
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew 3D graphical representation for RNA structure analysis and its application in the pre-miRNA identification of plants
Xiangzheng Fu ,
Bo Liao*,
Wen Zhu and
Lijun Cai*
,
Bo Liao*,
Wen Zhu and
Lijun Cai*
College of Information Science and Engineering, Hunan University, Changsha, Hunan 410082, China. E-mail: fxz326@hnu.edu.cn; Excelsior511@126.com
First published on 3rd September 2018
Abstract
MicroRNAs (miRNAs) are a family of short non-coding RNAs that play significant roles as post-transcriptional regulators. Consequently, various methods have been proposed to identify precursor miRNAs (pre-miRNAs), among which the comparative studies of miRNA structures are the most important. To measure and classify the structural similarity of miRNAs, we propose a new three-dimensional (3D) graphical representation of the secondary structure of miRNAs, in which an miRNA secondary structure is initially transformed into a characteristic sequence based on physicochemical properties and frequency of base. A numerical characterization of the 3D graph is used to represent the miRNA secondary structure. We then utilize a novel Euclidean distance method based on this expression to compute the distance of different miRNA sequences for the sequence similarity analysis. Finally, we use this sequence similarity analysis method to identify plant pre-miRNAs among three commonly used datasets. Results show that the method is reasonable and effective.
Introduction
MicroRNAs (miRNAs) are a family of short noncoding RNAs that play significant roles as post-transcriptional regulators.1 With extracellular miRNAs, hypothalamic stem cells partially control the aging rate.2 As such, miRNA is an important noncoding RNA involved in many important biological processes, including plant development, signal transduction, and protein degradation.3,4 miRNA prediction has constantly been an important issue in the miRNA research domain. The bases of single-stranded miRNAs in live cells are constantly folded to form an miRNA secondary structure rather than a linear form. The three-dimensional (3D) structure and function of miRNAs are determined by their secondary structures,3 and their functions are mainly determined by their structures.5 Thus, studies on RNA sequences and their secondary structures are essential for identifying and understanding the functional similarities between plant miRNAs. Precursor miRNAs (pre-miRNAs) of plants generally have a more complex secondary structure than those of animals, and existing prediction methods on animal pre-miRNA classification cannot be effectively applied to predict plant pre-miRNAs.6 Experimental methods, such as ChIP-sequencing for pre-miRNA identification, are expensive and time consuming, thereby presenting the need for computational methods. Computational methods, including machine learning (ML) and sequence analysis methods, should be developed to predict, analyze, and provide reliable miRNA candidates for subsequent biological experiments.7ML-based methods have been widely applied to identify plant miRNAs.1,8–15 ML-based methods have treated pre-miRNA identification as a binary classification task to discriminate between real and pseudo-pre-miRNAs. However, the performance of ML-based predictors mainly depends on ML algorithms or operation engines. Numerous classification prediction algorithms, which yield different results, have been utilized to recognize pre-miRNA. ML-based algorithms include support vector machines (SVM),1,8,16–26 back-propagation and self-organizing map (SOM) neural networks,27–29 and random forest (RF).30–32 Difficulties in using ML-based methods are attributed to the selection of representative samples that adequately describe the sample space of an entire positive dataset (pre-miRNA) and negative dataset counterexamples (pseudo pre-miRNA). Computational complexity in predicting large genome mass data is also high. These approaches involve a large number of false positive candidates. Therefore, miRNA classification prediction should be investigated and solved on the basis of ML prediction methods to improve sensitivity and specificity.
Sequence-based methods, including sequence alignment and distance analysis, are mainly used to analyze the similarities between miRNA sequences. T. Dezulian et al.33 used BLAST for sequence alignment to search for homologous sequences that are similar to known plant pre-miRNAs. The similarity of sequence distance is mainly transformed into the similarity between analysis sequences and secondary structures by graphical representation. Graphical representation has been widely applied to RNA sequence representation, especially for the analysis of RNA secondary structures. Y. H. Yao et al.34,35 proposed a graphical representation based on two-dimensionality (2D) to analyze the similarity of RNA secondary structures. On the basis of sequence and base physicochemical information, Jeffrey et al.36,37 proposed a 3D representation of RNA secondary structures. Liao et al.38,39 proposed four- to seven-dimensional graphical representation method for RNA secondary structures. This method can solve the problem of structural degradation and information loss of 2D graphical representation, but it is not conducive to graphic visualization. Zhang et al.40–42 developed a graphical representation for ncRNA secondary structures. To validate the aforementioned methods, researchers usually build phylogenetic trees based on the similarity between sequences to compare the reliability of the methods. In contrast to ML or other complex computing techniques, a graphical representation is an effective analysis method that can provide an intuitive and unique perspective in analyzing sequence similarity.
In this study, we propose a new 3D graphical representation of miRNA secondary structures. In this representation, an miRNA secondary structure is initially transformed into a characteristic sequence based on the frequency and physicochemical properties of nucleic acids. A numerical characterization of the 3D graph is then used to represent the miRNA secondary structure. On the basis of the proposed 3D graphical representation method, we utilize a novel Euclidean distance method to compute the distance of different miRNA secondary structures for similarity analysis. A small distance indicates a high similarity and vice versa. We use this similarity analysis method to identify plant pre-miRNAs among three commonly used datasets. Our results show that our method is reasonable, effective, simple to operate without training parameters, and more intuitive than several ML-methods.
