
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnti-tumor effect of HOTAIR–miR-613-SNAI2 axis through suppressing EMT and drug resistance in laryngeal squamous cell carcinoma
Jing-chun Zhoua,
Jing-jing Zhangb,
Wei Mac,
Wei Zhanga,
Zhao-yang Ke *a and
Ling-guo Maa
*a and
Ling-guo Maa
aDepartment of Otorhinolaryngology, The Second Clinical Medical College of Jinan University, Shenzhen People's Hospital, No. 1017 Dongmen North Road, Luohu District, Shenzhen, Guangdong 518020, China. E-mail: kezhaoyang2018@yeah.net; Tel: +86-755-25533018
bDepartment of Otorhinolaryngology, Peking University Shenzhen Hospital, Guangdong 518036, China
cTranslational Medicine Collaorative Innovation Center, The Second Clinical Medical College of Jinan University, Shenzhen People's Hospital, Guangdong 518020, China
First published on 23rd August 2018
Abstract
Laryngeal squamous cell carcinoma (LSCC) is the main pathological type of laryngeal cancer, which attacks the head and neck. Our present study aims to investigate the effect of long non-coding RNA (LncRNA) HOX transcript antisense RNA (HOTAIR) on epithelial mesenchymal transition (EMT) and drug resistance in LSCC. Firstly, the level of HOTAIR was found to be overexpressed in LSCC tissues compared with normal healthy tissues. Then, increased EMT and drug resistance were suppressed by specific HOTAIR shRNA effectively in LSCC cell lines. Besides, miR-613 was predicted to be a target of HOTAIR through bioinformatics analysis. Meanwhile, we found that a down-regulated level of miR-613 could be increased by HOTAIR shRNA and suppressed by LncRNA HOTAIR transfection in LSCC cells. The targeting relationship between miR-613 and HOTAIR was further demonstrated by a luciferase report assay. What is more, the inhibiting effect of HOTAIR shRNA on EMT and drug resistance was obviously abolished by the miR-613 inhibitor. Moreover, SNAI2, a critical regulator of EMT, was predicted as a target of miR-613 through bioinformatics analysis and luciferase report assays. As expected, the level of SNAI2 could be suppressed by HOTAIR shRNA and increased by the miR-613 inhibitor. Additionally, we discovered that SANI2 shRNA had similar inhibiting effect on EMT and drug resistance with HOTAIR shRNA in LSCC cells. Finally, the in vivo experiment further demonstrated that HOTAIR shRNA restricted tumor growth, EMT and drug resistance. Additionally, HOTAIR shRNA transfection could also increase the level of miR-613 and decrease the level of SNAI2 in vivo. Taken together, our research for the first time revealed the effect of the HOTAIR–miR-613-SNAI2 axis on EMT and drug resistance in LSCC, providing new targets for LSCC diagnosis and treatment.
1. Introduction
Laryngeal cancer (LC) is a common malignancy in the head and neck, which accounts for 5.7–7.6% of the malignant tumors in the whole world. According to statistics, LC has a higher incidence and mortality in rural areas than in the city and higher incidence and mortality in males than in females.1–3 Laryngeal squamous cell carcinoma (LSCC) is the main pathological type of LC and the incidence of LSCC is increasing year by year. What is more serious is that due to lack of suitable and specific biomarkers for early detection and monitoring of disease progress, 40% of the patients are diagnosed at stage III or IV.4,5 The traditional treatment for LSCC is total laryngectomy, which is accompanied by laryngeal function loss after operation, resulting in great harm to the quality of life. Advanced patients could just take chemotherapy and radiotherapy to kill cancer cells and prevent cancer from spreading, which bring increasing drug resistance as huge impediments to cancer treatment.6 Therefore, getting a better understanding of specific markers for early diagnosis and proper therapeutic targets is the top priority in LSCC research and treatment.Epithelial mesenchymal transition (EMT) refers to loss of cell polarity, tight junctions and adhesion between epithelial cells and transformation to stromal cells morphologically, endowing cancer cells stronger migration, invasion, proliferation and anti-apoptotic abilities. EMT has attracted much attention as a major inducer of cancer metastases during cancer progression. Besides that, large amount of studies have demonstrated that EMT is closely related to chemotherapy resistance.7,8 SNAI2, a zinc finger transcription factor, is a critical regulator of EMT. Previous studies have shown that SNAI2 is highly expressed in metastatic cancers and promotes the transformation of epithelium into interstitial cells, playing an essential role in the invasion and metastasis of epithelial tumor tissue.9 Additionally, as Hu F. Y. et al. reported, overexpressed miR-124 suppressed gefitinib resistance through down-regulating the expression of SNAI2 in non-small cell lung cancer.10 However, whether SNAI2 is associated with drug resistance in LSCC has not been explored.
Long non-coding RNAs (LncRNAs) are a large heterogeneous class of transcripts with more than 200 nucleotides but limited protein-coding potential.11 Up to now, only a small part of functional lncRNAs have been well characterized with diverse regulatory roles in cellular processes, such as cell proliferation and migration.12 Among them, LncRNA HOX transcript antisense RNA (HOTAIR) has been learned to play a vital role in carcinogenesis. As Xiao Z. et al. reported, LncRNA HOTAIR is a prognostic biomarker for the proliferation and chemoresistance of colorectal cancer by targeting miR-203a-3p via Wnt/β-Catenin signaling pathway.13 Additionally, HOTAIR has been demonstrated to be overexpressed in LSCC in previous reports.14 However, a full understanding the effect of overexpressed HOTAIR on drug resistance in LSCC is still far from being achieved.
MiRNAs are a class of non-coding RNA found in eukaryotes, and they act as important regulators in human cancer progression by binding their target mRNAs with the 3′un-translated region (3′UTR) of corresponding mRNAs.15 As Ding J. et al. reported, decreased expression of miR-223 enhanced doxorubicin sensitivity through suppressing EMT in colorectal cancer by targeting F-box and WD repeat domain-containing 7.16 Besides, up-regulated miR-613 was reported to weaken the resistance of triple-negative breast cancer cells against paclitaxel.17 Thus, in our present study, we explored the effect of miR-613 on drug resistance and associated regulation mechanism in LSCC.
In this study, we focused on HOTAIR, which was significantly up-regulated in LSCC tissues compared with the normal tissue. For the first time, we show that HOTAIR–miR-613-SNAI2 axis suppresses EMT-mediated drug resistance in LSCC in vitro and in vivo, providing new evidence that HOTAIR and miR-613 could function as key regulators in LSCC progression and hold great promise to be used as novel biomarkers and therapeutic targets in LSCC treatment.
2. Materials and methods
2.1 Tissue specimens and cell culture
Thirty LSCC samples and their corresponding no-tumor samples were obtained from The Second Clinical Medical College of Jinan University. The study was performed in accordance with the Helsinki Declaration and the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments were approved by the ethics committee of the Second Clinical Medical College of Jinan University. Informed consents were obtained from all the human participants of this study.LSCC cells Hep2 and TU686 cells were bought from the Cell Center of Life Science of Chinese Academy of Science (Shanghai, China).
Cisplatin (Selleckchem, Houston, TX, USA) was dissolved in DMSO and diluted to a final concentration of 0, 5, 10, 15, 20, 40 and 60 μM in cell viability detection. The cisplatin-resistant Hep2 cells (Hep2/R) and cisplatin-resistant TU686 cells (TU686/R) were established by stepwise exposure to increased concentrations of cisplatin as previously described.18 These cells were maintained in RPMI1640 medium (Gibco, NY, USA), supplemented with fetal bovine serum (FBS) (Hyclone, USA) and penicillin and streptomycin.
