
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of arrays containing porphyrin, chlorin, and perylene-imide constituents for panchromatic light-harvesting and charge separation†
Gongfang Hu a,
Hyun Suk Kangb,
Amit Kumar Mandalb,
Arpita Royb,
Christine Kirmaierb,
David F. Bocian*c,
Dewey Holten*b and
Jonathan S. Lindsey*a
a,
Hyun Suk Kangb,
Amit Kumar Mandalb,
Arpita Royb,
Christine Kirmaierb,
David F. Bocian*c,
Dewey Holten*b and
Jonathan S. Lindsey*a
aDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695-8204, USA. E-mail: jlindsey@ncsu.edu; Tel: +1-919-515-6406
bDepartment of Chemistry, Washington University, St. Louis, Missouri 63130-4889, USA. E-mail: holten@wustl.edu; Tel: +1-314-935-6502
cDepartment of Chemistry, University of California, Riverside, California 92521-0403, USA. E-mail: david.bocian@ucr.edu; Tel: +1-951-827-3660
First published on 29th June 2018
Abstract
Achieving solar light harvesting followed by efficient charge separation and transport is an essential objective of molecular-based artificial photosynthesis. Architectures that afford strong absorption across the near-UV to near-infrared region, namely panchromatic absorptivity, are critically important given the broad spectral distribution of sunlight. A tetrapyrrole–perylene pentad array was synthesized and investigated as a means to integrate panchromatic light harvesting and intramolecular charge separation. The pentad consists of three moieties: (1) a panchromatically absorbing triad, in which a porphyrin is strongly coupled to two perylene-monoimides via ethyne linkages; (2) a perylene-diimide electron acceptor; and (3) a chlorin hole-trapping unit. Integrating the three components with diphenylethyne linkers generates moderate electronic coupling for intramolecular energy and hole/electron transfer. The construction of the array relies on a stepwise strategy for incorporating modular pigment building blocks. The key building blocks include a trans-A2BC porphyrin, a chlorin, a perylene-monoimide, and a perylene-diimide, each bearing appropriate (halo, ethynyl) synthetic handles for Pd-catalyzed Sonogashira coupling reactions. One target pentad, three tetrads, four triads, and four monomeric benchmark compounds were synthesized from six building blocks (three new, three reported) and 10 new synthetic intermediates. Four of the tetrapyrrole-containing arrays are zinc chelated, and four others are in the free base form. Absorption and fluorescence spectra and fluorescence quantum yields were also measured. Collectively, investigations of the arrays reveal insights into principles for the design of novel reaction centers integrated with a panchromatic antenna for artificial photosynthetic studies.
Introduction
A chief objective of artificial photosynthesis is to create model systems that mimic native photosynthetic processes and through systematic modifications can be used to probe relevant physicochemical features. The processes encompass diverse phenomena including light capture, excited-state energy transfer, excited-state electron transfer, dark electron/hole transfer, proton pumping, carbon fixation, and water splitting. While early model systems were first reported decades ago,1–12 the ensuing half-century of research has not resolved fundamental synthetic limitations to the creation of molecular architectures that rival those in native photosynthesis. Such architectures require large numbers of pigments and redox cofactors organized in exacting 3-dimensional spatial arrangements over mesoscale dimensions. Given the challenges of creating such architectures, an alternative tack entails development of more compact structures with designs complementary to those in native photosynthesis.13–27As one case in point, we recently developed a design motif that engenders panchromatic absorption yet affords a lowest singlet excited-state of discrete energy.28,29 The motif relies on a perylene-monoimide attached to a porphyrin via an ethyne linker, where the linker joins the perylene 9-position and the porphyrin meso-position, both of which are sites of high electron density in the highest occupied molecular orbital (HOMO) of each constituent. Such a triad (PMI2TP) composed of two perylene-monoimides tightly coupled electronically (via ethyne linkers) to one porphyrin is shown in Fig. 1.29 The triad absorbs strongly across the 400–700 nm region (average peak ε ∼ 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1), yet exhibits excited-state features similar to those of the tetrapyrrole Indeed, the fluorescence emission spectrum generally resembles that of a free base tetraarylporphyrin, although shifted bathochromically, and the fluorescence quantum yield (Φf) is considerably enhanced versus that of the porphyrin alone. This latter effect arises from a dramatic shift in radiative probability from the normally intense porphyrin near-UV Soret band(s) to the normally weak porphyrin visible bands including the S0 → S1 absorption (and S1 → S0 fluorescence) transition.30 Because the triad exhibits tetrapyrrole-like lowest singlet excited-state properties we considered that the triad could be used as an antenna element in conjunction with an integrated charge-separation unit. The resulting integrated pentad array (C-T-PDI) contains a perylene-monoimide–porphyrin–perylene-monoimide triad unit (T), a perylene-diimide (PDI), and a chlorin (C) (Chart 1).