Methods
Framework of the proposed method
Fig. 1 illustrates the overall framework of our method, which consists of two main phases, namely, pre-miRNA similarity analysis and prediction. In the similarity analysis phase, the initial pre-miRNA sequences are extracted from the raw data. Then, homology bias is avoided by using the CD-HIT software43 (threshold set to 0.8) to filter samples with a similarity greater than the threshold in the initial dataset, and the secondary structure of the given benchmark dataset is predicted with the RNAfold software.44 We design a new 3D graphical representation to represent the miRNA secondary structure. On the basis of the proposed method, we utilize a novel Euclidean distance method to compute the distance of different miRNA secondary structures for similarity analysis. In the pre-miRNA prediction phase, the distance between any two sequences in the benchmark datasets is calculated using the proposed method. The smaller the distance is, the more similar the two pre-miRNA sequences will be. The jackknife method is applied to traverse the entire benchmark datasets and to predict whether a given sequence is a plant pre-miRNA.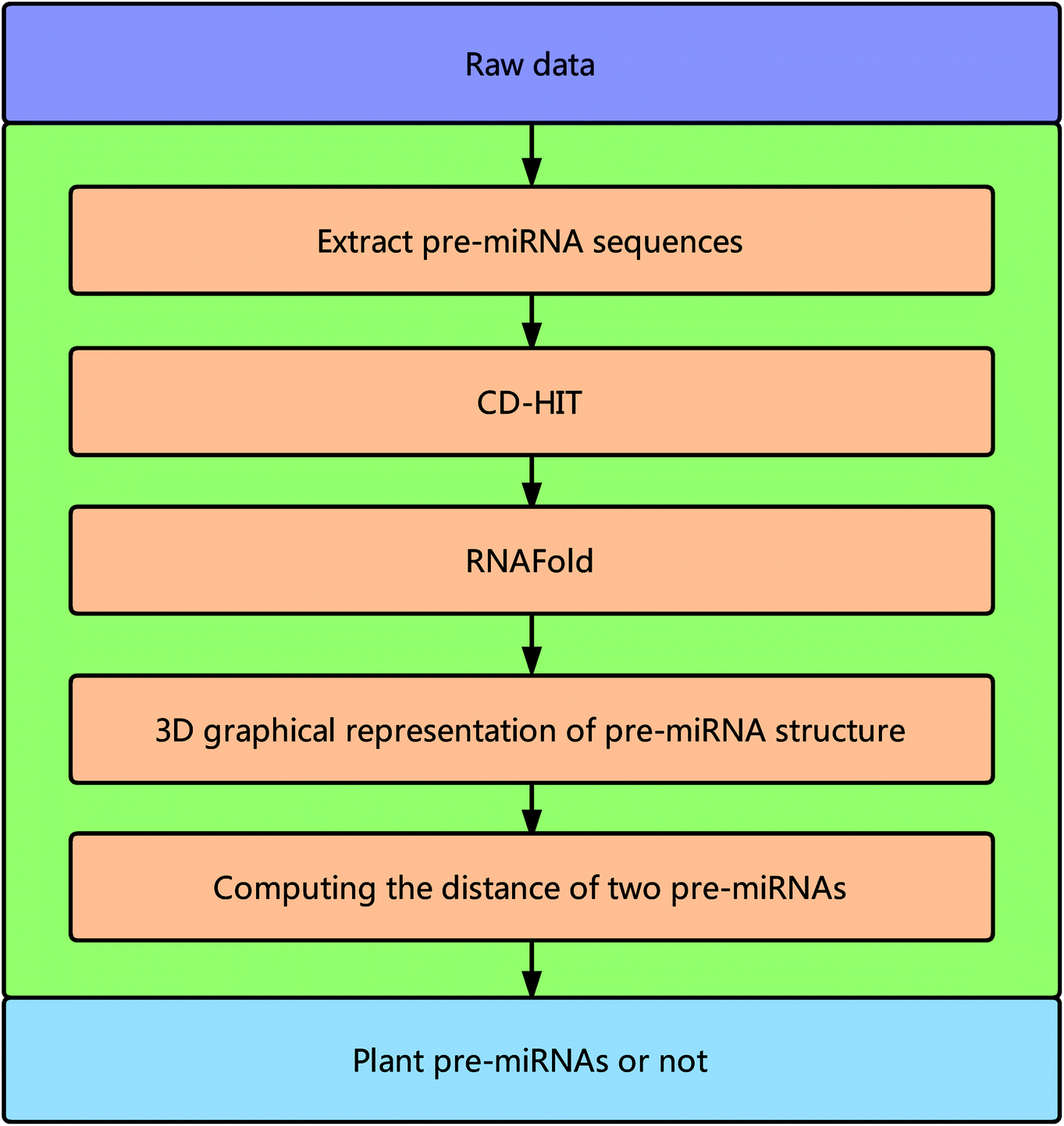 | ||
| Fig. 1 Overall framework of the proposed method. | ||
New 3D graphical representation of miRNA structure
The secondary structure of RNAs consists of a number of free bases (i.e., A, G, C, and U) and paired bases (i.e., A–U, G–C, and G–U). A total of 9 viral RNA base sequences are obtained from ref. 45. Fig. 2 shows the secondary structure of the RNA sequence of the obtained TSV-3 and AIMV-3 using the algorithm in ref. 46.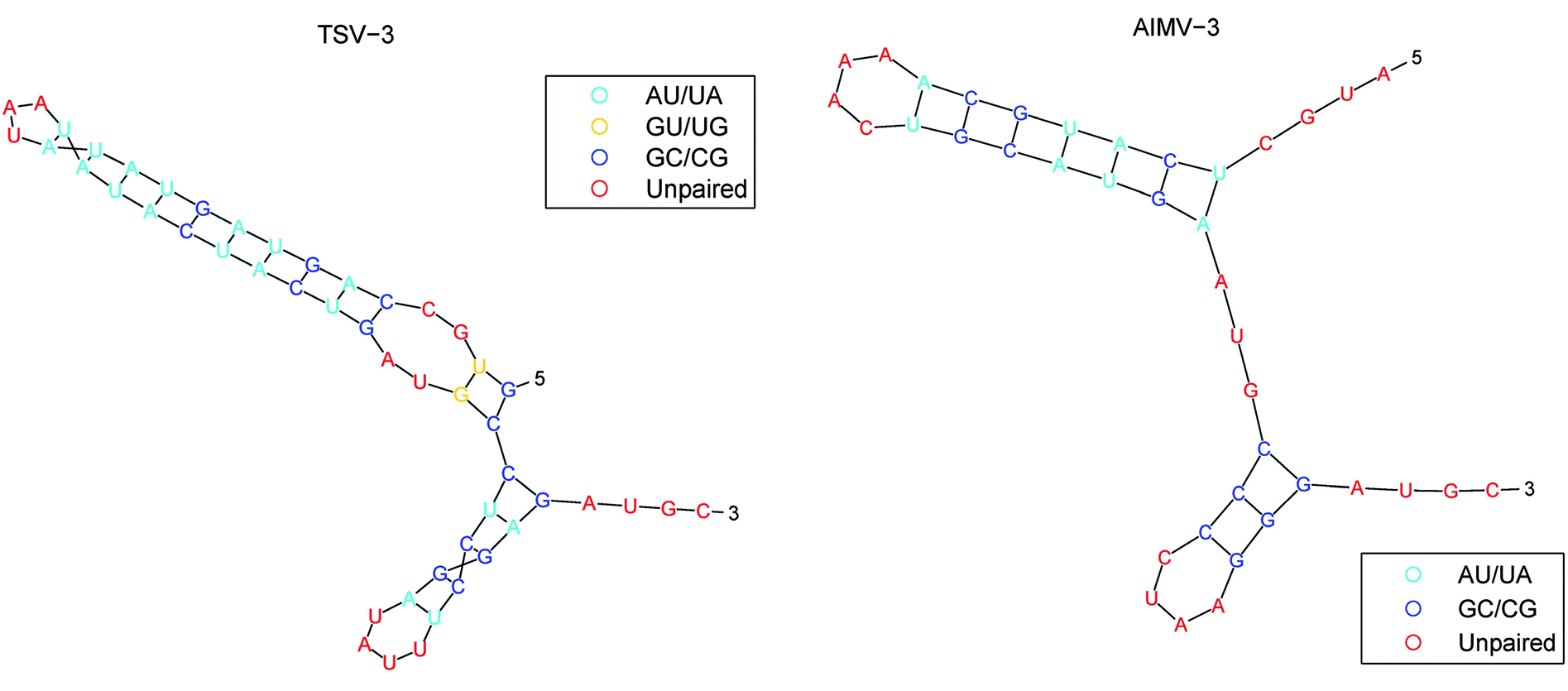 | ||
| Fig. 2 The secondary structure of the RNA sequence of the TSV-3 and AIMV-3. | ||
For research convenience, the base and unpaired bases should be distinguished. The bases of A, G, C, and U located in base pairs A–U, G–C, and G–U are denoted as a, g, c, and u, respectively. The RNA sequences of the 9 obtained viruses from ref. 45 are processed by RNAfold,44 and the RNA secondary structure sequence is shown in Table 1.
| Species | RNA secondary structure | Length |
|---|---|---|
| AIMV-3 | AUGCucaugcaAAACugcaugaAUGCcccUAAgggAUGC | 39 |
| APMV-3 | AAUGCccacaacGUGAAguuguggAUGCcccGUUAgggAAGC | 42 |
| AVII | AUGCcuaaUacucucucuCAGggagagaguuuagAUGCcuccAAAggagAUGC | 53 |
| CILRV | AUGCcuauauuuucucUCCUgagaaaauauagAUGCcuccAAAggagAUGC | 51 |
| CVV-3 | AUGCccaAAcucucucuCAUggagagagAAuggAUGCcuccGAAggagAUGC | 52 |
| EMV-3 | CcuaauUcucucucuCACggagagagauuagAUGCcucCAAGgagAUGC | 49 |
| LRMV-3 | UUCcuauucucucucUCAGgagagGagaauagAUGCcuccAAAggagUCGC | 51 |
| PDV-3 | AUGCccucaccGUAAggugaggAUGCcccuUAAagggAUGC | 41 |
| TSV-3 | GUGCcaguaguauaUAAuauacuacugAUGCcuccuUUAUaggagAUGC | 49 |
Let s = s1, s2, s3, …, sn represent an RNA secondary structure sequence, where n is the length of the sequence. Let point coordinates si(xi, yi, zi) be the i-th base of the secondary structure sequence of miRNA, which corresponds to the eqn (1).
 | (1) |
 | (2) |
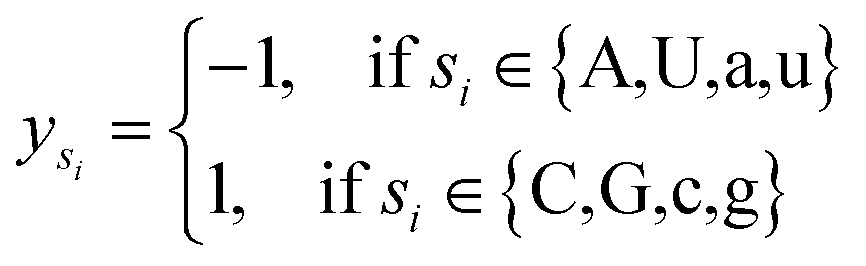 | (3) |
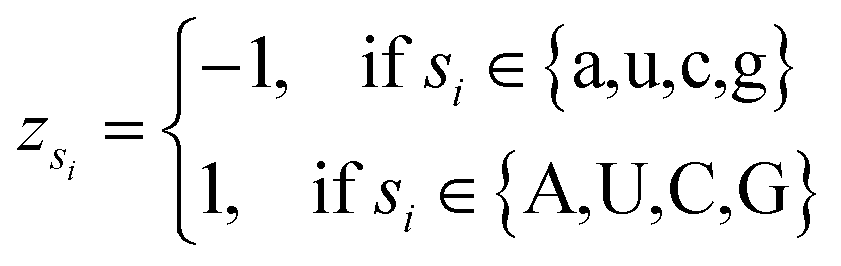 | (4) |
For every base in the RNA secondary structure, a new accumulative coordinate Si(Xi, Yi, Zi) can be obtained, which can be expressed as follows:
 | (5) |
Thus, every base can obtain another point Si(Xi, Yi, Zi). The advantages of the accumulative coordinate depend on the calculation where it contains a large amount of information, and the accuracy is good and computing the distance between sequences with different lengths is convenient. The RNA secondary structure sequences of TSV-3 and AIMV-3 are used as examples. Table 2 shows the accumulative coordinates of the 20 bases in front of the RNA secondary structures of TSV-3 and AIMV-3. Fig. 3 shows the 3D graphical representation of the RNA secondary structures of TSV-3 and AIMV-3.
