
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrasound-facilitated assembly and disassembly of a pH-sensitive self-assembly peptide†
Rong Ni ab,
Jianhui Liua and
Ying Chau*a
ab,
Jianhui Liua and
Ying Chau*a
aDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: keychau@ust.hk; Fax: +86-52-23580054; Tel: +86-52-23588935
bInstitute for Advanced Study, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 20th August 2018
Abstract
In this report, we investigated the impact of external stimulus (ultrasound) and internal stimulus (pH) on peptide assembly and disassembly. Two short rationally designed peptides K3C6SPD and F20H differing in the presence of a single pH-sensitive histidine residue were studied as the model peptides. The assembly kinetics studies demonstrated that the substitution of phenylalanine in K3C6SPD with histidine (F20H) significantly slowed down the peptide assembly rate at all three tested pHs (pH 9.5, pH 7.4 and pH 5.0). At the same time, this F to H substitution led to the increased pH-responsive assembly kinetics. By treating the peptide sample at the beginning of the assembly process at pH 9.5 for 5 min with the ultrasound power of 2.1 W cm−2, the assembly rate of peptide F20H was significantly accelerated with the lag phase being shortened from 10 days to 2 days. For the disassembly of F20H peptide nanofibrils preformed at pH 9.5, upon pH adjustment from pH 9.5 to pH 5.0, 5 min ultrasonication of the nanofibrils resulted in the nanofibril disassembly within 6 hours, instead of 5 days in the absence of ultrasound. On the contrary, similar ultrasound treatment of the peptide K3C6SPD did not produce any obvious effect on both assembly and disassembly processes. This study offers an effective strategy to modulate the stimuli-responsiveness of the peptide-based biomaterials.
Introduction
Peptide-based biomaterials, due to their biocompatibility, biodegradability and tuneable morphology/architecture, have been extensively investigated and well-oriented towards different applications.1–5 For some applications, control of the assembly/disassembly behaviours of peptide assemblies is essential. For example, the biological application of peptide-based biomaterials as drug carriers requires appropriate responsiveness towards different stimuli for controlled drug release.6–8 The strategies to enhance the stimuli-responsiveness have been broadly studied, and examples include the addition of stimuli-sensitive units (such as pH-sensitive histidine/hydrazine or redox-sensitive disulphide-bond) into the peptide assemblies to achieve intracellular stimuli-triggered drug release.6–8 External stimuli, such as light, ultrasound etc., have also been explored to control drug release.9,10 However, these internal and external strategies have usually been applied separately to the stimuli-responsive systems, which may limit their roles. Therefore, in this fundamental work, we aim to study the combination effect of internal and external stimuli, in order to explore a simple strategy to enhance the stimuli-responsiveness of peptide self-assemblies.Among different internal and external stimuli, we chose the well-established pH-trigger and ultrasound as the model representatives and investigated their combination effect on peptide assembly/disassembly behaviours. Trigger by pH is one of the well-explored internal stimuli, especially in the drug delivery field.11,12 For the intracellular delivery of therapeutics, upon endocytosis, cargoes and carriers usually experience a pH drop during intracellular trafficking, from neutral (∼pH 7.4) extracellular environment to slightly acidic (∼pH 6.5) in early endosome, then more acidic (∼pH 5.5) late endosome and finally lysosome (∼pH 4.5).13 By incorporation of pH-sensitive units into the assembled structures, acid-induced structure alteration may occur during this process, leading to the successful intracellular delivery through pH-triggered endosomal escape or cargo release.11,14 Ultrasound is one of the non-invasive external stimuli, which is able to enhance/disrupt or re-configure the peptide self-assemblies through adjustable mechanical perturbation. For example, short fibrils of 20–100 nm could be generated through ultra-sonication of mature fibrils,15 which is a routine method to make self-assembly seeds. On the other hand, ultrasound has also been found to accelerate the assembly of different peptides.16–19 These studies demonstrate that the ultrasound may initiate nucleation through intermolecular interaction by either breaking down the intra-molecular bonds or energizing the self-assembling peptide (SAP) into a metastable region. Besides influencing the peptide assembly kinetics, ultrasound could transform the morphology from nanofibrils to spherical aggregates, which was observed in the dipeptide Fmoc-YL assemblies.20 Therefore, both well-known stimuli (pH-trigger and ultrasound) will be studied together in this study to evaluate the combination effect of different stimuli. While ultrasound can generate both thermal and mechanical effects, we intend to explore the mechanical effects, which are maximized at low frequency ultrasound.21
Materials and methods
Reagents and chemicals
The rink amide resin and all the Fmoc-protected natural amino acids for peptide synthesis were supplied by GL Biochem (Shanghai, China). Fmoc-Hexacid-OH was purchased from Hanghong (China). Other chemicals (including those mentioned in the later sections) were purchased from Sigma-Aldrich unless otherwise specified.Peptide synthesis and purification
The peptide K3C6SPD and F20H were synthesized in our laboratory on microwave-assisted solid-phase peptide synthesizer (Biotage, Sweden) based on Fmoc-synthesis chemistry. The dry resin was treated with cocktail 1 (TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thioanisol:anisol 90:5:5) or cocktail 2 (TFA:TES: 95:5) for 1–2 h for the peptide cleavage. The filtrate was titrated into cold ether to produce the white peptide precipitate. The precipitate was washed with cold ether twice and collected through centrifugation and air-dried in the fume hood.
:thioanisol:anisol 90:5:5) or cocktail 2 (TFA:TES: 95:5) for 1–2 h for the peptide cleavage. The filtrate was titrated into cold ether to produce the white peptide precipitate. The precipitate was washed with cold ether twice and collected through centrifugation and air-dried in the fume hood.
The dry peptide pellet was dissolved in water/acetonitrile through sonication. After centrifugation, the supernatant was collected in a separate tube for HPLC purification. The peptides were purified by reverse-phase HPLC (Waters 1525 Binary HPLC pump and Waters 2487 Dual λ Absorbance Detector) using the Vydac protein & peptide C18 preparative column, 250 × 22 mm, 10–15 μm (218TP™ C18 Column. W. R. Grace & Co.). The mobile phase gradient is 2% min−1 and flow rate is 10 mL min−1. After acetonitrile was evaporated on rotavap, the peptide aqueous solution was frozen-dried on the freeze-drier to yield white peptide powder, which was stored in −20 °C for future usage. The peptide identity was confirmed by MALDI-TOF-mass (BRUKER ultraflectreme) analysis.
Peptide self-assembly and disassembly
To eliminate the preformed peptide seeds during the purification, concentration and drying process, the peptide powder was pre-treated with HFIP (1,1,1,3,3,3-hexafluoro-2-propanol) on ice for 1 h, followed by the evaporation of HFIP in the fume hood to generate peptide film. For peptide self-assembly, the treated peptide film was dissolved in ddH2O and the pH was adjusted with buffer of different pHs (acidic acetate buffer of pH 5.0, neutral HEPES buffer of pH 7.4 and CHES basic buffer of pH 9.5).The nanofibril disassembly was carried out by the pH adjustment of the mature peptide nanofibrils from pH 9.5 to pH 5.0 with the addition of 1 M HCl. The mature peptide assemblies was prepared under basic pH (pH 9.5) and incubated at room temperature for two weeks. The pH adjustment was preformed by the addition of 1 M HCl and the sample pH was measured with micro pH probe (Orion™ 9810BN Micro pH Electrode. Thermo Fisher Scientific Inc.), connected to a pH meter (Orion Star™ A111 pH Benchtop Meter, Thermo Fisher Scientific Inc.).
Peptide assembly/disassembly kinetics by ANS fluorescence assay
The peptide assembly and disassembly kinetics was monitored by ANS assay (8-anilinonaphthalene-1-sulfonic acid). The ANS stock (250 μM) was prepared and added into peptide solution with final ANS concentration of 25 μM. After 10 min incubation in the dark, the peptide/ANS solution was added into a black 96-well plate, followed by the fluorescence measurements on the plate reader (Varioskan LUX Multimode Microplate Reader, Thermo Fisher Scientific Inc.) with Ex = 350 nm. The maximal emission peak value was plotted versus the different time points, leading to the peptide assembly kinetic curve.Atomic force microscope (AFM)
At the pre-designated time point, the AFM samples were prepared by deposition of diluted peptide solutions (3-fold dilution) on freshly cleaned mica. After 5 minute incubation, the excess solution was wicked off with filter paper. After that, the mica was stored in a desiccator overnight before AFM characterization (Nanoscope-MultiMode/Dimension, Digital Instruments).Attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR)
Peptide secondary structure was characterized using FTIR (Bruker Vertex-70 spectrometer, USA), connected to an ATR accessory with diamond crystal (GladiATR Vision™, PIKE Technologies). The samples were scanned with an average of 32 scans in the range of 4000–400 cm−1 with 4 cm−1 resolution. 20 μL of the peptide solution was added on the diamond crystal and dried with air purge until a film was formed.Ultrasound experiment
To investigate the ultrasound effect on the peptide assembly, the peptide was dissolved into aqueous phase, followed by applying it on the top surface of the ultrasound transducer AptFlex F28 (Precision Acoustics, United Kingdom). To study the ultrasound effect on the peptide disassembly, after the pH of the preformed mature peptide nanofibril solution at pH 9.5 was adjusted to pH 5.0, the peptide nanofibril solution was applied on the top surface of the ultrasound transducer. Ultrasound at a frequency of 40 kHz with a spatial average temporal average intensity (ISATA) of 2.1 W cm−2 was applied to either peptide or peptide nanofibril solution for 5 min. Temperature change was negligible as confirmed by thermocouple measurement. After 5 min, the samples were collected back into the Eppendorf tubes, followed by the incubation at room temperature for the kinetics study or the structure characterization. The control experiments were performed by applying the samples on the ultrasound transducer and incubated for 5 min without ultrasonication.Results and discussion
Impact of histidine substitution on peptide assembly
In our previous study, we have studied a short model peptide K3C6SPD, which could self-assemble into typical nanofibrils with antiparallel β-sheet structure at neutral pH in an aqueous solution.22 The central β-sheet region (LVFFA) of K3C6SPD is derived from amyloid β-peptide, and two phenylalanine (F) residues are critical to the peptide assembly.23–25 Therefore, by the substitution of F20 with histidine (called F20H) (Fig. 1), we extended our previous study and have investigated the separate and combined effect of pH and ultrasound on the assembly and disassembly behaviours of K3C6SPD and F20H.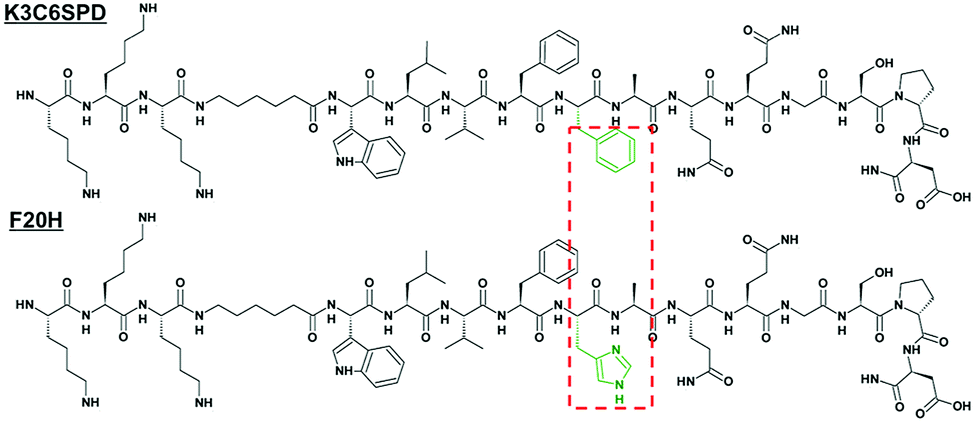 | ||
| Fig. 1 Primary structure of peptide K3C6SPD and the single histidine-substituted peptide F20H. The different residues between both peptide sequences were highlighted in the red dotted box. | ||
We firstly studied the pH-dependent self-assembly behaviour of both peptides without ultrasound treatment on three aspects, including (1) assembly kinetics (pH 5.0, pH 7.4 and pH 9.5); (2) assembly morphologies; and (3) peptide secondary structure within mature assemblies. We monitored the peptide assembly kinetics and assessed the peptide self-assembly propensity with ANS (8-anilinonaphthalene-1-sulfonic acid) assay.26 ANS is one of the popular probes for hydrophobicity. The binding of ANS in hydrophobic pockets induces significant increase in fluorescence intensity. The magnitude of ANS fluorescence intensity indicates the degree of the assembly. By following the ANS florescence intensity change along the peptide incubation process, the peptide assembly kinetics was tracked. Using this assay, we found that the assembly of K3C6SPD under all three-tested pHs reached the equilibrium immediately within a similar time frame, but their assembly propensity was slightly pH-dependent (Fig. 2). ANS fluorescence intensity of the K3C6SPD assemblies at neutral and acidic pHs was ∼25% and 50% magnitude lower than that of nanofibrils assembled at basic pH, respectively. This difference may be originated from the N-terminal free amine (pKa ∼ 8.9), the lysine side-chain amines (pKa ∼ 10.3), and the C-terminal glutamic acid side chain carboxylic acid group (pKa ∼ 4.3).
 | ||
| Fig. 2 Self-assembly kinetics of K3C6SPD and F20H assemblies under three different pHs (pH 9.5, pH 7.4 and pH 5.0) by following ANS (25 μM) fluorescence emission at 468 nm (Ex = 350 nm). The inset is the zoomed-in graph of F20H assembly kinetics at pH 7.4 and pH 5.0. | ||
Relative to the fast K3C6SPD assembly, the single histidine substitution of F20 (F20H) significantly slowed down the peptide assembly kinetics at all three tested pHs. F20H assembly at pH 9.5 reached the plateau in 10 days, instead of minutes for K3C6SPD (Fig. 2). The F20H assembly kinetic curve followed a typical sigmoid model: 1 day nucleation lag-phase, followed by 2 day propagation phase before attaining equilibrium in about 10 days (Fig. 2). The neutral pH (pH 7.4) further slowed down and suppressed the assembly process and the acidic pH (pH 5.0) completely prevented F20H from assembling (inset in Fig. 2). The single histidine substitution also decreases the peptide assembly propensity. The maximal ANS fluorescence intensity of F20H at basic, neutral and acidic pHs was about 11%, 1% and <1% of that of ANS in the K3C6SPD assemblies at the corresponding pHs, respectively. This remarkable decrease of the assembly kinetics and assembly propensity indicates the significant role of histidine substitution at F20 position (Fig. 2).
We next characterized the pH-dependent assembly morphology and the peptide secondary structure with atomic force microscope (AFM) and attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR), respectively. The AFM images in Fig. 3A show that K3C6SPD assembled into thin nanofibrils under acidic and neutral pHs. While at basic pH, K3C6SPD formed big bundles (Fig. 3A). Though the K3C6SPD nanofibrils displayed different dimensions, they were composed of the same antiparallel β-sheets, as indicated by the same amide I band of 1623 cm−1 and the shoulder band of 1693 cm−1 in their FTIR spectra (Fig. 3B). The data above indicate that K3C6SPD displays slight pH-sensitivity, which moderately influences the peptide assembly kinetics and propensity.
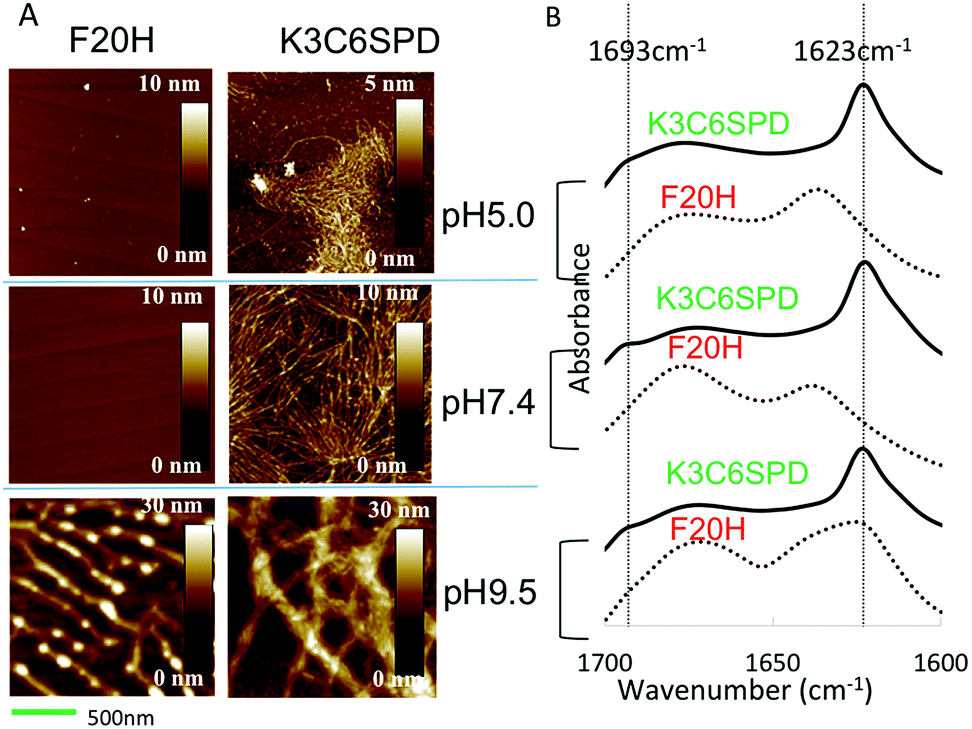 | ||
| Fig. 3 (A) AFM images of the matured K3C6SPD and F20H assemblies in an aqueous solution at pH 5.0, pH 7.4 and pH 9.5 from top to bottom (scale bar = 500 nm). (B) ATR-FTIR amide I spectra of K3C6SPD and F20H assemblies at different pHs. The β-sheet signals (1693 and 1623 cm−1) were highlighted with dashed lines. In each pH group, the curve of solid and dashed line represents K3C6SPD and F20H samples, respectively. | ||
Consistent with lower assembly propensity detected by ANS assay, F20H assemblies at acidic and neutral pHs revealed no obvious species, as shown in AFM images (Fig. 3A). At basic pH, F20H assembled into short proto-filaments with clear branches at early stage, which further matured into long nanofibrils (Fig. 3A). The peptide secondary structure defined by ATR-FTIR spectra is consistent with the morphologies. The acidic and neutral samples displayed random coil signal with the amide I band of 1645 cm−1 (Fig. 3B). While the F20H nanofibrils formed at basic pH showed the amide I band of 1623 cm−1, suggesting the β-sheets secondary structure. The dramatic decrease of the assembly kinetics and the assembly propensity indicates that the single histidine substitution is able to cause significant enhancement of the pH-sensitivity.
Therefore, K3C6SPD and F20H, differing by a single residue in the sequence and yet varying dramatically in the assembly behaviours, provide an ideal model system for us to further study the impact of ultrasound on the assembly and disassembly processes. Since F20H formed negligible species under neutral and acidic pHs, we mainly focused on the ultrasound effect on the assembly process at basic pH and disassembly process upon the pH adjustment from the basic to acidic pH. Ultrasound effects on peptide assembly systems were reported previously. The non-thermal effect of ultrasound at low frequency (80–120 kHz) was able to transform the morphologies (fibrils vs. spheres) by changing the thermodynamic state.20 Ultrasound at 43 kHz or 1.1 mHz (both at 300 mW cm−2) disrupted the ordered assembly of the coating of pHEMA (poly(hydroxylethyl(methacrylate))) hydrogel.27 This study indicates that ultrasound of low frequency can generate obvious effect on peptide assemblies.
Impact of ultrasound on peptide assembly and disassembly
In this work, we chose the ultrasound with frequency of 40 kHz and the average intensity of 2.1 W cm−2 and to mainly study its mechanical impact on the peptide assembly/disassembly kinetics and the assembled morphologies. The assembly kinetic curve in Fig. 4 showed that K3C6SPD assemblies being treated with 5 min ultrasonication reached the plateau immediately (Fig. 4A), similar as that of the non-ultrasonicated sample. The impact of ultrasound on K3C6SPD assembly kinetics was masked by the fast assembly process of K3C6SPD under basic pH. While ultrasound did enhance the assembly propensity, supporting by the increase of the maximal ANS fluorescence intensity with the magnitude of ∼20% (Fig. 4). The impact of ultrasound on the assembly morphology was studied with AFM. The AFM images of the K3C6SPD assemblies at early time point (3 h of incubation) displayed dense short well-dispersed fibrils, which is different from the bundled big nanofibrils observed in non-ultrasonicated samples (Fig. 5A). Since K3C6SPD has strong assembly propensity, it immediately assembles into large aggregates. The ultrasonication may just break down the large K3C6SPD bundles into small fibrils, which provides more accessible surface and promotes more assemblies. Ultrasound affects the peptide-assembled morphology, but does not alter the peptide β-sheet secondary structure. The FTIR spectra (Fig. 5B) of both K3C6SPD nanofibril samples show the same amide I band of 1625 cm−1, independent of ultrasonication.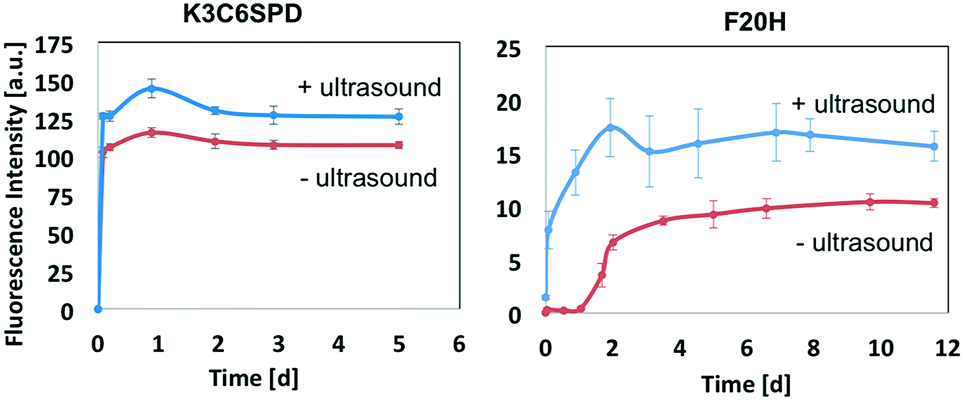 | ||
| Fig. 4 Ultrasound impact on the assembly kinetics and morphology. The assembly kinetics of K3C6SPD and F20H at basic condition with or without ultrasound treatment was monitored by following the ANS (25 μM, Ex = 350 nm) maximal fluorescence emission with time. | ||
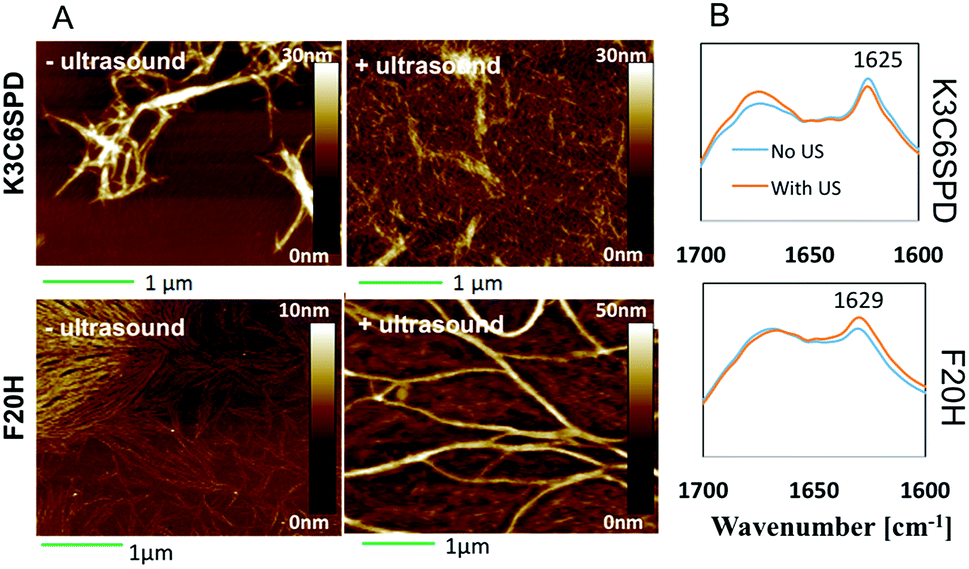 | ||
| Fig. 5 (A) AFM images of K3C6SPD and F20H assemblies at pH 9.5 with or without ultrasonication. 5 min ultrasonication was applied to the samples at beginning of the assembling process. The samples were incubated at room temperature for 30 min before AFM sample preparation (scale bar = 1 μm). (B) FTIR spectra of K3C6SPD and F20H assemblies at pH 9.5 with or without ultrasonication. The β-sheet signature bands of 1625 m−1 and 1629 cm−1 were labelled. | ||
Relative to the fast assembly kinetics of K3C6SPD with or without ultrasonication, the slower assembly process of F20H under basic pH highlights several key impacts of ultrasound on the peptide assembly. Firstly, ultrasound enhances F20H assembly kinetics. The assembly kinetic curve in Fig. 4 shows that ultrasonication of the F20H sample immediately drove the propagation phase by skipping the 1 day lag phase and let the assembly reach the plateau in 2 days, instead of 10 days in the absence of ultrasound. During this process, ultrasound may dissociate intra-molecular interaction and facilitate inter-molecular interaction, thus promoting the nucleation process and fastening the assembly process.16,19 Besides assembly kinetics, ultrasound also increases the assembly propensity of F20H assemblies. The ANS florescence intensity of ultrasonicated F20H sample was increased by a magnitude of ∼50% (Fig. 4). In addition, ultrasound also facilitates thin F20H filaments to align together into thick nanofibrils. The AFM images of ultrasonicated F20H sample at early time point (3 h incubation) displayed the co-existence of thin filaments and thick nanofibrils, instead of the merely thin filaments in the absence of ultrasound (Fig. 5A). This implies that ultrasound may facilitate the alignment of F20H filaments, driving the assembly equilibrium towards nanofibrils, which then leads to higher ANS fluorescence emission intensity. Ultrasonication of the peptide samples at the beginning of the assembly accelerates and enhances the peptide assembly process. Ultrasound only changes the F20H assembled morphology, but does not alter the peptide β-sheet secondary structure. The same amide I band of 1629 cm−1 was observed for both samples with or without ultrasound treatment (Fig. 5B), similar to the observation made for K3C6SPD nanofibrils (Fig. 5B).
Ultrasound did induce obvious impact on the peptide assembly process, and we continued to study its impact on the disassembly process of the pH-sensitive F20H nanofibrils upon pH adjustment (from pH 9.5 to pH 5.0). F20H nanofibrils were preformed and matured at pH 9.5 for around 2 weeks, followed by the pH adjustment into acidic solution (pH 5.0) with 1 M HCl. Disassembly kinetics was monitored with ANS assay. Fig. 6A shows that non-ultrasonicated F20H nanofibrils experienced two disassembly stages upon pH adjustment from pH 9.5 to pH 5.0. Specifically, ANS fluorescence intensity decreased by 10% within 6 hours, followed by 2 day to reach the equilibrium. Then another ANS fluorescence intensity decrease (∼55%) was observed from day 2 to day 5 before the plateau. Five-min ultrasonication of F20H nanofibrils, upon pH adjustment to pH 5.0 resulted in an immediate decrease of the ANS fluorescence emission intensity by 60% within 6 hours, followed by gradual equilibration in two days. This observation strongly supports that ultrasound accelerates peptide nanofibril disassembly. Once the disassembling process reached the equilibrium, no obvious β-sheet signal could be detected, as shown in the FTIR spectra of F20H disassembled samples at pH 5.0 (Fig. 6B). This suggests that F20H assembly is strongly pH-dependent. To understand the morphology change during this disassembly process, we imaged samples after 30 min of incubation at pH 5.0 with or without ultrasonication. AFM images in Fig. 6C show that without ultrasonication, majority of the nanofibrils maintained the bundles while a small population dissociated into thin filaments. With 5 min ultrasonication, only few thin filaments were observed, suggesting that ultrasound promoted the big nanofibril bundles to disassemble. The AFM images and the kinetics data both imply that ultrasound increases the pH-responsiveness of the pH-sensitive peptide self-assemblies.
 | ||
| Fig. 6 (A) Disassembly kinetics of the F20H assemblies upon the pH adjustment from basic to acidic pH with or without ultrasonication. (B) The FTIR spectra of F20H samples disassembled at pH 5.0 w/o ultrasonication. (C) AFM images of the samples upon pH adjustment w/o ultrasonication. The preformed F20H nanofibrils at pH 9.5 were acidified from pH 9.5 to 5.0 with 1 M HCL. Upon the pH adjustment, the sample was immediately treated with ultrasound of 40 kHz for 5 min, followed by the incubation at room temperature on the bench. | ||
Conclusion
In conclusion, we have systematically investigated the impacts of pH (via single histidine substitution) and ultrasound (5 min of 40 kHz ultrasonication) on the peptide assembly/disassembly process and made several new discoveries. (1) The single histidine substitution at F20 position dramatically slows down the assembly kinetics and decreases the peptide assembly propensity, but the response toward pH-triggered disassembly is slow (5 days). (2) Short ultrasound treatment of the pH-insensitive K3C6SPD sample results in negligible enhancement of peptide assembly and nanofibril disassembly. (3) The combination of pH-trigger and ultrasonication drastically accelerates the peptide nanofibril disassembly and enhances the stimuli-responsiveness of the peptide assemblies. Therefore, this study provides a simple but efficient strategy to regulate the stimuli-responsiveness of nanomaterials and these discoveries present valuable references for the combination of different internal and external stimuli towards designing smarter self-assembly systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the funding support from the Hong Kong Research Grant Council (GRF 16305815 and GRF 16306917) and the Hong Kong University of Science and Technology (IGN15IP01). We also thank Mr Chun Hang Yu for his contribution to this work.Notes and references
- D. M. Leite, E. Barbu, G. J. Pilkington and A. Lalatsa, Peptide Self-Assemblies for Drug Delivery, Curr. Top. Med. Chem., 2015, 15, 2277–2289 CrossRef PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Self-assembling peptide semiconductors, Science, 2017, 358, eaam9756 CrossRef PubMed.
- H. Hosseinkhani, P. D. Hong and D. S. Yu, Self-Assembled Proteins and Peptides for Regenerative Medicine, Chem. Rev., 2013, 113, 4837–4861 CrossRef PubMed.
- Y. Wang, A. G. Cheetham, G. Angacian, H. Su, L. S. Xie and H. G. Cui, Peptide-drug conjugates as effective prodrug strategies for targeted delivery, Adv. Drug Delivery Rev., 2017, 110, 112–126 CrossRef PubMed.
- Z. Q. Q. Feng, H. M. Wang, R. Zhou, J. Li and B. Xu, Enzyme-Instructed Assembly and Disassembly Processes for Targeting Downregulation in Cancer Cells, J. Am. Chem. Soc., 2017, 139, 3950–3953 CrossRef PubMed.
- C. Chang, P. Q. Liang, L. L. Chen, J. F. Liu, S. H. Chen, G. H. Zheng and C. Y. Quan, pH-responsive nanoparticle assembly from peptide amphiphiles for tumor targeting drug delivery, J. Biomater. Sci., Polym. Ed., 2017, 28, 1338–1350 CrossRef PubMed.
- Z. H. Song, X. Chen, X. R. You, K. Q. Huang, A. Dhinakar, Z. P. Gu and J. Wu, Self-assembly of peptide amphiphiles for drug delivery: the role of peptide primary and secondary structures, Biomater. Sci., 2017, 5, 2369–2380 RSC.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Self-assembled peptide-based nanostructures: Smart nanomaterials toward targeted drug delivery, Nano Today, 2016, 11, 41–60 CrossRef PubMed.
- H. P. Li, X. Yang, Z. W. Zhou, K. K. Wang, C. Z. Li, H. Z. Qiao, D. Oupicky and M. J. Sun, Near-infrared light-triggered drug release from a multiple lipid carrier complex using an all-in-one strategy, J. Controlled Release, 2017, 261, 126–137 CrossRef PubMed.
- T. Boissenot, A. Bordat, E. Fattal and N. Tsapis, Ultrasound-triggered drug delivery for cancer treatment using drug delivery systems: From theoretical considerations to practical applications, J. Controlled Release, 2016, 241, 144–163 CrossRef PubMed.
- Y. Y. Wang, P. F. Li, F. Chen, L. Q. Jia, Q. H. Xu, X. M. Gai, Y. B. Yu, Y. Di, Z. H. Zhu, Y. Y. Liang, M. Q. Liu, W. S. Pan and X. G. Yang, A novel pH-sensitive carrier for the delivery of antitumor drugs: histidine-modified auricularia auricular polysaccharide nano-micelles, Sci. Rep., 2017, 7, 4751 CrossRef PubMed.
- S. Lu, W. F. D. Bennett, Y. Ding, L. Zhang, H. Y. Fan, D. Y. Zhao, T. Zheng, P. K. Ouyang, J. Li, Y. Wu, W. Xu, D. F. Chu, Y. F. Yuan, H. Heerklotz, M. Karttunen and P. Chen, Design and Characterization of a Multifunctional pH-Triggered Peptide C8 for Selective Anticancer Activity, Adv. Healthcare Mater., 2015, 4, 2709–2718 CrossRef PubMed.
- Y. B. Hu, E. B. Dammer, R. J. Ren and G. Wang, The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration, Transl. Neurodegener., 2015, 4, 18 CrossRef PubMed.
- X. J. Zhang, D. W. Chen, S. Ba, J. Zhu, J. Zhang, W. Hong, X. L. Zhao, H. Y. Hu and M. X. Qiao, Poly(L-histidine) Based Triblock Copolymers: pH Induced Reassembly of Copolymer Micelles and Mechanism Underlying Endolysosomal Escape for Intracellular Delivery, Biomacromolecules, 2014, 15, 4032–4045 CrossRef PubMed.
- H. Yokoi, T. Kinoshita and S. G. Zhang, Dynamic reassembly of peptide RADA16 nanofiber scaffold, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef PubMed.
- K. Uesugi, H. Ogi, M. Fukushima, M. So, H. Yagi, Y. Goto and M. Hirao, Mechanisms of Ultrasonically Induced Fibrillation of Amyloid beta(1-40) Peptides, Jpn. J. Appl. Phys., 2013, 52, 07HE10 CrossRef.
- S. Maity, P. Kumar and D. Haldar, Sonication-induced instant amyloid-like fibril formation and organogelation by a tripeptide, Soft Matter, 2011, 7, 5239–5245 RSC.
- H. J. Kim, E. Chatani, Y. Goto and S. R. Paik, Seed-dependent accelerated fibrillation of alpha-synuclein induced by periodic ultrasonication treatment, J. Microbiol. Biotechnol., 2007, 17, 2027–2032 Search PubMed.
- K. Yamaguchi, T. Matsumoto and K. Kuwata, Proper calibration of ultrasonic power enabled the quantitative analysis of the ultrasonication-induced amyloid formation process, Protein Sci., 2012, 21, 38–49 CrossRef PubMed.
- C. G. Pappas, T. Mutasa, P. W. J. M. Frederix, S. Fleming, S. Bai, S. Debnath, S. M. Kelly, A. Gachagan and R. V. Ulijn, Transient supramolecular reconfiguration of peptide nanostructures using ultrasound, Mater. Horiz., 2015, 2, 198–202 RSC.
- C. K. Holland and R. E. Apfel, An improved theory for the prediction of microcavitation thresholds, IEEE Trans. Ultrason. Ferroelectr. Freq. Control, 1989, 36, 204–208 Search PubMed.
- R. Ni and Y. Chau, Structural Mimics of Viruses Through Peptide/DNA Co-Assembly, J. Am. Chem. Soc., 2014, 136, 17902–17905 CrossRef PubMed.
- E. Gazit, A possible role for pi-stacking in the self-assembly of amyloid fibrils, FASEB J., 2002, 16, 77–83 CrossRef PubMed.
- R. Cukalevski, B. Boland, B. Frohm, E. Thulin, D. Walsh and S. Linse, Role of Aromatic Side Chains in Amyloid beta-Protein Aggregation, ACS Chem. Neurosci., 2012, 3, 1008–1016 CrossRef PubMed.
- T. J. Measey and F. Gai, Light-Triggered Disassembly of Amyloid Fibrils, Langmuir, 2012, 28, 12588–12592 CrossRef PubMed.
- N. D. Younan and J. H. Viles, A Comparison of Three Fluorophores for the Detection of Amyloid Fibers and Prefibrillar Oligomeric Assemblies. ThT (Thioflavin T); ANS (1-Anilinonaphthalene-8-Sulfonic Acid); and bisANS (4,4′-Dianilino-1,1′-Binaphthyl-5,5′-Disulfonic Acid), Biochemistry, 2015, 54, 4297–4306 CrossRef PubMed.
- C. S. Kwok, P. D. Mourad, L. A. Crum and B. D. Ratner, Self-assembled molecular structures as ultrasonically-responsive barrier membranes for pulsatile drug delivery, J. Biomed. Mater. Res., 2001, 57, 151–164 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Peptide synthesis and purification, experimental procedures. See DOI: 10.1039/c8ra04391d |
| This journal is © The Royal Society of Chemistry 2018 |