2.2 Transfection
Mimics/inhibitors specific for miR-613 and LncRNA-HOTAIR were designed and purchased from Invitrogen (USA). The Hep2/Hep2/R and TU686/TU686/R cells were seeded in 24-well plates at 1 × 105 cells per well. LncRNA-HOTAIR and HOTAIR short hairpin RNA (shRNA) was amplified using Primer STAR premix (TaKaRa) and cloned into lentivirus plasmid according to the manufacturer's protocol. Hep2/Hep2/R and TU686/TU686/R cells were transfected with recombinant lentivirus as indicated. Mimics (50 nM)/inhibitors (120 nM) specific for miR-613 were transfected into TU686/R cells cells using Lipofectamine 3000 (Invitrogen, USA). Three pairs of SNAI2 shRNA specific for SNAI2 were also designed and purchased from Invitrogen (USA), from which the best efficient one was chosen and was also transfected into TU686/R cells cells using Lipofectamine 3000 (Invitrogen, USA). Cells were harvested for subsequent experiments after transfection for 24 hours.2.3 Quantitative real-time PCR
Total RNA from cell lines and tumor samples were extracted by using TRIzol (Invitrogen, CA, USA) following to the manufacturer's instruction. cDNA was synthesized from 500 ng of total RNA using the PrimeScript RT Reagent Kit (TaKaRa) for detecting the level of mRNA and lncRNA. The expression of HOTAIR, miR-613, MDR1 was determined by quantitative real-time PCR by using the ABI PRISM 7900 Detection System (Applied Biosystems, Carlsbad, USA). U6 and 18 s were used as the internal control for miR-613 and HOTAIR, respectively. GAPDH was used as the control for MDR1. The relative expression of HOTAIR, miR-613 and MDR1 was calculated by using the 2−ΔΔCt method.2.4 Western blot analysis
Total protein from cells or tissues was extracted with cell lysis kit (Sigma-Aldrich, MO, USA) according to instruction and the protein concentration was valued by BCA reagent (Pierce, IL, USA). Equal protein was separated by 10% SDS-PAGE and transferred to the nitrocellulose membranes. After blocking with 5% milk, the membrane was incubated with corresponding antibodies (Santa Cruz, CA, USA). The protein signal was detected using an enhanced chemiluminescence (ECL) commercial kit (Amersham Biosciences) following the manufacturer's instructions.2.5 Immunofluorescence
The cells seeded in co focal dishes were fixed with 4% paraformaldehyde and then kept stable in 0.2% Triton for 10 min to rupture the cell membranes. Following three PBS washing, non-specific antigen-binding sites were blocked by 2% BSA for 30 min. The cells were then incubated with anti-vimentin, E-cadherin (Abcam, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200); anti-β-catenin (Proteintech, 1:50) overnight at 4 °C.
:200); anti-β-catenin (Proteintech, 1:50) overnight at 4 °C.
After washing, cells were incubated with anti-rabbit antibody for 60 min and the nuclei were stained with DAPI for 2 min, which was washed with PBS later. Cells were kept from light before observed with a fluorescence microscope.
2.6 Dual-luciferase reporter assay
Dual-luciferase reporter assay was performed as described previously. Briefly, the RNA sequence of HOTAIR and SNAI2 mRNA 3′-UTR containing the putative binding sites of miR-613 was inserted into pGL3 (Promega, Madison, WI, United States), generating the pGL3-HOTAIR or pGL3-SNAI2 wild-type/mutant-type luciferase reporter vector. TU686/R cells were seeded onto 6-well culture plates in DMEM medium containing 10% fetal bovine serum and cultured overnight. Then the cells were co-transfected with pGL3 wild-type/mutant-type vector (100 ng) and miR-613 mimics or control (50 nmol L−1) using Lipofectamine 2000 (Invitrogen). Luciferase activity assays were detected by a dual-luciferase reporter system according to the manufacturer (Promega, E2920).2.7 Xenograft nude mice assay
Specific pathogen-free (SPF) athymic nude mice (male, six to eight weeks of age) were purchased from Animal Care and Use Committee of The Second Clinical Medical College of Jinan University. All animal experiments were performed according to relevant national and international protocols and were approved by the Experimental Animal Center of The Second Clinical Medical College of Jinan University. The mice were fed under sterile-specific pathogen-free conditions with free access to food and water. Cells used for tumor formation were harvested, counted, and resuspended in PBS to a final concentration of 2 × 108 cells per mL. Trypan blue exclusion testing showed that the cells were >95% viable before injection. The mice were divided into 4 groups as follows: (1) control group: about 1 × 107 TU686/R cells were injected