- X. Xin, K. Ito and Y. Kubo, ACS Appl. Mater. Interfaces, 2017, 9, 25976–25984 CrossRef PubMed.
- Z. Ma, X. Yuan, L. Li, Z.-F. Ma, D. P. Wilkinson, L. Zhang and J. Zhang, Energy Environ. Sci., 2015, 8, 2144–2198 RSC.
- B. M. Gallant, D. G. Kwabi, R. R. Mitchell, J. Zhou, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 2518–2528 RSC.
- R. R. Mitchell, B. M. Gallant, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 2952–2958 RSC.
- R. Black, S. H. Oh, J.-H. Lee, T. Yim, B. Adams and L. F. Nazar, J. Am. Chem. Soc., 2012, 134, 2902–2905 CrossRef PubMed.
- S. A. Freunberger, Y. Chen, N. E. Drewett, L. J. Hardwick, F. Bardé and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 8609–8613 CrossRef PubMed.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng, Y. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050–1056 CrossRef PubMed.
- H. Lim, E. Yilmaz and H. R. Byon, J. Phys. Chem. Lett., 2012, 3, 3210–3215 CrossRef PubMed.
- S. Ganapathy, B. D. Adams, G. Stenou, M. S. Anastasaki, K. Goubitz, X.-F. Miao, L. F. Nazar and M. Wagemaker, J. Am. Chem. Soc., 2014, 136, 16335–16344 CrossRef PubMed.
- M. M. Storm, R. E. Johnsen, R. Younesi and P. Norby, J. Mater. Chem. A, 2015, 3, 3113–3119 RSC.
- J.-L. Shui, J. S. Okasinski, P. Kenesei, H. A. Dobbs, D. Zhao, J. D. Almer and D.-J. Liu, Nat. Commun., 2013, 4, 2255–2261 CrossRef PubMed.
- J.-L. Shui, J. S. Okasinski, C. Chen, J. D. Almer and D.-J. Liu, ChemSusChem, 2014, 7, 543–548 CrossRef PubMed.
- Y. Katsuya, C. Song, M. Tanaka, K. Ito, Y. Kubo and O. Sakata, Rev. Sci. Instrum., 2016, 87, 016106 CrossRef PubMed.
- C. Liu, W. R. Brant, R. Younesi, Y. Dong, K. Edström, T. Gustafsson and J. Zhu, ChemSusChem, 2017, 10, 1592–1599 CrossRef PubMed.
- C. Liu, D. Rehnlund, W. R. Brant, J. Zhu, T. Gustafsson and R. Younesi, ACS Energy Lett., 2017, 2, 2440–2444 CrossRef.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2014, 7, 50–56 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: LAB cell assembly and experimental set-up for operando SR-XRD measurements, high quality XRD patterns and Rietveld refinement analysis for a discharge product, FWHM comparison of commercial bulk Li2O2 and discharge product Li2O2, FWHM and domain size of crystalline Li2O2 during LAB operation, integrated and normalised areas of crystalline Li2O2 during the discharge process, estimation of the magnitude of instrumental broadening, lattice constants of crystalline Li2O2 during the discharge/charge processes, and O2, CO2 evolution during the charge process (Fig. S1–S11 and Tables S1 and S2). See DOI: 10.1039/c8ra04855j |
| This journal is © The Royal Society of Chemistry 2018 |