| TSV-3 | X | Y | Z | AIMV-3 | X | Y | Z |
|---|---|---|---|---|---|---|---|
| G | 0.02 | 0.02 | 0.02 | A | −0.03 | −0.03 | 0.03 |
| U | 0 | 0 | 0.04 | U | −0.05 | −0.05 | 0.05 |
| G | 0.04 | 0.04 | 0.08 | G | −0.03 | −0.03 | 0.08 |
| C | 0.06 | 0.06 | 0.1 | C | 0 | 0 | 0.1 |
| c | 0.08 | 0.08 | 0.12 | u | 0.03 | −0.03 | 0.08 |
| a | 0.06 | 0.06 | 0.1 | c | 0.05 | 0 | 0.1 |
| g | 0.04 | 0.08 | 0.08 | a | 0.03 | −0.03 | 0.08 |
| u | 0.06 | 0.06 | 0.06 | u | 0.08 | −0.08 | 0.03 |
| a | 0.02 | 0.02 | 0.02 | g | 0.05 | −0.05 | 0 |
| g | −0.02 | 0.06 | −0.02 | c | 0.1 | 0 | 0.05 |
| u | 0.02 | 0.02 | −0.06 | a | 0.05 | −0.05 | 0 |
| a | −0.04 | −0.04 | −0.12 | A | 0 | −0.1 | 0.05 |
| u | 0.02 | −0.1 | −0.18 | A | −0.08 | −0.18 | 0.13 |
| a | −0.06 | −0.18 | −0.27 | A | −0.18 | −0.28 | 0.23 |
| U | −0.1 | −0.22 | −0.22 | C | −0.13 | −0.23 | 0.28 |
| A | −0.12 | −0.24 | −0.2 | u | −0.05 | −0.31 | 0.21 |
| A | −0.16 | −0.29 | −0.16 | g | −0.1 | −0.26 | 0.15 |
| u | −0.08 | −0.37 | −0.24 | c | −0.03 | −0.18 | 0.23 |
| a | −0.18 | −0.47 | −0.35 | a | −0.1 | −0.26 | 0.15 |
| u | −0.08 | −0.57 | −0.45 | u | 0 | −0.36 | 0.05 |
 | ||
| Fig. 3 The 3D graphical representation of the RNA secondary structure of viruses TSV-3 and AIMV-3. | ||
Cumulative coordinates or cumulative distances are widely used in many research areas because they show many advantages.48 However, the first residue may also be important, and the sequence space may be unbalanced. This study is different from the result of a previous cumulative coordinate study because the effects of sequence space imbalance are reduced in terms of the following aspects:
(1) The values of the cumulative coordinates are not monotonically increasing or decreasing. The coordinate value of each base may be positive or negative, and its positive and negative values depend on eqn (1)–(4). The cumulative coordinates are calculated by using eqn (5).
(2) The 3D coordinates of the constructed base are dynamically changed with the frequency of the base, reflecting the local characteristics of the sequence. For example, the initial sequence in Fig. 4 represents the first 20 bases of RNA “TSV-3”, which contains two g bases, and the coordinates of the two g bases are calculated using eqn (1). Re-routing from the beginning to the position of the base g is necessary to calculate the g base coordinate. Therefore, the coordinates of the base dynamically change with the position and number of bases, and the cumulative coordinates reflect the local characteristics of the pre-miRNA sequence to the base.
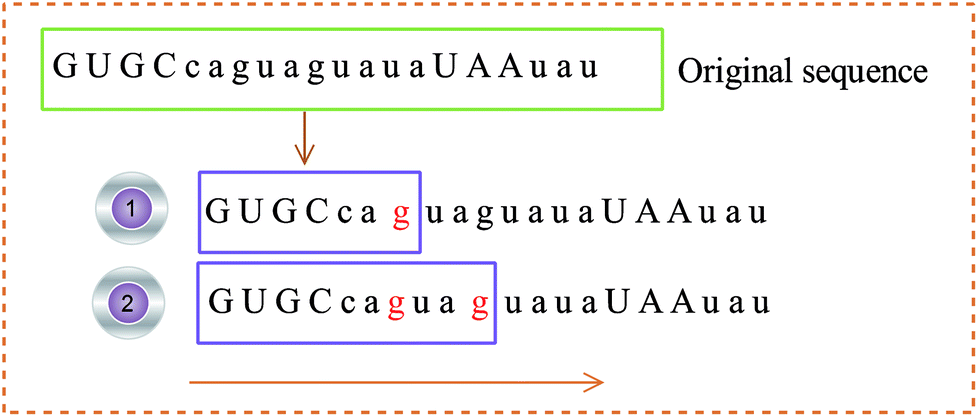 | ||
| Fig. 4 Example of the base coordinate calculation. | ||
(3) Table 2 shows that the coordinate values of the bases were not much different from the initial values, and the values gradually differed until the base position was about 10. Therefore, the cumulative coordinate values in this paper did not depend primarily on the first residue.
In summary, the cumulative coordinates are not monotonous, and they reflect the local characteristics of the sequence as the position and number of bases change dynamically. Therefore, the imbalance caused by the first residue in the sequence space has a slight effect.
A novel method for computing the distance of two sequences
To analyze the similarity between RNA sequences, a novel similarity calculation method for RNA secondary structure is proposed based on Euclidean distance. A smaller distance indicates more similarity, and vice versa.Let the secondary structures of two arbitrary RNA sequences be represented by Sa and Sb, where Na and Nb denote the lengths of the two sequences. The distance between Sa and Sb is calculated as follows:
(1) If the lengths of two sequences Sa and Sb are equal, that is, Na = Nb, then D(Sa, Sb) represents the distance between sequences Sa and Sb, and is defined as eqn (6)
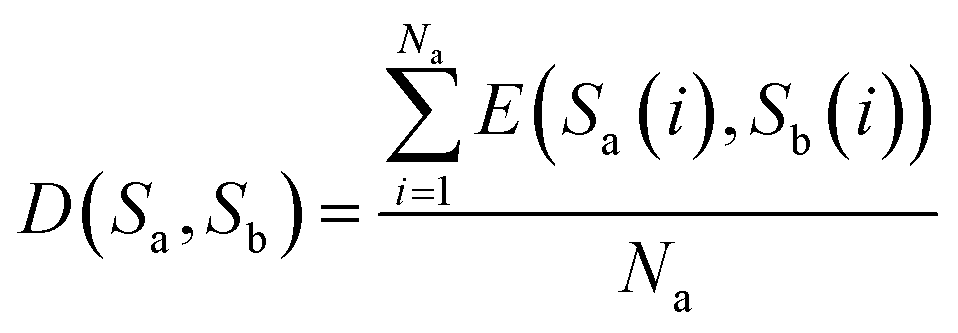 | (6) |
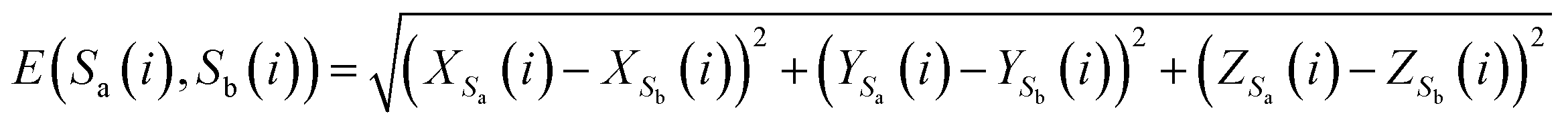 | (7) |
(2) If the lengths of two sequences are not equal, then the distance between sequences Sa and Sb are computed as follows to obtain considerable information of the sequences:
 | ||
| Fig. 5 Illustration of the steps of our method for calculating the distance between sequences. (A) shows the calculation steps for Pattern 1; (B) shows the calculation steps for Pattern 2. | ||
Step 1: use eqn (6) to calculate the distance between sequence Sa(1:Nb) or sequence “GUGCcagu” and sequence Sb;
Step 2: sequence Sb moves on the right by a base character. Use eqn (6) to calculate the distance between sequences Sa(2:Nb) and Sb, as shown in Step 2 of Fig. 5(a).
…
Step (Na − Nb + 1): sequence Sb moves on the right by a base character. Use eqn (6) to calculate the distance between sequences Sa((Na − Nb + 1):Na) and Sb.
Then, the average distance of every step is calculated (by dividing Na − Nb) as shown in eqn (8).
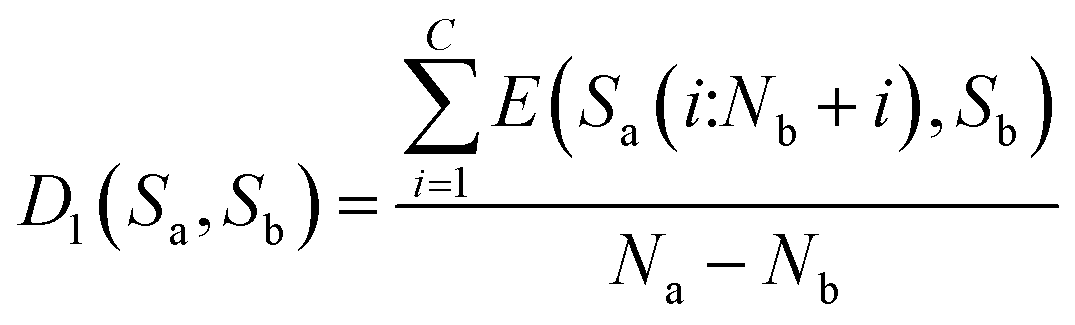 | (8) |
Step 1: exclude sequence Sa(1:Na − Nb), that is, sequence “GUGC”, and use eqn (6) to calculate the distance between Sa − Sa(1:Na − Nb) or sequence “caguagua” and sequence Sb, as shown in Step 1 of Fig. 5(b).
Step 2: use the sequence whose length is Na − Nb in sequence Sa, which moves one base character to the right. Use eqn (6) to calculate the distance between the remaining bases of sequences Sa and Sb, as shown in Step 2 of Fig. 5(b).
…
Step B: use eqn (6) to calculate the distance between Sa − Sa(Nb:Na) and Sb.
Then, calculate the average distance of each Step (B). The computational formula is demonstrated by eqn (9).
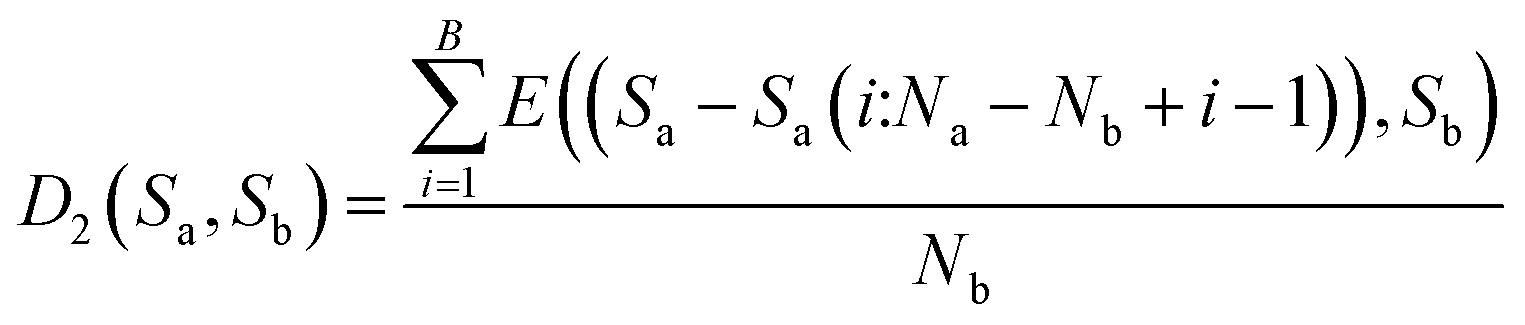 | (9) |
After synthesizing the aforementioned scenarios, the distance between sequences Sa and Sb is expressed as shown in eqn (10).
 | (10) |
We use the sequence similarity analysis method to compute the distances among 9 viruses.45 Table 3 shows the distance matrix of 9 RNA virus sequences. From the table, the three smallest values correspond to the RNA sequence pairs, namely, (AVII, LRMV-3), (LRMV-3, EMV-3), and (AVII, EMV-3), which indicate that they are the most similar. In addition, the large values in the table appear in the rows of APMV-3, AIMV-3, and PDV-3, which indicates that obvious differences exist between APMV-3, AIMV-3, and PDV-3 and other RNA sequences. In addition, the distances between APMV-3, AIMV-3, and PDV-3 22 are small, which indicate that the similarity among them is higher than the similarity among the other sequences. These results show that our method successfully captures the apparent similarity among the 9 RNA sequences. The results are similar to those of Liao et al.37,38,40,41 Our 3D graphical representation and sequence similarity analysis method extract some essential information on RNA secondary structure and can effectively analyze the similarity of RNA sequences.
| APMV-3 | AVII | CILRV | CVV-3 | EMV-3 | LRMV-3 | PDV-3 | TSV-3 | |
|---|---|---|---|---|---|---|---|---|
| AIMV-3 | 0.97 | 1.64 | 2.70 | 1.17 | 2.16 | 1.75 | 1.42 | 1.89 |
| APMV-3 | 0.00 | 2.14 | 3.33 | 1.09 | 2.58 | 2.18 | 0.76 | 2.69 |
| AVII | 0.00 | 1.57 | 1.64 | 0.69 | 0.58 | 2.31 | 1.40 | |
| CILRV | 0.00 | 2.92 | 1.47 | 1.89 | 3.62 | 1.14 | ||
| CVV-3 | 0.00 | 2.01 | 1.53 | 1.21 | 2.45 | |||
| EMV-3 | 0.00 | 0.70 | 2.67 | 1.79 | ||||
| LRMV-3 | 0.00 | 2.32 | 1.49 | |||||
| PDV-3 | 0.00 | 3.01 |
Results and discussions
MiRNAs are involved in a large number of biological processes, such as plant development and metabolism by either translational repression, RNA degradation, or through an RNA-induced silencing complex. Here, we apply our method to predict plant pre-miRNAs based on the similarity of pre-miRNA sequences.We divide the datasets of plant pre-miRNA sequences into sample and test datasets. In the test dataset, a test sequence can be classified as the category of the sequence in the sample dataset that has the smallest distance with the test sequence. For example, the sequence with the smallest distance from the sample dataset is the pseudo pre-miRNA (negative data), and this test sequence is also the pseudo pre-miRNA, and vice versa. We use the jackknife method to calculate the accuracy of our method.
Datasets
In this section, we use three datasets to evaluate the performance of the proposed method.Comparison of state-of-the-art algorithms
The following measures were used to assess the performance of the classifiers used in this study.To measure the effectiveness of identifying plant pre-miRNAs, the following equations are used to measure the experiment results, including the overall accuracy (ACC), sensitivity (SE), specificity (SP), and Mathews coefficient (MCC). The expressions are shown as follows:
 | (11) |
 | (12) |
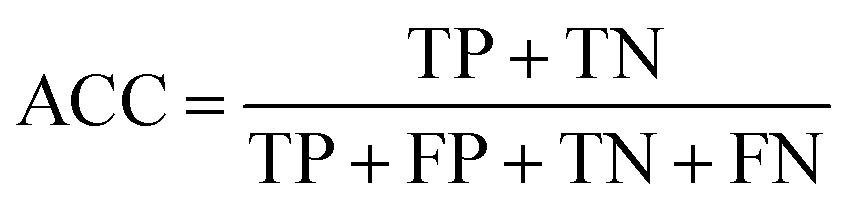 | (13) |
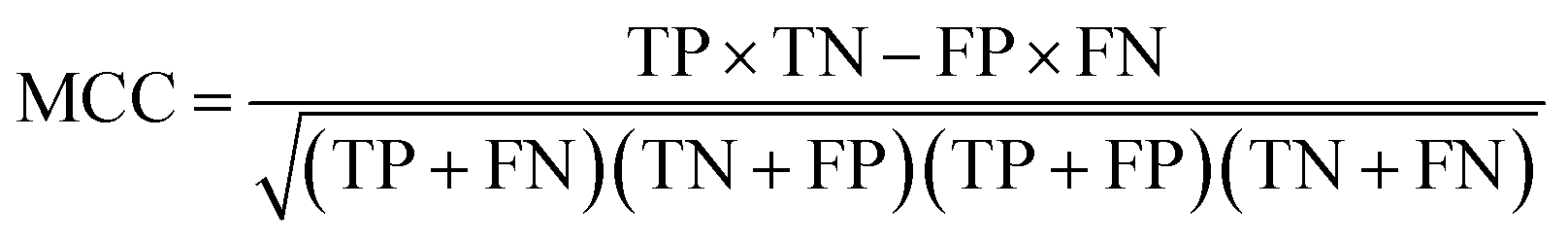 | (14) |
The results of the jackknife test for dataset 1, dataset 2, and dataset 3 are listed in Tables 4, 5, and 6, respectively. Table 4 shows the results of our method and of microPred 52, iMcRNA 24, TripletSVM 53, and miPlantPre 14 methods applied to dataset 1. From the table, the ACC and MCC achieve 89.74% and 79.67% using our method, respectively, which are higher than others. Table 5 shows that the accuracy of our method is lower than the miPlantPre 14 method in dataset 2.
| Methods | ACC | SE | SP | MCC |
|---|---|---|---|---|
| a The result based on the iMcRNA method.24b The result based on the miPlantPre method.14c The result based on the microPred method.52d The result based on the TripletSVM method.53 | ||||
| iMcRNAa | 85.88 | 87.83 | 83.31 | 71.86 |
| miPlantPreb | 82.68 | 97.59 | 75.18 | 68.48 |
| microPredc | 73.96 | 74.92 | 73.51 | 47.93 |
| TripletSVMd | 75.72 | 63.34 | 84.54 | 53.24 |
| Our method | 89.74 | 86.3 | 92.69 | 79.67 |
| Datasets | iMcRNAa | microPredb | miPlantPrec | Our method |
|---|---|---|---|---|
| a The result based on the iMcRNA method.24b The result based on the microPred method.52c The result based on the miPlantPre method.14 | ||||
| mtr_67 | 89.5 | 76.1 | 86.6 | 95.52 |
| osa_256 | 86.1 | 73.8 | 83.4 | 93.6 |
| ppt_184 | 76.9 | 68.8 | 84.5 | 96.5 |
| ath_153 | 86.2 | 67.6 | 85 | 96.1 |
| updated_aly_167 | 86.5 | 69.5 | 85 | 98.2 |
| ptc_133 | 78.6 | 72.2 | 82.7 | 91.4 |
| sbi_105 | 85.2 | 76.7 | 83.8 | 92.9 |
| updated_gma_105 | 88.1 | 82.4 | 83.8 | 92.4 |
| zma_74 | 85.8 | 74.3 | 85.1 | 96 |
| gma_69 | 88.4 | 71 | 85.5 | 92.8 |
In addition, our method did not use any machine learning classifiers, which can improve the accuracy by training and complicated computing. Thus, our method is easy to implement and requires a small amount of time. Table 6 shows the results of our method and of microPred 52, iMcRNA-PseSSC 24, and miPlantPre 14 methods applied to dataset 3. From the table, our method has the best ACC and MCC among the 10 plant pre-miRNA datasets (i.e., mtr, osa, ppt, ath, ptc, sbi, zma, gma, updated_aly, and updated_gma). This result indicates the effectiveness of our method.
In summary, our method obtains a good accuracy in identifying plant pre-miRNAs and has excellent stability based on the analysis of the aforementioned experiments. In comparison with existing machine learning algorithms, the proposed method is simple to operate and does not require training parameters.
Conclusions
Graphical representations based on sequences (e.g., DNA, RNA, and proteins) have been the focus of research.41,42,54–57 In this study, we proposed a 3D graphical representation of the secondary structure of the pre-miRNA in combination with the frequency and physicochemical properties of the base. We then subjected the pre-miRNA secondary structure to similarity analysis by calculating their Euclidean distance. The smaller the distance was, the higher the similarity between the two sequences would be and vice versa. Finally, the sequence similarity method proposed in this paper was used to identify plant pre-miRNA. The experimental results showed that the proposed method was reasonable and effective in the three common benchmark datasets.In future work, we will develop an enhanced representation of the pre-miRNA secondary structure by merging additional information and designing a more complete graphical model and more efficient similarity analysis methods to improve the performance of pre-miRNA prediction. In addition, our method for predicting and classifying other noncoding RNAs, such as Piwi-interacting RNA and long-noncoding RNA, is a key issue that should be further investigated.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the Program for New Century Excellent Talents in university (Grant No. NCET-10-0365), National Nature Science Foundation of China (Grant No. 11171369, 61272395, 61370171, 61300128, 61472127, 61572178 and 61672214).References
- J. Lei and Y. Sun, Bioinformatics, 2014, 30, 2837–2839 CrossRef PubMed.
- Y. Zhang, M. S. Kim, B. Jia, J. Yan, J. P. Zuniga-Hertz, C. Han and D. Cai, Nature, 2017, 548, 52 CrossRef PubMed.
- B. Zhang, X. Pan, G. P. Cobb and T. A. Anderson, Dev. Biol., 2006, 289, 3 CrossRef PubMed.
- C. C. Pritchard, H. H. Cheng and M. Tewari, Nat. Rev. Genet., 2012, 13, 358 CrossRef PubMed.
- I. T. Jr and C. Bustamante, J. Mol. Biol., 1999, 293, 271–281 CrossRef PubMed.
- P. Xuan, M. Guo, X. Liu, Y. Huang, W. Li and Y. Huang, Bioinformatics, 2011, 27, 1368 CrossRef PubMed.
- E. Berezikov, E. Cuppen and R. H. Plasterk, Nat. Genet., 2006, 38(suppl.), S2 CrossRef PubMed.
- A. Khan, S. Shah, F. Wahid, F. G. Khan and S. Jabeen, Mol. BioSyst., 2017, 13, 1640–1645 RSC.
- C. Paicu, I. Mohorianu, M. Stocks, P. Xu, A. Coince, M. Billmeier, T. Dalmay, V. Moulton and S. Moxon, Bioinformatics, 2017, 33, 2446–2454 CrossRef PubMed.
- B. Alptekin, B. A. Akpinar and H. Budak, Front. Plant Sci., 2016, 7, 2058 Search PubMed.
- Y. Yao, C. Ma, H. Deng, Q. Liu, J. Zhang and M. Yi, Mol. BioSyst., 2016, 12, 3124 RSC.
- M. Evers, M. Huttner, A. Dueck, G. Meister and J. C. Engelmann, BMC Bioinf., 2015, 16, 1–10 CrossRef PubMed.
- J. An, J. Lai, A. Sajjanhar, M. L. Lehman and C. C. Nelson, BMC Bioinf., 2014, 15, 275 CrossRef PubMed.
- J. Meng, D. Liu, C. Sun and Y. Luan, BMC Bioinf., 2014, 15, 423 CrossRef PubMed.
- L. Wei, M. Liao, G. Yue, R. Ji, Z. He and Z. Quan, IEEE/ACM Trans. Comput. Biol. Bioinf., 2014, 11, 192–201 Search PubMed.
- S. A. Helvik, S. O. Jr and P. Saetrom, Bioinformatics, 2007, 23, 142–149 CrossRef PubMed.
- T. H. Huang, B. Fan, M. F. Rothschild, Z. L. Hu, K. Li and S. H. Zhao, BMC Bioinf., 2007, 8, 341 CrossRef PubMed.
- C. Xue, F. Li, T. He, G. P. Liu, Y. Li and X. Zhang, BMC Bioinf., 2005, 6, 310 CrossRef PubMed.
- Y. Wang, X. Chen, W. Jiang, L. Li, W. Li, L. Yang, M. Liao, B. Lian, Y. Lv and S. Wang, Genomics, 2011, 98, 73–78 CrossRef PubMed.
- Y. Wu, B. Wei, H. Liu, T. Li and R. Simon, BMC Bioinf., 2011, 12, 107 CrossRef PubMed.
- J. W. Nam, K. R. Shin, J. Han, Y. Lee, V. N. Kim and B. T. Zhang, Nucleic Acids Res., 2005, 33, 3570–3581 CrossRef PubMed.
- L. Wei, M. Liao, Y. Gao, R. Ji, Z. He and Q. Zou, IEEE/ACM Trans. Comput. Biol. Bioinf., 2014, 11, 192–201 Search PubMed.
- I. D. O. Lopes, A. Schliep and A. C. D. L. D. Carvalho, BMC Bioinf., 2014, 15, 1–11 Search PubMed.
- B. Liu, L. Fang, F. Liu, X. Wang, J. Chen and K. C. Chou, PLoS One, 2015, 10, e0121501 CrossRef PubMed.
- B. Liu, L. Fang, S. Wang, X. Wang, H. Li and K. C. Chou, J. Theor. Biol., 2015, 385, 153–159 CrossRef PubMed.
- B. Liu, L. Fang, J. Chen, F. Liu and X. Wang, Mol. BioSyst., 2015, 11, 1194 RSC.
- T. Zhao, N. Zhang, Z. Ying, J. Ren, P. Xu, Z. Liu, C. Liang and H. Yang, J. Biomed. Semant., 2017, 8, 30 CrossRef PubMed.
- L. Jiang, J. Zhang, P. Xuan and Q. Zou, BioMed Res. Int., 2016, 2016, 9565689 Search PubMed.
- G. Stegmayer, C. Yones, L. Kamenetzky and D. H. Milone, IEEE/ACM Trans. Comput. Biol. Bioinf., 2016, 14, 1316–1326 Search PubMed.
- P. Jiang, H. Wu, W. Wang, W. Ma, X. Sun and Z. Lu, Nucleic Acids Res., 2007, 35, W339 CrossRef PubMed.
- K. K. Kandaswamy, K. C. Chou, T. Martinetz, S. Möller, P. N. Suganthan, S. Sridharan and G. Pugalenthi, J. Theor. Biol., 2011, 270, 56–62 CrossRef PubMed.
- W. Z. Lin, J. A. Fang, X. Xiao and K. C. Chou, PLoS One, 2011, 6, e24756 CrossRef PubMed.
- T. Dezulian, M. Remmert, J. F. Palatnik, D. Weigel and D. H. Huson, Bioinformatics, 2006, 22, 359–360 CrossRef PubMed.
- Y. H. Yao, X. Y. Nan and T. M. Wang, J. Comput. Chem., 2005, 26, 1339–1346 CrossRef PubMed.
- C. Li, L. Xing and X. Wang, Chem. Phys. Lett., 2008, 458, 249–252 CrossRef.
- H. J. Jeffrey, Nucleic Acids Res., 1990, 18, 2163 CrossRef PubMed.
- W. Zhu, B. Liao and K. Ding, J. Mol. Struct.: THEOCHEM, 2005, 757, 193–198 CrossRef.
- B. Liao, T. Wang and K. Ding, Mol. Simul., 2005, 22, 455 Search PubMed.
- B. Liao, W. Zhu and P. Li, J. Math. Chem., 2006, 42, 1015–1022 CrossRef.
- Y. Li, M. Duan and Y. Liang, BMC Bioinf., 2012, 13, 280 CrossRef PubMed.
- Y. Zhang, H. Huang, X. Dong, Y. Fang, K. Wang, L. Zhu, K. Wang, T. Huang and J. Yang, PLoS One, 2016, 11, e0152238 CrossRef PubMed.
- Y. Li, X. Shi, Y. Liang, J. Xie, Y. Zhang and Q. Ma, BMC Bioinf., 2017, 18, 51 CrossRef PubMed.
- W. Li and A. Godzik, Bioinformatics, 2006, 22, 1658 CrossRef PubMed.
- I. L. Hofacker, Nucleic Acids Res., 2003, 31, 3429 CrossRef PubMed.
- C. B. Reusken and J. F. Bol, Nucleic Acids Res., 1996, 24, 2660 CrossRef PubMed.
- D. H. Mathews, J. Sabina, M. Zuker and D. H. Turner, J. Mol. Biol., 1999, 288, 911 CrossRef PubMed.
- J. Feng and T. M. Wang, Chem. Phys. Lett., 2008, 454, 355–361 CrossRef.
- D. Xu, L. Theresa, N. L. Greenbaum and M. O. Fenley, Nucleic Acids Res., 2007, 35, 3836 CrossRef PubMed.
- J. Chen, X. Wang and B. Liu, Sci. Rep., 2016, 6, 19062 CrossRef PubMed.
- A. Kozomara and S. Griffithsjones, Nucleic Acids Res., 2011, 39, D152–D157 CrossRef PubMed.
- A. Kozomara and S. Griffithsjones, Nucleic Acids Res., 2014, 42, 68–73 CrossRef PubMed.
- R. Batuwita and V. Palade, Bioinformatics, 2009, 25, 989–995 CrossRef PubMed.
- G. P. Liu, T. He, F. Li, C. Xue, Y. Li and X. Zhang, BMC Bioinf., 2005, 6, 310 CrossRef PubMed.
- H. J. Yu and D. S. Huang, IEEE J. Biomed. Health Inform., 2013, 17, 503–511 Search PubMed.
- H. Hu, Z. Li, H. Dong and T. Zhou, IEEE/ACM Trans. Comput. Biol. Bioinf., 2017, 14, 182 Search PubMed.
- X. Watkins, L. J. Garcia, S. Pundir, M. J. Martin and U. Consortium, Bioinformatics, 2017, 33, 2040–2041 CrossRef PubMed.
- D. F. Thieker, J. A. Hadden, K. Schulten and R. J. Woods, Glycobiology, 2016, 26, 786 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |