
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfonated covalent triazine-based frameworks as catalysts for the hydrolysis of cellobiose to glucose†
Jens Artz a,
Irina Delidovicha,
Moritz Pilaskia,
Johannes Niemeiera,
Britta Maria Kübbera,
Khosrow Rahimib and
Regina Palkovits*a
a,
Irina Delidovicha,
Moritz Pilaskia,
Johannes Niemeiera,
Britta Maria Kübbera,
Khosrow Rahimib and
Regina Palkovits*a
aChair of Heterogeneous Catalysis & Chemical Technology Institut für Technische und Makromolekulare Chemie, RWTH Aachen University Worringerweg 2, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
bDWI Leibniz-Institut für Interaktive Materialien Forckenbeckstr. 50, 52074 Aachen, Germany
First published on 19th June 2018
Abstract
Covalent triazine-based frameworks (CTFs) were synthesized in large scale from various monomers. The materials were post-synthetically modified with acid functionalities via gas-phase sulfonation. Acid capacities of up to 0.83 mmol g−1 at sulfonation degrees of up to 10.7 mol% were achieved. Sulfonated CTFs exhibit high specific surface area and porosity as well as excellent thermal stability under aerobic conditions (>300 °C). Successful functionalization was verified investigating catalytic activity in the acid-catalyzed hydrolysis of cellobiose to glucose at 150 °C in H2O. Catalytic activity is mostly affected by porosity, indicating that mesoporosity is beneficial for hydrolysis of cellobiose. Like other sulfonated materials, S-CTFs show low stability under hydrothermal reaction conditions. Recycling of the catalyst is challenging and significant amounts of sulfur leached out of the materials. Nevertheless, gas-phase sulfonation opens a path to tailored solid acids for application in various reactions. S-CTFs form the basis for multi-functional catalysts, containing basic coordination sites for metal catalysts, tunable structural parameters and surface acidity within one sole system.
Introduction
Acids play an important key role in chemical synthesis. Numerous essential organic reactions such as esterification or addition reactions are carried out in the presence of acids. Due to its versatile applications, sulfuric acid is the chemical with the highest production output (170 Mio. t/a) worldwide.1 Therefore, this characteristic number was even utilized to describe the degree of industrialization of individual countries.2 Regardless of the inexpensiveness of these acids, their separation is a serious challenge in industrial processes and only possible via neutralization. The resulting non-recyclable salt load leads to high amounts of waste, thus causing a strong environmental impact.Substitution of homogeneous acids by solid acids facilitates the separation process strongly.3 Thus, solid acids can already be found in several industrial applications.1,4,5 The most commonly used solid acids are zeolites, crystalline microporous alumosilicates with extremely high thermal stability (700–1300 °C), tunable acidity and acid strength.4,6,7 Nevertheless, the zeolites microporosity can be an obstacle, resulting in pore diffusion limitation and thus, comparably low catalytic activity.6 To overcome such pore diffusion limitations, the sulfonation of macroporous polymers has been studied intensively. In this regard, especially polystyrene-based materials gained a lot of attention.8 Unfortunately, thermal stability in comparison to zeolites is relatively low and desulfonation takes place at 260–300 °C in the gas or at 120–150 °C in the liquid phase.9,10 Due to their ecological balance, solid acids are often used in “green” chemical processes, for example in the production of bio-diesel.11,12 Solid acids can be used as catalysts for both the transesterification of triglyceride in the classical bio-diesel production as well as in the hydrolysis of biomass to desired bio-diesel-like compounds.12–15
In this respect, hydrolysis of water-insoluble cellulose to soluble glucose attracts great attention as the most challenging step for the production of bio-ethanol, bio-HMF and other value-added products.16 Herein, we focus on a soluble dimer of glucose, cellobiose, representing a structural unit of cellulose. Vilcocq et al. has recently reviewed the activity of different solid catalysts for hydrolysis of cellobiose into glucose.17 Inorganic materials, such as zeolites17 or metal oxides,18 exhibited moderate catalytic activity and selectivity for glucose formation. The best catalytic performance was demontrated by sulfonated materials, including resins,19 carbons,20 silicas21 or organosilicas:22 up to quantitative yield of glucose can be attainable over these catalysts. However, stability of the materials against leaching of sulfur under reaction conditions appears to be the main challenge for reusability of the catalysts.21–23 At the same time, other structural changes of the materials were reported. For example, Zhao et al. observed elimination of oxygen-containing groups from the surface of the sulfonated graphite oxide used as catalyst for cellobiose hydrolysis.24 Therefore, water-tolerant acidic materials are of great interest for the hydrolysis reaction.
In this regard, covalent triazine-based frameworks (CTFs; Scheme 1) fulfil all necessary requirements for a solid acidic catalyst resulting after sulfonation.25,26 Thermally stable up to 400 °C and chemically robust under relatively harsh reaction conditions, these materials have drawn the interest of many researchers as solid catalyst supports.27–29 Additionally, they possess sufficient benzylic structure elements for a feasible sulfonation, while access to a large variety of commercial dinitrile monomers grants control of the resulting materials porosities and specific surface areas.29
 | ||
| Scheme 1 (a) Idealized synthesis of a CTF based on 1,4-DCB as a monomer (CTF-c). (b) Proposed sulfonation methodology resulting in the sulfonated CTF material (S-CTF-c). | ||
The sulfonation of aromatic systems is an electrophilic aromatic substitution which in general occurs in the liquid phase in presence of oleum or highly concentrated H2SO4.30–32 To perform sulfonation reactions of polymeric materials under milder conditions, the treatment of a solid sample with SO3 in the gas-phase is possible.33,34 In this manner, sole surface adsorption of the acid groups can be suppressed, while covalent bonding becomes favoured. First attempts to implement sulfuric species within CTF materials were described by Bai et al.35 Utilizing thiourea and thiosemicarbazide during a polymerization step with cyanuric chloride resulted in sulfur containing CTF materials with high sorption capacities and selectivities for uranium(VI). More recently, Talapaneni et al. substituted ZnCl2 by elemental polymeric sulfur to enable a solvent and catalyst-free polymerization of 1,4-dicyanobenzene to the corresponding CTF.36 This way, a homogeneous distribution of sulfur at high contents of 62 wt% was achieved. The resulting material exhibited excellent properties as a cathode material in Li–S batteries with superior initial coulombic efficiencies and cycle lifetimes. However, the porous character of the materials was entirely lost.