into the subcutaneous of mice. (2) HOTAIR shRNA group: about 1 × 107 TU686/R-LV-HOTAIR shRNA cells were injected into the subcutaneous of mice. (3) Cisplatin group: about 1 × 107 TU686/R cells were injected into the subcutaneous of mice. After the tumors had reached 100 mm3 in size, the mice of the cisplatin group were injected intraperitoneally twice per week with cisplatin (15 mg kg−1). (4) HOTAIR shRNA + cisplatin group: about 1 × 107 TU686/R-LV-HOTAIR shRNA cells were injected into the subcutaneous of mice. After the tumors had reached 100 mm3 in size, the mice of the cisplatin group were injected intraperitoneally twice per week with cisplatin (15 mg kg−1). Tumor volume was monitored every 5 days after the development of a palpable tumor and was assessed by measuring the 2 perpendicular dimensions using a caliper and the formula (a × b2)/2, where a is the larger and b is the smaller dimension of the tumor. At 30 days after inoculation, the mice were killed and tumor weights were assessed. Tumors from each mouse were randomly selected for immunohistochemical (IHC) analysis.2.8 Statistical analysis
All determinations were repeated in triplicate. All data are expressed as the mean ± standard deviation (SD) and assessed by using statistical software SPSS19.0. The Student's t test was used to compare means of two groups and One-Way ANOVA was used for comparing means of multiple samples. P < 0.05 was deemed to indicate significance.3. Results
3.1 HOTAIR is highly expressed in LSCC tissues and miR-613 is lowly expressed in LSCC tissues
To explore the role of HOTAIR and miR-613 in the progression of LSCC, the expression level of HOTAIR and miR-613 in LSCC tissues was detected through qPCR. As shown in Fig. 1A, relative expression of HOTAIR RNA was significantly increased (mean: 7-fold) in LSCC tissues compared with the normal tissues (***P < 0.001). At the same time, relative expression of miR-613 was largely decreased (mean: 2-fold) in LSCC tissues compared with the normal tissues (**P < 0.01) (Fig. 1B). These results above suggested that HOTAIR was highly expressed in LSCC tissues and miR-613 was lowly expressed in LSCC tissues.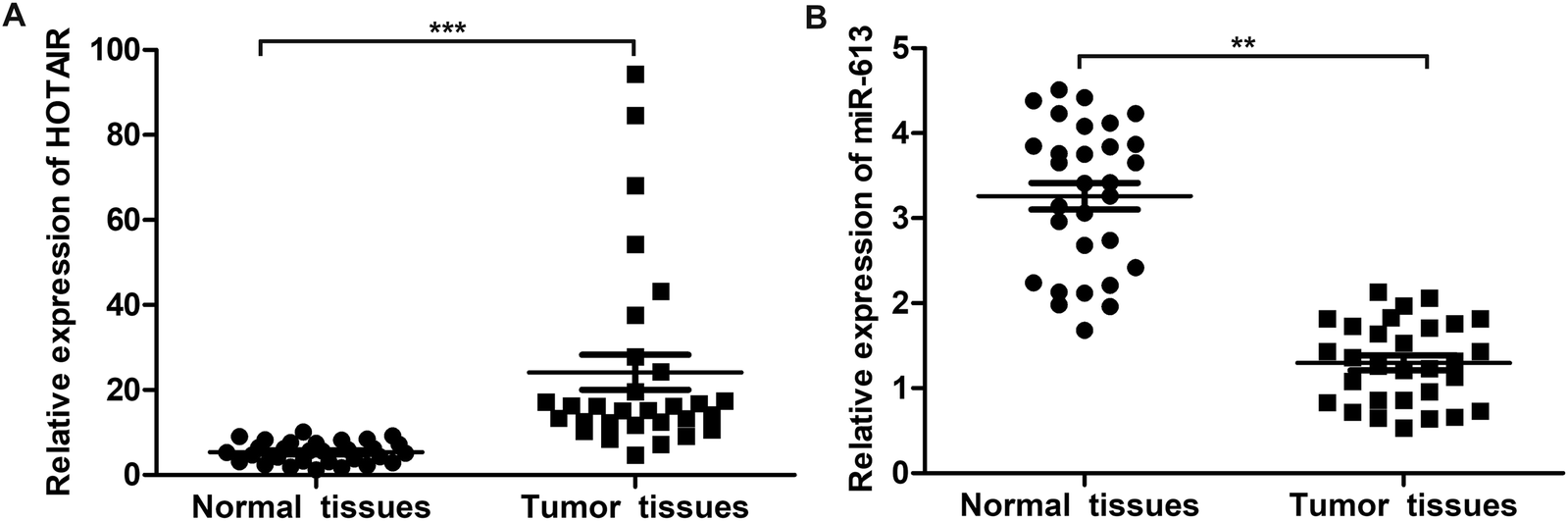 | ||
| Fig. 1 HOTAIR is highly expressed in LSCC tissues and miR-613 is lowly expressed in LSCC tissues. (A) The expression of HOTAIR in LSCC tissues and normal tissues was valued through qRT-PCR. (B) The expression of miR-613 in LSCC tissues and normal tissues was valued through qRT-PCR. The bars showed means ± SD of three independent experiments. **P < 0.01, ***P < 0.001 compared with normal tissues. | ||
3.2 HOTAIR shRNA suppresses EMT in LSCC cell lines
As shown in Fig. 2A, relative expression of HOTAIR was increased in Hep-2/R and TU686/R cells compared with normal Hep-2 and TU686 cells respectively, indicating that increased HOTAIR level was associated with elevated drug resistance. Then, specific HOTAIR shRNA was used to suppress HOTAIR expression in indicated cells (Fig. 2B). Then we found that EMT in Hep-2/R cells was induced accompanied by increased level of N-cadherin and decreased level of E-cadherin compared with the Hep-2 cells. However, the induced EMT was successfully suppressed by HOTAIR shRNA (Fig. 2C and D). The same phenomenon occurred in TU686 and TU686/R cells as shown in Fig. 2E and F. The result of immunocytochemical staining also showed that the expression of N-cadherin was elevated in Hep2/R cells compared with Hep2 cells but was then suppressed by HOTAIR shRNA in Hep2/R cells (Fig. 2G). The above results indicated the inhibiting effect of HOTAIR shRNA on EMT in LSCC cell lines.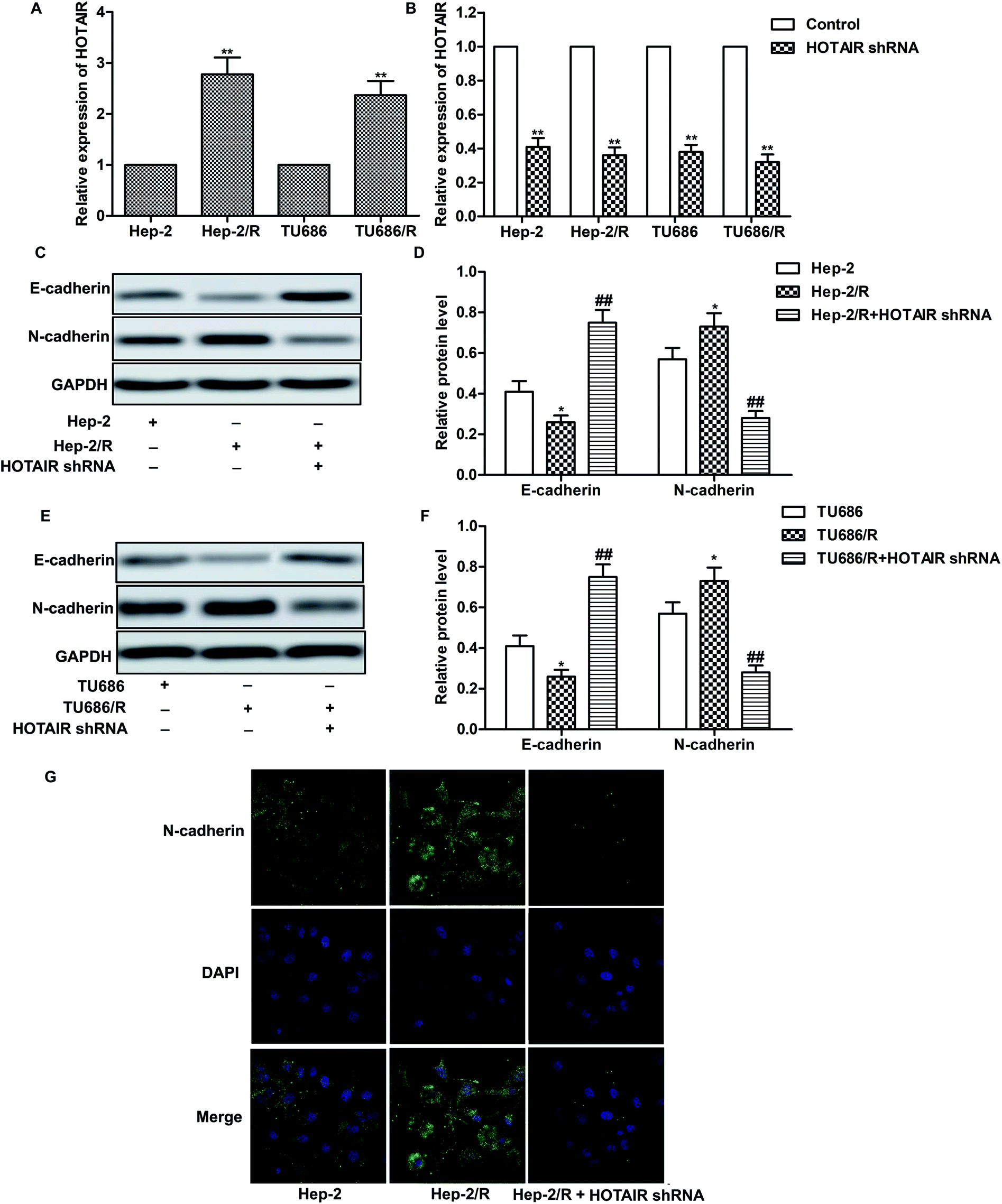 | ||
| Fig. 2 HOTAIR shRNA suppresses EMT in LSCC cell lines. (A) Relative expression of HOTAIR in Hep2/Hep2/R and TU686/TU686/R cells was valued through qRT-PCR (**P < 0.01 versus Hep2). (B) Hep2/Hep2/R and TU686/TU686/R cells were transfected with HOTAIR shRNA or left untreated, respectively. The level of HOTAIR was valued through qRT-PCR (**P < 0.01 versus control group). (C and D) The expression of E-cadherin and N-cadherin in Hep2 and Hep2/R cells or in HOTAIR shRNA treated Hep2/R cells was examined through western blot. Relative protein level of E-cadherin and N-cadherin was presented in the form of a histogram (*P < 0.05 versus Hep2 group, ##P < 0.01 versus Hep2/R group). (E and F) The expression of E-cadherin and N-cadherin in TU686 and TU686/R cells or in HOTAIR shRNA treated TU686/R cells was examined through western blot. Relative protein level of E-cadherin and N-cadherin was presented in the form of a histogram (*P < 0.05 versus TU686 group, ##P < 0.01 versus TU686/R group). GAPDH was used as a reference. (G) Immunocytochemical staining for N-cadherin and DAPI in Hep2 and Hep2/R cells or in HOTAIR shRNA treated Hep2/R cells. N-cadherin (green) and DAPI (blue) were observed. The bars showed means ± SD of three independent experiments. | ||
3.3 HOTAIR shRNA suppresses drug resistance in LSCC cell lines
A series of experiments were conducted to further explore the effect of HOTAIR shRNA on drug resistance in LSCC cell lines. As shown in Fig. 3A and C, the IC50 of Hep-2 and TU686 were both elevated from about 5 μM to more than 10 μM. The other way round, as shown in Fig. 3B and D, the IC50 of Hep-2/R and TU686/R was about 42 μM and 50 μM, respectively. Then, the transfection of HOTAIR shRNA obviously decreased the drug resistance to 18 μM and 14μM respectively, showing a strong inhibiting effect on drug resistance. Additionally, the results of western blot in Fig. 3E showed elevated expression level of MDR1 in both Hep-2/R and TU686/R cells compared with Hep-2 and TU686, but increased MDR1 was then largely suppressed by the transfection of HOTAIR shRNA. The above results indicated that HOTAIR shRNA could effectively suppress drug resistance in LSCC cell lines.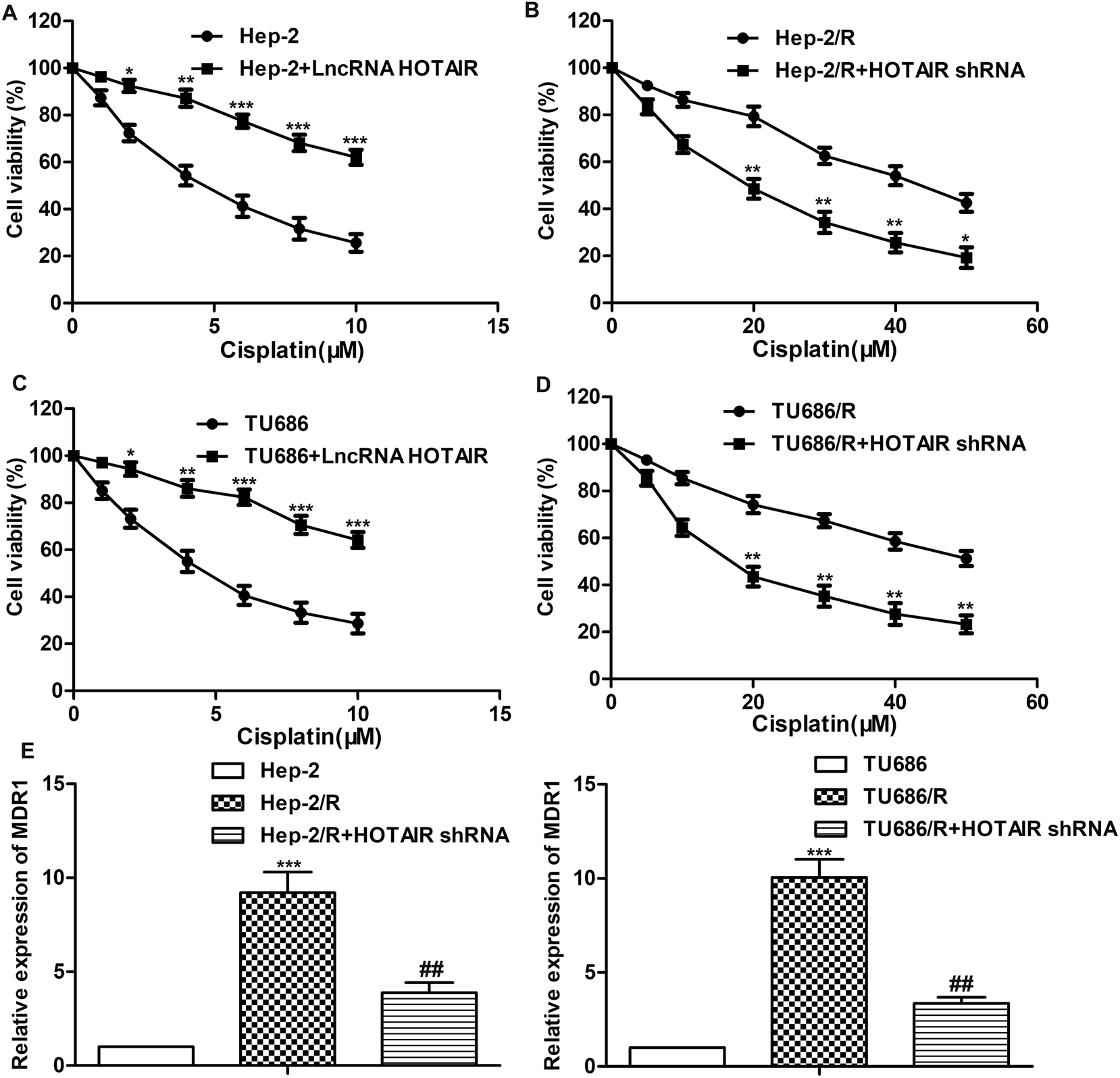 | ||
| Fig. 3 HOTAIR shRNA suppresses drug resistance in LSCC cell lines. (A) Cell viability in Hep2 cells and LncRNA HOTAIR transfected Hep2 cells was detected through MTT assay (*P < 0.05, **P < 0.01, ***P < 0.001 versus Hep2 group). (B) Cell viability in Hep2/R cells and HOTAIR shRNA treated Hep2/R cells was detected through MTT assay (*P < 0.05, **P < 0.01 versus Hep2/R group). (C) Cell viability in TU686 cells and LncRNA HOTAIR transfected TU686 cells was detected through MTT assay (*P < 0.05, **P < 0.01, ***P < 0.001 versus TU686 group). (D) Cell viability in TU686/R cells and HOTAIR shRNA treated TU686/R cells was detected through MTT assay (**P < 0.01 versus TU686/R group). (E) Relative expression of MDR1 in Hep2, Hep2/R cells and HOTAIR shRNA treated Hep2/R cells was detected through qRT-PCR (***P < 0.001 versus Hep2 group, ##P < 0.01 versus Hep2/R group). (F) Relative expression of MDR1 in TU686, TU686/R cells and HOTAIR shRNA treated TU686/R cells was detected through qRT-PCR (***P < 0.001 versus TU686 group, ##P < 0.01 versus TU686/R group). The bars showed means ± SD of three independent experiments. | ||
3.4 MiR-613 is a target of HOTAIR in LSCC cell lines
Having learned the effects of HOTAIR on EMT and drug resistance in LSCC cell lines, the associated regulatory mechanism was further explored. The result of bioinformatics analysis showed that there existed complementary sites of miR-613 in HOTAIR RNA (Fig. 4A). To further investigate the relationship between the two, specific miR-613 mimic and inhibitor were used in our following experiments. Firstly, the results of MTT assay showed that there was no significant cytotoxicity of miR-613 mimic and inhibitor in our indicated experiments (Fig. 4B). The expression of miR-613 was obviously elevated or suppressed by transfecting the Hep-2/R cells with miR-613 mimic or miR-613 inhibitor, respectively (Fig. 4C). As shown in Fig. 4D and E, relative expression of miR-613 was elevated by HOTAIR shRNA and was then suppressed by the co-transfection with miR-613 inhibitor (Fig. 4D). Contrarily, the level of miR-613 could also be suppressed by overexpression of HOTAIR and was then increased by adding miR-613 mimic in HOTAIR transfected cells (Fig. 4E). In general, the level of miR-613 was suppressed by HOTAIR and was elevated by HOTAIR shRNA. In addition, luciferase reporter assays further demonstrated the targeting relationship between HOTAIR and miR-613 by showing that relative luciferase activity in HOTAIR WT – miR-613 mimic recombinant vector was significantly decreased compared with control group (Fig. 4F). Beyond that, we found that the inhibiting effect of HOTAIR shRNA on EMT and MDR1 induction was abolished by the combination with miR-613 inhibitor (Fig. 4G–I). The above results revealed that miR-613 inhibitor had an opposite effect on EMT and drug resistance with HOTAIR shRNA and miR-613 was a target of HOTAIR in LSCC cell lines.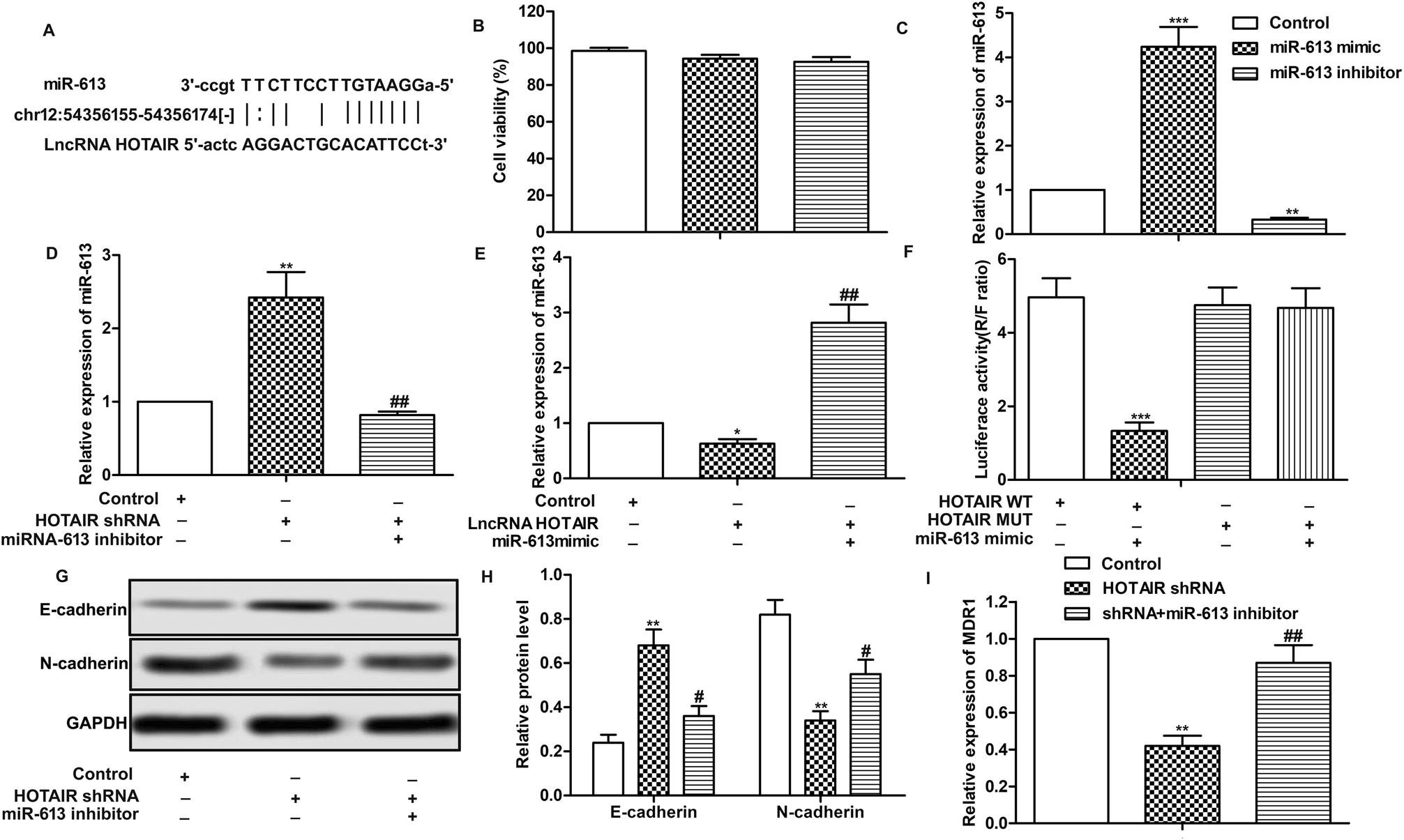 | ||
| Fig. 4 MiR-613 is a target of HOTAIR in LSCC cell lines. (A) Target sequences of miR-613 in HOTAIR mRNA were analysed through bioinformatics. (B) Hep2/R cells were transfected with miR-613 mimic or miR-613 inhibitor or just left untreated. Cytotoxicities of miR-613 mimic or miR-613 inhibitor were detected through MTT assay. (C) Relative expression of miR-613 was detected through qRT-PCR (**P < 0.01, ***P < 0.001 versus control group). (D) Hep2/R cells were transfected with HOTAIR shRNA or co-transfected with miR-613 inhibitor or left untreated. Relative expression of miR-613 was detected through qRT-PCR (**P < 0.01 versus control group,##P < 0.01 versus HOTAIR shRNA group). (E) Hep2/R cells were transfected with LncRNA HOTAIR or co-transfected with miR-613 mimic or left untreated. Relative expression of miR-613 was detected through qRT-PCR (**P < 0.01 versus control group, ##P < 0.01 versus HOTAIR group). (F) Luciferase reporter assay was used to detect the luciferase activity in TU686/R cells co-transfected with HOTAIR Wt/Mut and miR-138 mimics (***P < 0.001). (G and H) The expression of E-cadherin and N-cadherin in Hep2/R cells transfected with HOTAIR shRNA or co-transfected with miR-613 inhibitor was examined through western blot. Relative protein level of E-cadherin and N-cadherin was presented in the form of a histogram (**P < 0.01 versus control group, #P < 0.05 versus HOTAIR shRNA group). GAPDH was used as a reference. (I) Relative expression of MDR1 in Hep2/R cells transfected with HOTAIR shRNA or co-transfected with miR-613 inhibitor or the control cells was valued through qRT-PCR (**P < 0.01 versus control group, ##P < 0.05 versus HOTAIR shRNA group). The bars showed means ± SD of three independent experiments. | ||
3.5 SNAI2 is a target of miR-613 in LSCC cell lines
Further investigation was conducted to find the down-stream targets of miR-613. As shown in Fig. 5A, relative expression of SNAI2 was highly up-regulated in LSCC tissues compared with normal tissues. The results of bioinformatics analysis revealed that there existed complementary sites of SNAI2 in miR-613 RNA (Fig. 5B). As shown in Fig. 5C and D, the level of SNAI2 was suppressed by HOTAIR shRNA and was elevated by miR-613 inhibitor. Besides that, luciferase reporter assays further demonstrated the targeting relationship between SNAI2 and miR-613 as the combination of SNAI2 WT and miR-613 mimic brought an obvious decrease in luciferase activity compared with other groups (Fig. 5E). Moreover, trend lines in Fig. 5F–H displayed that the expression of SNAI2 was positively correlated with the level of HOTAIR in LSCC tissues. At the same time, the level of miR-613 was negatively correlated with HOTAIR and SNAI2. Thus, we concluded that SNAI2 was a target of miR-613 in Hep-2/R cells. Specific shRNA was used here to knock-down the expression of SNAI2 in Hep-2/R cells (Fig. 5I and J). As expect, we found that SNAI2 shRNA had the same inhibiting effect on EMT induction by reducing the transformation of E-cadherin into N-cadherin as HOTAIR shRNA (Fig. 5K and L). At the same time, the level of MDR1 was suppressed by SNAI2 shRNA (Fig. 5M). Drug resistance was also decreased by SNAI2 shRNA through decreasing the IC50 of Hep-2/R from 45 μM to 18 μM (Fig. 5N). Taken together, we concluded that SNAI2 shRNA had inhibiting effect on EMT and drug resistance and HOTAIR shRNA exerted its inhibiting effect on EMT and drug resistance through the HOTAIR/miR-613/SNAI2 axis in LSCC cell lines.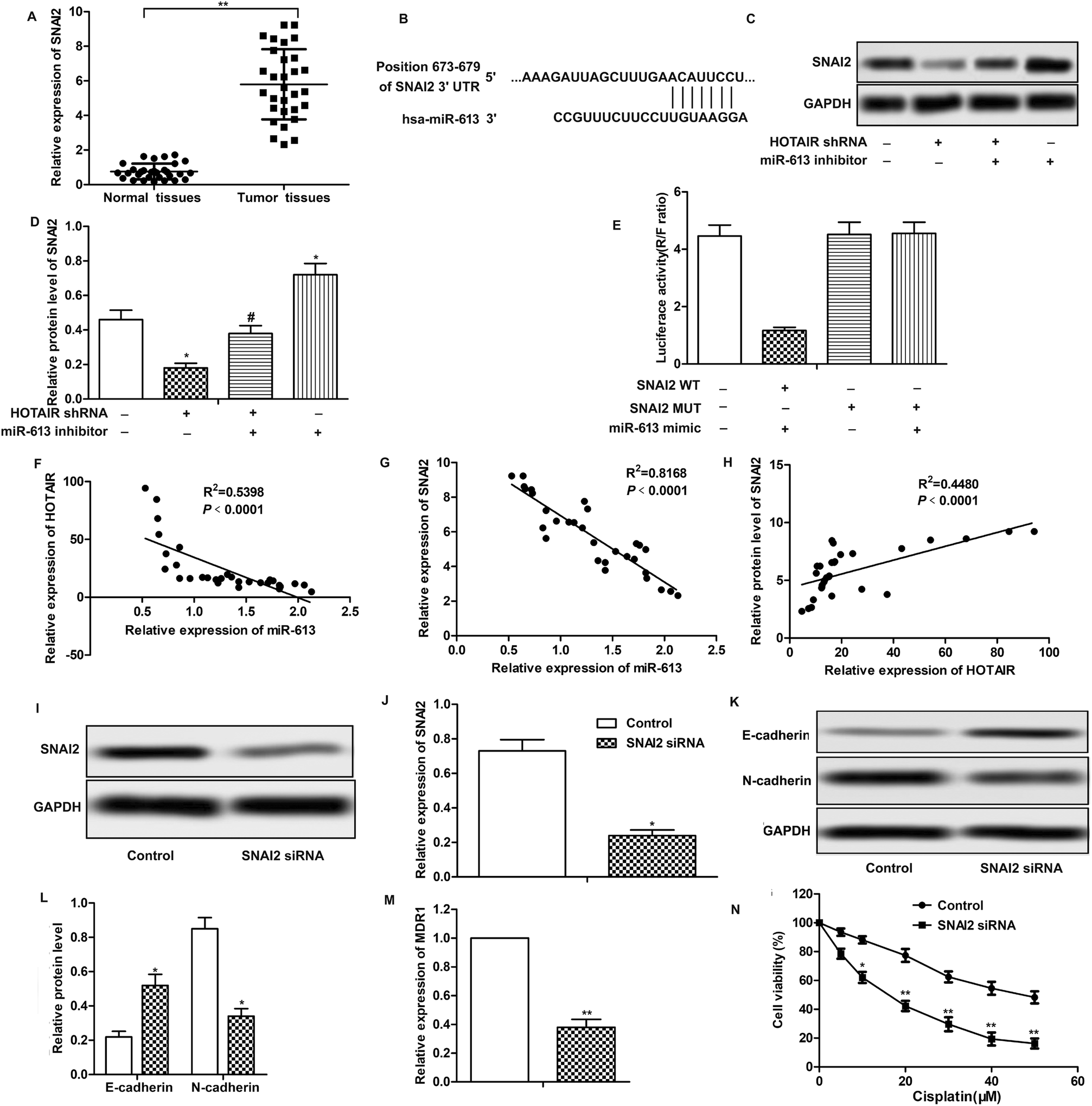 | ||
| Fig. 5 SNAI2 is a target of miR-613 in LSCC cell lines. (A) The expression of SNAI2 in LSCC tissues and normal tissues was valued through qRT-PCR (**P < 0.01 versus with normal tissues.). (B) Target sequences of SNAI2 in miR-613 mRNA were analysed through bioinformatics. (C and D) Specific shRNA targeting SNAI2 was transfected into Hep2/R cells. The expression of SNAI2 in TU686/R cells transfected with SNAI2 shRNA was examined through western blot. Relative protein level of SNAI2 was presented in the form of a histogram (*P < 0.05 versus control group). (E and F) The expression of SNAI2 in HOTAIR shRNA and/or miR-613 inhibitor transfected Hep2/R cells was detected through western blot. Relative protein level of SNAI2 was presented in the form of a histogram (*P < 0.05, **P < 0.01 compared with control group). GAPDH was used as a reference. (G–I) Trend lines between miRNA-613 and HOTAIR (G), miRNA-613 and SNAI2 (H), HOTAIR and SNAI2 (I) in 30 LSCC tissues samples used in our research. (J and K) The expression of E-cadherin and N-cadherin in Hep2/R cells transfected with SNAI2 shRNA or in the control cells was examined through western blot. Relative protein level of E-cadherin and N-cadherin was presented in the form of a histogram (*P < 0.05 versus control group). GAPDH was used as a reference. (L) Relative expression of MDR1 in Hep2/R cells transfected with SNAI2 shRNA or in the control cells was examined through qRT-PCR (**P < 0.01 versus control group). (M) Cell viability in Hep2/R cells or in SNAI2 shRNA treated Hep2/R cells was valued through MTT assay. The bars showed means ± SD of three independent experiments. | ||
3.6 HOTAIR shRNA suppresses tumor metastasis and EMT and drug resistance in vivo
Effects of HOTAIR shRNA and its related regulatory mechanism have been investigated in vitro, so corresponding in vivo experiments were conducted in our further study. As shown in Fig. 6A, there was no obvious inhibitory effect of cisplatin treatment on tumor growth compared with the control group. However, tumor growth was significantly suppressed by HOTAIR shRNA treatment compared with the control group. Besides that, a better inhibitory effect on tumor growth was exhibited by the combination of HOTAIR shRNA and cisplatin compared with the cisplatin group individually. The results of western blot showed that HOTAIR shRNA obviously inhibited EMT by elevating the level of E-cadherin and suppressing the level of N-cadherin compared with the control group (Fig. 6B and C). Moreover, the expression of VEGF in the HOTAIR shRNA group was also strongly limited compared with the control group detected through IHC analysis (Fig. 6D and E). In addition, the level of miR-613 was increased by HOTAIR shRNA and the level of SNAI2 was decreased by HOTAIR shRNA in vivo in accord with our in vitro experiments (Fig. 6F–H). Thus, we concluded that HOTAIR shRNA suppressed tumor metastasis and EMT-mediated drug resistance in vivo.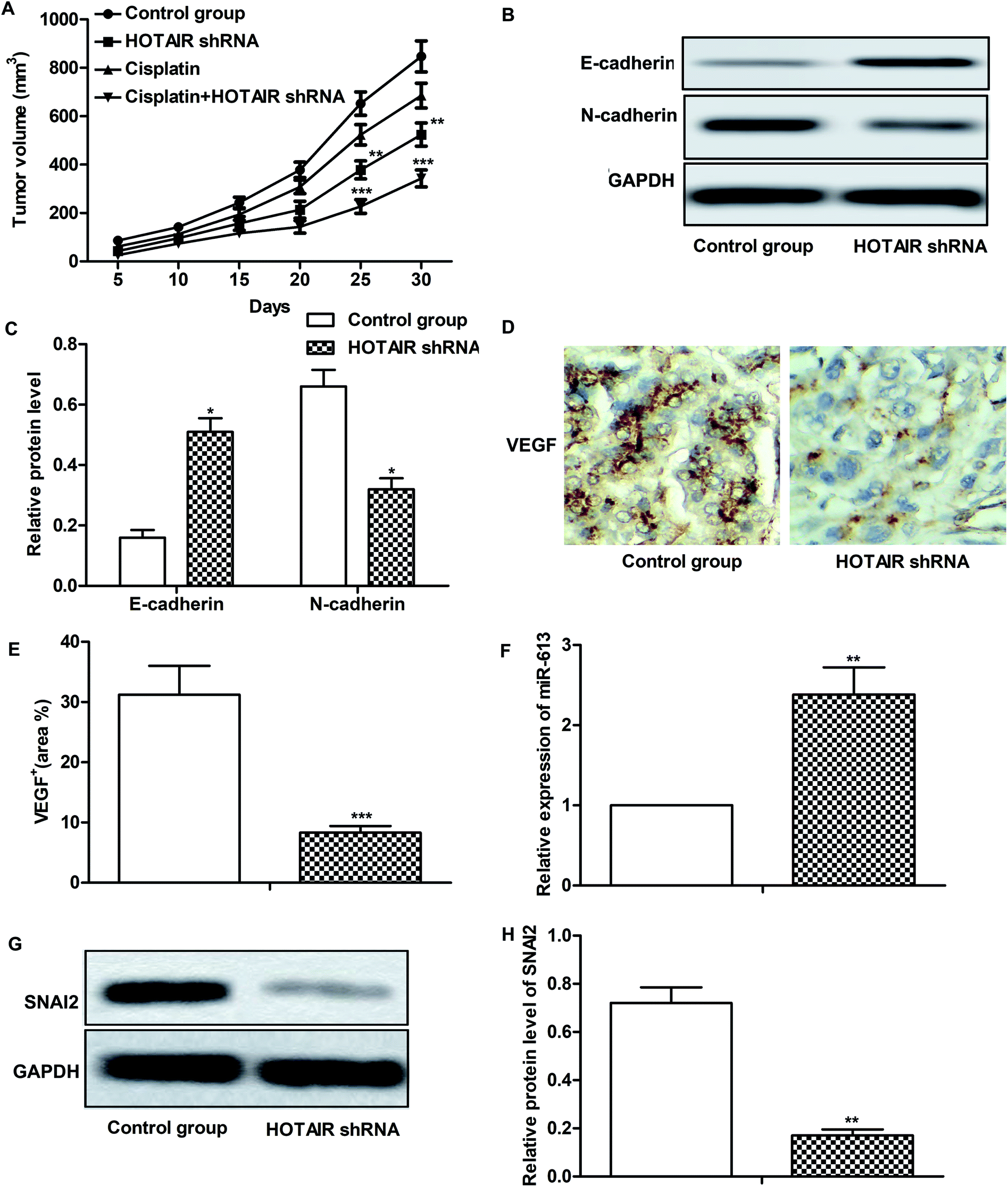 | ||
| Fig. 6 HOTAIR shRNA suppresses tumor metastasis and EMT-mediated drug resistance in vivo. The mice were divided into 4 groups as previously described. (A) Tumor volume and growth trend in mice from 4 groups was showed. (B and C) The expression of E-cadherin and N-cadherin in tumor tissues from control group and HOTAIR shRNA group was examined through western blot. GAPDH was used as a reference. Relative protein level of E-cadherin and N-cadherin was presented in the form of a histogram (*P < 0.05 versus control group). (D and E) Expression of migration markers VEGF in formalin-fixed, paraffin-embedded tumors from control group and HOTAIR shRNA group mice was detected through IHC analysis. The VEGF+ was counted and was represented in the form of a histogram (***P < 0.001 versus control group). (F) Relative expression of miR-613 in tumor tissues from control group and HOTAIR shRNA group was valued through qRT-PCR (**P < 0.01 versus control group). (G and H) The expression of SNAI2 in tumor tissues from control group and HOTAIR shRNA group was valued through western blot and was presented in the form of a histogram (**P < 0.01 versus control group). GAPDH was used as a reference. The bars showed means ± SD of three independent experiments. | ||
4. Discussion
In recent years, research on LSCC is becoming more and more deepened, but the present status of research progress is far from satisfactory with poor 5 year survival rate, which seriously affects postoperative quality of life. Besides that, tumor metastasis and increasing drug resistance have brought great difficulties for the treatment of LSCC. At present, the methods using in evaluating cancer cells invasion and metastasis are far off the required standards in clinical diagnosis and treatment. Therefore, it is necessary to find accurate molecular markers associated with cancer metastasis and drug resistance of LSCC, which are pretty important to guide operation and postoperative adjuvant therapy so as to improve the survival rate and quality of life of the patients.Amounts of research have demonstrated that LncRNAs have developmental and tissue-specific expression patterns, and their aberrant expression are closely associated with the progression of a variety of diseases, including cancer.19 HOTAIR, a 2.2 kb LncRNA binding to the PRC2 and the LSD1 complexes, is transcribed from the HOXC locus. HOTIR is found pervasively overexpressed in most human cancers compared with normal tissues, such as non-small cell lung cancer,20 breast cancer21 and gastric cancer22 et al. Elevated HOTAIR expression has been revealed to be associated with tumorigenesis, metastasis, and drug resistance.20 In addition, Zheng J. et al. reported in their study that suppressed HOTAIR expression increased the sensitivity to cis-platinum of the LSCC cells.23 However, the potential molecular mechanisms have not been explored completely clear till now. In our present study, high expression of HOTAIR was also detected in LSCC tissues compared with normal tissues. Besides that, higher HOTAIR level was found in Hep-2/R and TU686/R cells compared with normal Hep-2 and TU686 cells, identifying that higher HOTAIR expression is associated with drug resistance in LSCC. As expect, our further investigation revealed that suppressed HOTAIR by specific shRNA obviously inhibited EMT and drug resistance in LSCC cell lines.
Large amounts of studies have showed that EMT is closely associated with drug resistance. Thus, we speculated that HOTAIR suppressed drug resistance via inhibiting EMT in LSCC cells. Further study was conducted to explore the related regulation mechanism. As shown in our study, miR-613 was lowly expressed in LSCC tissues compared with the normal tissues. Besides that, the results of bioinformatics analysis showed that miR-613 was predicted to be a downstream target of HOTAIR. Just as Ma J. et al. reported, HOTAIR affected the progression of oesophageal squamous cell carcinoma by regulating HK2 expression through binding to endogenous miR-125 and miR-143.24 We also conjectured that HOTAIR played its regulation effect through binding to miR-613. Then we found that the level of miR-613 was suppressed by LncRNA HOTAIR and was elevated by HOTAIR shRNA. The results of luciferase assay showed that overexpressed miR-613 significantly restrained the intensity of fluorescence signal by binding with HOTAIR WT compared with other groups. Besides that, the inhibiting effect of HOTAIR shRNA on EMT and the expression of MDR1 were largely abolished by the combination of miR-613 inhibitor with HOTAIR shRNA. Thus, we concluded that miR-613 was a target of HOTAIR in LSCC cells.
The zinc finger transcription factor SNAI2 (also known as slug) is one of the members of SNAI/SLUG superfamily. SNAI2 implicates in the pathogenesis of different cancers to inhibit protein transcription and induce EMT by binding to the E-box of the target gene promoter, making the tumor cells migratory and invasive.25 Abnormal expression of SNAI2 is also found in prostate cancer,26 colorectal cancer27 and some other cancers. However, the effect of SANI2 has not been explored in LSCC up to now. In our present study, we for the first time revealed that SANI2 was highly expressed in LSCC tissues compared with normal tissues. Mounting researches have revealed that cytoplasmic lncRNAs function as ceRNAs to compete with miRNAs from their mRNA targets.28 Just as Yuan J. H. et al. reported, LncRNA-ATB elevated the expression of ZEB1 and ZEB2 by competitively binding the miR-200 family and induced EMT and invasion in hepatocellular carcinoma.29 Similarly, in our study, we found that there also existed target sequences of SANI2 3′-UTR in miR-613 mRNA. At the same time, the expression of SANI2 was positively correlated with HOTAIR level and negatively correlated with miR-613 level. Luciferase activity assay further demonstrated the targeting relationship between miR-613 and SANI2. What is more, SANI2 shRNA has the same inhibiting effect on EMT and drug resistance as HOTAIR shRNA. Taken together, we concluded that HOTAIR shRNA acted as a tumor suppressor in LSCC through the HOTAIR/miR-613/SNAI2 axis.
Inhibiting effect of HOTAIR on cell EMT and drug resistance in vitro has been identified, thus we further explored the effect of HOTAIR shRNA in vivo. In previous reports, decreased expression HOTAIR has been demonstrated to suppress the growth of neck squamous cell cancer (HNSCC) growth in vivo.30 Besides that, decreased HOTAIR level has the ability to sensitize HNSCC to cisplatin in vitro and in vivo.31 Similarly, in our present study, we found that HOTAIR shRNA largely suppressed LSCC growth and metastases by inhibiting EMT in vivo. As we demonstrated in vitro, HOTAIR shRNA decreased the level of miR-613 and increased the level of SNAI2 in vivo. These results above indicated that HOTAIR shRNA could inhibit metastases and drug resistance of LSCC by regulating miR-613 and SNAI2 in vivo.
In conclusion, our research found that HOTAIR was overexpressed in LSCC tissues and HOTAIR shRNA suppressed EMT and drug resistance of LSCC in vitro. Further researches revealed that HOTAIR shRNA down-regulated the expression of SNAI2 to suppress EMT and drug resistance of LSCC cells by targeting miR-613. Moreover, HOTAIR shRNA was identified to suppress LSCC growth, EMT and drug resistance in vivo. Our research is the first to establish the possible link between SNAI2 and EMT/drug resistance in LSCC. The HOTAIR-miR-613-SNAI2 axis will provide new targets for LSCC diagnosis and treatment.
Conflicts of interest
The authors have declared that no competing interest exists.Abbreviations
| LC | Laryngeal cancer |
| LSCC | Laryngeal squamous cell carcinoma |
| LncRNAs | Long non-coding RNAs |
| HOTAIR | HOX transcript antisense RNA |
| EMT | Epithelial mesenchymal transition |
| UTR | Un-translated region |
| HOTAIR | HOX transcript antisense RNA |
| HPV | Human papillomavirus |
Acknowledgements
This work was funded by Shenzhen People's Hospital (No: SYJY201802).References
- J. Perez-Escuredo, R. K. Dadhich, S. Dhup, A. Cacace, V. F. Van Hee, C. J. De Saedeleer, M. Sboarina, F. Rodriguez, M. J. Fontenille, L. Brisson, P. E. Porporato and P. Sonveaux, Cell Cycle, 2016, 15, 72–83 CrossRef PubMed.
- M. Adeva-Andany, M. Lopez-Ojen, R. Funcasta-Calderon, E. Ameneiros-Rodriguez, C. Donapetry-Garcia, M. Vila-Altesor and J. Rodriguez-Seijas, Mitochondrion, 2014, 17, 76–100 CrossRef PubMed.
- S. S. Zhang, Q. M. Xia, R. S. Zheng and W. Q. Chen, J. Cancer Res. Ther., 2015, 11(suppl 2), C143–C148 Search PubMed.
- D. G. Pfister, S. A. Laurie, G. S. Weinstein, W. M. Mendenhall, D. J. Adelstein, K. K. Ang, G. L. Clayman, S. G. Fisher, A. A. Forastiere, L. B. Harrison, J. L. Lefebvre, N. Leupold, M. A. List, B. O. O'Malley, S. Patel, M. R. Posner, M. A. Schwartz and G. T. Wolf, Am. J. Clin. Oncol., 2006, 24, 3693–3704 CrossRef PubMed.
- E. Morse and R. J. T. Fujiwara, Laryngoscope, 2018 DOI:10.1002/lary.27247.
- X. Lv, D. M. Song, Y. H. Niu and B. S. Wang, Apoptosis, 2016, 21, 489–501 CrossRef PubMed.
- Y. X. Zhao and Z. F. Sun, Journal of Clinical Otorhinolaryngology Head and Neck Surgery, 2017, 31, 888–891 Search PubMed.
- Z. Cai, Y. Cao, Y. Luo, H. Hu and H. Ling, Clinica Chimica Acta, 2018, 483, 156–163 CrossRef PubMed.
- G. Y. Li, W. Wang, J. Y. Sun, B. Xin, X. Zhang, T. Wang, Q. F. Zhang, L. B. Yao, H. Han, D. M. Fan, A. G. Yang, L. T. Jia and L. Wang, Theranostics, 2018, 8, 2846–2861 CrossRef PubMed.
- F. Y. Hu, X. N. Cao, Q. Z. Xu, Y. Deng, S. Y. Lai, J. Ma and J. B. Hu, J. Huazhong Univ. Sci. Technol., Med. Sci., 2016, 36, 839–845 CrossRef PubMed.
- J. L. Rinn and H. Y. Chang, Annu. Rev. Biochem., 2012, 81, 145–166 CrossRef PubMed.
- A. M. Schmitt and H. Y. Chang, Cancer Cell, 2016, 29, 452–463 CrossRef PubMed.
- Z. Xiao, Z. Qu, Z. Chen, Z. Fang, K. Zhou, Z. Huang, X. Guo and Y. Zhang, Cell. Physiol. Biochem., 2018, 46, 1275–1285 CrossRef PubMed.
- D. Li, J. Feng, T. Wu, Y. Wang, Y. Sun, J. Ren and M. Liu, Am. J. Pathol., 2013, 182, 64–70 CrossRef PubMed.
- J. Godlewski, M. O. Nowicki, A. Bronisz, S. Williams, A. Otsuki, G. Nuovo, A. Raychaudhury, H. B. Newton, E. A. Chiocca and S. Lawler, Cancer Res., 2008, 68, 9125–9130 CrossRef PubMed.
- J. Ding, Z. Zhao, J. Song, B. Luo and L. Huang, Acta Biochim. Biophys. Sin., 2018, 50(6) DOI:10.1093/abbs/gmy040.
- H. Xiong, T. Yan, W. Zhang, F. Shi, X. Jiang, X. Wang, S. Li, Y. Chen, C. Chen and Y. Zhu, Cell. Signalling, 2018, 44, 33–42 CrossRef PubMed.
- Z. Li, S. Hu, J. Wang, J. Cai, L. Xiao, L. Yu and Z. Wang, Gynecol. Oncol., 2010, 119, 125–130 CrossRef PubMed.
- A. Guffanti, M. Iacono, P. Pelucchi, N. Kim, G. Solda, L. J. Croft, R. J. Taft, E. Rizzi, M. Askarian-Amiri, R. J. Bonnal, M. Callari, F. Mignone, G. Pesole, G. Bertalot, L. R. Bernardi, A. Albertini, C. Lee, J. S. Mattick, I. Zucchi and G. De Bellis, BMC Genomics, 2009, 10, 163 CrossRef PubMed.
- M. Y. Liu, X. Q. Li, T. H. Gao, Y. Cui, N. Ma, Y. Zhou and G. J. Zhang, J. Thorac. Dis., 2016, 8, 3314–3322 CrossRef PubMed.
- H. He, Z. Wei, F. Du, C. Meng, D. Zheng, Y. Lai, H. Yao, H. Zhou, N. Wang, X. G. Luo, W. Ma and T. C. Zhang, Cell. Signalling, 2017, 31, 87–95 CrossRef PubMed.
- J. Yan, Y. Dang, S. Liu, Y. Zhang and G. Zhang, Tumor Biol., 2016 DOI:10.1007/s13277-016-5448-5.
- J. Zheng, X. Xiao, C. Wu, J. Huang, Y. Zhang, M. Xie, M. Zhang and L. Zhou, Acta Oto-Laryngol., 2017, 137, 90–98 CrossRef PubMed.
- J. Ma, Y. Fan, T. Feng, F. Chen, Z. Xu, S. Li, Q. Lin, X. He, W. Shi, Y. Liu, X. Cao, B. Zhu and Z. Liu, Oncotarget, 2017, 8, 86410–86422 Search PubMed.
- F. Tao, X. Tian and Z. Zhang, Oncotarget, 2018, 9, 12212–12225 CrossRef PubMed.
- X. Tian, F. Tao, B. Zhang, J. T. Dong and Z. Zhang, IUBMB Life, 2018, 70(3), 224–236 CrossRef PubMed.
- E. A. Pudova, A. V. Kudryavtseva, M. S. Fedorova, A. R. Zaretsky, D. S. Shcherbo, E. N. Lukyanova, A. Y. Popov, A. F. Sadritdinova, I. S. Abramov, S. L. Kharitonov, G. S. Krasnov, K. M. Klimina, N. V. Koroban, N. N. Volchenko, K. M. Nyushko, N. V. Melnikova, M. A. Chernichenko, D. V. Sidorov, B. Y. Alekseev, M. V. Kiseleva, A. D. Kaprin, A. A. Dmitriev and A. V. Snezhkina, BMC Genomics, 2018, 19, 113 CrossRef PubMed.
- F. A. Karreth and P. P. Pandolfi, Cancer Discovery, 2013, 3, 1113–1121 CrossRef PubMed.
- J. H. Yuan, F. Yang, F. Wang, J. Z. Ma, Y. J. Guo, Q. F. Tao, F. Liu, W. Pan, T. T. Wang, C. C. Zhou, S. B. Wang, Y. Z. Wang, Y. Yang, N. Yang, W. P. Zhou, G. S. Yang and S. H. Sun, Cancer Cell, 2014, 25, 666–681 CrossRef PubMed.
- S. Sun, Y. Wu, W. Guo, F. Yu, L. Kong, Y. Ren, Y. Wang, X. Yao, C. Jing, C. Zhang, M. Liu, Y. Zhang, M. Zhao, Z. Li, C. Wu, Y. Qiao, J. Yang, X. Wang, L. Zhang, M. Li and X. Zhou, Clin. Cancer Res., 2018, 24(11) DOI:10.1158/1078-0432.ccr-16-2248.
- J. Li, S. Yang, N. Su, Y. Wang, J. Yu, H. Qiu and X. He, Tumor Biol., 2016, 37, 2057–2065 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |