
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScope and advances in the catalytic propargylic substitution reaction
Rashmi Roy
a and
Satyajit Saha
 *b
*b
aIIT Kanpur, India
bDepartment of Dyestuff Technology, ICT Mumbai, N. P. Marg, Matunga-400019, Mumbai, Maharashtra, India. E-mail: ss.saha@ictmumbai.edu.in
First published on 5th September 2018
Abstract
Nucleophilic displacement of the propargylic alcohol is one of the sought-after methods in the current scenario. The highly nucleophilic alkyne functional moiety along with its considerably acidic terminal hydrogen atom allows the propargylic unit to play a crucial role in organic synthesis by offering a handle for further synthetic transformations. Until 2000, the most fundamental propargylic substitution reaction was the Nicolas reaction, a multi-step transformation, developed in 1972, which involved cobalt as a stoichiometric promoter. Therefore, the direct catalytic substitution of propargylic alcohols was a highly desirable method for development. The pioneering work on the Ru-catalyzed propargylic substitution reaction in 2000 encouraged many researchers to develop several novel catalytic propargylic substitution reactions, which have made rapid progress since then. The purpose of this review is to emphasise the involvement of diverse types of Lewis acid, transition metal and Brønsted acid catalysts in the propargylic substitution reaction and provide an updated summary of the recent developments in this field. The selected examples presented here are the most significant and relevant ones and we believe that this will help the readers to comprehend the scope of the propargylic substitution reaction with diverse types of catalysts and will envisage the scientific community for the future developments in this field.
 Rashmi Roy | Rashmi Roy was born in 1982 at Kolkata, West Bengal, India. After completing her B.Sc. in chemistry honours from the University of Calcutta in 2004 she moved to Bengal Engineering and Science University, Shibpur for pursuing a master's in applied chemistry. After master's study, she worked as a Junior Research Fellow at the Indian Institute of Chemical Biology, Kolkata for 1.5 years in the medicinal chemistry division and then moved to the Indian Institute of Technology, Kanpur for Ph.D study. In 2015 she completed her Ph.D in chemistry under the supervision of Prof. Yashwant D. Vankar. Her Ph.D study was mainly focused on carbohydrate synthesis and methodology development. After doing a Ph.D she worked in industry (Integral Biosciences Pvt. Ltd. India) and academia (Dept. of Mechanical Engineering, IIT Kanpur). Recently she has been working as a postdoctoral fellow at the University of Alberta, Canada with Prof. Todd L. Lowary. Her current research is focused on carbohydrate synthesis. |
 Satyajit Saha | Satyajit Saha received his Ph.D (Chemistry) in December 2010, from IIT Kanpur, India. Thereafter he moved to Belgium with a FWO Visiting Postdoctoral Fellowship for conducting postdoctoral research at the University of Antwerpen, Belgium and carried out his research until August 2012. He then worked as a Research Scientist at TCG Lifesciences Ltd for a brief period of 8 months. In April 2013, he went for his second postdoctoral research at the University of Leipzig, Germany and continued there until December 2014. In February 2015, he joined the Institute of Chemical Technology, Mumbai, India as a UGC-Assistant Professor under the Faculty Recharge Programme (FRP). His research interests are focused on synthetic organic chemistry related to their functional applications, organocatalytic enantioselective transformations, and green chemistry. |
1. Introduction
Direct nucleophilic displacement of the alpha-hydroxy of the propargylic alcohol is one of the desirable methods in the current scenario. The highly nucleophilic alkyne functional moiety along with the considerable acidic terminal hydrogen atom 1 allows the propargylic unit to play a crucial role in organic synthesis by providing access to saturated products by catalytic hydrogenation, as well as offering a handle for further synthetic transformations, Scheme 1.1 | ||
| Scheme 1 Various synthetic transformations of the alkyne moiety. | ||
Apart from the versatile applicability of the alkyne synthon in chemical transformations,2 many natural products,3 fine chemicals4 and synthetic pharmaceuticals5 are reported to possess the propargylic subunit as their structural component, Fig. 1.
 | ||
| Fig. 1 Some bioactive molecules with propargylic subunit. | ||
Until 2000, there were only a few intermittent reports of nucleophilic propargylic substitution reactions. The most fundamental being the Nicolas reaction, developed in 1972, involving a stoichiometric cobalt complex, Co2(CO)8 as the promoter for the reaction between a propargyl ether 15 and any nucleophile (Nu), Scheme 2.6–8
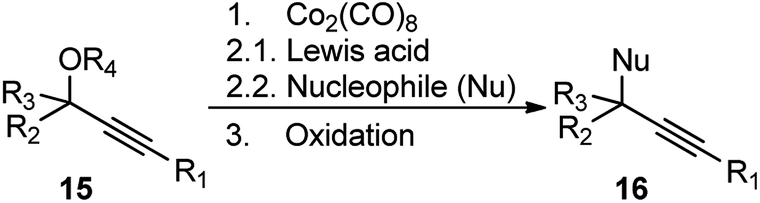 | ||
| Scheme 2 Propargylic substitution by Nicolas reaction. | ||
The reaction proceeds via the hexacarbonyldicobalt, Co2(CO)6 stabilized carbocationic pathway, prior to the encounter with the nucleophile. Subsequent oxidative demetallation gives the substitution product. Despite the broad scope of nucleophiles, this reaction did not gain popularity in the scientific community, due to the involvement of stoichiometric amount of Co2(CO)8 and multistep process. Another stoichiometric method, reported by Müller and Netz, for the propargylic substitution, uses ortho-substituted (arene)Cr(CO)3 17 in conjunction with Lewis acid like TiCl4 or TMSOTf, also has limited applicability, Scheme 3.9
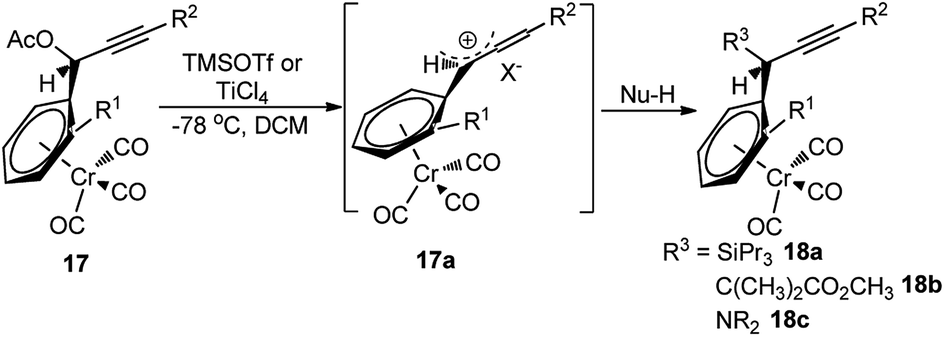 | ||
| Scheme 3 Stoichiometric propargylic substitution reaction. | ||
Therefore, the urge to develop an efficient catalytic propargylic substitution reaction become an inexorable task for the synthetic community. But unlike the catalytic allylic substitution reactions, propargylic substitution reactions are quite arduous due to the predominant formation of allene derivatives, thereby making the propargylic substitution difficult, Scheme 4.
 | ||
| Scheme 4 Formation of allene derivative during propargylic substitution reaction. | ||
The hydroxy being a poor leaving group is generally transformed into more reactive substituents in propargyl alcohol like halide, carboxylate, carbonate, phosphonate, sulfonate, etc. to facilitate the propargylic substitution reaction. However, this process unavoidably involves multiple reaction steps and bound to produce stoichiometric amounts of waste on the release of environmentally unfriendly leaving groups. Therefore, the direct catalytic substitution of propargylic alcohols is an appropriate method for development.
Essentially it was evident that apart from selecting the proper catalysts, several other factors including the nature of the nucleophiles, propargylic substrates and prior activation of the hydroxyl group, demands fine-tuning to selectively substitute the α-position of the propargylic moiety. Classical propargylic substitution reaction has had a great set-back until the Ru-catalyzed propargylic substitution reaction discovered in 2000 (ref. 10) which was instrumental in the rapid development in this area. Several pioneering works on transition-metal catalyzed propargylic substitution reactions have been reported by the groups of Toste,11 Campagne,12 Nishibayashi,13 and van Maarseveen14 involving rhenium, direhnium, gold, and copper-catalyzed nucleophilic substitution of propargylic alcohols, respectively. The very early review in this area mainly on the role of ruthenium–allenylidene complexes in propargylic substitution reaction was reported by Nishibayashi in 2006 (ref. 15) followed by another review in 2009 by van Maarseveen. Van Maarseveen gathered the early examples of catalytic propargylic substitution reaction that was focused on the role of diverse catalysts involved in the propargylic substitution reaction and early examples of enantioselective propargylic substitution reactions.16 In the same year 2009, Nishibayashi also summarized the advances in catalytic propargylic substitution reactions in terms of catalyst used, the mechanism involved and substrate scope.17 Ding and Hou in 2011 presented a review discussing the asymmetric propargylic substitution exemplified by selected enantioselective transformations using carbon and heteroatom nucleophiles.18 A detailed review on the copper-catalyzed propargylic substitution was published by Hu in 2015.19 Besides these reviews, Sakata and Nishibayashi recently reported a review in 2017 discussing the mechanism and reactivity of transition metal catalyzed propargylic substitution reactions from a theoretical perspective.20 Nishibayashi in 2011 presented a review describing the series of heterobimetallic complexes consisting of group IV metallocenyl diphosphines and Ru as catalysts for propargylic substitution reaction.21
2. Synthesis of propargylic alcohols
The propargylation reaction is one of the most fundamental reactions in organic synthesis featuring mild reaction conditions, diverse functional group tolerance leading to the easy construction of C–C bond. It can be easily accessed by the simple reaction between the corresponding aldehydes with in situ generated alkynyl lithium obtained from monosubstituted acetylene derivatives and n-BuLi. Several other practiced methods for the synthesis of propargyl alcohol are highlighted in the Scheme 5 and Scheme 6.22–30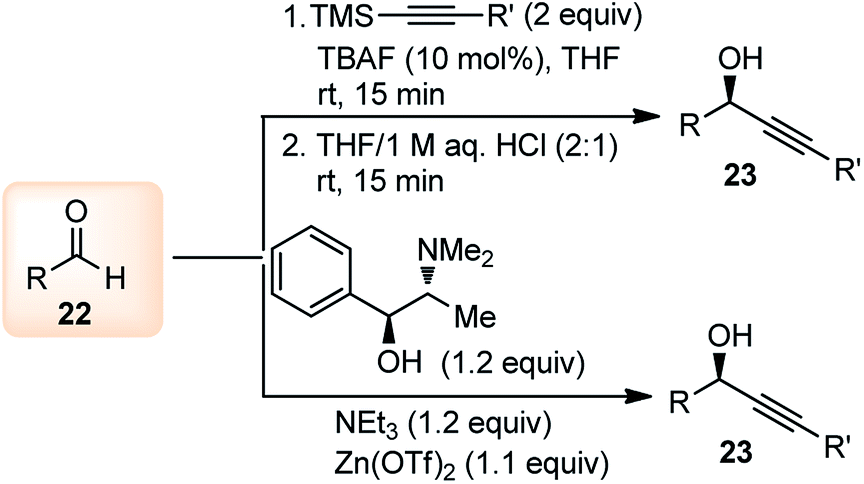 | ||
| Scheme 5 Different methods for the synthesis of propargylic alcohol. | ||
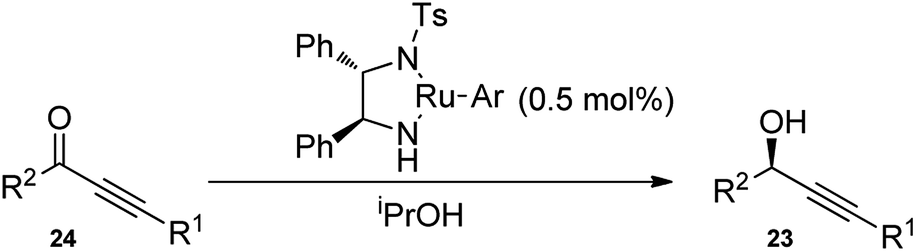 | ||
| Scheme 6 Different methods for the synthesis of propargylic alcohol. | ||
3. Structural modifications of the propargyl derivatives
The high electron density of the propargyl alcohols and their derivatives renders them exceptionally versatile synthetic intermediates.31,32 The π-nucleophilic character of the triple bond makes it a well-designed unit for further chemical transformations. Transition metal/Brønsted acid catalyzed intramolecular cycloisomerization of propargylic sub-unit offers a new dimension for the synthesis of extremely useful building blocks for organic synthesis as highlighted in Scheme 7 and Scheme 8.33–40 The nature of the catalysts and the structure of propargyl derivatives have great influences on the cyclization modes. These cyclization reactions, due to their ability to rapidly assemble into structurally complex molecules and good functional group compatibility, have emerged as an outstanding method for the synthesis of N-containing heterocycles.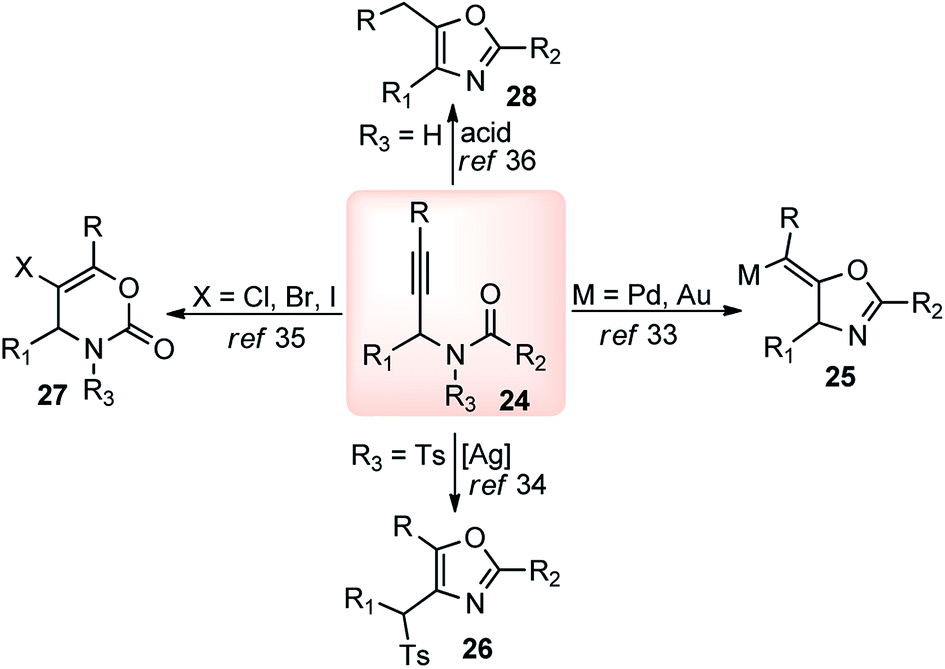 | ||
| Scheme 7 Synthesis of different heterocyclic building blocks from propargyl unit. | ||
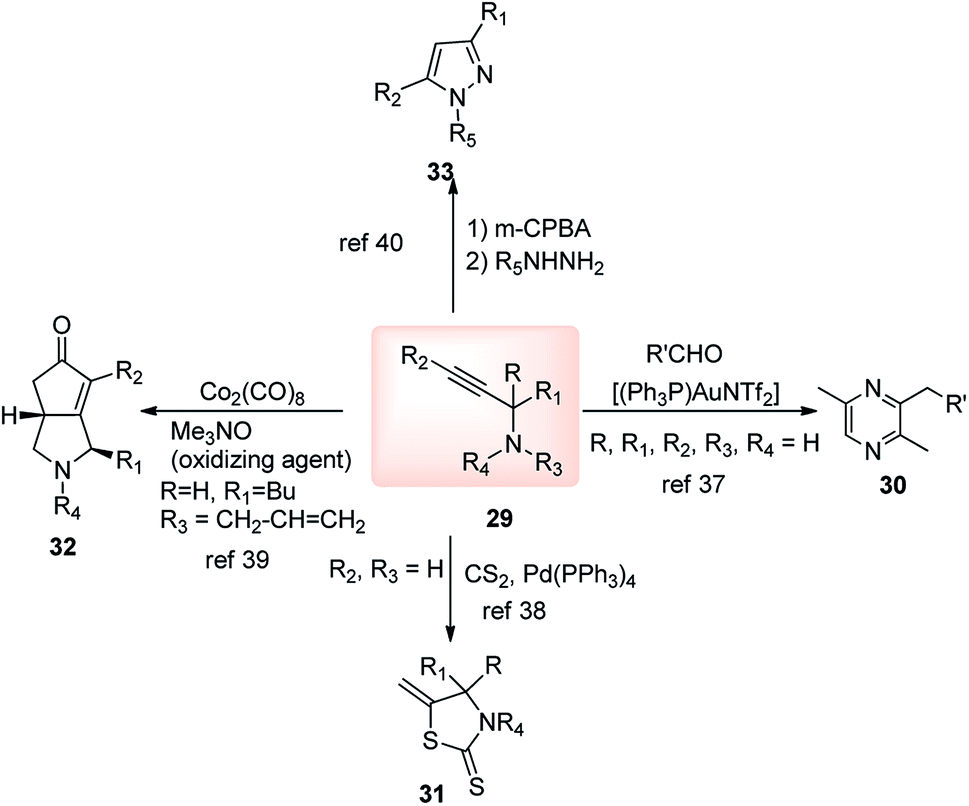 | ||
| Scheme 8 Synthesis of various heterocycles from propargylamine. | ||
4. The scope of this review
Since Nishibayashi and van Maarseveen's major review on the advances of catalytic propargylic substitution published in 2009, there has been a phenomenal development on catalytic propargylic substitution reactions. The reasons for the rapid progress of propargylic substitution reaction can be attributed to the development in the concept and in-depth knowledge of catalysis and design methodology. The assiduous interest in this domain of research is because of several reasons as highlighted below: (i) commercially available starting materials or easy availability of the starting materials; (ii) non-toxic H2O as the only side product; (iii) the synthetic utility of the propargyl subunits for further functional group transformations; (iv) involvement of mild reaction conditions. The gargantuan extent of work has been done in the last few years with diverse types of catalyst demands necessary documentation of different methods in a systematic manner.This review describes the latest developments on the synthetic methodologies of propargylic substitution reaction which has become a subject of burgeoning interest. Thus, the aim of this review is to provide an updated summary of the recent advancements in the catalytic propargylic substitution reaction with an emphasis on the role of different Lewis acid, transition metal catalysts as well as Brønsted acid catalysts in the propargylic substitution reaction and scope for further development. Initial attempts in early 2000 using ruthenium and soon after with copper as transition metal catalysts were consequently followed by a multitude of new methods employing a variety of Lewis- and Brønsted acids will be discussed systematically in terms of the use of catalysts, reaction conditions, substrates and mechanistic insights as we move across the periodic table, Fig. 2.
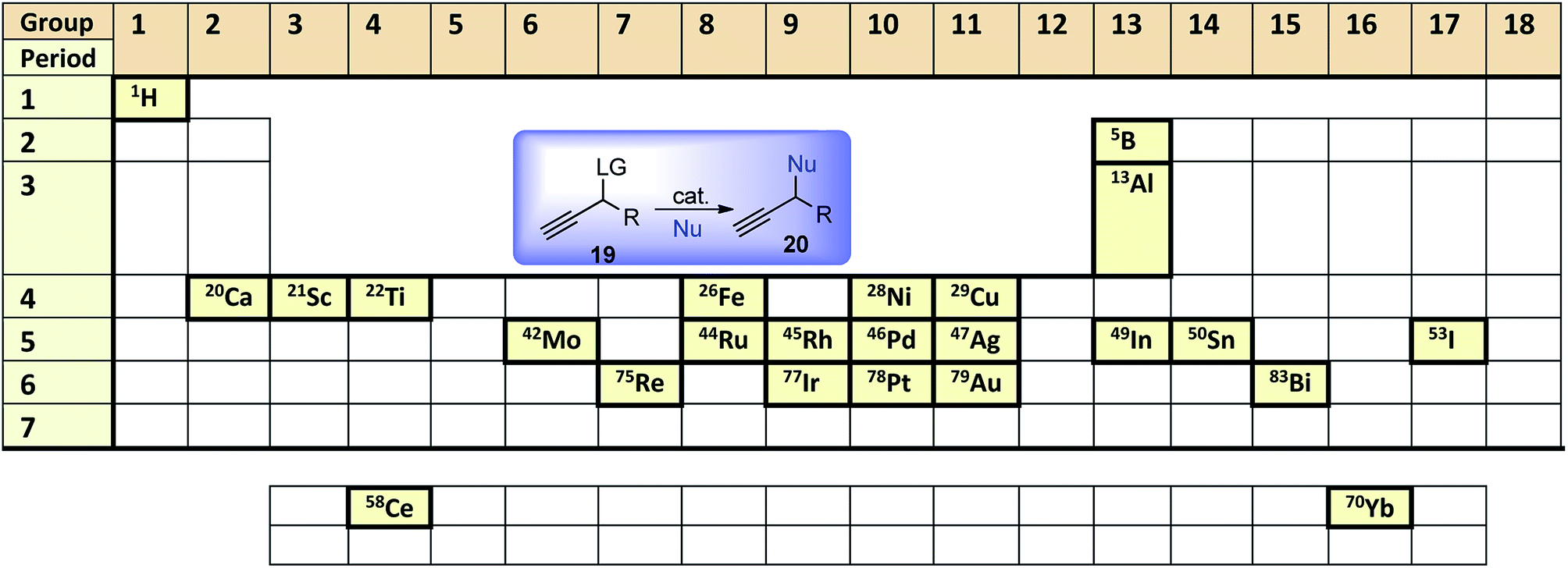 | ||
| Fig. 2 Frequently used elements as catalysts for catalytic propargylic substitution reaction in the periodic table. | ||
We hope this will help readers to grasp a general but in-depth impression of catalytic propargylic substitution reaction and will satisfy the expectations of readers who are interested in the advancement of the field and looking for an up-to-date. The selected examples presented here mostly are the significant and recent ones which will allow the readers to visualize the gradual progress in this field.
5. Different catalysts used in propargylic substitution reactions
5.1. Boron derived catalysts
Boron trihalides are electron deficient species acting as Lewis acids. The tendency to form back bonding is maximum with BF3 and falls rapidly on passing from BCl3 to BBr3 due to the increase in the size of the halogen 2p orbital. Use of boron halides as Lewis acid catalyst is well documented41 and several propargylic substitution reactions have also been carried out involving boron trihalide as a catalyst where propargylic carbocation 34a is formed as the reactive intermediate as shown in Scheme 9.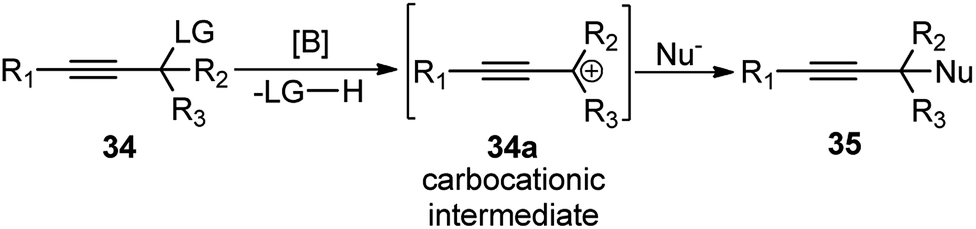 | ||
| Scheme 9 Propargylic substitution reaction via propargylic carbocation intermediate. | ||
Back in early 2000, the urge for the development of propargylic allylation using inexpensive reagents encouraged many researchers to develop several catalytic systems for this reaction. The use of B(C6F5)3 towards the coupling of allylsilanes with benzylic alcohols was first demonstrated by the group of Gevorgyan in the year 2001.42 The same group in the year 2004 described yet another application of B(C6F5)3 in the catalytic reaction of secondary propargylic alcohol derivatives 36 with allylsilane 37 in DCM at room temperature, Scheme 10.43
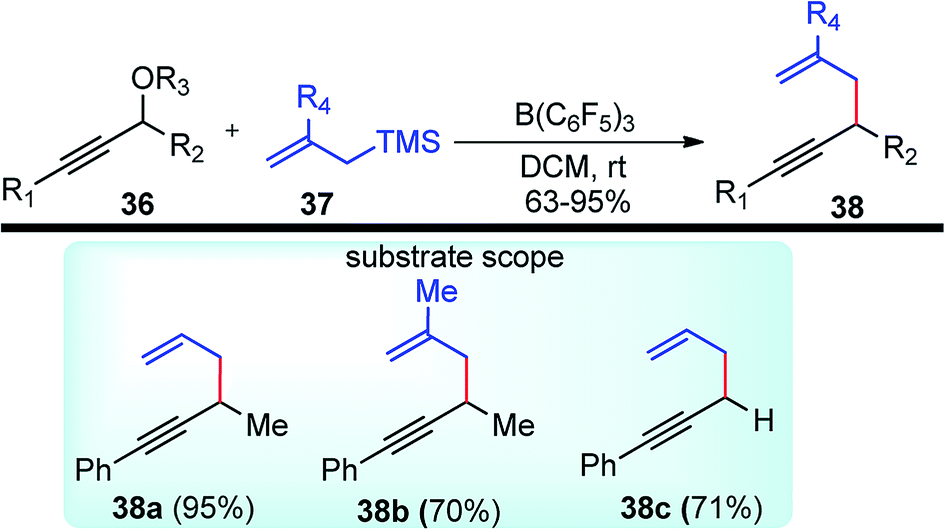 | ||
| Scheme 10 Allylation of propargylic alcohol derivatives with B(C6F5)3. | ||
The reaction on optimization with respect to several leaving groups like OH, OTMS, OCOCH3, OCOC(CH3)3, OCOCF3, OCOCCl3, OCOCH2Cl, OCO2Ph revealed that OH and OTMS were inefficient as leaving whereas OCOC(CH3)3, OCOCF3, OCOCCl3 were moderately good. The best results were achieved with OCOCH2Cl, OCO2Ph, and OCOCH3 with average reaction time ranging from 1 h to 5 days. With OCOCH2Cl, the reaction was completed within 1 h furnishing products in excellent yield. The product outcome is highly dependent on catalyst loading and typically 5 mol% catalyst loading was found optimum for this reaction.
Kabalka and co-workers in the year 2006 demonstrated yet another efficient allylation procedure with in situ generated lithium propargyl oxide using inexpensive and commercially available boron trichloride, Scheme 11.44
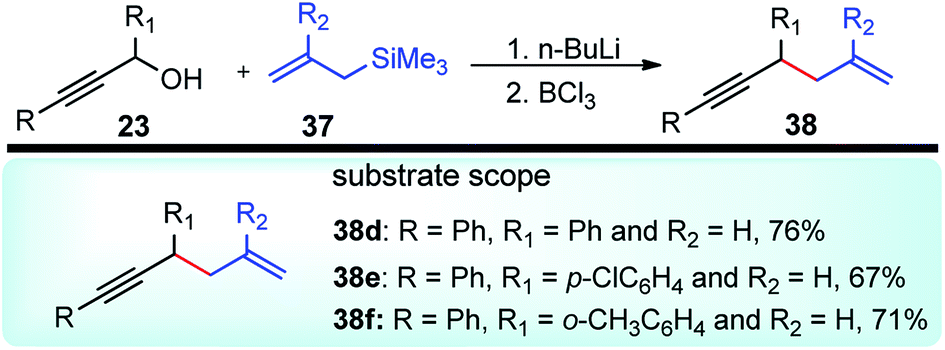 | ||
| Scheme 11 Regioselective allylation of propargyl alcohols with BCl3. | ||
The allylated products 38 were obtained in very high yield and excellent regioselectivity which is apparently due to the absence of Brønsted acids in the reaction medium which was known to reduce the regioselectivity. The mechanistic investigation revealed the role of steric hindrance between the substituents in the R group and chloride, as well as the high electronegativity of chlorine, are the two prime reasons for the weakening of the C–O bond and its subsequent cleavage.
Bi and co-workers in 2013 demonstrated an efficient and inexpensive approach for the synthesis of R-(1,3-dithiolan-2-ylidene) δ-lactam 42c and δ-lactone rings 42 using BF3·Et2O as the catalyst, involving propargylic substitution as the key step, Scheme 12.45
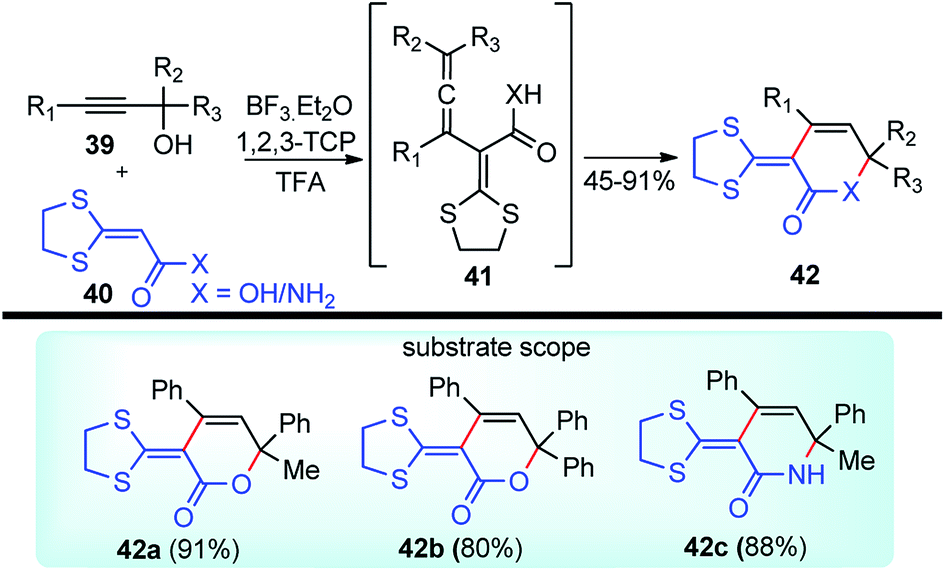 | ||
| Scheme 12 BF3·Et2O catalyzed propargylic substitution and synthesis of δ-lactone thereof. | ||
The maximum product yield of 93% was achieved in 1,2,3-trichloropropane (1,2,3-TCP) within 20 min in the presence of BF3·Et2O as a catalyst. A through solvent screening disclosed that other solvents like DMSO, 1,4-dioxane, and CH3CN were incapable of this transformation. The proposed mechanistic detail for this reaction is outlined in Scheme 13.
 | ||
| Scheme 13 Mechanism for the formation of δ-lactam involving propargylic substitution as the key step using BF3·Et2O as the catalyst. | ||
The OH of the propargyl alcohol 39 on polarization by the Lewis acid, BF3·Et2O, generates the incipient propargylic carbocation 39a by the loss of H2O followed by the attack of the α-carbon of ketene dithioacetal 40 and intramolecular nucleophilic addition to the tertiary carbocation to produce the δ-lactams 42. Particularly, cyclization did not occur with 4 equivalents of BF3·Et2O and TFA were essential for the final cyclization step. So, a dual catalytic system furnishes the delta-lactam 42 in high yield from propargyl alcohol and ketene dithioacetal.
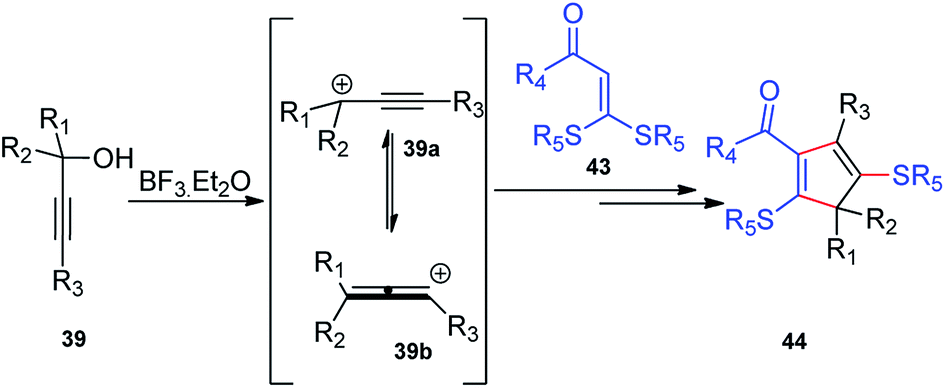
BF3·Et2O was again used as a catalyst by Bi and co-workers in the year 2014 for the synthesis of substituted 2,5-dialkylthiocyclopentadienes 44 in excellent yields. Regiospecific [3 + 2] cycloaddition of propargylic alcohols 39 and α-oxo ketene dithioacetals 43 in the presence of BF3·Et2O generates 2,5-dialkylthiocyclopentadienes 44 in good yields, Scheme 14.46
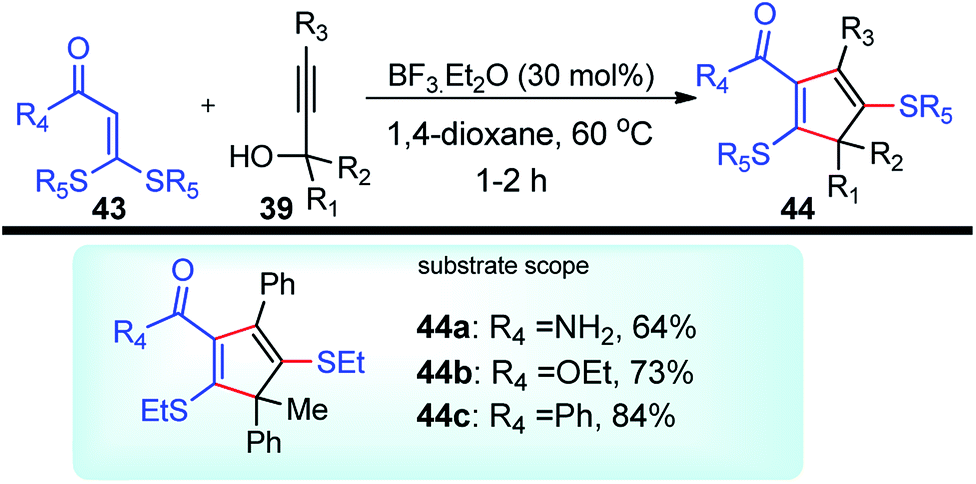 | ||
| Scheme 14 BF3·Et2O catalyzed propargylation and the synthesis of substituted 2,5-dialkylthiocyclopentadienes thereof. | ||
As detailed in the mechanism, the catalyst BF3·Et2O initiates the formation of propargylic carbocation 39a by the loss of the OH species from the propargylic alcohol 39 which isomerizes to the less sterically hindered allenic carbocation 39b. The preferable nucleophilic attack on the allenic carbocation 39b by the α-oxo ketene dithioacetals 43, followed by the cyclization, affords five-membered carbocycle, along with allylic carbocation. A subsequent attack of the sulfur atom to the allylic carbocation followed by the 1,4-alkylthio shift, elimination of a proton and cleavage of the C–S bond produces cyclopentadiene 44 in good yield, Scheme 15. The key step is the BF3·Et2O catalyzed generation of the propargylic cation.
 | ||
| Scheme 15 Mechanism of the propargylation of propargylic alcohols catalyzed by BF3·Et2O. | ||
Several other Lewis acid catalysts like FeCl3, TiCl4, and SnCl4 in different non-polar, polar and polar protic solvents like toluene, CH3CN and polar-protic (MeOH) were screened. Other than BF3·Et2O, all other catalysts afforded either trace amount of the desired product or no product in 1,4-dioxane. The reaction did not proceed in other solvents. Interestingly CF3SO3H was found efficient in catalyzing the reaction albeit with reduced yield. The reaction has a wide substrate scope and tolerates a variety of electron-donating and withdrawing (hetero)-aryl, fused aryl, alkyl, and ether amino, alkoxy, aryl, and styryl functional groups.
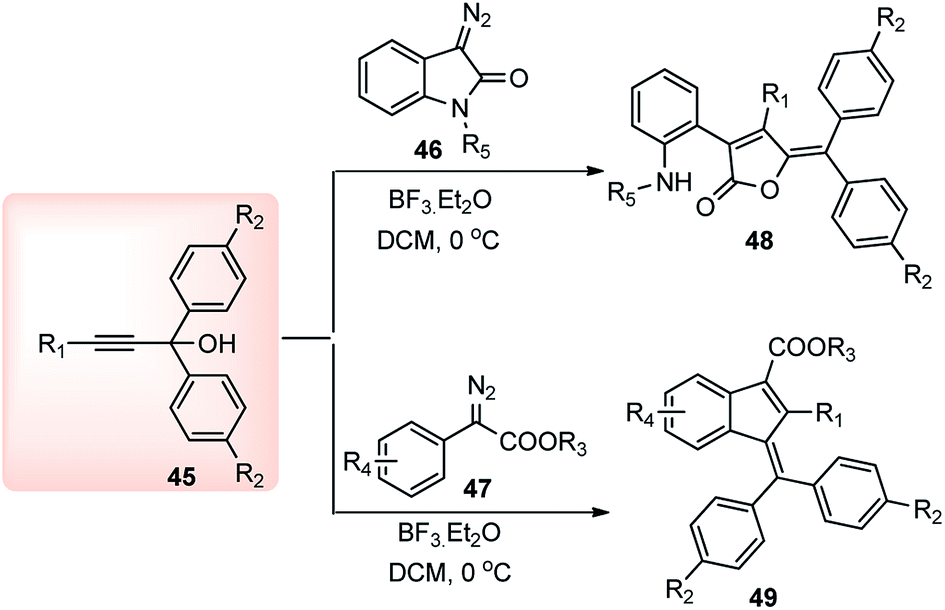
BF3·Et2O yet again proved to be an efficient catalyst in the propargylic substitution reaction for the facile synthesis of highly substituted furanone 48 and indene 49 systems. Muthusamy and co-workers in 2014 described a BF3·Et2O catalyzed tandem reaction of amides 46 and α-diazo-esters 47, and propargyl alcohols 39 towards the synthesis of substituted furanone 48 and indene 49, in high yield under mild conditions, Scheme 16.47
 | ||
| Scheme 16 BF3·Et2O catalyzed synthesis of substituted indene/furanone from propargyl alcohols. | ||
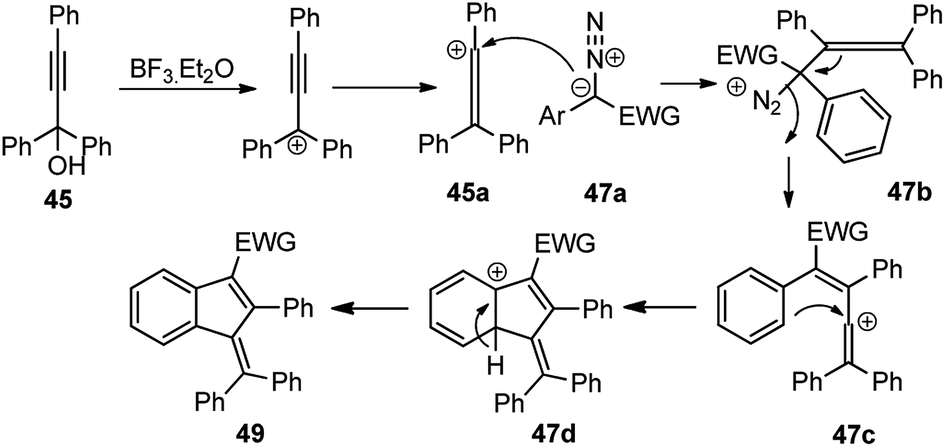
An equimolar mixture of α-diazoester 47 and propargyl alcohol 45 in presence of 10 mol% of BF3·Et2O at 0 °C afforded the product 49 in high yield within 10 min. Diminished product yield was observed when the reaction was carried out in DCE, hexane, THF and using catalysts like AlCl3, CuOTf, TfOH, Yb(OTf)3, Sc(OTf)3, FeCl3, or InCl3. The proposed mechanistic pathway for the above reaction is outlined in Scheme 17.
 | ||
| Scheme 17 Mechanism for the indene formation catalyzed by BF3·Et2O. | ||
In the above-depicted mechanism, BF3·Et2O catalyzes the formation of allene carbocation 45b via Meyer–Schuster rearrangement from propargyl carbocation followed by the nucleophilic attack of the diazo compound to generate aryl allene 47b followed by the extrusion of nitrogen to produce the vinylic cationic species 47c. Electrocyclic ring closure of the vinylic cationic species 47c furnishes the desired product 49 after proton elimination. The reaction is independent of the nature of the substituents on both α-diazoesters 47 and propargyl alcohols 45 and offered the highly substituted and conjugated indene 49 in excellent yields with both electrons donating and withdrawing groups as substituents.
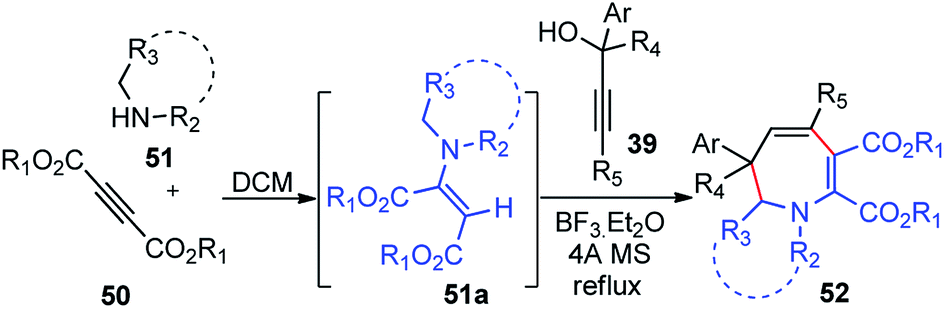
The use of BF3·Et2O as a Lewis acid catalyst for propargylic substitution was again demonstrated by Wang and co-workers in the year 2011 for the synthesis of functionalized dihydroazepines 52 by a three-component reaction between propargylic alcohols 39, 2-butynedioates 50 and secondary amines 51, Scheme 18.48
 | ||
| Scheme 18 BF3·Et2O catalyzed synthesis of functionalized dihydroazepines from propargyl alcohols. | ||
The key step is ascribed to the reaction of the in situ generated enamine 51a with allenic intermediate formed from the propargylic alcohol. Several catalysts like ZnCl2, FeCl3, or I2 were tested and BF3·Et2O gave the best result yield among all the catalysts. Changing the solvent from DCM to toluene, CH3CN, THF or DMF diminished the product yield. Enhancing the reaction temperature and exclusion of moisture from the reaction mixture by addition of molecular sieves had a positive effect on the reaction yield.
A novel catalytic method using BF3·Et2O to synthesize a quinoline 55 backbone by uniting a propargylic alcohol unit across an electron-rich aromatic center to undergo intramolecular propargylic substitution reaction was described by Saito and co-workers in the year 2004, Scheme 19.49
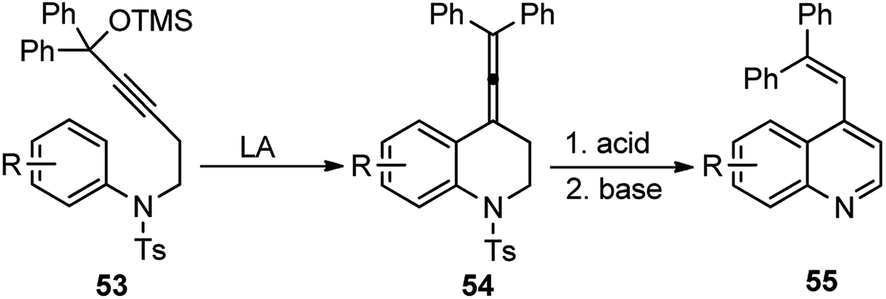 | ||
| Scheme 19 BF3·Et2O catalyzed synthesis of quinoline derivatives from propargyl alcohol. | ||
The key step is the intramolecular Friedel–Crafts alkylation of the electron-rich aromatics to the in situ generated allenyl cation generated from the propargyl unit under the action of a catalytic amount of Lewis acid BF3·Et2O.
Talbot and co-workers in the year 2017 reported an efficient one-pot procedure for the conversion of propargylic silyl ethers 56 to cyclopropylboronic acid pinacol esters 57 using BF3·Et2O as a catalyst, Scheme 20.50
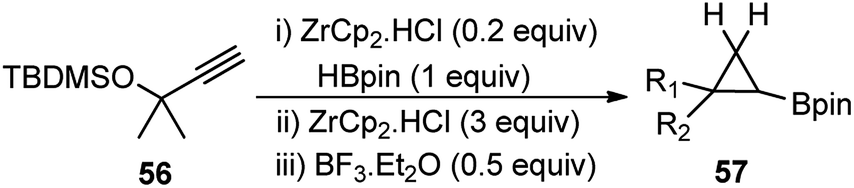 | ||
| Scheme 20 BF3·Et2O catalyzed synthesis of cyclopropylboronic acid pinacol esters from propargyl silyl ethers. | ||
The mechanistic pathway is illustrated in Scheme 21 where Schwartz's reagent (ZrCp2HCl) catalyzes the hydroboration of the terminal acetylenes followed by further addition of Schwartz's reagent and subsequent cyclization leading to a range of cyclopropylboronic acid pinacol ester 57.
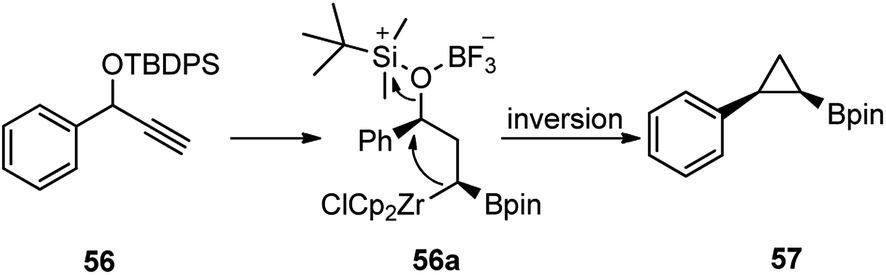 | ||
| Scheme 21 Mechanism for the BF3·Et2O mediated synthesis of synthesis of cyclopropylboronic. | ||
Muthusamy and co-workers in the year 2018 described the synthesis of highly-substituted indole-3-carbinols 60 in good yields by from 1 equiv. of nitrosobenzene 58 and 2 equiv. of propargylic alcohols 39 in the presence of BF3·Et2O as a catalyst, Scheme 22.51
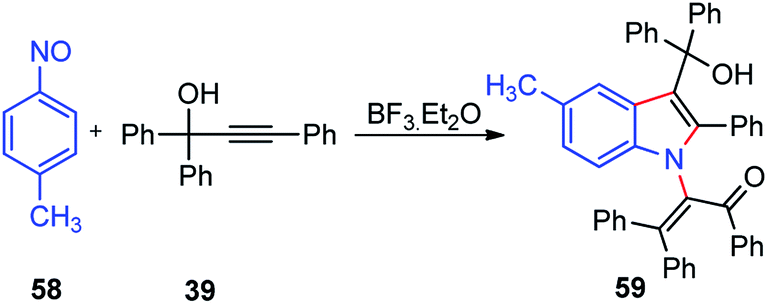 | ||
| Scheme 22 BF3·Et2O catalyzed synthesis of highly-substituted indole-3-carbinols from propargylic alcohols. | ||
The mechanism for the propargylation is highlighted in Scheme 23. The key step is the formation of allene carbocation 39b under the influence of the Lewis acid catalyst, BF3·Et2O. Subsequent addition of the nitrosobenzene 58 to the allenyl carbocation intermediate followed by the Friedel–Crafts cyclization generates 3-alkylidene-3H-indole N-oxides 58b. Indole N-oxide 58c on reaction with the second equivalent of allene carbocation followed by the addition of water and concomitant N–O bond cleavage leads to the formation of indole-3-carbinols 59 in good yield.
 | ||
| Scheme 23 Mechanism for the BF3·Et2O mediated synthesis of substituted indole-3-carbinols. | ||
Several other Lewis acids catalysts like AlCl3, FeCl3, InCl3, and SnCl4, did not furnish the desired product in good yield. Even Brønsted acids like p-TSA were found inefficient. Apart from DCM, poor product yield was obtained when investigated with other organic solvents, such as dichloroethane, 1,4-dioxane, and toluene. The reaction has a wide substrate scope and its operational simplicity allows for scaling up.
5.2. Aluminium derived catalysts
Propargylic and benzyl alcohols are known to form stabilized carbocations in the presence of Lewis acids like aluminium and gallium triflate salts. Especially, aluminium triflate is a highly active yet selective catalyst for a diverse set of reactions. Importantly, it tolerates protic conditions and so can be readily recycled by simply extracting it in water and recovered by subsequent removal of the water.52A water-tolerant reusable Al(OTf)3 Lewis acid catalyst for the propargylation of indoles 61 in refluxing acetonitrile was reported by Bezuidenhoudt and co-workers in the year 2012, Scheme 24.53
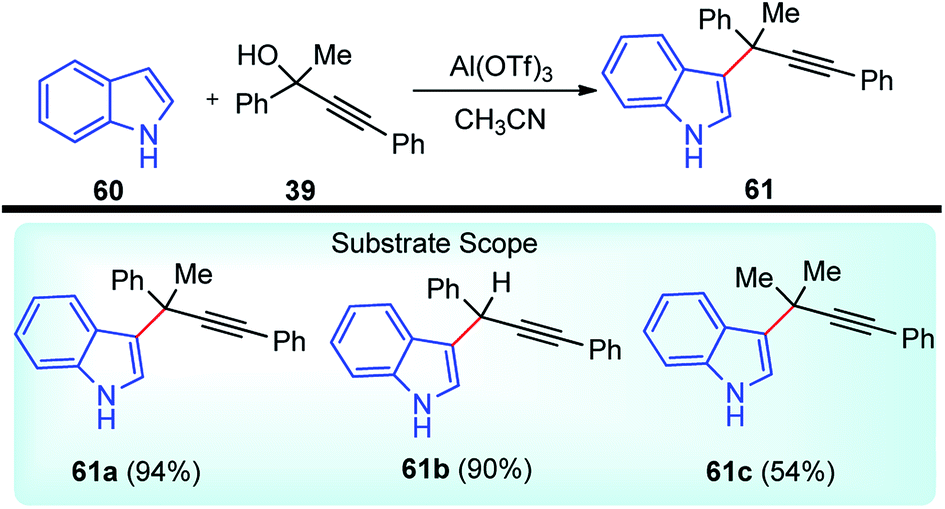 | ||
| Scheme 24 Al(OTf)3 catalyzed propargylation of indoles. | ||
From the environmental point of view, water-tolerant Lewis acids are quite attractive due to their recyclability and reusability attributes and the by-product from the reaction is also non-hazardous H2O. The catalyst was recycled and reused with minimum loss of activity over three consecutive cycles. This methodology has a wide substrate scope and was successfully applied to both 2° and 3° propargylic alcohols for the preparation of propargylated indoles in a very high yield. The reaction is believed to pass through propargylic carbocationic species followed by a nucleophilic attack on the propargyl carbocation.
Use of Al(OTf)3 in the direct nucleophilic displacement of OH in ‘activated’ systems with several nucleophiles were reported by Williams and co-workers in 2017.54 This efficient process allows the ready synthesis of a wide range of benzylic and allylic substituted products in very high yields with water as the only by-product.
5.3. Calcium derived catalysts
Use of precious metals in homogeneously catalyzed reactions becomes increasingly expensive as we continuously deplete the earth's natural resources.55 The potential application of early main group metals as catalysts has remained a widely underexplored research field, despite the apparent ecological and economic benefits. Among other alkaline earth metals, calcium is an ideal main group metal catalyst, essentially free of toxicity, very cheap, and the fifth most abundant element of the earth crust. Recently, calcium-based Lewis acid catalyst has been used by the group of Niggemann for the nucleophilic substitution of benzylic and allylic alcohols under very mild reaction conditions.56,57In further pursuit towards the cost-effective methodology for propargylic substitution reaction avoiding costly transition metals, Niggemann and co-workers in the year 2011 developed an efficient calcium-catalyzed allylation of π-activated alcohols 23 with diverse silyl-based carbanions 63 under very mild reaction conditions, Scheme 25.58
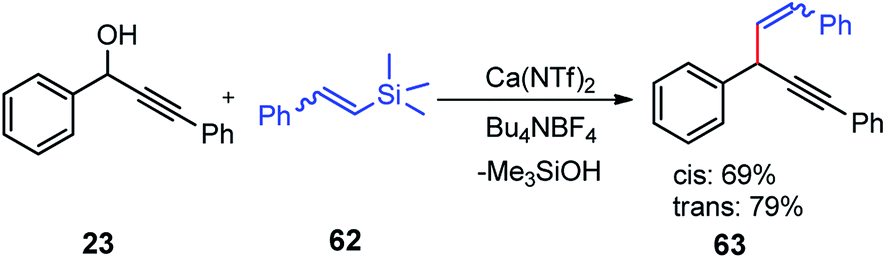 | ||
| Scheme 25 Calcium-catalyzed direct substitution of π-activated alcohols with allyltrimethylsilane. | ||
The reaction of propargylic alcohols 23 and allyltrimethylsilane 63 in presence of 5 mol% each of Ca(NTf2)2 and Bu4NPF6 proceeded smoothly at room temperature in dichloromethane to give the desired allylated product 64 after 1 h as a 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of allylated regioisomers in good yield. Incomplete transformation was observed with strongly coordinating solvents like ether, THF whereas dichloromethane was found to be the most suitable solvent for the allylation of propargyl alcohols. Toluene could replace DCM as a solvent although with slightly poorer product yield. Typical reactions proceed at room temperature, with no added strong acids or bases, and special precautions for the exclusion of moisture or air are not necessary.
:1 mixture of allylated regioisomers in good yield. Incomplete transformation was observed with strongly coordinating solvents like ether, THF whereas dichloromethane was found to be the most suitable solvent for the allylation of propargyl alcohols. Toluene could replace DCM as a solvent although with slightly poorer product yield. Typical reactions proceed at room temperature, with no added strong acids or bases, and special precautions for the exclusion of moisture or air are not necessary.
Yaragorla and co-workers in the year 2017 described a highly regioselective annulation reaction between propargyl alcohols 39 and several enols 65 using calcium triflate catalyst, under a solvent-free condition at 110 °C, Scheme 26.59
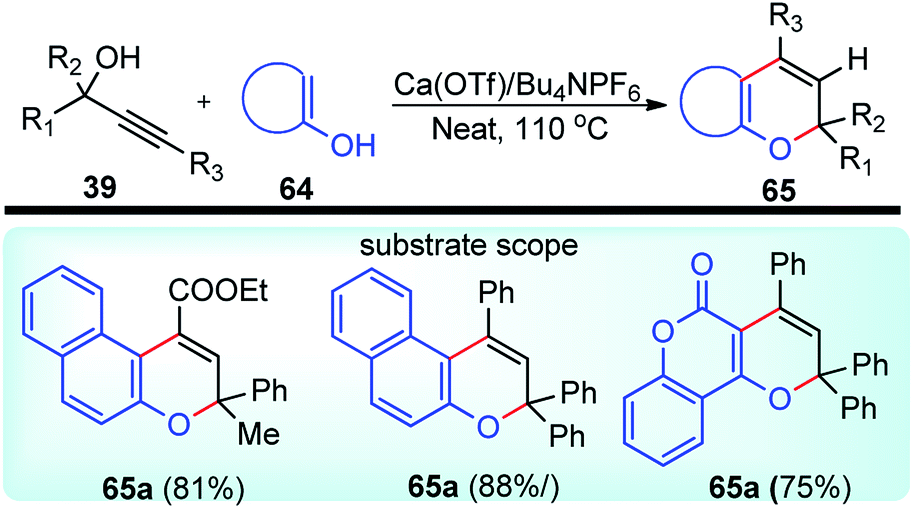 | ||
| Scheme 26 Calcium triflate catalyst synthesis of 2H-chromene from propargylic alcohols. | ||
The propargyl alcohol 39 and 2-naphthol 65 in the presence of 10 mol% of Ca(OTf)2, 10 mol% of Bu4NPF6 when heated at 110 °C furnished the desired product in good yield. The temperature had a pronounced effect on the reaction yield. Lowering the temperature reduced the product yield and the presence of additive like Bu4NPF6 enhanced the product yield possibly due to the formation of Ca[(OTf)(PF6)] by ligand metathesis which has an enhanced Lewis acidity. Other additives like KPF6, NH4BF4, and Bu4NF provided moderate yields with Ca(OTf)2. Moderate yields of the product were achieved with metal catalysts, such as Mg(OTf)2 and FeCl3. The reaction did not occur in water as well as in other protic solvents and gave poor yield in aprotic non-polar solvents. The reaction proceeds via a cascade annulation between propargyl alcohols 39 and 4-hydroxy coumarin 64a involving a calcium promoted vinyl-propargyl ether 64b formation followed by [3,3]-Claisen type rearrangement to furnish the allene 64c. Subsequent 6-endo regioselective cyclization furnishes the desired product 65, Scheme 27.
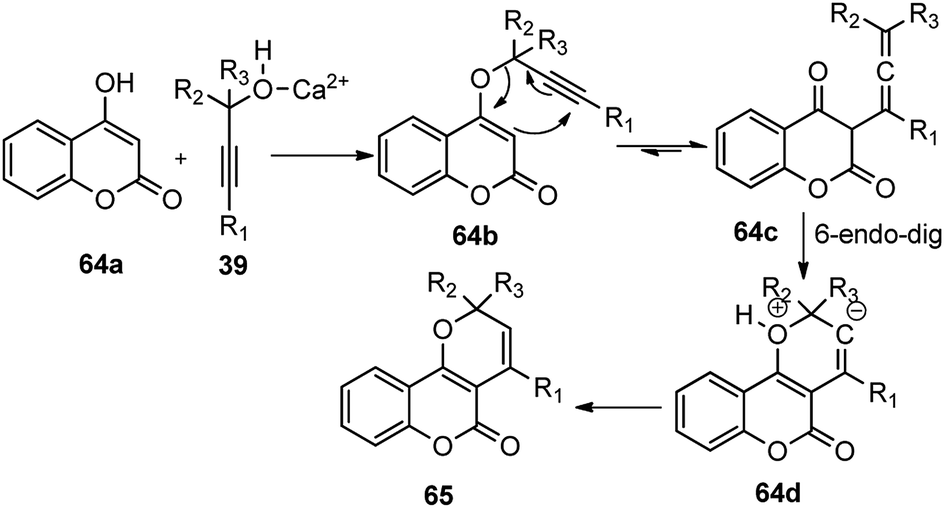 | ||
| Scheme 27 Mechanism for the Ca-catalyzed synthesis of 2H-chromenes form propargylic alcohols. | ||
5.4. Scandium derived catalysts
Scandium chemistry is almost completely dominated by the trivalent ion, Sc(III). The chemical properties of scandium ions have more in common with yttrium ions than with aluminium ions but have been rarely used as a catalyst in the propargylic substitution reaction.In pursuit of the catalytic uses of scandium salts as water-tolerant and recyclable Lewis acid catalyst, Yadav and co-workers in the year 2007 reported a scandium triflate catalyzed efficient alkylation of indoles 60 with propargylic alcohols 23, Scheme 28.60
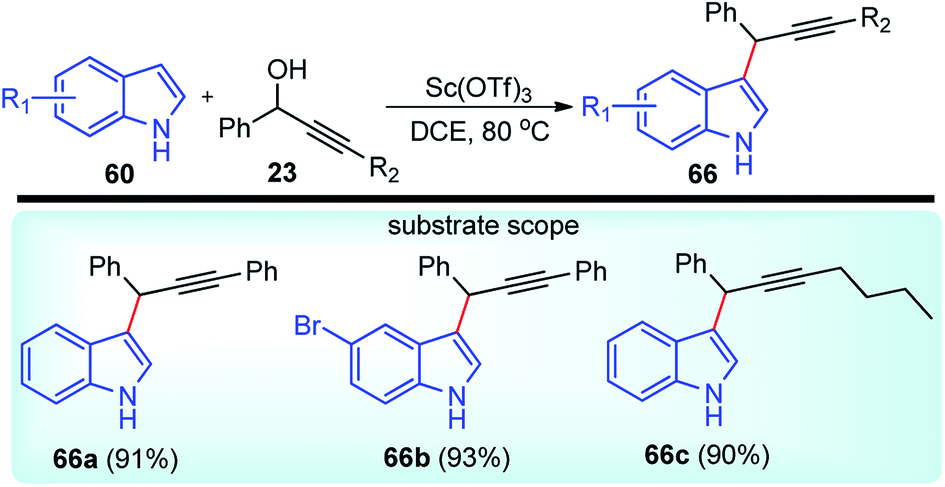 | ||
| Scheme 28 Sc(OTf)3 catalyzed propargylation of indoles. | ||
Within a short time-span, the hydroxyl group was replaced by the indole in an SN2 manner to furnish products in more than 90% yield. However, in absence of scandium triflate, no product was observed. Reaction in dichloroethane provided the best yield among several other solvents screened. The reaction is equally competent with several other Lewis acids like Bi(OTf)3, In(OTf)3, Sm(OTf)3 and Yb(OTf)3.
Sc(OTf)3 was yet again used for the regioselective propargylic substitution reactions of the phenylsulfanyl and phenylselanyl propargyl alcohols 67/68 in very high yields by Yoshimatsu and co-workers in the year 2008, Scheme 29.61
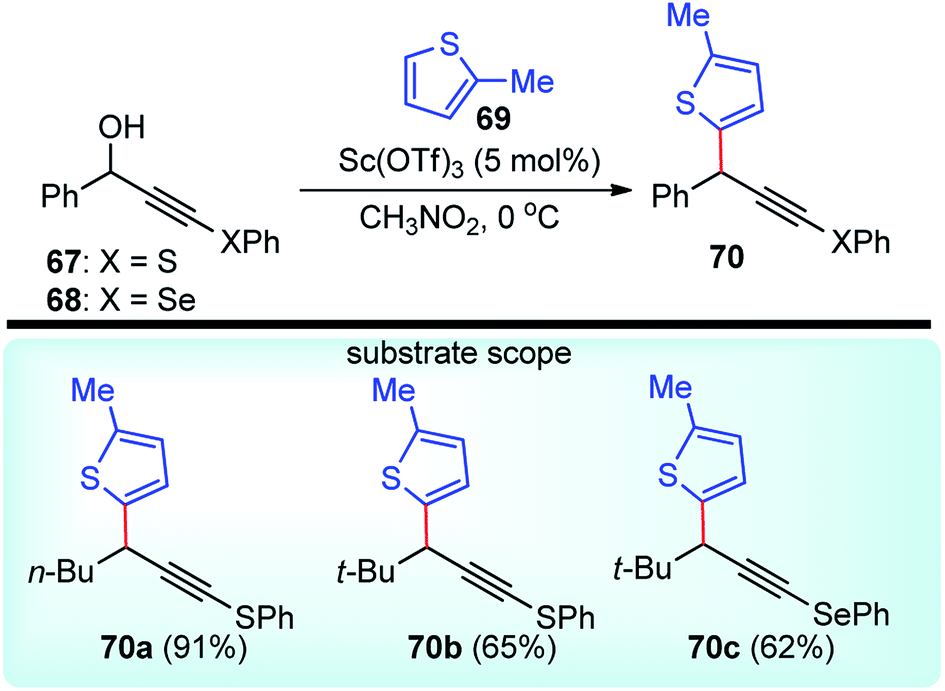 | ||
| Scheme 29 Scandium catalyzed propargylic substitution reaction. | ||
A thorough screening of various Lewis acids like Yb(OTf)3, La(OTf)3, BF3·Et2O, as well as the solvent (e.g., dichloromethane, 1,2-dichloroethane, DMF, and THF), disclosed scandium triflate-nitromethane as the ideal combination for the highly regioselective substitution reaction.
5.5. Titanium derived catalysts
The relevance of Ti(IV) Lewis acidity is due to low-lying empty d orbitals which are responsible to play a crucial role in catalysis. But not much has been explored in the domain of propargylic substitution reaction.TiCl4 has been utilized as an efficient Lewis acid catalyst by Mahrwaid and co-workers in the year 1999 for the direct displacement of propargylic esters 71 with diverse oxygen-centered nucleophiles, Scheme 30.62
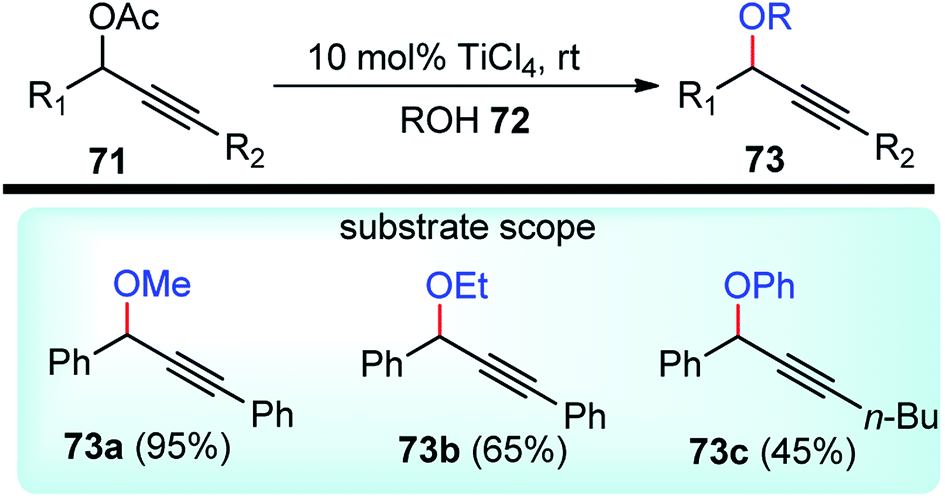 | ||
| Scheme 30 TiCl4 catalyzed propargylic substitution with diverse oxygen-centered nucleophiles. | ||
Alcoholic solvents like MeOH, EtOH, iPrOH, tBuOH resulted in decreased product yield with concomitant formation of hydrolyzed products. Even with aliphatic substituents hydrolyzed products were mainly obtained probably due to the decreasing stabilization of the formed cation during the reaction. The challenge was overcome by using catalytic amounts of trifluoromethane sulfonates as a Lewis-acid catalyst. By using 10 mol% of trimethylsilyl triflate, the propargylic methyl ether was obtained in about 40% yield for deactivated propargylic esters.
5.6. Iron derived catalysts
Transition metal catalyzed methods for organic synthesis often use expensive and transition metals such as platinum, palladium, gold, nickel, and mercury has led to significant concern about possible metal residues in the final product and waste, especially in pharmaceuticals and food additives. This, in turn, will increase the expenses in the process and waste management. In comparison with cobalt, rhenium, ruthenium, and gold complexes, which are usually used to catalyze the nucleophilic substitution of propargylic alcohols, FeCl3 as the catalyst offers several relevant advantages including cheapness and commercial availability, broad scope, and mild reaction conditions of this transformation.63 Partly due to these reasons, iron has only recently received growing interest in the field of catalysis. On the one hand, it often suffers from poor reactivity or selectivity in comparison to the more “noble” metals.Zhan and co-workers in 2006 reported an efficient FeCl3-catalyzed propargylic substitution reaction of propargylic alcohols 39 with various carbon and heteroatom-centered nucleophiles, Scheme 31.64
 | ||
| Scheme 31 FeCl3-catalyzed propargylic substitution reaction of propargylic alcohols. | ||
The following methodology provides an easy access to the construction of C–S bonds by the nucleophilic substitution of propargylic alcohols with a series of nucleophiles. Transition metal-catalyzed substitution of propargylic alcohols with thiols has been considered difficult to achieve, probably due to the reason of the strong co-ordinating ability of sulfur which causes catalyst poisoning.
The experimental results suggest that the mechanism proceeds via the formation of propargylic cation intermediate. Both electron-rich and electron-poor aromatic substrates reacted efficiently to afford the corresponding products in high yield. The primary aliphatic alcohol 3-phenylprop-2-yn-1-ol failed to furnish propargylated product. Instability of the propargylic cation intermediate clearly made the substitution reaction less favorable.
A new method for the Friedel–Crafts propargylation of several electron-rich aromatics 82 catalyzed by FeCl3 was developed by Zhan and co-workers in the year 2006, Scheme 32.65
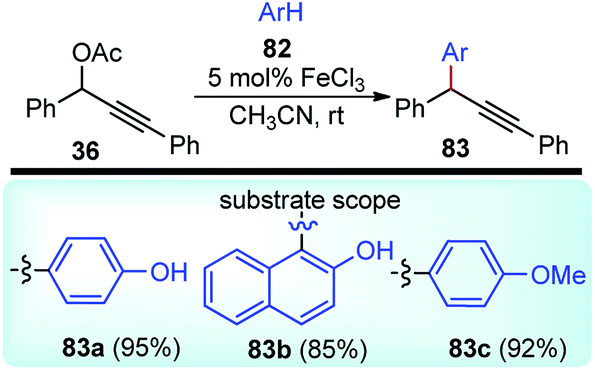 | ||
| Scheme 32 FeCl3 catalyzed propargylic substitution reaction of electron-rich aromatics. | ||
A facile aromatic electrophilic substitution of the electron-rich aromatics at room temperature in acetonitrile without exclusion of moisture or air resulted in substituted products with high yield and excellent regioselectivity.
Jana and co-workers in the year 2007 have developed an efficient and atom economical method for the direct alkylation of indoles 60 with various propargyl alcohols 23 in the presence of the inexpensive and non-toxic FeCl3, under mild conditions, Scheme 33.66
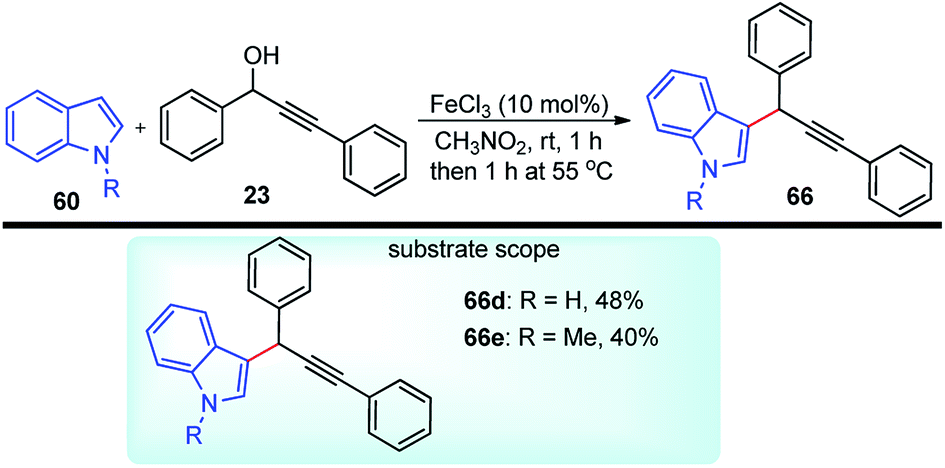 | ||
| Scheme 33 Propargylic substitution of indoles with FeCl3 catalyst. | ||
The authors found that the reaction proceeded in the presence of FeCl3 (10 mol%) at room temperature in nitromethane affording the propargylated product in moderate yield. Other solvents didn't afford the product in good yield. The reaction was clean and furnished the C3-substitution product with high regioselectivity within 2 h without the need for an inert atmosphere.
Zhan and co-workers in the year 2007 developed an efficient method for the synthesis of γ-alkynyl ketones 85 in high yield via FeCl3-catalyzed substitution reaction of propargylic acetates 36 with enoxysilanes 84 under mild conditions. Subsequent cyclization by p-TsOH without purification of the γ-alkynyl ketone intermediates resulted into tri- or tetra substituted furans 86 in high yield, Scheme 34.67
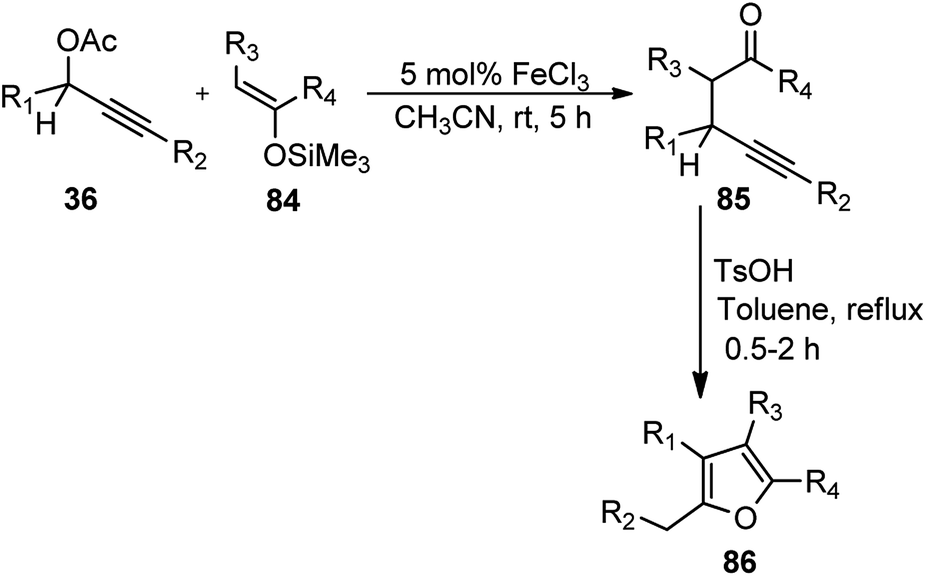 | ||
| Scheme 34 FeCl3 catalyzed synthesis of γ-alkynyl ketones from propargylic acetates with enoxysilanes. | ||
The reaction proceeded smoothly without exclusion of moisture or air from the reaction mixture. Both electron-donating and electron-withdrawing aromatic substrates reacted smoothly with enoxysilane 84 affording the corresponding alkylated products in high yields. Additionally, the methodology provided a straight forward access to polysubstituted furans 86 starting from easily available starting materials utilizing cheap catalysts in an open atmosphere.
Chan and co-workers in the year 2010 described an efficient method to prepare highly conjugated indenes 88 by iron(III) chloride-catalyzed dimerization of trisubstituted propargylic alcohols 87 under very mild conditions at room temperature, Scheme 35.68
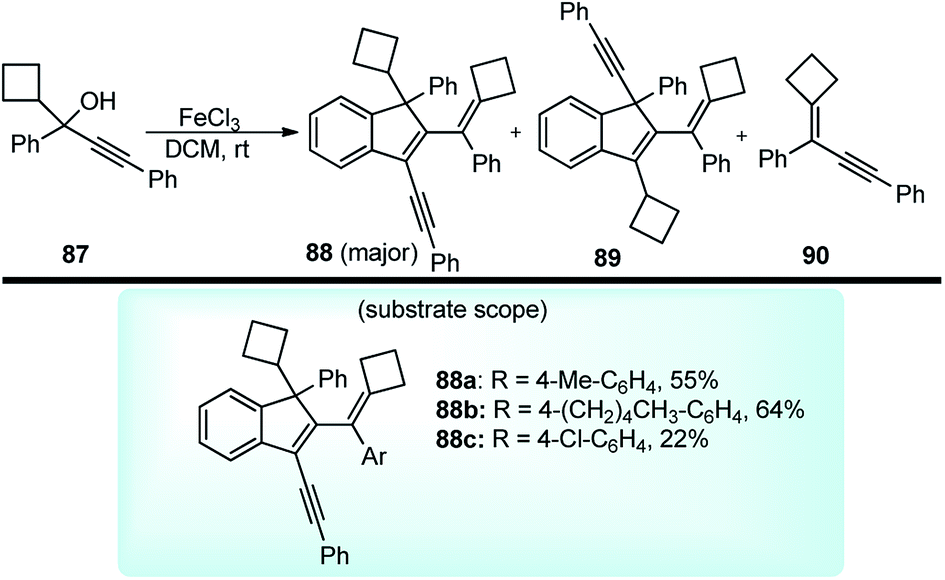 | ||
| Scheme 35 Synthesis of highly conjugated indenes by iron(III) chloride-catalyzed dimerization of trisubstituted propargylic alcohols. | ||
After careful screening of the reaction conditions, the best result was observed with 5 mol% of FeCl3 and 4 Å MS at room temperature in DCM. Products with reasonably good yield were also achieved with catalysts like ZnCl2 and AuCl3. However, in the absence of a catalyst, or switching to the metal triflates like Cu(OTf)2 and Yb(OTf)3, or Brønsted acids like p-TsOH and TfOH, or changing the substitution pattern in the propargyl derivatives did not furnish any product or resulted in products with low chemo and regioselectivity. The mechanism outlined in Scheme 36 involves the activation of the alcohol substrate 87 through coordination of the hydroxyl group to the FeCl3 catalyst resulting in the formation of a Fe(III)-coordinated intermediate 87a which on the elimination of water furnishes the alkenyl cation 87b or its resonance analogue allenic cation 87c. Alkoxylation of carbocation 87c at the sterically less hindered carbon center by another molecule of the propargyl alcohol followed by the intramolecular Friedel–Crafts reaction affords the dimer 88.
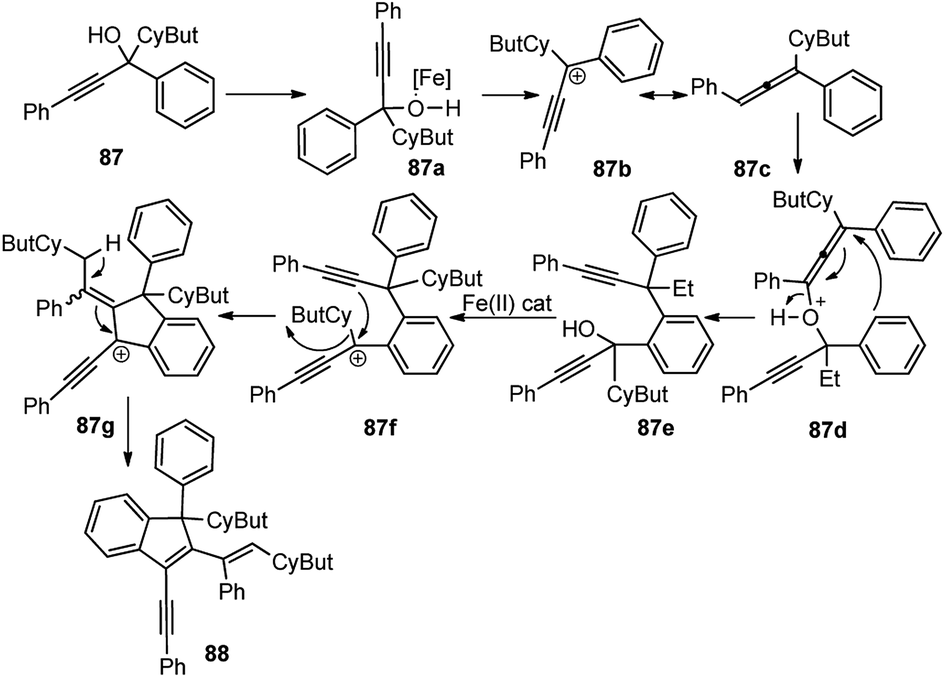 | ||
| Scheme 36 Mechanism for the formation of highly conjugated indenes by iron(III) chloride-catalyzed dimerization of trisubstituted propargylic alcohols. | ||
Xu and co-workers in the year 2014 developed a FeCl3 catalyzed intermolecular Friedel–Crafts alkylations of aryloxy and arylamino-substituted propargylic alcohols 89/90 towards the synthesis of dihydrobenzofurans 91 and dihydroindoles 92 in high yields, Scheme 37.69
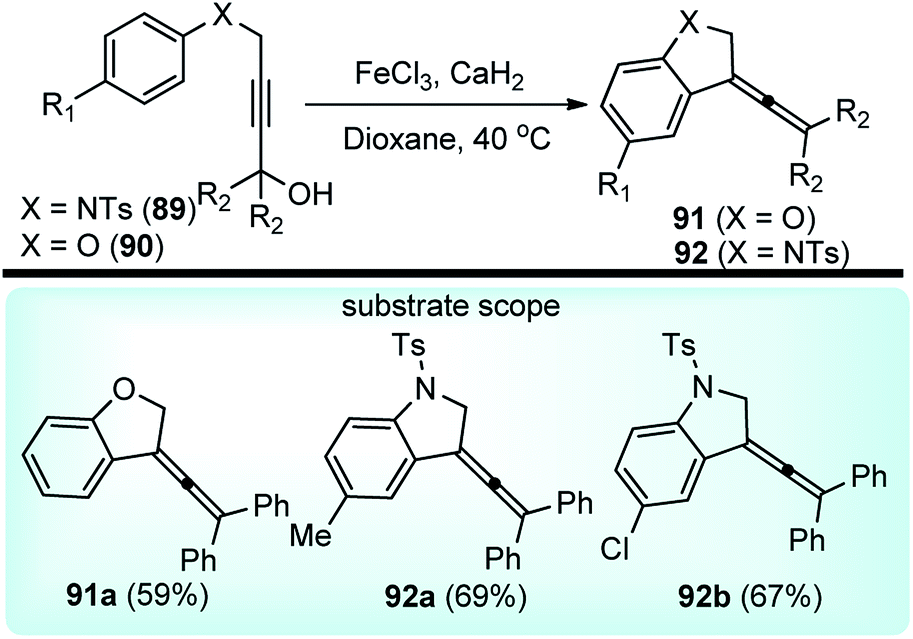 | ||
| Scheme 37 FeCl3 catalyzed intermolecular Friedel–Crafts cyclizations of aryloxy and arylamino-substituted propargylic alcohols. | ||
This transformation provides a versatile and direct strategy for the construction of dihydrobenzofurans 91 (when X = O) and dihydroindoles 92 (when X = NTs) from easily accessible starting materials. The reaction was optimized by screening several Lewis acid catalysts like TiCl4, BF3·Et2O, AlCl3, Sc(OTf)3, ZnBr2 and Brønsted acids, such as p-TsOH and TFA. Unfortunately, none of them furnished product in good yield. Desiccants had a role on reaction yield, as higher efficiency was observed with CaH2 compared to MgSO4 when tested in solvents like MeNO2, toluene, MeCN, and DCM. Catalyst loading significantly influenced the reaction yield. The product yield reduced to only 14% when 20 mol% catalyst was used. The probable mechanistic pathway depicted in Scheme 38 portrays that propargyl alcohol is first converted into the allenylic cations 89b/90b in the presence of Lewis acid FeCl3 via a Meyer–Schuster rearrangement followed by an intramolecular Friedel–Crafts type alkylation to trap the cation and form the corresponding dihydrobenzofuran 91 and dihydroindole derivatives 92.
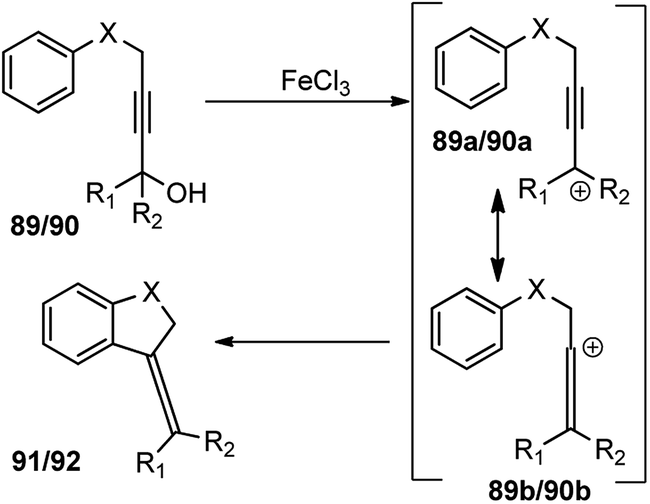 | ||
| Scheme 38 Mechanism of FeCl3 catalyzed synthesis of dihydrobenzofurans and dihydroindoles from propargylic alcohols. | ||
Substituents have a definite role to play in the reactivity. No product was observed for substrates having aliphatic substituents, probably due to the lesser stabilization of the formed carbocation 89a/90a. Further, a mixture of unidentified side-products was obtained for propargylic alcohol bearing p-Me group, while the p-Cl substituted propargylic alcohol afforded product in 34% yield only.
Bauer and co-workers in 2015 reported an efficient protocol for the propargylic etherification of the terminal, tertiary propargylic alcohols 93 with primary and secondary alcohols 72 under ambient temperature in CH2Cl2 using commercial ferrocenium hexafluorophosphate ([FeCp2]PF6), Scheme 39.70
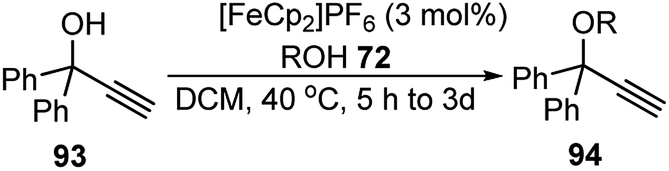 | ||
| Scheme 39 Ferrocenium hexafluorophosphate ([FeCp2]PF6) catalyzed etherification of the terminal, tertiary propargylic alcohols. | ||
Shang and co-workers in 2015 described a FeCl3-catalyzed domino reaction of N-cyclohexyl propargylamines 95 with 1,3-diketones 96 towards the synthesis of β-alkynyl ketones 97 in good to excellent yields, Scheme 40.71
 | ||
| Scheme 40 Iron-catalyzed domino reaction of N-cyclohexyl propargyl amines and 1,3-diketones. | ||
Several Lewis acid catalysts like CuBr2, CuI, Sc(OTf)3 and Brønsted acid like H2SO4 were tested but the highest yield was achieved using FeCl3. Even altering the catalyst loading did not have any positive impact on the reaction yield. Further solvent screening revealed that solvents like CH2Cl2, THF, DMF, DMSO, H2O, and 1,4-dioxane were inappropriate for this reaction. The highest yield was achieved when the reaction was carried out with 50 mol% catalyst FeCl3 in CH3CN at 80 °C for 24 h. The mechanistic detail of this process is highlighted in Scheme 41.
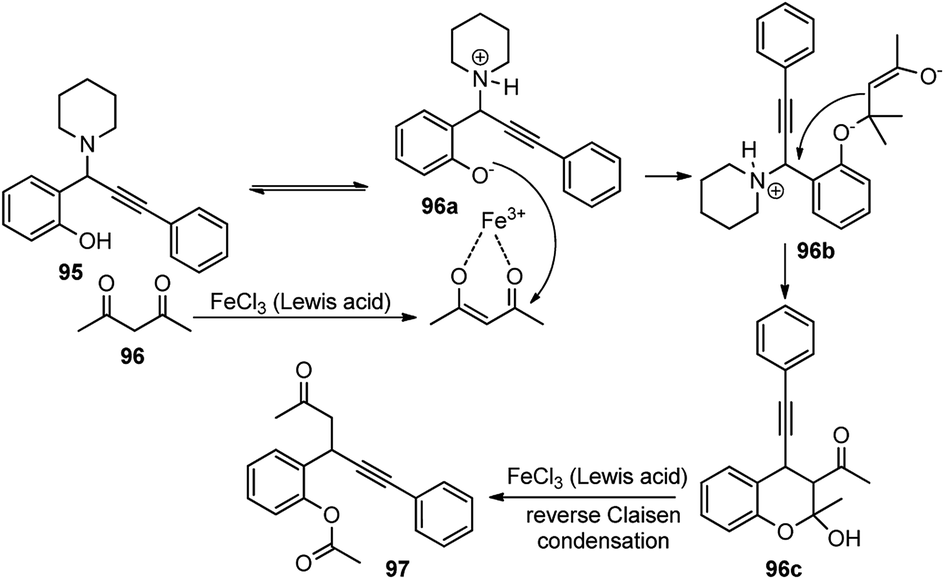 | ||
| Scheme 41 Mechanism of the Fe(III)-catalyzed propargylation of propargyl amines. | ||
The reaction initiates with the proton transfer from the hydroxyl group of phenol to the nitrogen of the piperidine resulting in the formation of phenoxide intermediate 96a under the influence of FeCl3 catalyst. Subsequently, phenoxide 96a attacks the 1,3-dicarbonyl compound to form the β-keto ester 96b followed by the propargylic substitution reaction to release piperidine and eventually cyclizes to form 96c. Finally, a reverse Claisen condensation reaction of intermediate 96c in the presence of FeCl3, followed by a proton transfer process, generates the desired product 97 in good yield.
Chen and co-workers in the year 2018 developed a FeCl3 catalyzed straightforward protocol to synthesize complex indene-based polycyclic compounds 99 via the cascade cyclization of propargylic alcohols 39 with alkenes 98, Scheme 42.72
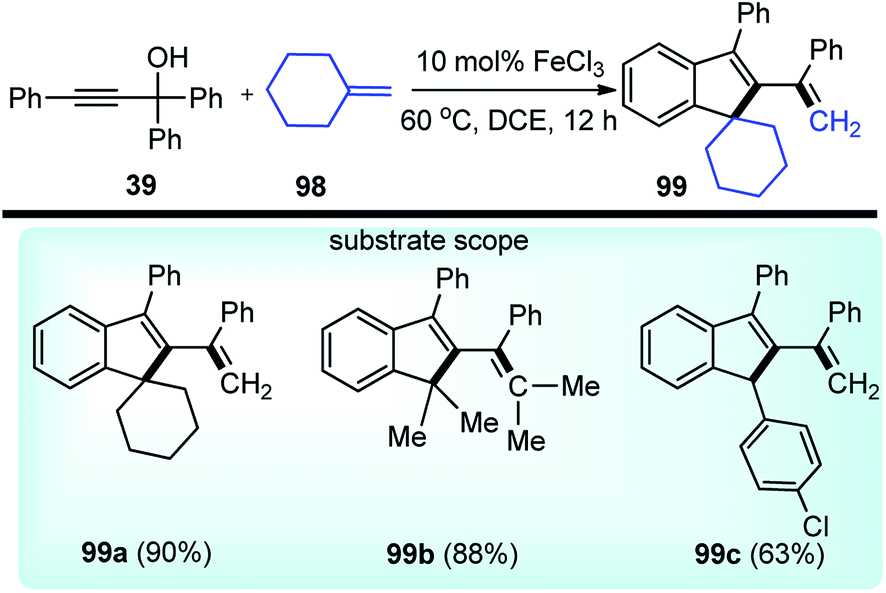 | ||
| Scheme 42 FeCl3 catalyzed indene-based polycyclic compounds. | ||
The authors investigated a series of Lewis acids and observed that except for AgSbF6, InCl3 or Cu(OTf)2 all other catalysts like Zn(OTf)2, Sc(OTf)3, and FeCl3 were effective for the formation of the desired product and FeCl3 was the best among them in terms of yield and selectivity. Solvent screening revealed that better result was achieved in DCE. The mechanism for this reaction is highlighted in Scheme 43.
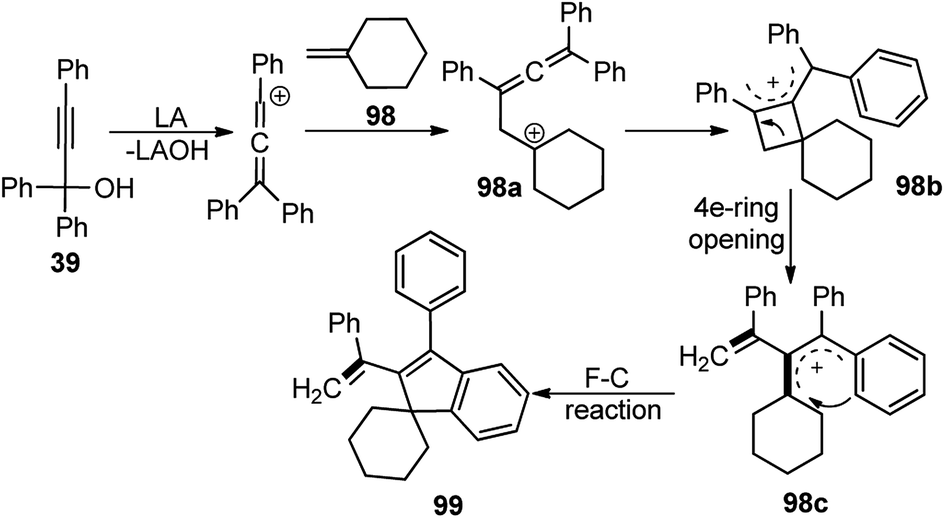 | ||
| Scheme 43 A proposed mechanism for the FeCl3-catalyzed synthesis of indenes. | ||
The starting material gets converted into the triphenylallenyl cation 39a under the influence of FeCl3 which underwent a stepwise [2 + 2]-cycloaddition to form the intermediate 98a. Subsequent ring opening and intramolecular Friedel–Crafts reaction delivered the product 99 in good yield.
5.7. Nickel derived catalysts
Nickel lies just above palladium in the periodic table and can perform many of the same elementary reactions as palladium or platinum. Practically speaking, the cost of nickel in its elemental form is roughly 2000 times lower than palladium and 10000 times lower than platinum on a mole for mole basis which tremendously appeals as a catalyst. As a first-row transition metal, nickel has a small atomic radius, and Ni–ligand bond lengths are often relatively short. The field of nickel catalysis has made tremendous advances in the past decade.73 NiCl2 and its hydrate are occasionally useful in organic synthesis mainly as a mild Lewis acid. Researchers have taken the advantages of all the above features to develop nickel catalyzed propargylic substitution reaction apart from several other reactions.
Fu and co-workers in the year 2008 reported the nickel catalyzed Negishi cross-coupling reactions of propargylic electrophile with secondary alkyl and aryl nucleophiles, Scheme 44.74 Later, asymmetric version of this reaction was also reported, Scheme 45.75
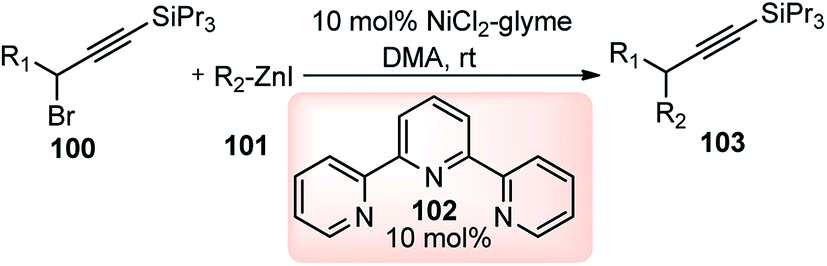 | ||
| Scheme 44 The nickel catalyzed propargylic substitution reaction. | ||
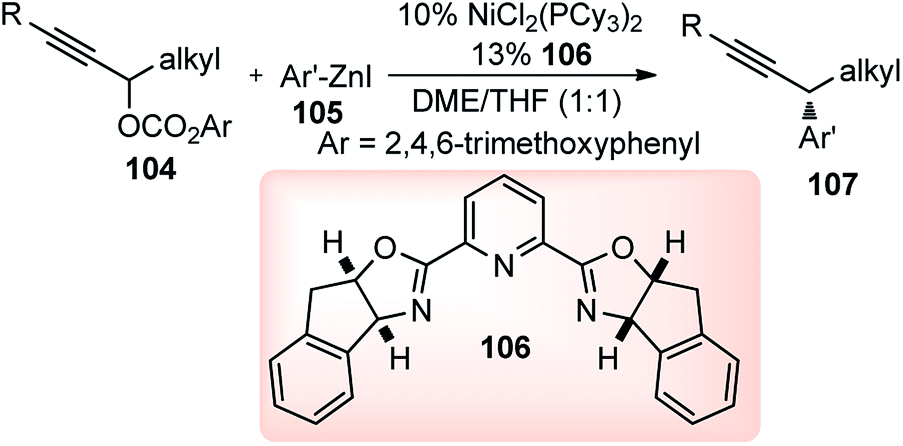 | ||
| Scheme 45 Nickel catalyzed enantioselective propargylic substitution reaction. | ||
This methodology disclosed a base free novel approach for the coupling of propargylic halides 100 with alkylzinc reagents 101 under mild reaction conditions at room temperature. The authors have observed that both propargylic bromides and propargylic chlorides were equally compatible with the reaction. Alkylzincs were readily coupled with R-bromoesters, 1-haloindanes, and allylic chlorides in good yield and ee but the identical reaction condition was inefficient in furnishing the desired product when propargylic halides were reacted with alkyl zinc reagent.76
In the year 2012, Fu and co-workers developed a method that can utilize oxygen leaving groups for the nickel catalyzed asymmetric Negishi reactions of racemic propargylic carbonates 104 with aryl electrophiles 105, Scheme 45.75
The development of a versatile nickel-catalyzed enantioselective cross-coupling process for electrophiles that bear a leaving group other than a halide adds a new dimension to the scope of nickel-catalyzed propargylic substitution reaction.
Zhang and co-workers in the year 2017 described an efficient nickel-catalyzed cross-coupling of deactivated alkylzinc reagents 108 with gem-difluoropropargyl bromides 109, Scheme 46.77
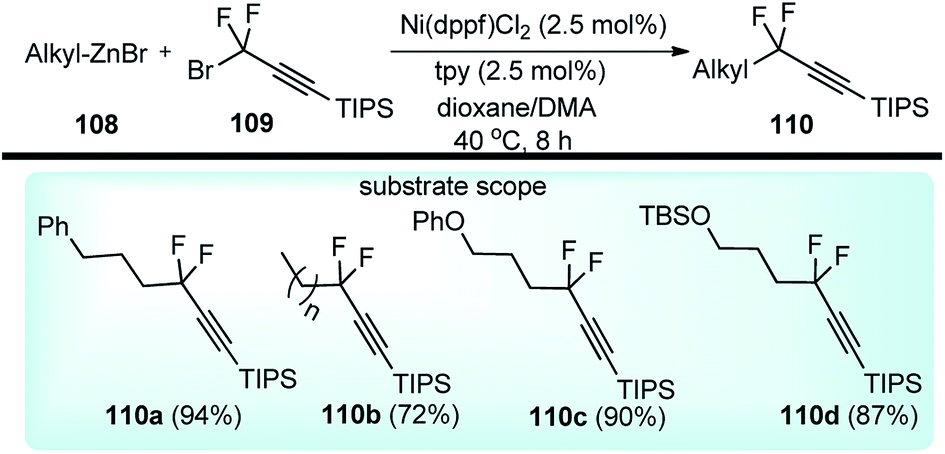 | ||
| Scheme 46 Nickel catalyzed substitution of propargyl bromides with deactivated alkyl zinc compounds. | ||
The reaction proceeded under mild reaction conditions with high efficiency and excellent regioselectivity. Low yields were only observed when other conventional diamine ligands (111–117) were used along with the nickel catalyst, Fig. 3.
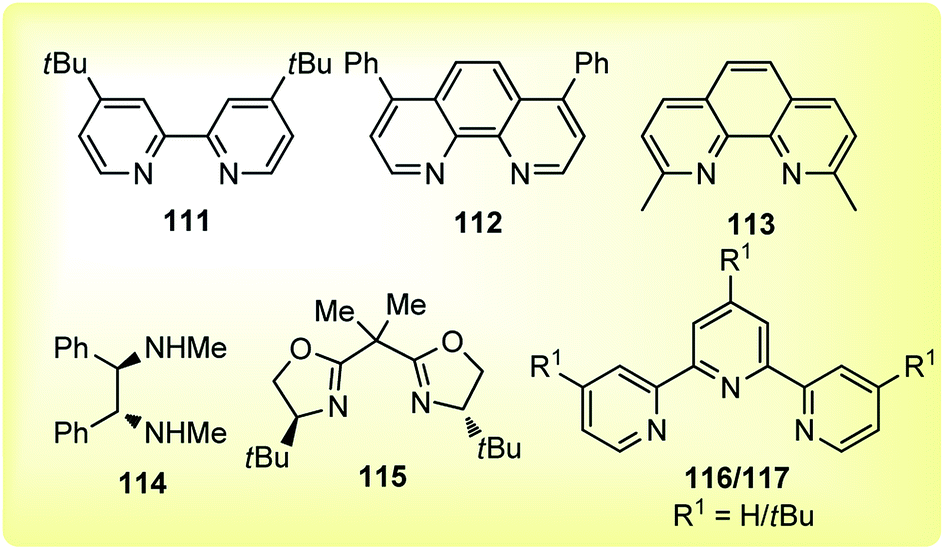 | ||
| Fig. 3 Conventional ligands used in the nickel catalyzed reaction. | ||
Triamine ligand (tpy) significantly improved the product yield without any observable formation of an allenic side product. The reaction was independent of the nature of nickel catalysts and among all the screened nickel salts, Ni(dppf)Cl2 (2.5 mol%) performed best, providing product in 94% yield. No product was obtained in the absence of nickel catalyst and only 9% yield of product was produced when the reaction was carried out in absence of ligand. A plausible reaction mechanism involving a Ni(I/III) catalytic cycle is shown in Scheme 47.
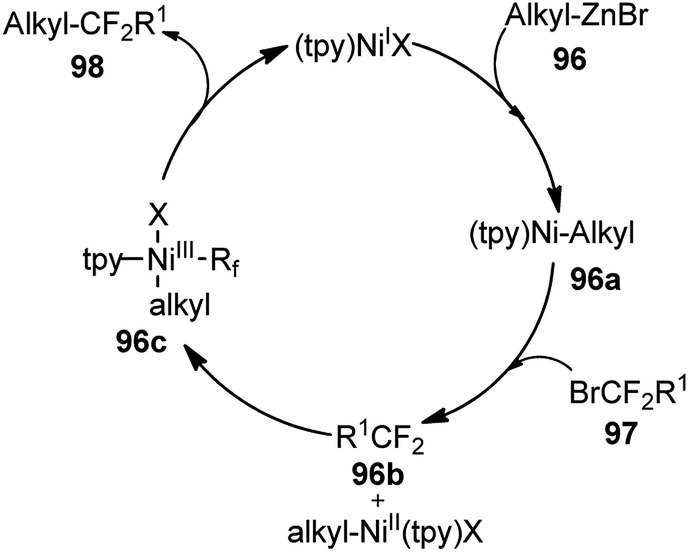 | ||
| Scheme 47 Ni(I/II) catalytic cycle for the nickel catalyzed substitution of propargyl bromides with alkylzinc compounds. | ||
Preliminary mechanistic studies revealed that the reaction was initiated by the transmetalation reaction between Ni(I) and alkylzinc to generate alkyl nickel complex [alkylNi(tpy)] 96a followed by a Ni(I/III) catalytic cycle. The proposed mechanism is different from that of the reported nickel-catalyzed Negishi arylation of propargylic bromides, which has been suggested through a bimetallic pathway.
5.8. Copper derived catalysts
The copper-catalyzed propargylic substitution reaction has become a powerful synthetic method to prepare the compounds containing the propargylic subunit. Compared with the other transition-metals applied in the propargylic substitution, copper has many obvious advantages, such as much more inexpensive, easier to handle, milder reaction condition, and higher selectivity which has resulted in to the development of several Cu-catalyzed propargylic substitution reactions, Scheme 48.19 The astounding progress of the Cu-catalyzed propargylic substitution reaction itself deserves an individual account and Hu has extensively reviewed the progress of the Cu catalyzed propargylic substitution reaction till 2014.19 Significant advances have been achieved in the copper-catalyzed propargylic substitutions after 2014. The Cu-catalyzed propargylic substitution, especially its asymmetric version, which was in a dormant stage, has bloomed to the fullest. Diverse nucleophiles such as nitrogen, carbon, oxygen, sulfur nucleophiles have been successfully applied in the reaction. Many kinds of propargylic compounds have been prepared in satisfactory yields, regioselectivities, and enantioselectivities under very mild conditions. The general pathway for the copper-catalyzed propargylic substitution reaction is highlighted in Scheme 48.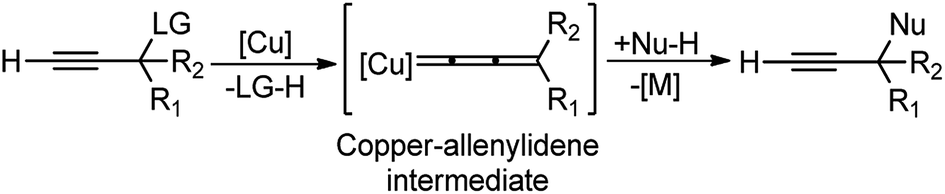 | ||
| Scheme 48 General mechanism for the copper-catalyzed propargylic substitution reaction. | ||
A novel approach for the propargylic allylation of propargyl alcohols with allylsilanes to afford corresponding 1,5-enynes 119 was demonstrated by Yadav and co-workers in 2008 using copper(II) tetrafluoroborate as the catalyst, Scheme 49.78
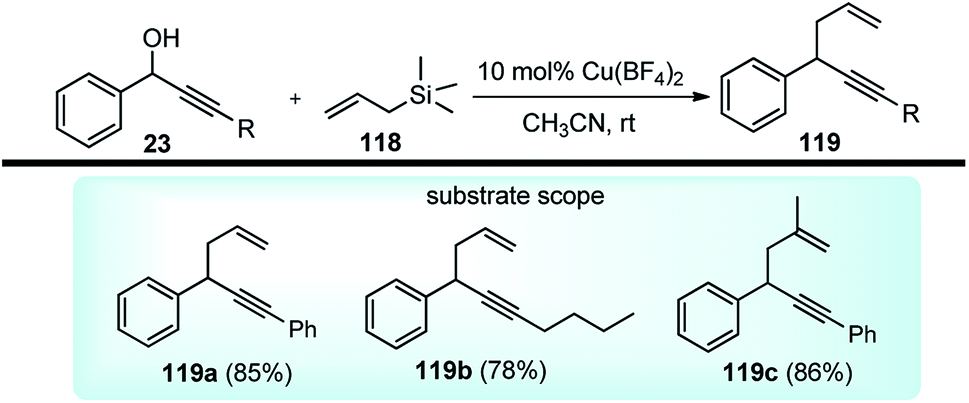 | ||
| Scheme 49 Allylation of propargylic alcohols catalyzed by Cu(BF4)2 catalyst. | ||
This method is compatible with halides, aryl alkyl ethers, alkynes, and alkenes present in the molecule. Among the various copper(II) salts such as Cu(BF4)2, Cu(OTf)2, Cu(acac)2, and Cu(ClO4)2 screened for this transformation, Cu(BF4)2 was found to be most effective. The reaction has a broad substrate scope and works well with both electron donating and electron withdrawing substituents.
With the rapid attractiveness of the catalytic propargylic substitution reactions over the Nicholas reaction, several examples of propargylic substitution reaction have appeared. As an expansion of these studies, their asymmetric versions were also studied and the first examples of enantioselective copper-catalyzed propargylic amination reactions were described by both van Maarseveen's14 and Nishibayashi's79 groups independently in 2008. Maarseveen's group has reported the propargylic substitutions from a variety of readily available propargylic acetates 36 using optically active 2,6-bis(oxazolinyl)pyridines (pybox) as chiral ligands in very high yield and high optical purity, Scheme 50.14
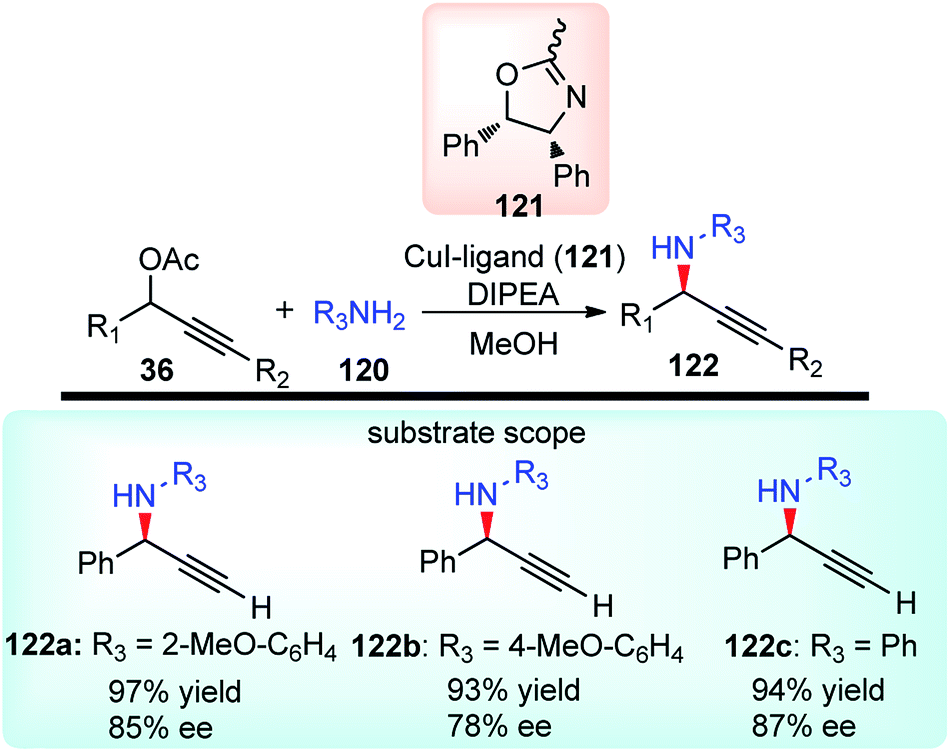 | ||
| Scheme 50 Propargylic amination of various propargylic acetates. | ||
The optimization study revealed the superiority of CuI over other copper salts, such as [Cu(CH3CN)4]PF6, CuOTf·benzene, and Cu(OAc)2, in terms of yield and selectivity. High enantioselectivity and a high reaction rate were only observed in polar protic solvents, especially in methanol. The addition of a base seemed to have a positive impact on both the yield as well as enantioselectivity of the reaction. However, both the yield and enantioselectivity decreased in presence of stronger bases, like 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or cesium carbonate. The best results were achieved with tertiary amines, such as diisopropylethylamine (DIPEA). At lower temperature, there was further enhancement of the enantioselectivity although at the expense of an increased reaction time. The reaction has a wide substrate scope except for aliphatic substrates, where low enantioselectivities were observed. The mechanistic pathway for this reaction as illustrated in Scheme 51 was in accordance to Nishibayashi and co-workers postulated mechanism. The very first step is the formation of the pi-complex between Cu-catalyst and the alkyne followed by the formation of Cu–allenylidene complex 36b with the concomitant removal of the OAc group. The amine nucleophile then attacks the Cu–allenylidene complex 36b. Selective blocking of one side of the cationic intermediate by the copper–pybox complex directs the amine to selectively attack from one face. After proteolysis, the product 122 is released to complete the catalytic cycle.
 | ||
| Scheme 51 Mechanism for the propargylic amination of propargyl alcohols. | ||
Nishibayashi and co-workers on the other hand, described the reaction between 1-phenyl-2-propynyl acetate and N-methylaniline to form the substituted product N-methyl-N-(1-phenyl-2-propynyl) aniline with good enantioselectivity using Cu(OTf)2.1/2C6H6 and (R)-Cl-MeO-BIPHEP ligand under basic condition.79
Followed by the seminal work by Maarseveen's and Nishibayashi's groups, in the year 2010, Nishibayashi and co-workers have reported a copper-catalyzed enantioselective propargylic amination of propargylic esters 123 with various secondary amines 124 using optically active diphosphines such as BINAP and BIPHEP as chiral ligands, Scheme 52.80
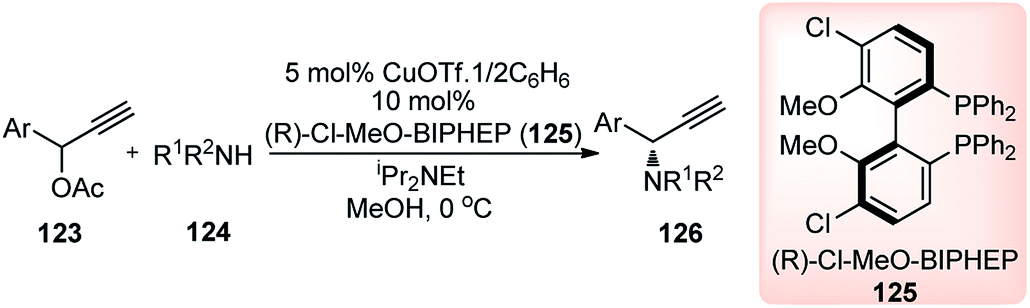 | ||
| Scheme 52 Copper-catalyzed enantioselective propargylic amination of propargylic esters. | ||
The reaction is believed to pass through a copper–allenylidene complex followed by the attack of the amine. A transition state consisting of copper–allenylidene complex bearing a chiral ligand BIPHEP was proposed to account for the high enantioselectivity of the product, Fig. 4.
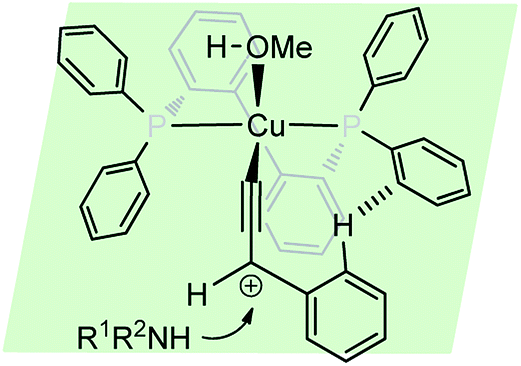 | ||
| Fig. 4 Probable transition state for the copper-catalyzed propargylic substitution reaction. | ||
The re-facial attack of the N-methyl aniline, due to the edge-to-face aromatic interaction between the two phenyl groups is considered to play a significant role in achieving the high enantioselectivity.81
Introduction of a better leaving group than acetate, in the propargylic system, might promote the propargylic amination of propargylic esters bearing an alkyl group at the propargylic position. With this hypothesis, Nishibayashi and co-workers in 2011 reported the propargylic amination of propargylic pentafluorobenzoates 127 with amines 128 in the presence of catalytic amounts of a copper complex and (R)-BINAP in good yields and high enantioselectivity, Scheme 53.82
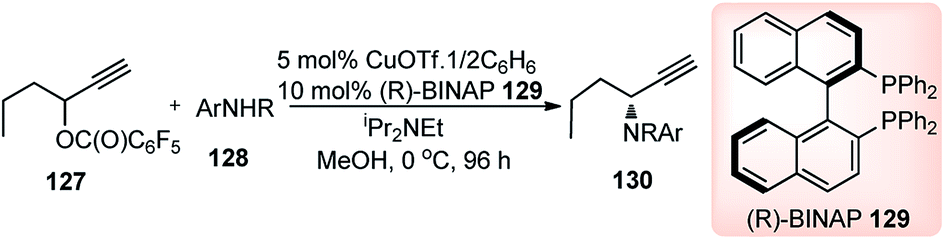 | ||
| Scheme 53 Copper catalyzed propargylic amination of propargyl esters. | ||
Like all other Cu-catalyzed propargylic reaction the above propargylic amination proceeded via copper–allenylidene complexes as the key intermediate.
Rapid assembly of structural complexity for the synthesis of N-containing heterocycles via intramolecular cycloisomerization emerged as an exceptionally powerful tool for the synthesis of various heterocyclic structures. To achieve such tandem methodologies, Zhan and co-workers in the year 2011 developed a Cu(OTf)2 catalyzed synthesis of 2,4,6-trisubstituted pyrimidines 132 from propargylic alcohols 23 with amidine 131, Scheme 54.83
 | ||
| Scheme 54 Cu(OTf)2 catalyzed synthesis of 2,4,6-trisubstituted pyrimidines from propargylic alcohols. | ||
Several other catalysts were tested, like, BiCl3, InCl3, FeCl3, Bi(OTf)3, Zn(OTf)2, and AgOTf. Regrettably, all of them were found either inefficient or inferior as compared to Cu(OTf)2. Solvents like acetonitrile, 1,2-dichloroethane, nitromethane, and toluene were also tested and were found to facilitate the reaction. However, the best result was achieved with chlorobenzene. Surprisingly, dichloromethane failed to promote this reaction which clearly implied that the reaction rate was influenced by the boiling point and polarity of the solvent used. The proposed mechanism for the pyrimidine ring 132 formation via a propargylation–cyclization–oxidation tandem sequence is represented in Scheme 55.
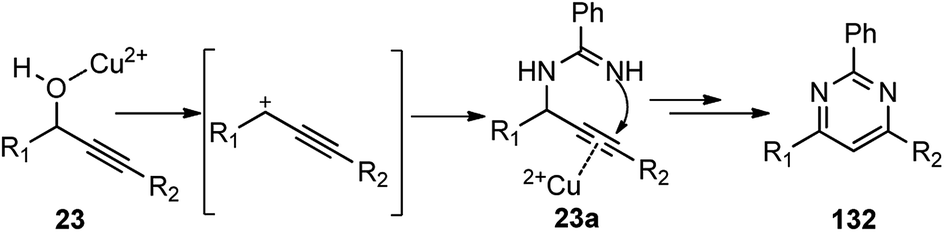 | ||
| Scheme 55 Mechanism for the Cu(OTf)2 catalyzed synthesis of 2,4,6-trisubstituted pyrimidines from propargylic alcohols. | ||
Cu(OTf)2 acts as a bifunctional catalyst, by promoting an SN1 substitution of propargylic alcohol 23 by the amidine 131, followed by facile intramolecular cyclization via 6-endo-dig process via the triple bond activation to produce cyclic dihydropyrimidine intermediate. Subsequent air oxidation and aromatization of the cyclic dihydropyrimidine intermediate leads to the pyrimidine 132.
Cooperative catalysis using organocatalysts and transition metals has become a new tool to access compounds with excellent enantioselectivities which were otherwise difficult to prepare and therefore has gained significant interest in the chemical community in the recent years.84,85
Nishibayashi and co-workers in 2011 described an enantioselective propargylic substitution reaction of propargyl acetates 133 with α-branched aldehydes 134, using both the transition metal catalyst (copper complex) and organocatalyst (secondary amine), Scheme 56.86
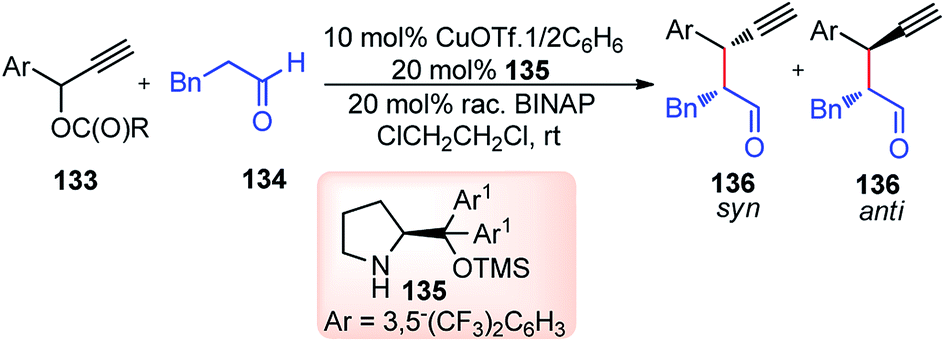 | ||
| Scheme 56 Propargylic substitution employing cooperative catalysis. | ||
It was envisioned that both the copper complex and organocatalyst will simultaneously activate the propargylic ester 133 and aldehyde 134 to promote the enantioselective propargylic alkylation. In fact, the authors have found that reactions of propargylic esters 133 with aldehydes 134 in the presence of a copper complex bearing racemic diphosphine and an optically active secondary amine 135 as co-catalysts furnished the corresponding propargylic alkylated products 136 in good yields as a mixture of two diastereomers with a high enantioselectivity.
A proposed reaction pathway is presented in Scheme 57. The initial step is the formation of a copper–allenylidene complex 133a followed by the attack of an enamine, generated in situ from aldehyde and chiral amine 135, resulting in the formation of alkyne complex 133b via an acetylide complex. Subsequent ligand exchange with another propargylic ester furnishes the desired alkylated product 136.
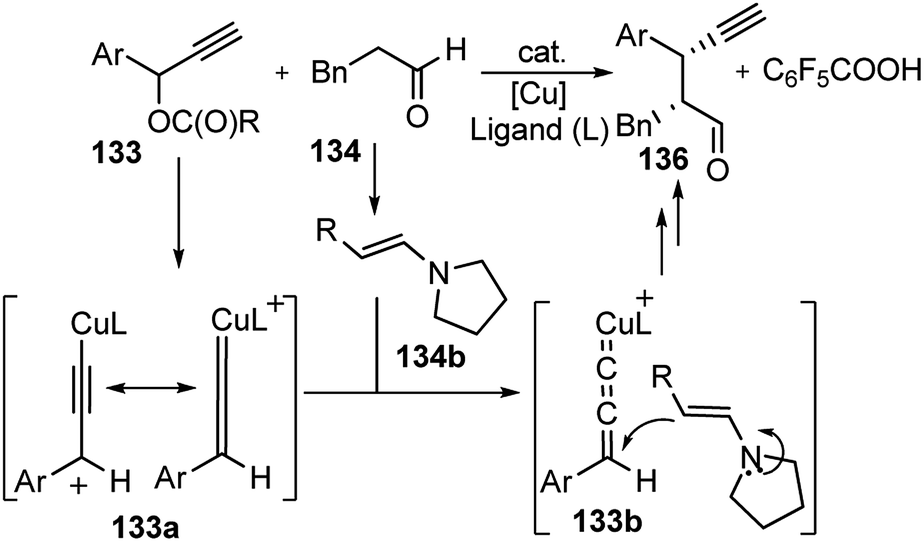 | ||
| Scheme 57 Mechanism for the propargylation of aldehydes by cooperative catalysis. | ||
In 2014, Hu and co-workers developed a facile approach for the synthesis of β-ethynyl ketones 139 in a highly enantioenriched form by copper-catalyzed decarboxylative propargylic alkylation of propargyl β-ketoesters 137, Scheme 58.87
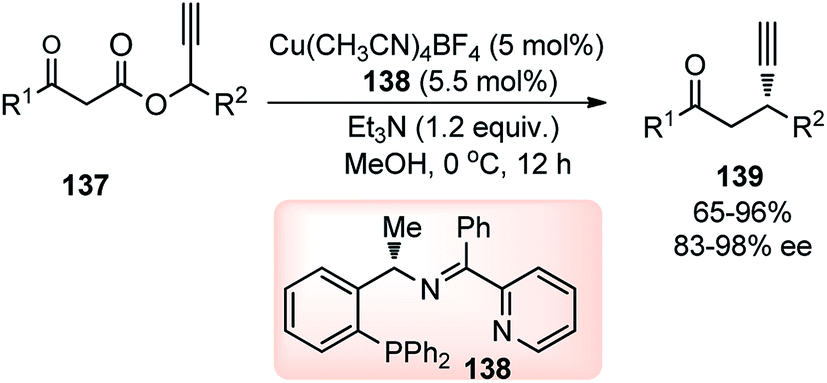 | ||
| Scheme 58 Copper-catalyzed decarboxylative propargylic alkylation of propargyl β-ketoesters. | ||
A new chiral tridentate ketimine P,N,N-ligand was designed to provide a facile access to a variety of β-ethynyl ketones 139 in good yields and with high enantioselectivities. Careful evaluation of several ligands and copper catalysts disclosed chiral tridentate P,N,N-ligand in conjunction with [Cu(CH3CN)4BF4] as the most promising combination. A reaction pathway is highlighted in Scheme 59 where asymmetric decarboxylative propargylic alkylation occurs when propargyl β-ketoesters 137, containing propargyl moiety and a nucleophile together with an ester functional group, is subjected to a chiral copper catalyst.
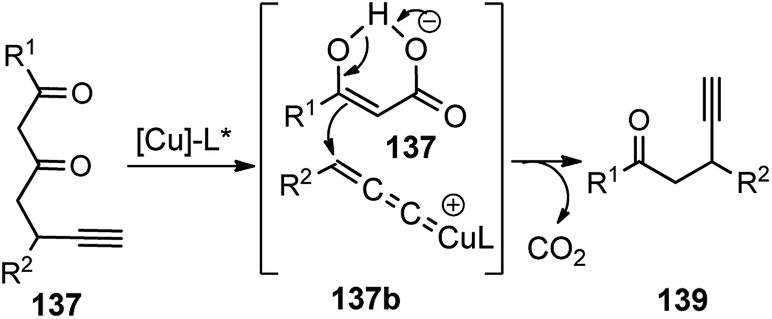 | ||
| Scheme 59 Mechanism for the copper-catalyzed decarboxylative propargylic alkylation of propargyl β-ketoesters. | ||
A broad range of 1-phenyl-2-propynyl 3-oxo-3-arylpropanoates 137 worked efficiently to provide the desired β-ethynyl ketones in good to excellent yields and with very high enantioselectivities. Electron-donating and electron-withdrawing substituents in the para or meta position of the phenyl ring were compatible with this transformation. The reaction works well even for heteroaromatic and aliphatic β-ketoesters although with a higher catalyst loading to reach complete conversion. However, the stereoselectivity reduces for substrates having a substituent at the ortho position of the phenyl ring.
Hu and co-workers in the year 2014 utilized their newly designed tridentate P,N,N ligand in conjunction with Cu(OTf)2 catalyst for the enantioselective synthesis of highly functionalized dihydrofurans 141 by a [3 + 2] cycloaddition of β-ketoesters 140 with propargylic ester 123, Scheme 60.88
 | ||
| Scheme 60 Enantioselective synthesis of highly functionalized dihydrofurans from propargyl acetates and β-ketoesters. | ||
Through ligand and copper salts screening identified chiral tridentate P,N,N ligand and Cu(OTf)2 as the best combination, furnishing the product in 90% yield with 95% ee. No reaction occurred in the absence of a base and interestingly poor regioselectivity was observed when iPr2NEt was used in place of Et3N. Among the solvents that were tested, MeOH was the only suitable one, and no reaction occurred in DCM and toluene. The reaction proceeds via catalytic sequential propargylation/cycloisomerization reactions of propargylic alcohols 123 with 1,3-dicarbonyl compounds 140 in presence of a chiral copper catalyst and ligand 138, Scheme 61.
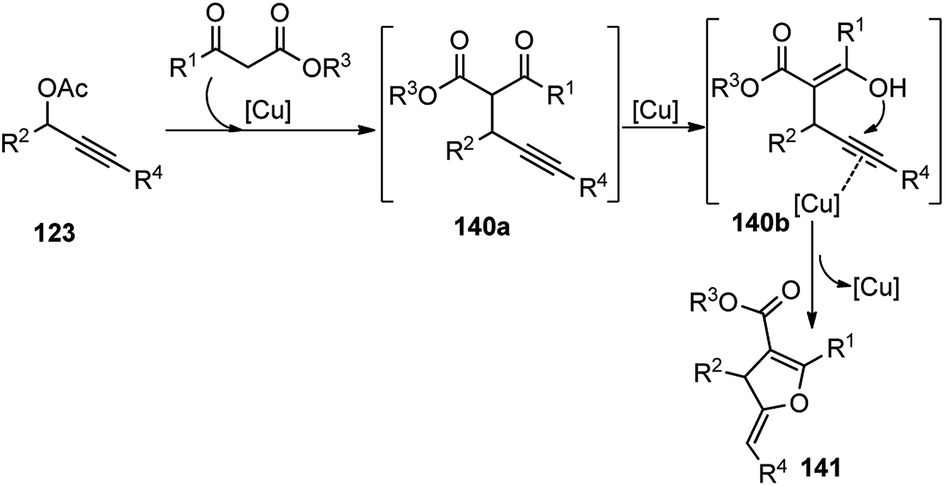 | ||
| Scheme 61 Mechanism for the synthesis of highly functionalized dihydrofurans from propargyl acetates and β-ketoesters. | ||
Bulky and structurally rigid tridentate ketimine-type P,N,N ligand was responsible for the high stereoinduction during the propargylation step. The reaction has a wide substrate scope and tolerates all kinds of electron donating and withdrawing groups. This method allows obtaining optically active 2,3-dihydrofurans bearing an exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in high yield and enantioselectivity.
C bond in high yield and enantioselectivity.
Hou and co-workers in 2009 demonstrated the use of enamines as a source of carbon nucleophiles for the first time in the copper-catalyzed enantioselective propargylic substitution reactions of propargylic acetates to afford ethynyl-substituted ketones in high yield and excellent enantioselectivities.89
Followed by their initial work on the Cu-catalyzed substitution of propargylic esters with enamines, Hu and co-workers in the year 2014 once again demonstrated the use of chiral P,N,N ligand in the copper-catalyzed propargylic alkylation of enamines derived from acyclic ketones 142 with propargylic esters 123 in excellent enantio- and diastereoselectivities, Scheme 62.90
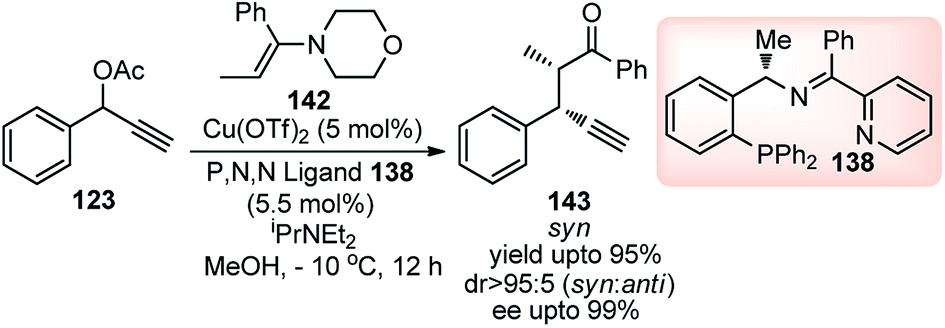 | ||
| Scheme 62 Copper catalyzed propargylic alkylation of enamines derived from acyclic ketones with propargylic esters. | ||
High diastereo and enantioselectivities observed is probably due to the interaction of the Cu–allenylidene complex with the chiral P,N,N-ligand shown in the transition state, Fig. 5. The edge-to-face aromatic interaction81 and the steric hindrance directs the attack of the enamine at the propargylic cation favorably from the si-face.
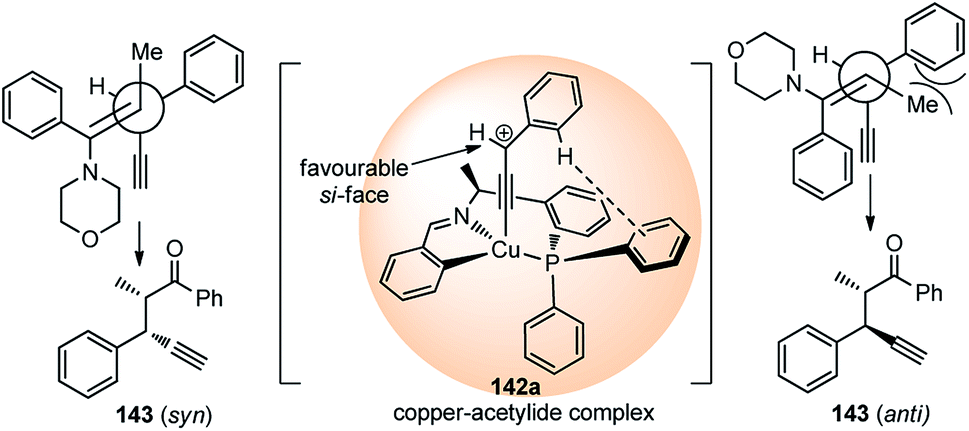 | ||
| Fig. 5 Proposed transition state for the Cu-catalyzed substitution of propargylic esters with enamines. | ||
The final cyclization step leading to the alkylation products in the above methodology is greatly suppressed by the morpholine-derived cyclic enamines because of the increased stability of the resulting morpholinium ions.90 This observation encouraged the authors to extend the application of the above-mentioned transformation towards ketone enamines.
The same group have developed a Cu-catalyzed [3 + 3] cycloaddition of propargylic acetates 123 with cyclic N,N-diethyl-1-enamines 144 for the facile access of optically active bicyclo[n.3.1] frameworks 147 bearing three stereocenters with highly diastereo- and enantioselectivity, Scheme 63.91
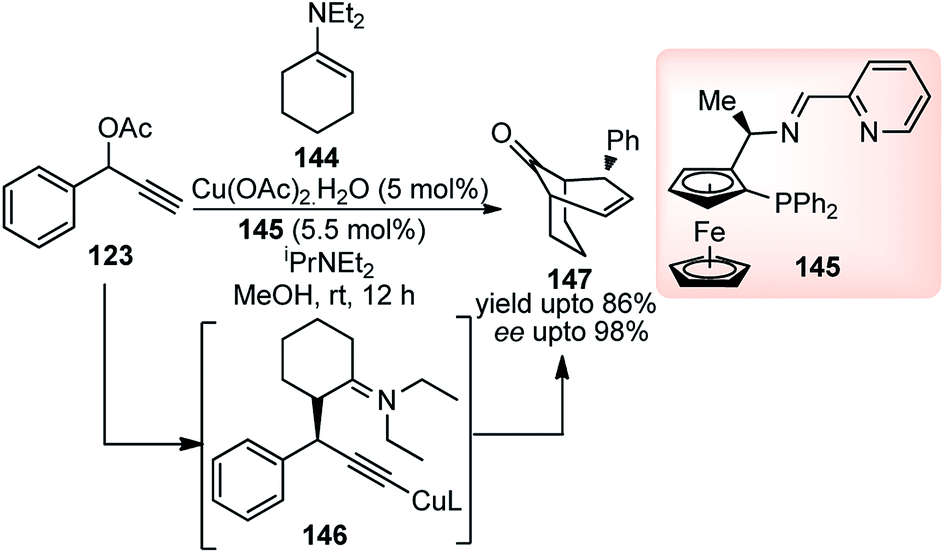 | ||
| Scheme 63 Cu-catalyzed [3 + 3] cycloaddition of propargylic acetates with cyclic N,N-diethyl-1-enamines. | ||
Among various other ligands screened, ferrocenyl ligand displayed the best performance, affording the cycloadduct 147 in good yield (86%) with high enantioselectivity. The reaction proceeds via the formation of Cu–allenylidene complex as a key intermediate. Subsequent nucleophilic attack of the enamine furnishes the corresponding Cu–acetylide complex which is the key step for the stereoselection. It was further observed by the authors that the presence of cyclic enamine, derived from morpholine, greatly reduced the selectivity for cycloaddition and predominantly formed the alkylation product.
Analogous to the above-reported work, Hu in 2016 has developed a highly diastereo- and enantioselective copper-catalyzed propargylic alkylation of propargylic acetates with morpholine-derived cyclic enamines for the synthesis of two vicinal tertiary stereocenters.92 Using chiral 1-phenylethylamine-derived tridentate P,N,N-ligand, good to excellent diastereo- and enantioselectivities were achieved for a wide range of substrates. Less sterically hindered P,N,N-ligand showed better performance in the propargylic alkylation of cyclic enamines compared to the acyclic enamines in terms of chemoselectivity, because of its lower reactivity which suppressed the cyclization step.
In 2014 Hu and co-workers reported the first enantioselective Cu-catalyzed propargylic amination of propargylic carbamates 148 through decarboxylation in presence of Cu(OAc)2·H2O and a chiral ketimine ligand 138, Scheme 64.93
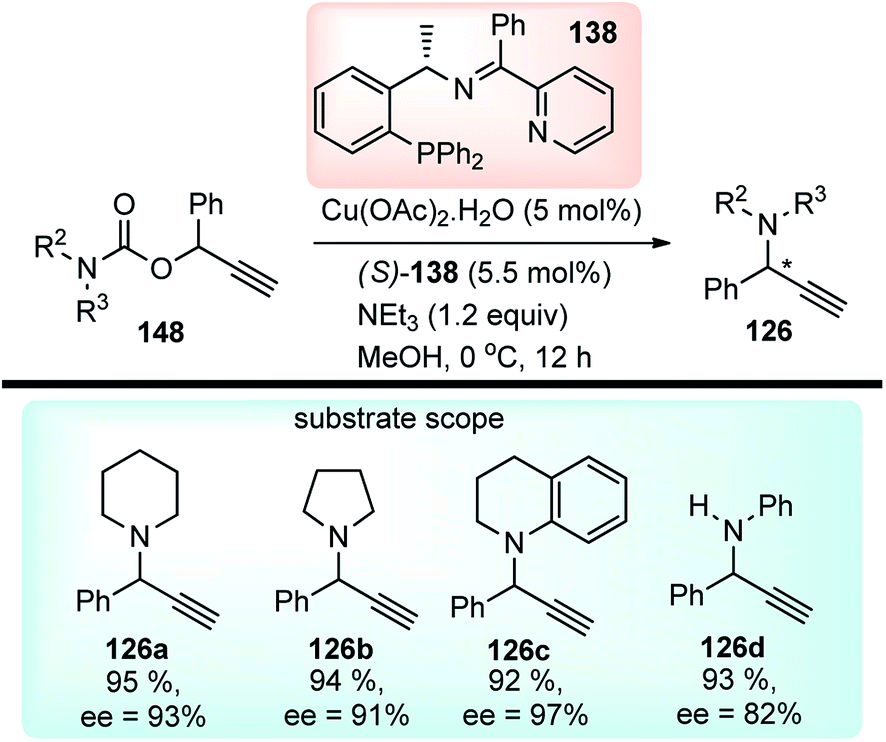 | ||
| Scheme 64 Copper catalyzed propargylic amination of propargyl carbamates. | ||
Other copper salts like CuI, CuCl, Cu(OTf)2, Cu(CH3CN)4BF4, Cu(CH3CN)4ClO4, in combination with bases like NEt3, DBU, iPr2NEt were not efficient in terms of product yields and selectivity. The polarity of the solvent had a significant impact on the reaction outcome as the use of solvents like THF and toluene furnished only 10% and 15% ee respectively although with superior product yield. The transition state consisting of copper–allenylidene complex bearing a ketimine ligand 138 like the earlier reported TS (Fig. 5) was proposed to account for the high enantioselectivity of the product. The reaction has a broad substrate scope and tolerates tertiary as well as secondary carbamates and with the substitutions in the propargyl moiety.
With the advent of Cu-catalyzed enantioselective propargylic substitution reaction, it was realized that phenyl hydrazine could be an interesting substrate to access N-containing heterocycles by intramolecular tandem cycloaddition. Since both the nitrogen atoms of hydrazines are suitable nucleophiles, one major concern in the implementation of this cycloaddition was the possible formation of two regioisomeric cycloadducts via the competitive attack of N-atoms of hydrazines at the Cγ-atom of Cu–allenylidene complex. Hu and co-workers in the year 2015 have reported a highly regio- and enantioselective Cu-catalyzed [3 + 2] cycloaddition of propargylic acetates 123 with monosubstituted hydrazines 149, followed by a spontaneous 1,3-proton migration, for the stereoselective construction of optically active 2-pyrazolines, Scheme 65.94
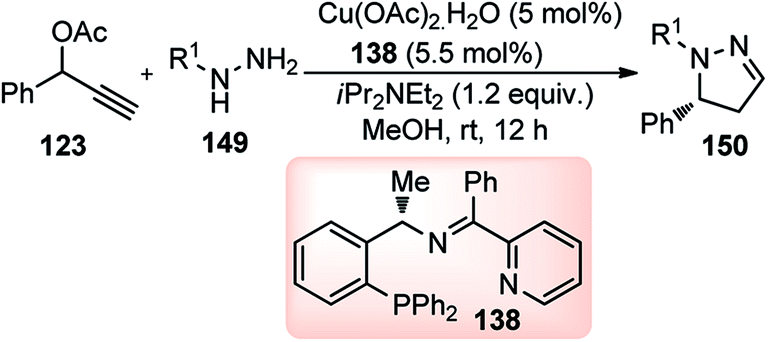 | ||
| Scheme 65 Cu-catalyzed [3 + 2] cycloaddition of propargylic acetates with monosubstituted hydrazines. | ||
Interestingly, instead of the expected 3-pyrazoline the thermodynamically more stable 2-pyrazoline 131 was obtained, suggesting a cycloaddition via “pathway-a” took place, followed by a spontaneous 1,3-proton shift under the reaction condition, Scheme 66.
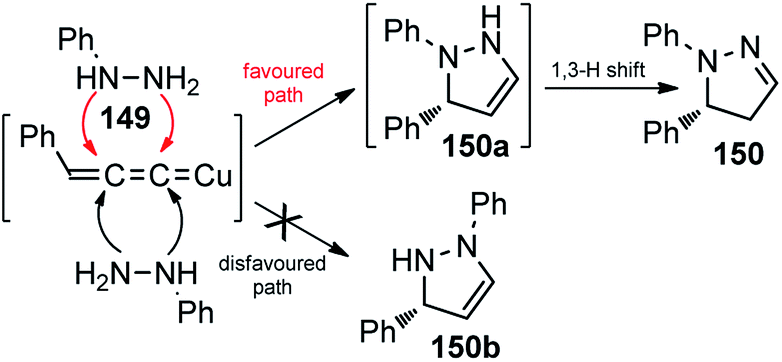 | ||
| Scheme 66 Competitive pathway for the Cu-catalyzed [3 + 2] cycloaddition of propargylic acetates with monosubstituted hydrazines. | ||
Tridentate P,N,N-ligand was reported to furnish regioselectively 2-pyrazoline derivatives in high yields (90%) and with excellent enantioselectivity (94% ee) irrespective of the electronic property of the phenyl ring. However, the reaction was sensitive to the substitution pattern on the phenyl ring in the hydrazine. The reaction with the substrates bearing an ortho-substituent resulted in the significantly decreased yield presumably due to the steric hindrance. Remarkably, non-aromatic hydrazine also turned out to be a suitable reaction partner, providing the cycloadduct in 70% yield and with 92% ee. However, unsubstituted hydrazine did not furnish any desired product but resulted in the decomposition of the starting material.
The high regio- and enantioselectivity was rationalized via the favoured si-facial attack of the hydrazine to the Cu–allenylidene complex with chiral P,N,N ligand as represented in the transition state structure in Fig. 6. The combined effects of edge-to-face aromatic interaction as well as the steric hindrance of ligand directs the approach of phenylhydrazine 149 from the si-face, thus facilitating the formation of (R)-cycloadduct.
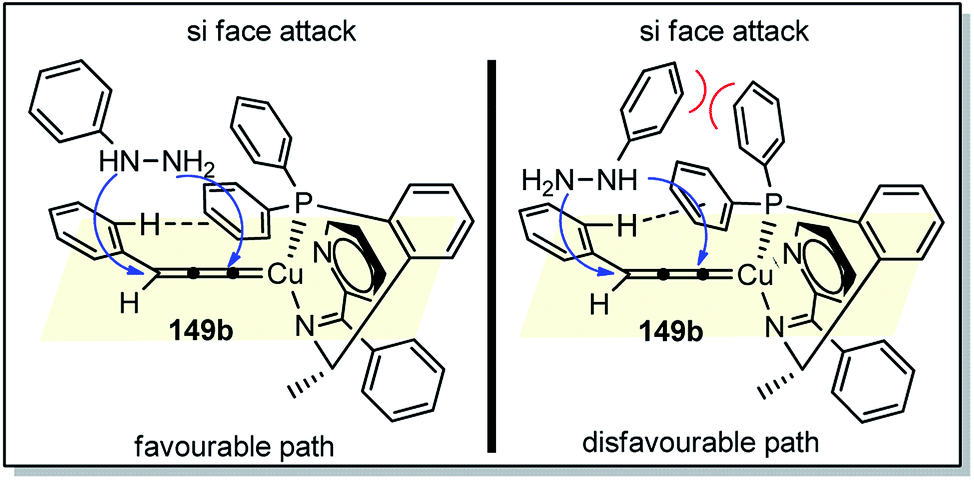 | ||
| Fig. 6 Proposed transition state for the Cu-catalyzed [3 + 2] cycloaddition of propargylic acetates with monosubstituted hydrazines showing favoured and disfavoured cycloaddition pathway. | ||
The methodology represented a novel strategy for the stereoselective access to enantioenriched 2-pyrazolines 150 without a substituent at the 3-position.
Based on the proficiency on the copper-catalyzed propargylic transformation for the synthesis of heterocyclic compounds, Hu and co-workers envisioned that copper catalyzed cyclization of propargylic carbonates with β-enamino esters should also be a suitable approach to construct fully substituted pyrroles which were earlier reported by Yoshida and co-workers using costly palladium.95
Therefore, Hu and co-workers in the year 2015 developed the Cu-catalyzed [3 + 2] cycloaddition of propargylic esters 123 with β-enamino esters 152 for the synthesis of highly functionalized pyrroles 153, Scheme 67.96
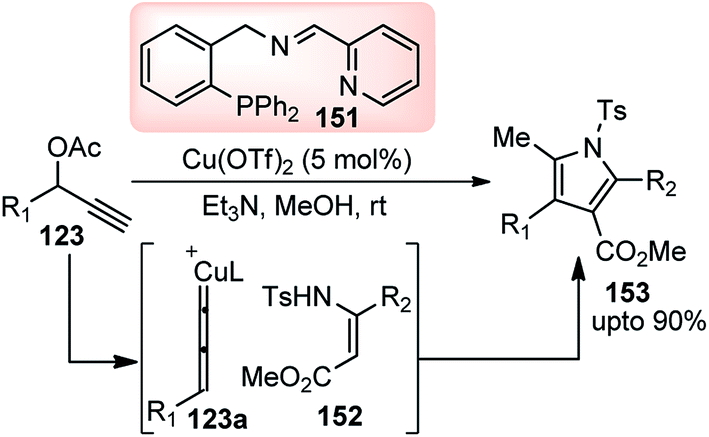 | ||
| Scheme 67 Cu-catalyzed [3 + 2] cycloaddition of propargylic esters with β-enamino esters. | ||
Except for the P,N,N-ligands all other ligands like DPPF, DPPP, and BINAP, as well as bidentate nitrogen ligands such as biPy and 1,10-phenanthroline yielded the [3 + 2] cycloaddition product 153 in moderate yield. Among several copper salts tested, Cu(OTf)2 proved to be the best choice, which gave the corresponding product in 87% yield. The reaction outcome was highly dependent on the presence and nature of the base. No reaction occurred in the absence of base and Et3N provided a better result than i-Pr2NEt and DBU. Among different solvents tested, MeOH was only found to be suitable. The plausible mechanism for the formation of the product is depicted in Scheme 68. The key step is the formation of the Cu–allenylidene complex form the propargyl acetate which was subsequently attacked by the β-enamino ester 152 to generate Cu–acetylide complex. Then, intramolecular nucleophilic attack of N at the Cβ atom of 152a would, at last, afford the 2,3-dihydropyrrole 153 bearing an exocyclic double bond at the 2-position.
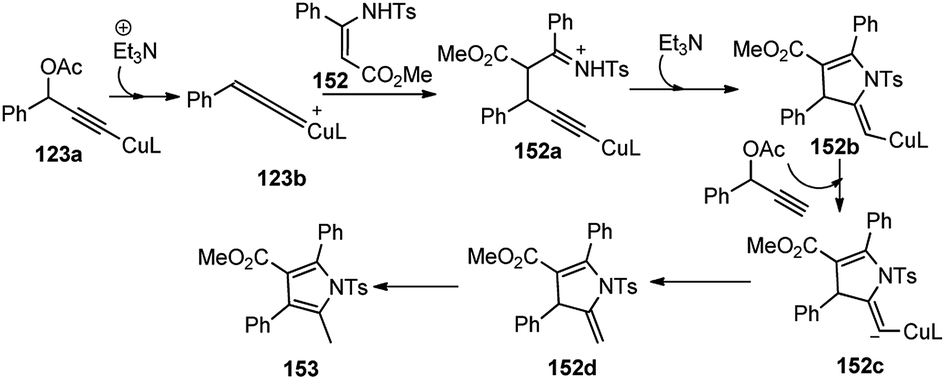 | ||
| Scheme 68 Plausible mechanism for the Cu-catalyzed [3 + 2] cycloaddition of propargylic esters with β-enamino esters. | ||
Wu and co-workers in the year 2014 demonstrated the use of pybox as a ligand in the copper-catalyzed reaction between 2-substituted benzofuran-3(2H)-one and propargyl acetate for the highly diastereo and enantioselective synthesis of 2,2-disubstituted benzofuran-3(2H)-one.97
Nishibayashi and co-workers in the year 2015, developed first transition-metal catalyzed enantioselective propargylic etherification of propargylic esters 154 with aliphatic alcohols 155 as well as phenols in the presence of a catalytic amount of copper–pybox complex 156 in high yields with excellent enantioselectivity, up to 99% ee, Scheme 69.98
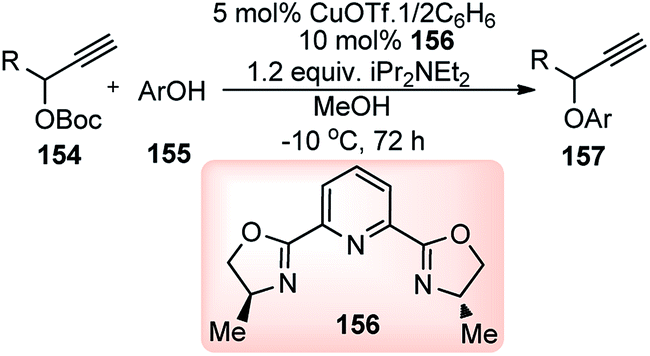 | ||
| Scheme 69 Enantioselective propargylic etherification of propargylic esters with aliphatic alcohols. | ||
After careful screening, MePybox was found to be the best chiral ligand and the copper-catalyzed enantioselective propargylic etherification of propargylic carbonates works efficiently not only with simple alcohols but also with phenols also.
Hu and co-workers in 2014 reported for the first time an efficient copper-catalyzed propargylic substitution of propargylic acetates with a variety of β-dicarbonyl compounds in moderate yield and excellent enantioselectivities using ketamine P,N,N ligand.99 The same reaction was also reported by the research group of Maarseveen employing a Cu–Pybox complex as the catalyst affording the desired product in moderate yield and low ee.100 Unfortunately, the reaction of propargylic acetates with dimethyl malonate resulted in a mixture of unidentified products.
In the search for a suitable malonate surrogate for this type of reaction, trialkyl methanetricarboxylate, with pKa approximately equals that of Meldrum's acid (ca. 7.3) provided an impressive solution. The success of copper–Pybox complexes for enantioselective propargylic substitution reactions prompted Wu and co-workers in the year 2015 for the first time developed enantioselective propargylation of trialkyl methane tricarboxylate 159 with propargylic alcohol derivatives 158, using pybox ligand 160, Scheme 70.101
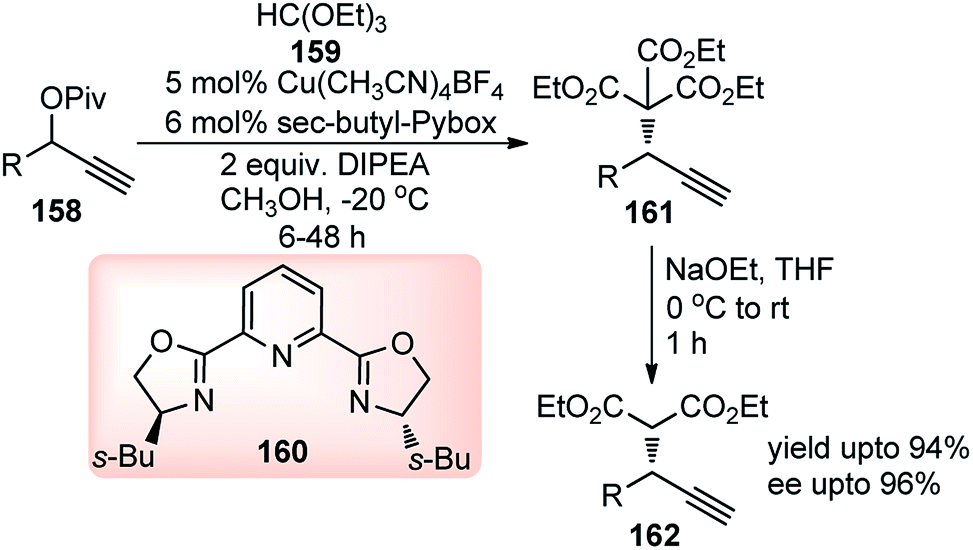 | ||
| Scheme 70 Enantioselective propargylation of trialkyl methantricarboxylate with propargylic alcohol derivatives. | ||
The reaction proceeded via the formation of copper allenylidene complexes as key intermediates following the attack of the trialkyl methane tricarboxylate. The temperature shift from room temperature to 0 °C drastically improved the enantioselectivity from 31% ee to 68% ee. The ee further improved to 76%, when the reaction was performed at −20 °C although required a longer reaction time for completion.
Sakai and co-workers in the year 2016 developed novel preparation of 2,3-disubstituted indole derivatives 165 form propargylic acetates 164 and anilines 163 using a co-operative catalytic system composed of Cu(OTf)2 and Me3SiCl via a [3 + 2] annulation reaction, Scheme 71.102
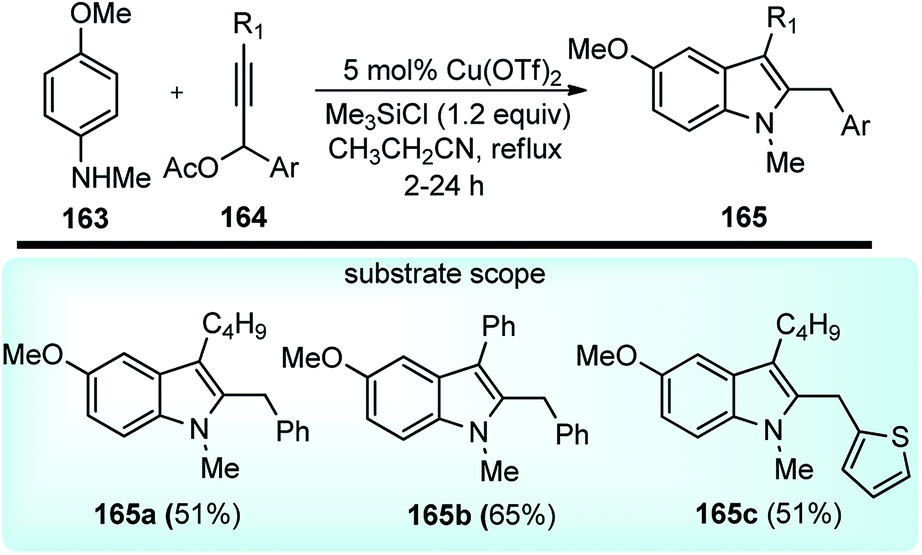 | ||
| Scheme 71 Synthesis of 2,3-disubstituted indole derivatives from propargylic acetates and anilines using a copper catalyst. | ||
Several Lewis acids, when tested for the coupling of N-methyl-p-anisidine 163 and propargylic acetate 164 and the screening, revealed that 20 mol% of either AlCl3, Cu(OTf)2 and ZnCl2 doped with Me3SiCl would furnish products in good yield. Catalysts like CuCl and InBr3 were reported to reduce the chemical yield. Interestingly 5 mol% of Cu(OTf)2 preserved the catalytic activity, producing the corresponding indole in a 90% isolated yield. Control experiment clearly pointed out the role of a cooperative catalytic system composed of Cu(OTf)2 and Me3SiCl to promote the annulation of anilines with propargylic acetates. The plausible reaction pathway for the formation of 2,3-disubstituted indoles is shown in Scheme 72.
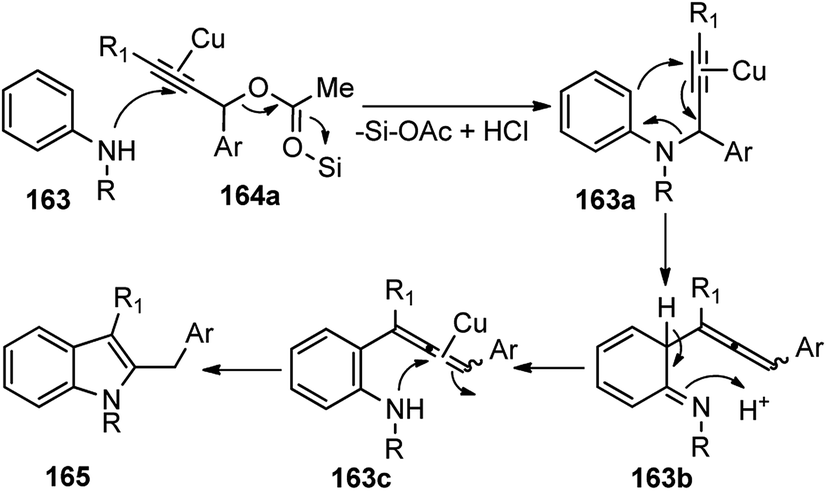 | ||
| Scheme 72 Mechanism for the synthesis of 2,3-disubstituted indole derivatives from propargylic acetates and anilines using cooperative catalysis. | ||
Attack of the amino group on the propargylic carbon center generates the propargylic amine intermediate 163a. Then further transformation via an aza-Claisen rearrangement through activation of the alkyne 163a by the copper catalyst produces allene intermediate 163b. Subsequent aromatization, followed by the intramolecular cyclization produces indole derivative 165.
Hu and co-workers in the year 2016, have reported a highly enantioselective copper-catalyzed formal [3 + 2] cycloaddition of 3-trimethylsilyl propargylic acetates 144 with either β-naphthols 145, Scheme 73.103
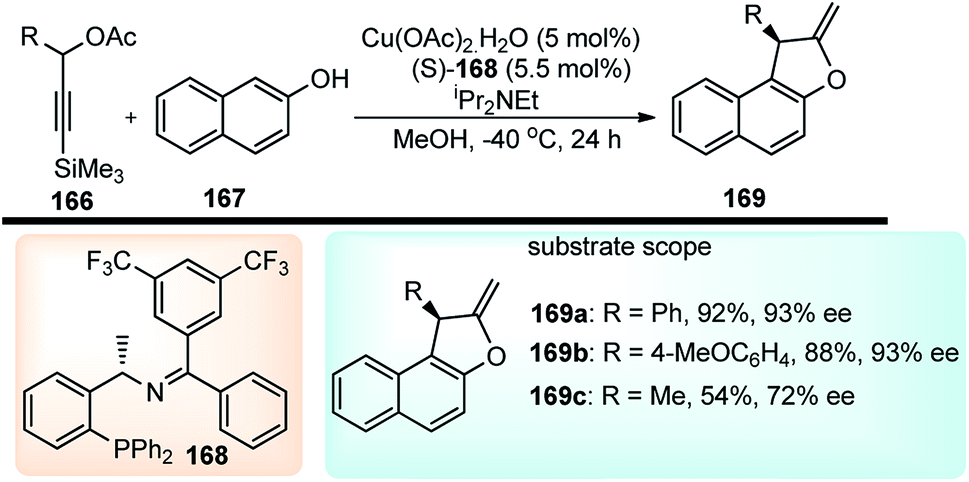 | ||
| Scheme 73 Copper-catalyzed [3 + 2] cycloaddition of 3-trimethyl-silylpropargylic acetates with either β-naphthols or electron rich phenols. | ||
Benzofurans were obtained in good yields and with high enantioselectivities under the influence of Cu(OAc)2·H2O and ketimine P,N,N-ligand. The reaction proceeds via a desilylation followed by the nucleophilic attack by the naphthol, as highlighted in Scheme 74.
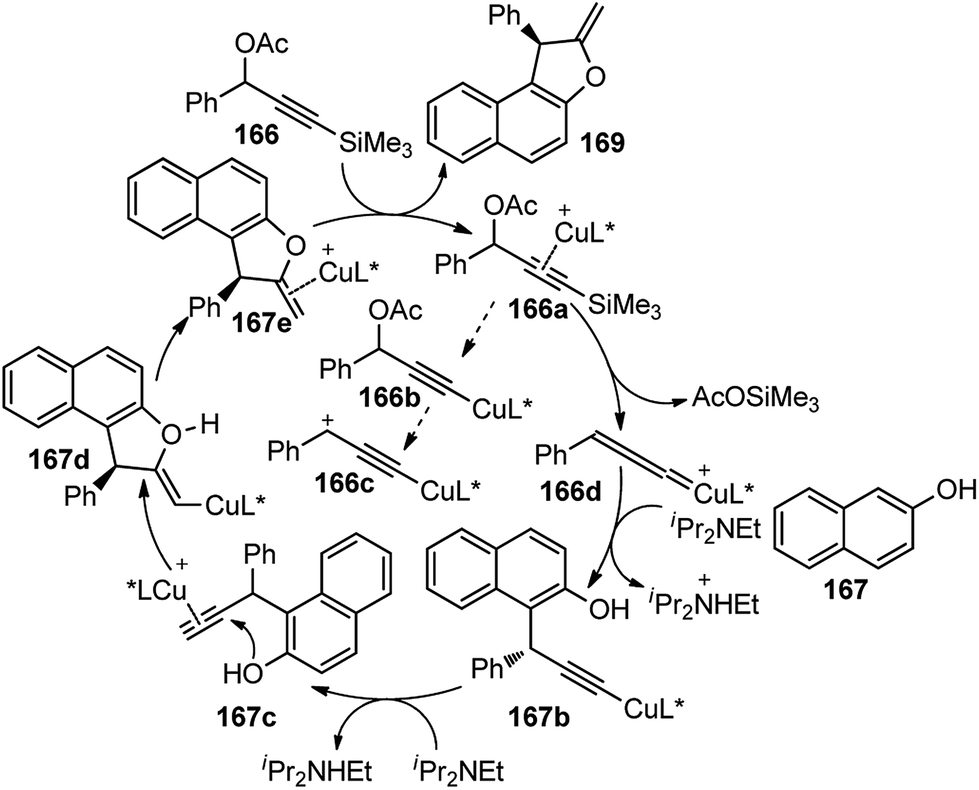 | ||
| Scheme 74 Mechanism for the copper-catalyzed formal [3 + 2] cycloaddition of 3-trimethyl-silylpropargylic acetates with either β-naphthols. | ||
The reaction has a wide substrate scope and represented the first example of a catalytic propargylic transformation via desilylation strategy where copper allenylidene complex was efficiently generated from 3-trimethylsilyl propargylic esters by a copper promoted Si–C(sp) bond cleavage. The high enantioselectivity of the products was explained probably due to the favorable si-face attack by the β-naphthol to the gamma carbon of the Cu–allenylidene complexes, as shown in the transition state diagram, Fig. 7.
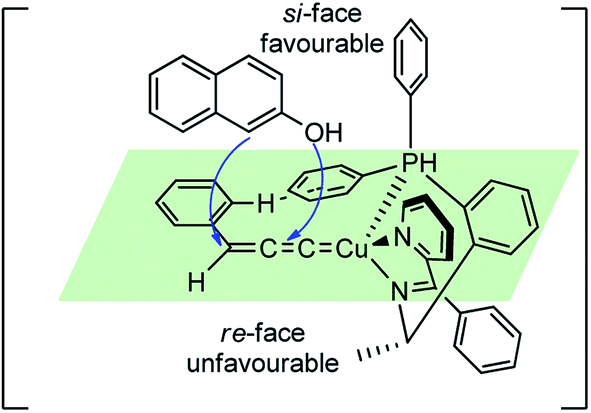 | ||
| Fig. 7 Probable transition state structure for the copper-catalyzed enantioselective synthesis of benzofurans. | ||
In 2016, Hu et al. has reported an interesting synthesis of chiral phosphonylated 2,3-dihydrofurans 171 with good enantioselectivity using copper triflate and chiral P,N,N ligand 138 via a [3 + 2] cycloaddition pathway, Scheme 75.104
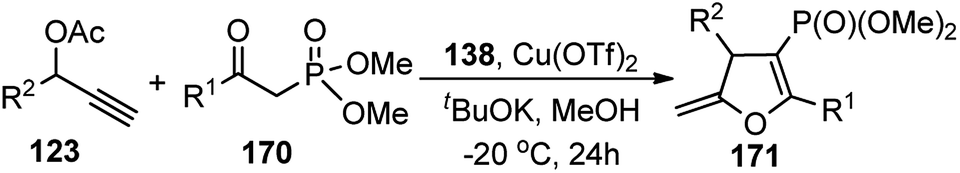 | ||
| Scheme 75 Copper catalyzed synthesis of chiral phosphonylated 2,3-dihydrofurans. | ||
Hu and co-workers in the year 2016 disclosed the first copper-catalyzed asymmetric formal [4 + 2] cycloaddition reaction o-aminophenol derivatives 172 and propargylic esters 123 to generate the chiral 2,3,4-trisubstituted 2H-1,4-benzoxazines 173 in high yields and good to excellent enantioselectivities, Scheme 76.105
 | ||
| Scheme 76 Copper-catalyzed asymmetric synthesis of 2,3,4-trisubstituted 2H-1,4-benzoxazines via formal [4 + 2] cycloaddition reaction. | ||
A wide range of ortho-aminophenol derivatives and propargylic esters efficiently reacted to generate the chiral 2,3,4-trisubstituted 2H-1,4-benzoxazines 173 in high yields and good to excellent enantioselectivities. The reaction mechanism is believed to proceed via the sequential propargylic alkylation/intramolecular hydroalkoxylation process, leading to highly functionalized chiral 3,4-dihydro-2H-1,4-benzoxazines 173 using a structurally rigid and finely modified ketimine P,N,N-ligand. The electronic property of the substituents at the para-position of the phenyl ring also affected the reaction outcome. The decrease in the reactivity and enantioselectivity were observed with the 4-CF3-substituted substrate as well as with heteroaromatic propargylic ester. Very low conversions were observed with aliphatic substrates also.
In 2015, Nishibayashi et al. reported the first copper-catalyzed asymmetric propargylic etherification of propargylic carbonates with not only alcohols but also with phenols as oxygen nucleophiles to generate the corresponding propargylic ethers 175 in good to high yields with a high to excellent enantioselectivity.98 However, only propargylic carbonates bearing an alkyl substituent at the propargylic position served as suitable substrates for the reaction. In search of an alternate catalytic system with broad substrate scope, Hu and co-workers in the year 2016 developed an efficient and highly enantioselective copper-catalyzed propargylic etherification of both aliphatic and aromatic propargylic esters with phenols 174 as oxygen nucleophiles, Scheme 77.106
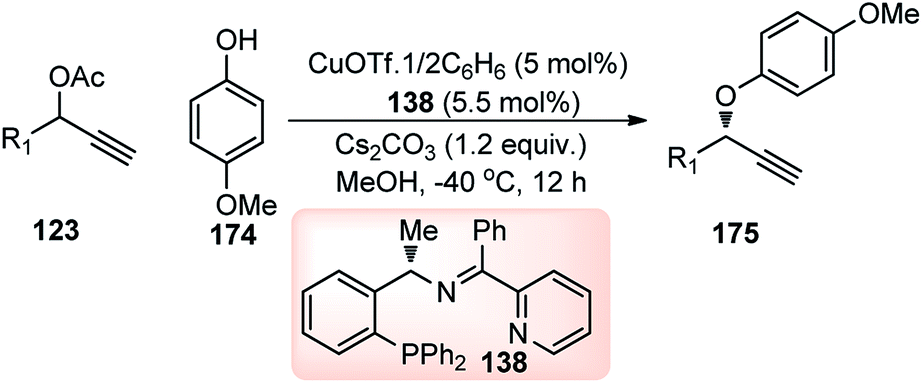 | ||
| Scheme 77 Highly enantioselective copper-catalyzed propargylic etherification of both aliphatic and aromatic propargylic esters with phenols. | ||
Addition of Cs2CO3 was found to significantly promote the reaction rate as well as enhance the enantioselectivity of the reaction. With the use of structurally rigid tridentate ketimine P,N,N-ligand 138 variety of aryl propargylic ethers 175 could be prepared in good to high yields and excellent enantioselectivities.
Based on the commonly explained edge-to-face aromatic interaction a transition state model was proposed between the Cu–acetylide complex with chiral P,N,N-ligand to explain the observed stereochemistry, Fig. 8. Due to the steric hindrance of the ligand, the attack of the γ-carbon atom by the oxygen atom of hydroxy group happened favorably from the si-face.
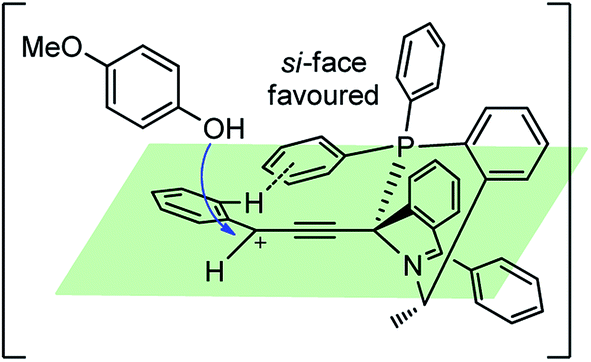 | ||
| Fig. 8 Probable transition state for the copper-catalyzed enantioselective propargylic etherification reaction. | ||
Hu and co-workers in 2015 have further extended their Cu catalyzed enantioselective nucleophilic propargylic substitutions methodology for the synthesis of N-substituted indoles 178 started from propargylic acetates 123, Scheme 78 (Path A).107
 | ||
| Scheme 78 Copper catalyzed propargylation of indoles, coumarin derivatives, and enamines. | ||
Optimization of the reaction conditions revealed that Cu(OAc)2·H2O in presence of chiral ketimine P,N,N-ligand 138 and iPr2NEt was the best reagent system for the chiral induction. Other bases like NEt3 even K2CO3 were also equally effective for this reaction. However, the reaction is very much solvent selective, and the reaction did not work well in CH2Cl2, DMSO, DMF, toluene, and THF. The methodology worked well with a variety of substituted indolines like 2-, 3-, 4-methyl indolines, 6-fluoro indolines etc. and the enantioselectivity was found to be substrate dependent. Chloro substitution at the C-2 position in the phenyl ring of the propargylic acetate gave poor yield and decreased enantioselectivity. However, 3-Cl or 4-Cl substitution in the same ring gave products with good yields and higher selectivity.
A few examples of products obtained by changing the propargylic side chains and by changing the coumarin rings are shown in Fig. 9.
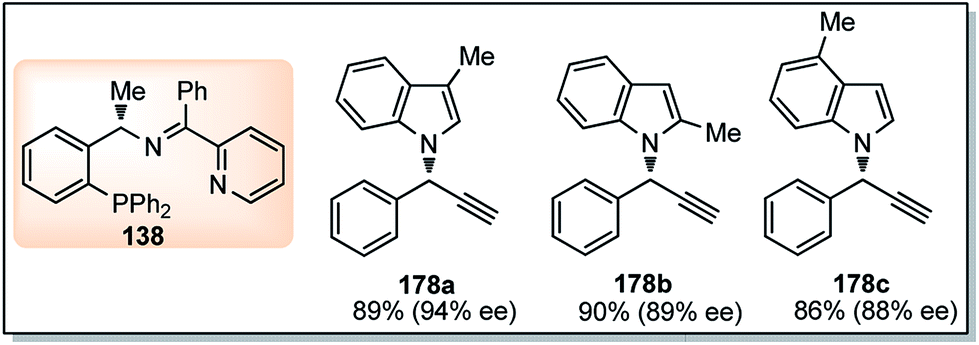 | ||
| Fig. 9 Structures of P,N,N ligand and propargylated indoles with corresponding yield and enantioselectivities. | ||
Antonchick and Waldmann's group has carried out an asymmetric vinylogous propargylation of substituted coumarins 174 using Cu(OTf)2-138 catalyst system, in 2017, Scheme 78 (Path B).108
Screening of various chiral ligands like Cl–MeO-BIPHEP, iPr-PyBOX, Fesulphos, iPr-PHOX along with a copper catalyst gave poor yields and insignificant enantioselectivity. Finally, (S)-138-Cu(OTf)2 catalyst combination in DIPEA medium at −40 °C gave the excellent yields and good enantioselectivity. A few examples of products obtained using the above methodology are shown in Fig. 10.
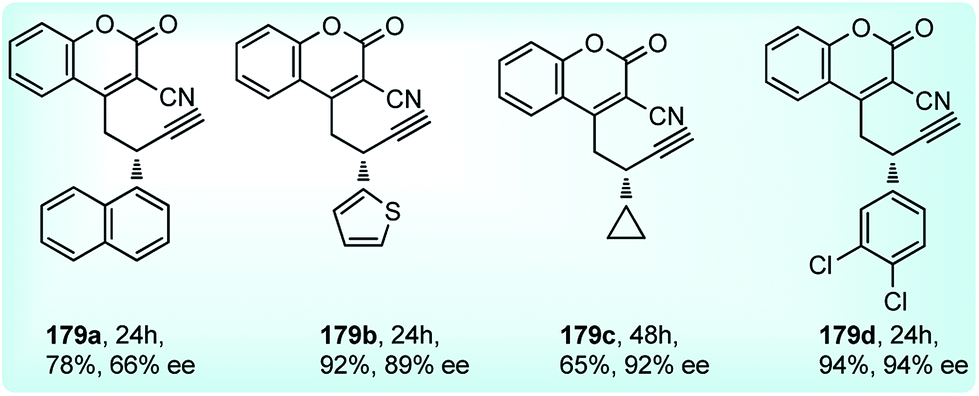 | ||
| Fig. 10 Structures of propargylated coumarins with corresponding yields and enantioselectivities. | ||
Propargylic esters having aliphatic substitution containing a heteroatom underwent smooth reaction, however; tertiary-butyl substituted propargylic ester resulted in sluggish reaction with only 32% yields. Poor enantioselectivity was found with alkynyl substitution in the propargylic substrate (91% yield, 41% ee).
Guo and co-workers in the year 2014 have introduced a chiral tridentate ferrocenyl P,N,N-ligand 145 along with a copper catalyst for the enantioselective propargylic substitution of propargylic acetates, Scheme 78, (Path C).109 Enamines were selected as the nucleophiles to deliver optically active propargylic ketones. Initially, ferrocenyl ligand 145 was combined with a copper salts such as Cu(OAc)2, Cu(OTf)2, CuCl, CuI, CuClO4, Cu(CH3CN)4ClO4 etc. in either MeOH, THF, toluene, CH2Cl2, acetone or CH3CN medium to pursue the substitution reaction. However, Cu(CH3CN)4ClO4-145 reagent system in the methanolic medium at room temperature turned out to be the best with the product yield 75% and 93% ee. Next, the same reaction when carried out by lowering the temperature to 0 °C both yield and enantioselectivity increased to 90% and 97% respectively. Subsequent study was carried out with several substituted acetates and enamines to obtain highly functionalized propargylic ketones with excellent yields and enantiomeric purity as shown in Fig. 11.
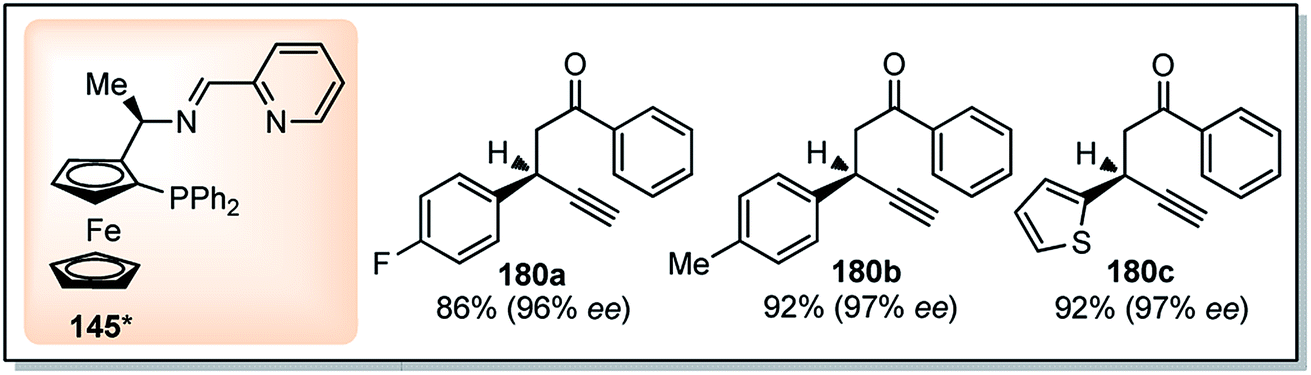 | ||
| Fig. 11 Structures of propargylated enamines with corresponding yields and enantioselectivities. | ||
Han and co-workers in the year 2016 developed a novel Cu-catalysed substitution reaction of propargyl acetates 181 with P(O)H compounds 182 to afford allenylphosphoryl compounds 183 via C–P bond coupling in high yields under mild conditions, Scheme 79.110
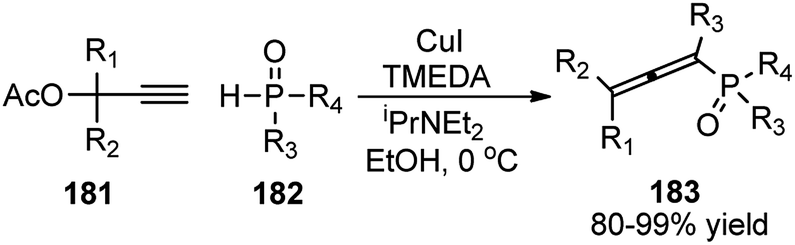 | ||
| Scheme 79 Copper catalyzed substitution reaction of propargyl acetates with P(O)H compounds. | ||
Several bidentate N,N-ligands such as bpy, 1,10-Phen, and ethylenediamine, N,P-ligands like 2-(2-(diphenylphosphino) ethyl)pyridine and P,P-ligand dppe were evaluated and all were proved to be less effective except TMEDA. Screening of the catalysts revealed that CuBr, CuCl, and Cu(MeCN)4PF6 were all equally efficient catalysts, giving the product with comparable yields and selectivity. However, other metal salts such as CoCl2, FeCl3, RuCl3 and Pd(OAc)2 showed no reactivity. A proposed mechanistic pathway is shown in Scheme 80.
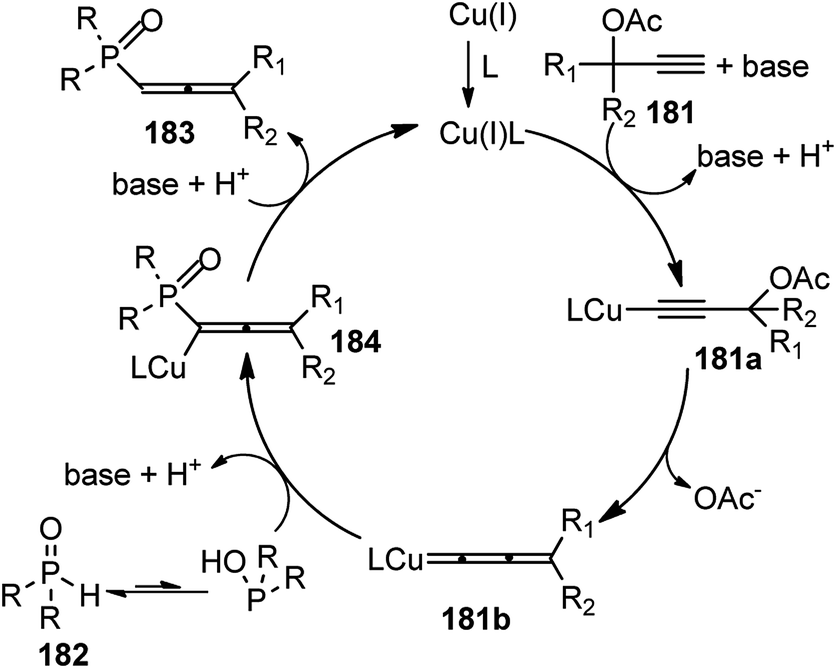 | ||
| Scheme 80 Mechanism for the novel Cu-catalysed substitution reaction of propargyl acetates with P(O)H compounds. | ||
The active Cu(I) species generated from the reaction of the copper salt with TMEDA in presence of base reacted first with the propargyl acetate to produce the copper acetylide complex which then generates the allenylidene complex 181b with the loss of ester group. The nucleophilic reaction of allenylidene complex 181b with P(O)H compounds 182 produce an allenylcopper intermediate 181c, the protonation of which gives the product 183 and regenerates the catalyst, Scheme 80.
Nishibayashi and co-workers in the year 2016 developed a copper-catalyzed enantioselective propargylation of indoles 61 with propargylic esters 185, Scheme 81.111
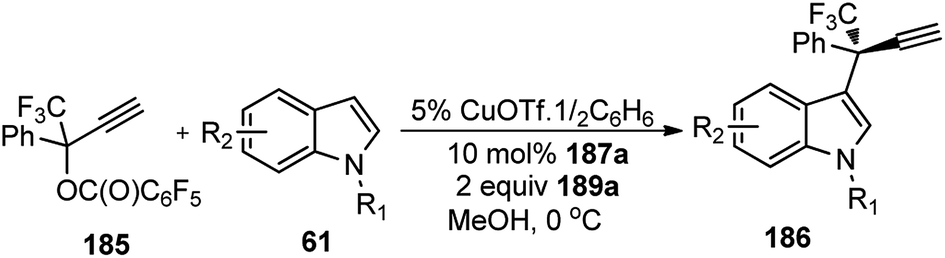 | ||
| Scheme 81 Copper-catalyzed enantioselective propargylation of indoles with propargylic esters. | ||
Treatment of 1,1,1-trifluoro-2-phenylbut-3-yn-2-yl perfluorobenzoate 185 with indole 61 in the presence of 5 mol% of CuOTf·1/2C6H6 and 10 mol% of (4S,5R)-diPh-Pybox 187a and N,N-diisopropylethylamine in methanol at room temperature for 1 hour furnished the desired propargyl substituted product 186 in 41% yield with 47% ee. Propargylic esters such as acetate and tert-butyl carbonate were unable to react under this reaction condition. The base had a pronounced effect on both the reactivity and enantioselectivity and it was observed that cyclic amines like 4-methyl morpholine worked as more effective bases to promote the catalytic reactions smoothly. The previously known ligands for the copper-catalyzed enantioselective propargylic amination and etherification like (S)-Ph-Pybox, (S)-Me-Pybox, and (R)-Cl-MeO-BIPHEP were found unsatisfactory (Fig. 12). Lowering the reaction temperature to 0 °C improved the yield and enantioselectivity drastically. Also, it was observed that using a larger amount of base was effective in leading to the reaction completion in a shorter reaction time. Reactions of other propargylic perfluorobenzoates having substituents like fluoro, chloro, bromo, methoxy, phenyl, and a methyl group at the para-position of the benzene ring of the propargylic perfluorobenzoates were also equally efficient. Unfortunately, no reaction occurred at all with the ortho-methyl-substituted benzene ring.
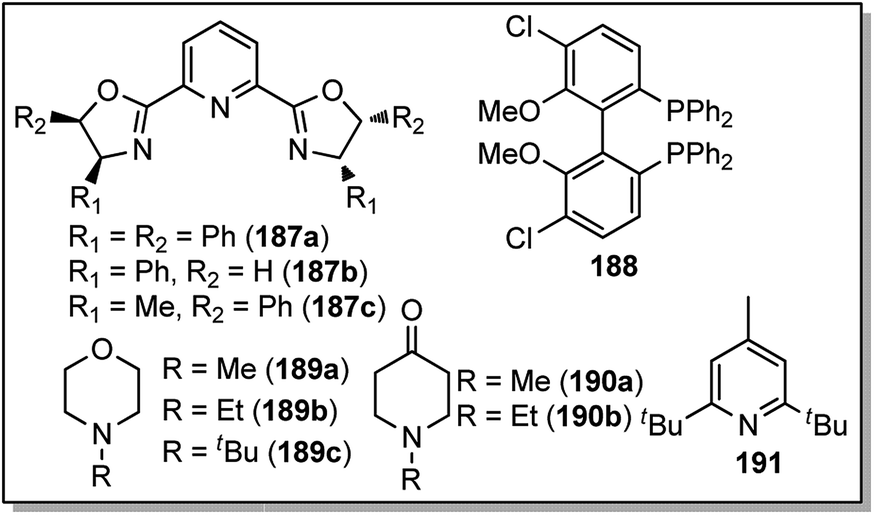 | ||
| Fig. 12 Structures of different ligands and additives screened in the copper-catalyzed enantioselective propargylation of indoles with propargylic esters. | ||
The propargylated product can be efficiently converted to chiral triarylmethanes, bearing a quaternary carbon center, with high to excellent enantioselectivities via sequential Huisgen cycloaddition with azides.
In the year 2016, Gao and co-workers also successfully developed similar Cu-catalyzed direct coupling of the unprotected terminal and internal propargylic alcohols 39 with P(O)H compounds 182, Scheme 82.112
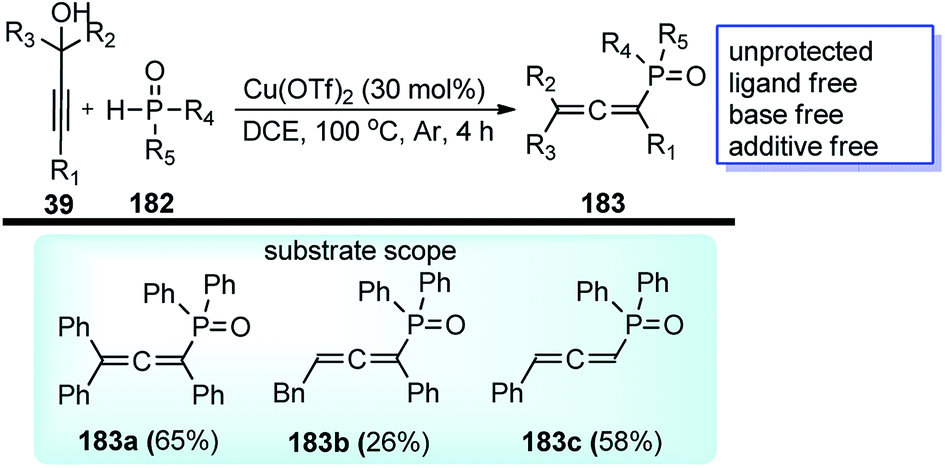 | ||
| Scheme 82 Cu-catalyzed direct coupling of the unprotected terminal and internal propargylic alcohols with P(O)H compounds. | ||
The reaction does not require any base, ligand, or an additive and proceeds only in presence of catalytic amount of Cu(OTf)2. Additionally, the use of inexpensive Cu(OTf)2 as a catalyst, H2O as the only by-product, remarkable functional group tolerance, and the high atom-economy associated with this method makes it a highly prospective methodology for the construction of important allenylphosphoryl frameworks in organic synthesis and pharmaceutical research.
Cordier and co-workers in the year 2017 have developed the first enantioselective Cu-catalyzed rearrangement of electron-deficient 2-propargyloxy-pyridines 192 in high enantioselectivity within a brief time, Scheme 83.113
 | ||
| Scheme 83 Enantioselective Cu-catalyzed rearrangement of electron-deficient 2-propargyloxy-pyridines. | ||
The nature of copper catalyst had a strong influence on both conversion and enantioselectivity. Ligands like diphenyl- and di-4-methylphenyl-phosphines (188, 193, 193a–f) Fig. 13, resulted in high conversion and stereoinduction. However, under the identical conditions, bis-3,5-dimethylphenyl analogs led to low product enantiomeric ratios and the highly bulky ligand 183f resulted in no conversion. Performing the transformation in THF offered results like toluene. The authors noted that base is not required for this process. Employing lower catalyst loading did not impact enantioselectivity but did influence yield and reaction time. Significantly, the rearrangement was complete in less than 10 minutes at ambient temperature.
 | ||
| Fig. 13 Structures of the ligands used for the copper-catalyzed propargylic substitution reaction. | ||
A proposed mechanistic pathway is shown in Scheme 84. Bimetallic intermediate is involved, and it was speculated that the Cu-coordination to the pyridyl nitrogen is responsible for the C–O bond cleavage.
 | ||
| Scheme 84 Mechanism for the enantioselective Cu-catalyzed rearrangement of electron-deficient 2-propargyloxy-pyridines. | ||
Hu and co-workers in the year 2017 developed a copper-catalyzed intermolecular propargylic dearomatization of phenol derivatives 195, with the support of a sterically hindered ketimine P,N,N-ligand leading to optically active cyclohexadienone derivatives 197 in high yield and excellent enantioselectivities with up to 99% ee, Scheme 85.114
 | ||
| Scheme 85 Propargylic dearomatization of phenols. | ||
Screening of the ligand structure showed their considerable influence on the reactivity and enantioselectivity. Chiral tridentate ketimine P,N,N-ligand was identified as the most promising ligand in terms of yield and enantioselectivity. Base additives proved to be crucial for the reaction since no dearomatization product was detected in its absence. The best base additive was Et3N, which furnished products in 59% yield and 95% ee. Subsequent screening of Cu salts identified Cu(OTf)/C6H6 as the best choice. The solvent screening disclosed that the protic solvent was favorable for the reaction, and MeOH proved to be the best one, presumably facilitating the formation of Cu–allenylidene intermediates.
Lowering the reaction temperature significantly inhibits the competitive Friedel–Crafts reactions, thereby dramatically improving the reaction yield and enantioselectivity. Replacing the OAc group with OCOCF3 or TMS didn't result in the dearomatization product. A plausible transition state of the Cu–allenylidene complex with ligand 138 is shown in Fig. 14 to account for the observed stereochemistry.
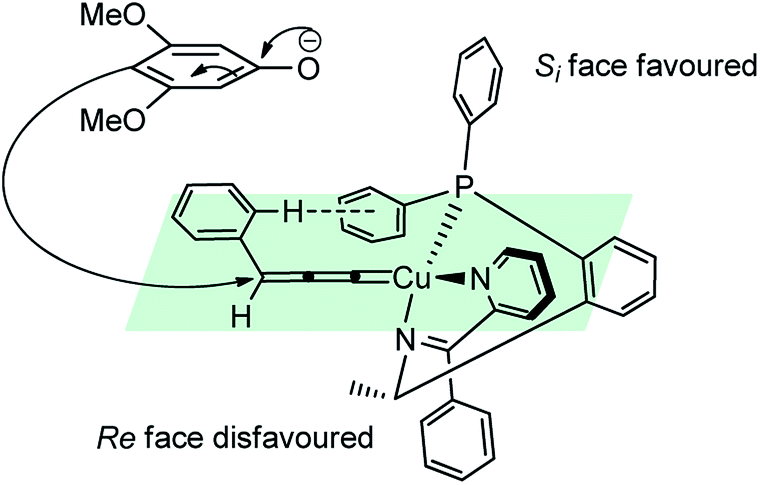 | ||
| Fig. 14 Probable transition state structure for the copper-catalyzed Propargylic dearomatization of phenols. | ||
Unlike the dearomatization reaction, nucleophilic attack preferably occurs from the si-face due to the steric hindrance surrounding the re-face. It was also observed that, unsubstituted phenol predominantly produced O-alkylated product instead of the desired Friedel–Crafts product. Also, meta-methoxy phenol gave the desired product with only 13% yields. However, 3,5-dimethoxy phenol with various propargylic esters furnished the Friedel–Crafts products in reasonably good to excellent yields.
An asymmetric catalytic decarboxylative [4 + 2] annulation of 4-ethynyl dihydrobenzooxazinones 196 and carboxylic acids 197 was established by Gong and co-workers in 2017 using cooperative copper and nucleophilic Lewis base catalysis, Scheme 86.115
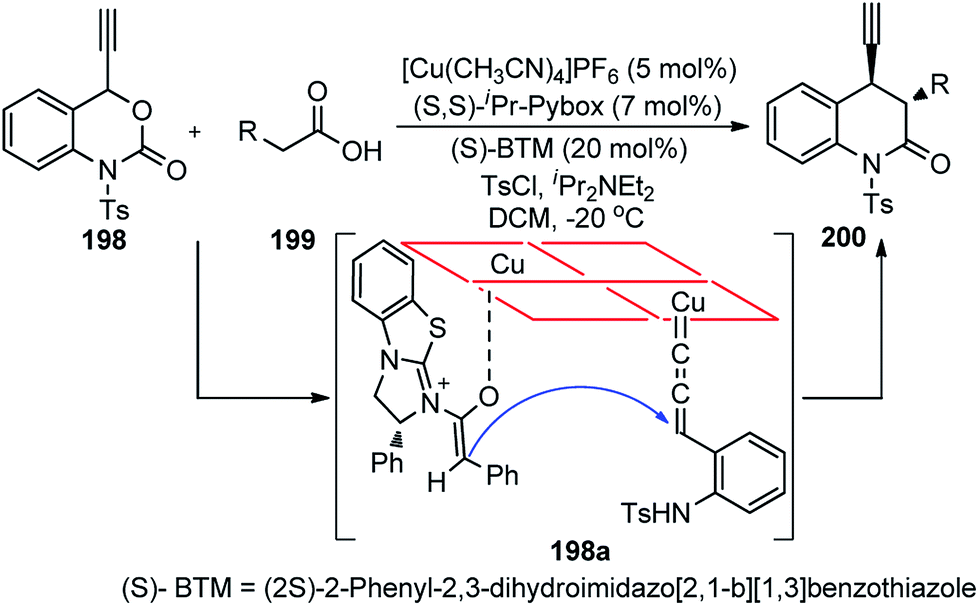 | ||
| Scheme 86 Decarboxylative [4 + 2] annulation of 4-ethynyl dihydrobenzooxazinones and carboxylic acids via cooperative catalysis. | ||
Substituted benzoxazinanones were also excellent reaction partners affording products with excellent enantioselectivities ranging from 94 to 99% ee. Furthermore, excellent enantioselectivities ranging from 95–99% ee were achieved with both electron-donating and electron-withdrawing substituents. The dinuclear copper complex plays a dual role by participating in the decarboxylation process to generate the copper–allenylidene complex of 196 and acting as a counter ion to bind the ammonium Z-enolate. Following the activation of the reacting partners, propargylation proceeds via nucleophilic attack to the si-face of the copper–allenylidene complex followed by lactonization to furnish the (R,R) product 198 as the major isomer, Scheme 86.
Despite the enthusiastic studies by several groups, enantioselective propargylic substitution reactions with alcohols as nucleophiles seemed to be elusive till 2015. Nishibayashi's group reported the first successful example of the enantioselective propargylic etherification with the use of copper-pybox system.98
Nishibayashi and co-workers in the year 2018 developed a copper- and borinic acid-catalyzed propargylic etherification strategy with aliphatic alcohols as nucleophiles in good yields with excellent enantioselectivities, Scheme 87.116 However, the nucleophiles were limited to phenol derivatives. Initial investigations were carried out with tert-butyl 1-phenyl-2-propyn-1-yl carbonate 154 and benzyl alcohol 155/155a as typical substrates. Under the optimized condition employing copper(I) trifluoromethanesulfonate benzene complex, (S)-Ph-pybox ligand 156 (10 mol%), 3,5-bis(trifluoromethyl)phenylboronic acid (5 mol%), and Hünig's base (30 mol%) the desired product was obtained in 68% yield with 89% ee. Other boron compounds listed in Fig. 15 were also screened but none of them offered products with better enantioselectivity than dibenzo-1,4-oxaborine-derived borinic acid 201. Several other pybox ligands like (S)-Me-pybox, diPh-pybox were tested and the best result was achieved by (S)-Ph-pybox ligand.
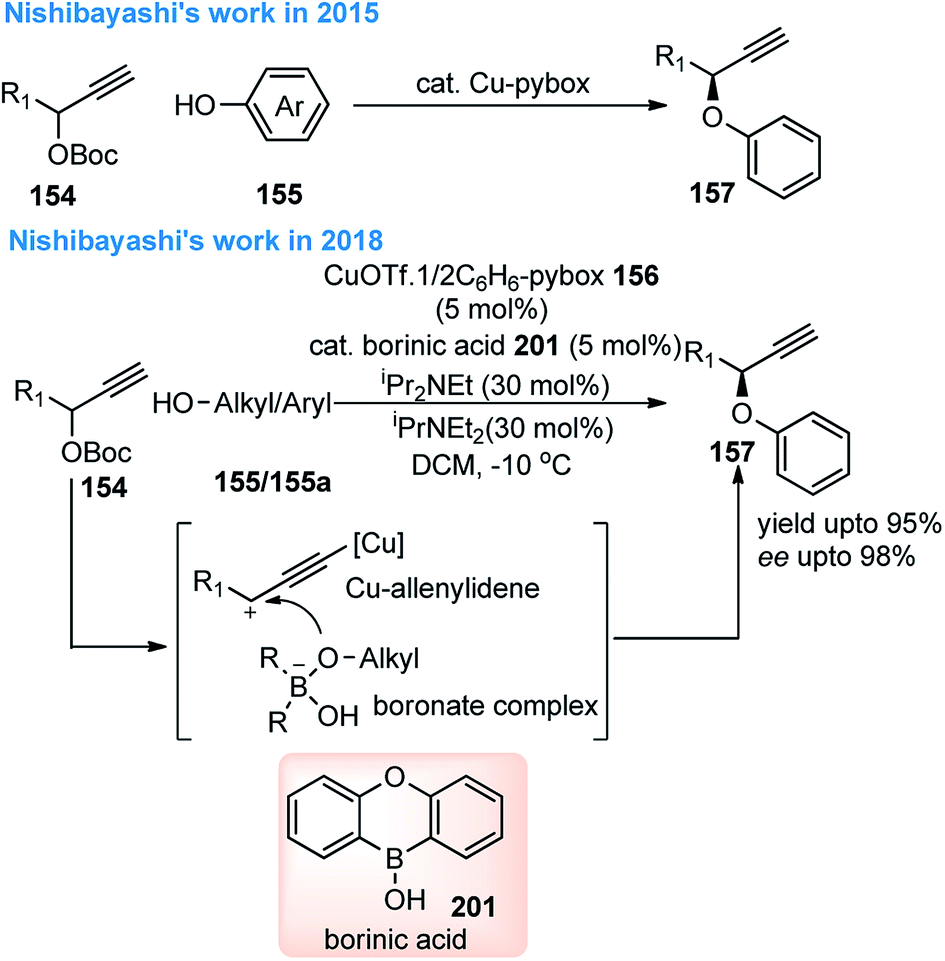 | ||
| Scheme 87 Copper catalyzed propargylic etherification reaction. | ||
 | ||
| Fig. 15 Structures of the different borinic acid used. | ||
Propargylic carbonates bearing electron donating as well as withdrawing substituents at the para-position of the benzene ring were successfully transformed into the corresponding propargylic etherified products in moderate to high yields with an excellent enantioselectivity.
The present etherification proceeds via a nucleophilic attack of an alcohol toward a dimetallic copper–allenylidene complex from si-face of the allenylidene ligand, Fig. 16.98
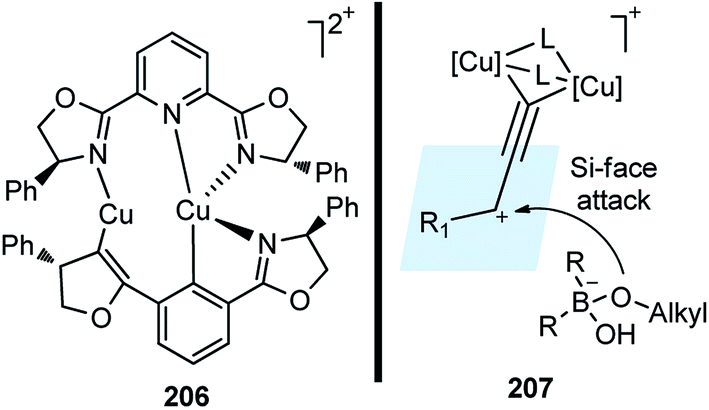 | ||
| Fig. 16 Transition state structure for the copper-catalyzed propargylic etherification reaction. | ||
A similar report of copper and borinic acid-catalyzed propargylic etherification reactions was also independently carried out by the group of Niu in 2017.117,118 But the substrate scope is limited and has been limited to diols and polyols, which may coordinate the boron center in a bidentate fashion.
5.9. Molybdenum derived catalysts
Molybdenum although falls under the category of ‘heavy metal’ its properties are very different from those of the typical heavy metals like mercury, thallium, and lead. It is much less toxic than these and other heavy metals which makes it an attractive substitute for several other toxic heavy metals. The oxidation states ranging from (−II) to (VI), coordination numbers expanding from 4 to 8 and their ability to form compounds with most inorganic and organic ligands makes the chemistry of molybdenum extremely versatile.119,120Molybdenum(VI) was first used as a catalyst for the direct substitution of propargylic alcohol 23 with several O, N and C-centered nucleophiles by Zhu and co-workers in the year 2010, Scheme 88.121
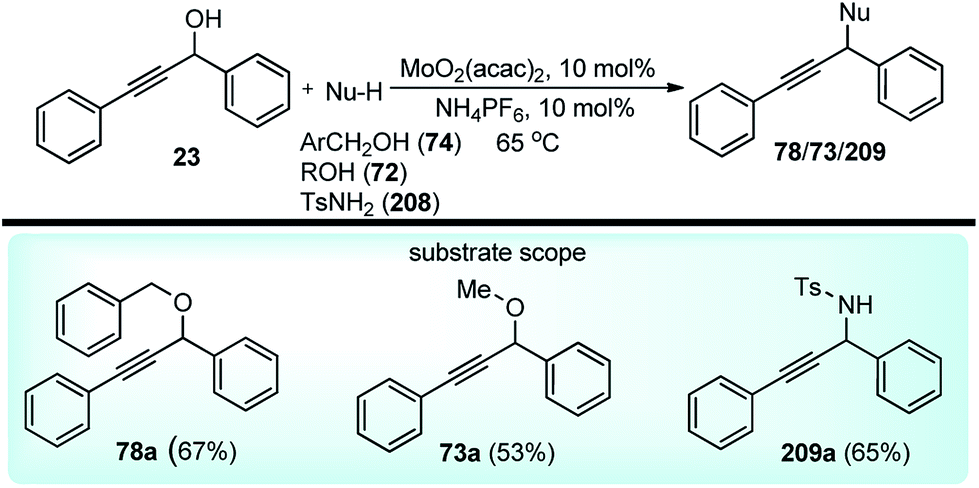 | ||
| Scheme 88 Direct substitution of propargylic alcohol with oxygen, nitrogen, and carbon nucleophiles catalyzed by molybdenum. | ||
When 1,3-diphenylprop-2-yn-1-ol 23 was treated with an excess of phenylmethanol 74 or other nucleophiles like alcohol 72 or tosylamine 208 in the presence of 10 mol% of MoO2(acac)2 and 10 mol% of NH4PF6 in acetonitrile at 65 °C for 6 h the desired product (3-(benzyloxy)prop-1-yne-1,3-diyl)dibenzene 73/78/209 was obtained in good yield. Reduced yield was obtained when NH4PF6 was not used as an additive, whereas NH4PF6 alone showed no catalytic activity.
The presence of air and moisture did not have any role in the reaction outcome. The reaction is solvent dependent and yields considerably reduced when the reaction was carried out in toluene, nitromethane, dioxane or DMF. The mechanistic pathway for this Mo catalyzed reaction is illustrated in Scheme 89.
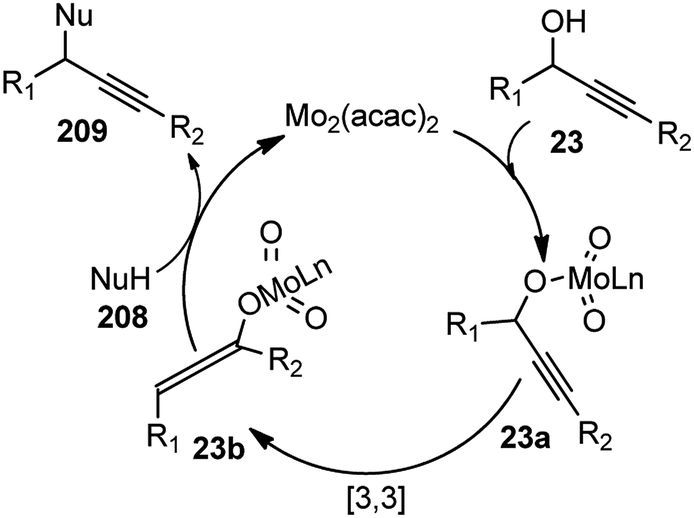 | ||
| Scheme 89 Molybdenum(VI) catalyzed direct substitution of propargylic alcohols. | ||
It was proposed that the transition state involves propargyl alcohol combined with molybdenum complex to form intermediate, which was followed by [3,3] rearrangement to form allenolate intermediate 23b. Subsequent nucleophilic attack by 206 on allenolate 23b delivered the desired product 198.
5.10. Ruthenium derived catalysts
Ruthenium can exist in various valences (0 to 8 valence) and is not so expensive transition metal which prompted the scientific community to explore ruthenium for various useful catalytic reactions. Various substrates bearing heteroatoms are activated by these catalysts and undergo reactions with either nucleophiles or electrophiles under neutral conditions. Ruthenium catalysts react with propargyl alcohols to generate metal allenylidene complexes, which have attracted a great deal of attention in recent years as a new type of organometallic intermediate. The Cα and Cγ carbon atoms of allenylidene ligands are electrophilic centers, while the Cβ carbon atom is nucleophilic. In general, reactions of catalytic allenylidene ruthenium complexes with a variety of nucleophiles proceed via the nucleophilic attack either at the Cβ or Cγ carbon atom in allenylidene complex. Only a few examples have been reported where propargylation occurs via the coordination of the alkyne bond, thereby enhancing the nucleofugacity of the hydroxide, Scheme 90. | ||
| Scheme 90 General mechanism for the ruthenium catalyzed propargylic substitution reaction. | ||
Hidai and co-workers in the year 2000 described a series of novel propargylic substitution reactions catalyzed by thiolate-bridged diruthenium complexes, Scheme 91.10 They have demonstrated the facile ruthenium-catalyzed substitution reactions of propargyl alcohol with various nucleophiles like amine, amides, thiophenols, and diphenylphosphines to produce the corresponding propargylic derivatives 211–214 in high yields with complete regioselectivity.
 | ||
| Scheme 91 Ruthenium catalyzed propargylic substitution reaction. | ||
A mechanism for this novel catalytic reaction is highlighted in Scheme 92. The initial step is the formation of a vinylidene complex 209a in the presence of NH4BF4 followed by conversion of vinylidene complex into an allenylidene complex 209b. Subsequent nucleophilic attack of an alcohol on the Cγ atom in the allenylidene ligand resulted in the formation of another vinylidene complex 209c. The complex 209c is then transformed into the η2-coordinated propargylic ether tautomer 209d, which furnishes propargylic ether by reaction with a propargylic alcohol to regenerate the complex.
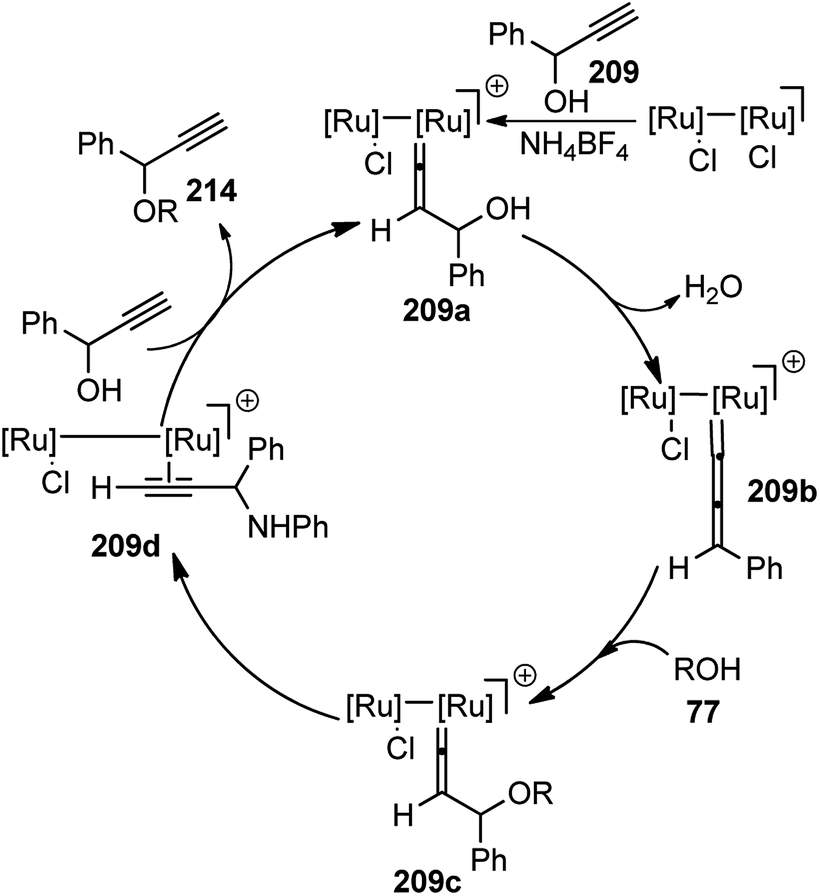 | ||
| Scheme 92 Mechanism for the ruthenium catalyzed propargylic substitution reaction. | ||
Until 2002, the most reliable method for the propargylic substitution was the Nicholas reaction. However, there were only a few studies on the substitution by thiols by the Nicholas reaction and the preparative methods for propargylic sulfides by other methods were quite limited. So, to device propargylic substitution reactions with thiols, Uemura and co-workers in the year 2002 reported a highly selective and efficient propargylic substitution reaction of propargylic alcohols 39 with thiols 76 catalyzed by the cationic diruthenium complex, Scheme 93. That was the initiation of the subsequent propargylic reactions carried using diruthenium complex.122
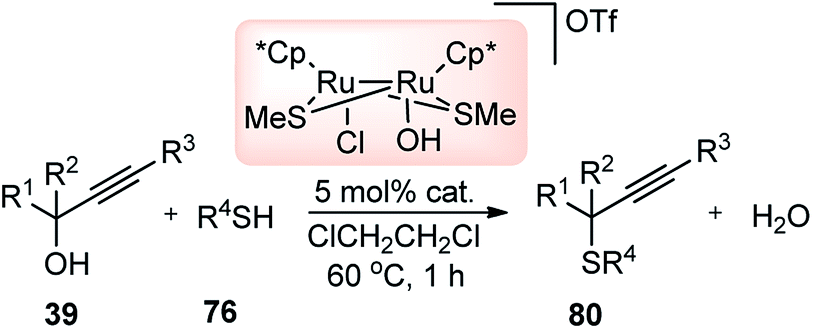 | ||
| Scheme 93 Cationic diruthenium complex catalyzed propargylic thio-etherification reaction. | ||
Tyrrell and co-workers in the year 1997 described a series of intramolecular cyclisation reactions of trisubstituted alkene 215 promoted by dicobalt hexacarbonyl complex, Scheme 94. Although a variety of activated carbon nucleophiles have been used to quench a dicobalt hexacarbonyl stabilized cation in the Nicholas reaction the use of deactivated alkenes has received little or no attention in the chemistry literature.123
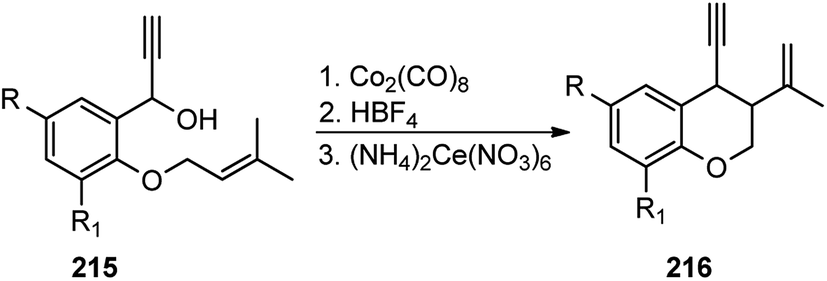 | ||
| Scheme 94 Co2(CO)8 catalyzed propargylic substitution reaction of deactivated alkenes. | ||
Although the intramolecular cyclization reaction of the trisubstituted alkene was accomplished to afford a series of benzopyran derivatives 216, the reaction suffered from a serious drawback of involving stoichiometric amount of dicobalt hexacarbonyl complex. This limitation prompted researchers to develop new methodologies in the subsequent years.
To devise a new methodology to construct C–C bond, Hidai, and Uemura in the year 2003 reported a ruthenium-catalyzed inter- and intramolecular carbon–carbon bond forming reactions between propargylic alcohols 209 and alkenes 217 via an allenylidene-ene type pathway, Scheme 95.124
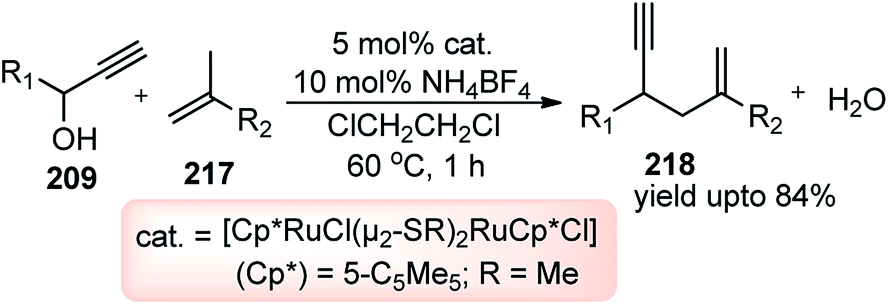 | ||
| Scheme 95 Ruthenium-catalyzed inter- and intramolecular carbon–carbon bond forming reactions between propargylic alcohols and alkenes. | ||
The authors have developed a simple method involving both Ru and Pt catalyst for the preparation of fused polycyclic compounds by an intramolecular cyclization of propargylic alcohols bearing an alkene moiety 218 at a suitable position, Scheme 96.125
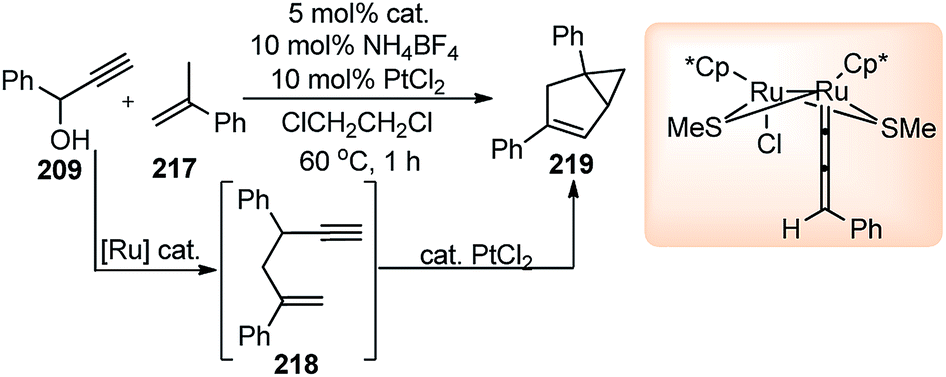 | ||
| Scheme 96 Mechanism for the ruthenium catalyzed the synthesis of fused polycyclic compounds. | ||
This one-pot efficient synthetic method provides a new type of skeleton of fused polycyclic compounds with a potential for biological activity. The thiolate-bridged diruthenium complexes promoted the catalytic propargylic substitution reaction between propargylic alcohols and alkenes in the first step, while PtCl2 catalyzed the cycloisomerization of the enynes in the same medium in the second step.
Hidai and Uemura in the year 2003 have found a combination of ruthenium- and platinum-catalyzed sequential reactions to afford the corresponding tri- and tetra-substituted furans or pyrroles from propargylic alcohols 209 with ketones 220 and anilines 221, respectively, in moderate to good yields with high regioselectivities, Scheme 97.126
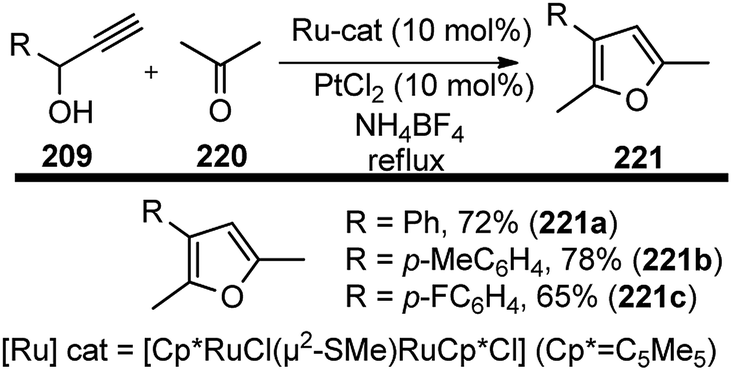 | ||
| Scheme 97 Ruthenium- and platinum-catalyzed sequential reactions towards the synthesis of substituted furans from propargylic alcohols and ketones. | ||
Treatment of 1-phenyl-2-propyn-1-ol 209 in acetone 220 in the presence of Ru-catalyst, NH4BF4 (20 mol%), and PtCl2 (20 mol%) at reflux temperature for 36 h afforded 2,5-dimethyl-3-phenylfuran 221 in 75% yield. The reaction proceeded even in the presence of smaller quantities of Ru-cat (5 mol%) and PtCl2. Other transition-metal complexes, such as Pd(OAc)2, [PdCl2(MeCN)2] and PdCl2 were found inefficient. Although AuCl3 was found to furnish the desired product, an intermediate diketone was also formed. The reaction has a wide substrate scope and the use of various propargylic alcohols resulted in the formation of the corresponding 3-aryl and 3-alkenyl substituted 2,5-dimethylfurans in moderate to high yields with a complete regioselectivity.
The mechanistic pathway for this reaction is highlighted in Scheme 98.
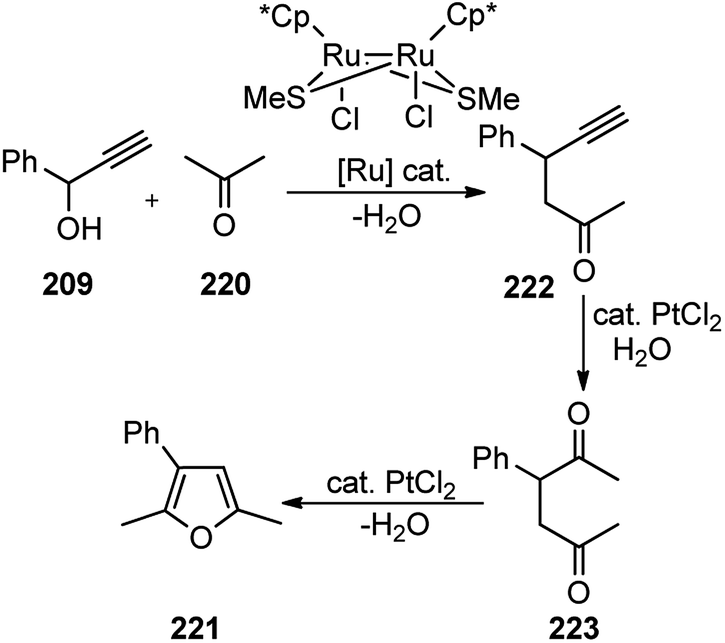 | ||
| Scheme 98 The mechanism for the ruthenium- and platinum-catalyzed sequential reactions towards the synthesis of substituted furans from propargylic alcohols and ketones. | ||
At first, under the influence of Ru-catalyst 209 is transformed rapidly into γ-ketoalkyne 222. Then, hydration of the alkyne moiety slowly delivers the 1,4-diketone 223, under the influence of PtCl2. Intramolecular cyclization of the produced diketone resulted in the formation of the furan 221, catalyzed by PtCl2.
In 2003, Nishibayashi and co-workers reported catalytic enantioselective propargylic alkylation of propargylic alcohols 209 with acetone 220, using diruthenium complex in good yields, but only with moderate enantioselectivity (up to 35% ee), Scheme 99.127
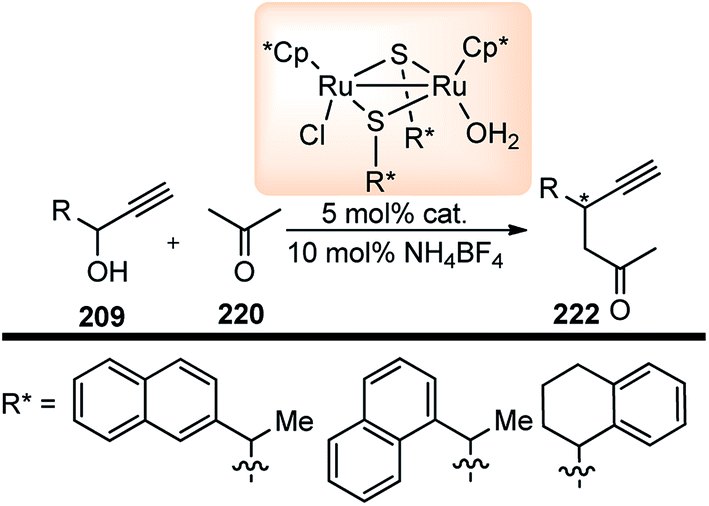 | ||
| Scheme 99 Ruthenium catalyzed enantioselective propargylic alkylation of propargylic alcohols with acetone. | ||
The authors envisaged that edge to face steric repulsion between substrates and chiral ligands would not be enough to achieve high enantioselectivity. Therefore, a new type of chiral ligands with a phenyl ring that might interact with a phenyl ring of ruthenium–allenylidene complexes by p–p interaction was designed.128 Also the chiral thiolate-bridged ligands work to control the chiral environment around the diruthenium site.
Nucleophilic attack at the Cγ atom of the allenylidene ligand from the less hindered side accounts for the high enantioselectivity. Here, pi–pi interaction of phenyl rings between the ligand and allenylidene moieties is considered to play a crucial role in achieving such a high selectivity.129,130 The method reported was the first of its kind compared to the so-far-known highly diastereoselective and enantioselective propargylic substitution reaction methods, which used a stoichiometric amount of transition metal complexes.131–133 for the enantioselective reactions with a ketone.
Reactions of allenylidene complexes with various heteroatom and carbon-centered nucleophiles at the α- and γ-carbons were already known but the direct reaction of the allenylidene ligand with aromatic compounds was not explored. This prompted several research groups to investigate the catalytic reaction of aromatic compounds with propargylic alcohols in the presence of the thiolate-bridged diruthenium complex [Cp*RuCl(μ2-SMe)2-RuCp*Cl] as a catalyst.
Nishibayashi and Uemura in between 2004 and 2005 have found several interesting ruthenium-catalyzed reactions between propargylic alcohols and cyclic 1,3-dicarbonyl compounds134 aromatic compounds135,136 and ketones134,137 and 1,3-dicarbonyls134a,138 to afford the corresponding chromen-5-ones and pyran-5-ones, propargylated aromatic products and related products respectively in high to excellent yields with high regioselectivity, Scheme 100. Although the ruthenium-catalyzed propargylic alkylation of propargylic alcohols with ketones 210 was disclosed in 2001 by Nishibayashi and co-workers134 but the highly enantioselective propargylic substitution reaction of propargylic alcohols with acetone with up to 82% ee was reported only in 2005.137 The diruthenium complex with a new class off chiral disulfides acted as catalyst where the π–π interaction of phenyl rings between the ligand and allenylidene moieties played a crucial role in achieving high selectivity.
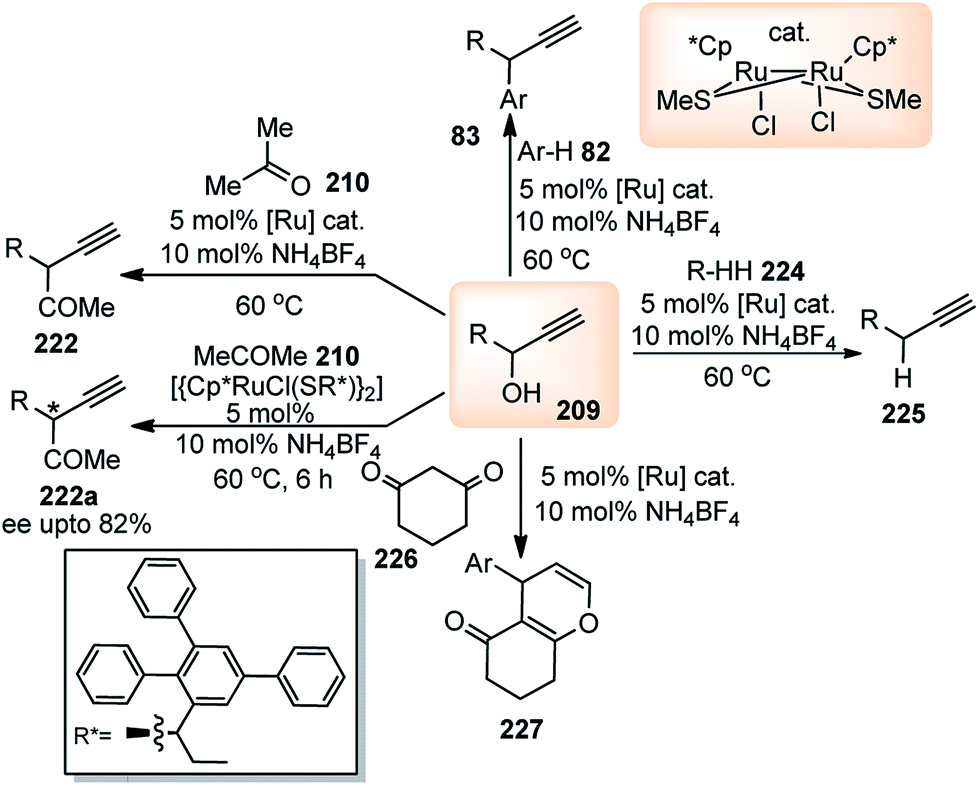 | ||
| Scheme 100 Ruthenium catalyzed propargylic substitution reaction of ketones and arynes. | ||
The mechanistic studies indicated that the catalytic cycloaddition proceeds via allenylidene intermediate.139 The electrophilic aromatic substitution reaction occurs at the γ-carbon of an allenylidene ligand.
Nishibayashi and Uemura in the year 2004 developed a convenient and straightforward methodology for the synthesis of substituted oxazoles 228 in good yields with a complete regioselectivity, Scheme 101.140 The reaction proceeds via a one-pot reaction of propargylic alcohols bearing a terminal alkyne moiety 209 with amides 77 by the sequential action of ruthenium and gold catalysts.
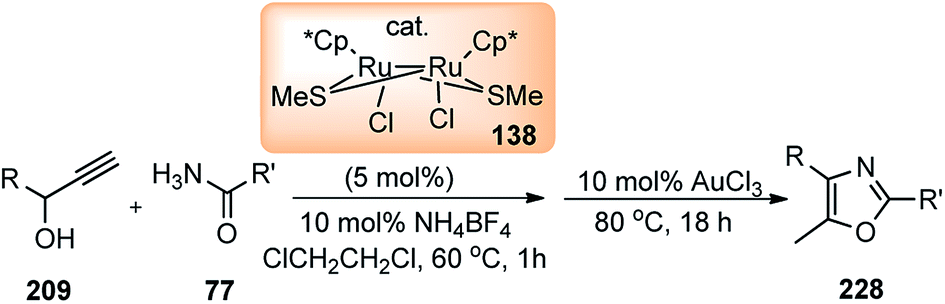 | ||
| Scheme 101 Ruthenium and gold catalyzed synthesis of substituted oxazoles from propargyl alcohols with terminal alkyne moiety with amides. | ||
When PtCl2 and PdCl2 were used separately in the presence of methane thiolate-bridged diruthenium complex and NH4BF4 (10 mol%) in 1,2-dichloroethane (ClCH2CH2Cl) at 80 °C for 18 h, the reaction of 1-phenyl-2-propyn-1-ol 209 with acetamide did not afford any desired product. Interestingly, improved yield of the product was observed when AuCl3 (10 mol%) was used as a catalyst in place of PtCl2. Marginal improvement in the product yield was observed when the amount of AuCl3 was increased to 20 mol% whereas the reduction of the amount of ruthenium catalyst (2.5 mol%) improved the product yield. Also prolonging the reaction time did not improve the yield under the same reaction conditions.
The best result was obtained with 5 mol% of Ru catalyst and NH4BF4 (10 mol%) in ClCH2CH2Cl at 60 °C for 1 h followed by the addition of AuCl3 (10 mol%). Reactions of a variety of propargylic alcohols with various amides were carried out by the stepwise addition of diruthenium complex 2a (5 mol%) and AuCl3 (10 mol%) which furnished the expected 2-isopropyl-5-methyloxazoles in good to high isolated yields. Unfortunately, no reaction of alkyl-substituted propargylic alcohols proceeded at all. As expected, no reaction occurred when N-methylacetamide, 2-pyrrolidone, and methanesulfonamide were used as substrates. An elegant route for the synthesis of a variety of substituted oxazoles 228 in good yields with a complete regioselectivity was achieved by the sequential action of ruthenium and gold catalysts from simple and readily available starting materials such as propargylic alcohols and amides.
Dixneuf and co-workers in 2005 have earlier demonstrated the role of bisoxazoline–ruthenium complex precursors in the direct propargylation of weak nucleophiles with propargyl alcohols141 Thereafter the same group reported that a mononuclear (bisoxazoline)(arene)ruthenium(II) complex catalyses the direct propargylation of furan 230 by propargyl alcohols 229 with C–C bond formation in moderate yields, Scheme 102.142
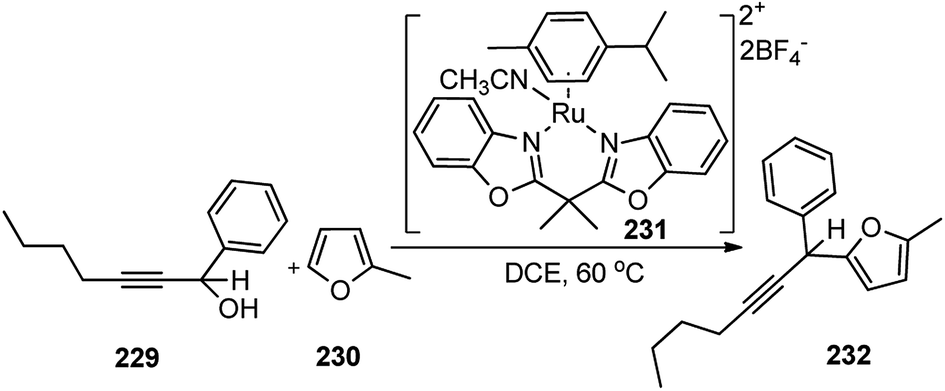 | ||
| Scheme 102 Ruthenium(II)-catalysed direct propargylation of furan and arene with propargyl alcohols. | ||
Although the yields were modest the scope to modify the bisoxazolidine ligand by steric and electronic modifications to provide higher catalytic activity and play a crucial role in enantioselective catalysis was encouraging. A mechanistic detail was proposed for the above reaction, Scheme 100, where propargylation took place via coordinating the CC bond, thereby enhancing the nucleofugality of the hydroxide leading to the stabilization of the resulting a carbenium ion and allowing further nucleophilic addition at the α-carbon of the coordinated CC bond, (Scheme 103).
 | ||
| Scheme 103 Mechanism for the ruthenium(II)-catalysed direct propargylation reaction. | ||
Nishibayashi in the year 2005 described a general procedure for the catalytic propargylic substitution reactions of propargylic alcohols 209 with heteroatom-centered nucleophiles like alcohols 72, amines 210, amides 77, and phosphine oxide 180 using di-ruthenium catalyst, Scheme 104.143
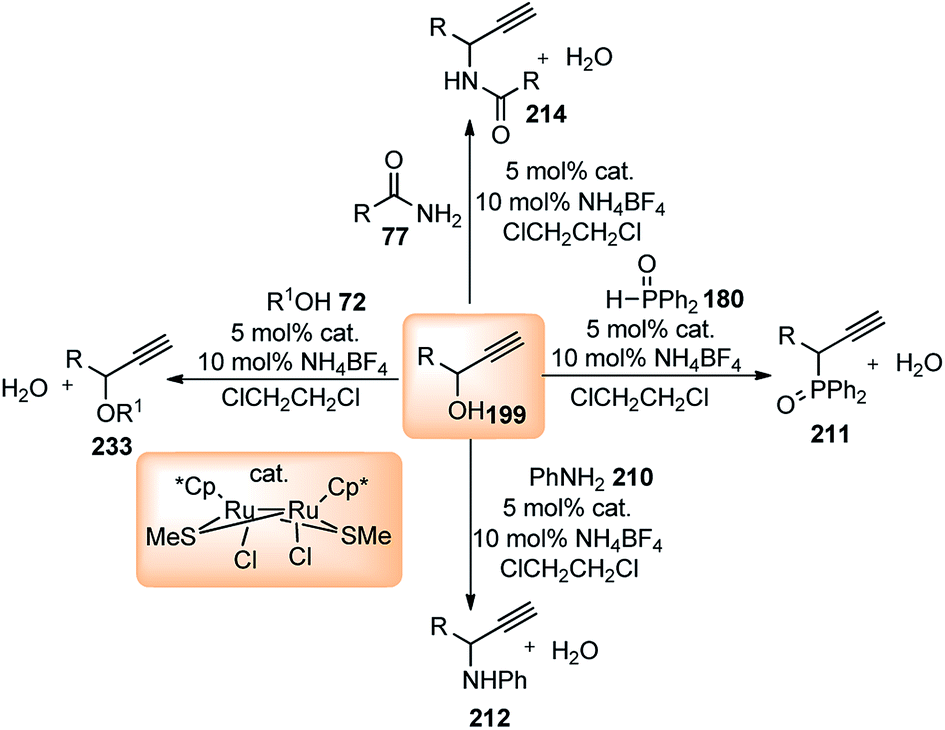 | ||
| Scheme 104 Ruthenium catalyzed propargylic substitution reaction with diverse nucleophiles. | ||
The catalytic cycle for the propargylic substitution reaction is illustrated in Scheme 105 which proceeds via the allenylidene intermediate. The attack of nucleophiles to the Cγ atom of the allenylidene complex is the key step for the formation of the propargylated product.
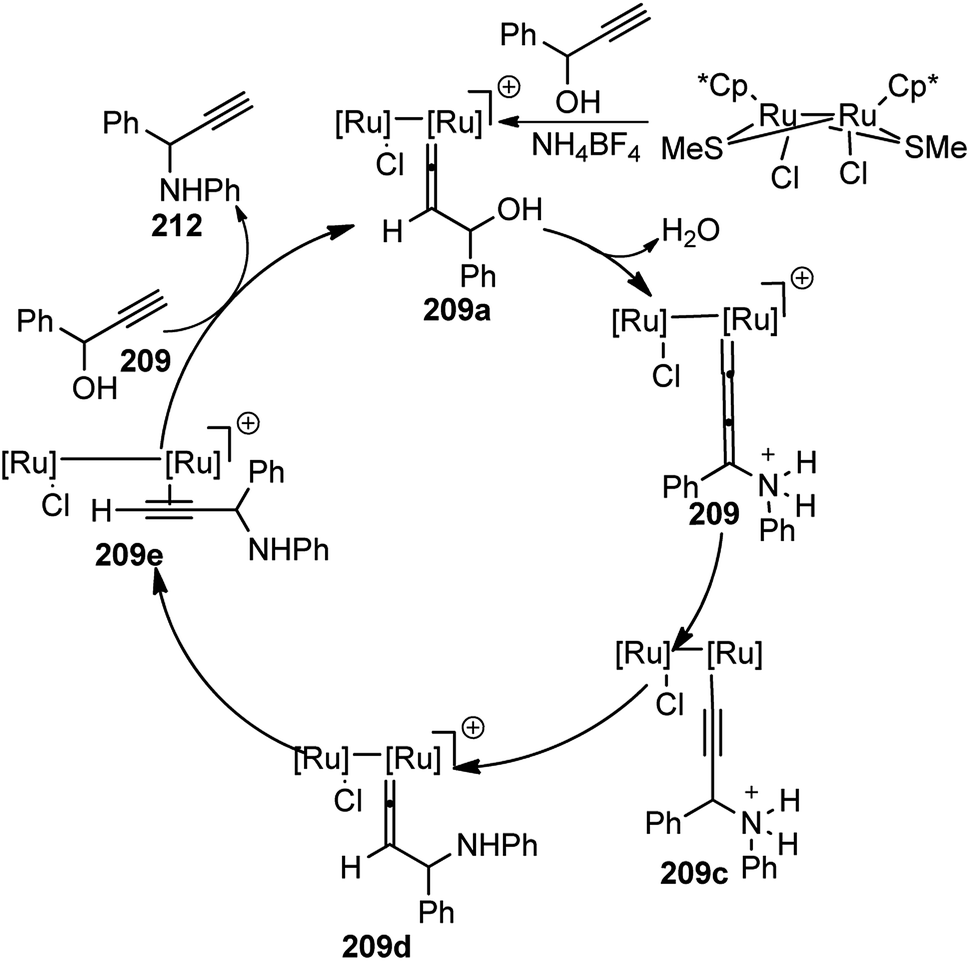 | ||
| Scheme 105 Mechanism for the ruthenium catalyzed propargylic substitution reaction. | ||
Nishibayashi and co-workers in the year 2006 developed a novel method for the reduction of propargylic alcohols 23 with triethylsilane 234 using ruthenium catalyst in good to high yields and complete selectivity, Scheme 106.144
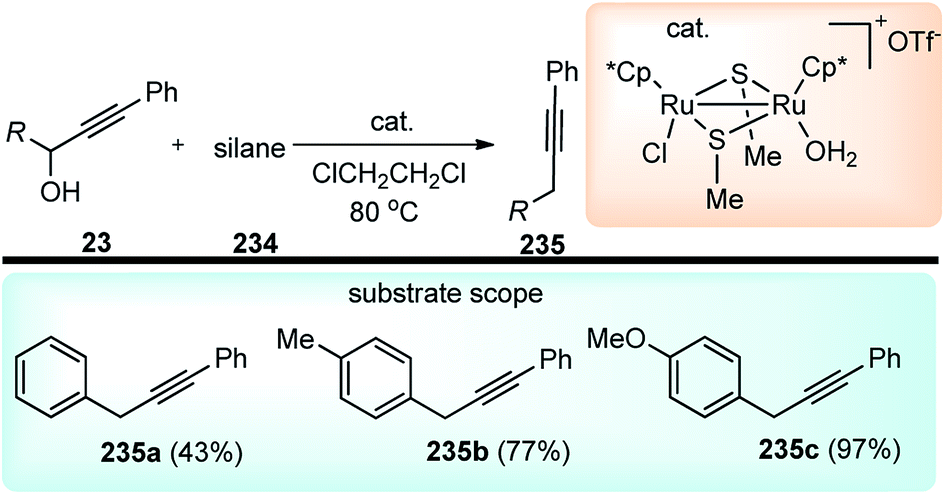 | ||
| Scheme 106 Ruthenium-catalyzed propargylic reduction of propargylic alcohols with triethylsilane. | ||
5 mol% of diruthenium complex in ClCH2CH2Cl at 80 °C was enough to afford the reduced product 235 in 77% yield. Although, there were several reports of gold-catalyzed propargylic substitution reactions145 but AuCl3 was inefficient in catalyzing the propargylic reduction.
Several other silanes such as tert-butyldimethylsilane, dimethylphenylsilane, and triisopropylsilane performed well as reducing agents but triphenylsilane, diphenylsilane, methylphenylsilane, phenylsilane, and poly(methylhydrosiloxane) were not effective. Effect of the substituent on the phenyl ring had a marked influence on the reaction yield. Higher product yield was observed in the presence of an electron-donating group whereas electron withdrawing groups resulted in reduced yield suggesting the involvement of propargylic cation as the reactive intermediate. The proposed reactive intermediate is shown in Fig. 17 where the alkyne moiety interacts with the ruthenium centers of the thiolate diruthenium complex followed by a nucleophilic attack of hydride from the triflate-activated silane to furnish the corresponding reduction product.
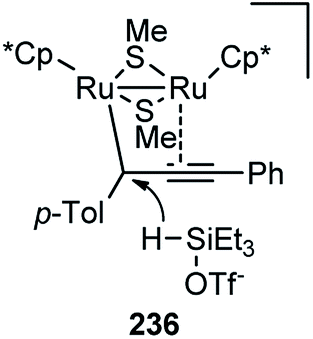 | ||
| Fig. 17 Proposed reactive intermediate for the propargylic reduction with triethysilane. | ||
Analogous to this reaction, etherification of propargylic alcohols was reported using ruthenium catalyst.146 Furthermore, the authors have also investigated the ruthenium-catalyzed reactions of propargylic alcohols 237 with organosilicon compounds 238–240 other than trialkylsilanes under the same reaction conditions. Interestingly, the reactions with allyltrimethoxysilane 238 and triethoxysilane 239 proceeded to the corresponding propargylic ethers whereas allyldimethylsilyl hydride provided the corresponding allylated products in high yield at 80 °C within one hour, Scheme 107.144
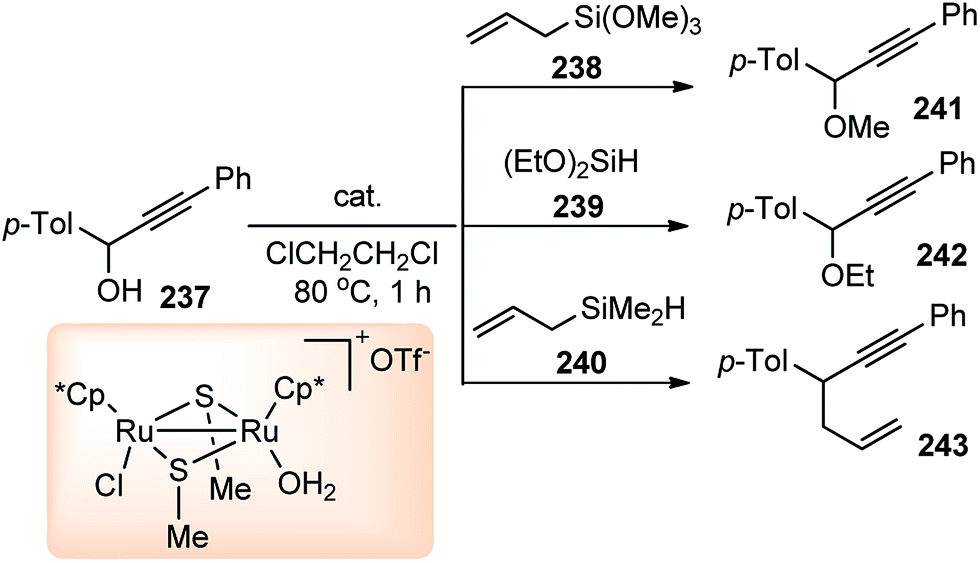 | ||
| Scheme 107 Ru-catalyzed propargylic substitution with different nucleophiles. | ||
Nishibayashi and co-workers in 2008 have extended the application of Ru catalyzed propargylic substitution reaction for the preparation of a variety of optically active heterocycles like chromanes 216, thiochromanes 246, and 1,2,3,4-tetrahydroquinolines 247 in good to high yields with up to 99% ee, Scheme 108.147
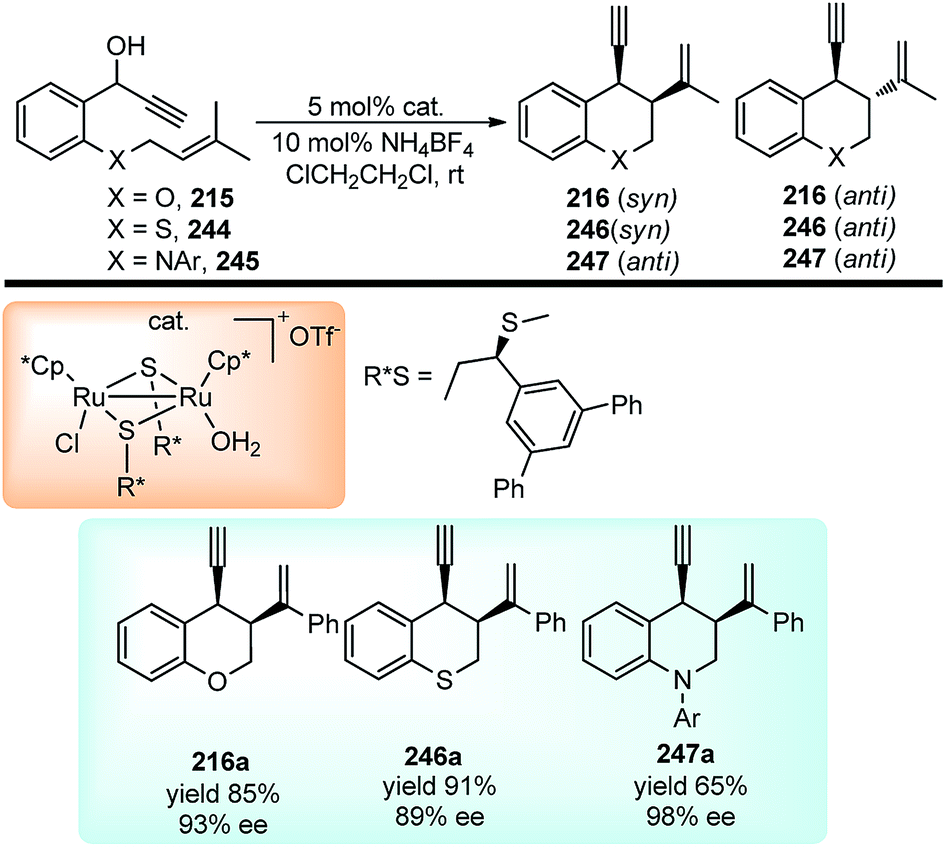 | ||
| Scheme 108 Intramolecular cyclization of propargylic alcohols towards the synthesis of chiral chromanes, thiochromanes, and 1,2,3,4-tetrahydroquinolines. | ||
The reaction occurs via a ruthenium catalyzed intramolecular cyclization of propargylic alcohols 215 bearing an alkene moiety at a suitable position. Probable mechanistic pathway for the reaction is represented in Scheme 109. The intramolecular attack of an alkene on the cationic γ-carbon of the intermediate occurs from the si-face to provide the corresponding alkynyl complex 215b, followed by the facile proton transfer into the alkynyl moiety to give the corresponding vinylidene complex 215c.
 | ||
| Scheme 109 Probable mechanistic pathway and transition state structure for the ruthenium catalyzed intramolecular cyclization of propargylic alcohols bearing an alkene moiety. | ||
Nishibayashi and co-workers in the year 2009 developed a ruthenium-catalyzed enantioselective cyclization of propargylic alcohols 248 bearing a thiophene moiety to afford the corresponding propargylated thiophenes 249 in good to high yields with a high enantioselectivity (up to 97% ee), Scheme 110.148
 | ||
| Scheme 110 Ruthenium-catalyzed enantioselective cyclization of propargylic alcohols bearing a thiophene. | ||
The reaction proceeds via ruthenium–allenylidene complexes as key intermediates where the thiophene intermolecularly attacks the alkynyl complex selectively from the si-face because of the π–π interaction between the chiral ligand and allenylidene complex as depicted in Scheme 111. The π–π interaction of the phenyl rings between the chiral ligand and allenylidene moieties was instrumental in achieving high enantioselectivity.
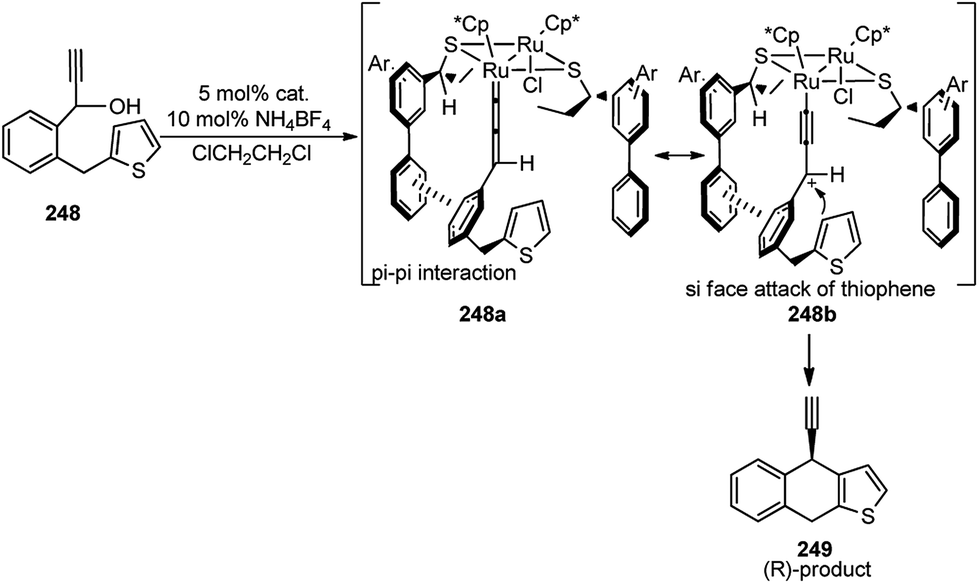 | ||
| Scheme 111 Mechanistic pathway for the ruthenium-catalyzed enantioselective cyclization of propargylic alcohols bearing a thiophene. | ||
Nishibayashi and co-workers in the year 2009 developed a ruthenium-catalyzed oxypropargylation of alkenes 250 with propargylic alcohols 209 and simple alcohols 72 using Ru-catalyst, Scheme 112.149
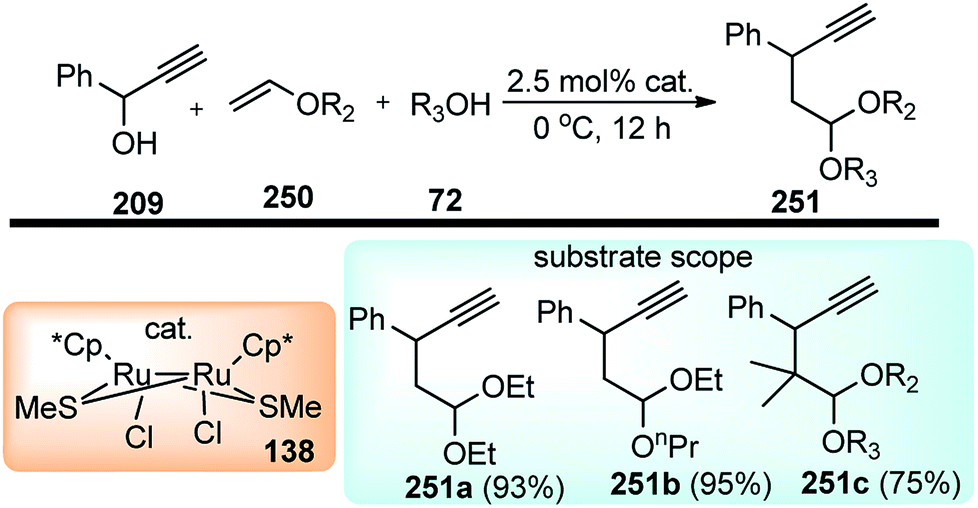 | ||
| Scheme 112 Ruthenium-catalyzed oxypropargylation of alkenes with propargylic alcohols. | ||
A proposed reaction pathway is shown in Scheme 108. The propargylic alcohol gets converted to the ruthenium–allenylidene complexes under the influence of the ruthenium catalyst. Subsequent attack of a vinyl ether on the electron-deficient γ-carbon of ruthenium–allenylidene complexes generated the corresponding alkynyl complex 250a, followed by the attack of an alcohol on the cationic-carbon to afford the corresponding oxypropargylated product via the ruthenium vinylidene complex 250c, Scheme 113.
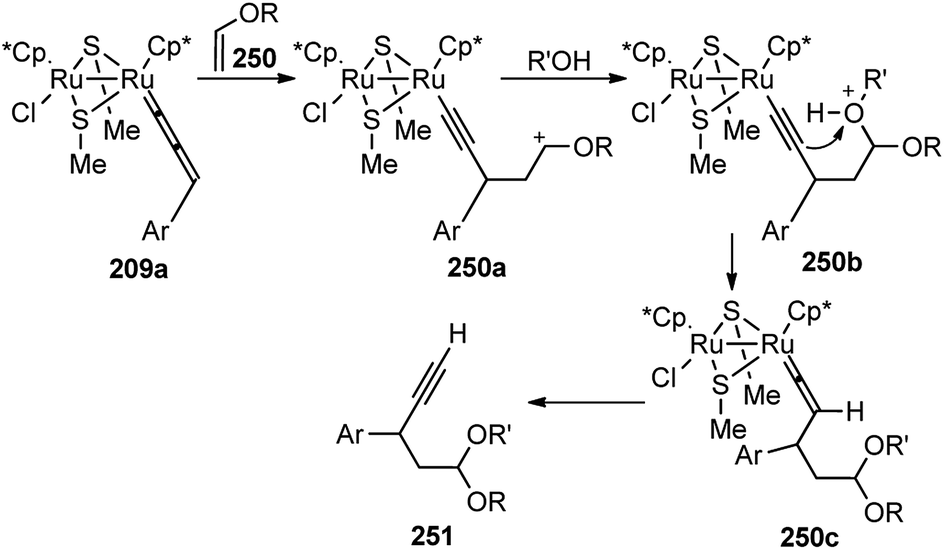 | ||
| Scheme 113 Mechanism for the ruthenium-catalyzed oxypropargylation of alkenes with propargylic alcohols. | ||
Nishibayashi and co-workers in the year 2010 reported a ruthenium-catalyzed enantioselective [3 + 3] cycloaddition of propargylic alcohols 209 with 2-naphthols 167 to afford the corresponding naphthopyran derivatives 253 in moderate to good yields with excellent enantioselectivity up to 99% ee, Scheme 111.150
This cycloaddition reaction proceeds via stepwise reactions of propargylation and intramolecular cyclization, where ruthenium–allenylidene and vinylidene complexes work as key reactive intermediates, as highlighted in Scheme 114.
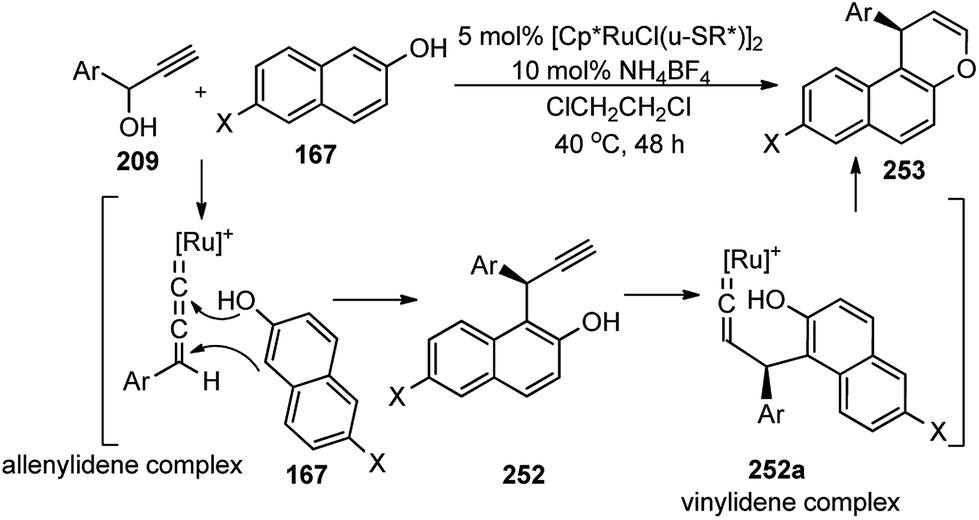 | ||
| Scheme 114 Ruthenium-catalyzed enantioselective synthesis of naphthopyrans by [3 + 3] cycloaddition of propargylic alcohols with 2-naphthols. | ||
In 2010, Nishibayashi and co-workers reported an enantioselective propargylic alkylation of propargylic alcohols 209 with aldehydes 255 in the presence of a thiolate-bridged diruthenium complex and chiral secondary amine 258 as the co-catalysts to give the corresponding propargylic alkylated products in excellent yield and enantioselectivity (up to 98% ee) as a mixture of two diastereoisomers, (Path B) Scheme 115.151 This catalytic reaction developed was a new type of enantioselective propargylic substitution reaction, wherein the enamines generated in situ from aldehydes stereoselectively attack the ruthenium–allenylidene complexes. In the present reaction system, both the transition metal catalyst (ruthenium complex) and organocatalyst (secondary amine) activated propargylic alcohols and aldehydes, respectively, and cooperatively work to promote the enantioselective propargylic alkylation.
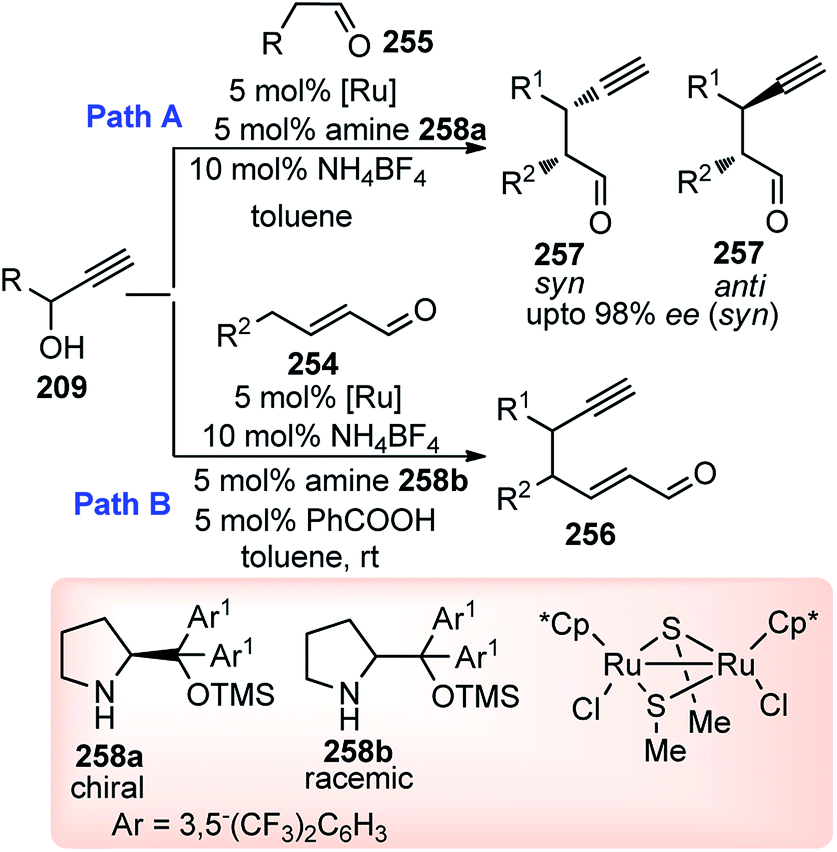 | ||
| Scheme 115 Ruthenium catalyzed enantioselective alpha-propargylation of aldehydes (Path A) and gamma-propargylation of α,β-unsaturated aldehydes (Path B) by cooperative catalysis. | ||
In the year 2012, Nishibayashi and co-workers further extended the scope of cooperative catalysis. They reported a propargylic allylation of propargylic alcohols 209 with α,β-unsaturated aldehydes 254 in the presence of the same thiolate-bridged diruthenium complex and racemic secondary amine 247 as co-catalysts to furnish the corresponding propargylic allylated products 256, (Path B) Scheme 115, where the γ-propargylation occurred selectively at α,β-unsaturated aldehydes, in moderate to high yields as a mixture of two diastereoisomers.152 The reaction system worked via a co-operative catalytic system where both the catalysts simultaneously worked together to promote the propargylic allylation.152
The initial step for the reaction is the formation of the allenylidene complex. Subsequent attack of the enamine E generated in situ from aldehyde 254/255 and amine 258 resulted in the formation of the vinylidene complex followed by ligand exchange to generate the alkylate product. The key step followed by both the reactions (Path A and Path B) are shown in Fig. 18.
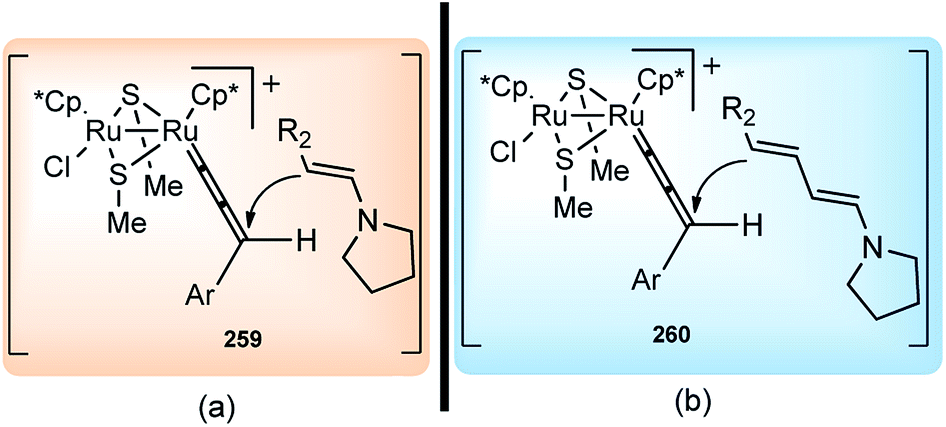 | ||
| Fig. 18 (a) The key step for the α-propargylation of aldehydes with propargyl alcohols (Path A); (b) the key step for the gamma propargylation of α,β-unsaturated aldehydes with propargylic alcohols (Path B). | ||
Combination of ruthenium and copper as catalyst leads to the propargylic alkylated products from propargylic alcohols and β-keto esters in high yields with excellent enantioselectivities.
Nishibayashi and co-workers in the year 2012 developed the enantioselective propargylic alkylation of propargylic alcohols with β-keto phosphonates in the presence of a thiolate-bridged diruthenium complex and a copper complex as co-catalyst in excellent yields with high diastereo- and enantioselectivities, Scheme 116.153
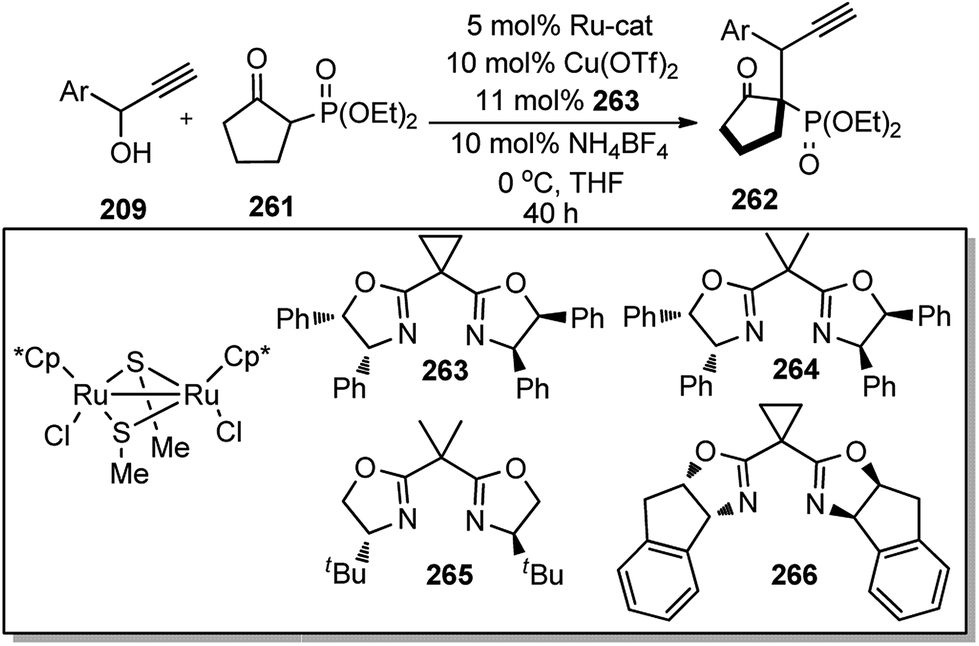 | ||
| Scheme 116 Ruthenium and copper-catalyzed enantioselective propargylic alkylation of propargylic alcohols with β-keto phosphonates. | ||
Treatment of propargyl alcohol 209 with 3 equiv. of diethyl 2-oxocyclopentylphosphonate 261 in the presence of catalytic amounts of the ruthenium catalyst, Cu(OTf)2, (4R,4′R,5S,5′S)-2,2′-(cyclopropane-1,1-diyl)bis(4,5-diphenyl-4,5-dihydrooxazole) 263 and NH4BF4 in tetrahydrofuran (THF) at room temperature for 40 h furnished the desired product in 63% isolated yield as a mixture of two diastereoisomers (anti/syn = 15/1) with 89% ee of the major diastereomer. Unsatisfactory yields were received when the reaction was carried out in other solvents like dichloromethane, 1,4-dioxane, diethyl ether, and toluene. With ligands like bis(oxazoline) was used although yields were good but the diastereoselectivity as well as enantioselectivity deteriorated. Other diruthenium complexes such as the complex bearing the sterically more demanding SiPr moiety [Cp*RuCl-(μ2-SiPr)]2 exhibited a lower enantioselectivity. Also, the use of only ruthenium catalyst or copper complex did not promote the propargylic alkylation which indicates that both ruthenium catalyst and copper complex worked cooperatively as catalysts to promote the catalytic reaction enantioselectively.
A proposed reaction pathway is highlighted in Scheme 117. The initial step is the formation of an allenylidene complex 209a by the reaction of propargylic alcohol 209 with 261 via a vinylidene complex. Subsequent attack of an enolate 261a, on the γ carbon of 209a resulted in the formation of another vinylidene complex. After transformation of the vinylidene complex into the corresponding π-alkyne complex, the alkylated product 262 is formed by ligand exchange with another molecule of propargylic alcohol 209.
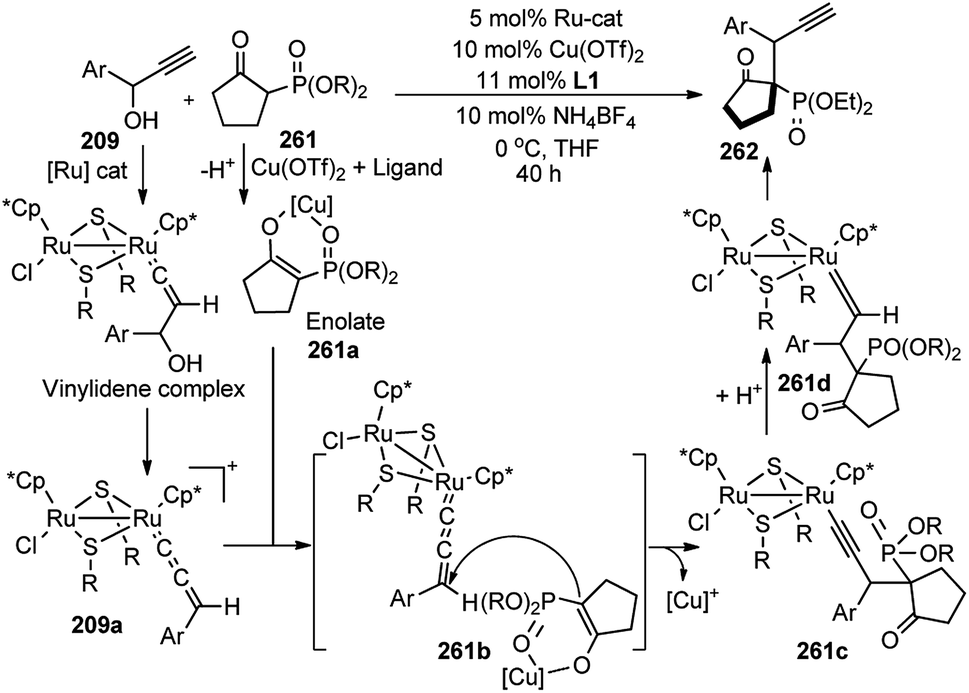 | ||
| Scheme 117 Mechanistic pathway for the ruthenium and copper-catalyzed enantioselective propargylic alkylation of propargylic alcohols with β-keto phosphonates. | ||
Both ruthenium complex and copper complex activate propargylic alcohols and β-keto phosphonates, respectively, and both catalysts cooperatively and simultaneously work to promote the propargylic alkylation with high stereoselectivity.
Nishibayashi and co-workers in the year 2012 developed an efficient enantioselective propargylic alkylation of propargylic alcohols 209 with β-ketoesters 267 in the presence of a thiolate-bridged diruthenium complex 138 and a copper complex as co-catalyst in high enantioselectivity, Scheme 118.154
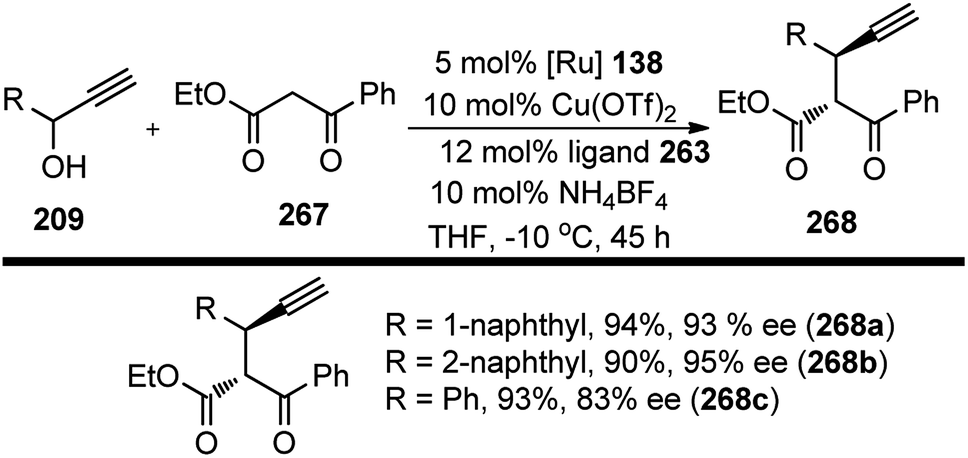 | ||
| Scheme 118 Ruthenium and copper-catalyzed enantioselective propargylic alkylation of propargylic alcohols with β-ketoesters. | ||
In the present reaction system, the transition metal catalysts (ruthenium and copper complexes) activate propargylic alcohols and β-ketoesters, respectively, thereby cooperatively promoting the propargylic alkylation enantioselectively. Treatment of 1-(1-naphthyl)-2-propyn-1-ol with three equivalents of ethyl 3-oxo-3-phenylpropanoate in the presence of catalytic amounts of thiolate-bridged diruthenium complex, Cu(OTf)2 with (4R,4′R,5S,5′S)-2,2′-(cyclopropane-1,1-diyl)-bis(4,5-diphenyl-4,5-dihydrooxazole) 263, and NH4BF4 in THF at room temperature for 120 h gave ethyl 2-benzoyl-3-(naphthalen-1-yl)-4-pentynoate 268 in 96% isolated yield as a mixture of two diastereoisomers (anti/syn = 4:1) with 86% ee of the major diastereomer. Other bis(oxazoline) ligands were not efficient in terms of product selectivity. Use of either ruthenium catalyst or the copper complex alone did not promote the propargylic alkylation which indicated that both the catalysts acted cooperatively as catalysts to promote the catalytic reaction enantioselectively. The reaction has a wide substrate scope and a wide variety of propargylic alcohols efficiently reacted under the optimized conditions.
Nishibayashi and co-workers in the year 2013 described ruthenium-catalyzed [3 + 2] cycloaddition of ethynylcyclopropanes 269, bearing two carboxy groups at the homopropargylic position, with aldehydes 270 and aldimines 271 leading to the corresponding 2-ethynyltetrahydrofurans 272 and pyrrolidines 273 in high to excellent yields, Scheme 119.155
 | ||
| Scheme 119 Ruthenium-catalyzed [3 + 2] cycloaddition of ethynylcyclopropanes. | ||
2-Ethynyltetrahydrofurans 272 and pyrrolidines 273 were also achieved in high to excellent yields using different co-catalysts like BF3·OEt2 and Sc(OTf)3. But when BF3·OEt2 was used in place of Sc(OTf)3, the yield of pyrrolidine decreased dramatically, probably due to the difference in the coordination ability of BF3 over Sc(OTf)3. A plausible reaction pathway is shown in Scheme 120.
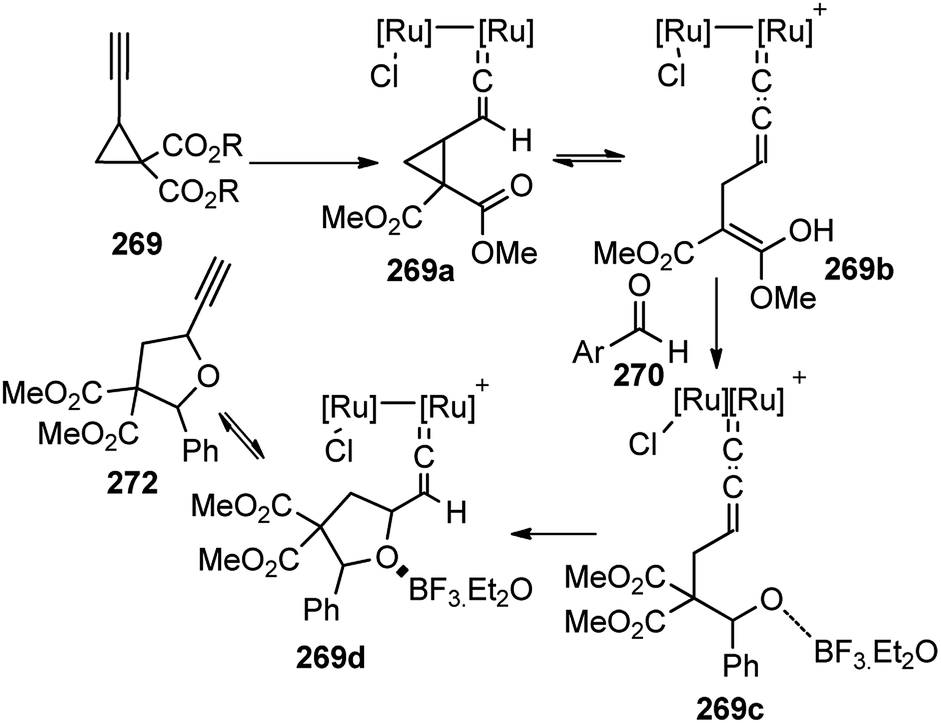 | ||
| Scheme 120 Mechanism for the ruthenium-catalyzed [3 + 2] cycloaddition of ethynylcyclopropanes. | ||
The initial step is the formation of the ruthenium vinylidene complex 269a followed by the formation of allenylidene complex bearing an enol moiety 269b. Allenylidene complex bearing an enol moiety, on activation by BF3·OEt2, afford a new allenylidene complex 269c by the nucleophilic attack to the corresponding aldehyde followed by intramolecular cyclization to give a vinylidene complex 269d. Finally, a ligand exchange reaction furnishes the corresponding cycloaddition product 272.
Nishibayashi and co-workers in the year 2015 have reported a highly diastereo and enantioselective propargylic alkylation of propargylic alcohols 209 with E-ene-carbamates 274 in the presence of a catalytic amount of a thiolate-bridged diruthenium complex, bearing an optically active phosphoramide moiety, Scheme 121.156
 | ||
| Scheme 121 Highly diastereo- and enantioselective propargylic alkylation of propargylic alcohols with E-enecarbamates. | ||
The authors have designed a hybrid catalyst consisting of a thiolate-bridged diruthenium complex tagged with (R)-BINOL based phosphoramide moiety with the rationale that the ruthenium complex will activate the allenediene intermediate and the phosphoramide will guide the ene-carbamate to control the nucleophilic attack, Fig. 19. The phosphoramide moiety (NH) was found to be essential for achieving high enantioselectivity and the introduction of two methyl groups at the 3,3′-positions of the naphthyl moiety in the chiral ligand dramatically increased the enantioselectivity of the product. However, the introduction of more bulky substituents at the 3,3′ position had a negative effect on the reaction selectivity. The reaction proceeded via ruthenium–allenylidene complex and the probable transition state is depicted in Fig. 19.
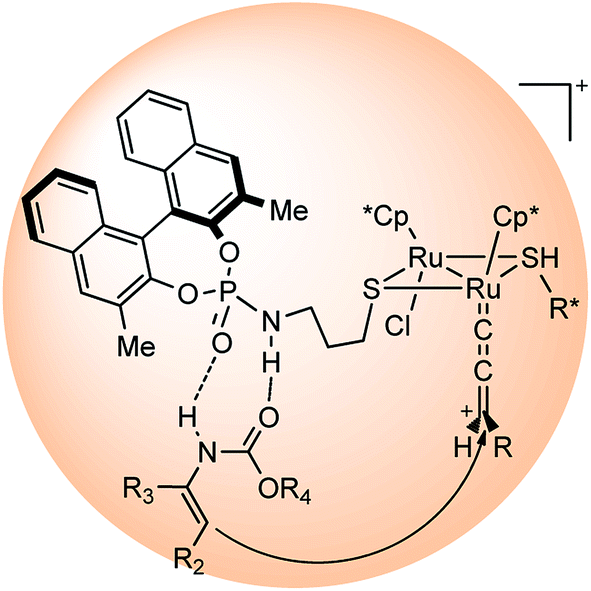 | ||
| Fig. 19 Probable transition state structure for the highly diastereo and enantioselective propargylic alkylation of propargylic alcohols with E-ene-carbamates. | ||
It was also observed that phosphoramide catalyst added externally did not work as an effective organocatalyst; the intramolecular phosphoramide moiety in the ruthenium complex played an essential role in promoting the propargylic alkylation with high diastereo- and enantioselectivities.
5.11. Rhodium derived catalysts
Compared to the commonly used nickel, palladium, and platinum catalysts, rhodium presents the chemist with new and interesting catalytic possibilities.157 Generally nickel, palladium, and platinum typically operate within catalytic cycles shuttling between the (0) and (II) oxidation states whereas rhodium typically shuttles between the (I) and (III) oxidation states. As a consequence, transmetalation can theoretically occur at two points in the catalytic cycle and opened up immense opportunity in catalysis.158 But rhodium has rarely been utilized as a catalyst for the propargylic substitution reaction. A general mechanistic pathway followed by rhodium catalyzed propargylic substitution reaction is illustrated in Scheme 122.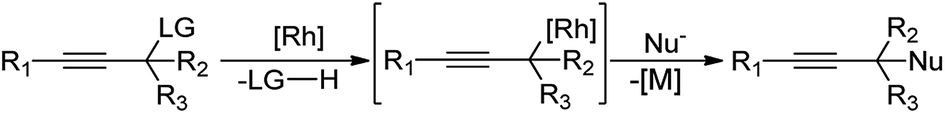 | ||
| Scheme 122 General mechanistic pathway for the rhodium catalyzed propargylic substitution reaction. | ||
Rhodium-catalyzed propargylic amination of propargylic carbonates 276 with sulphonamides 277 was reported by Evans and Lawler in 2006, Scheme 123.159
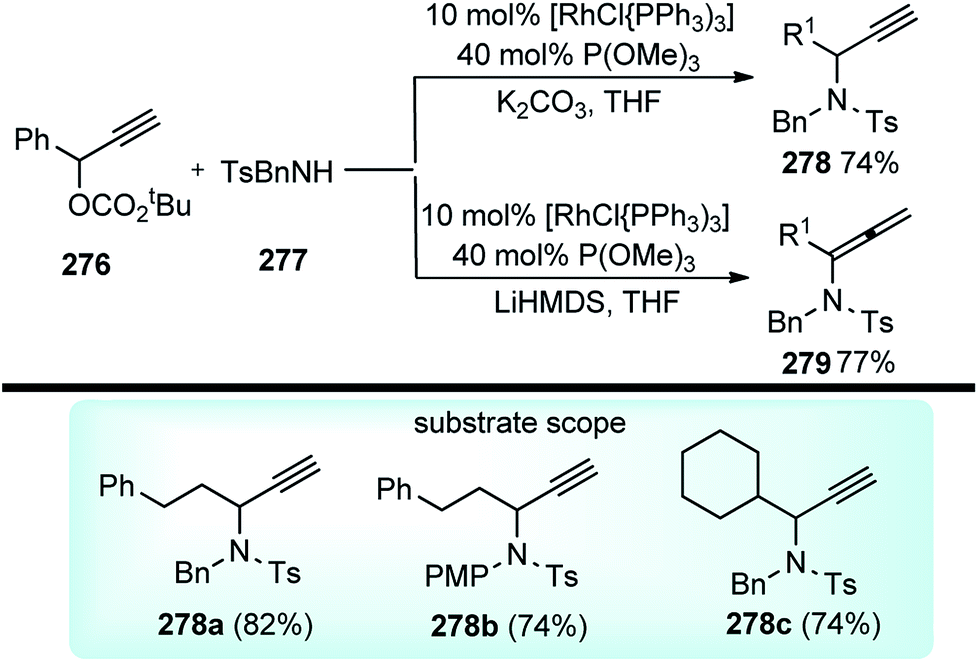 | ||
| Scheme 123 Rhodium-catalyzed propargylic amination of propargylic carbonates. | ||
A range of alkyl-substituted propargylic carbonates reacted smoothly with sulfonamides in the presence of catalytic amounts of [RhCl(PPh3)2] and P(OMe)3 to furnish the corresponding propargylated amides in good yields. The base has a pronounced effect on the regioselectivity of the reaction. When a powerful base was used, the propargylic sulphonamide 278 isomerized to the corresponding allenyl compounds 279. Unfortunately, this rhodium-catalyzed reaction did not allow chirality transfer.
5.12. Palladium derived catalysts
The earliest example of palladium-catalyzed propargylic substitution reaction was the hydrogenolysis of propargylic substrates with triethylammonium formate reported by Dixneuf and co-workers, in which the reduction at the propargylic position occurred in the presence of a combination of Pd(dba) and PnBu3 as a catalyst.160But no systematic studies were carried out till the report by Marshall and Wolf in 1996 on the palladium-catalyzed propargylic amination which they accidentally discovered during the attempted amidocarbonylation of the racemic mesylate with diethylamine, in the presence of CO and catalytic Pd(PPh3)4, Scheme 124.161
 | ||
| Scheme 124 Palladium catalyzed enantioselective propargylic amination reaction. | ||
Desired propargylated amines were obtained in higher ratio when the more nucleophilic amines were used and vice versa. In the absence of CO, amines were obtained from the nonracemic propargyl mesylate 275 as the sole products of high enantioselectivity. Treatment of propargyl mesylate with aniline in the absence of Pd-catalyst afforded the propargylamine of opposite configuration to that of amine which provided a direct evidence that the Pd(0)-catalyzed aminations proceed with retention of configuration.
A novel approach for the synthesis of allenylphosphonates 183 and related compounds from the reaction between propargylic derivatives 39 with H-phosphonate 182, H-phosphonothioate, H-phosphonoselenoate, and H-phosphinate esters utilizing palladium(0) as catalyst catalyzed was reported by Stawinski and co-workers in the year 2010, Scheme 125.162,163
 | ||
| Scheme 125 Palladium-catalyzed synthesis of allenylphosphonates from propargylic derivatives. | ||
Ligand screening revealed that bidentate ligands like DPEPhos with large bite angles were able to promote the conversion into the allenylphosphonate. Several catalytic systems were evaluated, and the most efficient combination was found to consist of Pd2(dba)3·CHCl3 as a palladium source and bis(2-diphenylphosphino)diphenyl ether (DPEPhos) as a supporting ligand. A mechanistic pathway for the Pd-catalyzed synthesis of allenylphosphonates from propargyl alcohol is illustrated in Scheme 126.
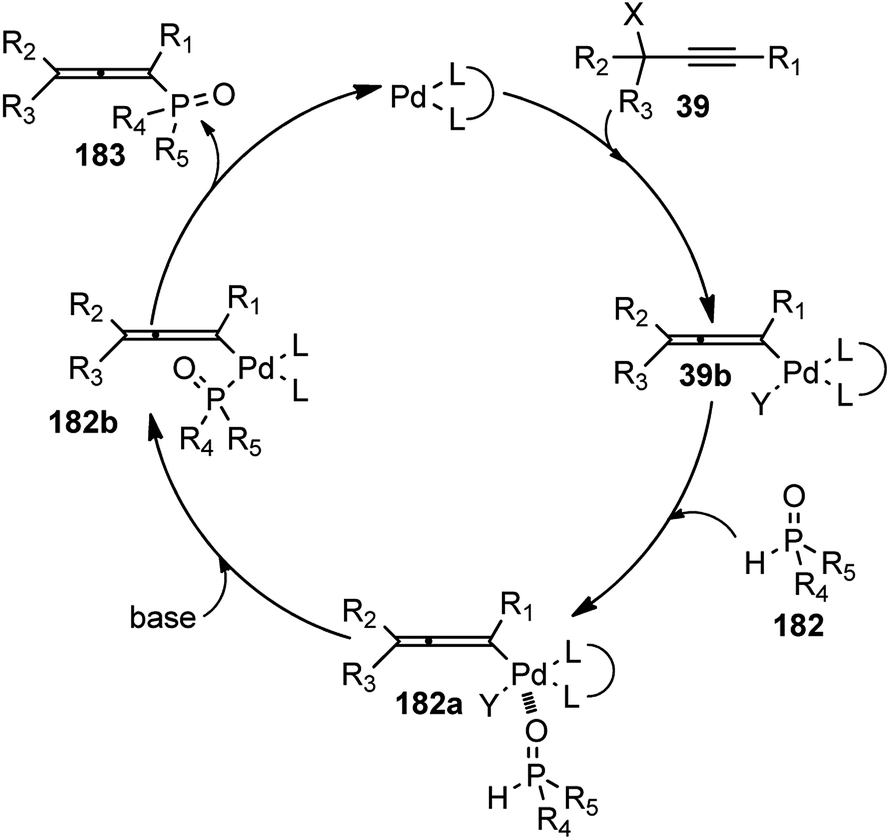 | ||
| Scheme 126 Catalytic cycle for the palladium-catalyzed synthesis of allenyl phosphonates from propargylic derivatives. | ||
Iazzetti and co-workers in 2015 designed a palladium-catalyzed reaction of propargylic carbonates 282 with Meldrum's acid derivatives 283 to give the substitution products 284 in very good yield and high regioselectivity, Scheme 127.164
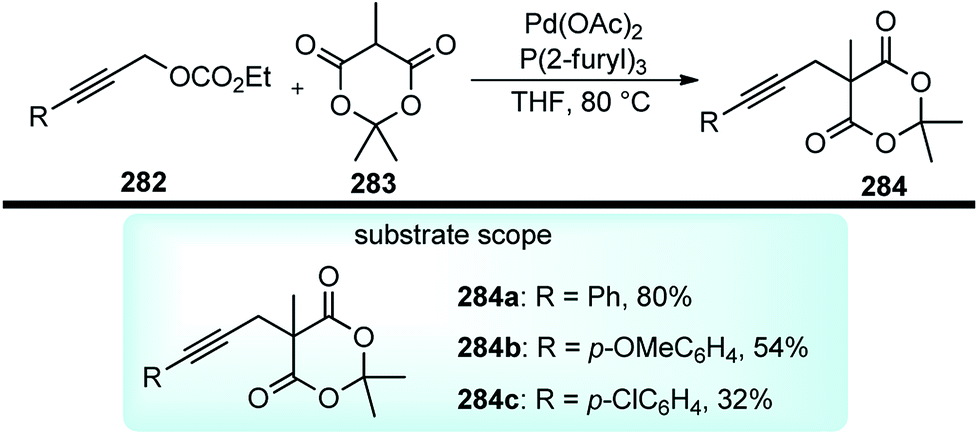 | ||
| Scheme 127 Palladium-catalyzed reaction of propargylic carbonates with Meldrum's acid derivatives. | ||
The reaction of propargylic carbonates with Meldrum's acid derivatives proceeded smoothly in the presence of Pd(OAc)2 and P(2-furyl)3 in THF at 80 °C to offer the propargyl substitution products in good yields.
Among the several other palladium catalysts screened, Pd(OAc)2 appeared to be the best in terms of reaction yield. The methodology has wide functional group tolerance and tolerates a variety of functional groups like ester, keto, ether, amido, and chloro substituents. The reaction is highly dependent on the nature of the phosphine ligand as well as the basicity/polarizability of the anion derived from the 1,3-dicarbonyl compound. The reaction mechanism is outlined in Scheme 128.
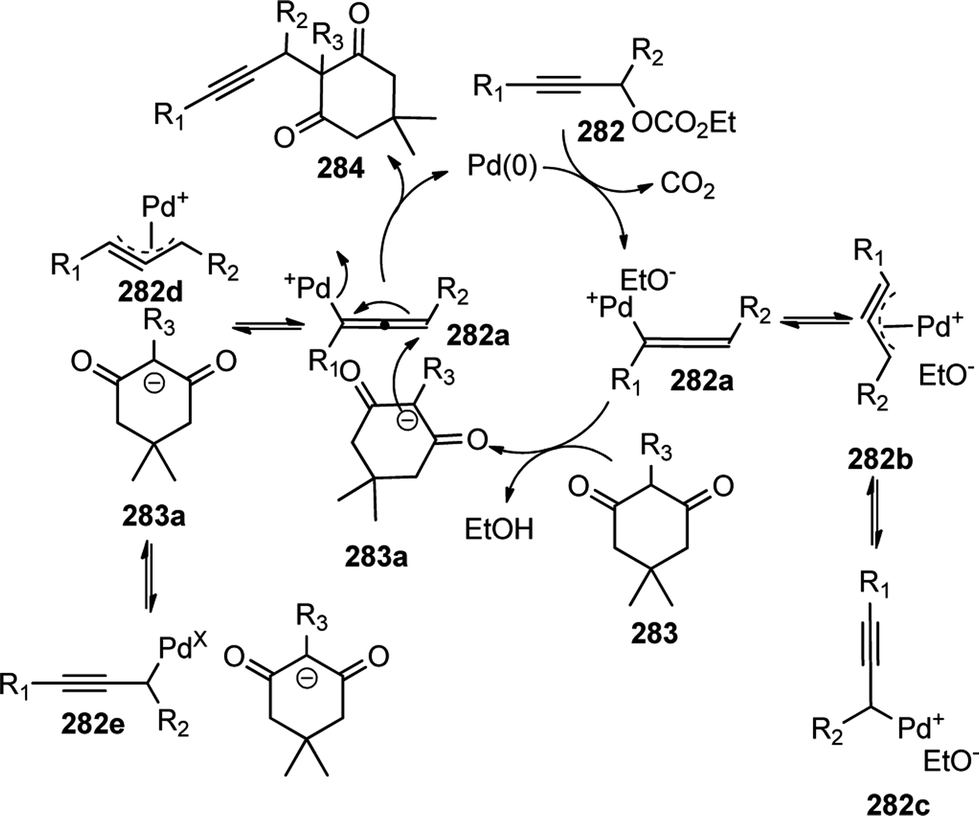 | ||
| Scheme 128 Mechanistic pathway for the palladium-catalyzed reaction of propargylic carbonates with Meldrum's acid derivatives. | ||
Lattenzi's report in 2015 about the propargylic alkylation using propargylic carbonates was limited to highly stabilized Meldrum's acid-like nucleophiles. Tunge and co-workers in the year 2016 reported a regioselective substitution of diaryl acetonitrile pronucleophiles 285 to propargylic carbonates 286 in good yields, Scheme 129.165
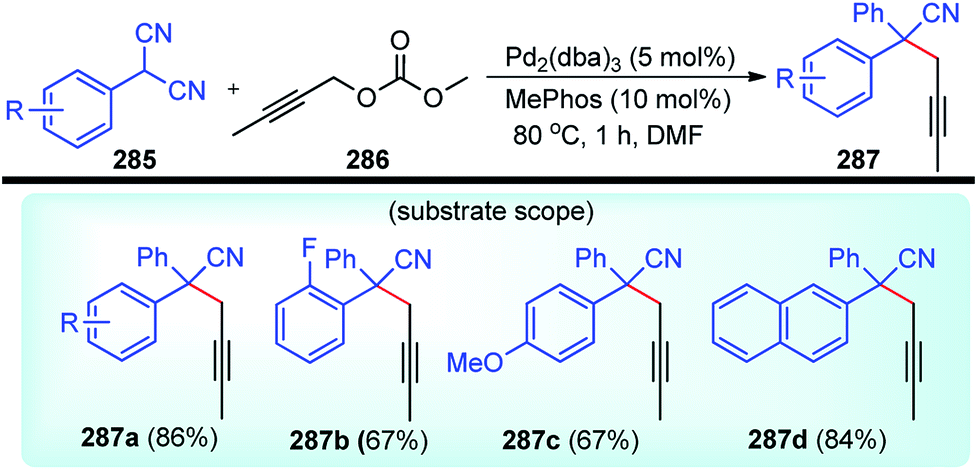 | ||
| Scheme 129 Palladium catalyzedregioselective substitution of diaryl acetonitrile to propargylic carbonates. | ||
The method described by the authors has expanded the scope of palladium-catalyzed propargylation to weakly acidic α,α-diarylacetonitrile motifs that resulted into the functionalized quaternary diarylmethane products. The reaction course is controlled by the nature of the ligand. With bidentate ligand like dppe, substitution of methyl propargyl carbonate with α,α-diarylacetonitrile resulted in 1,3-dienyl products whereas using a monodentate ligand like MePhos leads to the formation of propargyl isomer under identical reaction conditions. Regioselective nucleophilic substitution occurs via the inner sphere and outer sphere mechanism that is controlled by the denticity of the ligand. Bidentate ligands blocked the coordination of the nitrile nucleophile, favoring outer-sphere mechanism, leading to dienylation. Whereas, a monodentate ligand allowed coordination of the nucleophile to palladium, resulting in propargylation through an inner sphere nucleophilic attack as illustrated in Scheme 130.
 | ||
| Scheme 130 Mechanistic pathway for the palladium-catalyzed regioselective substitution of diaryl acetonitrile to propargylic carbonates. | ||
5.13. Silver derived catalysts
Silver(I) salts have been used in several organic transformations. The high oxidation potentials associated with Ag ions allowed silver catalysis, complexes, and ligands to enjoy significant importance in chemical synthesis. The mechanistic pathway for the silver catalyzed propargylic substitution reaction is illustrated in Scheme 131 where the silver salt polarizes the hydroxy group sufficiently to generate the propargylic carbocation intermediate, which was subsequently attacked by the approaching nucleophile. | ||
| Scheme 131 General mechanism for the silver catalyzed propargylic substitution reaction. | ||
A AgOTf catalyzed cascade and chemoselective approach towards the synthesis of 3,5/1,3-disubstituted pyrazoles 289 from propargylic alcohols 23 and para-tolylsulfonylhydrazide 288 was reported by Zhan and co-workers in the year 2013, Scheme 132.166
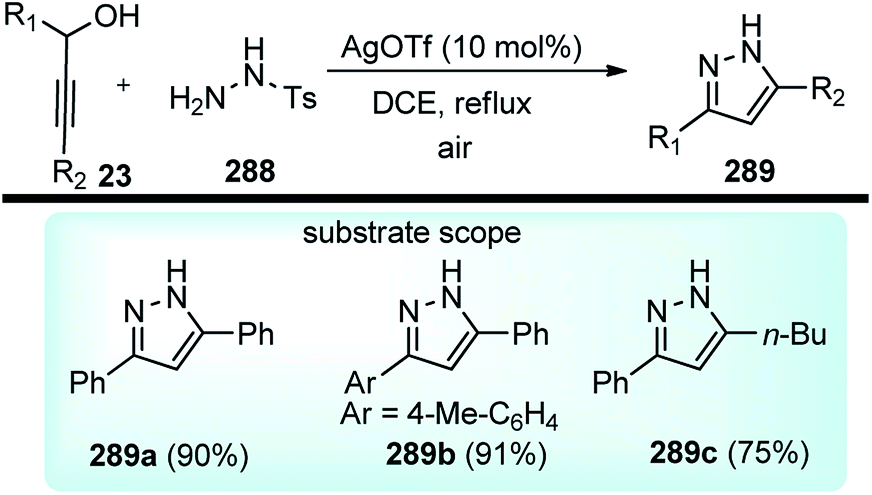 | ||
| Scheme 132 AgOTf-catalyzed chemoselective approach to 3,5/1,3-disubstituted pyrazoles from propargylic alcohols. | ||
Several other catalysts like FeCl3, AgOTf, AgBF4 InCl3, In(OTf)3, Zn(OTf)2, Cu(OTf)2, BiCl3 were also tested for this reaction but the best result in terms of yield was achieved with AgOTf. Solvent study evaluation revealed that the reaction, when performed in 1,2-dichloroethane at reflux, afforded the product in highest yield. With other silver catalysts like AgOAc and AgBF4, reaction yield decreased significantly clearly indicating the role of counter anion to facilitate this cascade reaction. Even no desired product was observed when the reaction was carried out under a Brønsted acid condition which clearly ruled out the effect of “hidden of Brønsted acid” as proposed by several other research groups.167 It was further observed that other substituted hydrazines under standard conditions did not furnish the products due to the suppression of the catalytic activity of the AgOTf by the stronger alkalinity of those substituted hydrazines. Electron-donating functional groups were found to enhance the reaction rate and furnished products in good yields whereas electron-withdrawing substituents slowed down the transformation.
A plausible mechanistic pathway is illustrated in Scheme 133 involved the formation of propargylic cation intermediate under the influence of silver catalyst followed by the regioselective attack of the terminal nitrogen atom, resulting in the substituted product 288a. Then, a series of reactions involving 5-endo-dig cyclization, proton exchange, and aromatization finally leads to the desired product 289, Scheme 133.
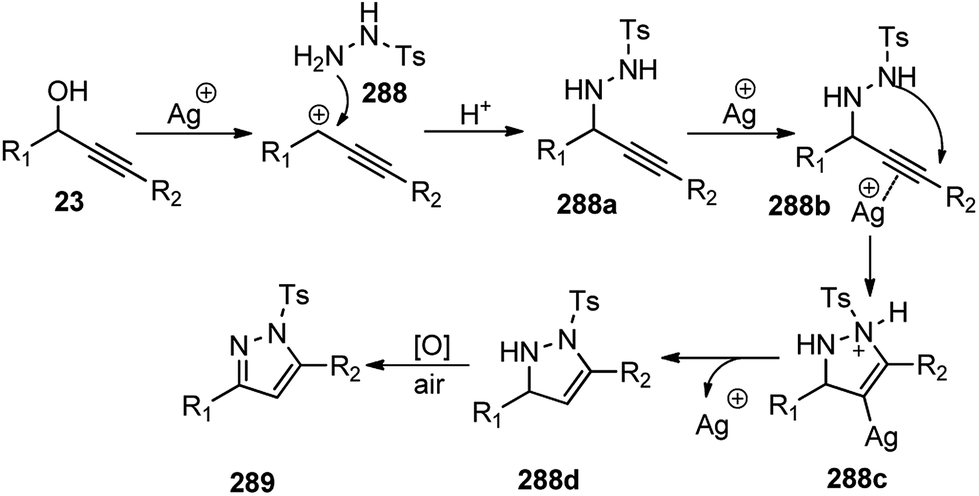 | ||
| Scheme 133 Mechanism for the AgOTf-catalyzed synthesis of 3,5/1,3-disubstituted pyrazoles from propargylic alcohols. | ||
Sahoo and co-workers in the year 2013 reported a silver(I) catalyzed propargylation of pyrazole 290 with propargyl acetates 123 towards the synthesis of (E)-allyl-gem-dipyrazoles 291, Scheme 134.168
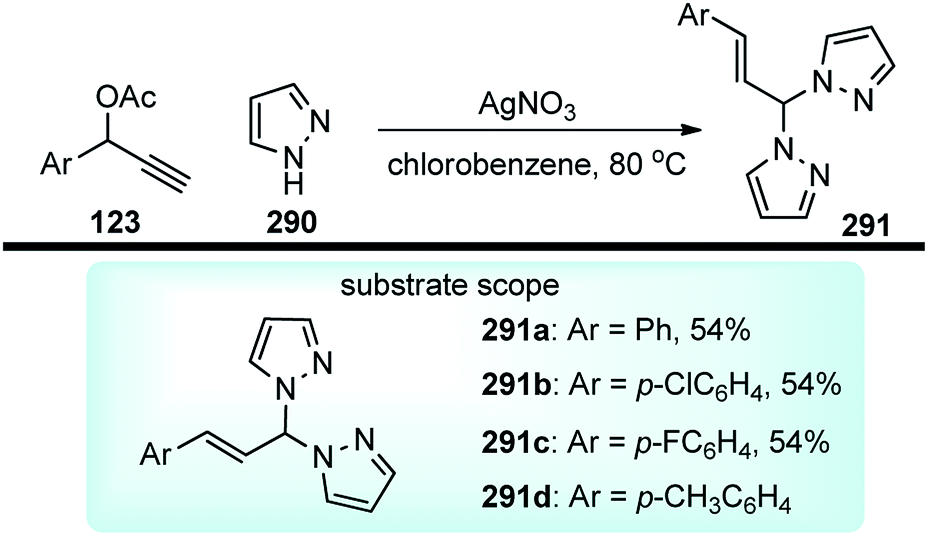 | ||
| Scheme 134 Silver(I) catalyzed propargylation of pyrazole with propargyl acetates. | ||
Several catalysts were tested in different solvents like chlorobenzene, THF, CH3CN, DMF, DCE. Incomplete consumption of the starting material, complex reaction mixture and poor overall yield of the products were noticed when Cu(OTf)2, In(OTf)3, and CuNO3·H2O, AuCl3, AgNO3, AgCl, Ag2CO3, AgNO3, AgBF4 were independently employed. AgNO3 was found to be the most effective among the various Ag-salts examined. Surprisingly, the reaction was incomplete in polar aprotic solvents like THF, CH3CN, and DMF. The product yield in DCE was poor. The probable mechanism for the propargylation reaction is shown below, Scheme 135.
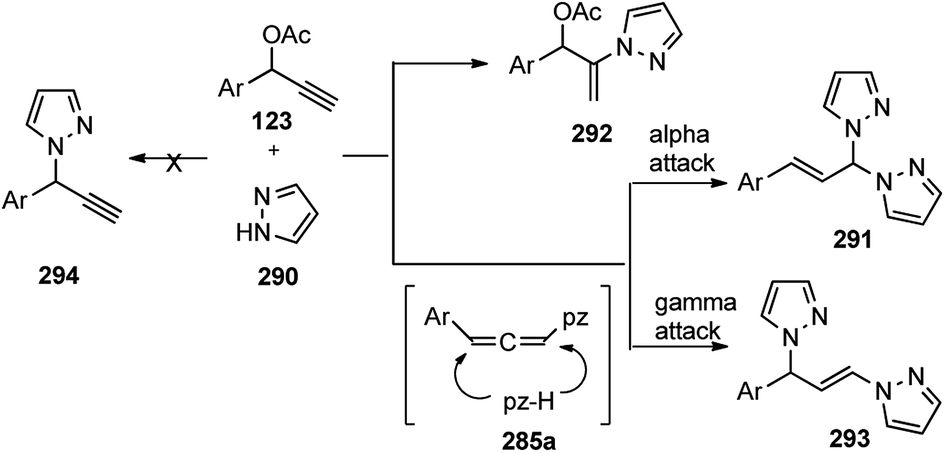 | ||
| Scheme 135 Probable mechanistic pathway for the silver(I) catalyzed propargylation of pyrazole with propargyl acetates. | ||
The above methodology provided a quick access to a wide array of (E)-allyl-gem-dipyrazoles (ADPs) derivatives 291, whose two adjacent N atoms offered a strong chelating ability to the metal. Their capability to act as the bidentate ligand was nicely demonstrated in the well-known Suzuki–Miyaura biaryl cross-coupling reaction. Both the electron-rich, electron poor as well as sterically hindered, and heteroaryl iodides were effectively coupled with PhB(OH)2 under the influence of 0.1 mol% of catalyst in aqueous K2CO3 at 80 °C.
Yang and co-workers in the year 2013 reported a silver-catalyzed direct phosphorylation of propargyl alcohols 39 with readily available and stable P(O)H compounds 182 in an efficient manner, Scheme 136.169
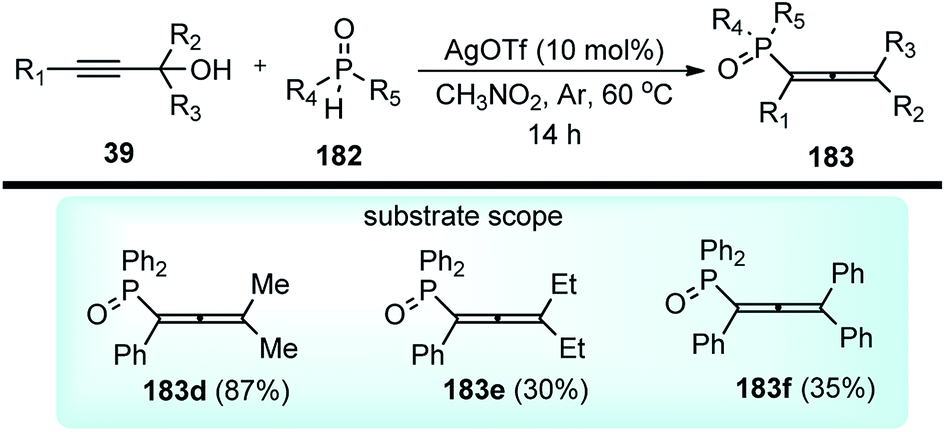 | ||
| Scheme 136 Silver catalyzed synthesis of synthesis of allenylphosphoryl compounds from propargylic alcohol. | ||
The desired product was obtained in a moderately low yield in the presence of 10 mol% of AgOTf as a catalyst in DCE at 60 °C for 14 h under an argon atmosphere. Product yield did not improve when other catalysts like Cu(OTf)2, LiOTf, CuOTf, Sm(OTf)3, AgNTf2, Cu(ClO4)2 and Ni(OAc)2 were used. Subsequently, the effect of different solvents such as CH3NO2, MeCN, and THF was tested and it was found that CH3NO2 was the optimal solvent and could enhance the yield up to 47%.
A proposed reaction mechanism is shown in Scheme 137. The OH is polarized by the coordination of the AgOTf with the alkyne resulting in the formation of propargylic carbocation intermediate which is resonance stabilized to the allenylidene intermediate 39b. Consequent attack by the nucleophile furnishes the product 183 with the concomitant loss of H+.
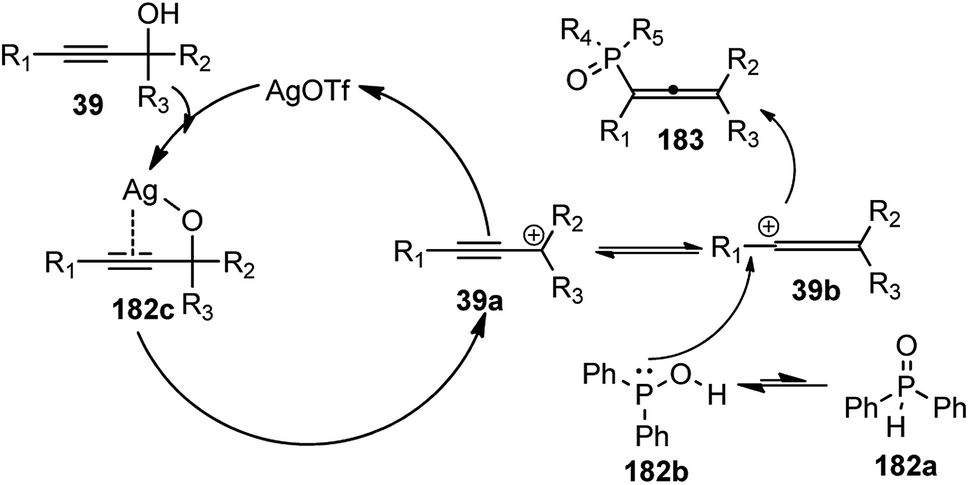 | ||
| Scheme 137 Silver catalyzed synthesis of allenyl-phosphoryl compounds from propargylic alcohols. | ||
5.14. Indium derived catalysts
Recently, indium(III) salts have received a great deal of attention due to its water-tolerant green Lewis acid for performing chemo and regioselective organic transformations.170 Compared to conventional Lewis acids, it has the advantages of water stability, recyclability, operational simplicity and strong functional group tolerance. In general, indium catalyzed propargylic substitution reaction using indium catalyst proceeds via propargylic carbocation intermediate formed by the loss of water, Scheme 138.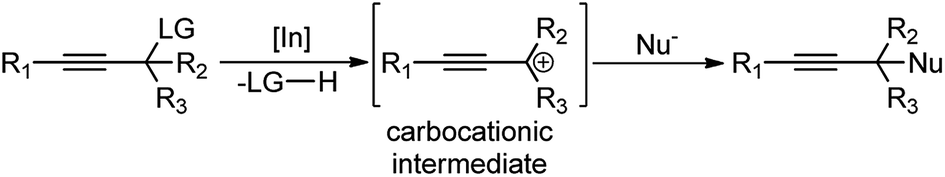 | ||
| Scheme 138 General mechanism for the indium catalyzed propargylic substitution reaction. | ||
Yadav and co-workers in the year 2007 described propargylation of heteroaromatic systems 61 in high yields and high selectivity by propargylic alcohols 23 within a short reaction time using indium(III) bromide as a catalyst, Scheme 139.171
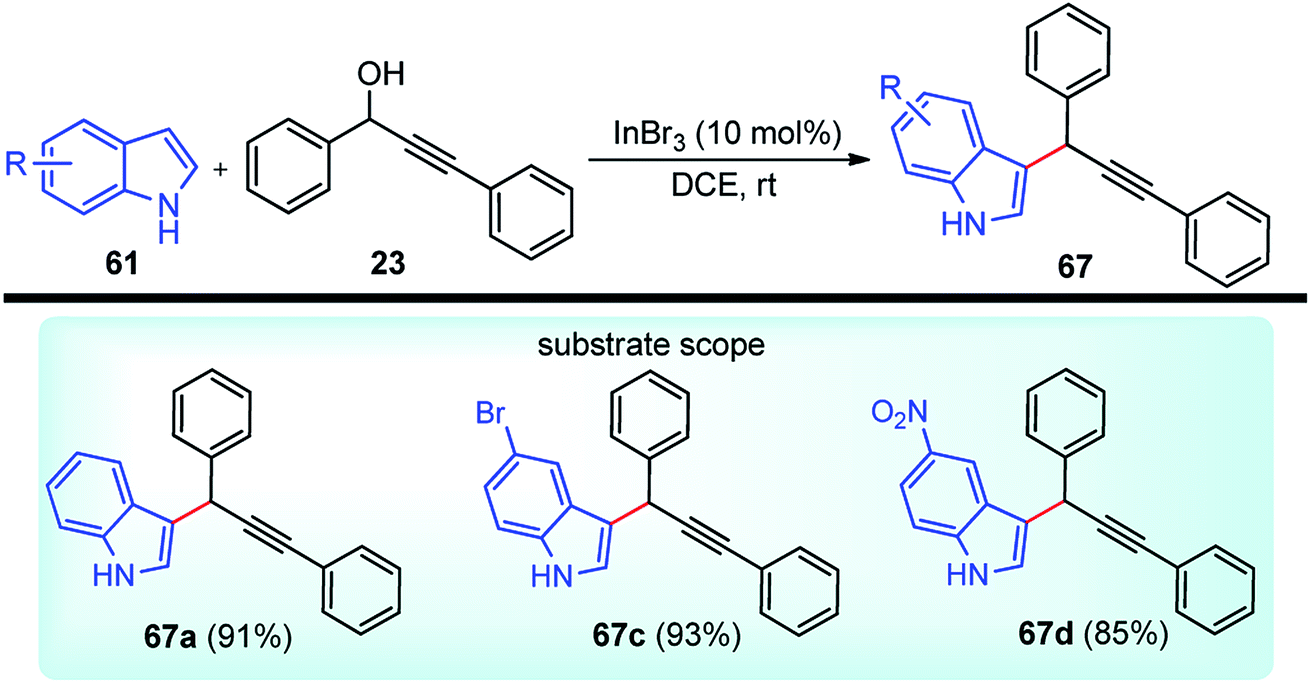 | ||
| Scheme 139 InBr3 catalyzed C3-propargylation of indoles. | ||
The reaction proceeded under the influence of the catalytic amount of InBr3 and a wide range of heteroaromatic compounds such as indole, carbazole, pyrrole, and furan underwent facile reaction under mild reaction conditions. No product was obtained in the absence of indium(III) bromide. This methodology has a wide substrate scope and tolerates functional groups like alkenyl, alkynyl, halo, nitro, cyano, and free amino groups as well as acid sensitive substrates like pyrrole and furan.
In the same year 2007, Chen and co-workers, demonstrated a similar propargylic substitution reaction of propargylic acetates 71 with indoles 61 utilizing InCl3 as the catalyst, Scheme 140.172
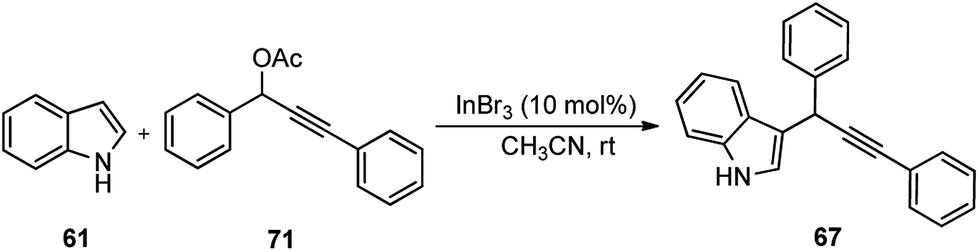 | ||
| Scheme 140 InBr3 catalyzed C3-propargylation of indoles. | ||
Catalyst screening revealed that indium(III) chloride was the most efficient Lewis acid catalyst for the reaction. The methodology has a wide substrate scope and tolerates both electrons rich and electron-poor substrates.
Recently, the concept of combining organocatalysis with transition metal complexes has led to exciting strategies for the development of dual catalysis for several innovative transformations.173–175 Nishibayashi in the recent past has described highly enantioselective propargylation of aldehydes by combining the Hayashi–Jørgensen organocatalyst with a ruthenium complex.153 The possibility to induce the formation of carbenium ion in the presence of water by merging an organocatalytic process with a Lewis acid was believed to open several new opportunities in the field of organocatalysis.
Cozzi and co-workers in the year 2011 described the first catalytic stereoselective addition of aldehydes 295 to propargylic alcohols 23, promoted by a combination of an organocatalyst and indium triflate, Scheme 141.176
 | ||
| Scheme 141 In(OTf)3 catalyzed the stereoselective addition of aldehydes to propargylic alcohols. | ||
Indium(III)triflate is known to generate less stabilized carbocations form secondary alcohols and are not deactivated by aldehydes, secondary amines or water. Therefore, the authors have selected alcohol as model substrates and performed the reaction in the presence of different MacMillan catalysts 297–301, Fig. 20.
 | ||
| Fig. 20 Different organocatalysts screened. | ||
Propargylic alcohol which can form a stabilized carbocation did not furnish any product in the absence of indium salts. Quite remarkably, the reaction tolerated a wide range of functional groups including thio, amides, silyl ether, and even acetals in the alkyne moiety. The probable mechanistic pathway along with the transition state for this dual catalytic enantioselective propargylic substitution reaction is shown in Fig. 21.
 | ||
| Fig. 21 Probable transition state for the stereoselective addition of aldehydes to propargylic alcohols. | ||
Nishibayashi and co-workers in the year 2011 developed the enantioselective propargylic alkylation of propargylic alcohols bearing an internal alkyne moiety with aldehydes in the presence of InBr3 and an optically active secondary amine in excellent yields and high enantioselectivity (up to 98% ee), Scheme 142.177
 | ||
| Scheme 142 Enantioselective propargylic alkylation of propargylic alcohols with aldehydes in the presence of InBr3 and an optically active secondary amine. | ||
This catalytic reaction provided a new type of enantioselective propargylic substitution reaction, where the enamines generated in situ from the aldehydes enantioselectively attacked the propargylic cations as reactive intermediates. In the present reaction system, both the Lewis acid catalyst (InBr3 or FeCl3) and the organocatalyst (secondary amine) activate the propargylic alcohols 23 and aldehydes 302, respectively, and both catalysts cooperatively and simultaneously work to promote the propargylic alkylation enantioselectively.
Similar product yield was also achieved when InCl3 was used as the Lewis acid. The catalytic reaction did not proceed smoothly in the presence of a catalytic amount of BiCl3 as well as TsOH did not work at all as a catalyst for the cooperative catalytic reaction. A longer reaction time was necessary when the amounts of InBr3 (10 mol%) and secondary amine catalyst (10 mol%) were decreased under the same reaction conditions. It was also observed that the use of only InBr3 or the secondary amine catalyst did not promote the propargylic alkylation. The reaction has a wide substrate scope and furnished products with various substituents in good yields and enantioselectivities.
5.15. Stannous derived catalysts
Stannous chloride is known to be an inexpensive mild Lewis acid has been used in various organic synthetic operations and has recently expanded the scope of its applications in organic synthesis.178Masuyama's group in 2013 demonstrated the use of weak Lewis acid, tin(II) chloride as an efficient catalyst for the propargylic substitution of secondary propargylic alcohols 23 with different carbon nucleophiles 82/304, Scheme 143.179
 | ||
| Scheme 143 Tin(II) chloride as an efficient catalyst for the propargylic substitution of secondary propargylic alcohols. | ||
Different electron-rich arenes, heteroarenes, 1,3-dicarbonyl compounds and nitrogen nucleophiles, like sulfonamides, carbamates, and carboxamides easily underwent propargylic substitution reaction at 40–80 °C in CH3NO2 solvent under air in presence of catalytic amount of SnCl2. The authors optimized the reaction condition by screening several other Lewis acid catalysts like tin(II) bromide, tin(II) iodide, tin(II) fluoride in different solvents like 1,2-dichloroethane, toluene, acetonitrile, THF, and DMF. Nitromethane was found superior to other solvents such as 1,2-dichloroethane, toluene, and acetonitrile. No reaction was observed either in THF or DMF and even in the absence of a catalyst. Tin(II) chloride exhibited a higher catalytic activity in CH3NO2 than other tin halides like tin(II) bromide or tin(II) iodide. With tin(II) fluoride no propargylic substitution reaction occurred because of the extremely low solubility of tin(II) fluoride in CH3NO2. The temperature did have a positive impact on the rate of the reaction but furnishes products with lower yield due to by-product formation. A probable mechanism for the tin(II) chloride catalyzed propargylic substitution is described in Scheme 144.
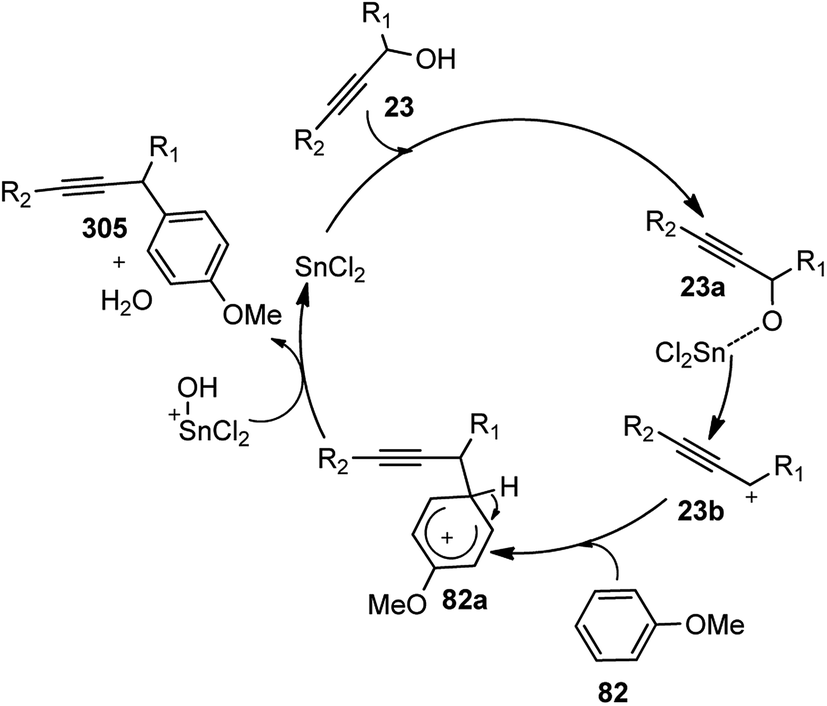 | ||
| Scheme 144 Mechanistic pathway for the SnCl2 catalyzed propargylic substitution of propargylic alcohols. | ||
Another example of Sn-catalyzed propargylic substitution for the synthesis of 1,5-ene-yne derivative was disclosed by Mukaiyama and co-workers in the year 1987, Scheme 138. When propargylic ether was treated with allyltrimethylsilane 118 in the presence of catalytic amounts of SnCl4 and ZnCl2, 1,5-ene-yne derivative 287 was obtained in a reasonably good yield, Scheme 145.180
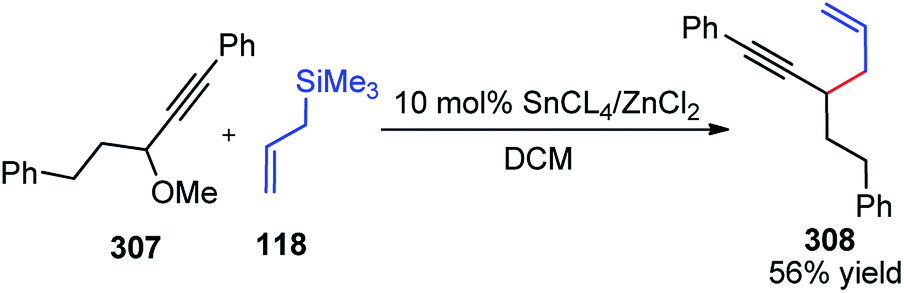 | ||
| Scheme 145 Synthesis of 1,5-ene-yne using catalytic amounts of SnCl4 and ZnCl2. | ||
5.16. Iodine derived catalysts
Recently, molecular iodine has gained a huge interest in organic synthesis because of its low cost and ready availability. The mild Lewis acidity associated with iodine has enhanced its use in organic synthesis to perform several organic transformations using stoichiometric levels to catalytic amounts which are able to activate the hydroxyl group.181Srihari and co-workers the year 2007 demonstrated the role of elemental iodine as a catalyst in mediating the nucleophilic propargylic substitution reactions, Scheme 146.182
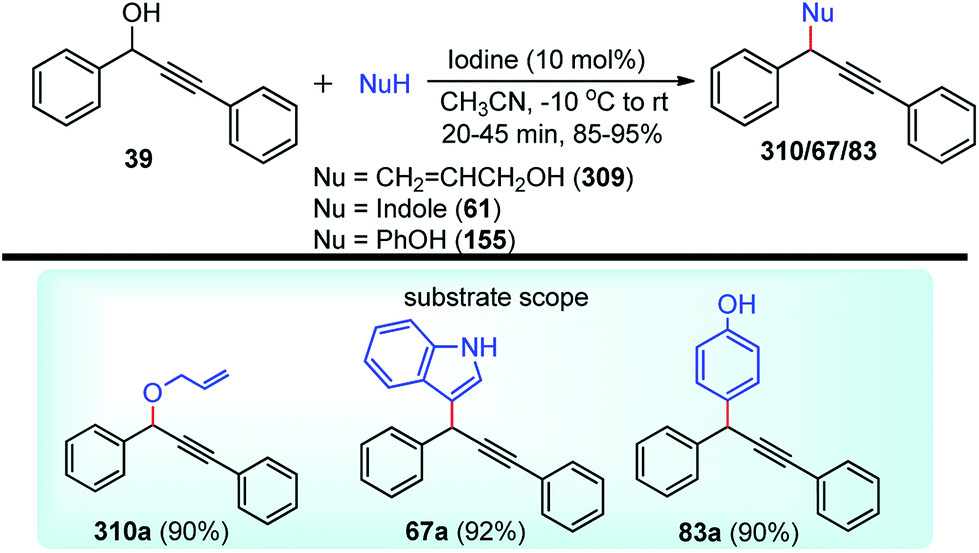 | ||
| Scheme 146 Iodine mediated propargylic substitution reaction. | ||
The reaction was screened with varied concentrations of iodine and it was observed that the reaction progressed well with even 1 mol% of the catalyst. But the highest yield was achieved with 5% iodine. The reaction proceeds via direct displacement of the hydroxy group with nucleophiles. Substrates with electron donating groups on the aromatic ring underwent the reaction much faster than unsubstituted aryl propargylmethanols.
Chen and co-workers in the year 2007 reported a similar type of iodine catalyzed efficient propargylation of indoles 61 under a mild condition in high regioselectivity and excellent yields, Scheme 147.183
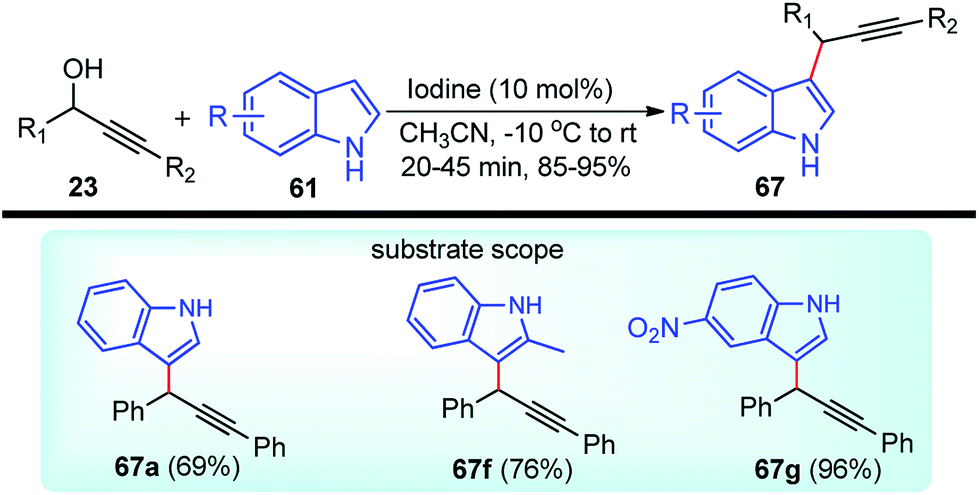 | ||
| Scheme 147 Iodine mediated propargylic substitution reaction. | ||
Operational simplicity, excellent yields, and chemoselectivity are major advantages of this protocol compared to other catalyzed propargylic substitution reactions reported in the literature.
Subba Reddy and co-workers in the year 2010 developed a novel and efficient catalytic process for the synthesis of substituted indenes 311 from aryl-substituted propargylic alcohols 23 by means of a series of cascade reactions involving tandem isomerization of the propargylic cations, reduction followed by intramolecular Friedel–Crafts type cyclization, Scheme 148.184
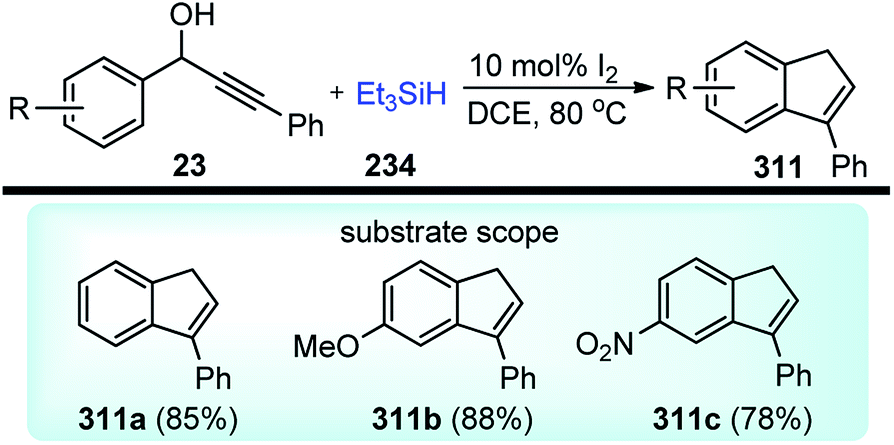 | ||
| Scheme 148 Iodine mediated propargylic substitution reaction. | ||
The catalytic efficiency of various Lewis acids such as BiCl3, ZnCl2, and FeCl3·6H2O was tested and none of them were found efficient. Among the several solvents screened, dichloroethane appeared to give the best results.
Hypervalent iodine acting as Lewis acid catalyst in the absence of an extra oxidant or other acidic activator have been developed and received much attention in recent years.185 PIFA is known to behave as a Lewis acid and its utility towards the propargylic substitution reactions in absence of any acid activator or oxidants has been nicely demonstrated by Weng and co-workers.186
Weng and co-workers in the year 2015 reported PhI(OCOCF3)2 as an efficient catalyst for the reaction of propargyl alcohols 23 with allyl trimethylsilane 118 and several electron-rich arenes 75/312 in very good yield, Scheme 149.186
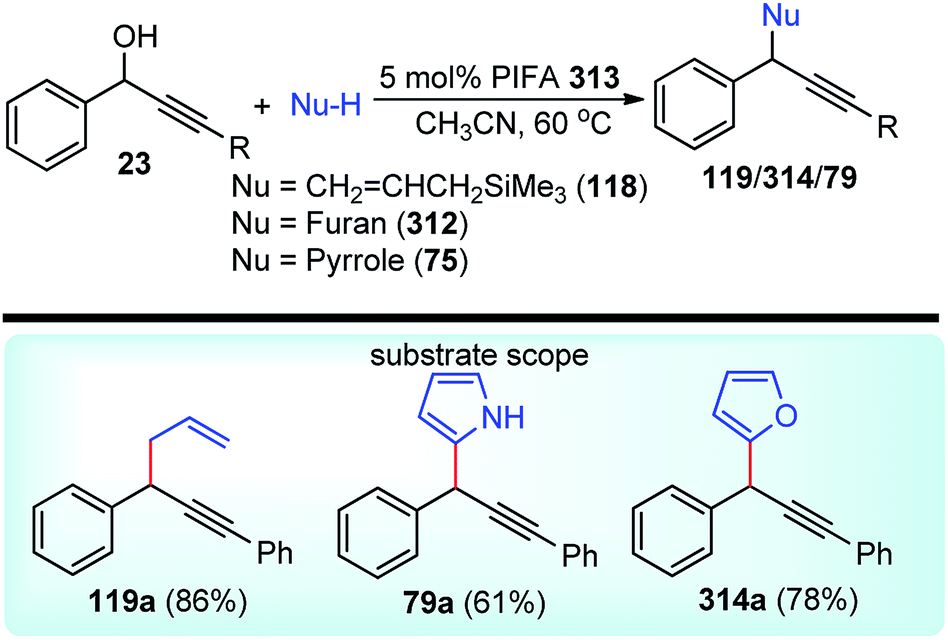 | ||
| Scheme 149 PIFA catalyzed propargylic substitution reaction. | ||
The methodology is highly substrate dependent and electron donating substrates furnished products in higher yield compared to electron withdrawing substrates. Even, trimethylsilyl substituted and terminal propargyl alcohols were also well tolerated although with longer reaction time and reduced yields. Sterically hindered substrates were also found to reduce the reaction rate. The mechanism for the reaction is demonstrated in Scheme 150.
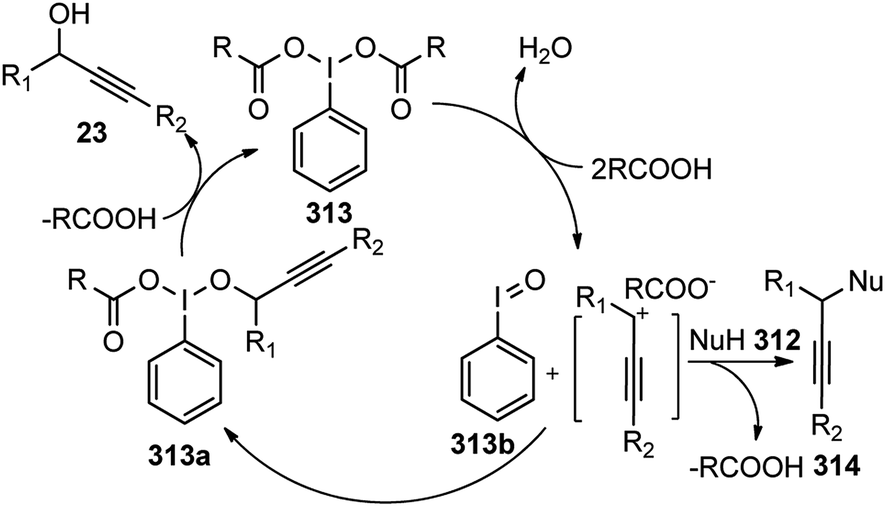 | ||
| Scheme 150 Mechanism for the hypervalent iodine mediated propargylic substitution reaction. | ||
Muthusamy and co-workers in the year 2016 reported a iodine mediated reaction of propargyl alcohols 39 and isatin hydrazones 315 under open air condition to synthesize highly substituted as well as conjugated unsymmetrical azines 316 in good yield, Scheme 151.187
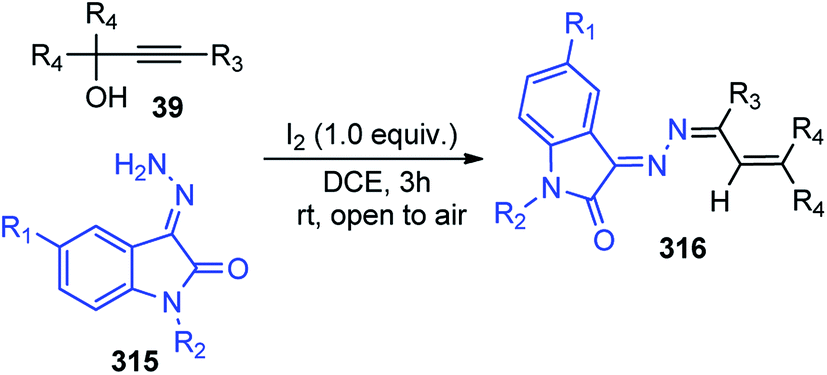 | ||
| Scheme 151 Iodine mediated reaction of propargyl alcohols and isatin hydrazones for the synthesis of azines. | ||
Several other Lewis acids like BF3·OEt2, Yb(OTf)3, Sc(OTf)3, FeCl3 and InCl3, were found inefficient in catalyzing the reaction. Surprisingly, only DCE was found to the suitable solvent, among several other solvents screened, for this reaction. A mechanistic detail of the formation of unsymmetrical azines is shown in Scheme 152.
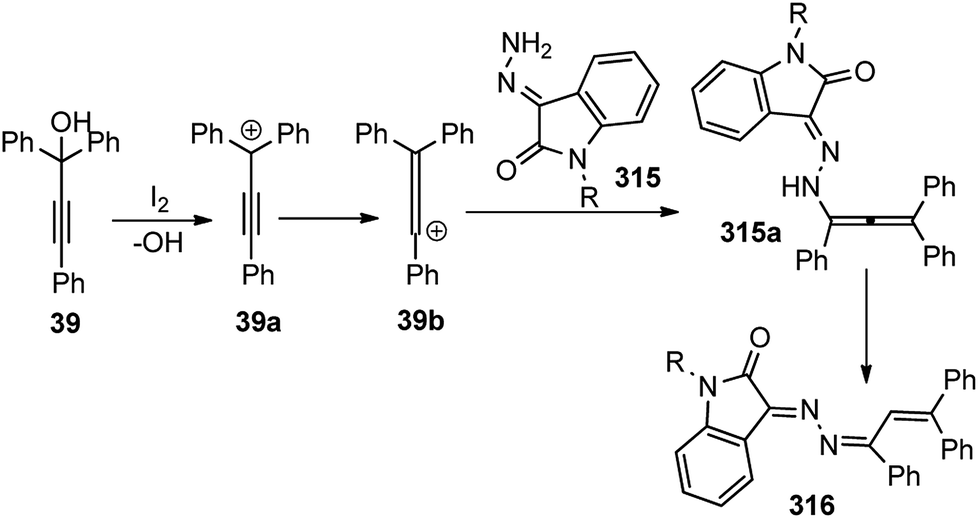 | ||
| Scheme 152 Mechanism for the iodine mediated synthesis of azines from propargyl alcohols and isatin hydrazones. | ||
Under the influence of iodine, the propargylic alcohol is converted to the allene carbocation intermediate 39b which is subsequently trapped by isatinhydrazones followed by a [1,3]-shift of the N-group through aza-Meyer–Schuster rearrangement resulting into an allene intermediate 315a. Isomerization of the allene intermediate 315a furnished the unsymmetrical azines 316.
5.17. Cerium derived catalysts
Lanthanide salts are often employed as catalysts in organic synthesis, mainly due to their low toxicity, affordability, stability, and ease of handling. Cerium(III) chloride has emerged as a very useful Lewis acid imparting high regio- and chemoselectivity in various chemical transformations over the past few years. It is an inexpensive, nontoxic and water-tolerant catalyst and has been used in several different forms, alone as heptahydrate, anhydrous, and in combination with NaI. The salt has also been used in solid supports which modify their reactivity. Even organocerium compounds also found extensive applications in organic synthesis.188Silveira and co-workers in the year 2010 employed anhydrous CeCl3 as a catalyst for the propargylation of indoles 61 in good yield and high regioselectivity Scheme 153.189
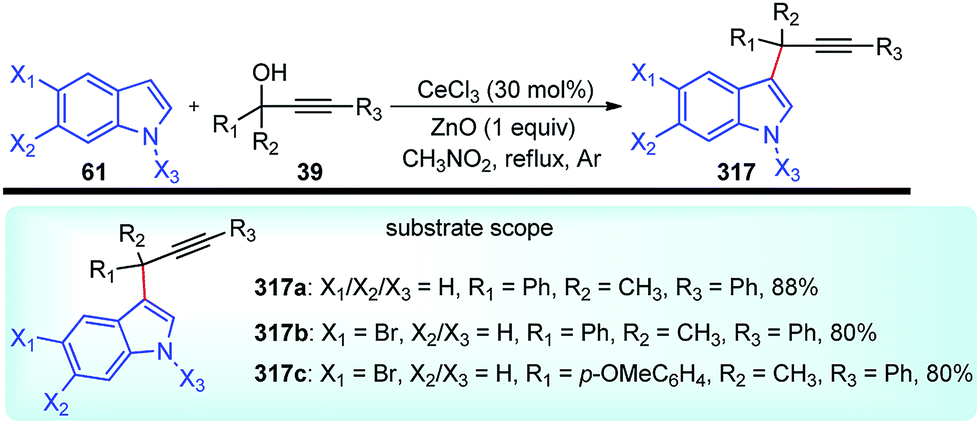 | ||
| Scheme 153 CeCl3 catalyzed propargylic substitution reaction. | ||
The authors found that stirring propargylic alcohols and indole with 30 mol% of CeCl3 in CH3NO2 under reflux condition furnished 3-propargyl indole in 60% yield. Other solvents such as glycerine, DMA, iPrOH were also employed but the best yield was achieved with MeNO2. The use of larger amounts of CeCl3 neither improved the reaction yield nor reduced the reaction time. However, when 0.1 equiv. of anhydrous CeCl3 was used, product yield reduced to 25%. Replacing anhydrous CeCl3 with CeCl3·7H2O, gave no desired product. The reported method was simple, general and the products were obtained in good yields.
Silveira and co-workers in the year 2012 demonstrated that Ce(OTf)3 could be successfully employed as a catalyst for the propargylation of aromatic compounds 82, Scheme 154.190
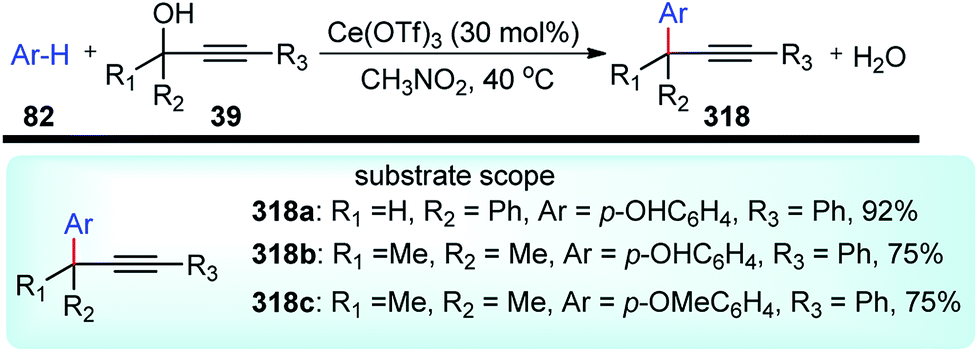 | ||
| Scheme 154 Ce(OTf)3 catalyzed propargylic substitution reaction. | ||
The effect of solvent, the amount of catalyst, and reaction temperature were thoroughly screened, and the best condition was established with 30 mol% of Ce(OTf)3 in nitromethane at 40 °C. Other solvents like CH3CN, MeNO2, glycerol, DMA, and 2-propanol were found inefficient in furnishing products in good yield. When the reaction was carried out at 80 °C, a decrease in selectivity was observed. The protocol demonstrated the application of CeCl3 a useful alternative to triflic acid and other usual catalysts for the propargylation reaction.
5.18. Rhenium catalyzed reactions
Rhenium(V) is known to form a variety of stable octahedral complexes with multiple bonds to oxygen.191Re-catalyzed methodologies remained mostly overlooked as a tool for organic transformations. Recently, high oxidation-state Re complexes has been employed in a variety of organic transformations. 2Re–oxo complexes offer several advantages in metal-mediated catalysis, because of the (1) high stability against moisture due to the high oxidation state of the metal and (2) mild conditions which allow for the activation of sensitive substrates.
Toste and co-workers in the year 2003 have reported the formation of propargylic ethers 73 by the coupling of simple alcohols 72 and propargyl alcohols 39 using a robust air- and moisture-tolerant rhenium(V)–oxo complex, Scheme 155.192
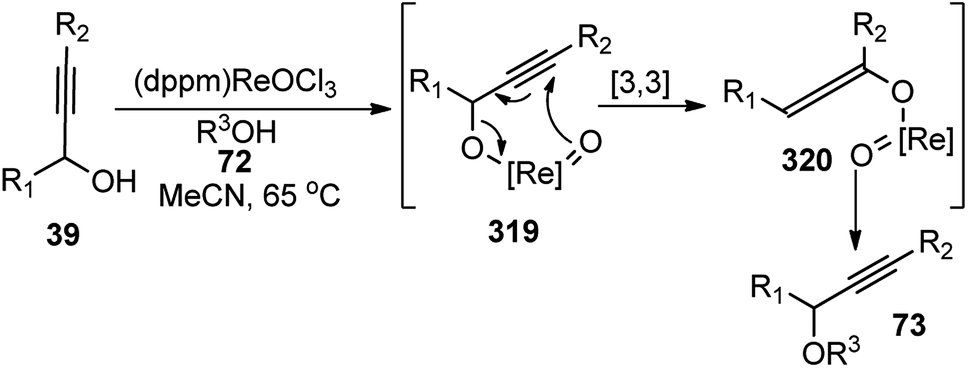 | ||
| Scheme 155 Mechanism for the Re-catalyzed etherification of propargylic alcohols. | ||
Following their discovery, the same group in 2003 described the application of that catalyst system to the formation of C–C bonds by the coupling of allylsilanes 37 and propargyl alcohols 39, Scheme 156.193
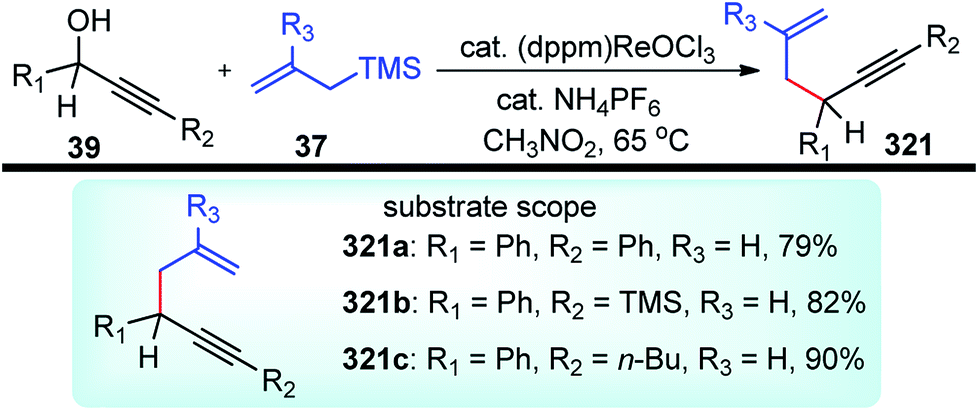 | ||
| Scheme 156 Re-catalyzed propargylic substitution with allyl trimethyl silane. | ||
The reaction was even carried out with lower catalyst loadings without any significant deterioration in yield by increasing the reaction temperature to 80 °C. The reaction tolerated a wide range of electron donating and electron withdrawing substrates. The catalyst being both air and moisture stable was easily recovered and reused several times which exemplifies its obvious advantages in organic transformations.
Toste and co-workers in the year 2005 reported the use of air and moisture-tolerant rhenium–oxo complex for the regioselective synthesis of propargylamine derivatives 324 from propargyl alcohols 323 and carbamates 322, Scheme 157.194
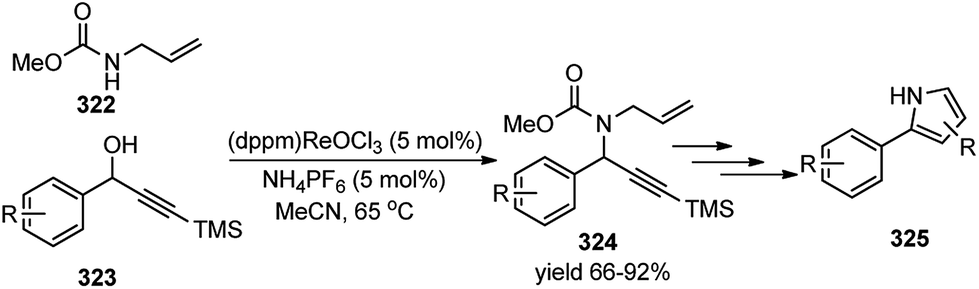 | ||
| Scheme 157 Rhenium catalyzed propargylic substitution reaction. | ||
The reaction has a wide substrate scope and interestingly, both sterically hindered ortho-disubstituted and heteroaromatic substrates furnished products in good yield. The substrate scope, mild reaction conditions, and operational simplicity have made it a valuable method for construction of C–N bonds.
Recently, dual catalysis has received considerable attention because they lead to products that cannot be synthesized with a single catalyst in one-pot. Takai and co-workers in 2007 report rhenium-and gold-catalyzed synthesis of diethynylmethanes 327 by reactions of propargylic alcohols 39 with trimethyl(phenylethynyl)silane 326, Scheme 158.195
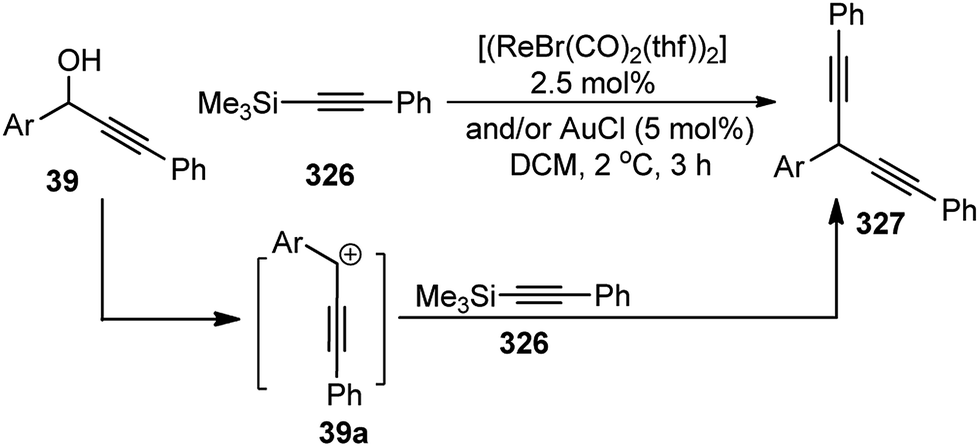 | ||
| Scheme 158 Rhenium-and gold-catalyzed synthesis of diethynylmethanes by reactions of propargylic alcohols with trimethyl(phenylethynyl)silane. | ||
The authors have demonstrated the rhenium-catalyzed transformations of propargyl and benzyl alcohols using organosilanes, as well as the rhenium- and gold-catalyzed synthesis of diethynylmethanes. This reaction proceeded via propargylic carbocation as a reactive intermediate. A combination of the rhenium and gold complexes promoted the first ethynylation step, and the rhenium complex accelerated the second ethynylation ultimately producing the desired product.
Kuninobu and co-workers in the year 2008, described rhenium-catalyzed coupling reactions between 2-propynyl alcohols 39 and several 1,3-diketone nucleophiles 304 to construct C–C bond efficiently, Scheme 159.196
 | ||
| Scheme 159 Rhenium catalyzed propargylic substitution reaction. | ||
The dehydrative coupling reactions between 2-propynylalcohol and several carbon and heteroatom nucleophiles furnished substituted propargylic derivatives 306 efficiently. The reaction mechanism proceeds via the formation of a 2-propynyl cation via dihydroxylation followed by a nucleophilic attack on the propargyl cation and finally deprotonation to produce the desired product 285.
Ghorai and co-workers in the year 2012 described the direct catalytic amination of π-activated alcohols 39, producing water as the only by-product using Re2O7 as a catalyst, Scheme 160.197
 | ||
| Scheme 160 Synthesis of various π-activated amines from propargylic alcohols. | ||
Propargylic substituted products 329 were observed with high regioselectivity. Optically active propargylic alcohol with 50% ee, when reacted under this reaction condition, resulted in racemic product formation suggesting that the substitution probably proceeded through an SN1-like mechanism.
5.19. Ytterbium derived catalysts
Ytterbium belongs to a low cost and commercially available metal salt family of the lanthanide series that has a high tolerance to air and moisture. These salts have been shown to be versatile in mediating a wide variety of organic transformations that make use of alcohol pro-electrophiles in excellent yields and with high selectivity. Taking account of the versatile reactivity of the ytterbium, several interesting propargylic substitution reactions have been carried out.Zhou and co-workers in the year 2007 developed a highly efficient coupling reaction of 1,3-dicarbonyl compounds 304 and 4-hydroxycoumarins 64a with propargylic alcohols 23 in presence mildly Lewis acidic Yb(OTf)3, Scheme 161.198
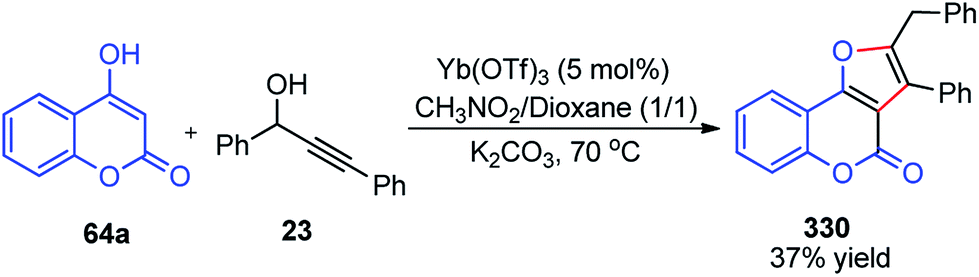 | ||
| Scheme 161 Yb(OTf)3 catalyzed propargylic substitution of 1,3-dicarbonyls and 4-hydroxycoumarin. | ||
The reaction mechanism proceeds via the propargylic carbocationic intermediate. Broad scope, mild conditions, and easy handling are the advantages of this method. This method provided a mild and straightforward route to multi-substituted furocoumarin.
Different methodologies to synthesize indenes employing environmentally benign Lewis acid catalysts from propargylic alcohols have been reported recently.199–201 Following those work, Chan and co-workers in the year 2009 developed a method to prepare indenols 333 efficiently by ytterbium(III) triflate catalyzed tandem Friedel–Crafts alkylation/hydroarylation of propargylic alcohols 39 with phenols 331, Scheme 162.202
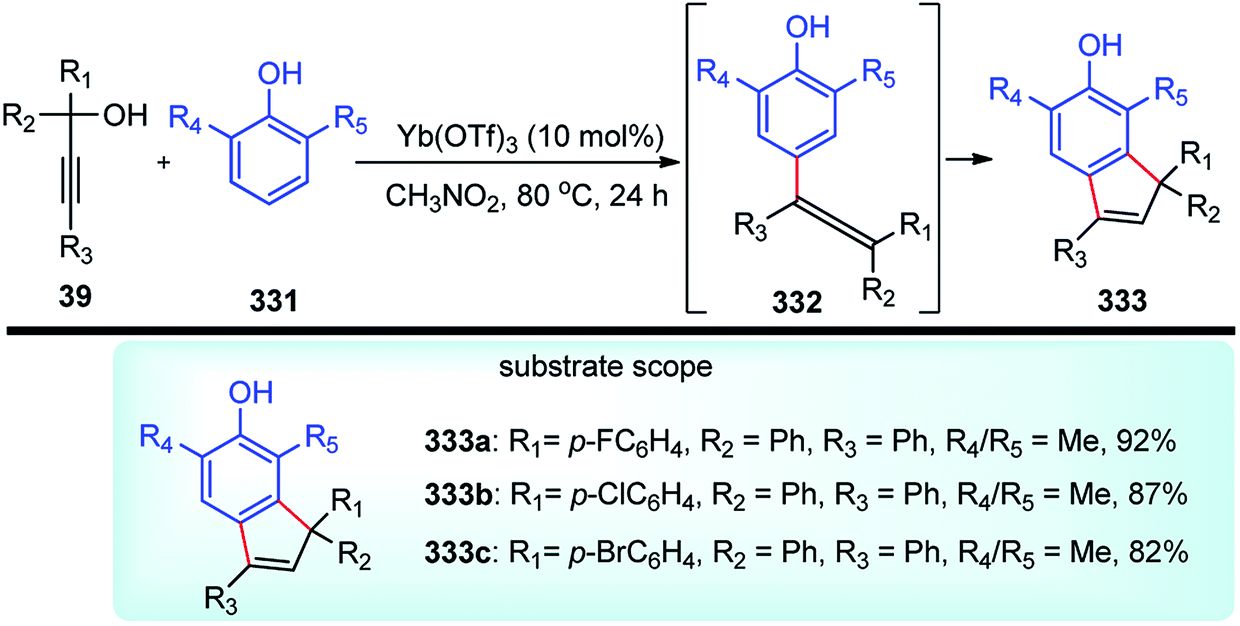 | ||
| Scheme 162 Yb-catalyzed synthesis of 3-phenyl-1H-indenes form propargyl alcohols. | ||
The reaction furnished products in moderate to excellent yields with high regioselectivity under mild conditions. It offered a straightforward and convenient one-step access to bioactive indenols and its derivatives. Different Lewis acid catalysts like Yb(OTf)3 Cu(OTf)2, InCl3 FeCl3·6H2O, AlCl3, AuBr3, PtCl2 and even Brønsted acid like TfOH were tested to establish the reaction conditions. AuBr3 and PtCl2 were found to be equally effective as catalyst whereas Cu(OTf)2, InCl3 and AlCl3 furnished products with diminished yield. Both electrons donating and electron withdrawing substituents on the carbinol or alkyne carbon delivered the desired indenols 333 in good to excellent yields. For aliphatic substituents, longer reaction time and lower yields were obtained. The reaction is believed to proceed by a mechanism outlined in Scheme 163, involving the activation of the alcohol substrate through coordination of the ytterbium triflate followed by the Friedel–Crafts type reaction at the allenic carbocation center to generate the intermediate 334. Subsequent Yb(III)-mediated intramolecular hydroarylation, re-aromatization and protodemetalation deliver the desired product 333 in good yield.
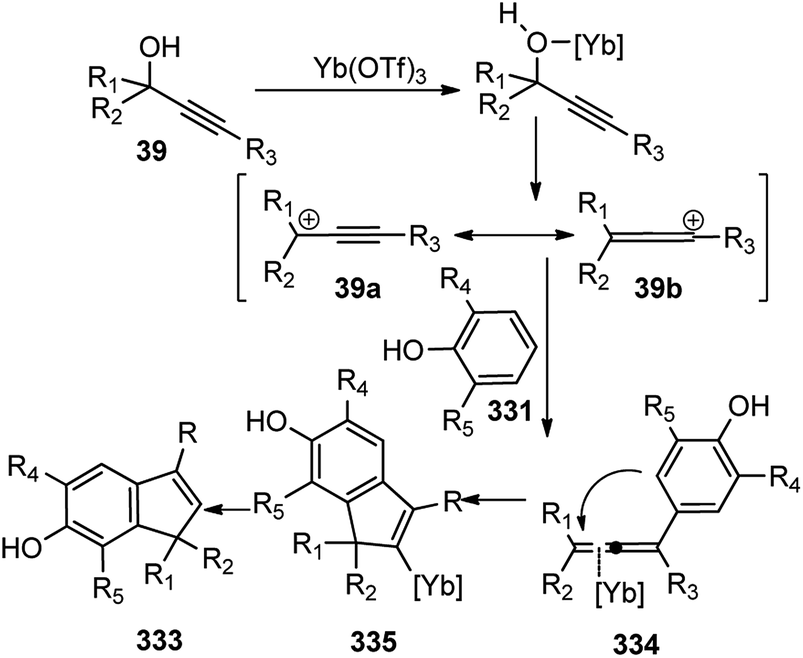 | ||
| Scheme 163 Mechanism for the synthesis of 3-phenyl-1H-indenes form propargyl alcohols promoted by Yb-catalyst. | ||
Constructing aromatic heterocycle remains the continuous pursuit of several research groups using more efficient and flexible approaches. Uemura and co-workers first reported the synthesis of oxazole by employing a dual diruthenium (II,III) and gold(III) catalytic system.140,203
Following this work, Liu and Kumar demonstrated a dual ruthenium(III) and zinc(II) catalyst combination could also mediate the cyclization of propargylic alcohols with amides that afforded the oxazole product in excellent yields.204 More recently, Zhan and co-workers described a Brønsted acid-mediated synthesis of oxazole that could be achieved in the presence of a stoichiometric amount of p-TsOH·H2O.205 Although both these methodologies were shown to be highly efficient, the need for two metal catalysts is a serious limitation. In addition to this, the cost of the catalysts and the limited substrate scope restricted to terminal propargylic alcohols poses a serious challenge to this reported method.
Chan and co-workers in the year 2011 reported a Yb(OTf)3 catalyzed cyclization of trisubstituted propargylic alcohols 39 with aryl amides 77 towards the synthesis of di- and trisubstituted 2-aryloxazoles 336, Scheme 164.203
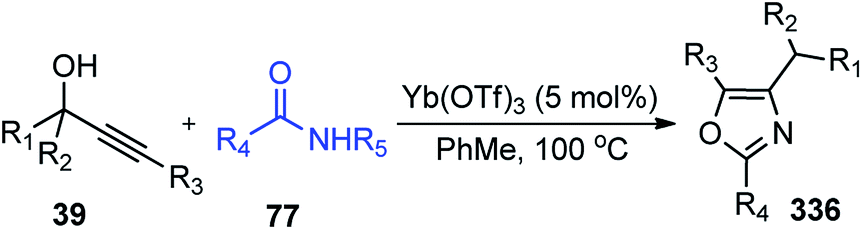 | ||
| Scheme 164 Yb(OTf)3 catalyzed efficient method for the synthesis of di- and trisubstituted 2-aryloxazoles from propargylic alcohols. | ||
Moderate to excellent product yields were obtained along with complete regioselectivity. Both toluene and 1,2-dichloroethane were found to be equally competent for this reaction. Lower product yields were obtained when the reaction was carried out in MeCN or MeNO2 as well as on switching the catalyst from Yb(OTf)3 to either AgOTf, CuBr or FeCl3·6H2O. The mechanism highlighted in Scheme 165 involves the activation of the starting alcohol by the metal catalyst that resulted in its ionization. Subsequent cyclization of this newly generated carbocationic species 39b with the aryl amide 77 finally afforded the oxazole 336, Scheme 165.
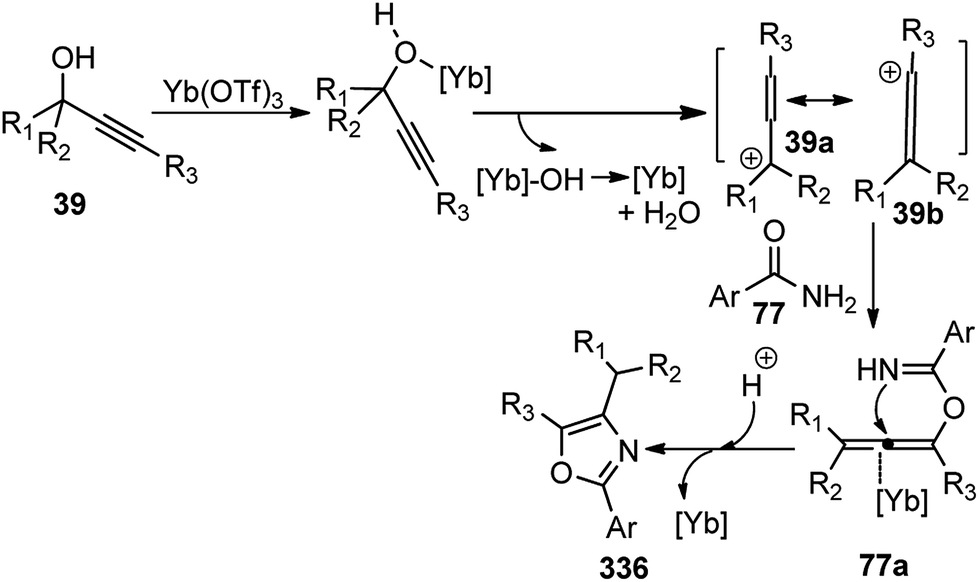 | ||
| Scheme 165 Mechanism for the Yb(OTf)3 catalyzed synthesis of di- and trisubstituted 2-aryloxazoles from propargylic alcohols. | ||
In view of the mild reaction conditions along with the low cost, commercially availability of Yb(OTf)3 and its high tolerance to air and moisture, the above synthetic approach offered an operationally simplistic and convenient approach for the synthesis of oxazole derivatives.
5.20. Iridium derived catalysts
Iridium has been rarely used as a catalyst for the propargylic substitution reaction. In one of the early examples, Matsuda and co-workers reported [Ir(cod)(PR)]OTf modified by H2 as an active catalyst for the Mukaiyama-type aldol reaction, Michael addition, and the allylic substitution reaction. It was concluded that enoxysilane behaved as a nucleophile in all these reactions.206Subsequently, Matsuda and co-workers in the year 2002 reported the use of Ir-catalyst for the substitution reactions of a propargylic-type alcohol 337 with an enoxysilane 338, Scheme 166.207
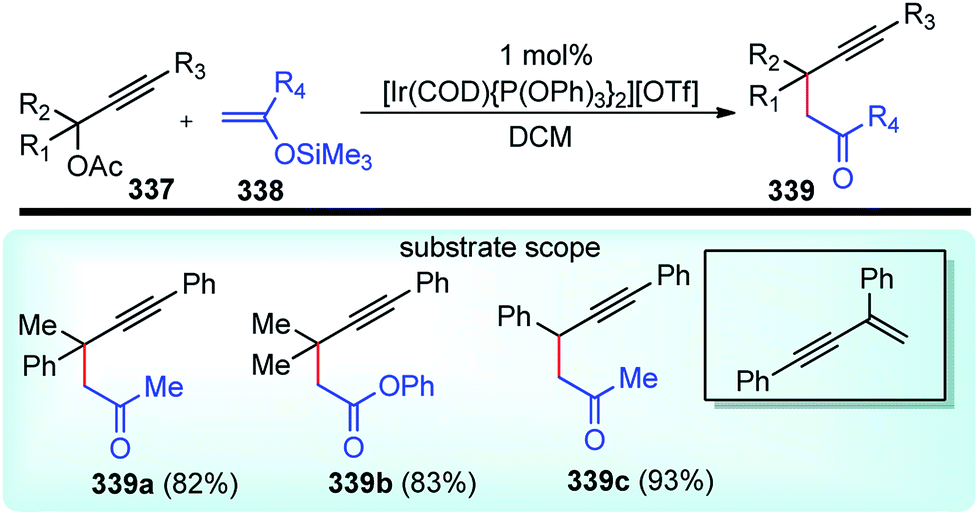 | ||
| Scheme 166 Iridium catalyzed propargylic substitution with enoxysilane. | ||
The authors found that enoxysilanes worked as a good nucleophile to form β-alkynyl carbonyl compounds in the presence of a catalytic amount of Ir catalyst. Other cationic complexes such as [Ir(cod)(PPh3)2]OTf and [Ir(cod)(binap)]OTf, were also effective without any change in regioselectivity for this substitution product, except that they required a longer reaction time.
5.21. Platinum derived catalysts
Platinum has also been used, although not so extensively, as catalyst for the propargyic substitution reaction. The reaction proceeds via the Pt-allene intermediate where the nucleophile normally attacks in an SN2 fashion to furnish the product, Scheme 167. | ||
| Scheme 167 General mechanism for the platinum catalyzed propargylic substitution reaction. | ||
Brabander and co-workers in the year 2008 reported a platinum-catalyzed propargylic substitution of ω-hydroxy 2-propargylic esters 340 to furnish substituted tetrahydropyrans 341 in good yield, Scheme 168.208
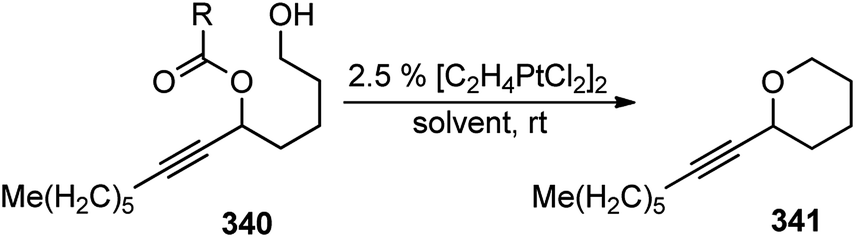 | ||
| Scheme 168 Pt-catalyzed synthesis of tetrahydrofurans from a propargylic ester by an intramolecular substitution reaction. | ||
Use of Zeise's dimer ω-([CH2CH2–PtCl2]2) did not induce cycloisomerization but instead catalyzed the propargylic substitution pathway. Various control experiments ruled out the mechanism via propargylic carbocation intermediates. Strong Lewis acids (cat. or equivalent TiCl4, BF3·OEt2, FeCl3) did not furnish the product. Also, sterically hindered substrates that prevent coordination to the alkyne but not the ester did not furnish the desired products. Overall, the authors reported a novel platinum-catalyzed propargylic substitution reaction that functions with internal alkynes lacking a carbocation-stabilizing substituent.
Platinum-catalyzed nucleophilic propargylic substitutions are relatively rare. One classic example reported in 2008 was Zeise's dimer [(CH2CH2)Cl2Pt]2 catalyzed intramolecular cyclization of ω-substituted propargylic acetates to furnish heterocycles.209 Despite the therapeutic importance of the cis-2,6-disubstituted morpholines there are only a few synthetic methodologies available for them.
Brabander and co-workers in 2011 demonstrated that ω-hydroxy propargylic acetates 342 with a side chain containing Boc-protected nitrogen atom underwent intramolecular substitution in presence of 2.5 mol% Zeise's dimer in THF medium to form 2,6-disubstituted morpholines 343 with the predominance of the cis-isomer in good yields at room temperature, Scheme 169.209
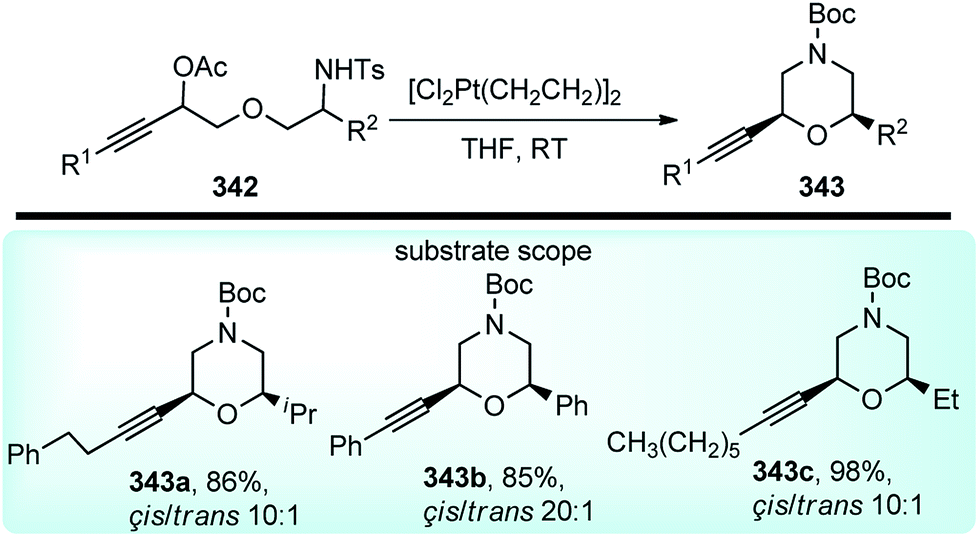 | ||
| Scheme 169 Pt-catalyzed synthesis of cis-2,6-disubstituted morpholines. | ||
The methodology has been extended to synthesize cis-2,6-disubstituted-1,4-dioxanes 344 from oxygen tethered ω-hydroxy propargylic acetates under similar reaction conditions in good yield and high diastereoselectivity. Other classes of molecules synthesized using this methodology included 3-substituted morpholines 345 and cyclic sulfamates 346 from various heteroatom tethered propargylic acetates, Fig. 22.
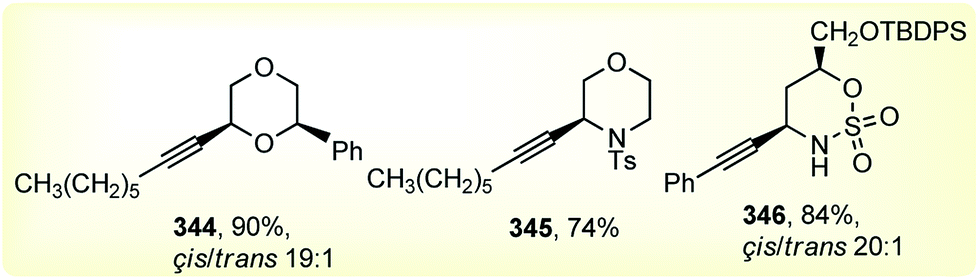 | ||
| Fig. 22 Representative examples of disubstituted-1,4-dioxanes, 3-substituted morpholines, and cyclic sulfamates. | ||
An SN2′ kind of mechanism was proposed for the intramolecular cyclization of the propargylic acetates. In the reaction mechanism, [1,3] shift or double [1,2] shift of the acetate group followed by the metal complex coordination resulted in the formation of intermediate 342a. Finally, intramolecular attack from the nucleophilic residue in an SN2′ manner furnished the desired heterocycle with the liberation of acetate, Scheme 170.
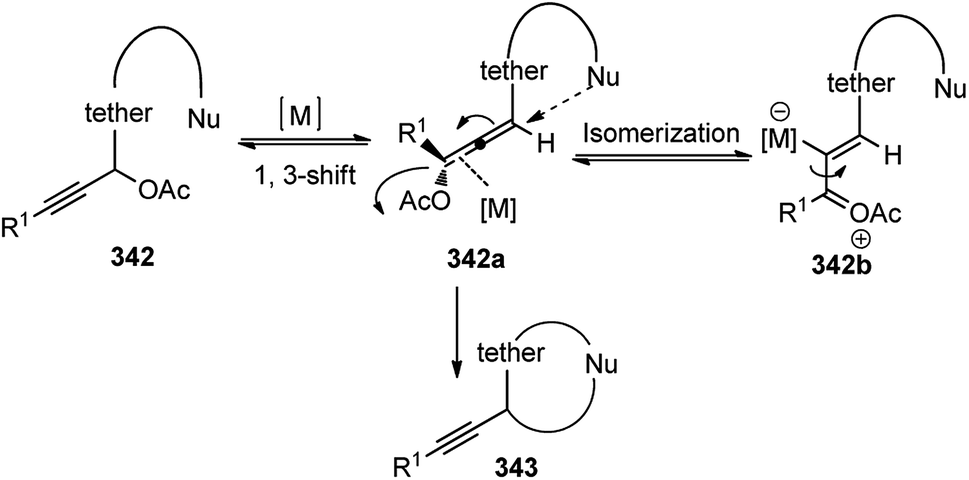 | ||
| Scheme 170 Proposed mechanism for the intramolecular cyclization. | ||
Kim and co-workers in the year 2014 developed a Pt(II)-catalyzed synthesis of phenanthrenes 348 from readily accessible biaryl propargyl alcohol substrates 347, Scheme 171.210
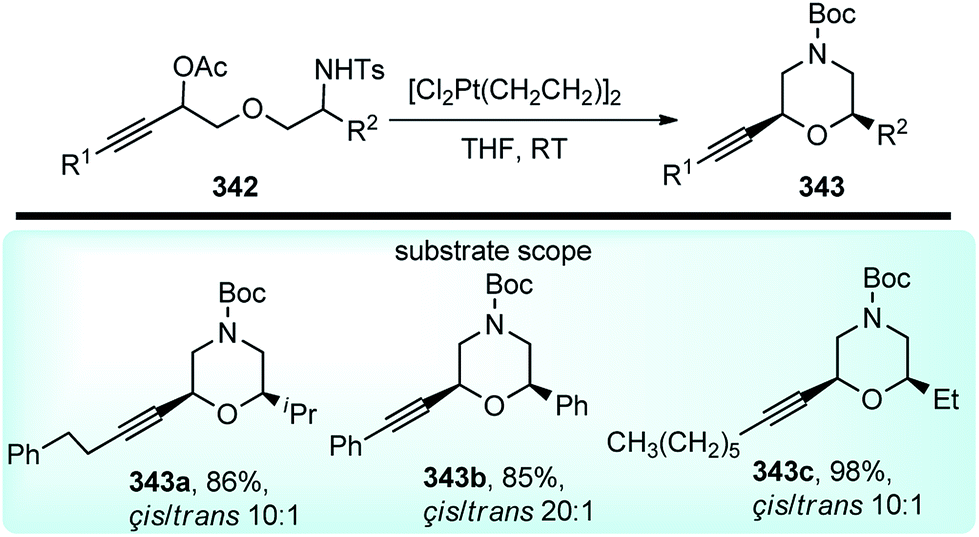 | ||
| Scheme 171 Pt(II)-catalyzed synthesis of phenanthrenes from propargylic alcohols. | ||
The reaction tolerated a wide range of functional groups and afforded products in excellent yields. Catalysts like Pd(OAc)2, Ru complex, InCl3, and AgOTf were found ineffective in promoting the reaction, apart from PtCl2, other Pt salts, such as PtCl4 and PtBr2, although afforded the desired product, but were less efficient than PtCl2 in terms of reaction yield and selectivity. The mechanism for the synthesis of substituted vinyl phenanthrenes is shown in Scheme 172.
 | ||
| Scheme 172 Mechanism of the Pt-catalyzed synthesis of phenanthrenes from propargylic alcohols. | ||
In the presence of a catalytic amount of PtCl2, intramolecular cyclization and subsequent dehydration resulted into phenanthrene with a carbene functional group which rapidly underwent 1,2-H migration to afford a vinylphenanthrene system 348.
5.22. Gold derived catalysts
Gold catalysis has gained huge popularity and becomes an efficient tool to activate triple bonds for various nucleophilic addition reactions.211 Gold stands out among the other conventional Lewis acids since it possesses a unique hard/soft Lewis acid nature which allows the simultaneous activation of both alcohols and π-bonds that could possibly take the advantage to perform new domino processes, Scheme 173.212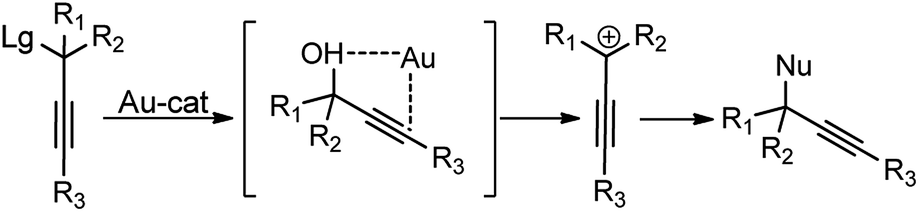 | ||
| Scheme 173 Gold-catalyzed propargylic substitution reaction. | ||
Campagne and co-workers described an excellent methodology for the gold-catalyzed allylation of propargylic alcohols 39 at room temperature, Scheme 174213
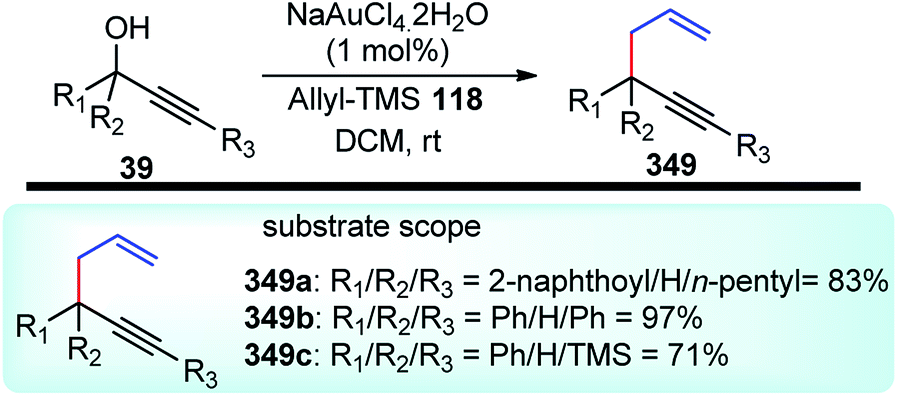 | ||
| Scheme 174 Au-catalyzed allylation of propargylic alcohols. | ||
The reaction was found to furnish products in moderate to good yield in the range of 30–68% with several catalysts like AuBr3, AuCl3, HAuCl4·3H2O, and AuCl. Whereas no product formation was observed with Ph3PAuCl, PdCl2(PhCN)2, PtCl2. The reaction displayed a wide substrate scope and the reaction found equally efficient with both electron-rich and moderately electron-poor aromatic substrates.
Allylation of optically pure propargylic alcohol (96% ee) resulted in the formation of the racemic product, Scheme 175, suggesting the reaction mechanism proceeded via a carbocationic intermediate.
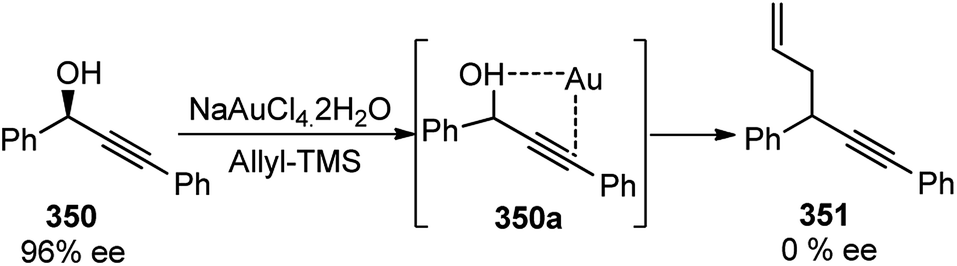 | ||
| Scheme 175 Mechanism of the gold-catalyzed propargylic substitution reaction. | ||
Dyker and co-workers in the year 2006, demonstrated the powerful strategy for the Friedel–Crafts propargylation of electron rich arenes 352 using gold chloride catalyst, Scheme 176.214
 | ||
| Scheme 176 Au-catalyzed propargylation of electron rich arenes. | ||
In the process of evaluating the catalytic activities and selectivities for the multi-fold Friedel–Crafts propargylation of electron rich arenes, the authors compared the reactivities of gold(III) chloride with the classical Friedel–Crafts catalyst, i.e. BF3·Et2O.
Several electron-rich arenes like 1,3-dimethoxybenzene, 1,3,5-trimethoxybenzene and azulene when tested with just 0.3 to 1% of the gold catalyst, monosubstituted products were obtained in 96% isolated yield. Whereas, BF3·Et2O resulted in either the mono- or the dipropargylated product proving that gold chloride is a very mild, yet highly reactive Friedel–Crafts catalyst, which works at rather low concentration and ensures a high selectivity for propargylation reactions.
Campagne and co-workers in the year 2009, described a gold(III)-catalyzed direct nucleophilic substitution of propargylic alcohols 23 with various nucleophiles like allylsilane and electron-rich aromatics like alcohols/thiols/hydrides/1,3-dicarbonyl derivatives/sulphonamides in DCM at room temperature, Scheme 177.12
 | ||
| Scheme 177 Au-catalyzed propargylic substitution reactions. | ||
Disappointing results were obtained when Au(I) catalysts were used. Even PtCl2 and PdCl2(PhCN)2 catalysts were found inefficient with no observable product formation. An excellent result was achieved when Au(III) catalysts were used at room temperature in DCM solvent. Mechanistic investigations by the authors suggested a mechanism through a carbocationic intermediate.
Kirsch and co-workers in the year 2009, developed a gold(III)-catalyzed regioselective nucleophilic substitution reaction of propargylic enol silyl ether 355 with alcohols 72, Scheme 178.215
 | ||
| Scheme 178 Gold(III)-catalyzed regioselective nucleophilic substitution reaction. | ||
Gold(III) complex acted as a pre-catalyst to activate propargyl silyl ether. An extensive catalyst screening with various gold(I) and gold(III) catalysts revealed K[AuCl4] to be the best choice for the conversion of 3-trimethylsilyloxy-1-phenylhex-4-en-1-yne 355 into (5-methoxyhex-3-en-1-ynyl)benzene 356. Other gold catalysts such as HAuCl4·4H2O, AuCl, and [(Me3P)AuSbF6] resulted in markedly reduced product yield. Interestingly, Brønsted and Lewis-acid catalysts like Bi(OTf)3 (67%), p-TsOH (61%), and HBF4 (52%) furnished products in moderate yield.
Morita and co-workers in the year 2015 purposefully used hard gold(III) and soft gold(I) catalysts for the facile access of two types of 2-substituted piperidines 358 from propargylic alcohols 357, Scheme 179 and Scheme 180.216
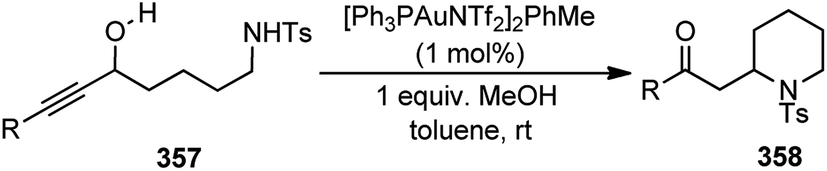 | ||
| Scheme 179 Facile synthesis of 2-substituted piperidines from propargylic alcohols. | ||
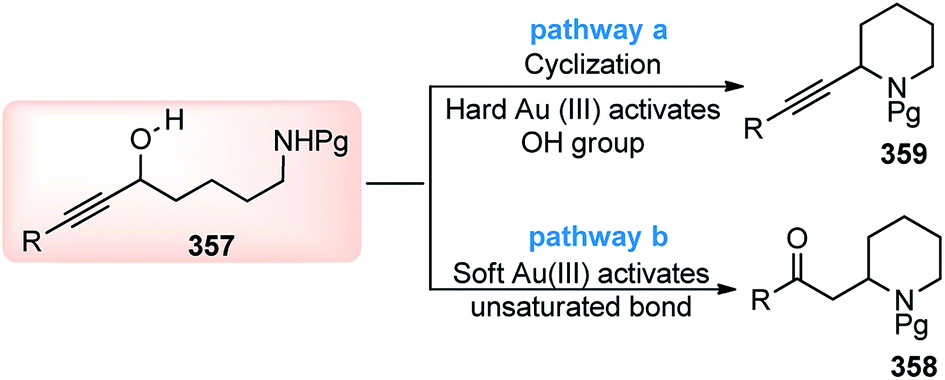 | ||
| Scheme 180 Facile synthesis of two types of 2-substituted piperidines from propargylic alcohols using hard and soft gold catalysts. | ||
Screening of the reaction with different catalyst combinations like AuCl (15 mol%), Ph3PAuCl (15 mol%) or Ph3PAuCl (5 mol%)/AgSbF6 (5 mol%) afforded the desired product in poor to moderate yield except for [Ph3PAuNTf2]2PhMe (1 mol%) which afforded piperidine in 81% yield.
The mechanistic pathway for gold-catalyzed synthesis of the two types of piperidines is illustrated in Scheme 181. In both cases, complex 357a would be formed as a common reaction intermediate, whose character would play a pivotal role in determining the reaction pathway. Hard gold(III) in complex 357a strongly activated the hydroxyl group to induce cyclization by intramolecular nucleophilic substitution, to afford piperidine having an acetylenic moiety (pathway a). On the other hand, soft gold(I) in complex 357a promoted the addition of methanol to generate an allenyl ether, which hydrolyzed to furnish α,β-unsaturated ketone (pathway b) which smoothly cyclized to afford the desired product.
 | ||
| Scheme 181 Mechanism for the synthesis of two types of 2-substituted piperidines from propargylic alcohols using hard and soft gold catalysts. | ||
5.23. Bismuth derived catalysts
Bismuth(III) compounds have gained extensive interest in organic synthesis due to their low toxicity, low cost and relative insensitivity to air and traces of moisture. Due to the pronounced lanthanide contraction, bismuth(III) compounds exhibits Lewis acidity and they have been used in many chemical transformations.217–220 Bismuth trichloride is particularly attractive because it not only is commercially available and inexpensive but also highly stable.Zhan and co-workers in the year 2006, demonstrated a BiCl3-catalyzed propargylic substitution reaction of propargylic alcohols 39 with C-, O-, S- and N-centered nucleophiles 77/118/312, Scheme 182.221
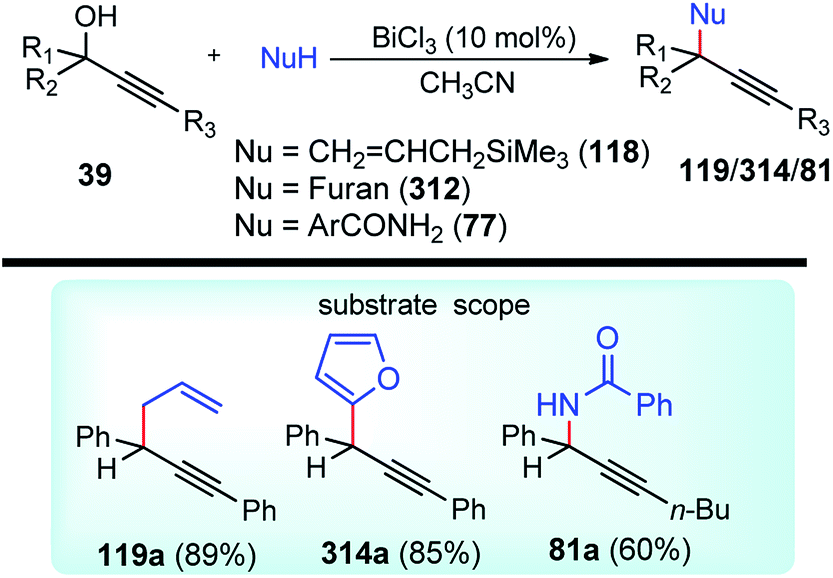 | ||
| Scheme 182 BiCl3 catalyzed nucleophilic substitution of propargylic alcohols. | ||
The reaction proceeded smoothly with various aryl- and alkyl-substituted propargylic alcohols. Electron-rich or moderately electron-poor aromatic substrates reacted smoothly with allyl trimethylsilane affording the corresponding allylated products in high yields.
Because of the strong co-ordinating properties of sulfur-containing compounds, transition metal-catalyzed coupling of propargylic alcohols with thiols was rarely reported. Gratifyingly, this methodology offered the construction of sp3 C–S bonds by the nucleophilic substitution of propargylic alcohols with a series of thiols employing 10 mol% BiCl3 as the catalyst.
Several studies on coupling of allylic and benzylic alcohols with allylsilanes have appeared, using various Lewis and Brønsted acids, namely BiCl3, ZrCl4, ion-exchanged montmorillonite, phosphomolybdic acid, FeCl3·6H2O, and HN(SO2F)2.222,223 Despite the availability of these earlier methods, development of environmentally benign efficient processes that employ readily available catalysts in combination with non-volatile solvents that can be recycled and reused is highly desirable.
In search of a greener protocol for the propargylic substitution reaction, Laali and co-workers in the year 2012 described the use of ionic liquid as the reaction medium for the efficient allylation, alkynylation, and deoxygenation of propargylic alcohols 350 using Bi(OTf)3, Scheme 183.224
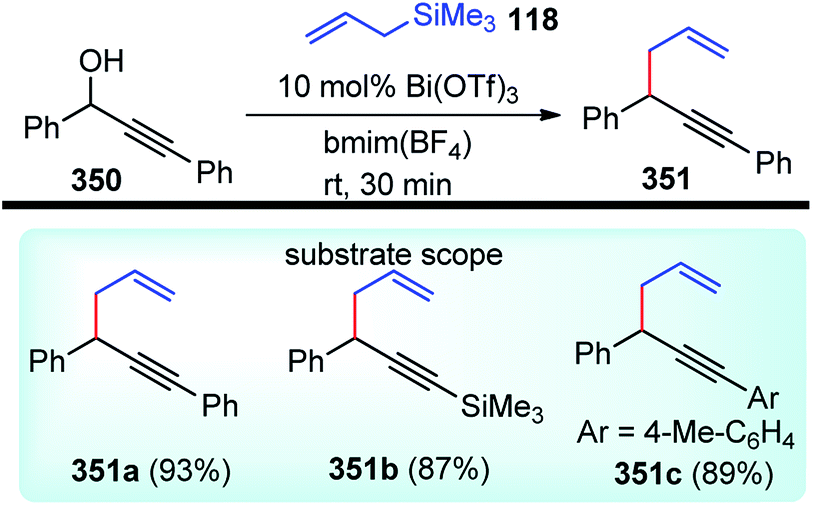 | ||
| Scheme 183 Bi(OTf)3 in [BMIM][BF4] as an efficient catalytic system for allylation, alkynylation, and deoxygenation of propargylic alcohols. | ||
Reactions were performed at room temperature and furnished products in good yield within a brief period. These attributes coupled with the recycling and reusability of the ionic liquids made this a superior method for the synthesis of various propargylic substituted products over several other reported methods.
Bach in the year 2014 developed a bismuth(III) triflate-catalyzed synthesis of substituted 2-alkenylfurans 361 via a successive 2-fold SN′-type substitution reaction at methoxy-substituted propargylic acetates 360 with enolsilylethers 75, Scheme 184.225
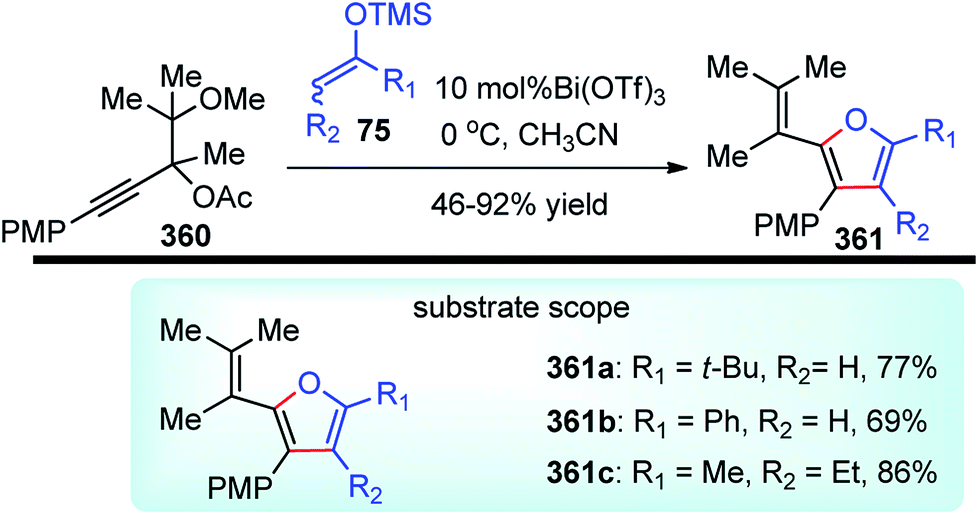 | ||
| Scheme 184 Bi-catalyzed synthesis of 2-alkenyl furans from propargylic alcohols. | ||
The reaction progressed in a similar fashion like the FeCl3-catalyzed substitution reaction of secondary propargylic acetates with silyl enol ethers affording the corresponding γ-alkynyl ketones. The mechanistic pathway is highlighted in Scheme 185. The tertiary group adjacent to the α-position prevents the usual SN1-type attack and directs the nucleophile to form allene intermediate 360b. Deprotonation at the resulting cationic intermediate generates the observed product 361.
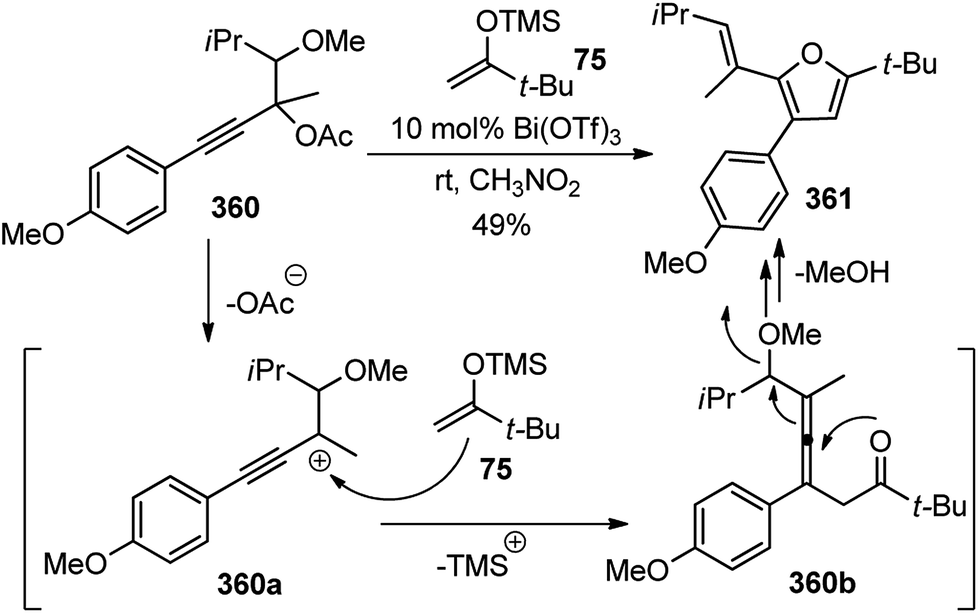 | ||
| Scheme 185 Mechanism for the Bi-catalyzed synthesis of 2-alkenyl furans from propargylic alcohols. | ||
The reaction appeared in general for several substitution patterns at both the substrates and allow for the construction of a diverse array of 2-alkenylfurans.
6. Brønsted acid catalyzed reactions
Several catalytic methodologies for the of substitution of propargylic alcohols have been reported in the presence of metal catalysts like copper, ruthenium, rhenium, platinum, palladium, gold, as well as other metal salts like BiCl3, FeCl3, AlCl3 etc. However, all these strategies were limited by the syntheses and costs of the catalysts, the need for anhydrous conditions, elevated temperatures, use of costly, corrosive and toxic catalysts. In general, harsh reaction conditions, costly catalysts and the chromatographic purification of products are not the desirable parameters due to the high operational cost associated with them. Thus, the development of efficient substitution reactions of propargylic alcohols using inexpensive air and moisture tolerant catalyst under mild reaction conditions was inevitable. Use of Brønsted acid in the propargylic substitution reaction could have been a solution to the problem. Since the reaction implies the use of very simple starting materials and catalysts under mild conditions which could be easily scalable, this methodology represents a clean, an environmentally friendly alternative to the existing protocols. Based on this concept several Brønsted acids catalyzed propargylic substitution reactions has evolved in the last couple of decades.Most of the Brønsted acid-mediated propargylic substitution mechanistically proceeded via a propargylic cation intermediate followed by the nucleophilic substitution reaction, Scheme 186.
 | ||
| Scheme 186 General mechanism for the Brønsted acid catalyzed propargylic substitution reaction. | ||
In 2006 Sanz and co-workers demonstrated the use of simple organic acids as catalysts for the direct propargylic substitution of hydroxy groups by different heteroatom and carbon-centered nucleophiles, Scheme 187.226
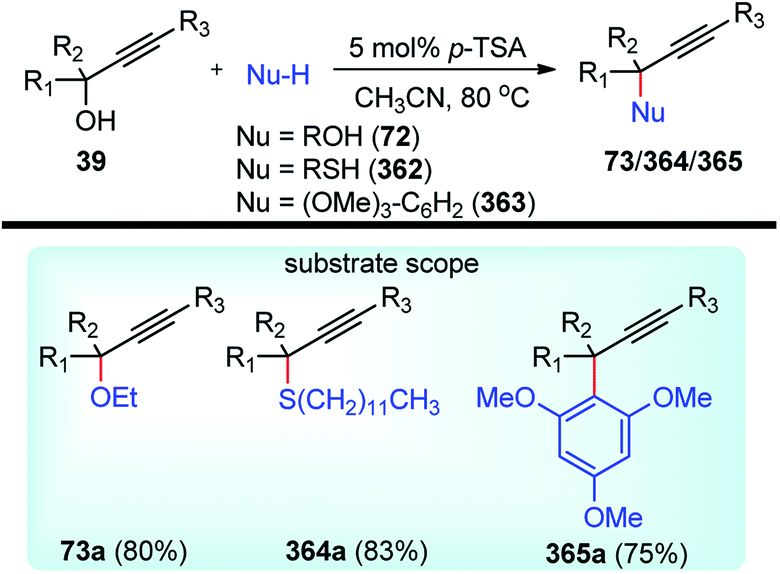 | ||
| Scheme 187 p-TSA catalyzed propargylic substitution by a heteroatom and carbon-centered nucleophiles. | ||
Various other Lewis acid catalysts like as InCl3, AlCl3, and CeCl3 also afforded the desired product but propargylic substituted products are obtained in short times and in essentially quantitative yields with organic acids like p-TSA or (±)-CSA. The reaction progressed efficiently with various electron-rich arenes like 1,3,5-trimethoxybenzene, phenols and heteroaromatic compounds like furan and N-methylindole with complete regioselectivity.
Sanz and co-workers in 2007 also demonstrated the use of p-toluenesulfonic acid as a catalyst in the preparation of 1,5-enynes 38 from propargylic alcohols 23 and allylsilanes 368, Scheme 188.227
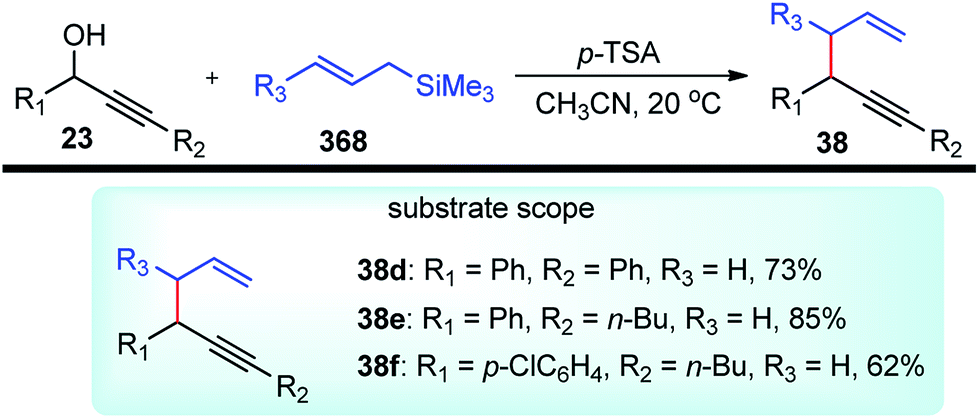 | ||
| Scheme 188 p-TSA catalyzed preparation of 1,5-enynes from propargylic alcohols. | ||
To explore the synthetic potential of this methodology, the authors developed a practical sequential dual catalytic protocol for the synthesis of bicyclo[3.1.0]hexenes 369 from the same starting materials, Scheme 189. The one-pot tandem sequential reaction reported by Sanz et al. involved both p-TSA and gold-catalyzed reaction, Scheme 189.227
 | ||
| Scheme 189 p-TSA catalyzed synthesis of bicyclo[3.1.0]hexanes. | ||
This metal-free, air and moisture tolerant methodology represented a greener protocol compared to the existing protocols employing metal catalysts.
A similar type of p-TSA catalyzed propargylic substitution reaction by a series of heteroatom- and carbon-centered nucleophiles for the synthesis of propargylic ethers, thioethers, amines, and amides were reported by Rodríguez and co-workers in the year 2007, Scheme 190.227
 | ||
| Scheme 190 Nucleophilic substitution of propargylic alcohols catalyzed by p-TSA. | ||
The reaction was found to be air and moisture tolerant and released water as the only by-product. The reaction also proceeded effectively with terminal alkynols albeit in longer durations. Enantiomerically enriched propargylic alcohol (94% ee) furnished racemic ether after the reaction indicating that the reaction followed an SN1 pathway.
KumaraSwamy and co-workers in 2014 developed a novel one-pot methodology for the synthesis of highly conjugated cyclopenta[c]quinolines 371 by the reaction of indoles 61 with propargyl alcohols 39, Scheme 191.228 The key step being the propargylic substitution of propargyl alcohols 39 with indoles 61.
 | ||
| Scheme 191 Synthesis of highly conjugated cyclopenta[c]quinolines from propargylic alcohols catalyzed by p-TSA. | ||
The mechanism for this reaction is illustrated in Scheme 192. The Brønsted acid catalyzed Friedel–Crafts alkenylation and isomerization, followed by copper-catalyzed dehydrogenation/oxidative ring expansion leads to the desired product.
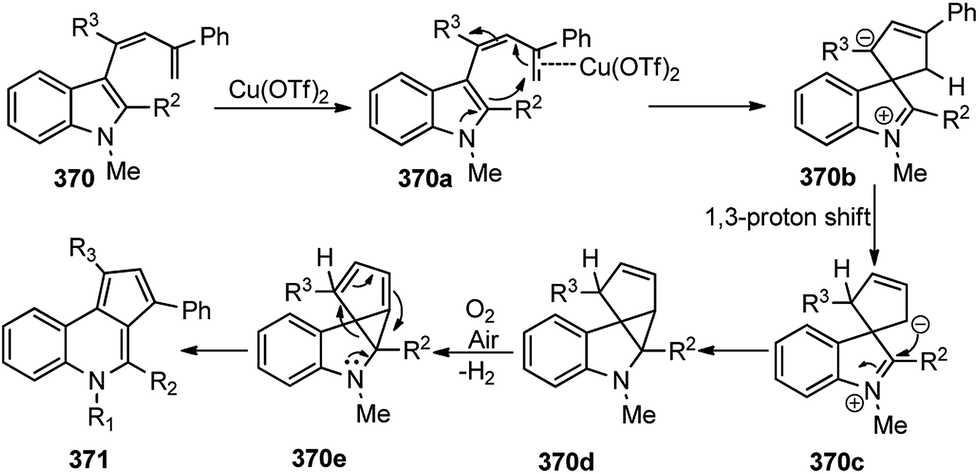 | ||
| Scheme 192 Mechanistic pathway for the p-TSA/Cu(OTf)2 catalyzed synthesis of highly conjugated cyclopenta[c]quinolines from propargylic alcohol. | ||
Electron-withdrawing groups on propargyl alcohols furnished better yields than those containing an electron donating substituents. Alkyl chain containing propargyl alcohol also led to good yields of the products. Even alkyl substituent on indole furnished the desired products in moderate yields.
Chan and co-workers in the year 2010 described an efficient p-TsOH·H2O-catalyzed protocol to prepare 2,4-di-and trisubstituted thiazoles 373 from trisubstituted propargylic alcohols 39 and thioamides 372, Scheme 193.229
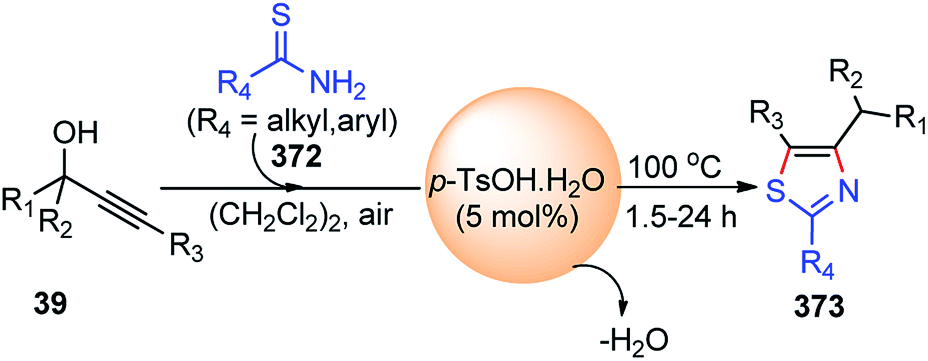 | ||
| Scheme 193 Efficient synthesis of 2,4-di- and trisubstituted thiazoles via p-TsOH·H2O-catalyzed cyclization of trisubstituted propargylic alcohols. | ||
The reaction was found to be temperature dependent and poor yield was obtained when the reaction was carried out at room temperature with 10 mol% of p-TSOH. Solvent screening revealed that 1,2-dichloroethane, MeNO2, MeCN, 1,4-dioxane, or toluene were inefficient. Poor yield was observed when stronger Brønsted acid catalysts like TfOH, TFA, and H3PO4 were examined.
The proposed mechanistic pathway is illustrated in Scheme 194 which involved Brønsted acid-mediated dehydration of the propargyl alcohol to generate allenic carbocation 39b. Subsequent nucleophilic attack by the thioamide 372 at the sterically less hindered allenic carbocation center followed by 5-exo-trig cyclization resulted in the thiazole product 373.
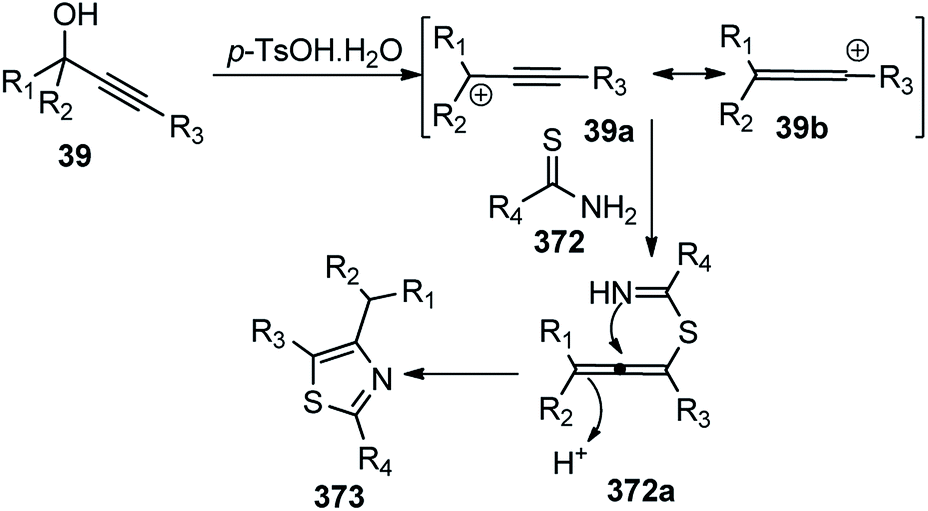 | ||
| Scheme 194 Mechanism for the synthesis of trisubstituted thiazoles. | ||
Sanz and co-workers in the year 2010 developed direct alkylation of indoles 61 with a wide variety of tertiary propargylic alcohols 39 using p-TSA as a catalyst, Scheme 195.230
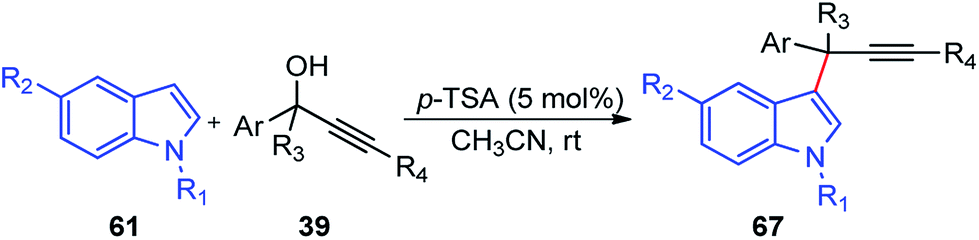 | ||
| Scheme 195 Propargylation of indoles catalyzed by p-TSA. | ||
Several Lewis acids were also found to catalyze the process, however; the substitution reactions were significantly slower and/or less efficient. Propargylic substitution reaction did not take place in the absence of a catalyst.
Earlier reports for the synthesis of substituted oxazoles by sequential propargylation/cycloisomerization of propargylic alcohols with amides were reported using [Cp*RuCl(μ2-SMe)2RuCp*Cl]/AuCl3/NH4BF4 or Zn(OTf)2/TpRuPPh3(CH3CN)2PF6, catalysts.140,204 These methodologies were only compatible with terminal propargylic alcohols and two different catalysts were involved in the process making it economically less feasible.
Therefore, in the process to design a more economical and environmentally acceptable methodology, Zhan and co-workers in the year 2009 described the synthesis of substituted oxazole 374 by an efficient one-pot tandem propargylation/cycloisomerization process from propargylic alcohols 23 and amides 77 using p-toluenesulfonic acid as a bifunctional catalyst, Scheme 196.205
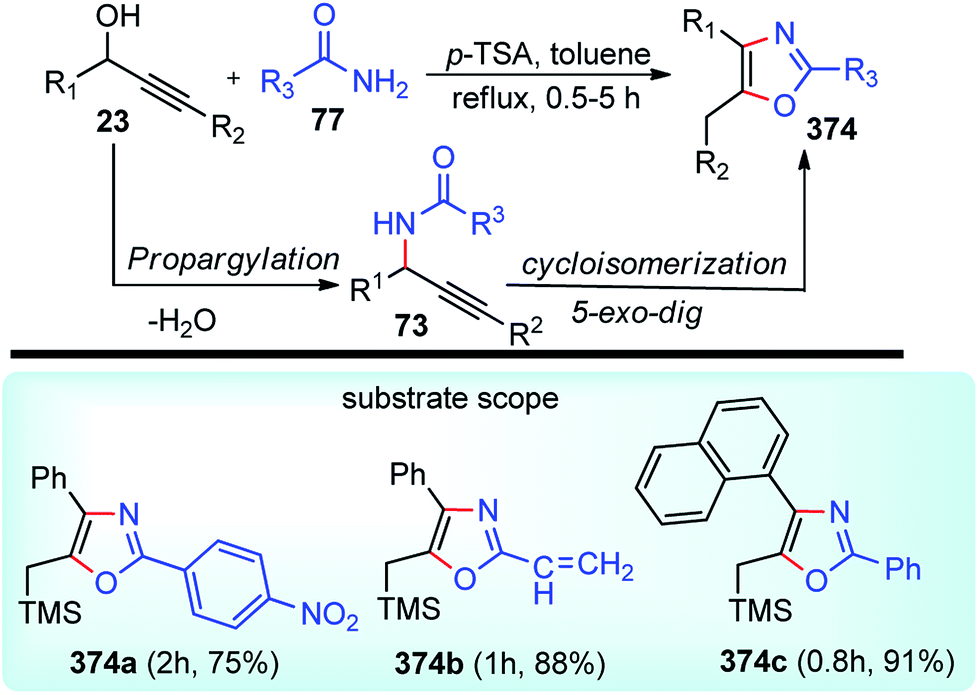 | ||
| Scheme 196 p-TSA catalyzed synthesis of oxazoles from propargylic alcohols. | ||
Several other Lewis acids like Cu(OTf)2, FeCl3, and InCl3, although furnished the propargylated product but did not catalyze the intramolecular cycloisomerization step to afford the cyclized product. A very slow reaction rate was observed when the amount of p-TSA decreased from 1 equiv. to 50 mol%. With 50 mol% of p-TSA, the propargylation proceeded rapidly but subsequent intramolecular cycloisomerization was sluggish. The results showed that stoichiometric amount of p-TSA was essential for cycloisomerization probably by counteracting the basicity of the oxazole and making the reaction medium acidic. Other Brønsted acids like oxalic acid and hydrochloric acid failed to catalyze the reaction. Reaction in solvents like acetonitrile and 1,2-dichloroethane were also sluggish. However, toluene had a positive impact on the reaction rate and obviously reduced the reaction time from 24 to 0.8 h. The reaction pathway proceeded via a nucleophilic attack by the amide 77 followed by a 5-exo-dig cyclization in a one-pot manner to furnish the desired product. The reaction has a wide substrate scope and many functional groups like bromo, chloro, vinyl, TMS, methoxy are well tolerated under this reaction condition.
Although the stoichiometric amount of p-TSA was necessary, the methodology provided an easy access to the substituted oxazoles from simple starting materials and reagents using an inexpensive catalyst.
Cycloisomerizations of alkynyl- and allenyl functionalized compounds is particularly an attractive strategy to synthesize polysubstituted furans and pyrroles. However, these strategies mostly utilize noble metal catalysis, multiple synthetic steps, or highly functionalized substrates. As a result, the development of efficient synthetic routes allowing the facile assembly of polysubstituted furans and pyrroles from readily available precursors will be a challenging process.
Zhan and co-workers in the year 2011, reported a novel efficient synthetic method for the synthesis of polysubstituted furans 376 as well as pyrroles from propargyl alcohols 23 and terminal alkynes 375 using TfOH as the catalyst, Scheme 197.231
 | ||
| Scheme 197 TfOH catalyzed synthesis of polysubstituted furans/pyrroles from propargyl alcohols and terminal alkynes. | ||
With 10 mol% of TfOH in refluxing CH3CN, the reaction of propargyl alcohols and terminal alkynes leads to the selective formation of 1,4-diynes which eventually cyclized to polysubstituted furans/pyrroles. The reaction is highly solvent dependent and maximum yield of the 1,4-diynes was reported in CH3CN. Notably, removal of H2O using 4 Å molecular sieves improved the product yield.
The mechanism for the reaction is highlighted in Scheme 198. The activation of propargyl alcohol 23 by the Brønsted acid affords the propargylic carbocation followed by a nucleophilic attack of the alkyne 375 leading to the formation of alkenyl cation 375a. Subsequent hydrolysis results in the formation of γ-alkynyl ketones 339 which then transforms to furan 377 under the reaction condition.
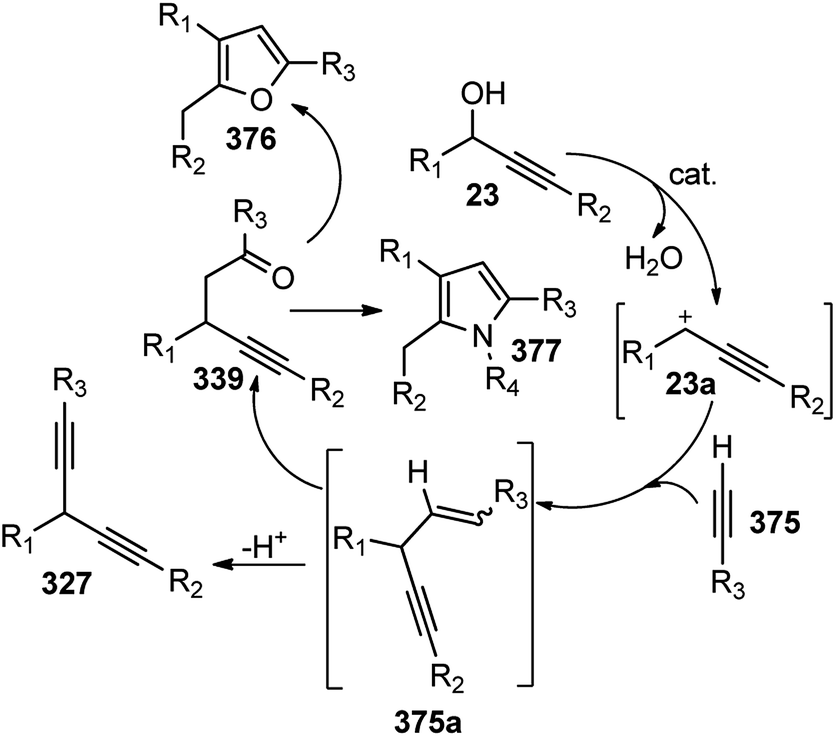 | ||
| Scheme 198 Mechanism for the synthesis of polysubstituted furans/pyrroles from propargyl alcohols and terminal alkynes. | ||
Silvia Díez-González and co-workers in the year 2015 demonstrated the use of HBF4 as a catalyst for the substitution of propargyl alcohols 23 with several nucleophiles in excellent yields, towards the formation of C–O, C–N and C–C bonds, Scheme 199.232
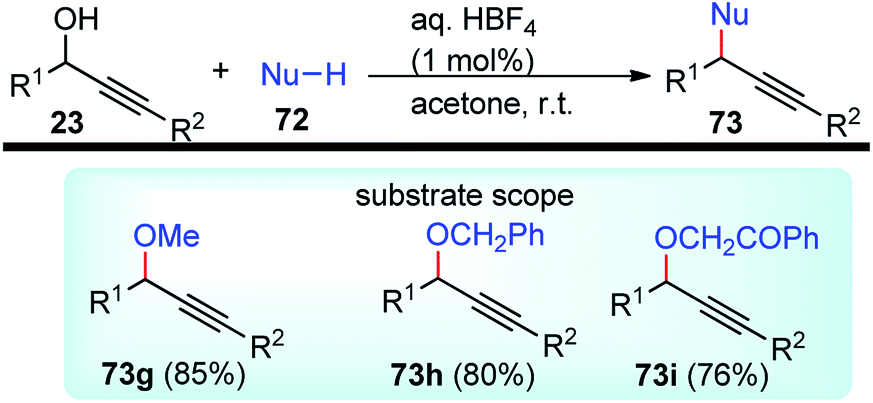 | ||
| Scheme 199 HBF4 as a practical catalyst for propargylation reactions. | ||
The high regioselectivity without any formation of allene intermediate and simple aqueous workup without any chromatographic purification are the advantages of this process. Electron-poor propargylic alcohols or acid-sensitive indoles furnished the products, although with little forcing conditions. A through solvent screening revealed that reactions were sluggish in THF and water whereas high conversions were obtained in solvents like DCM, acetonitrile, and acetone.
Savarimuthu and co-workers in 2014 employed NBSA as a cost-effective catalyst for the nucleophilic substitution of propargyl alcohols 23 with several nucleophiles like alcohols, amines, and heterocycles, Scheme 200.233
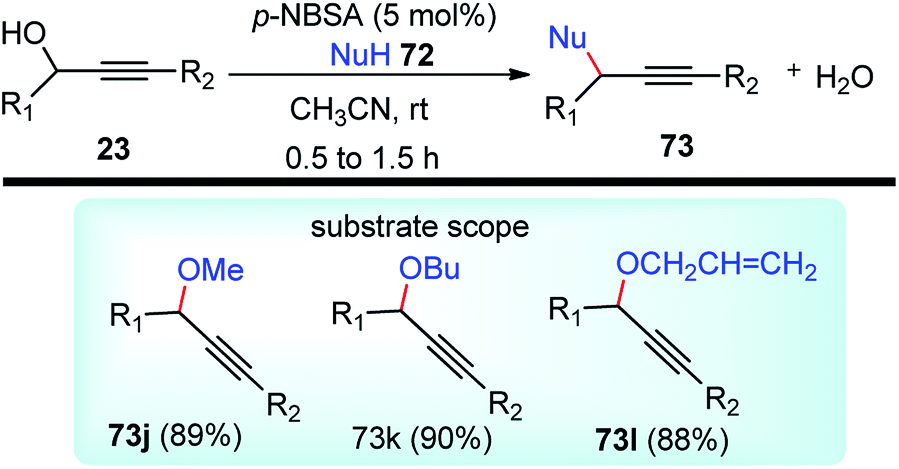 | ||
| Scheme 200 NBSA catalyzed nucleophilic substitution of propargyl alcohols. | ||
The reaction was devoid of any corrosive and costly metal catalysts, toxic solvents and does not involve any column chromatographic purification. The authors screened several protic acids like p-toluenesulfonic acid (p-TSA), benzene sulfonic acid (BSA), p-nitrobenzene sulfonic acid (NBSA) in acetonitrile, tetrahydrofuran and dichloromethane solvents. But the use of 5 mol% of 4-nitrobenzenesulfonic acid in CH3CN provided very good yields of the desired products with several O, N and C centered nucleophiles. The reaction proceeds via the protonation of the hydroxyl group of 1,3-diphenyl-prop-2-yn-1-ol by p-NBSA followed by the removal of water to form a secondary carbocation, which is in equilibrium with allene carbocation. Afterward, nucleophilic substitution via SN1 reaction by several nucleophiles furnishes the product in good to excellent yields.
Hamada and co-workers in 2014 reported the efficient synthesis of fused heterocycles 379 by an acid-promoted intramolecular ipso-Friedel–Crafts alkylation of phenol derivatives 378, Scheme 201.234
 | ||
| Scheme 201 Acid-promoted intramolecular ipso-Friedel–Crafts alkylation of phenol derivatives involving propargylic substitution reaction. | ||
The use of other catalysts like TsOH·H2O, Sc(OTf)3, In(OTf)3, Yb(OTf)3 and B(C6F5)3 did not furnish any desired product in considerable yield. A plausible reaction pathway of this cascade cyclization process is shown in Scheme 202 where the Brønsted acid promotes the intramolecular ipso-Friedel–Crafts alkylation of phenol derivatives, followed by the formation of an iminium cation 378a via a rearomatization promoted C–C bond cleavage. Then an aza-Prins reaction and subsequent 6-membered ring formation in the presence of TFA affords the fused-tricyclic dihydroquinoline derivatives 379.
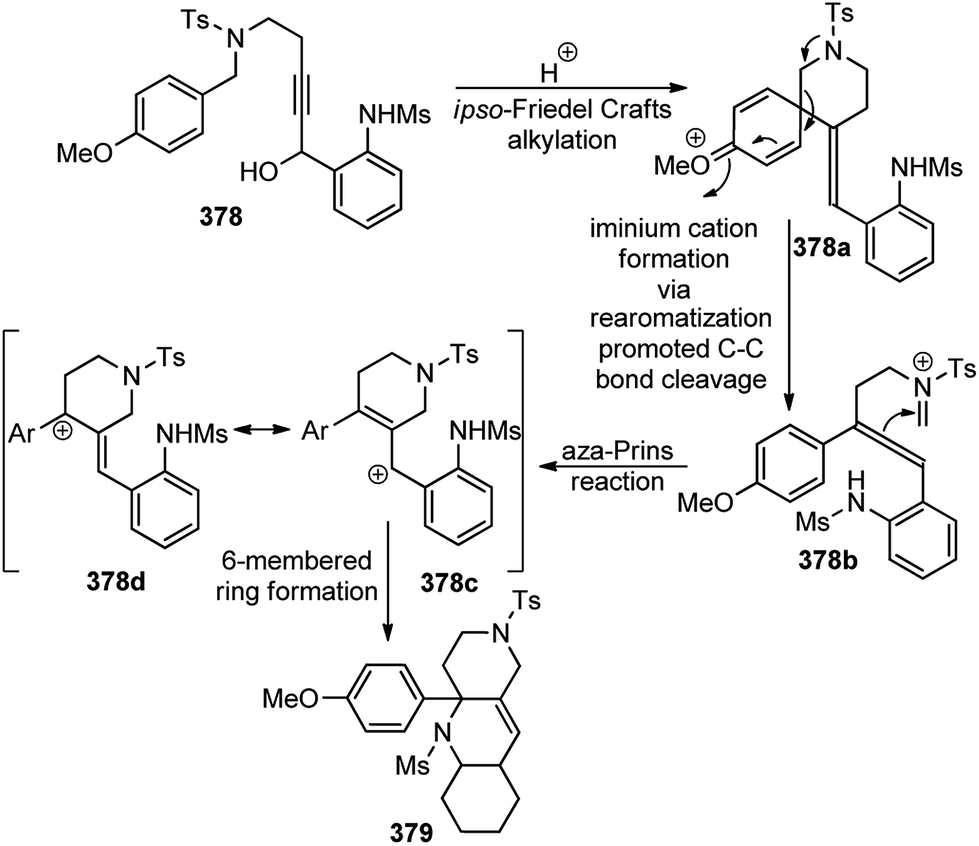 | ||
| Scheme 202 Mechanism for the acid-promoted fused heterocycles from propargylic alcohols. | ||
Liu and co-workers in the year 2012 described a facile synthesis of substituted dihydro-β-carbolines via Brønsted acid promoted cascade reactions of α-indolyl propargylic alcohols 380 with nitrones 381, Scheme 203.235
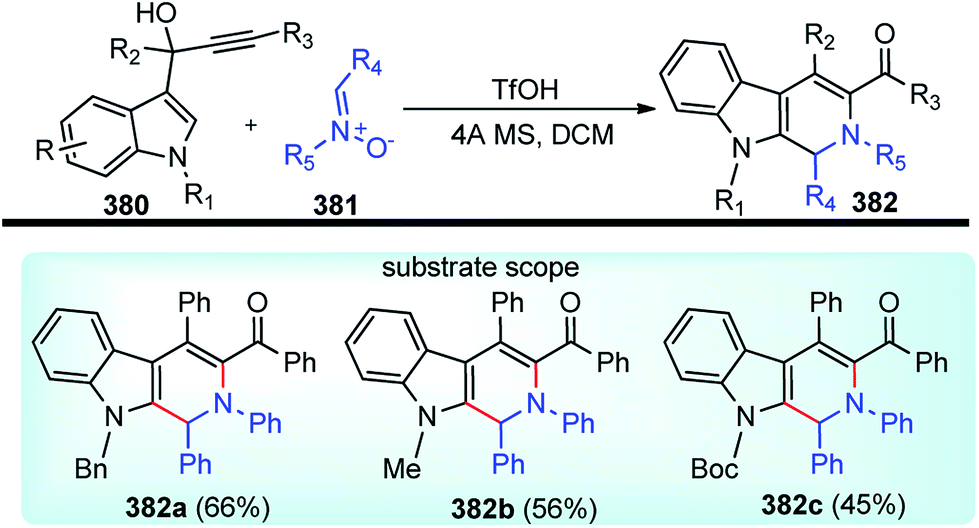 | ||
| Scheme 203 Synthesis of substituted dihydro-β-carbolines promoted by Brønsted acid. | ||
A plausible reaction pathway of this cascade cyclization process is illustrated in Scheme 204. Initially, allenyl cation is generated via a Meyer–Schuster rearrangement, which is attacked by nitrone 381 to generate the intermediate 380b which undergoes intramolecular cyclization leading to eight-membered N–O heterocycle 380c. Subsequent 1,3-rearrangement with cleavage of the N–O bond delivers highly substituted β-carboline 382.
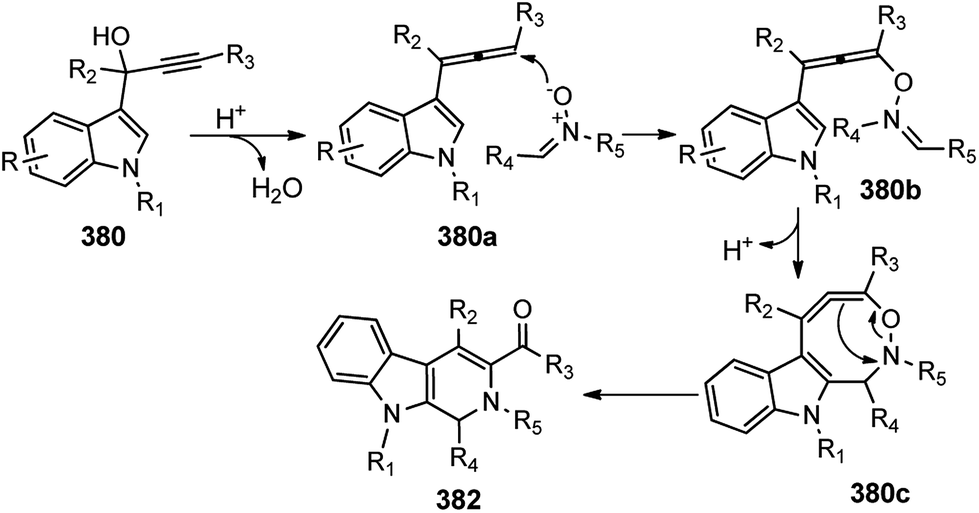 | ||
| Scheme 204 Mechanism for the synthesis of substituted dihydro-β-carbolines promoted by p-TSA. | ||
The authors observed that elevating the reaction temperature to 150 °C did not improve the reaction, and the product was isolated in 24% yield. Advantageously, the use of 30 mol% of Sc(OTf)3 enabled the reaction to proceed at 0 °C and improved the yield of the product to 62%. Notable variation in the reaction yield was not observed on changing the amounts of the catalyst. Other Brønsted acids, such as TsOH·H2O, MsOH, or CF3COOH, were found less effective.
Heterogeneous catalysis has played a significant role in various organic transformations.236 Among several heterogeneous catalysts, ion-exchange resins are widely used owing to their low cost, reusability, wide range of acid/base strength, ease of handling, environmental compatibility, and low toxicity.236 Moreover, they can be easily recovered from reaction mixtures by filtration and can be reused after activation or even without activation, making the process economically viable. Aluminosilicates are solid acids with industrial applications for ion-exchange, gas separation, and catalysis.237 Between them, zeolites are by volume the most used catalysts worldwide, with an important impact in both petrochemical and fine chemical industries. Since zeolite with larger external surface area can efficiently generate carbocation and stabilize them, they become subject of synthetic interest as several catalytic transformations using zeolite would increase significantly, Scheme 205.
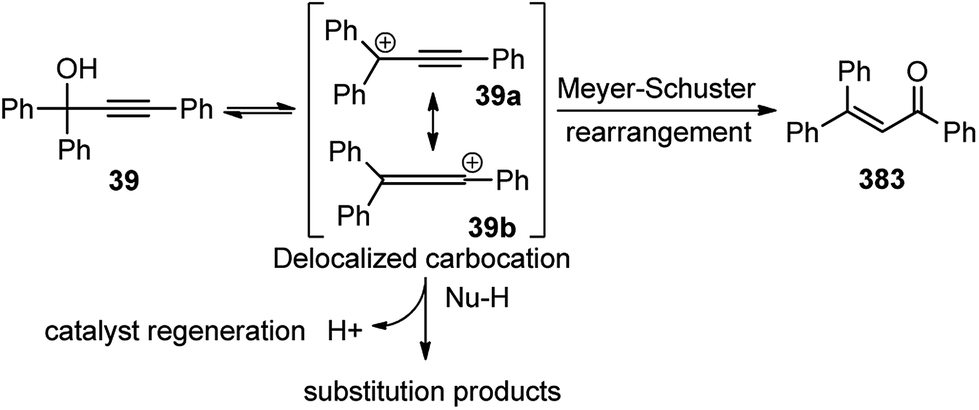 | ||
| Scheme 205 Zeolite catalyzed propargylic substitution reaction. | ||
The activity of heterogeneous catalysts occurs at the surface atoms. Therefore, significant effort is made to maximize the surface area of a catalyst by distributing it over the support. Typical supports include various kinds of carbon, alumina, and silica etc. Several propargylic substitution reactions were also reported with the solid supported catalyst. These clean and operationally simple methods with an easy work-up procedure, involving simple filtration of the catalyst, made those protocols attractive.
Perchloric acid supported on silica gel has already been shown to be a promising catalyst for numerous synthetically relevant organic transformations.238 Chen and co-workers in 2011 demonstrated HClO4-supported on silica gel as a very mild and efficient catalyst for the allylation of propargyl alcohol 23, Scheme 206.239
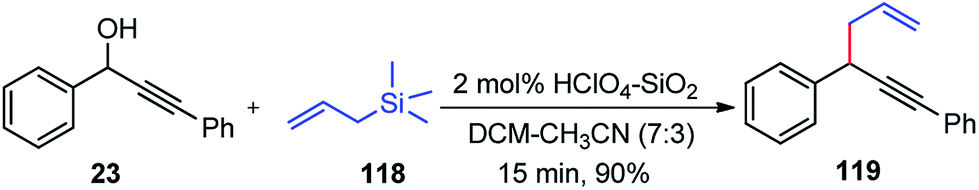 | ||
| Scheme 206 Silica supported HClO4 catalyzed allylation of propargylic alcohols. | ||
Allylated products were obtained in excellent yields, and allyltrimethylsilane was only used in a 1.5 equivalent. The reusability of the catalyst also enhances the synthetic applicability of this process.
Recently, heteropoly acids, HPAs, has gained much attention as an environmentally friendly alternative to harsh inorganic acids due to their ease of handling and high catalytic activities and reactivities.242 These compounds possess unique properties, such as well-defined structure, Brønsted acidity, redox modulating property, ability to accept and release electrons, and high proton mobility. In view of green chemistry principles, the replacement of harmful liquid acids by reusable solid HPAs as catalysts is a promising application of these acids.240 Among them, phosphomolybdic acid (PMA, H3PMo12O40) is one of the less expensive and commercially available catalysts.
Yadav and co-workers in the year 2008, described a mild environmentally friendly protocol involving of phosphomolybdic acid supported on silica gel (PMA/SiO2) catalyst for the synthesis of 2-propargylic-1,3-dicarbonyl compounds 306 in excellent yields and with high selectivity, Scheme 207.241
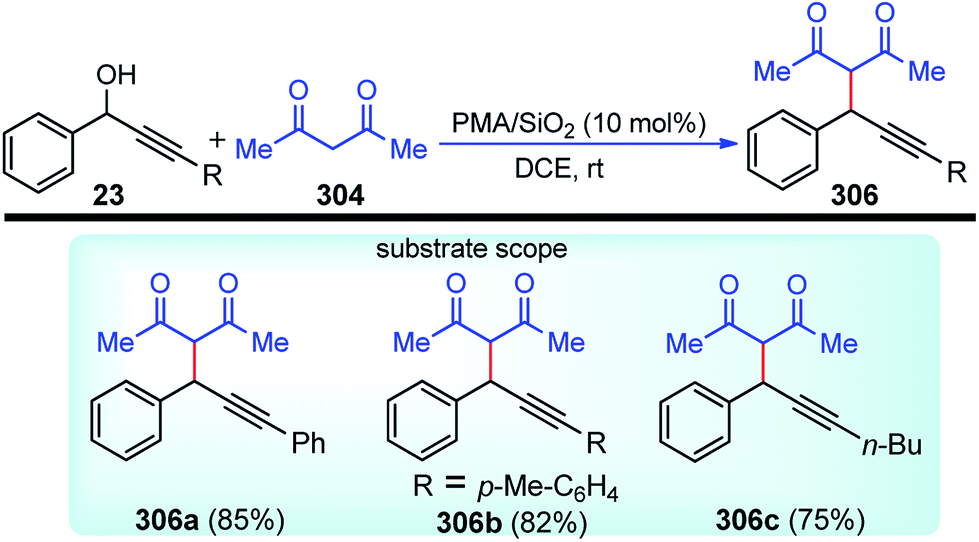 | ||
| Scheme 207 Phosphomolybdic acid supported on silica gel (PMA/SiO2) for propargylic substitution. | ||
Several silica-supported acid catalysts such as HClO4/SiO2, H2SO4/SiO2 and NaHSO4/SiO2 were screened for this reaction. PMA/SiO2 was found to give the best results in terms of conversion and reaction time. The advantages of this method are the ready availability of alcohols, high atom efficiency, no salt formation and water as the only by-product.
Yadav and co-workers have also demonstrated the use of PMA-SiO2 as a heterogeneous catalyst for O-, S-, and N-nucleophilic substitution reactions of aryl propargyl alcohols 23, Scheme 208.242
 | ||
| Scheme 208 Silica supported PMA in the nucleophilic propargylic substitution reaction. | ||
The authors demonstrated the use of phosphomolybdic acid supported on silica gel (PMA/SiO2) as an excellent heterogenous solid supported catalytic system, for the nucleophilic propargylic substitution reaction.
Srihari and co-workers in the year 2008, utilized PMA–silica to catalyze efficiently the propargylation of aromatic compounds with arylpropargyl alcohols 23 in the absence of solvent under environmentally benign conditions, Scheme 209.243
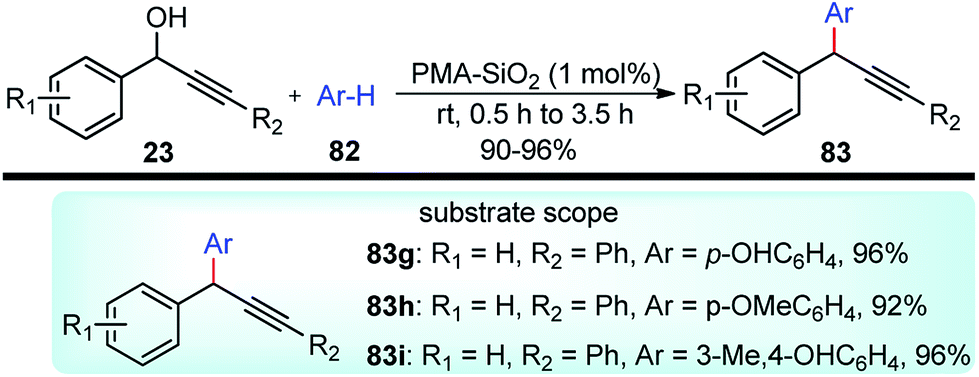 | ||
| Scheme 209 Silica supported PMA for the propargylation of electron rich arenes. | ||
Different acids such as 4-toluenesulfonic acid, phosphomolybdic acid, silica gel, silica gel supported sodium bisulfate, and silica gel supported perchloric acid and heterogeneous catalysts such as Amberlyst and montmorillonite K-10 were also tested for propargylation. It was found that PMA–silica gel and perchloric acid were the best in terms of yields and reaction time, while others including phosphomolybdic acid without silica gel took longer for complete conversion. The solvent study revealed that reaction proceeded faster in absence of solvent than in solvents like nitromethane, dichloromethane, 1,2-dichloroethane, polyethylene glycol (PEG), and water. The mechanism for the propargylation is shown in Scheme 210.
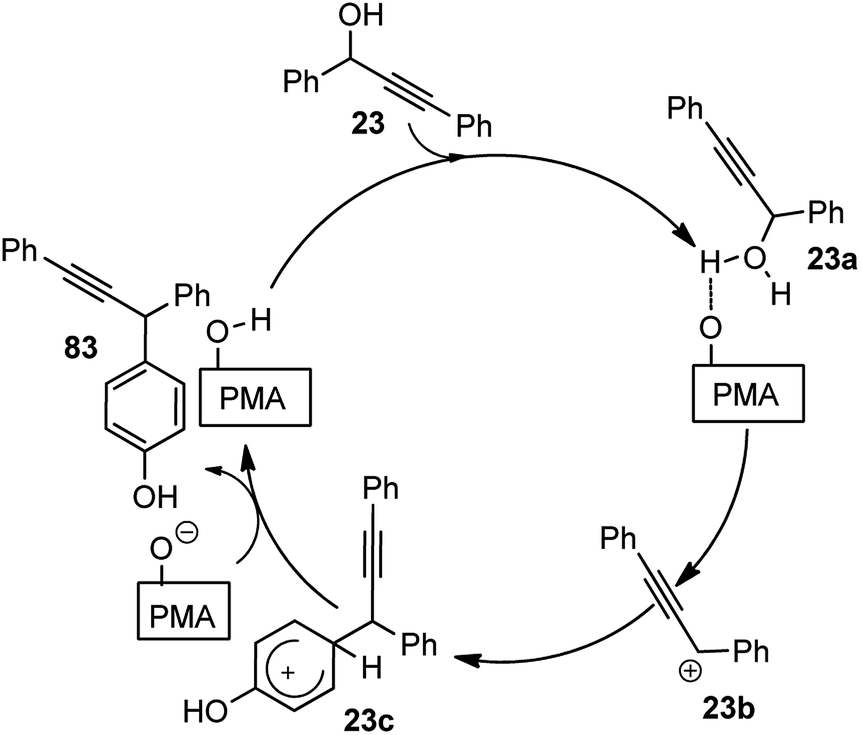 | ||
| Scheme 210 Mechanism for the PMA catalyzed propargylic substitution reaction. | ||
First, the hydroxyl group of the propargylic alcohol is protonated by the H+ active site to generate propargylic carbenium ion through dehydration followed by the nucleophilic attack by the electron-rich arene to the carbenium compound. Finally, the removal of a proton from the intermediate regenerates the H+-active site of PMA and furnished the product.
Park and co-workers in 2015 used mesoporous silica spheres embedded with Fe–Co/graphitic shell nanocrystals as recyclable catalysts for the propargylic substitution reactions with various electron rich arenes 155, Scheme 211.244
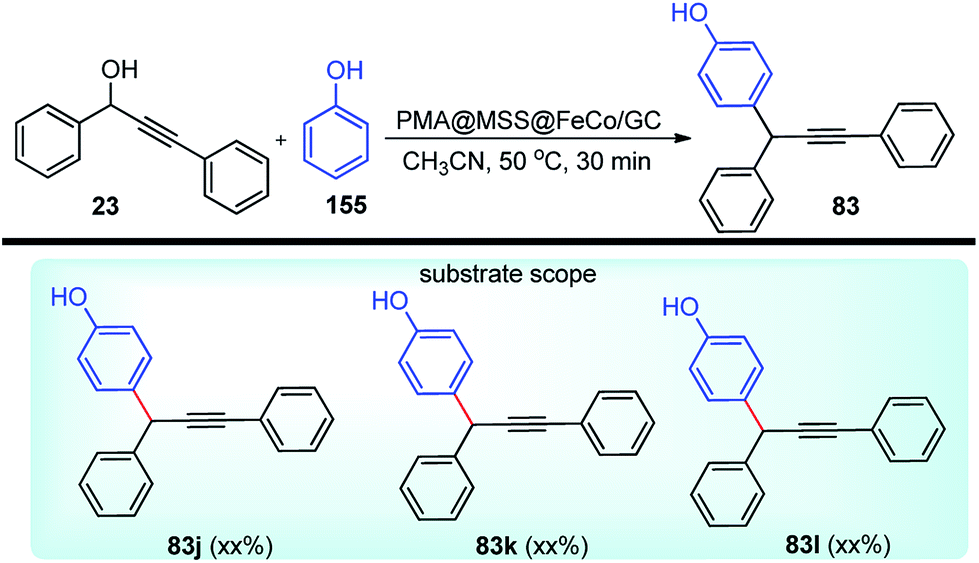 | ||
| Scheme 211 Propargylic substitution using recyclable mesoporous silica spheres embedded with Fe–Co/graphitic shell nanocrystals. | ||
The authors studied the solvent scope for this propargylic substitution reaction and observed that hydrophilic solvents such as water and PEG-400 showed low conversion, whereas hydrophobic solvents such as acetonitrile, dichloromethane, and dichloroethane showed high conversion. The highest yield was observed in acetonitrile. The reaction yield has a positive effect on the variation of catalyst loading, reaction temperature and reaction time. The authors also demonstrated the recyclability of the PMA@FeCo/GC@MSS catalyst by a magnetic separation which could be recycled for atleast five times without any change in the catalyst morphology. The propargylic substitution reactions for PMA@FeCo/GC@MSS followed the SN1 mechanism.
Zhan and coworkers reported HCl treated K10 montmorillonite for the nucleophilic substitution of propargylic alcohols by terminal TMS-alkynes to form 1,4-diynes under solvent-free condition. In the beginning, the reaction was studied by commercial virgin K10 under a neat condition which led to the formation of the desired product with only 48% yield, Scheme 212.245
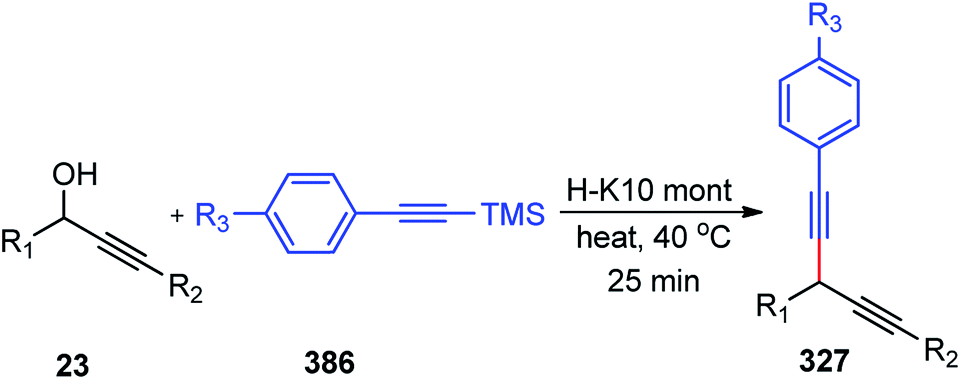 | ||
| Scheme 212 K-10 catalyzed propargylic substitution reaction. | ||
It was anticipated that due to lack of adequate exchangeable protons in the catalyst the reaction did not work well. Thus, K10 mont was treated with different concentrations of hydrochloric acids in different temperature. Finally, K10 mont obtained after the treatment of the catalyst in 1 M HCl at 80 °C proved to be the most efficient one. Acid-treated K10 mont catalyzed the substitution of propargylic alcohols by trimethyl(phenylethynyl)silane to form 1,4-diynes with 89% yields in 25 min.
Treatment of the substrates with other Brønsted acids like HCl or p-TsOH resulted in trace amount of product after 24 h of stirring. On the other hand, TfOH resulted in only 37% product after 24 h. However, the reaction did not proceed at all in the absence of a catalyst. Several 1,4-diynes were synthesized using this methodology by varying the substituents in the propargylic alcohols as well as in the nucleophile. A positive Sheldon test confirmed that the reaction was occurring on the surface of the catalyst which led to the conclusion that K-10 mont catalyzed reactions were heterogeneous in nature.246 The proposed mechanism for the catalytic pathway is shown in Scheme 213.
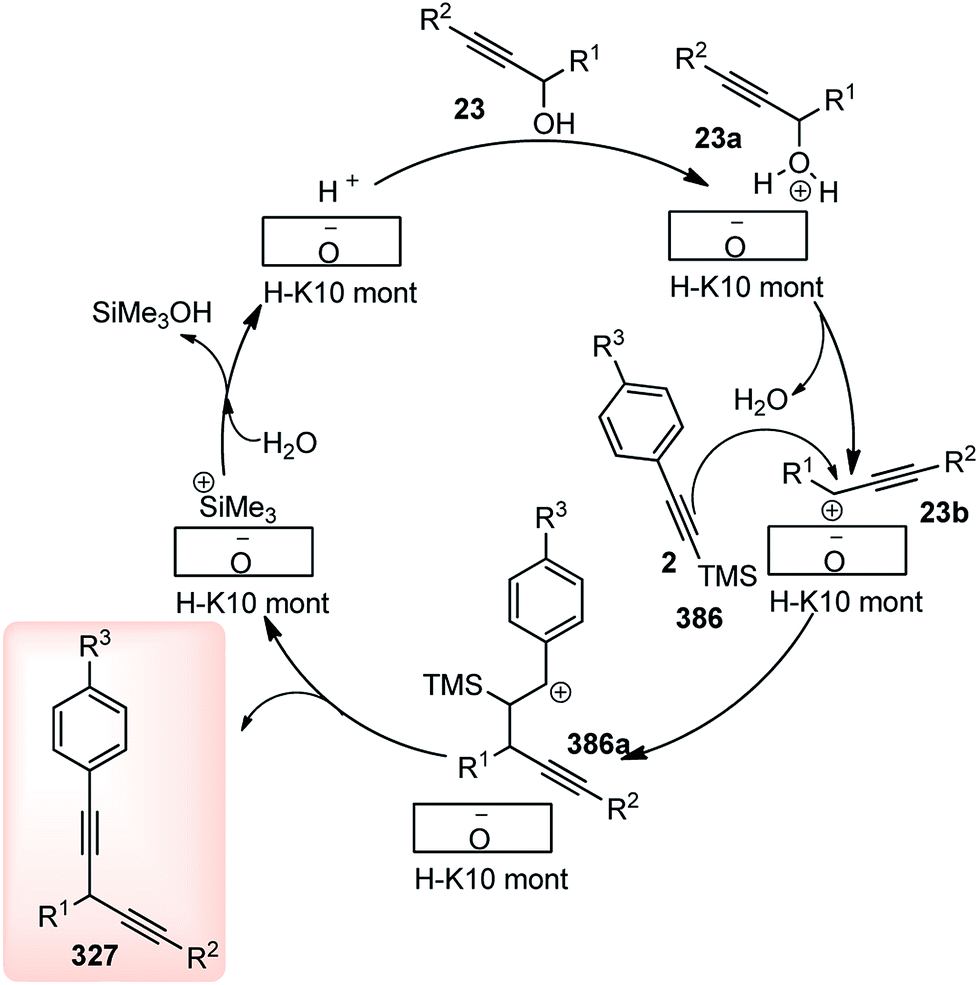 | ||
| Scheme 213 Mechanism for the K-10 catalyzed propargylic substitution reaction. | ||
Corma and co-workers in the year 2015 showed the application of zeolites under mild reaction conditions for propargylic substitution reaction towards the synthesis of a variety of heterocycles like oxazoles 389, thiazoles 389, and indenols 390 with high yield, and good selectivity, Scheme 214.247
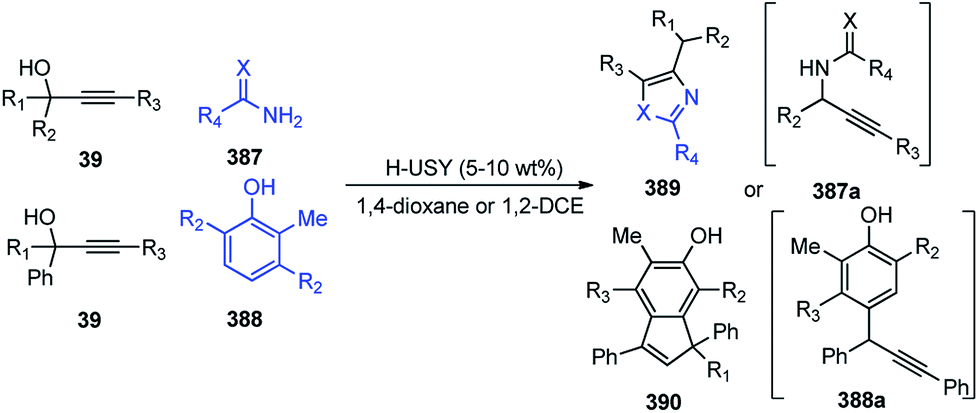 | ||
| Scheme 214 Zeolite as a catalyst for propargylic substitution reaction. | ||
Zeolites generate and stabilize delocalized carbocations after dehydration of propargyl alcohols 39, under mild reaction conditions, and then catalyze the synthesis. Following previously proposed mechanisms for homogeneous acid catalysts, the first step is the formation of the carbocation on the acid sites, followed by nucleophilic attack and cyclization.
Yadav and co-workers in the year 2007 described an efficient alkylation of indoles 61 with propargylic acetates 71 using Amberlyst-151 as a novel promoter, Scheme 215.248
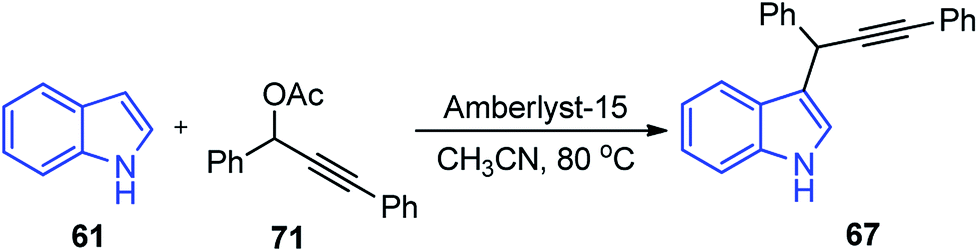 | ||
| Scheme 215 Efficient alkylation of indoles with propargylic acetates using Amberlyst-151. | ||
The reaction proceeds via an SN2 pathway and the operationally simple and efficient methodology with mild reaction conditions makes it a useful and attractive process for the alkylation of indoles. Interestingly, a highly acid sensitive pyrrole and furan gave the desired propargylated derivatives without the formation of any side products arising from the polymerization.
Another direct nucleophilic substitution of propargylic alcohols 23 with various nucleophiles using Amberlite IR-120H-resin as a catalyst was reported by Zheng and co-workers in the year 2013, Scheme 216.249
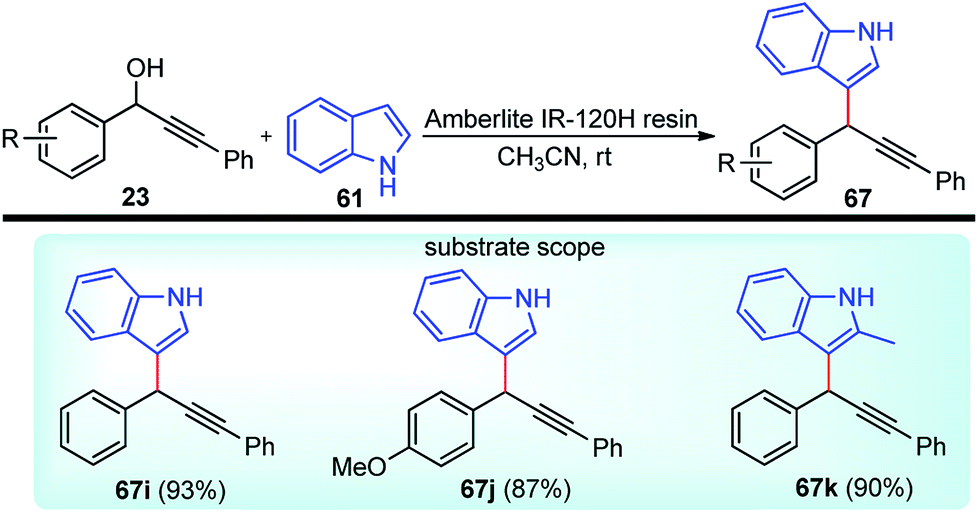 | ||
| Scheme 216 Propargylation of indoles by amberlite resin catalyst. | ||
The rate of the reaction was found to be dependent on the amount of resin used. With 300 mg mmol−1 of resin, the reaction completed within 30 min at room temperature to give 3-(1-(4-methoxyphenyl)-3-phenylprop-2-yn-1-yl)-1H-indole 67j in 93% yield. The method features short reaction times, mild reaction conditions, simplicity in operation, zero aqueous waste generation, complete regioselectivity.
Recently quinone methides (QMs), featuring a unique assembly of carbonyl and olefinic moieties, have gained considerable interest as a highly reactive intermediate. A wide range of catalytic, enantioselective transformations has been successfully developed for o-QM chemistry using Brønsted acid catalyst.250–252 Schneider and co-workers in 2015 reported a highly enantioselective conjugate addition of enamides and ene-carbamates to in situ generated ortho-quinone methides, towards the synthesis of acetamido-substituted tetrahydroxanthenes.253
In continuation of their research on ortho-quinone methide chemistry, the same group in 2015 demonstrated a highly enantioselective BINOL-based, chiral phosphoric acids catalyzed propargylic substitution of 1-(o-hydroxyphenyl)propargylic alcohols 391 with enamides 392 towards the synthesis of substituted benzo[c]xanthenes 394 and related heterocycles in a one-pot operation, Scheme 217.253
 | ||
| Scheme 217 Chiral phosphoric acid catalyzed highly enantioselective synthesis of 7-alkynyl-12a-acetamido-substituted benzo[c]xanthenes in very good yields form propargylic alcohols. | ||
Various chiral BINOL based catalysts were screened for this reaction and the results revealed that ortho-substitution within the 3,3′-aryl substituents in the BINOL backbone of the phosphoric acid catalyst gave superior enantioselectivities and the products were obtained in good yields irrespective of the substitution pattern within the alkynyl or the aryl group. A probable mechanism along with the proposed transition state is shown below, Fig. 23.
 | ||
| Fig. 23 Transition state structure for the formation of 7-alkynyl-12a-acetamido-substituted benzo[c]xanthenes in high enantioselectivity. | ||
The phosphoric acid catalyzes the generation of the intermediate orthro-quinone methide from the ortho-hydroxy propargylic alcohol derivatives which is then subsequently attacked by the enamide 392 with high facial selectivity guided by the chiral BINOL based phosphoric acid catalyst.
7. Conclusions
Catalytic propargylic substitution continues to be a very active field of research due to the alkyne handle which provides ample opportunity for the synthesis of various heterocyclic structures which are important building blocks in organic synthesis. There has been an incredible progress in the design and development of catalysts for propargylic substitution reaction. In this account, we have attempted to document comprehensively the developments and evolution of catalysts in the last few years that has witnessed an exponential growth. Some reactions involved newer methods, safer catalysts, environmentally friendly conditions, significantly contributing to simplifying the methodology, towards the synthesis of novel molecules. Despite these advancements, further efforts toward the development of better and safer catalysts are necessary. The appearance of new catalysts and chemical transformations is expected to provide avenues for the propargylic substitution reaction, leading to the discovery of molecules with new properties and biological activities. We conclude this review by hoping that it will stimulate researchers to develop new and creative methods for the propargylic substitution reaction which will be instrumental in the advancement of many branches of chemistry. Despite the impressive progress that has been made in this area, it is still highly desirable to explore more efficient and practical, as well as sustainable, catalytic systems for the propargylic substitution reactions. Thus, we hope this review will promote continued interests in developing new catalytic methodologies and encourage chemists to employ these valuable methodologies in heterocyclic, medicinal chemistry as well as in other branches of chemistry.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
R. R. is thankful to Prof. Todd Lowary for valuable discussions and Dr Anurag Gupta for her fellowship. S. S. is grateful to CSIR (02(0253)/16/EMR-II)), India, DST-SERB, India (YSS/2015/000934) UGC Start-Up Grant (2015) and ICT Golden Jubilee Research grant (2018) for the financial support and UGC-FRP (F.4-5(128)/2014(BSR), India for the position.References
- J. Tsuji and T. Mandai, Angew. Chem., Int. Ed., 1996, 34, 2589–2612 CrossRef.
- A. S. Thompson, E. G. Corley, M. F. Huntington and E. J. J. Grabowski, Tetrahedron Lett., 1995, 36, 8937–8940 CrossRef.
- N. Fusetani, M. Sugano, S. Matsunaga and K. Hashimoto, Tetrahedron Lett., 1987, 28, 4311–4312 CrossRef.
- M. Konishi, H. Ohkuma, K. Matsumoto, T. Tsuno, H. Kamei, T. Miyaki, T. Oki, H. Kawaguchi, G. D. VanDuyne and J. Clardy, J. Antibiot., 1989, 42, 1449–1452 CrossRef PubMed.
- J. L. Wright, T. F. Gregory, S. P. Kesten, P. A. Boxer, K. A. Serpa, L. T. Meltzer, L. D. Wise, S. A. Espitia, C. S. Konkoy, E. R. Whittemore and R. M. Woodward, J. Med. Chem., 2000, 43, 3408–3419 CrossRef PubMed.
- R. F. Lockwood and K. M. Nicholas, Tetrahedron Lett., 1977, 48, 4163–4165 CrossRef.
- K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207–214 CrossRef.
- K. M. Nicholas, J. Organomet. Chem., 1972, 21, 44–48 Search PubMed.
- T. J. J. Müller and A. Netz, Tetrahedron Lett., 1999, 40, 3145–3148 CrossRef.
- Y. Nishibayashi, I. Wakiji and M. Hidai, J. Am. Chem. Soc., 2000, 122, 11019–11023 CrossRef.
- J. J. K. Smith, L. A. Young and F. D. Toste, Org. Lett., 2004, 6, 1325–1328 CrossRef PubMed.
- M. Georgy, V. Boucard, O. Debleds, C. Dal Zotto and J.-M. Campagne, Tetrahedron, 2009, 65, 1758–1762 CrossRef.
- Y. Nishibayashi, Y. Inada, M. Yoshikawa, M. Hidai and S. Uemura, Angew. Chem., Int. Ed., 2003, 42, 1495–1498 CrossRef PubMed.
- R. J. Detz, M. M. E. Delville, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2008, 47, 3777–3780 CrossRef PubMed.
- Y. Nishibayashi and S. Uemura, Curr. Org. Chem., 2006, 10, 135–150 CrossRef.
- R. J. Detz, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2009, 6263–6276 CrossRef.
- Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342–356 CrossRef.
- C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914–1937 CrossRef PubMed.
- D.-Y. Zhang and X.-P. Hu, Tetrahedron Lett., 2015, 56, 283–295 CrossRef.
- K. Sakata and Y. Nishibayashi, Catal. Sci. Technol., 2018, 8, 12–25 RSC.
- Y. Nishibayashi, Synthesis, 2012, 4, 489–503 CrossRef.
- J. S. Yadav, P. K Deshpande and V. M. Sharma, Tetrahedron Lett., 1990, 46, 7033–7046 CrossRef.
- E. R. Graham and R. R. Tykwinski, J. Org. Chem., 2011, 76, 6574–6583 CrossRef PubMed.
- G. E. Keck, D. Krishnamurthy and X. Chen, Tetrahedron Lett., 1994, 35, 8323–8326 CrossRef.
- L. C. Hirayama, K. K. Dunham and B. Singaram, Tetrahedron Lett., 2006, 47, 5173–5176 CrossRef.
- N. Minowa and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1987, 60, 3697–3700 CrossRef.
- C.-M. Yu, J.-M. Kim, M.-S. Shin and D. Cho, Tetrahedron Lett., 2003, 44, 5487–5490 CrossRef.
- S. E. Denmark and T. Wynn, J. Am. Chem. Soc., 2001, 123, 6199–6203 CrossRef PubMed.
- A. M. Cook and C. Wolf, Chem. Commun., 2014, 50, 3151–3154 RSC.
- K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef.
- E. B. Bauer, Synthesis, 2012, 44, 1131–1151 CrossRef.
- Y. Hu, X. Xin and B. Wan, Tetrahedron Lett., 2015, 56, 32–52 CrossRef.
- (a) P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Chem.–Eur. J., 2012, 18, 5460–5465 CrossRef PubMed; (b) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937–2977 CrossRef PubMed; (c) A. Palisse and S. F. Kirsch, Org. Biomol. Chem., 2012, 10, 8041–8048 RSC.
- V. A. Peshkov, O. P. Pereshivko, A. A. Nechaev, A. A. Peshkov and E. V. Van der Eycken, Chem. Soc. Rev., 2018, 47, 3861–3898 RSC.
- A. Monleón, G. Blay, L. R. Domingo, M. C. Muñoz and J. R. Pedro, Chem.–Eur. J., 2013, 19, 14852–14856 CrossRef PubMed.
- P. Wipf, Y. Aoyama and T. E. Benedum, Org. Lett., 2004, 6, 3593–3595 CrossRef PubMed.
- J. Su, H. Liu and R. Hua, Int. J. Mol. Sci., 2015, 16, 3599–3608 CrossRef PubMed.
- M. Shi and Y.-M. Shen, J. Org. Chem., 2002, 67, 16–21 CrossRef PubMed.
- C. Koradin, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2002, 41, 2535–2538 CrossRef PubMed.
- J. Chen, R. Properzi, D. P. Uccello, J. A. Young, R. G. Dushin and J. T. Starr, Org. Lett., 2014, 16, 4146–4149 CrossRef PubMed.
- Q. Zhang, S. Sun, J. Hu, Q. Liu and J. Tan, J. Org. Chem., 2007, 72, 139–143 CrossRef PubMed.
- M. Rubin and V. Gevorgyan, Org. Lett., 2001, 3, 2705–2709 CrossRef PubMed.
- T. Schwier, M. Rubin and V. Gevorgyan, Org. Lett., 2004, 6, 1999–2001 CrossRef PubMed.
- G. W. Kabalka, M.-L. Yao and S. Borella, J. Am. Chem. Soc., 2006, 128, 11320–11321 CrossRef PubMed.
- Y. Liu, B.-D. Barry, H. Yu, J. Liu, P. Liao and X. Bi, Org. Lett., 2013, 13, 2608–2611 CrossRef PubMed.
- Z. Fang, J. Liu, Q. Liu and X. Bi, Angew. Chem., Int. Ed., 2014, 53, 7209–7213 CrossRef PubMed.
- S. Muthusamy and M. Sivaguru, Org. Lett., 2014, 16, 4248–4251 CrossRef PubMed.
- G. Yin, Y. Zhu, P. Lu and Y. Wang, J. Org. Chem., 2011, 76, 8922–8929 CrossRef PubMed.
- T. Ishikawa, S. Manabe, T. Aikawa, T. Kudo and S. Saito, Org. Lett., 2004, 6, 2361–2364 CrossRef PubMed.
- J. A. Spencer, C. Jamieson and E. P. A. Talbot, Org. Lett., 2017, 19, 3891–3894 CrossRef PubMed.
- S. Muthusamy, A. Balasubramani and E. Suresh, Org. Biomol. Chem., 2018, 16, 756–764 RSC.
- S. B. Simelane, H. H. Kinfe, A. Muller and D. B. G. Williams, Org. Lett., 2014, 16, 4543–4545 CrossRef PubMed.
- M. Gohain, C. Marais and B. C. B. Bezuidenhoudt, Tetrahedron Lett., 2012, 53, 4704–4708 CrossRef.
- A. Cullen, A. J. Muller, D. Bradley and G. Williams, RSC Adv., 2017, 7, 42168–42171 RSC.
- G. C. Bond, Advances in Chemistry, Homogeneous Catalysis, 1974, vol. 70, ch. 2, pp. 25–34 Search PubMed.
- M. Niggemann and N. Bisek, Chem.–Eur. J., 2010, 16, 11246–11252 CrossRef PubMed.
- M. Niggemann and M. J. Meel, Angew. Chem., Int. Ed., 2010, 49, 3684–3687 CrossRef PubMed.
- V. J. Meyer and M. Niggemann, Eur. J. Org. Chem., 2011, 3671–3674 CrossRef.
- S. Yaragorla, A. Pareek and R. Dada, Tetrahedron Lett., 2017, 58, 4642–4647 CrossRef.
- J. S. Yadav, B. V. Subba Reddy, K. V. Raghavendra Rao and G. G. K. S. Narayana Kumar, Tetrahedron Lett., 2007, 48, 5573–5576 CrossRef.
- M. Yoshimatsu, T. Otani, S. Matsuda, T. Yamamoto and A. Sawa, Org. Lett., 2008, 10, 4251–4254 CrossRef PubMed.
- A. Barrels, R. Mahrwaid and S. Quint, Tetrahedron Lett., 1999, 40, 5989–5990 CrossRef.
- A. A. O. Sarhanw and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC.
- Z.-P. Zhan, J.-l. Yu, H.-J. Liu, Y.-Y. Cui, R.-F. Yang, W.-Z. Yang and J.-P. Li, J. Org. Chem., 2006, 71, 8298–8301 CrossRef PubMed.
- Z.-P. Zhan, Y.-Y. Cui and H.-J. Liu, Tetrahedron Lett., 2006, 47, 9143–9146 CrossRef.
- U. Jana, S. Maiti and S. Biswas, Tetrahedron Lett., 2007, 48, 7160–7163 CrossRef.
- Z.-P. Zhan, X.-B. Cai, S.-P. Wang, J.-l. Yu, H.-J. Liu and Y.-Y. Cui, J. Org. Chem., 2007, 72, 9838–9841 CrossRef PubMed.
- W. Rao, P. Wai and H. Chan, Org. Biomol. Chem., 2010, 8, 4016–4025 RSC.
- X. Xu, Y. Lu, G. Hong, Z. Zhao and X. Li, ARKIVOC, 2014, 237–246 Search PubMed.
- M. J. Queensen, J. M. Rabus and E. B. Bauer, J. Mol. Catal. A: Chem., 2015, 407, 221–229 CrossRef.
- Y.-J. Shang, X.-Q. Hu, X.-W. He, J.-J. Tao, G. Han, F.-L. Wu and J. Wang, J. Org. Chem., 2015, 80, 4760–4765 CrossRef PubMed.
- X. Su, P. Wu, W. Liu and C. Chen, Org. Chem. Front., 2018, 5, 1165–1169 RSC.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef PubMed.
- S. W. Smith and G. C. Fu, Angew. Chem., Int. Ed., 2008, 47, 9334–9336 CrossRef PubMed.
- A. J. Oelke, J. Sun and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 2966–2969 CrossRef PubMed.
- C. Fischer and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 4594–4595 CrossRef PubMed.
- L. An, C. Xu and X. Zhang, Nat. Commun., 2017, 8, 1460–1469 CrossRef PubMed.
- J. S. Yadav, B. V. Subba Reddy, T. Srinivasa Rao and K. V. Raghavendra Rao, Tetrahedron Lett., 2008, 49, 614–618 CrossRef.
- G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2008, 47, 3781–3783 CrossRef PubMed.
- G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef PubMed.
- E. C. Lee, B. H. Hong, J. Y. Lee, J. C. Kim, D. Kim, Y. Kim, P. Tarakeshwar and K. S. Kim, J. Am. Chem. Soc., 2005, 127, 4530–4537 CrossRef PubMed.
- A. Yoshida, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 2460–2463 CrossRef PubMed.
- M. Lin, Q.-Z. Chen, Y. Zhu, X.-l. Chen, J.-j. Cai, Y.-m. Pan and Z.-p. Zhan, Synlett, 2011, 8, 1179–1183 Search PubMed.
- R. Mitra and J. Niemeyer, ChemCatChem, 2018, 10, 1221–1234 CrossRef.
- S. Afewerki and A. Cordova, Chem. Rev., 2016, 116, 13512–13570 CrossRef PubMed.
- A. Yoshida, M. Ikeda, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 592–595 CrossRef PubMed.
- F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 1410–1414 CrossRef PubMed.
- F.-L. Zhu, Y.-H. Wang, D.-Y. Zhang, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 10223–10227 CrossRef PubMed.
- P. Fang and X.-L. Hou, Org. Lett., 2009, 11, 4612–4615 CrossRef PubMed.
- D.-Y. Zhang, F.-L. Zhu, Y.-H. Wang, X.-H. Hu, S. Chen, C.-J. Hou and X.-P. Hu, Chem. Commun., 2014, 50, 14459–14462 RSC.
- C. Zhang, X.-H. Hu, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, J. Am. Chem. Soc., 2012, 134, 9585–9588 CrossRef PubMed.
- C. Zhang, Y.-Z. Hui, D.-Y. Zhang and X.-P. Hu, RSC Adv., 2016, 6, 14763–14767 RSC.
- Y. Zou, F.-L. Zhu, Z.-C. Duan, Y.-H. Wang, D.-Y. Zhang, Z. Cao, Z. Zheng and X.-P. Hu, Tetrahedron Lett., 2014, 55, 2033–2036 CrossRef.
- D.-Y. Zhang, L. Shao, J. Xu and X.-P. Hu, ACS Catal., 2015, 5, 5026–5030 CrossRef.
- M. Yoshida and C. Sugimura, Tetrahedron Lett., 2013, 54, 2082–2086 CrossRef.
- Q. Li, C.-J. Hou, Y.-Z. Hui, Y.-J. Liu, R.-F. Yang and X.-P. Hu, RSC Adv., 2015, 5, 85879–85883 RSC.
- L. Zhao, G. H. B. Guo, L. Xu, J. Chen, W. Cao, G. Zhao and X. Wu, Org. Lett., 2014, 16, 5584–5587 CrossRef PubMed.
- K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472–2475 CrossRef PubMed.
- F.-Z. Han, F.-L. Zhu, Y.-H. Wang, Y. Zou, X.-H. Hu, S. Chen and X.-P. Hu, Org. Lett., 2014, 16, 588–591 CrossRef PubMed.
- R. J. Detz, Z. Abiri, R. le Griel, H. Hiemstra and J. H. van Maarseveen, Chem.–Eur. J., 2011, 17, 5921–5924 CrossRef PubMed.
- G. Huang, C. Cheng, L. Ge, B. Guo, L. Zhao and X. Wu, Org. Lett., 2015, 17, 4894–4897 CrossRef PubMed.
- N. Sakai, K. Enomoto, M. Takayanagi, T. Konakahara and Y. Ogiwara, Tetrahedron Lett., 2016, 57, 2175–2178 CrossRef.
- L. Shao, Y.-H. Wang, D.-Y. Zhang, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2016, 55, 5014–5018 CrossRef PubMed.
- X. Chen, C. Hou, Q. Li, Y. Liu, R. Yang and X. Hu, Chin. J. Catal., 2016, 37, 1389–1395 CrossRef.
- Z.-T. Liu, Y.-H. Wang, F.-L. Zhu and X.-P. Hu, Org. Lett., 2016, 18, 1190–1193 CrossRef PubMed.
- L. Shao, D.-Y. Zhang, Y.-H. Wang and X.-P. Hu, Adv. Synth. Catal., 2016, 358, 2558–2563 CrossRef.
- F. Zhu and X. Hu, Chin. J. Catal., 2015, 36, 86–92 CrossRef.
- H. Xu, L. Laraia, L. Schneider, K. Louven, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 11232–11236 CrossRef PubMed.
- B. Wang, C. Liu and H. Guo, RSC Adv., 2014, 4, 53216–53219 RSC.
- R. Shen, B. Luo, J. Yang, L. Zhang and L.-B. Han, Chem. Commun., 2016, 52, 6451–6454 RSC.
- K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 CrossRef PubMed.
- G. Hu, C. Shan, W. Chen, P. Xu, Y. Gao and Y. Zhao, Org. Lett., 2016, 18, 6066–6069 CrossRef PubMed.
- L.-J. Cheng, A. P. N. Brown and C. J. Cordier, Chem. Sci., 2017, 8, 4299–4305 RSC.
- L. Shao and X.-P. Hu, Chem. Commun., 2017, 53, 8192–8195 RSC.
- J. Song, Z.-J. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 5212–5216 CrossRef PubMed.
- K. Tsuchida, M. Yuki, K. Nakajima and Y. Nishibayashi, Chem. Lett., 2018, 47, 671–673 CrossRef.
- R.-Z. Li, H. Tang, K. R. Yang, L.-Q. Wan, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2017, 56, 7213–7216 CrossRef PubMed.
- R.-Z. Li, H. Tang, L. Wan, X. Zhang, Z. Fu, J. Liu, S. Yang, D. Jia and D. Niu, Chem, 2017, 3, 834–838 Search PubMed.
- M. V. Dias, M. S. Saraiva, P. Ferreira and M. J. Calhorda, Organometallics, 2015, 34, 1465–1478 CrossRef.
- B. M. Trost, J. R. Miller and C. M. Hoffman, J. Am. Chem. Soc., 2011, 133, 8165–8167 CrossRef PubMed.
- M. Zhang, H. Yang, Y. Cheng, Y. Zhu and C. Zhu, Tetrahedron Lett., 2010, 51, 1176–1179 CrossRef.
- Y. Inada, Y. Nishibayashi, M. Hidai and S. Uemura, J. Am. Chem. Soc., 2002, 124, 15172–15173 CrossRef PubMed.
- J. Berge, S. Claridge, A. Mann, C. Muller and E. Tyrrell, Tetrahedron Lett., 1997, 38, 685–686 CrossRef.
- Y. Nishibayashi, Y. Inada, M. Hidai and S. Uemura, J. Am. Chem. Soc., 2003, 125, 6060–6061 CrossRef PubMed.
- Y. Nishibayashi, M. Yoshikawa, Y. Inada, M. Hidai and S. Uemura, J. Am. Chem. Soc., 2004, 126, 16066–16072 CrossRef PubMed.
- Y. Nishibayashi, M. Yoshikawa, Y. Inada, M. D. Milton, M. Hidai and S. Uemura, Angew. Chem., Int. Ed., 2003, 42, 2681–2684 CrossRef PubMed.
- Y. Nishibayashi, G. Onodera, Y. Inada, M. Hidai and S. Uemura, Organometallics, 2003, 22, 873–878 CrossRef.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1214 CrossRef PubMed.
- K. Kanao, Y. Tanabe, Y. Miyake and Y. Nishibayashi, Organometallics, 2010, 29, 2381–2384 CrossRef.
- K. Sakata, Y. Miyake and Y. Nishibayashi, Chem.–Asian J., 2009, 4, 81–88 CrossRef PubMed.
- A. J. M. Caffyn and K. M. Nicholas, J. Am. Chem. Soc., 1993, 115, 6438–6442 CrossRef.
- V. Cadierno, S. Conejero, M. P. Gamasa and J. Gimeno, Dalton Trans., 2003, 3060–3066 RSC.
- Y. Nishibayashi, H. Imajima, G. Onodera and S. Uemura, Organometallics, 2005, 24, 4106–4110 CrossRef.
- (a) Y. Nishibayashi, M. Yoshikawa, Y. Inada, M. Hidai and S. Uemura, J. Org. Chem., 2004, 69, 3408–3412 CrossRef PubMed; (b) Y. Nishibayashi, I. Wakiji, Y. Ishii, S. Uemura and M. Hidai, J. Am. Chem. Soc., 2001, 123, 3393–3396 CrossRef PubMed.
- Y. Nishibayashi, M. Yoshikawa, Y. Inada, M. Hidai and S. Uemura, J. Am. Chem. Soc., 2002, 124, 11846–11847 CrossRef PubMed.
- Y. Inada, M. Yoshikawa, M. D. Milton, Y. Nishibayashi and S. Uemura, Eur. J. Org. Chem., 2006, 881–890 CrossRef.
- Y. Inada, Y. Nishibayashi and S. Uemura, Angew. Chem., Int. Ed., 2005, 44, 7715–7717 CrossRef PubMed.
- S. Berger and E. Haak, Tetrahedron Lett., 2010, 51, 6630–6634 CrossRef.
- C. Bruneau, Ruthenium Vinylidenes and Allenylidenes in Catalysis, in Ruthenium Catalysts and Fine Chemistry-Topics in Organometallic Chemistry, ed. C. Bruneau and P. H. Dixneuf, Springer, 2004, vol. 11, pp. 125–153 Search PubMed.
- M. D. Milton, Y. Inada, Y. Nishibayashi and S. Uemura, Chem. Commun., 2004, 2712–2715 RSC.
- H. Ben Ammar, J. Le Nôtre, M. Salem, M. T. Kaddachi, L. Toupet, J.-L. Renaud, C. Bruneau and P. H. Dixneuf, Eur. J. Inorg. Chem., 2003, 4055–4060 CrossRef.
- C. Fischmeister, L. Toupet and P. H. Dixneuf, New J. Chem., 2005, 29, 765–768 RSC.
- Y. Nishibayashi, M. D. Milton, Y. Inada, M. Yoshikawa, I. Wakiji, M. Hidai and S. Uemura, Chem.–Eur. J., 2005, 11, 1433–1451 CrossRef PubMed.
- Y. Nishibayashi, A. Shinoda, O. Miyake, H. Matsuzawa and M. Sato, Angew. Chem., Int. Ed., 2006, 45, 4835–4839 CrossRef PubMed.
- Y. Fukuda and K. J. Utimoto, J. Org. Chem., 1991, 56, 3729–3731 CrossRef.
- D. F. Alkhaleeli, K. J. Baum, J. M. Rabus and E. B. Bauer, Catal. Commun., 2014, 47, 45–48 CrossRef.
- K. Fukamizu, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2008, 130, 10498–10499 CrossRef PubMed.
- K. Kanao, Y. Miyake and Y. Nishibayashi, Organometallics, 2009, 28, 2920–2926 CrossRef.
- Y. Yamauchi, Y. Miyake and Y. Nishibayashi, Organometallics, 2009, 28, 48–50 CrossRef.
- K. Kanao, Y. Miyake and Y. Nishibayashi, Organometallics, 2010, 29, 2126–2131 CrossRef.
- M. Ikeda, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2010, 49, 7289–7293 CrossRef PubMed.
- M. Ikeda, Y. Miyake and Y. Nishibayashi, Organometallics, 2012, 31, 3810–3813 CrossRef.
- K. Motoyama, M. Ikeda, Y. Miyake and Y. Nishibayashi, Organometallics, 2012, 31, 3426–3430 CrossRef.
- M. Ikeda, Y. Miyake and Y. Nishibayashi, Chem.–Eur. J., 2012, 18, 3321–3328 CrossRef PubMed.
- Y. Miyake, S. Endo, T. Moriyama, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2013, 52, 1758–1762 CrossRef PubMed.
- Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2015, 54, 4060–4064 CrossRef PubMed.
- P. Andrew Evans, Modern Rhodium-Catalyzed Organic Reactions, Wiley Online Library, 2005, DOI:10.1002/3527604693.
- K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169–196 CrossRef PubMed.
- P. A. Evans and M. J. Lawler, Angew. Chem., Int. Ed., 2006, 45, 4970–4972 CrossRef PubMed.
- C. Darcel, S. Bartsch, C. Bruneau and P. H. Dixneuf, Synlett, 1994, 457–460 CrossRef.
- J. A. Marshall and M. A. Wolf, J. Org. Chem., 1996, 61, 3238–3239 CrossRef.
- M. Kalek, T. Johansson, M. Jezowska and J. Stawinski, Org. Lett., 2010, 12, 4702–4704 CrossRef PubMed.
- M. Kalek and J. Stawinski, Adv. Synth. Catal., 2011, 353, 1741–1755 CrossRef.
- I. Ambrogio, S. Cacchi, G. Fabrizi, A. Goggiamani and A. Iazzetti, Eur. J. Org. Chem., 2015, 3147–3151 CrossRef.
- T. M. Locascio and J. A. Tunge, Chem.–Eur. J., 2016, 22, 18140–18146 CrossRef PubMed.
- S.-X. Xu, L. Hao, T. Wang, Z.-C. Ding and Z.-P. Zhan, Org. Biomol. Chem., 2013, 11, 294–298 RSC.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353–9356 CrossRef PubMed.
- M. Bhanuchandra, M. R. Kuram and A. K. Sahoo, J. Org. Chem., 2013, 78, 11824–11834 CrossRef PubMed.
- L.-L. Mao, Y.-H. Lia and S.-D. Yang, Org. Chem. Front., 2017, 4, 608–611 RSC.
- G. F. Christopher and J. P. Hartley, Mini-Rev. Org. Chem., 2004, 1, 1–7 CrossRef.
- J. S. Yadav, B. V. S. Reddy, K. V. R. Rao and G. G. K. S. Narayana Kumar, Synthesis, 2007, 20, 3205–3210 CrossRef.
- Z. Liu, L. Liu, Z. Shafiq, Y.-C. Wu, D. Wang and Y.-J. Chen, Synthesis, 2007, 13, 1961–1969 CrossRef.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2009, 38, 2745–2755 RSC.
- S. Belot, K. A. Vogt, C. Besnard, N. Krause and A. Alexakis, Angew. Chem., Int. Ed., 2009, 48, 8923–8926 CrossRef PubMed.
- A. S. Hashmi and C. Hubbert, Angew. Chem., Int. Ed., 2010, 49, 1010–1012 CrossRef PubMed.
- R. Sinisi, M. V. Vita, A. Gualandi, E. Emer and P. G. Cozzi, Chem.–Eur. J., 2011, 17, 7404–7408 CrossRef PubMed.
- K. Motoyama, M. Ikeda, Y. Miyake and Y. Nishibayashi, Eur. J. Org. Chem., 2011, 2239–2246 CrossRef.
- O. M. Singh and L. R. Devi, Mini-Rev. Org. Chem., 2013, 10, 84–96 CrossRef.
- Y. Masuyama, M. Hayashi and N. Suzuki, Eur. J. Org. Chem., 2013, 2914–2921 CrossRef.
- M. Hayashi, A. Inubushi and T. Mukaiyama, Chem. Lett., 1987, 1975–1978 CrossRef.
- Y.-M. Ren, C. Cai and R.-C. Yang, RSC Adv., 2013, 3, 7182–7204 RSC.
- P. Srihari, D. C. Bhunia, P. Sreedhar, S. S. Mandal, J. S. S. Reddy and J. S. Yadav, Tetrahedron Lett., 2007, 48, 8120–8124 CrossRef.
- Z. Liu, L. Liu, Z. Shafiq, Y.-C. Wu, D. Wang and Y.-J. Chen, Tetrahedron Lett., 2007, 48, 3963–3967 CrossRef.
- B. V. Subba Reddy, B. B. Reddy, K. V. R. Rao and J. S. Yadav, Tetrahedron Lett., 2010, 51, 5697–5700 CrossRef.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef PubMed.
- S.-S. Weng, K.-Y. Hsieh and Z.-J. Zeng, Tetrahedron, 2015, 71, 2549–2554 CrossRef.
- S. Muthusamy, K. Selvaraj and E. Suresh, Tetrahedron Lett., 2016, 57, 4829–4833 CrossRef.
- R. Properzi and E. Marcantoni, Chem. Soc. Rev., 2014, 43, 779–791 RSC.
- C. C. Silveira, S. R. Mendes, L. Wolf and G. M. Martins, Tetrahedron Lett., 2010, 51, 4560–4562 CrossRef.
- C. C. Silveira, S. R. Mendes and G. M. Martins, Tetrahedron Lett., 2012, 53, 1567–1570 CrossRef.
- G. Rouschias, Chem. Rev., 1974, 74, 531–566 CrossRef.
- B. D. Sherry, A. T. Radosevich and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 6076–6080 CrossRef PubMed.
- M. R. Luzung and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 15760–15761 CrossRef PubMed.
- R. V. Ohri, A. T. Radosevich, K. J. Hrovat, C. Musich, D. Huang, T. R. Holman and F. D. Toste, Org. Lett., 2005, 7, 2501–2504 CrossRef PubMed.
- Y. Kuninobu, E. Ishii and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 3296–3299 CrossRef PubMed.
- Y. Kuninobu, H. Ueda and K. Takai, Chem. Lett., 2008, 37, 878–879 CrossRef.
- B. G. Das, R. Nallagonda and P. Ghorai, J. Org. Chem., 2012, 77, 5577–5583 CrossRef PubMed.
- W. Huang, J. Wang, Q. Shena and X. Zhou, Tetrahedron, 2007, 63, 11636–11643 CrossRef.
- A. O. Shchukin and A. V. Vasil'ev, Russ. J. Org. Chem., 2007, 43, 784–787 CrossRef.
- W. Huang, P. Z. Zheng, Z. X. Zhang, R. Liu, Z. X. Chen and X. G. Zhou, J. Org. Chem., 2008, 73, 6845–6850 CrossRef PubMed.
- S. Y. Wang, Y. X. Zhu, Y. G. Wang and P. Lu, Org. Lett., 2009, 11, 2615–2618 CrossRef PubMed.
- X. Zhang, W. T. Teo and P. W. H. Chan, Org. Lett., 2009, 21, 4990–4993 CrossRef PubMed.
- X. Zhang, W. Teng, T. Philip and W. H. Chan, J. Organomet. Chem., 2011, 696, 331–337 CrossRef.
- M. P. Kumar and R.-S. Liu, J. Org. Chem., 2006, 71, 4951–4955 CrossRef PubMed.
- Y. M. Pan, F. J. Zheng, H. X. Lin and Z. P. Zhan, J. Org. Chem., 2009, 74, 3148–3151 CrossRef PubMed.
- I. Matsuda, S. Wakamatsu, K. Komori, T. Makino and K. Itoh, Tetrahedron Lett., 2002, 43, 1043–1046 CrossRef.
- I. Matsuda, K. Komori and K. Itoh, J. Am. Chem. Soc., 2002, 124, 9072–9073 CrossRef PubMed.
- J. K. D. Brabander, B. Liu and M. Qian, Org. Lett., 2008, 10, 2533–2536 CrossRef PubMed.
- Q. Liang and J. K. D. Brabander, Tetrahedron, 2011, 67, 5046–5053 CrossRef PubMed.
- Y. Kwon, I. Kim and S. Kim, Org. Lett., 2014, 16, 4936–4939 CrossRef PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef PubMed.
- O. Debleds, E. Gayon, E. Vrancken and J.-M. Campagne, Beilstein J. Org. Chem., 2011, 7, 866–877 CrossRef PubMed.
- M. Georgy, V. Boucard and J.-M. Campagne, J. Am. Chem. Soc., 2005, 127, 14180–14181 CrossRef PubMed.
- J. Liu, E. Muth, U. Flörke, G. Henkel, K. Merz, J. Sauvageau, E. Schwake and G. Dyker, Adv. Synth. Catal., 2006, 348, 456–462 CrossRef.
- T. T. Haug, T. Harschneck, A. Duschek, C.-U. Lee, J. T. Binder, H. Menz and S. F. Kirsch, J. Organomet. Chem., 2009, 694, 510–514 CrossRef.
- N. Morita, T. Tsunokake, Y. Narikiyo, M. Harada, T. Tachibana, Y. Saito, S. B. Yoshimitsu, H. Okamoto and O. Tamura, Tetrahedron Lett., 2015, 56, 6269–6272 CrossRef.
- H. Suzuki, T. Ikegami and T. Matano, Synthesis, 1997, 249–252 CrossRef.
- N. M. Leonard, L. C. Wieland and R. S. Mohan, Tetrahedron, 2002, 58, 8373–8378 CrossRef.
- M. Postel and E. Dunach, Coord. Chem. Rev., 1996, 155, 127–144 CrossRef.
- T. Ollevier, Org. Biomol. Chem., 2013, 11, 2740–2755 RSC.
- Z.-P. Zhan, W.-Z. Yang, R.-F. Yang, J.-l. Yu, J. P. Li and H.-J. Liu, Chem. Commun., 2006, 3352–3354 RSC.
- M. Saito, N. Tsuji, Y. Kobayashi and Y. Takemoto, Org. Lett., 2015, 17, 3000–3003 CrossRef PubMed.
- J. C. T. Reddel, W. Wang, K. Koukounas and R. J. Thomson, Chem. Sci., 2017, 8, 2156–2160 RSC.
- G. G. K. S. Narayana Kumar and K. K. Laali, Org. Biomol. Chem., 2012, 10, 7347–7355 RSC.
- D. Nitsch and T. Bach, J. Org. Chem., 2014, 79, 6372–6379 CrossRef PubMed.
- R. Sanz, A. Martínez, J. M. Á. Gutiérrez and F. Rodríguez, Eur. J. Org. Chem., 2006, 1383–1386 CrossRef.
- R. Sanz, A. Martínez, D. Miguel, J. M. Álvarez-Gutiérrez and F. Rodríguez, Synthesis, 2007, 20, 3252–3256 CrossRef.
- G. Rao, A. Uruvakilli and K. C. KumaraSwamy, Org. Lett., 2014, 16, 6060–6063 CrossRef PubMed.
- X. Zhang, W. Teng, T. Sally, P. Wai and H. Chan, J. Org. Chem., 2010, 75, 6290–6293 CrossRef PubMed.
- R. Sanz, D. Miguel, A. Martínez, M. Gohain, P. García-García, M. A. F. Rodríguez, E. Álvarez and F. Rodríguez, Eur. J. Org. Chem., 2010, 7027–7039 CrossRef.
- T. Wang, X.-l. Chen, L. Chen and Z.-P. Zhan, Org. Lett., 2011, 13, 3324–3327 CrossRef PubMed.
- E. Barreiro, A. S. Vidal, E. Tan, S.-H. Lau, T. D. Sheppard and S. D. González, Eur. J. Org. Chem., 2015, 7544–7549 CrossRef PubMed.
- A. Savarimuthu, D. G. L. Prakash and S. A. Thomas, Tetrahedron Lett., 2014, 55, 3213–3217 CrossRef.
- T. Yokosaka, N. Shiga, T. Nemoto and Y. Hamada, J. Org. Chem., 2014, 79, 3866–3875 CrossRef PubMed.
- L. Wang, X. Xie and Y. Liu, Org. Lett., 2012, 23, 5848–5851 CrossRef PubMed.
- R. Schlőgl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- J. H. Clark, Acc. Chem. Res., 2002, 35, 791–797 CrossRef PubMed.
- H. R. Shaterian, H. Yarahmadi and M. Ghashang, Tetrahedron, 2008, 64, 1263–1269 CrossRef.
- K. Murugan and C. Chen, Tetrahedron Lett., 2011, 52, 5827–5830 CrossRef.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef PubMed.
- J. S. Yadav, B. V. S. Reddy and A. S. Reddy, J. Mol. Catal. A: Chem., 2008, 280, 219–223 CrossRef.
- P. Srihari, J. S. S. Reddy, D. C. Bhunia, S. S. Mandal and J. S. Yadav, Synth. Commun., 2008, 38, 1448–1455 CrossRef.
- P. Srihari, J. S. S. Reddy, S. S. Mandal, K. Satyanarayana and J. S. Yadav, Synthesis, 2008, 12, 1853–1860 CrossRef.
- S. Jang, A. Y. Kim, W. S. Seo and K. H. Park, Nanoscale Res. Lett., 2015, 10, 2–8 CrossRef PubMed.
- T. Wang, R.-d. Ma, L. Liu and Z.-p. Zhan, Green Chem., 2010, 12, 1576–1579 RSC.
- T. Subramanian and K. Pitchumani, Catal. Sci. Technol., 2012, 2, 296–300 RSC.
- J. R. C. Antonino, A. L. Prez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 5658–5661 CrossRef PubMed.
- J. S. Yadav, B. V. S. Reddy, G. G. K. S. NarayanaKumar and K. V. R. Rao, Chem. Lett., 2007, 36, 942–943 CrossRef.
- S. Gujarathi, H. P. Hendrickson and G. Zheng, Tetrahedron Lett., 2013, 54, 3550–3553 CrossRef.
- O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923 CrossRef PubMed.
- S. Saha and C. Schneider, Chem.–Eur. J., 2015, 21, 2348–2352 CrossRef PubMed.
- S. Saha, S. K. Alamsetti and C. Schneider, Chem. Commun., 2015, 51, 1461–1464 RSC.
- S. Saha and C. Schneider, Org. Lett., 2015, 17, 648–651 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |