
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring weak intermolecular interactions in thiocyanate-bonded Zn(II) and Cd(II) complexes with methylimidazole: crystal structures, Hirshfeld surface analysis and luminescence properties†
Alejandro Di Santoa,
Hiram Pérez*b,
Gustavo A. Echeverría‡
c,
Oscar E. Piro‡c,
Rodrigo A. Iglesias‡d,
Raúl E. Carbonio‡d,
Aida Ben Altabef‡ a and
Diego M. Gil‡*a
a and
Diego M. Gil‡*a
aINQUINOA (CONICET-UNT). Instituto de Química Física. Facultad de Bioquímica, Química y Farmacia, Universidad Nacional de Tucumán, San Lorenzo 456, T4000CAN, San Miguel de Tucumán, Argentina. E-mail: dmgil@fbqf.unt.edu.ar
bDepartamento de Química General e Inorgánica, Facultad de Química, Universidad de La Habana, Habana 10400, Cuba. E-mail: alinaca@infomed.sld.cu
cDepartamento de Física, Facultad de Ciencias Exactas, Universidad Nacional de La Plata e IFLP (CONICET, CCT-La Plata), C.C. 1900, La Plata, Argentina
dINFIQC (CONICET-UNC), Departamento de Fisicoquímica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, X5000HUA, Córdoba, Argentina
First published on 29th June 2018
Abstract
Four new thiocyanate-Zn(II) and -Cd(II) complexes with 1-methylimidazole (1-MeIm) and 2-methylimidazole (2-MeIm), namely, Zn(1-MeIm)2(SCN)2 (1), Zn(2-MeIm)2(SCN)2 (2), Cd(1-MeIm)4(SCN)2 (3) and polymeric [Cd(2-MeIm)2(SCN)2]n (4), have been synthesized and characterized by IR, Raman and UV-Vis spectroscopy. The thermal behavior for all complexes was evaluated by thermo-gravimetric analysis and differential thermal analysis. The crystal structures of complexes 1–4 were solved by single-crystal X-ray diffraction methods. A study of intermolecular interactions in the solid state compounds revealed that molecules are linked by weak N–H⋯S and C–H⋯S hydrogen bonds and also by C–H⋯π interaction in the case of structures 2–4, which are responsible for the formation and stability of the molecular assemblies. Hirshfeld surfaces and 2D-fingerprint plots allowed us to visualize the intermolecular contacts and their relative contributions to the total surface for each compound. A comparative analysis against similar halogen-bonded complexes was carried out to investigate the tendency of inter-molecular interactions to form contacts in crystals by using the enrichment ratio descriptor. The emission spectra of the free imidazole derivatives and their Zn(II) and Cd(II) complexes were recorded in acetonitrile solutions. The emissions observed in the spectra of complexes were ascribed to the intra-ligand transitions and ligand-to-metal charge transfer and we have observed an interesting correlation between the fluorescence intensities and C–H⋯π interactions.
1. Introduction
The design and synthesis of metal complexes with structural diversities such as metal–organic frameworks are topics of great interest for the possible applications in molecular separation, sensing, luminescent materials and nonlinear optical devices, and also in the production of micro-porous materials.1–5Pseudohalides, especially thiocyanate, azide and dicyanamide anions, have attracted attention due to the variety of their bonding modes, including various bridging modes (see Scheme 1 for the thiocyanate ligand).6 The observed coordination modes of the SCN− anion are largely influenced by the nature of the co-ligands. The synthesis and structural characterization of thiocyanate-metal(II) complexes using diverse organic ancillaries as co-ligands have been recently reported.7 Among them there are a few crystalline complexes with metals of the group 12 and imidazole ligands, which present exclusively conventional hydrogen bonds in their respective supra-molecular assemblies.8,9 However, weak intermolecular hydrogen bonds and C–H⋯π interactions in our thiocyanate-linked complexes provide further stability to the crystal structures. In the complexes under study, we have found an interesting correlation between fluorescence and the C–H⋯π interactions, indicating that longer values of H⋯Cg (inter-centroid) distances are not significant but in complexes with lower distances, higher fluorescence intensity are obtained. To the best of our knowledge, C–H⋯π interactions are not common in the referred complexes, and that is why this work is of particular interest. The role and importance of C–H⋯π interactions has been extensively observed in diverse organic/bioorganic systems,10 receiving a special attention in supra-molecular chemistry and crystal engineering.11 On the other hand, it is well-known that C–H⋯π interaction is a type of hydrogen bond characterized by a shortening of the C–H bond and a corresponding blue shift of the C–H stretching frequency, when compared to conventional hydrogen bonding.12 In the studied complexes, the C–H⋯π contacts are afforded through the methyl and aromatic C–H groups, which are capable to interact with the imidazole ring leading a shift of the C–H stretching mode corresponding to the imidazole ring observed in the IR spectra with respect to the free ligand. The steric and electronic effects of the ligands (methylimidazole) are found to be capable to modify the chain structure. With mono-dentate ligands having the capacity to form additional hydrogen bonding interactions, the chain structure has been extended to higher dimensional hydrogen bonded architectures.
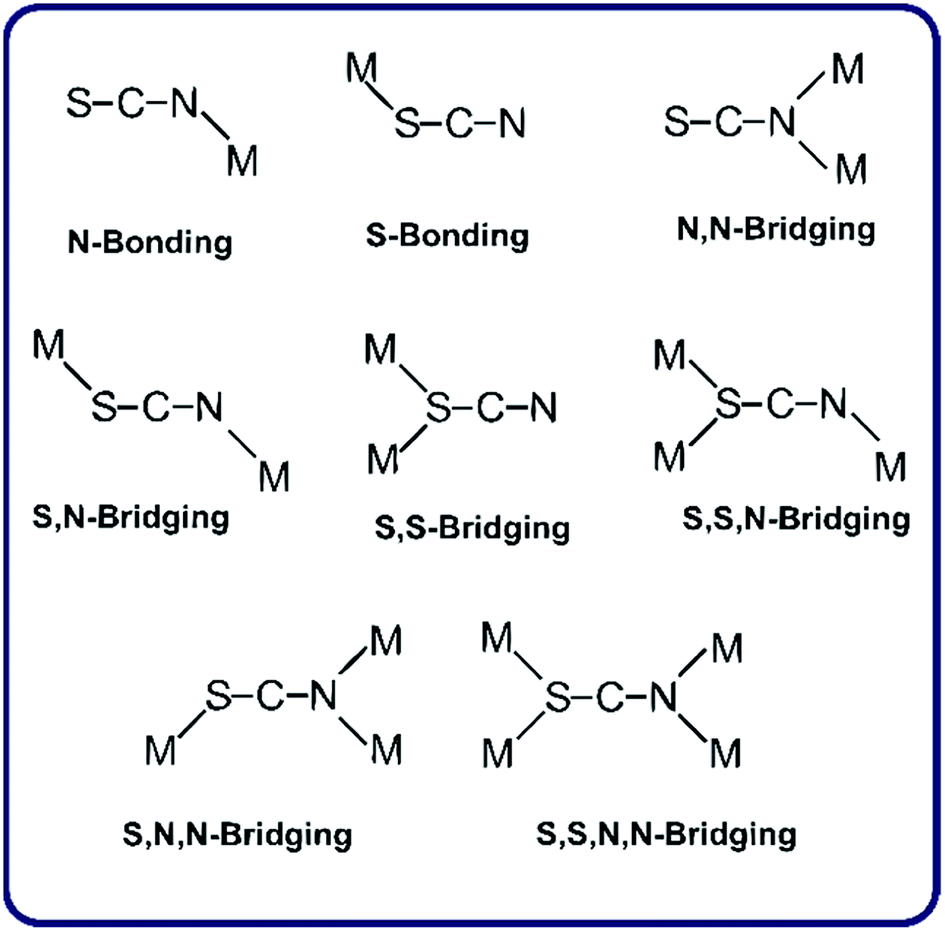 | ||
| Scheme 1 Possible coordination modes of thiocyanate. | ||
As a part of our continued interest in Zn(II) and Cd(II) complexes containing imidazole ligands, we report here the synthesis, spectroscopic studies and crystal structures of two Zn(II) and two Cd(II) thiocyanate-bonded complexes with methylimidazole ligands. A detailed analysis of the intermolecular interactions in these complexes was performed by the Hirshfeld surface analysis. Hirshfeld surface (HS) based tools, such as dnorm and shape index surface properties13,14, have been used for exploration of the packing modes and visualization of interactions. Full 2D-fingerprint plots have been derived from HSs to calculate the relative percentage of each type of interaction.15,16 The intermolecular contacts were further assessed by using the enrichment ratio parameter,17 which is a relatively new descriptor, based on Hirshfeld surface analysis, to quantify the likelihood of occurrence of intermolecular contacts in crystals. This magnitude has been recently computed for cadmium complexes.18
2. Experimental
Commercially available 1-methylimidazole, 2-methylimidazole, ZnSO4·7H2O, KSCN and CdBr2 were used without further purification.2.1. Synthesis of metal complexes 1–4
2.2. Instrumentation
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a CarloErba elemental analyzer. The Infrared (IR) absorption spectra were measured at room temperature in KBr pellets, with a resolution of 2 cm−1 on a FTIR Perkin Elmer GX1 in the frequency range 4000–400 cm−1. The Raman spectra of the solids were recorded in a 3500–50 cm−1 interval with a Thermo scientific DXR Raman microscope. The Raman dispersion data were collected using a diode-pump, solid state laser of 780 nm (at 5 cm−1 spectral resolution), a con-focal aperture of 25 μm pinhole and a 10× objective lens. Thermo-gravimetric (TG) and differential thermal analysis (DTA) were performed with a Shimadzu DTG-50 thermo-balance in the 25–800 °C range at a heating rate of 5° min−1 under air flow. The electronic absorption spectra of 1-MeIm, 2-MeIm and 1–4 complexes were recorded on a Beckman/DU 7500 spectrophotometer in the 200–800 nm spectral range on samples dissolved in acetonitrile at 1 × 10−3 M concentration. Fluorescence spectra of the ligands and complexes dissolved in acetonitrile at a concentration of 1 × 10−4 M were recorded with a Deltaflex Horiba spectrofluorometer using a diode laser (λ = 267 nm) for the excitation of the samples.2.3. X-ray structure determination
The measurements were performed on an Oxford Xcalibur, Eos, Gemini CCD diffractometer with graphite-monochromated MoKα (λ = 0.7107 Å) radiation. X-ray diffraction intensities were collected (ω scans with θ and κ-offsets), integrated and scaled with CrysAlisPro suite of programs.19 The unit cell parameters were obtained by least-squares refinement (based on the angular settings for all collected reflections with intensities larger than seven times the standard deviation of measurement errors) using CrysAlisPro. Data were corrected empirically for absorption employing the multi-scan method implemented in CrysAlisPro. The structures were solved by intrinsic phasing with SHELXT of the SHELX suit of programs.20 Molecular models were refined by full-matrix least-squares procedure with SHELXL of the same package. The hydrogen atoms were positioned on stereo-chemical bases and refined with the riding model. The methyl H-atoms were refined as rigid groups allowed to rotate around their corresponding C–C or N–C bond such as to maximize the sum of the observed residual electron density at their calculated positions. Crystal data, data collection procedure, and refinement results for all four complexes are summarized in Table 1.| Complex 1 | Complex 2 | Complex 3 | Complex 4 | |
|---|---|---|---|---|
| Empirical formula | C10H12N6S2Zn | C10H12N6S2Zn | C18H24CdN10S2 | C10H12CdN6S2 |
| Formula weight | 345.75 | 345.75 | 556.99 | 392.78 |
| Temperature/K | 297(2) | 293(2) | 293(2) | 293(2) |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Orthorhombic |
| Space group | P21/c | Pnma | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Cmc21 |
| Unit cell dimensions | a = 11.9876(6) Å | a = 8.4322(3) Å | a = 8.1914(3) Å | a = 13.9249(4) Å |
| b = 11.6755(4) Å | b = 12.5613(5) Å | b = 8.7859(6) Å | b = 14.7811(4) Å | |
| c = 11.8909(5) Å | c = 13.9647(5) Å | c = 9.4685(6) Å | c = 6.9732(2) Å | |
| — | — | α = 90.962(6)° | — | |
| β = 109.447(5) | — | β = 112.629(5)° | — | |
| — | — | γ = 104.257(5)° | — | |
| Volume/Å3 | 1569.3(1) | 1479.13(10) | 604.85(7) | 1435.26(7) |
| Z | 4 | 4 | 1 | 4 |
| ρ calc./mg mm−3 | 1.463 | 1.553 | 1.529 | 1.818 |
| μ/mm−1 | 1.826 | 1.937 | 1.101 | 1.807 |
| F(000) | 704 | 704 | 282 | 776 |
| Crystal size/mm3 | 0.282 × 0.134 × 0.098 | 0.209 × 0.143 × 0.048 | 0.441 × 0.337 × 0.138 | 0.377 × 0.260 × 0.211 |
| θ-range for data collection (°) | 3.43 to 28.82 | 2.92 to 29.04 | 3.16 to 28.63 | 2.93 to 28.95 |
| Index ranges | −16 ≤ h ≤ 9 | −7 ≤ h ≤ 11 | −9 ≤ h ≤ 10 | −17 ≤ h ≤ 15 |
| −15 ≤ k ≤ 13 | −16 ≤ k ≤ 15 | −11 ≤ k ≤ 7 | −13 ≤ k ≤ 19 | |
| −15 ≤ l ≤ 14 | −18 ≤ l ≤ 17 | −12 ≤ l ≤ 11 | −9 ≤ l ≤ 6 | |
| Reflections collected | 7733 | 4502 | 4285 | 2057 |
| Independent reflections | 3397 [R(int) = 0.0272] | 1742 [R(int) = 0.045] | 2584 [R(int) = 0.0251] | 1166 [R(int) = 0.0218] |
| Data/restraints/parameters | 3397/0/198 | 1742/0/98 | 2584/0/144 | 1166/1/99 |
| Goodness-of-fit on F2 | 1.006 | 1.063 | 1.063 | 1.032 |
| Final R indexes [I > 2σ(I)] | R1 = 0.0393, | R1 = 0.0386, | R1 = 0.0256, | R1 = 0.0199, |
| wR2 = 0.0803 | wR2 = 0.0727 | wR2 = 0.0632 | wR2 = 0.0444 | |
| Largest diff. peak/hole/e Å−3 | 0.326/−0.277 | 0.302/−0.319 | 0.248/−0.632 | 0.545/−0.367 |
2.4. Hirshfeld surface calculations
Hirshfeld surfaces and the associated 2D-fingerprint plots24–27 are generated using CrystalExplorer3.0 software.28 The normalized contact distance (dnorm) enables the identification of the regions of particular importance to the intermolecular interactions, being dnorm a symmetric function of distances to the surface from nuclei inside and outside the Hirshfeld surface (di and de, respectively), relative to their respective van der Waals radii. Hirshfeld surfaces for the title structures were also mapped with the shape index property. The 2D-fingerprint plot provides decomposition of Hirshfeld surfaces into relative contribution of different intermolecular interactions present in the crystal structures.29 A color scale of red (shorter than vdW separation)-white (equal to vdW separation)-blue (longer than vdW separation) is used to visualize the intermolecular contacts in the dnorm plot. The 3D dnorm surfaces are mapped over a fixed color scale of −0.085 (red) to 1.090 (blue), and shape index mapped in the color range of −1.0 a.u. (concave) to 1.0 a.u. (convex) Å.3. Results and discussion
3.1. Description of the crystal structures
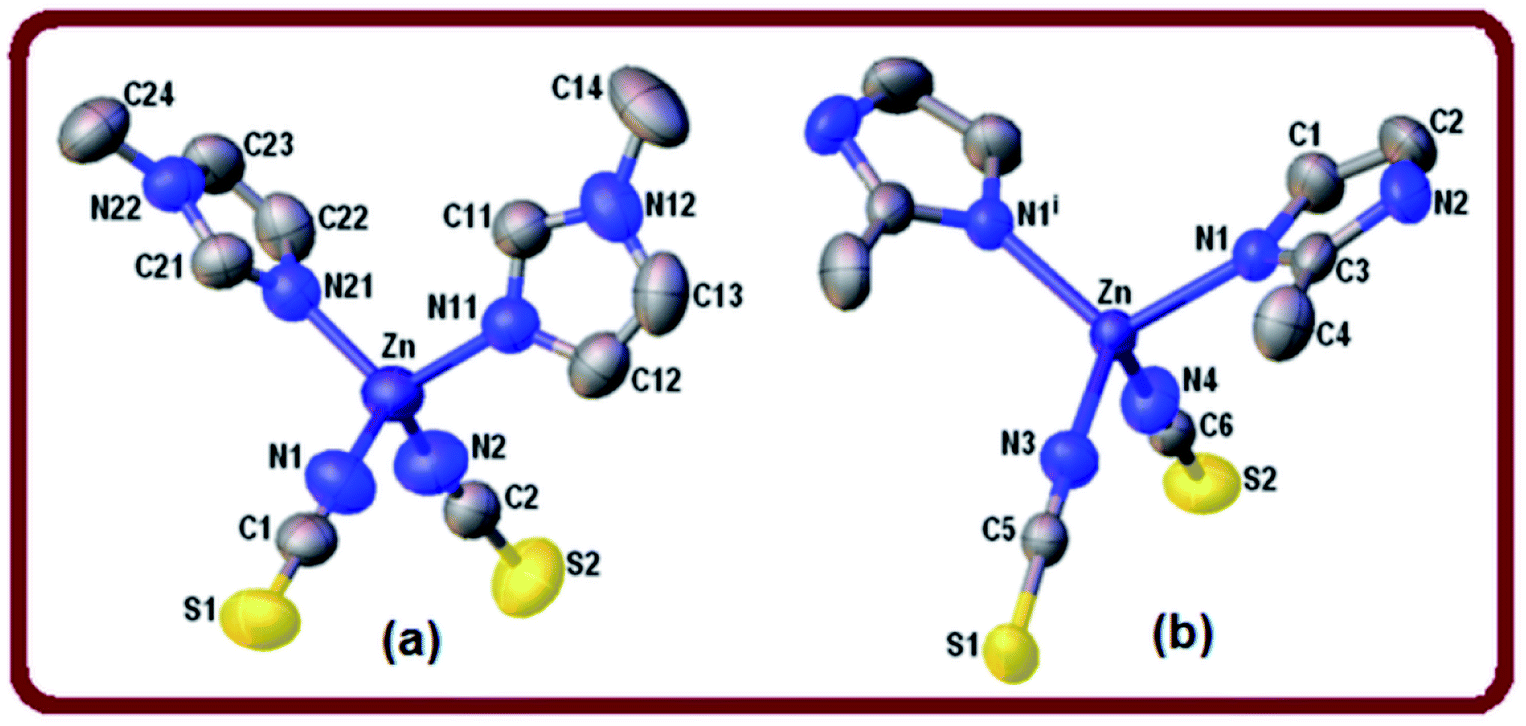 | ||
| Fig. 1 View of the zinc complexes 1 (a) and 2 (b) showing the labeling of the atoms and their displacement ellipsoids at the 50% probability level. H-atoms are omitted for clarity. Symmetry operation for 2 (i): x, 1/2 − y, z. | ||
| Compound 1 | Compound 2 | Compound 3 | Compound 4 | ||||
|---|---|---|---|---|---|---|---|
| a Symmetry codes: x, −y + 1/2, z for 2.b Symmetry codes: −x + 1, −y, −z + 1 for 3.c Symmetry codes: −x + 1, y, z for 4. | |||||||
| Zn–N1 | 1.953(3) | Zn–N1 | 1.992(2) | Cd–N1 | 2.353(2) | Cd–S1 | 2.605(2) |
| Zn–N2 | 1.943(3) | Zn–N3 | 1.954(4) | Cd–N11 | 2.358(2) | Cd–N1 | 2.377(4) |
| Zn–N11 | 1.988(3) | Zn–N4 | 1.988(3) | Cd–N21 | 2.334(2) | Cd–N2 | 2.348(5) |
| Cd–N3 | 2.241(3) | ||||||
| N11–Zn–N21 | 107.5(1) | N1–Zn–N1a | 110.8(1) | N1–Cd–N11 | 88.1(1) | N1–Cd–N2 | 166.3(2) |
| N1–Zn–N11 | 108.2(1) | N1–Zn–N3 | 113.8(1) | N1–Cd–N21 | 91.4(1) | N3–Cd–N3c | 146.2(2) |
| N1–Zn–N21 | 108.1(1) | N1–Zn–N4 | 104.3(1) | N1–Cd–N1b | 180.0(1) | N3–Cd–S1 | 106.8(1) |
| Zn–N1–C1 | 173.6(3) | Zn–N3–C5 | 174.0(4) | Cd–N1–C1 | 144.7(2) | Cd–N1–C1 | 157.0(5) |
| Zn–N2–C2 | 163.4(3) | Zn–N4–C6 | 177.3(4) | Cd–N2–C2 | 156.3(5) | ||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D⋯A) | ∠(D–H⋯A) |
|---|---|---|---|---|
| a Symmetry operations: 1 − x, 1 − y, 1 − z for 1.b Symmetry operations: 3/2 − x, 1 − y, 1/2 + z for 2.c Symmetry operations: 1 − x, 1 − y, 1 − z for 3.d Symmetry operations: x, y, −1 + z for 3.e Symmetry operations: 1/2 − x, 1/2 − y, 1/2 + z for 4.f Symmetry operations: 1 − x,1 − y, −1/2 + z for 4. | ||||
| Compound 1 | ||||
| C11–H11⋯S1a | 0.93(4) | 2.93(3) | 3.749(4) | 148 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Compound 2 | ||||
| N2–H2A⋯S1b | 0.860(3) | 2.676(1) | 3.469(3) | 154 |
|
||||
| Compound 3 | ||||
| C24–H24C⋯S1c | 0.960(3) | 2.823(1) | 3.740(3) | 160 |
| C13–H13⋯S1d | 0.960(3) | 3.004(1) | 3.878(2) | 157 |
|
||||
| Compound 4 | ||||
| N4–H4A⋯S2e | 0.860(3) | 2.583(1) | 3.404(3) | 160 |
| C5–H5⋯S1f | 0.930(4) | 2.936(1) | 3.731(4) | 144 |
Parameter τ4 calculated for complex 2 is equal to 0.96 which confirms almost ideal tetrahedral geometry around the zinc ion. The shortest metal–metal separation is 6.314(1) Å. The crystal packing of complex 2 is directed by weak32 intermolecular N–H⋯S hydrogen bonds [d(H⋯S) = 2.676(1) Å, ∠(N–H⋯S) = 154°] (Table 3) forming R34 (26) ring motifs (see Fig. S2, ESI†).
Two different π-stacking contacts also contribute to the crystal stability of 2. On one hand, a weak C–H⋯π interaction involves the methyl C4–H4C group donor and the imidazole ring [Cg1 centroid, H⋯Cg1 = 3.00 Å] (Table 4). By the other hand, an unusual lone-pair (l.p.)⋯π interaction involves the thiocyanate sulfur S2 lone pair(s) and the aromatic ring. This interesting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S(l.p.)⋯π interaction is characterized by a short S2⋯Cg1 distance of 3.831(1) Å, and the angular distribution [deviation of the angle α (α is the angle CS⋯Cg) from 120°] of 18° is in the range of the mean value of 30.6° reported for CO(l.p.)⋯π interactions.33,34 In addition, these contacts are significant through the dihedral angle ω = 60.6° between the N4–C6S2 moiety and imidazole ring mean planes indicating that the thiocyanate group takes an angular approach towards the ring (25 ≤ ω ≤ 64°).33
S(l.p.)⋯π interaction is characterized by a short S2⋯Cg1 distance of 3.831(1) Å, and the angular distribution [deviation of the angle α (α is the angle CS⋯Cg) from 120°] of 18° is in the range of the mean value of 30.6° reported for CO(l.p.)⋯π interactions.33,34 In addition, these contacts are significant through the dihedral angle ω = 60.6° between the N4–C6S2 moiety and imidazole ring mean planes indicating that the thiocyanate group takes an angular approach towards the ring (25 ≤ ω ≤ 64°).33
| C–H⋯Cg(j)b | Symmetry | H⋯Cg | H-perpc | γd | C–H⋯Cg | H⋯Ce |
|---|---|---|---|---|---|---|
| a (H⋯Cg < 3.0 Å, γ < 30.0°).b Centroid of aromatic rings.c Perpendicular distance of H to ring plane J.d Angle between the Cg–H vector and ring J normal.e Distance between H-atom and the nearest carbon atom in the aromatic ring. | ||||||
| Compound 2 | ||||||
| C4–H4C⋯Cg(1) | 1 − x, 1 − y, 1 − z | 3.00 | 2.82 | 19.79 | 146 | 2.832(3) |
|
||||||
| Compound 3 | ||||||
| C14–H14B⋯Cg(1) | −x, −y, −z | 2.86 | 2.79 | 13.26 | 135 | 2.992(2) |
| C22–H22⋯Cg(1) | 1 − x, 1 − y, 1 − z | 2.58 | 2.58 | 3.99 | 170 | 2.852(2) |
| C24–H24B⋯Cg(2) | 2 − x, 1 − y, 1 − z | 2.86 | 2.78 | 13.60 | 123 | 2.815(2) |
|
||||||
| Compound 4 | ||||||
| C6–H6C⋯Cg(1) | x, 1 − y, 1/2 + z | 2.91 | 2.87 | 9.32 | 113 | 3.052(5) |
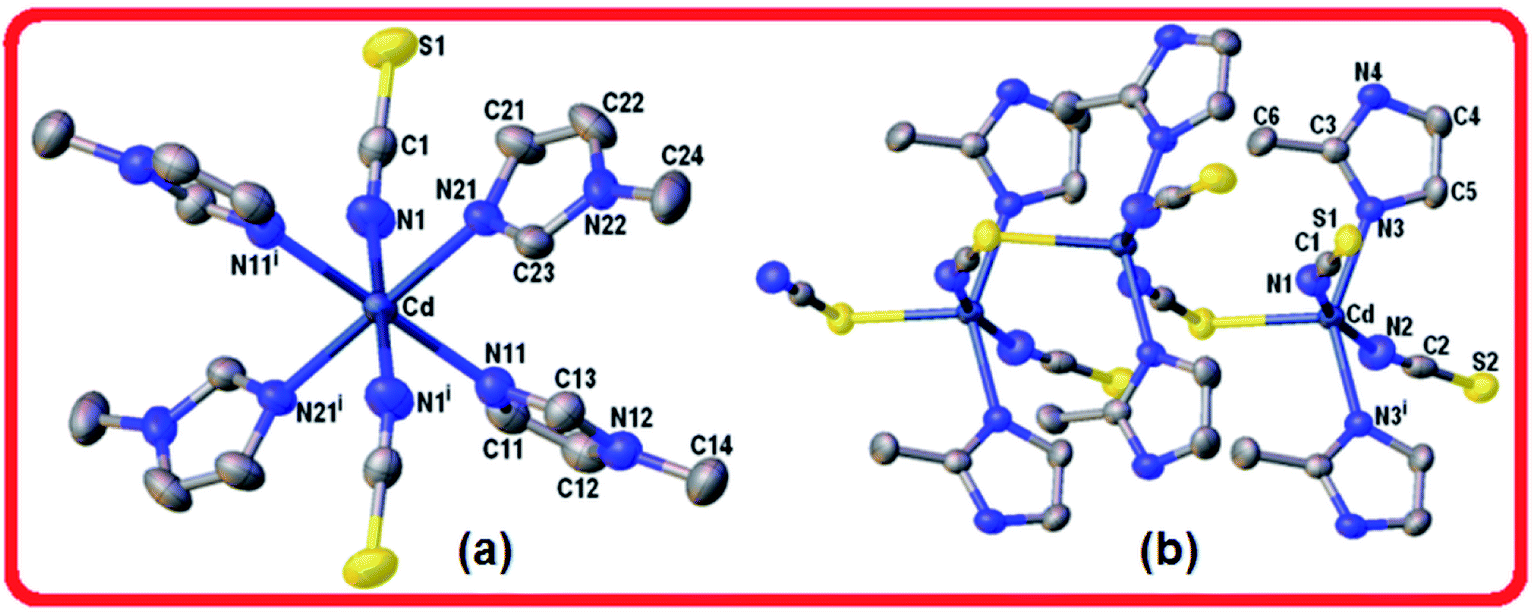 | ||
| Fig. 2 (a) View of the cadmium complex 3, and (b) view of the zig-zag chain for the polymeric complex 4 showing one μ-SCN anion linking two CdL2 units (L = 2-methylimidazole). Displacement ellipsoids are shown at the 50% probability level. H-atoms are omitted for clarity. Symmetry operations: for 3 (i) 1 − x, −y, 1 − z; for 4 (i) 1 − x, y, z. | ||
In the CdN4S coordination core the metal departs from the mean plane of nitrogen atoms at the basis in 0.466(3) Å towards the apical sulfur atom. The coordination bonding gives rise to a zig-zag chain arrangement of Cd(2-MeIm)2(SCN)2 monomers that extends along the crystal c-axis (Fig. 2b). Alternatively, the chain can be described as an helix wound around the crystallographic two-fold screw axis with a pitch equal to the unit cell length c and having two symmetry related Cd(2-MeIm)2(SCN)2 monomers per turn. The bent and zigzagged conformations in this complex explain the observed very short inter-metallic distance of 5.673(1) Å in the polymeric chain when compared with those for similar compounds.8 Hydrogen bond interactions in coordination polymers play an important role in the crystal packing.37 Neighboring chains in the lattice are linked to each other through a weak N–H⋯S [d(H⋯S) = 2.583(1) Å, ∠(N–H⋯S) = 160°] and longer C–H⋯S hydrogen bonds [d(H⋯S) = 2.936(1) Å, ∠(N–H⋯S) = 144°] leading to the formation of a 3D network (Table 3, Fig. S4, ESI†). The crystal packing appears to be also controlled by weak C–H⋯π interactions involving the methyl C6–H6C donor (Table 4).
3.2. Hirshfeld surface analysis
Hirshfeld surface analysis have been carried out to get a better comprehension on the nature of packing motifs and the contribution of the main intermolecular interactions directing the molecular architecture in crystalline complexes 1–4. Fig. 3 shows Hirshfeld surfaces (HSs) mapped over the dnorm property in two orientations (columns 1 and 2), and the corresponding full two-dimensional fingerprint plots (FPs) are displayed in column 3. The surfaces are shown as transparent to allow visualization of the molecules. Contacts with distances equal to the sum of the van der Waals radii are represented as white regions and the contacts with distances shorter than and longer than van der Waals radii are shown as red and blue colors, respectively. The relative contributions to the Hirshfeld surface area due to the main intermolecular interactions are shown as histogram in Fig. 4.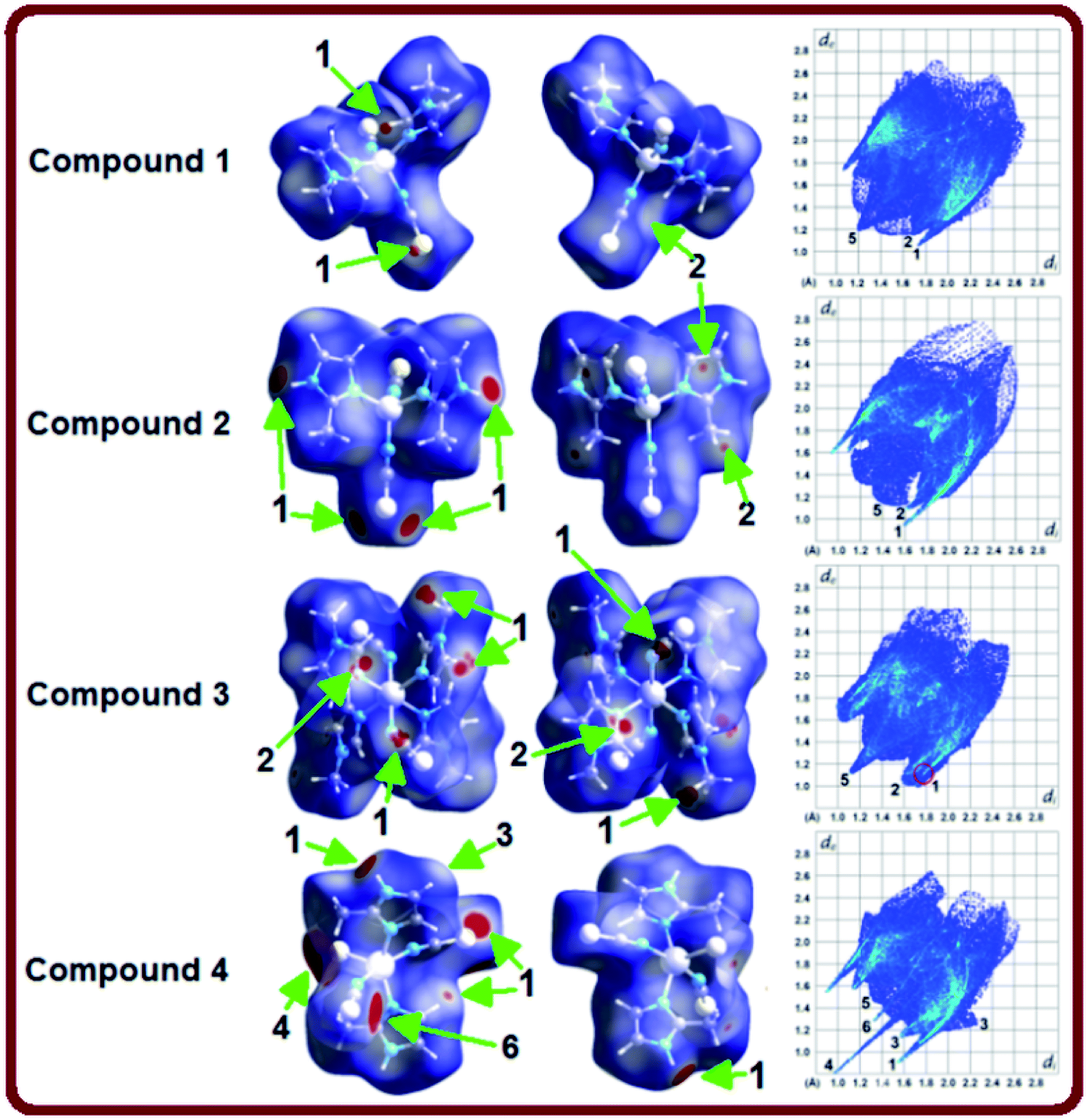 | ||
| Fig. 3 Views of the Hirshfeld surfaces of compounds 1–4 (columns 1–2) mapped with dnorm in two orientations: front view and back view (180° rotated around the vertical axis of the plot), and full 2D-fingerprint plots (column 3) derived from the surfaces. Close contacts are labeled as follows: (1) S⋯H, (2) C⋯H, (3) N⋯H, (4) C⋯S, (5) H⋯H and (6) Cd⋯S. | ||
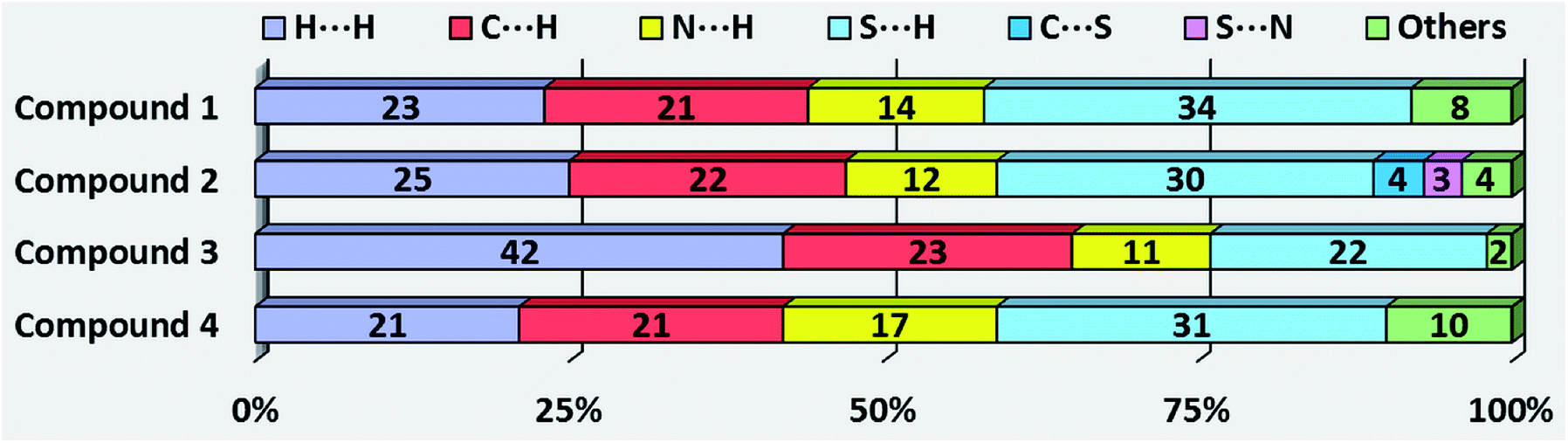 | ||
| Fig. 4 Relative contributions of the main intermolecular contacts to the Hirshfeld surface area in compounds 1–4. | ||
The large red regions labeled 1 in Fig. 3 represent H⋯S/S⋯H contacts, which are relevant in the dnorm maps for all the four compounds. In the zinc complexes (1 and 2), these contacts are attributed to C11–H11⋯S1 and N2–H2A⋯S1 hydrogen bonds (Fig. 3), respectively, which can also be seen in the FPs as a pair of symmetrical spikes at (de + di) ≅ 2.8 Å for the former, and (de + di) ≅ 2.6 Å for the later interaction, in agreement with the expected higher strength for the N2–H2A⋯S1 hydrogen bond. The H⋯S/S⋯H contacts are dominant for complexes 1 and 2, with highest contributions of 33.7% and 30.1%, respectively, of the total Hirshfeld surface area (see Fig. 4).
Four small red spots labeled 2 for compounds 1 and 2 (Fig. 3, column 2) represent H⋯C/C⋯H contacts with large and similar area fractions of 20.7 and 22.2%, respectively. However, unlike of 1, where no significant C–H⋯π interactions are observed, the H⋯C/C⋯H contacts in 2 appear in a characteristic way for C–H⋯π interactions, i.e. in the form of pronounced “wings” on the sides of the FP with the shortest (di + de) ≅ 2.7–2.8 Å, and comprising a 22.2% of the total Hirshfeld surface area.38 Two upper spots are associated to the acceptor region of the FP (lower right) corresponding to a 12.6% contribution from C⋯H contacts (C inside the surface), while the other two spots are related to the donor region in the FP (upper left) with a lower contribution of 9.6% from H⋯C contacts (H inside the surface). A view of shape index surface (Fig. 5a) confirms the existence of point-to-face C–H⋯π interactions (Table 4) showing a large red depression above the electron system and a blue region surrounding the C–H donor, both the regions highlighted by black circles. Finally, the H⋯H interactions labeled 5 in the middle of scattered points in FP, are showed as two symmetrical broad regions with minimum (de + di) contact distance around 2.6 Å (longer than the sum of van der Waals radii), comprising a 24.5% contribution to the Hirshfeld surface area.
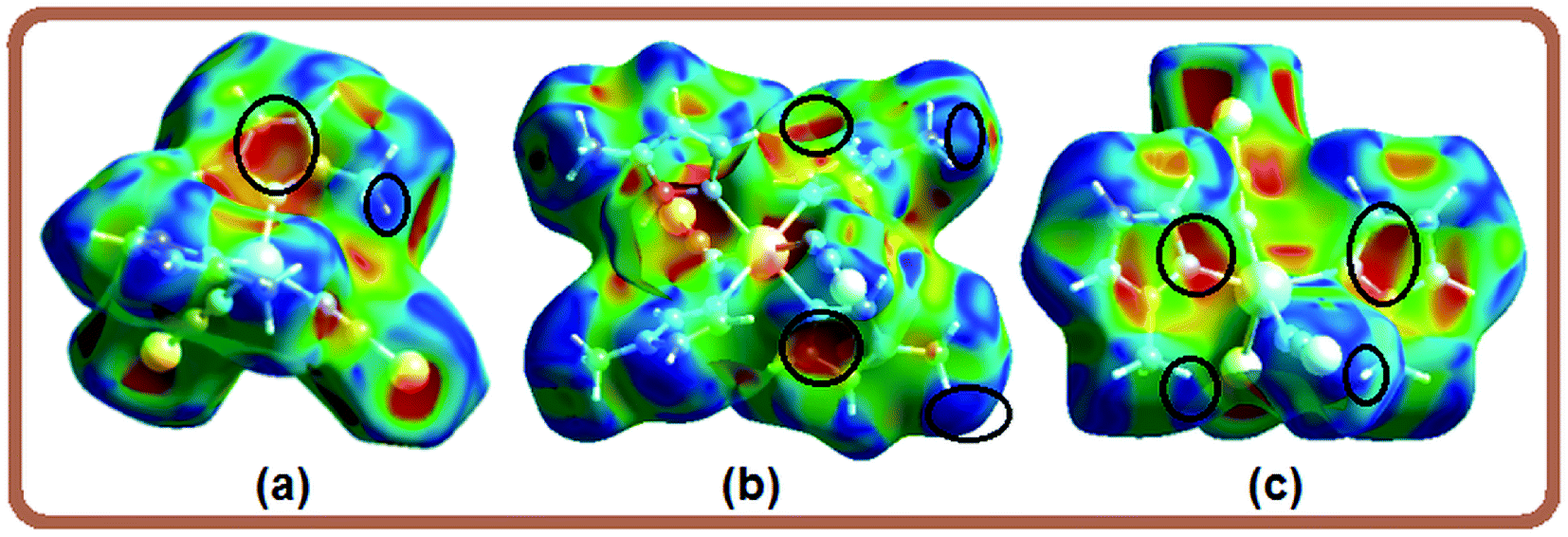 | ||
| Fig. 5 Hirshfeld surfaces mapped over shape index for compounds 2–4. | ||
In compound 3, the tiny red regions labeled 1 in the dnorm surfaces (Fig. 3) are attributed to C24–H24C⋯S1 hydrogen bonds (Fig. 2a), and represented as a pair of short spikes highlighted in red at (de + di) ≅ 2.6 Å in FP, with a high 21.6% contribution. Like in structure 2, the small red spots labeled 2 indicate weak H⋯C/C⋯H contacts corresponding to point-to-face C–H⋯π interactions (Table 4), which also appear as two broad spikes at (de + di) ≅ 2.7 Å in the FP with 22.8% contribution to the Hirshfeld surface area, and in shape index surface (Fig. 5b). The H⋯H contacts (labeled 5) are visible showing a single broad peak in the middle region of FP, and represent the dominant interactions (41.9% contribution) in this Cd complex.
In compound 4, the H⋯S/S⋯H contacts labeled 1 in Fig. 3 are again dominant, appearing as two larger deep-red spots at the upper (around H4A atom) and right (around S2 atom) sides in the dnorm surface (left) attributed to strongest N4–H4A⋯S2 hydrogen bonds. The smaller red spot labeled 1 at right is associated to weaker C5–H5⋯S1 hydrogen bonds. The former interactions are also observed as a sharp spike at the acceptor region in FP with short (de + di) ≅ 2.5 Å and higher contribution of 22.4% to the total Hirshfeld surface, in comparison with a sharp spike at the donor region representing H5⋯S1 contacts, and minor contribution of 8.8%. The presence of point-to-face C–H⋯π interactions in the crystal assembly of this polymeric Cd complex is evident in shape index surface (Fig. 5c).
The white spot labeled 3 in the dnorm map show weak H⋯N/N⋯H contacts attributed to C4–H4⋯N2 hydrogen bonds, which are viewed as a pair of wings at (de + di) ≅ 2.8 Å in FP with notable contribution of 16.6% to the Hirshfeld surface area. The broad peak labeled 5 in FP is characteristic of H⋯H interactions providing a significant contribution of 21.2%. It is worthwhile to indicate that the large deep-red spots labeled 4 (at left) and 6 in dnorm surface of structure 4 represent inter-atomic C⋯S/S⋯C (4.3%) and Cd⋯S/S⋯Cd (1.4%) contacts, respectively, connecting two monomeric species (see Fig. S4, ESI†).
The EXY values for all the six complexes are listed in Table 5, while complete information is provided in Table S1 given as ESI.† The largest contributions to the Hirshfeld surfaces are from S⋯H/H⋯S, N⋯H/H⋯N and C⋯H/H⋯C contacts, and their ER values (1.11–1.54) are significantly higher than unity for compounds 1–4, showing high propensity to form C–H⋯S and C–H⋯N hydrogen bonds, as well as C–H⋯π interactions. The corresponding ER values (0.98–1.13) for related structures 5–7 are smaller than those for structures 1–4 indicating that the replacement of thiocyanate by halogen ligand in the three complexes decreases the propensity to form X⋯H (X = N, C) contacts, apart from S⋯H contacts which evidently disappear as expected.
| Interaction | SCN-bonded | Halogen-bonded | |||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| a EXY values for random contacts RXY lower than 0.6% (Table S1, ESI) were not calculated, as they are not meaningful. | |||||||
| H⋯H | 0.70 | 0.76 | 0.86 | 0.68 | 0.90 | 0.85 | 1.02 |
| C⋯H/H⋯C | 1.25 | 1.39 | 1.29 | 1.41 | 1.00 | 1.04 | 0.98 |
| N⋯H/H⋯N | 1.34 | 1.11 | 1.28 | 1.46 | 1.04 | 1.13 | 1.11 |
| S⋯H/H⋯S | 1.54 | 1.39 | 1.38 | 1.45 | — | — | — |
| O⋯H/H⋯O | — | — | — | — | — | 1.44 | — |
| Cl⋯H/H⋯Cl | — | — | — | — | 1.37 | — | — |
| Br⋯H/H⋯Br | — | — | — | — | — | 1.46 | 1.12 |
| C⋯C | 0.76 | 0.25 | 0.63 | 0.00 | 1.86 | 2.67 | 3.33 |
| C⋯S/S…C | — | 0.74 | — | 0.83 | — | — | — |
| N⋯S/S⋯N | 0.44 | 0.89 | — | — | — | — | — |
| Cl⋯C/C⋯Cl | — | — | — | — | 0.53 | — | — |
| C⋯N/N⋯C | 1.04 | — | — | — | — | — | 1.25 |
It is also observed similar EXY values from the three above-mentioned contacts in compounds 3 and 4, whereas the likelihood of N⋯H/H⋯N contacts (ENH = 1.11) is reduced in compound 2 related to those for compounds 3 (1.28) and 4 (1.46). This difference is presumably attributed to the existence of nitrogen atoms which are also involved in enriched N⋯S/S⋯N contacts for structure 2 as reflected by its relatively high ENS value of 0.89. On the other hand, the slightly high likelihood to form C⋯S contacts in compounds 2 (ECS = 0.74) and 4 (ECS = 0.83) is due to higher proportion of sulfur (around 19.2%) on the corresponding molecular surfaces, in comparison to 11.3% for structure.
ER values close to, but slightly less than unity, for the H⋯H contacts are in accordance with the highest proportion SH of hydrogen atoms (55.9–69.7%) at the molecular surfaces, indicating a significant contribution from dispersive forces in the crystal packing of most of structures. The likelihood to form H⋯H contacts in the polymeric Cd(II) complexes mediated by thiocyanate (4) and halogen (7) ions is lowest (EHH = 0.68) for the former and highest for compound 7 (EHH = 1.02), consistent with the smallest (CHH = 21.4) and longest (CHH = 43.5) contact surfaces, respectively, as showed in Table S1, ESI.†
The propensity of imidazole rings to form π⋯π stacking interactions is much more extensive in compounds 6 and 7 as reflected by the highly increased ECC values of 2.67 and 3.33, respectively, which are associated to C⋯C contacts according to the literature.19,38 These values explain the relatively low tendency observed for C⋯H contacts in the two Cd(II) halogen-complexes (ECH = 1.04 for 6, and 0.98 for 7) when compared to remaining structures (ECH = 1.29–1.41), due to both C⋯C and C⋯H contacts are presumably in competition.41 However, the ECC value (1.86) is greatest than unity in compound 5 showing notable likelihood to form C⋯C contacts, but remarkably lower than those above indicated for 6 and 7, despite the very similar ECH values we found in the three halogen-bonded complexes. According to crystal structure of 5, imidazole carbon atoms also weakly interact with chloride atoms [Cl1⋯C21 = 3.510(3), Cl2⋯C23 = 3.615(3) Å], and though we obtained a relative low propensity to form Cl⋯C contacts (EClC = 0.53), it may be enough to explain the reduced ECC value. The absence of significant π⋯π stacking for compounds 1–4 is reflected in ECC values lower than 0.63 (Table 5).
3.3. Vibrational results
The solid state IR and Raman spectra for all complexes (Fig. 6a and b, respectively) are consistent with their crystal structures. The assignment of the bands observed in the IR and Raman spectra are shown in the ESI.† The assignment of the bands referred to the thiocyanate moiety is shown in Table 6. The spectra of 1–4 complexes show strong intensity bands in the 3147–3111 cm−1 spectral range which are attributed to C–H stretching modes of the imidazole ring. These particular frequencies are slightly shifted because of the presence of intermolecular C–H⋯π interactions, as was discussed in the analysis of the crystal structures. The absorption bands of the νa(CH3) and νs(CH3) group in the complexes are observed in the frequency range 2984–2933 cm−1, and are blue-shifted as compared with the free ligand. The vibrations of C–N and C–C bonds of the imidazole ring generally appear coupled and can be observed in the 1671–1495 cm−1 range in the complexes spectra (see Tables S2 and S3, ESI†).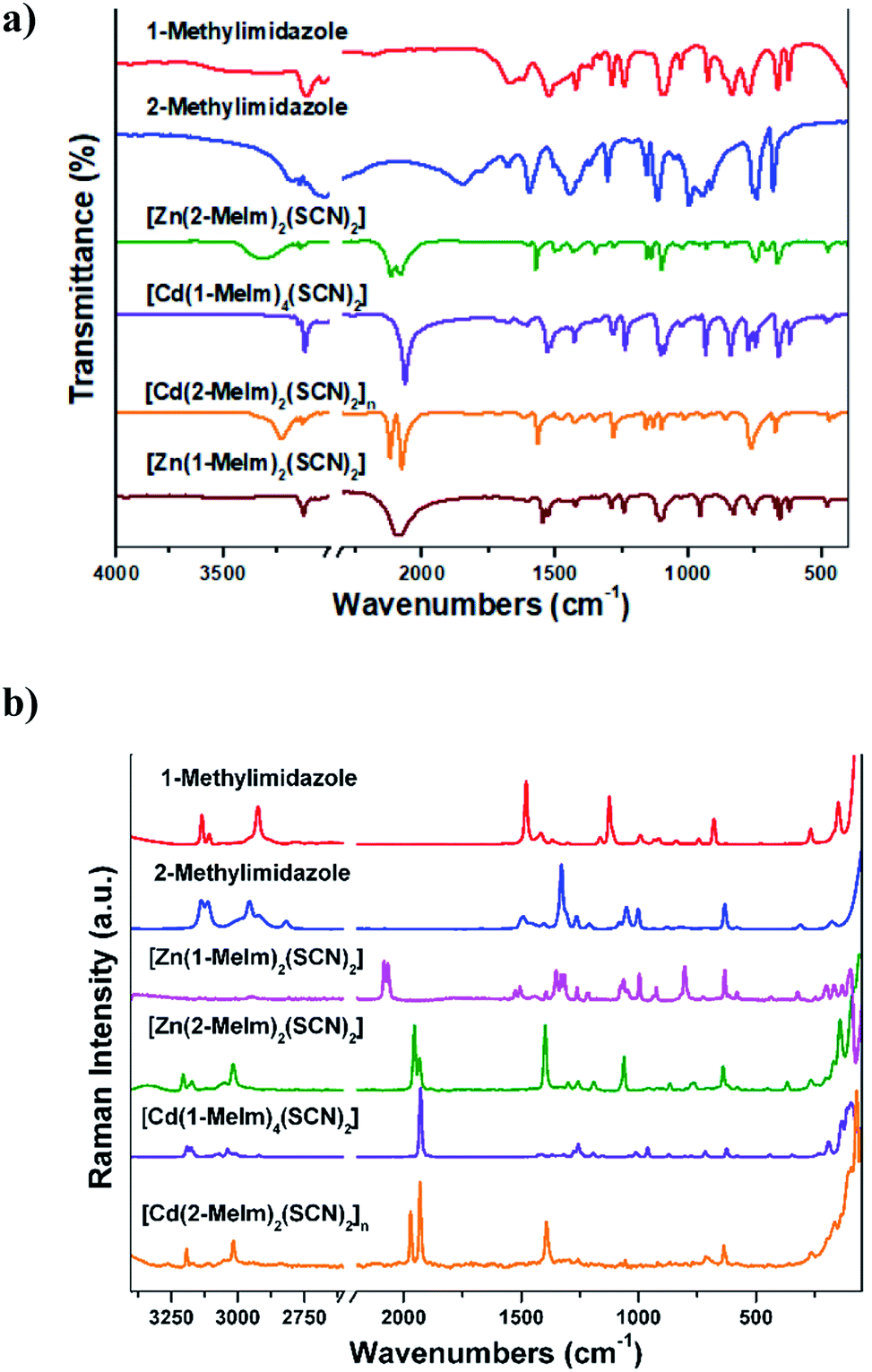 | ||
| Fig. 6 (a) IR and (b) Raman spectra of 1-methylimidazole, 2-methylimidazole and their metal complexes. | ||
| Complex | ν(CN) | ν(CS) | δ(NCS) |
|---|---|---|---|
| 1 | 2098 | 768 | 478 |
| 2083 | 753 | 473 | |
| 2 | 2100 | 757 | 478 |
| 2077 | 747 | 470 | |
| 3 | 2083 | 769 | 473 |
| 2066 | 764 | 464 | |
| 4 | 2119 | 770 | 469 |
| 2076 | 765 | 451 |
The IR spectra of 1–3 monomeric complexes show two absorption bands in the 2100–2066 cm−1 range, assignable to the CN stretching mode of the thiocyanate moiety. In the complexes, these vibrations are below 2100 cm−1 hence suggesting the presence of terminal SCN group bounded through the N atom to the metal center.42 The CN stretching mode appears split, the spectral signature of not equivalent NCS groups. Thus, the band corresponding to the CS stretching mode appears as doublets in the 769–747 cm−1 frequency range. The bands observed between 478 and 464 cm−1 in the IR spectra of complexes 1–3 are assigned to the δ(NCS) bending mode and correspond to a N-bonded NCS group.42 The IR spectra of the polymeric complex 4 show two intense absorption bands at 2119 and 2076 cm−1. The high value of the CN stretching mode for this complex is consistent with either S-bonding or N,S-bridging mode (bands expected near 2100 cm−1). The bands corresponding to the CS stretching mode appear as doublets at 770 and 765 cm−1, confirming that we are dealing with N-bonded (860–760 cm−1) rather than S-bonded (720–690 cm−1) thiocyanate complexes.42
3.4. Thermal studies of compounds 1–4
TGA and DTA were used to investigate the thermal stability and the decomposition process of complexes 1–4. The curves for the thermal decomposition of complexes 1–4 are shown in Fig. S5–S8, ESI.† The thermal behavior of all the complexes is similar. They decompose in two consecutive steps. The first one corresponds to the removal of 2 molecules of methylimidazole to form Zn(SCN)2 (exp. mass loss: 47.0%, theoretical: 47.5%), in agreement with the endothermic peaks located at 350 and 375 °C in the DTA curves of 1 and 2, respectively. The second step finishes at 750 °C, with a mass loss of 76.0% for 1 and 2 and it corresponds to the evolution of thiocyanate groups to form ZnO as final product. The observed mass loss is in very good agreement with the calculated ones (see Table S4, ESI†). Similar results were obtained for Cd(II) complexes.The endothermic peaks located at 100, 167, 94 and 144 °C in the DTA curves (without mass loss in TGA) are attributed to the melting point of complexes 1, 2, 3 and 4, respectively.
3.5. Electronic spectra and photoluminescence properties
The electronic spectra of the ligands and its complexes were collected in acetonitrile (Fig. S9, ESI†). The spectra of Zn(II) and Cd(II) complexes with d10 electronic configuration do not present any d–d electronic transitions. The intense band at 240 nm in the electronic spectra of the ligands and complexes are attributed to intra-ligand π → π* transitions. All complexes display bands in the region 380–550 nm attributable to charge transfer (CT) from the metal to ligand (M–L) transitions expected for these types of systems.43Photoluminescence properties of 1-MeIm, 2-MeIm and 1–4 complexes have been studied in acetonitrile solution (1 × 10−3 M). The emission spectra of 1-MeIm, 2-MeIm and their Zn(II) and Cd(II) complexes are shown in Fig. 7(a) and (b). The main chromosphere studied in these compounds is the aromatic imidazole ring. The free ligands display the main emission peaks at 404 and 327 nm (λexc = 270 nm) for 1-MeIm and at 327 and 355 nm for 2-MeIm. The emission bands of the free ligands are probably assigned to π → π* or n → π* transitions.44
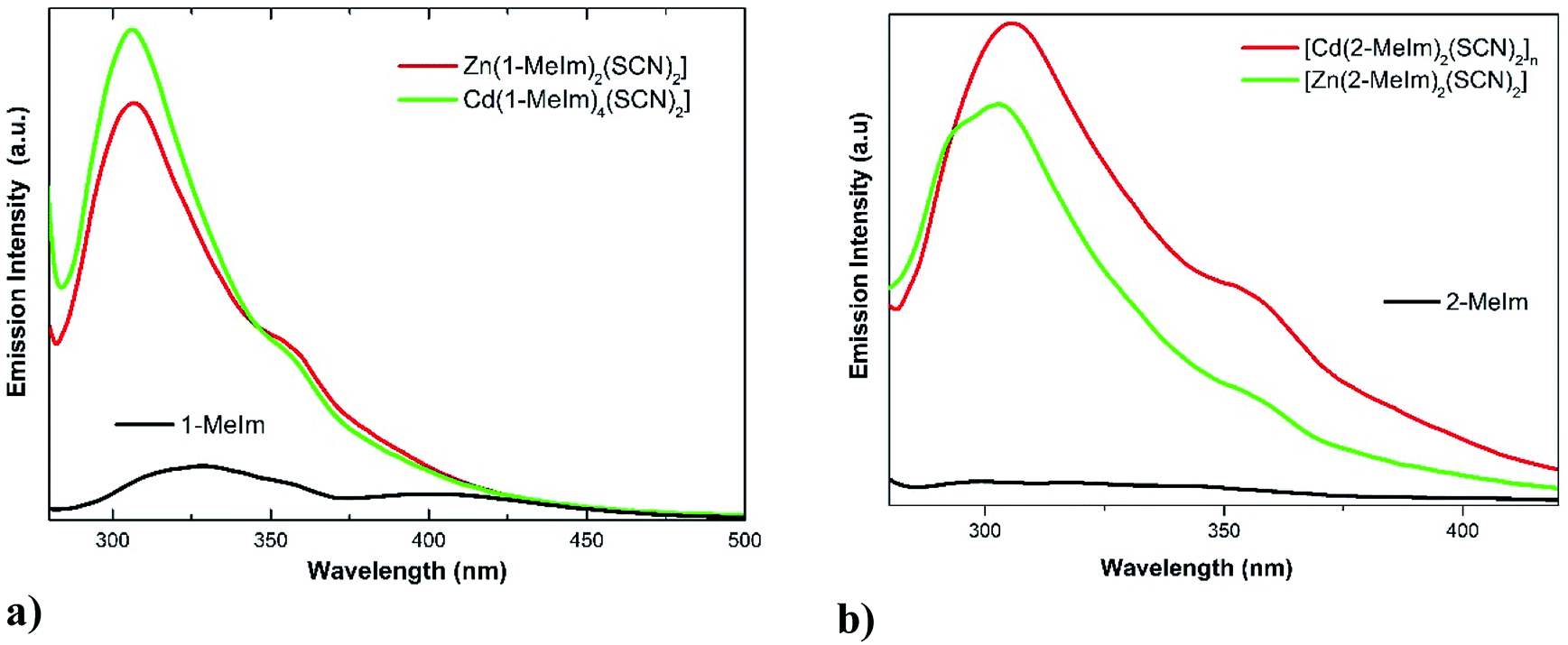 | ||
| Fig. 7 Emission spectra of (a) 1-MeIm free ligand and its Zn(II) and Cd(II) thiocyanate complexes; (b) 2-MeIm and its complexes. | ||
As shown in Fig. 7a, the emission spectra of complexes 1 and 3 display two similar bands located at 306 and 360 nm (λexc = 270 nm), which suggest that the emission properties are closely related. Interestingly, the positions of the first emission bands of the complexes are hypsochromically shifted compared with the free ligand. The observed blue shift could be explained by considering that the excited state originating from Zn(II) and Cd(II) complexes are mainly intra-ligand transitions modified by metal coordination. A similar behavior was observed for 2-MeIm and its Zn(II) and Cd(II) thiocyanate complexes. The enhancement of the photoluminescence observed in the complexes 1–4 with respect to the free ligands are attributed to coordination effect of ligands to Zn(II) and Cd(II) ions, which increases the rigidity of the ligand and reduce the loss of energy by non-radiative decay pathways.45 The emission band observed at around 360 nm in complexes 1–4 may be assigned to ligand to metal charge transfer (MLCT).46 Average luminescence lifetimes (τ) were measured for both ligands and complexes 1–4. For the emission band at around 360 nm in complexes 1–4, the average luminescence lifetime of 11.0 ns were measured at room temperature, much longer than those observed in free ligands. The decay of the luminescence intensity integrated over the whole spectral range of the ligands and complexes 1–4 are shown in Fig. S10, ESI.†
It has been reported that the π⋯π stacking occurring between methylimidazole rings may control the luminescence efficiency of metal complexes in both solid and solution state.2 In the series of compounds 1–4, the inter-centroid Cg⋯Cg distances (4.212–4.644 Å) are longer than the typical 3.3–3.8 Å for face-to-face π⋯π interactions,29 and the observed values have an unclear relationship with the fluorescence intensities (see Fig. 7). Instead, we have found an interesting correlation between fluorescence and the C–H⋯π interactions. In compound 1, the H24A⋯Cg value of 3.543 Å is out the limit (H⋯Cg < 3.0 Å) set by PLATON,15 and hence the C–H⋯π interaction is not significant. However, in structures 2–4 with significant C–H⋯π contacts (Table 4), we have found that with lower H⋯Cg distances (stronger contacts), higher fluorescence intensities are obtained, which reveal that at least in the three mentioned structures the existence of C–H⋯π interactions may be responsible for fluorescence. In comparison to structures 2 and 4 with large Cg⋯Cg distances of 4.396(1) and 4.644(1) Å, respectively, the higher luminescence intensity in 1 may be due to the presence of a stronger π⋯π contact (shorter Cg⋯Cg distance = 4.220(1) Å).47
4. Conclusions
Four new thiocyanate-Zn(II) and Cd(II) complexes with 1-methylimidazole (1-MeIm) and 2-methylimidazole (2-MeIm), namely, Zn(1-MeIm)2(SCN)2 (1), Zn(2-MeIm)2(SCN)2 (2), Cd(1-MeIm)4(SCN)2 (3) and [Cd(2-MeIm)2(SCN)2]n (4), have been synthesized and characterized by IR, Raman and UV-Vis spectroscopy and thermal analysis. The crystal structures of complexes 1–4 have been solved by single-crystal X-ray diffraction methods. The 3D Hirshfeld surface analysis and 2D fingerprint maps analysis have shown that weak intermolecular S⋯H hydrogen bonds and C–H⋯π stacking interactions play dominant roles in stabilizing the lattice of the three structures, revealing also different packing modes. The enrichment ratios of different intermolecular interactions in the thiocyanate-bonded complexes when compared for similar halogen-bonded complexes lead us to conclude that the substitution of halogen by thiocyanate in the coordination sphere increases the tendency to form C⋯H contacts attributed to C–H⋯π interactions. In addition, the luminescent properties of the free ligands and complexes 1–4 were studied in acetonitrile solution at room temperature. The emissions observed in the spectra of complexes were ascribed to the intra-ligand transitions and ligand-to-metal charge transfer. This work reveals that the C–H⋯π and π⋯π intermolecular interactions are also responsible of the fluorescence observed.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by ANPCyT (PICT 2016-0226 and PICT 2013-0697), SCAIT-UNT (PIUNT D542/2), CONICET (PIP 11220130100651CO and PIP 11220150100002CO) and UNLP (Grant 11/X709) of Argentina. A. D. S. thanks CONICET for the doctoral fellowship. G. A. E and O. E. P. are Research Fellows of CONICET. R. E. C. thanks support from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), PIP # 11220120100360, the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), PICT-2013-2149 and the Secretaría de Ciencia y Tecnología de la Universidad Nacional de Córdoba (SECyT-UNC), Project 203/14. We thank Lic. Alejandro Menzaque for taking the PXRD and TGA-DTA data.References
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC
.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Kupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef PubMed
- J. Rocha, L. D. Carlos, F. A. Almeida Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC
- O. R. Evans and W. Lin, Chem. Mater., 2001, 13, 3009–3017 CrossRef
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef PubMed
- N. Upadhayay, Chem. Sci. Trans., 2013, 2, 455–460 CrossRef
-
(a) F. A. Mautner, M. Scherzer, C. Berger, R. C. Fischer, R. Vicente and S. S. Massoud, Polyhedron, 2015, 85, 20–26 CrossRef
- K. S. Banu, S. Mondal, A. Guha, S. Das, T. Chattopadhyay, E. Suresh, E. Zangrando and D. Das, Polyhedron, 2011, 30, 163–168 CrossRef
-
(a) N. Sarkar, K. Harms, A. Frontera and S. Chattopadhyay, New J. Chem., 2017, 41, 8053–8065 RSC
- R. M. Kumar, M. Elango, R. Parthasarathi, D. Vijay and V. Subramanian, J. Chem. Sci., 2012, 124, 193–202 CrossRef
-
(a) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef PubMed
- P. Hobza and Z. Havlas, Chem. Rev., 2000, 100, 4253 CrossRef PubMed
- S. K. Seth, I. Saha, C. Estarellas, A. Frontera, T. Kar and S. Mukhopadhyay, Cryst. Growth Des., 2011, 11, 3250–3265 CrossRef
- A. Saeed, M. Bolte, M. F. Erben and H. Pérez, CrystEngComm, 2015, 17, 7551–7563 RSC
- M. Montazerozohori, A. Masoudiasl and T. Doert, Inorg. Chim. Acta, 2016, 443, 207–217 CrossRef
- M. Owczarek, I. Majerz and R. Jakubas, CrystEngComm, 2014, 16, 7638–7648 RSC
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef PubMed
- S. Soudani, V. Ferretti, C. Jelsch, F. Lefebvre and C. B. Nasr, J. Solid State Chem., 2016, 237, 7–13 CrossRef
- CrysAlisPro, Oxford Diffraction Ltd., version 1.171.33.48 (release 15-09-2009 CrysAlis171.NET) Search PubMed
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed
- A. Spek, Acta Crystallogr., 2009, D65, 148–155 CrossRef PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef
-
(a) J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., 2004, B60, 627–668 CrossRef PubMed
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC
- M. A. Spackman, Chem. Rev., 1992, 92, 1769–1797 CrossRef
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.0), University of Western Australia, 2012 Search PubMed
- M. A. Spackman, Phys. Scr., 2013, 87, 048103 CrossRef
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su and T. L. Windus, J. Comput. Chem., 1993, 14, 1347 CrossRef
-
(a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef
- M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197–205 CrossRef PubMed
- S. K. Seth, CrystEngComm, 2013, 15, 1772–1781 RSC
-
(a) C. Janiak, S. Temizdemir, S. Dechert, W. Deck, F. Girgsdies, J. Heinze, M. J. Kolm, T. G. Scharmann and O. M. Zipffel, Eur. J. Inorg. Chem., 2000, 1229–1241 CrossRef
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- D. Bose, J. Banerjee, S. H. Rahaman, G. Mostafa, H.-K. Fun, R. D. Bailey Walsh, M. J. Zaworotko and B. K. Ghosh, Polyhedron, 2004, 23, 2045–2053 CrossRef
- E. Rozycka-Sokolowska, B. Marciniak, J. Ławeck, B. Bujnicki, J. Drabowicz and A. Rykowski, J. Sulfur Chem., 2013, 34, 651–660 CrossRef
- S. Syed, M. M. Jotani, S. N. A. Halim and E. R. T. Tiekink, Acta Crystallogr., 2016, E72, 391–398 CrossRef PubMed
- A. Di Santo, G. A. Echeverría, O. E. Piro, H. Pérez, A. B. Altabef and D. M. Gil, J. Mol. Struct., 2017, 1134, 492–503 CrossRef
- C. Jelsch, S. Soudani and C. B. Nasr, IUCrJ, 2015, 2, 327–340 CrossRef PubMed
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley Interscience, New York, 5th edn, 1997 Search PubMed
- Z. Leka, S. A. Gruji, Z. Tesic, S. Lukic, S. Skuban and S. Trifunovi, J. Serb. Chem. Soc., 2004, 69, 137–143 CrossRef
- D. F. Weng, Z. M. Wang and S. Gao, Chem. Soc. Rev., 2011, 40, 3157–3181 RSC
- R. Boča, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef
- M. Saber and K. R. Dunbar, Chem. Commun., 2014, 50, 12266 RSC
-
(a) G. Jin, R. Wang, Y. Xiu, Y. Yang and X. Meng, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 596–602 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC Any request to the Cambridge Crystallographic Data Centre for these material should quote the full literature citation and the reference number CCDC 1588816 [Zn(1-MeIm)2(SCN)2], CCDC 1588817 [Zn(2-MeIm)2(SCN)2], CCDC 1588818 [Cd(1-MeIm)4(SCN)2] and CCDC 1588819 ([Cd(2-MeIm)2(SCN)2]n). The geometry of the molecules was calculated using Platon for Windows Taskbar v1.17.21. The drawings were made with Olex2 (ref. 22) and Mercury.23 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra04452j |
| ‡ Members of the Research Career of CONICET |
| This journal is © The Royal Society of Chemistry 2018 |