Herein, we present a novel approach to obtain solid acid catalysts with controllable structural properties based on CTFs. CTFs obtained from a variety of dinitrile monomers (Scheme 2) have been synthesized in large scale, resulting in materials with the desired structural properties, namely porosity, specific surface area and nitrogen content. The influence of these structural properties on the gas-phase sulfonation method has been studied intensively. Subsequently, the catalytic activity in the hydrolysis reaction of cellobiose to glucose was investigated with regard to the catalysts acidity, porosity and specific surface area.
 | ||
| Scheme 2 Monomers applied in the CTF preparation. | ||
Experimental section
Preparation of the CTF materials
For the synthesis of the CTF-a material, 1,3-dicyanobenzene (3.00 g, 23.4 mmol, 1 eq.) and ZnCl2 (15.96 g, 117.1 mmol, 5 eq.) were mixed and ground together within a glovebox, transferred into a quartz ampule (12 cm height and 3 cm diameter) and dried in vacuum for at least 3 hours. The ampule was then flame-sealed and placed inside a furnace for 10 hours of heat treatment at 400 °C and further 10 hours at 600 °C (heating rate: 10 K min−1). After cooling to room temperature, the ampule was broken open (CAUTION: the ampules are under pressure, which is released during opening) and the solid product was ground and washed thoroughly with water and diluted HCl (0.1 M). The solid material was then ground in a ball mill (Fritsch Pulverisette 23, 5 min, 30 Hz) to obtain a black powder, which was finally washed successively with water, diluted HCl, diluted NaOH, water and THF. Afterwards, it was dried in vacuum for at least 12 h at 60 °C. Materials based on 2,6-pyridinedicarbonitrile (CTF-b), 1,4-dicyanobenzene (CTF-c) and 4,4′-biphenyldicarbonitrile (CTF-d) were synthesized as described for 1,3-DCB (3.00 g of monomer, 5 eq. of ZnCl2). As the filling degree of the ampule apparently has no effect on the physical parameters of the resulting polymer, the ampule was charged to a maximum of half ampule volume to prevent bursting within the furnace.Gas-phase sulfonation of the materials
Gas-phase sulfonation reactions of all materials have been carried out in a glass reactor equipped with a glass frit according to literature (Fig. S8†).34 After evacuation of the sample for at least 30 min (tap 1 closed), the CTF material (1.0 g) is exposed to SO3 gas (tap 1 opened). The SO3 gas is withdrawn from a reservoir (equipped with a stirring bar, 300 rpm) filled with 20% oleum (10 mL) via dynamic vacuum (p < 0.035 mbar). Within the reactor, the SO3 reacts with the CTF sample at ambient temperature. After 6 h tap 1 is closed and vacuum is applied to the sulfonated sample for further 30 min. The material is suspended carefully in water, filtered and washed thoroughly with water until a neutral pH value of the washing solution is attained. The S-CTF sample is then dried within a furnace (at 80 °C, ambient pressure) and a vacuum furnace (at 60 °C) for at least 12 h.Hydrolysis of cellobiose to glucose with S-CTF
In a typical experiment cellobiose (0.5 g, 1.5 mmol), catalyst (0.05 g) and 4.5 mL water were charged into a stainless steel autoclave with a gas inlet. The autoclave was sealed, pressurized to 30 bar of nitrogen and heated to 110–150 °C under stirring at 750 rpm. After the experiment, the autoclave was cooled down in an ice bath and depressurized. The catalyst was filtered off using a syringe filter (CHROMAFIL® Xtra, PA-20/25, 0.20 μm) and the reaction solution was analyzed via HPLC (Shimadzu 2020) using two successfully connected organic acid resin columns (100 mm × 8.0 mm and 300 × 8.0 mm) as previously reported.45 For recycling studies (after 2 h reaction time) the catalyst (0.075 mg) was filtered off via a Whatman™ filtration system equipped with Anodisc 25 (0.20 μm) membranes, thoroughly washed with water and dried overnight in vacuum at 60 °C.Characterization of the CTF and S-CTF materials
CHNS elemental analysis was performed on a PerkinElmer® 2400 Series II CHNS/O Elemental Analyzer and supported by measurements carried out at the microanalytical laboratory Kolbe (Mülheim an der Ruhr, Germany). ICP-OES analysis was conducted with a SPEKTROFLAME ICP-D instrument from SPEKTRO-Analytical Instruments. The samples (0.03 g) were molten in the presence of KNO3 (0.12 g) and KOH (1 g) within a porcelain crucible and dissolved in aqueous HCl. For titration of the sulfonated CTFs, samples (0.05 g) were dispersed in 10 mL of saturated NaCl solution (aq.) and stirred (500 rpm) for 1 week before titration to guarantee complete ion exchange. The titrations were then carried out using a HI 9124 pH-meter from HANNA INSTRUMENTS and 0.005 M NaOH (aq.). Thermogravimetric analysis (TGA) was carried out in air (60 mL min−1) on a Netzsch Simultaneous Thermal Analyzer Type STA449G Jupiter instrument at a heating rate of 10 K min−1. Nitrogen physisorption experiments were conducted on a Micromeritics ASAP 2010 instrument. Samples were degassed for at least 15 h at 150 °C using a FloVacDegasser. Static volumetric measurements were carried out at −195.8 °C. The empty volume of the cell was determined with helium. The specific surface area was determined with the Brunauer–Emmet–Teller method (BET) using data points at a relative pressure p/p0 between 0.05 and 0.3. The total pore volume was determined at a relative pressure of 0.98. The pore size distribution was calculated via MicroActive (version 1.01) using the density functional theory (DFT) N2-model for slit geometry at optimal Goodness of Fit vs. Regularization (0.01) values for both RMS Error of Fit and Roughness of Distribution. The cumulative pore volume at the pore width of 2 nm was used to determine the micropore volume of the samples. Scanning electron microscopy (SEM) images were recorded on a Hitachi SU9000 Ultra-high resolution microscope equipped with a cooled Si(Li) X-ray detector Oxford Inca. The acceleration voltage was set to 20 kV. SEM samples were prepared by adsorbing dry powder on a Cu/lacey carbon grid and shaking off any loose material. Transmission electron microscopy (TEM) images were recorded on a Zeiss Libra 120 microscope. TEM samples were prepared by adsorbing dry powder on a Cu/lacey carbon grid and shaking off any loose material.Results and discussion
CTF preparation
The synthesis of various CTF materials with different structural properties was conducted using 1,3-dicyanobenzene (1,3-DCB), 2,6-dicyanopyridine (2,6-DCP), 1,4-dicyanobenzene (1,4-DCB) and 4,4′-biphenyldicarbonitrile (4,4′-DCBP) as monomers (Scheme 2). As described elsewhere, bimodal microporous and mesoporous materials containing numerous N moieties are accessible via ionothermal synthesis in molten ZnCl2 acting as a solvent and a Lewis-acidic catalyst.25 Since ZnCl2 furthermore acts as a porogen, an excess ZnCl2/monomer molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was applied. Sequential heating of the monomer/salt mixture for 10 h each to 400 and 600 °C in quartz ampules leads to the formation of an amorphous and partially carbonized framework containing tunable amounts of N with exceptionally high porosity and specific surface areas.
:1 was applied. Sequential heating of the monomer/salt mixture for 10 h each to 400 and 600 °C in quartz ampules leads to the formation of an amorphous and partially carbonized framework containing tunable amounts of N with exceptionally high porosity and specific surface areas.
In prior studies, quartz ampules of small volumes (l = 12 cm, ∅ = 1.5 cm) have been used to guarantee sufficient heat transfer during the polymerization reaction.29 Unfortunately, the small ampule volume restricts to small quantities of the desired material and therefore the synthesis becomes both cost and time intensive. To tackle this challenge, ampules of larger volume (l = 12 cm, ∅ = 3.0 cm) were used in the present study and result in approximately the 5-fold quantity of the desired product (4-fold ampule volume increase combined with increasing filling height). This enabled increasing the CTF yield from 0.6 g to approximately 3.0 g per batch. Properties of the CTFs synthesized in small and large ampules are summarized in Table 1. The resulting CTF materials were investigated via N2 physisorption as well as elemental and thermogravimetric analysis (Table 1, Fig. S1–S5†). The materials obtained show the characteristic N2 physisorption isotherms comparable to the previously described CTFs synthesized in small scale.29 In this regard, N2 physisorption isotherms of CTFs based on 1,3-DCB (CTF-a, Fig. S1†), 1,4-DCB (CTF-c, Fig. S3†) and 4,4′-DCBP (CTF-d, Fig. S4†) correspond to type IV isotherms, emphasizing the mesoporous structure of all materials. While the hysteresis for both CTF-a and -c is barely existent, CTF-d exhibits a strongly pronounced hysteresis according to the significantly increased porosity, as it would be expected from the structural properties of the monomer. Regardless of the synthesis scale, a material based on 2,6-DCP (CTF-b, Fig. S2†) presents a type I isotherm characteristic for completely microporous materials. The most drastic decrease in porosity and specific surface area when applying large scale synthesis is found for CTF-a based on 1,3-DCB (Table 1, entry 1 and 2). This might be correlated to the incontrollable orientation of the meta-substituted nitrile-function during polymerization, thus leading to unregulated structural parameters. In contrast, only a slight decrease in specific surface area and microporosity is observed for CTF-b (Table 1, entry 4 and 5). This can be assigned to the dense coordination sphere during polymerization, which is caused by coordination of the pyridine backbone to the Lewis-acidic ZnCl2 regardless of the synthesis scale. A similar trend can be observed for CTF-c based on 1,4-DCB, which exhibits a nearly unchanged specific surface area and pore volume regardless of the production scale (Table 1, entry 8 and 9). Interestingly, the specific surface area for CTF-d increases while the total pore volume decreases slightly in large scale synthesis, which can be assigned to favored partial carbonization as reflected by the lower amount of N within the resulting polymer (Table 1, entry 11 and 12). In both cases, the rigid structure of the para-substituted monomers seems to play an important role during the formation of the porous framework. All four materials have been synthesized repeatedly to investigate the reproducibility of the large scale synthesis method (Table S1 and Fig. S7†). Only minor changes in specific surface areas and total pore volume could be detected and the course of the isotherms was unchanged.
| No. | Material | Ca [wt%] | Ha [wt%] | Na [wt%] | SBETb [m2 g−1] | VP/VP(micro)c [cm3 g−1] | Sa,d [wt%] |
|---|---|---|---|---|---|---|---|
| a Determined with elemental analysis.b Specific surface area identified by Brunauer–Emmet–Teller (BET) method.c Total pore volume determined at p/p0 = 0.98 and micropore volume determined via N2-DFT model.d Determined via EA after sulfonation with SO3. A reference of non-sulfonated CTF contained <0.01 wt% of sulfur.e Material obtained via small scale synthesis as described in prior studies.29 With S-CTF-b* as reproduction of S-CTF-b. | |||||||
| 1 | CTF-ae | 72.7 | 2.8 | 9.5 | 2439 | 1.96/0.47 | — |
| 2 | CTF-a | 71.2 | 2.2 | 7.9 | 1840 | 1.15/0.44 | — |
| 3 | S-CTF-a | — | — | — | 1509 | 0.86/0.40 | 1.18 |
| 4 | CTF-be | 48.8 | 3.9 | 17.2 | 1179 | 0.64/0.64 | — |
| 5 | CTF-b | 59.1 | 3.1 | 17.6 | 972 | 0.54/0.38 | — |
| 6 | S-CTF-b | — | — | — | 603 | 0.33/0.24 | 2.67 |
| 7 | S-CTF-b* | — | — | — | 699 | 0.37/0.27 | 0.72 |
| 8 | CTF-ce | 73.4 | 2.2 | 10.4 | 2071 | 1.36/0.43 | — |
| 9 | CTF-c | 62.1 | 2.5 | 6.6 | 2001 | 1.37/0.44 | — |
| 10 | S-CTF-c | — | — | — | 1206 | 0.65/0.37 | 1.27 |
| 11 | CTF-de | 84.9 | 1.7 | 3.7 | 1683 | 2.63/0.30 | — |
| 12 | CTF-d | 75.5 | 4.3 | 1.3 | 1859 | 1.54/0.30 | — |
| 13 | S-CTF-d | — | — | — | 1561 | 1.24/0.26 | 1.57 |
The N content of the resulting CTFs is slightly lower for all materials synthesized in larger scale, except for CTF-b. This seems to correspond to a slightly favored carbonization under these reaction conditions. Nevertheless, reproducibility tests for material synthesis (Table S1†) delivered comparable results for CHN-analysis, confirming the N contents reported in prior studies.29 In summary, N contents within the resulting materials follow the same trend as reported for small scale ampule synthesis and can be correlated to the amount of nitrogen available in the used monomers with slight deviations due to partial carbonization.
Sulfonation in the gas-phase
Via sulfonation, acid sites can be introduced into the solid material, resulting in an acidic catalyst. By introducing acid sites, both polarity and hence hydrophilicity will be increased. Generally, the sulfonation of materials can be carried out in the liquid or the gas phase with sulfuric acid or oleum as a sulfonation agent.30–34 Prior studies could not confirm the sulfonation of CTF materials in the liquid phase.27 Therefore, the sulfonation of the CTFs investigated in the present study was carried out in the gas-phase as it was presented by Richter et al.,34 using 20% oleum (20 wt% of SO3 in sulfuric acid). Applying a dynamic vacuum, SO3 is evaporated from a reservoir, passing through the material exhibited within a glass reactor. The apparatus for the sulfonation is depicted in the ESI (Fig. S8†). The sulfonation in the gas-phase takes place under mild conditions at room temperature and enables easy work-up, while sole adsorption of H2SO4 from the liquid phase on the materials surface is suppressed.The sulfonation of all CTF materials leads to a decrease in both porosity as well as specific surface area (Table 1, entries 3, 6, 7, 10 and 13), which cannot only be explained by the increasing weight after introduction of the sulfonic acid group. More likely, the introduction of sterically demanding –SO3H groups after sulfonation is responsible for the blockage of the pore system. The course of N2 physisorption isotherms of the sulfonated materials (referred to as S-CTF) shows a comparable trend to the untreated materials, indicating that the porous structure stays intact even after treatment (Fig. S1–S4†). The pore size distribution calculated for all S-CTF materials reveals a loss of porosity mainly in the mesoporous range for bimodal samples, while the micropores are mostly unaffected. These structural findings could be reproduced excellently for all four CTFs (Table S1 and Fig. S7†).
TEM images of a sulfonated S-CTF-d in comparison to untreated CTF-d do not reveal any signs of degradation or decomposition (Fig. 1). Furthermore, SEM/EDX mapping images of S-CTF-d confirm sulfur evenly distributed throughout the whole CTF-d material (Fig. 2).
 | ||
| Fig. 1 TEM images of (a) untreated CTF-d and (b) S-CTF-d after sulfonation with SO3 in the gas-phase. | ||
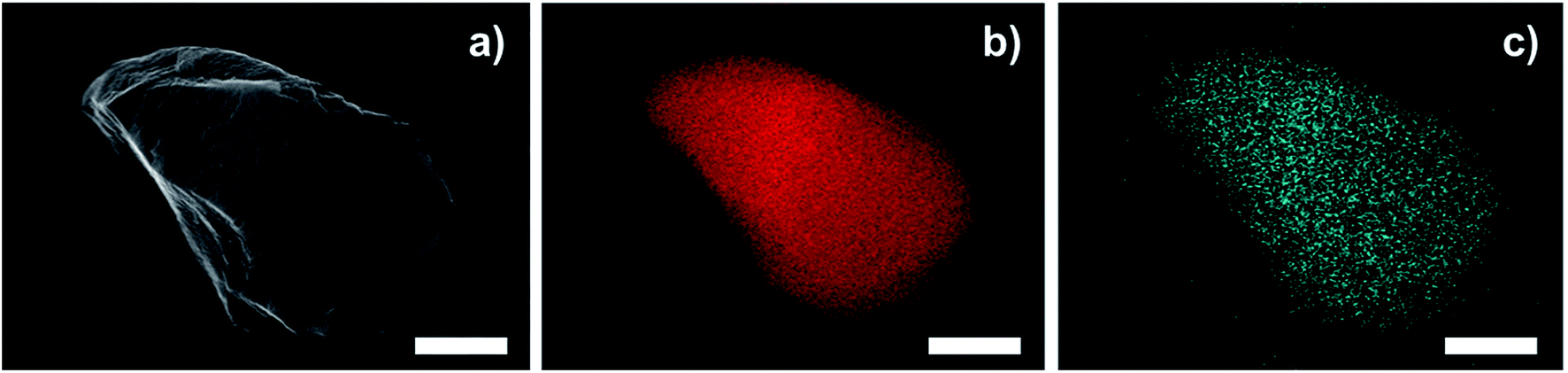 | ||
| Fig. 2 SEM/EDX mapping images of S-CTF-d. (a) SEM of S-CTF-d particle. (b) C mapping (red). (c) S mapping (blue). The scale bars represent 1 μm. | ||
For all S-CTF samples a similar loading of sulfur around 1.2 to 1.6 wt% has been determined via elemental analysis (Table 1). Interestingly, S-CTF-b based on the pyridinic 2,6-DCP monomer contains nearly twice the amount of sulfur after sulfonation (2.67 wt%, Table 1, entry 6). This might be assigned to a SO3-pyridine adduct formation after treatment with SO3 in the gas-phase. Reproducibility tests confirm this assumption, as the reproduced S-CTF-b contained only 0.72 wt% S, while the other S-CTF materials showed comparable loadings as found for the first experiments after extensive washing with an excess of water for a prolonged time period (Table 1, entry 7).
The acidity of all samples was determined via titration, ICP-OES analysis after digestion of the S-CTF and elemental analysis (EA). All analysis techniques give comparable results for the acidity of the S-CTFs being approximately between 0.4 and 0.8 mmol g−1 (Table 2). The results of all the methods were in line with each other. The S-content determined via EA was used to calculate the sulfonation degree for all samples. The degree of sulfonation was calculated for the entire material, assuming complete and ideal polymerization of the corresponding monomer.
| No. | Material | Titrationa [mmol g−1] | ICP-OESb [mmol g−1] | EAc [mmol g−1] | S-degreed [%] |
|---|---|---|---|---|---|
| a Ion exchange in saturated NaCl solution followed by titration with NaOH (0.005 M).b Derived from sulfur content determined via ICP-OES analysis.c Calculated from sulfur content determined via elemental analysis.d Degree of sulfonation calculated as amount of sulfur (mmol) to amount of monomer (mmol) for entire material assuming complete and ideal polymerization. With S-CTF-b* as reproduction of S-CTF-b. | |||||
| 1 | S-CTF-a | 0.38 | 0.36 | 0.37 | 4.7 |
| 2 | S-CTF-b | 0.59 | 0.60 | 0.83 | 10.7 |
| 3 | S-CTF-b* | — | — | 0.23 | 2.8 |
| 4 | S-CTF-c | 0.53 | 0.34 | 0.40 | 5.1 |
| 5 | S-CTF-d | 0.61 | 0.46 | 0.49 | 10.1 |
In general, the sulfur loading does not follow a particular trend but all S-CTF materials contain around 1.0–1.5 wt% of sulfur (∼0.4 mmol g−1) at a sulfonation degree in-between 5–10% on average. This could also be shown via reproducibility tests, which confirm only small discrepancies when sulfonating the same CTF type under identical conditions (Table S1†). The degree of sulfonation for CTF materials is relatively low when compared to commercially available solid acids. E.g. the degree of sulfonation for an amberlyst resin with ion-exchange capacities of 4.8 meq g−1 is 10-fold higher in comparison to the S-CTF materials presented in the present study. On the one hand, this might reflect the high amount of triazine and pyridine moieties and their electronic properties hindering the sulfonation of every single monomer. Especially the high degree of carbonization leads to a reduction of the aromatic functionalities suitable for sulfonation. Hence, there is no idealized polymer structure available as it is for defined polymers such as polystyrene in the case of Amberlyst® resins. On the other hand, the partially carbonized regions of the CTF materials might have been sulfonated, meaning that the amount of fixated SO3H-functionalities does not correspond to the ideal aromatic functions within the material, but rather to the expected carbonized units. However, distinction between both functionalizations is rather difficult and even via 13C-NMR of different sulfonated carbons,20 no clear evidence of the incorporated SO3H-function could be verified.
Thermogravimetric analysis of sulfonated S-CTF-a, -c and -d indicate the loss of the sulfonic acid functionalities at temperatures above 300 °C (Fig. S5 and S6†). Decomposition of the porous material is initiated at temperatures above 400 °C. In the case of CTF-b, desulfonation takes place at somewhat lower temperatures. This can be attributed to the assumed adduct formation to the pyridinic function of the 2,6-DCP monomer.
Additionally, CTF-a was sulfonated in the liquid phase to compare the materials obtained by different procedures (Table S1 and Fig. S7†). Interestingly, the liquid phase sulfonation resulted in higher sulfur loadings of 3.0 wt% leading to an acidity of 0.96 mmol g−1 and thus a sulfonation degree of approximately 12.2%. The amount of acid sites is 2-fold higher compared to the gas-phase sulfonation method, but can mostly be attributed to adsorption effects of H2SO4 on the materials surface and adduct formation to the triazine moieties, which cannot be suppressed in the liquid phase. This assumption can further be confirmed by thermogravimetric analysis as the desulfonation begins at relatively low temperatures in comparison to the S-CTF-a sulfonated in the gas-phase (Fig. S5†).
It is well-known that sulfonated carbons can catalyze the hydrolysis of cellulose and cellobiose to glucose at high conversions and yields.20 Since the CTF materials presented in this work can be allocated to sulfonated N-doped carbon systems, we decided to investigate them within the same catalytic reaction in order to verify their successful functionalization.
Catalytic hydrolysis of cellobiose to glucose
The synthesized S-CTF materials were tested for the hydrolysis of 10 wt% aqueous cellobiose solution at 150 °C. The results of the tests are shown in Table 3. All the sulfonated materials were catalytically active for the hydrolysis. Very high cellobiose conversions up to 88% are attainable over mesoporous catalysts, whereas only 31–55% were reached over microporous catalysts for 2 h of reaction (Table 3, entry 5 and 11). Prolongation of the reaction time for another 2 h did not bring a significant increase of the conversion (Table 3, entry 6 and 12). This points at deactivation of the microporous catalysts under the reaction conditions, probably due to adsorption of substrate/product in the micropores. Additionally, leaching of sulfur corresponding to 20–40% of its initial content in the materials was observed (Table 3). Leaching of sulfonic groups into the aqueous medium led to formation of sulfuric acid under reaction conditions. As a result, acidification took place during the experiments, and slightly acidic solutions were obtained after filtration of the reaction mixtures.| No. | Catalyst | t [h] | X [%] | Y [%] | pH | Leached sulfur [%] | khydr [h−1] | khomo [H+]/khydr [%] | khetero [L mol−1 h−1] |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1–4 h, 150 °C, 30 bar of N2 at 750 rpm. Acid:cellobiose molar ratio (given in brackets): S-CTF-a (1.7 mol%), S-CTF-b (0.7 mol%), S-CTF-c (1.8 mol%) and S-CTF-d (1.4 mol%). |
|||||||||
| 1 | S-CTF-a | 1 | 32.9 | 33.3 | 2.5 | 34.0 | 0.58 | 53 | 7.2 |
| 2 | S-CTF-a | 2 | 59.2 | 59.7 | 2.5 | 42.4 | |||
| 3 | S-CTF-a | 4 | 88.1 | 85.6 | 2.2 | 48.4 | |||
| 4 | S-CTF-b | 1 | 3.4 | 10.9 | 3.3 | 38.1 | 0.33 | 24 | 5.0 |
| 5 | S-CTF-b | 2 | 30.6 | 24.7 | 3.2 | 43.8 | |||
| 6 | S-CTF-b | 4 | 34.6 | 27.8 | 3.2 | 29.8 | |||
| 7 | S-CTF-c | 1 | 41.3 | 42.7 | 2.7 | 29.5 | 0.62 | 44 | 8.6 |
| 8 | S-CTF-c | 2 | 49.6 | 51.5 | 2.6 | 24.7 | |||
| 9 | S-CTF-c | 4 | 85.4 | 83.9 | 2.6 | 46.2 | |||
| 10 | S-CTF-d | 1 | 20.0 | 23.5 | 2.7 | 27.8 | 0.59 | 46 | 6.5 |
| 11 | S-CTF-d | 2 | 55.8 | 62.3 | 2.6 | 32.0 | |||
| 12 | S-CTF-d | 4 | 66.2 | 66.0 | 2.6 | 32.9 | |||
Hydrolysis of cellobiose can proceed both homogeneously and heterogeneously catalyzed, as shown in Fig. 3. The first orders on cellobiose and H+ active species were assumed.37 The parallel processes yield glucose with the rate constants of hydrolysis khomo and khetero, respectively. Apparently, the overall rate constant for the hydrolysis can be expressed as khydr = (khomo[H+] + khetero[H+]surfCcat). The values of khydr were obtained from the results of cellobiose hydrolysis in the presence of the sulfonated CTF materials (Table 3).
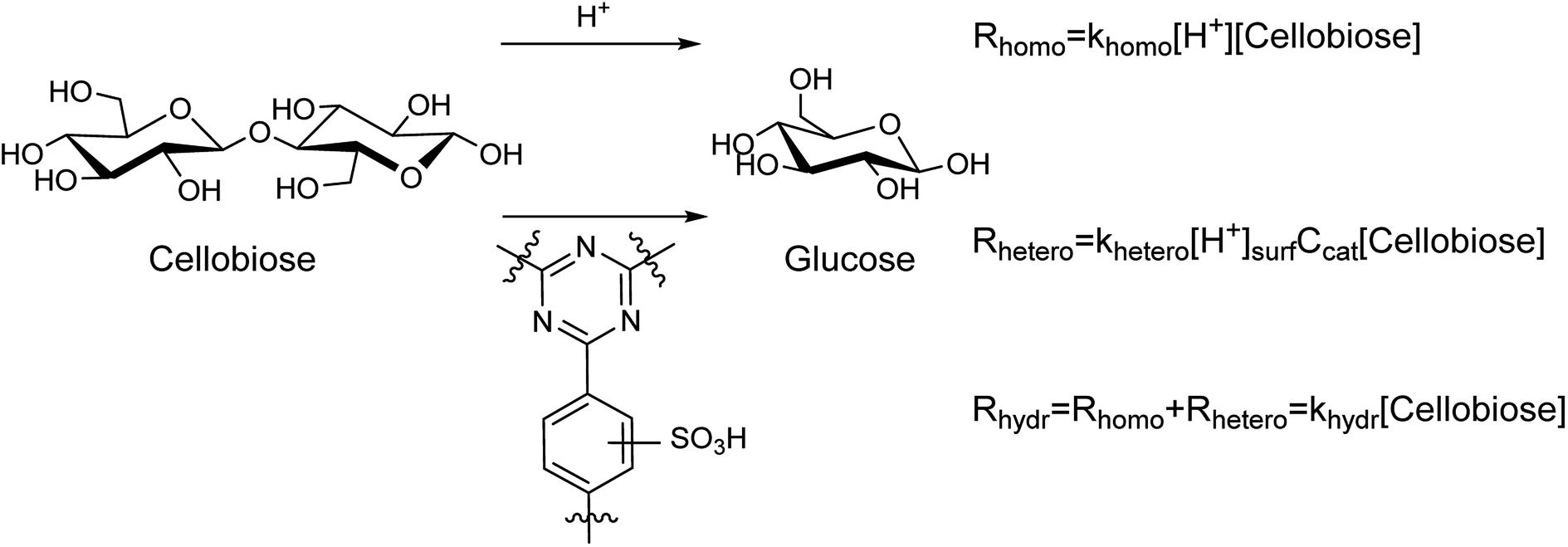 | ||
| Fig. 3 Parallel homogeneous and heterogeneous routs for cellobiose hydrolysis in the presence of S-CTF. Rhomo, Rhetero, and Rhydr denote the rate of homogeneous hydrolysis, the rate of heterogeneous hydrolysis, and the apparent reaction rate (mol L−1 h−1). The rate constants of homogeneous (khomo, L mol−1 h−1) and heterogeneous (khetero, L mol−1 h−1) hydrolysis as well as overall rate constant khydr (h−1) are considered, [H+] and [cellobiose] denote concentrations (mol L−1) of these species in aqueous phase, Ccat is concentration of the catalyst (g L−1). [H+]surf is a surface concentration of acidic groups (mol g−1). | ||
Homogeneous hydrolysis of cellobiose under the applied conditions was investigated. A blank test of cellobiose hydrolysis demonstrated that hydrolysis takes place even in the absence of a catalyst. The hydrolysis of cellobiose accelerated during the course of the experiment (Fig. S9†). This is in line with acidification of the aqueous medium from pH 5.2 till pH 3 for the first hour of the hydrolysis. Further prolongation of the reaction did not lead to dramatic change of the pH value (Fig. S9†). The observed pH change can be explained by formation of acidic by-products upon dehydration of saccharides. It must be noted that the reaction mixture turned brownish upon hydrolysis in pure water. Clearly, despite rather high selectivity for glucose (88–95%), the saccharides partly underwent degradation forming humins. Next, H2SO4 was tested for hydrolysis of cellobiose. In these tests, the concentration of sulfuric acid (ca. 1.6 mM, pH 2.4) was close to that obtained over S-CTFs due to leaching of sulfur (Table 3). Again, darkening of the reaction solution took place owing to formation of humins. Finally, we studied catalytic activity of non-sulfonated CTF-a and a mixture of CTF-a with H2SO4. Interestingly, filtration of the reaction mixture after the experiments gave transparent solutions. This indicates efficient adsorption of humins onto CTF under the reaction conditions. Furanic compounds, mainly HMF, are products of consecutive dehydration of glucose under acidic conditions. A generally accepted mechanism of humin formation comprehends condensation of the furanics with saccharides.38 Apparently, adsorption of humins onto CTF is favorable due to high affinity of HMF towards aromatic functionalities of CTF.39 Furthermore, the pH value of the reaction mixture after the reaction over CTF + H2SO4 (2.9) was slightly higher compared to the pH0 value of 2.4. This can be explained by adsorption of sulfuric acid onto basic centers of the CTF material. We performed a control experiment to investigate adsorption of H2SO4 onto CTF at room temperature. Stirring of 1.6 mM H2SO4 with CTF-a led to total neutralization of the solution, apparently due to interaction of the strong acid with nitrogen of triazine rings (Fig. S10†). The values of khomo were estimated based on the obtained data (Table 4). Interestingly, the rate constants appear to be similar for different reaction conditions and independent of the nature of acid (H2SO4 or organic acids produced due to dehydration of saccharides). The presence of the CTF-a material does not influence the rate constant of cellobiose hydrolysis. Data on pH of reaction mixture (Table 3), overall hydrolysis constants khydr (Table 3), concentration of surface acidic sites (Table 2), and khomo (ca. 123 L mol−1 h−1, according to the data from Table 4) enable estimation of khetero values according to the equations presented in Fig. 3. The results of these calculations are listed in Table 3, indicating that the homogeneous reaction makes a contribution of 24–53% to the apparent rate of cellobiose hydrolysis. The apparent khetero appeared to be similar for all the materials, though a somewhat lower value was observed for CTF-b.
| No. | Catalyst | khomo, L mol−1 h−1 |
|---|---|---|
| 1 | No | 134 |
| 2 | H2SO4 | 117 |
| 3 | CTF-a | 127 |
| 4 | H2SO4 + CTF-a | 114 |
Activation energy of the hydrolysis equals ca. 115 kJ mol−1 in the presence of the most active catalyst S-CTF-a (Fig. S11†), similar to the previously obtained data for molecular acids.37,40 Importantly, share of the leached out sulfur of ca. 20% does not dramatically depend on the reaction temperature (Fig. 4). This indicates the presence of weakly bound sulfur-containing groups, which are easily removed upon contact with hot water. Previous investigations suggest that sulfonated carbon materials also exhibit weakly- and strongly bound sulfonic groups.41,42 Pre-treatment of the sulfonated carbons with hot water results in removal of the weakly bound sulfur, whereas strongly bound groups were stable under the reaction conditions of cellobiose hydrolysis.41,43 This was also indicated via a hot filtration test resulting in only slight increase of conversion and yield after filtration of the solid catalyst and processing the liquid phase for additional 2 hours (Fig. 4b).
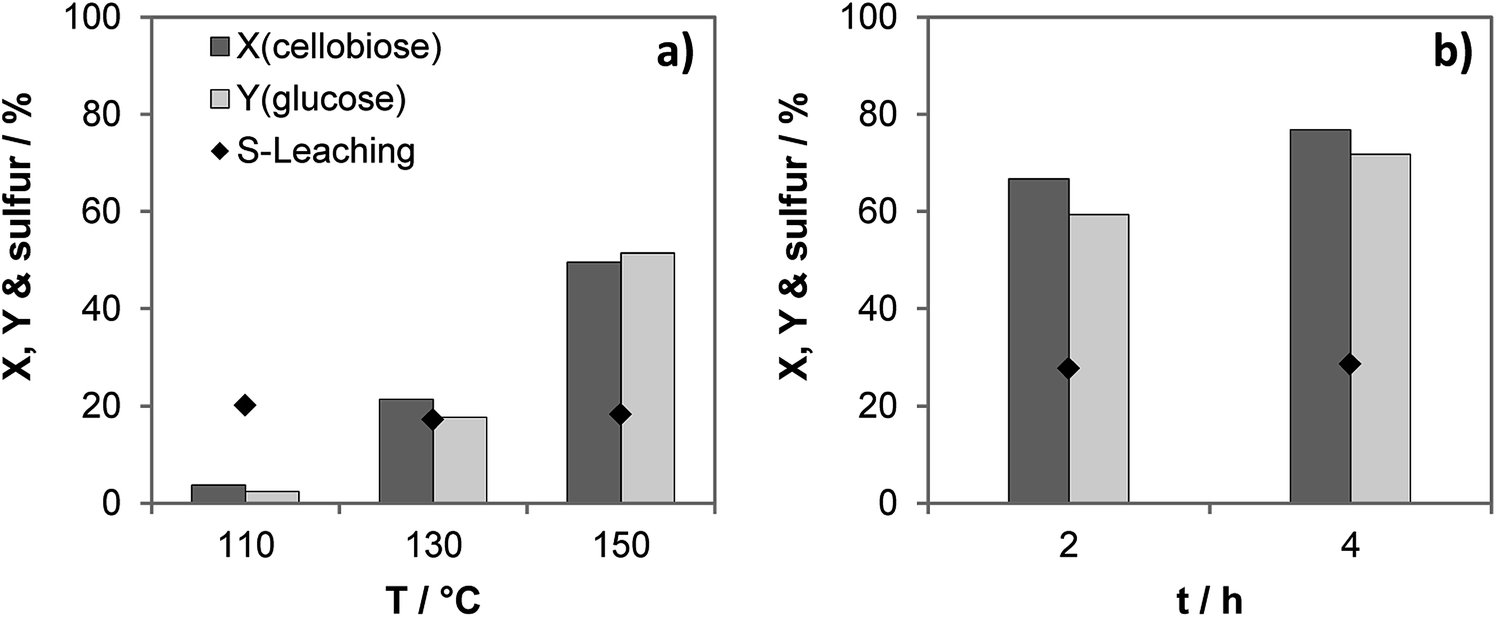 | ||
| Fig. 4 (a) Temperature dependency of the cellobiose conversion (X), glucose yield (Y) and sulfur leaching; (b) hot filtration test. Reaction conditions: 2 h, 110–150 °C, 30 bar of N2 at 750 rpm, 5 mL of 10 wt% cellobiose, 50 mg of S-CTF-a. | ||
We studied the recycling of the S-CTF-a catalyst (Fig. 5). As expected, high amounts of sulfur, corresponding to 43% content in solid catalyst, were only leached in the first run. In the following two runs, only 4 and 2% of the initial S concentration were detected in the product solution. In total, ca. 49% of the initial sulfur content was leached out of the catalyst during the three consecutive runs, which was confirmed by ICP-OES analysis of the liquid after reaction and the elemental analysis of the used catalyst. A significant drop of the cellobiose conversion from 78 to 36% was observed for the first and the second run, which can be explained by considerable loss of sulfur during the first run. Despite very low leaching of sulfur during the second and the third runs, further decrease of the cellobiose conversion to 12% occurred in the third run. Therefore, catalyst deactivation cannot only be attributed to the leaching of the catalytically active species. Our results on molecular H2SO4 physically mixed with CTF-a (see above) suggest that the materials are very prone to adsorption of partly dehydrated by-products, which probably brings about blockage of active sites and drop of catalytic activity.
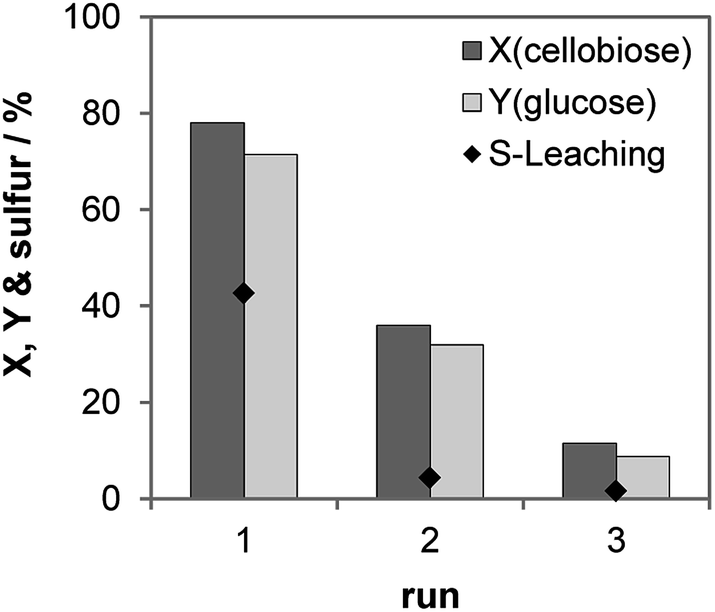 | ||
| Fig. 5 Recycling study of S-CTF-a. Reaction conditions: 2 h, 150 °C, 30 bar of N2 at 750 rpm, 5 mL 10 wt% cellobiose, 75 mg S-CTF-a. | ||
Further improvements of the catalytic activity can possibly be achieved by optimization of the procedure of catalyst synthesis and pretreatment,41,42 as well as conducting the reaction under continuous conditions to facilitate the removal of (by)-products from the reactor.43,44
Conclusions
Sulfonation of CTF materials was conducted for the first time by treating the materials with gas-phase SO3. This procedure was successfully applied to obtain four S-CTFs which vary in composition of the monomers. Sulfonation degrees in the range of 5–10% and an acid surface concentration of around 0.4–0.8 mmol g−1 were reached. The acid sites are evenly distributed within the CTF materials, while the material itself does not show any signs of decomposition after sulfonation. Thermal stability of the sulfonated S-CTFs suggests a covalent binding of the introduced acid sites. Notably, the CTFs preserve their structural properties and slight losses in specific surface area as well as porosity can be assigned mostly to the space demanding acid sites.Catalytic testing provides evidence of the successful acid functionalization of the polymers. The S-CTF materials were active when applied in the catalytic hydrolysis of cellobiose to glucose yielding up to 86% glucose in 4 hours at a rather high substrate-to-catalyst weight ratio of 10. Very high selectivity of glucose formation over 90% was observed for hydrolysis in the presence of S-CTF. Leaching of sulfur took place during the course of hydrolysis, as it is expected for any sulfonated materials under the hydrothermal reaction conditions. Additionally, co-formation of acidic by-products was observed. Ca. 24–53% of cellobiose were hydrolyzed over S-CTFs due to contribution of homogeneous catalysis. The leaching along with strong adsorption of (by)-products on the surface of the S-CTF appears to be a reason for catalyst deactivation. Nevertheless, the successful sulfonation of CTFs in combination with afore published coordination approaches of molecular catalysts within the frameworks enables new synthesis routes for multi-functional catalysts. In this manner, the design of a catalyst containing basic coordination sites for molecular or nanoparticulate species, tunable structural parameters as well as surface acidity opens the path to numerous applications, enabling the rational development of multi-step reactions at one sole catalyst system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the DFG (PA1689/1–1) and funding from the Excellence Initiative of the German federal and state governments. SEM/EDX mapping and TEM imaging was performed at the Center for Chemical Polymer Technology CPT, which is supported by the EU and the federal state of North Rhine-Westphalia (grant no. EFRE 30 00 883 02).Notes and references
- M. Baerns, A. Behr, A. Brehm, J. Gmehling, H. Hofmann, U. Onken and A. Resch, in Technische Chemie, Wiley-VCH, Weinheim, 1st edn, 2006 Search PubMed.
- M. B. Hocking, in Handbook of Chemical Technology and Pollution Control, Academic Press, London, 2nd edn, 1998 Search PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef.
- K. Tanabe and W. F. Hölderich, Appl. Catal., A, 1999, 181, 399–434 CrossRef.
- A. Corma and H. Garciam, Catal. Today, 1997, 38, 257–308 CrossRef.
- S. M. Auerbach, K. A. Carrado and P. K. Dutta, in Handbook of Zeolithe Science and Technology, Marcel Dekker Inc., Basel, 1st edn, 2003 Search PubMed.
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef PubMed.
- M. A. Harmer and Q. Sun, Appl. Catal., A, 2001, 221, 45–62 CrossRef.
- V. V. Ordomsky, J. C. Schouten, J. van der Schaaf and T. A. Nijhuis, Chem. Eng. J., 2012, 207, 218–255 CrossRef.
- Dow Chemical Company, Form No. 177-02436-0511, 2011.
- J. H. Clark, Acc. Chem. Res., 2002, 35, 791–797 CrossRef PubMed.
- E. Lotero, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin Jr, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef.
- P. A. Russo, M. M. Antunes, P. Neves, P. V. Wiper, E. Fazio, F. Neri, F. Barreca, L. Mafra, M. Pillinger, N. Pinna and A. A. Valente, J. Mater. Chem. A, 2014, 2, 11813–11824 RSC.
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef.
- P. Lanzafame, D. M. Temi, S. Perathoner, G. Centi, A. Macario, A. Aloise and G. Giordano, Catal. Today, 2011, 175, 435–441 CrossRef.
- (a) J.-P. Lange, Angew. Chem., Int. Ed., 2015, 54, 13186–13197 CrossRef PubMed; (b) A. T. To, P.-W. Chung and A. Katz, Angew. Chem., Int. Ed., 2015, 54, 11050–11053 CrossRef PubMed.
- L. Vilcocq, P. C. Castilho, F. Carvalheiro and L. C. Duarte, ChemSusChem, 2014, 7, 1010–1019 CrossRef PubMed.
- H. Wang, Y. Guo, C. Chang, X. Zhu, X. Liu, J. Han and Q. Ge, Appl. Catal., A, 2016, 523, 182–192 CrossRef.
- (a) Y. Kim, R. Hendrickson, N. Mosier and M. R. Ladisch, Energy Fuels, 2005, 19, 2189–2200 CrossRef; (b) A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363–5365 RSC; (c) L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889–6894 RSC.
- (a) S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef PubMed; (b) M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima, H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25, 5068–5075 CrossRef PubMed; (c) G. S. Foo, A. H. Van Pelt, D. Krötschel, B. F. Sauk, A. K. Rogers, C. R. Jolly, M. M. Yung and C. Sievers, ACS Sustainable Chem. Eng., 2015, 3, 1934–1942 CrossRef.
- J. A. Bootsma and B. H. Shanks, Appl. Catal., A, 2007, 327, 44–51 CrossRef.
- (a) M. Karaki, A. Karout, J. Toufaily, F. Rataboul, N. Essayem and B. Lebeau, J. Catal., 2013, 305, 204–216 CrossRef; (b) L. Sandbrink, T. Lazaridis, M. Rose and R. Palkovits, Microporous Mesoporous Mater., 2018, 267, 198–202 CrossRef.
- R. Ormsby, J. R. Kastner and J. Miller, Catal. Today, 2012, 190, 89–97 CrossRef.
- X. Zhao, J. Wang, C. Chen, Y. Huang, A. Wang and T. Zhang, Chem. Commun., 2014, 50, 3439–3442 RSC.
- (a) P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef PubMed; (b) P. Kuhn, A. Thomas and M. Antonietti, Macromolecules, 2009, 42, 319–326 CrossRef.
- (a) J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef PubMed; (b) P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef.
- (a) R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef PubMed; (b) M. Soorholtz, R. J. White, T. Zimmermann, M.-M. Titirici, M. Antonietti, R. Palkovits and F. Schüth, Chem. Commun., 2013, 49, 240–242 RSC.
- (a) C. E. Chan-Thaw, A. Villa, G. M. Veith, K. Kailasam, L. A. Adamczyk, R. R. Unocic, L. Prati and A. Thomas, Chem.–Asian J., 2012, 7, 387–393 CrossRef PubMed; (b) S. Armenise, L. Roldán, Y. Marco, A. Monzón and E. García-Bordejé, J. Phys. Chem. C, 2012, 116, 26385–26395 CrossRef.
- (a) J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef PubMed; (b) J. Artz and R. Palkovits, ChemSusChem, 2015, 8, 3832–3838 CrossRef PubMed; (c) A. K. Beine, A. J. D. Krüger, J. Artz, C. Weidenthaler, C. Glotzbach, P. J. C. Hausoul and R. Palkovits, Green Chem., 2018, 20, 1316–1322 RSC; (d) J. Artz, ChemCatChem, 2018, 10, 1753–1771 CrossRef.
- R. Brückner, in Reaktionsmechanismen, Spektrum Akademischer Verlag, Berlin, 3rd edn, 2007 Search PubMed.
- F. Kucera and J. Jancar, Polym. Eng. Sci., 1998, 38, 783–792 CrossRef.
- F. H. Richter, K. Pupovac, R. Palkovits and F. Schüth, ACS Catal., 2013, 3, 123–127 CrossRef.
- G. Schäfer, P. Neumann and G. Blinne, German patent, DE3732539 A1, BASF, 1989.
- F. H. Richter, Y. Meng, T. Klasen, L. Sahraoui and F. Schüth, J. Catal., 2013, 308, 341–351 CrossRef.
- C. Bai, M. Zhang, B. Li, Y. Tian, S. Zhang, X. Zhao, Y. Li, L. Wang, L. Ma and S. Li, J. Hazard. Mater., 2015, 300, 368–377 CrossRef PubMed.
- S. N. Talapaneni, T. H. Hwang, S. H. Je, O. Buyukcakir, J. W. Choi and A. Coskun, Angew. Chem., Int. Ed., 2016, 55, 3106–3111 CrossRef PubMed.
- (a) O. Bobleter, W. Schwald, R. Concin and H. Binder, J. Carbohydr. Chem., 1986, 5, 387–399 CrossRef; (b) N. S. Mosier, C. M. Ladisch and M. R. Ladisch, Biotechnol. Bioeng., 2002, 79, 610–618 CrossRef PubMed.
- (a) M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281 CrossRef; (b) M. Sevilla and A. B. Fuertes, Chem.–Eur. J., 2009, 15, 4195–4203 CrossRef PubMed; (c) I. van Zandvoort, Y. Wang, C. B. Rasrenda, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef PubMed.
- C. Detoni, C. H. Gierlich, M. Rose and R. Palkovits, ACS Sustainable Chem. Eng., 2014, 2, 2407–2415 CrossRef.
- L. Negahdar, J. U. Oltmanns, S. Palkovits and R. Palkovits, Appl. Catal., B, 2014, 147, 677–683 CrossRef.
- (a) A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC; (b) M. Karaki, A. Karout, J. Toufaily, F. Rataboul, N. Essayem and B. Lebeau, J. Catal., 2013, 305, 204–216 CrossRef.
- A. H. Van Pelt., O. A. Simakova, S. M. Schwimming, J. L. Ewbank, G. S. Foo, E. A. Pidko, E. J. M. Hensen and C. Sievers, Carbon, 2014, 77, 143–154 CrossRef.
- G. S. Foo, A. H. Van Pelt, D. Krötschel, B. F. Sauk, A. K. Rogers, C. R. Jolly, M. M. Yung and C. Sievers, ACS Sustainable Chem. Eng., 2015, 3, 1934–1942 CrossRef.
- I. Delidovich and R. Palkovits, Catal. Sci. Technol., 2014, 4, 4322–4329 RSC.
- I. Delidovich and R. Palkovits, Microporous Mesoporous Mater., 2016, 219, 317–321 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04254c |
| This journal is © The Royal Society of Chemistry 2018 |