000 M−1 cm−1), yet exhibits excited-state features similar to those of the tetrapyrrole Indeed, the fluorescence emission spectrum generally resembles that of a free base tetraarylporphyrin, although shifted bathochromically, and the fluorescence quantum yield (Φf) is considerably enhanced versus that of the porphyrin alone. This latter effect arises from a dramatic shift in radiative probability from the normally intense porphyrin near-UV Soret band(s) to the normally weak porphyrin visible bands including the S0 → S1 absorption (and S1 → S0 fluorescence) transition.30 Because the triad exhibits tetrapyrrole-like lowest singlet excited-state properties we considered that the triad could be used as an antenna element in conjunction with an integrated charge-separation unit. The resulting integrated pentad array (C-T-PDI) contains a perylene-monoimide–porphyrin–perylene-monoimide triad unit (T), a perylene-diimide (PDI), and a chlorin (C) (Chart 1).
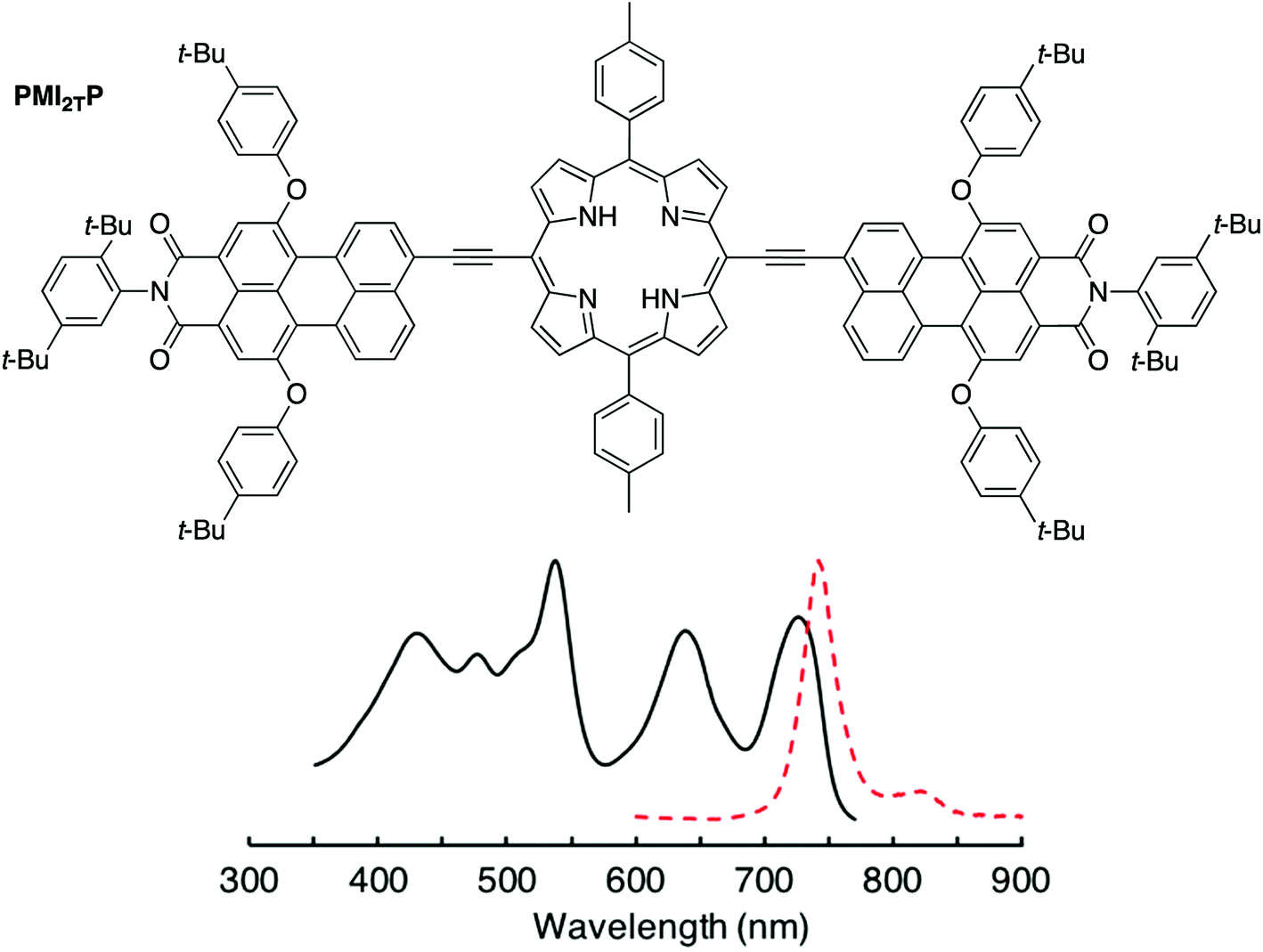 | ||
| Fig. 1 The absorption (black solid) and fluorescence (red dashed) spectra of a prior panchromatic triad PMI2TP. | ||
 | ||
| Chart 1 Target pentad array. | ||
The processes envisaged for light harvesting and charge separation in the pentad C-T-PDI are illustrated in Fig. 2. The central triad acts as a supermolecule with panchromatic absorption (see e.g. Fig. 1) as a result of the substantial electronic interactions between the porphyrin and perylene-monoimides afforded by the direct ethyne linkers. Excitation in any of the absorption bands of the triad is followed by rapid, quantitative energy flow to the tetrapyrrole-like lowest energy singlet excited state (S1) of this array, analogous to internal conversion from upper to lower energy excited states of a simple chromophore. The core porphyrin is of a trans-A2BC design. The perylene-diimide and chlorin are each attached to the core porphyrin via a diphenylethyne linker, which affords electronic interactions that while weaker than a direct ethyne linkage, enable electronic coupling sufficient to support rapid energy and hole/electron transfer with only slight perturbation of energy levels and electronic spectra. The perylene-diimide serves as an acceptor upon excited-state electron transfer from the excited triad (T*), whereas the chlorin functions as a hole trap via a ground-state process involving the oxidized triad (T+). The overall light-driven process is thus expected to afford the chlorin cation radical and perylene-diimide anion radical upon charge separation across the pentad (forming C+-T-PDI−). While the chief functions of the perylene-diimide and the chlorin are for charge separation, both also absorb light and are expected to afford additional light-harvesting capacity to the pentad by funneling harvested excitation energy to the S1 excited state of the central triad.
 | ||
| Fig. 2 Illustration of the expected light-harvesting and charge-separation processes in pentad C-T-PDI. | ||
Herein, we report the synthesis of the pentad array as well as all companion arrays for full photophysical characterization. The synthesis relies on a building block approach31–34 wherein constituent chromophores are synthesized separately and then joined together through Pd-mediated coupling reactions. The companion arrays include three tetrads (Chart 2), four triads (Chart 3), and four monomeric compounds (Chart 4). The 12 targets (one pentad and 11 benchmark compounds) were prepared from six building blocks (three new, three reported) and 10 new synthetic intermediates. Absorption and fluorescence studies are also reported as a first step toward in-depth analysis of integrated light-harvesting and charge-separation processes in the arrays.
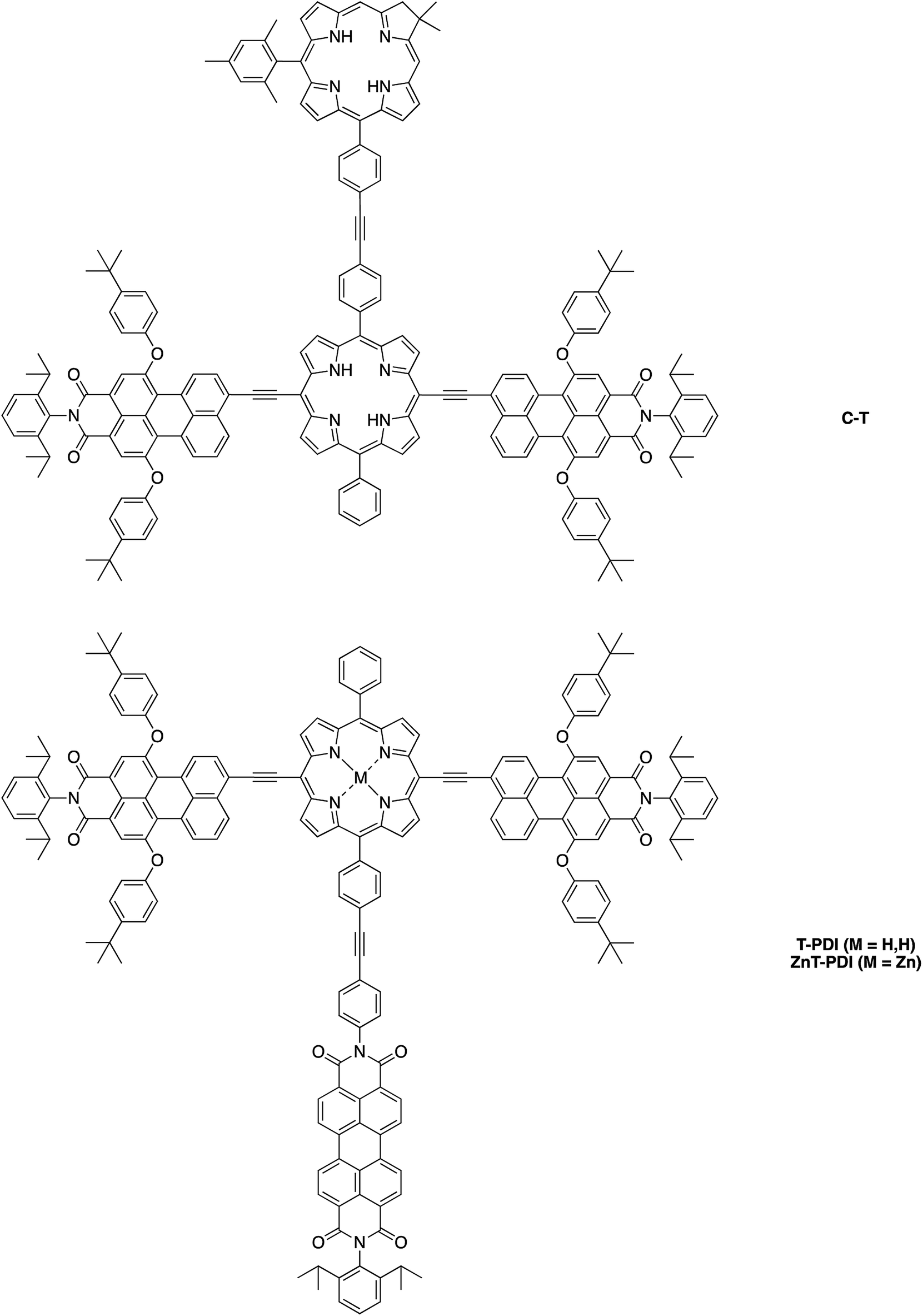 | ||
| Chart 2 Constituent tetrad benchmarks. | ||
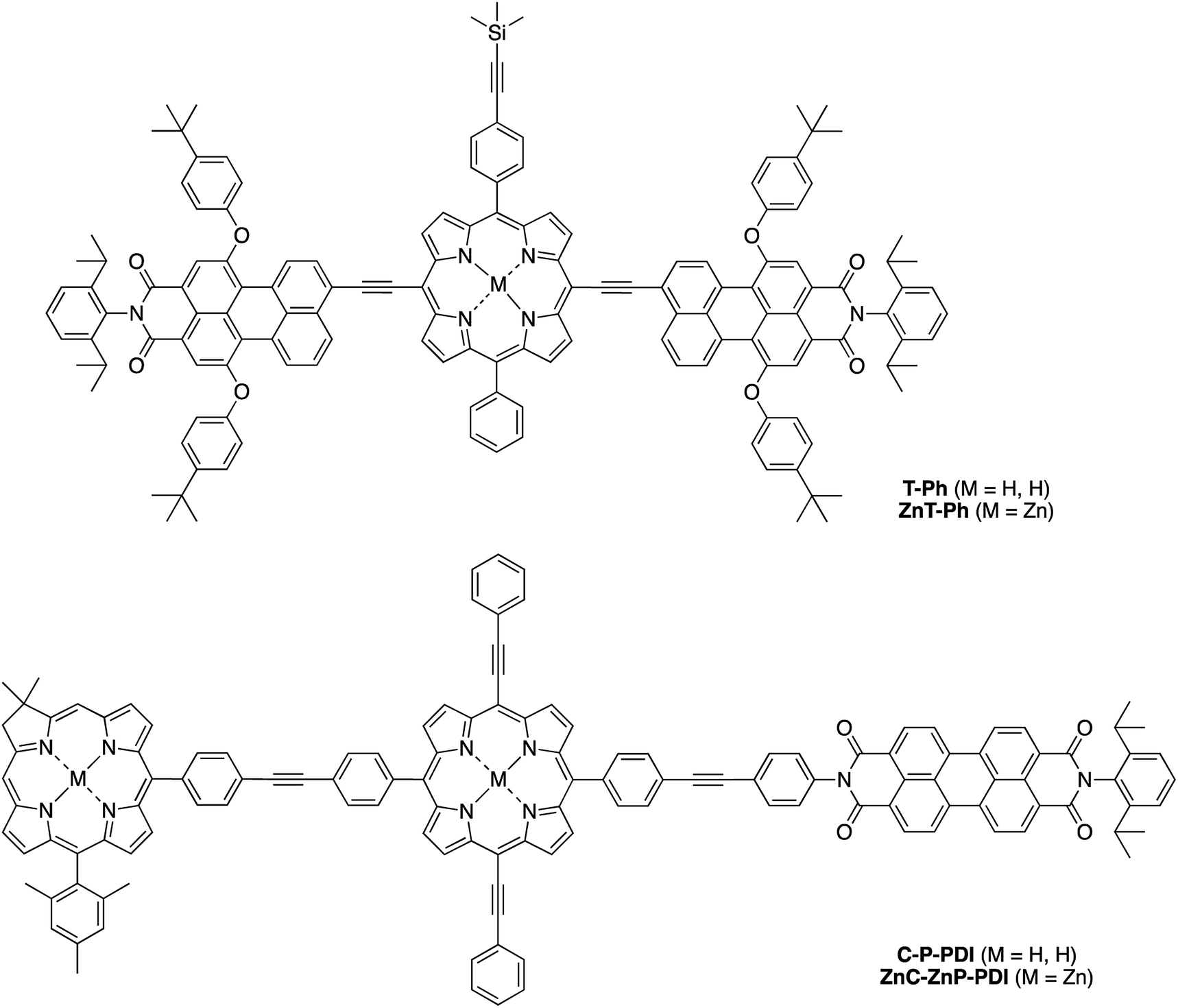 | ||
| Chart 3 Constituent triad benchmarks. | ||
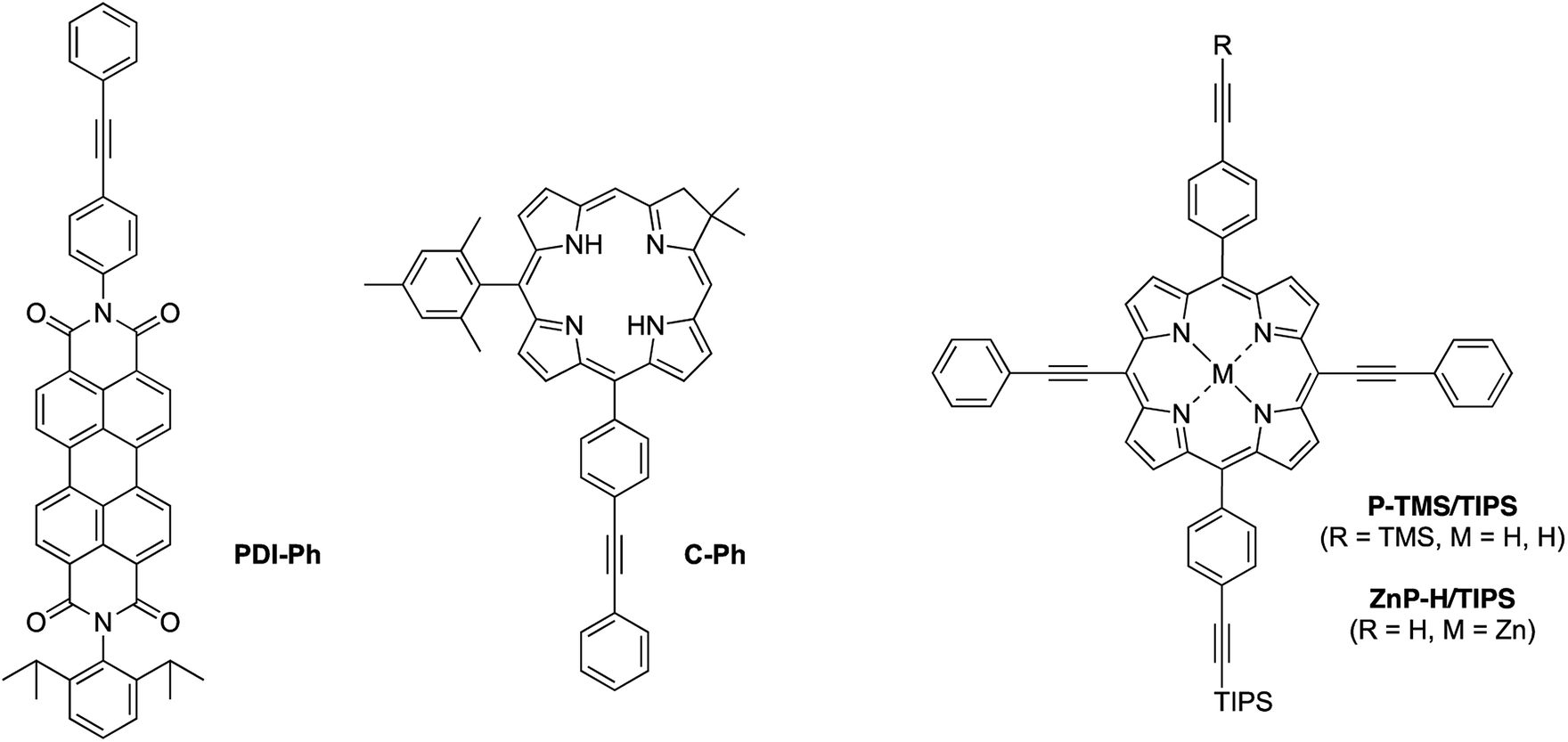 | ||
| Chart 4 Constituent monomer benchmarks. | ||
Results and discussion
Molecular design
The rationale for the selection of components of the pentad was based on optical and redox properties. In particular, a chlorin with sparse substitution35 was chosen given that such chlorins exhibit a wavelength absorption band at shorter wavelength, and thus an S1 excited-state at higher energy, than that of the central bis(perylene-monoimide)porphyrin triad. The lower S1 energy of the central panchromatic porphyrin-based triad unit versus that of a standard chlorin is one of the functionally important features of the C-T-PDI pentad. Redox potentials of reported benchmark compounds with similar structures of the chlorin, panchromatic bis(perylene-monoimide)porphyrin triad, and the perylene-diimide constituents28,36–38 were considered in the design of an array to achieve photoinduced charge separation. The envisaged processes based on the choices are illustrated in Fig. 2: the central panchromatic triad (in both the ground and excited state) is more readily oxidized compared to the perylene-diimide and more readily reduced relative to the chlorin moiety, whereupon a charge-separated state is expected with the electron localized at the perylene-diimide and the hole trapped at the chlorin.One slight structural modification in the arrays reported here compared with the previously made triad PMI2TP is the N-substituents of the imido groups in the perylenes.29,39 The prior 2,5-di-tert-butylphenyl substituents are replaced with 2,6-diisopropylphenyl groups, because the former substituents can give rise to atropisomers,40–42 thereby complicating NMR analysis. The use of 2,6-diisopropyl versus 2,5-di-tert-butyl groups for solubilization has insignificant effects on perylene photophysics and energetics,43 in large part due to the presence of a node at the perylene-imide nitrogen atom in both the HOMO and the lowest unoccupied molecular orbital (LUMO).41
Synthesis
Eleven target compounds (Charts 2–4) were prepared for comparison with the pentad, including two free base tetrads (T-PDI and C-T, omitting either the chlorin or the perylene-diimide moiety in the pentad) and a zinc chelate (ZnT-PDI); two free base triads (T-Ph and C-P-PDI) and their zinc chelates (ZnT-Ph and ZnC-ZnP-PDI); a monomeric perylene-diimide (PDI-Ph); a monomeric chlorin (C-Ph); and a monomeric porphyrin (P-TMS/TIPS) bearing ethynes with trimethylsilyl and triisopropylsilyl protecting groups, and related zinc chelate (ZnP-H/TIPS). Zinc chelates were prepared to tune the electrochemical properties (because zinc chelates are more easily oxidized than the free base forms), providing more insights to the intramolecular electron–transfer processes in the arrays.The pentad C-T-PDI contains four functional entities attached to the core porphyrin via ethynyl and ethynylphenyl linkers. The building blocks include a trans-A2BC-porphyrin,44 which constitutes the core of the pentad and establishes a star-shaped architecture; an iodophenyl–chlorin,35 an ethynyl-perylene-monoimide,45 and an iodophenyl-perylene-diimide.37,42 The preparation of tetrapyrrole arrays with direct ethynyl linkages was first reported by Arnold,46 next developed by Anderson47 and Therien,48 and has since found widespread use.18,26,49–53 The chief reaction for joining building blocks to construct either direct ethynyl linkers or longer diphenylethyne linkers entails Pd-mediated Sonogashira coupling of halo-(iodo or bromo) and ethynyl-substituted reactants. Such reactions with free base tetrapyrroles must be carried out in the absence of copper to avoid unwanted metalation.54,55
 | ||
| Scheme 1 “2 + 2” condensation to prepare porphyrin building block 2-Br2/TMS. | ||
Porphyrin building block 4-Br2/TMS/TIPS (Scheme 2) bears two distinct ethynylphenyl units (with TMS and TIPS protecting groups for successive coupling reactions) and was synthesized in a strategy similar to that for porphyrin 2-Br2/TMS. A dibutyltin-chelated diformyldipyrromethane 3,56 which was more readily purified than the free base counterpart, was synthesized according to the literature. The zinc trans-AB-porphyrin Zn4-I/TMS, which bears one iodophenyl group and one TMS-protected ethynylphenyl group, was prepared therefrom with 1-TMS in 15% yield. Sonogashira coupling of Zn4-I/TMS and TIPS-acetylene in the presence of catalytic CuI (1 mol% of the porphyrin) produced the zinc porphyrin Zn4-TMS/TIPS in 80% yield. Subsequent bromination59 and demetalation43 generated the free base dibromoporphyrin 4-Br2/TMS/TIPS in a yield of 27% for two steps. The zinc-chelated benchmark compound ZnP-H/TIPS was also synthesized from the trans-AB-porphyrin Zn4-TMS/TIPS in 44% yield, through three successive steps of bromination, coupling with phenylacetylene, and cleavage of the TMS group.
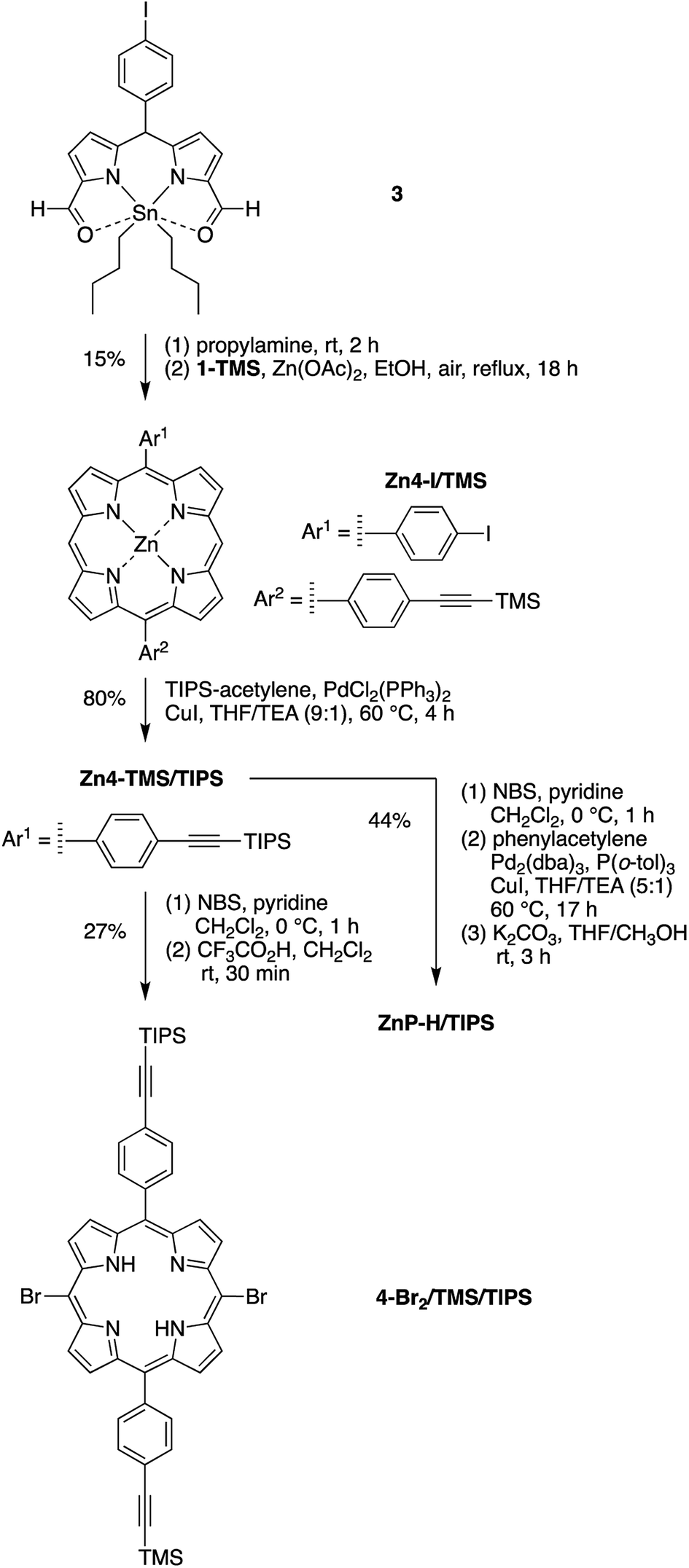 | ||
| Scheme 2 Preparation of the porphyrin building block 4-Br2/TMS/TIPS and benchmark ZnP-H/TIPS. | ||
 | ||
| Scheme 3 The chlorin building block 5 and the preparation of benchmark compound C-Ph. | ||
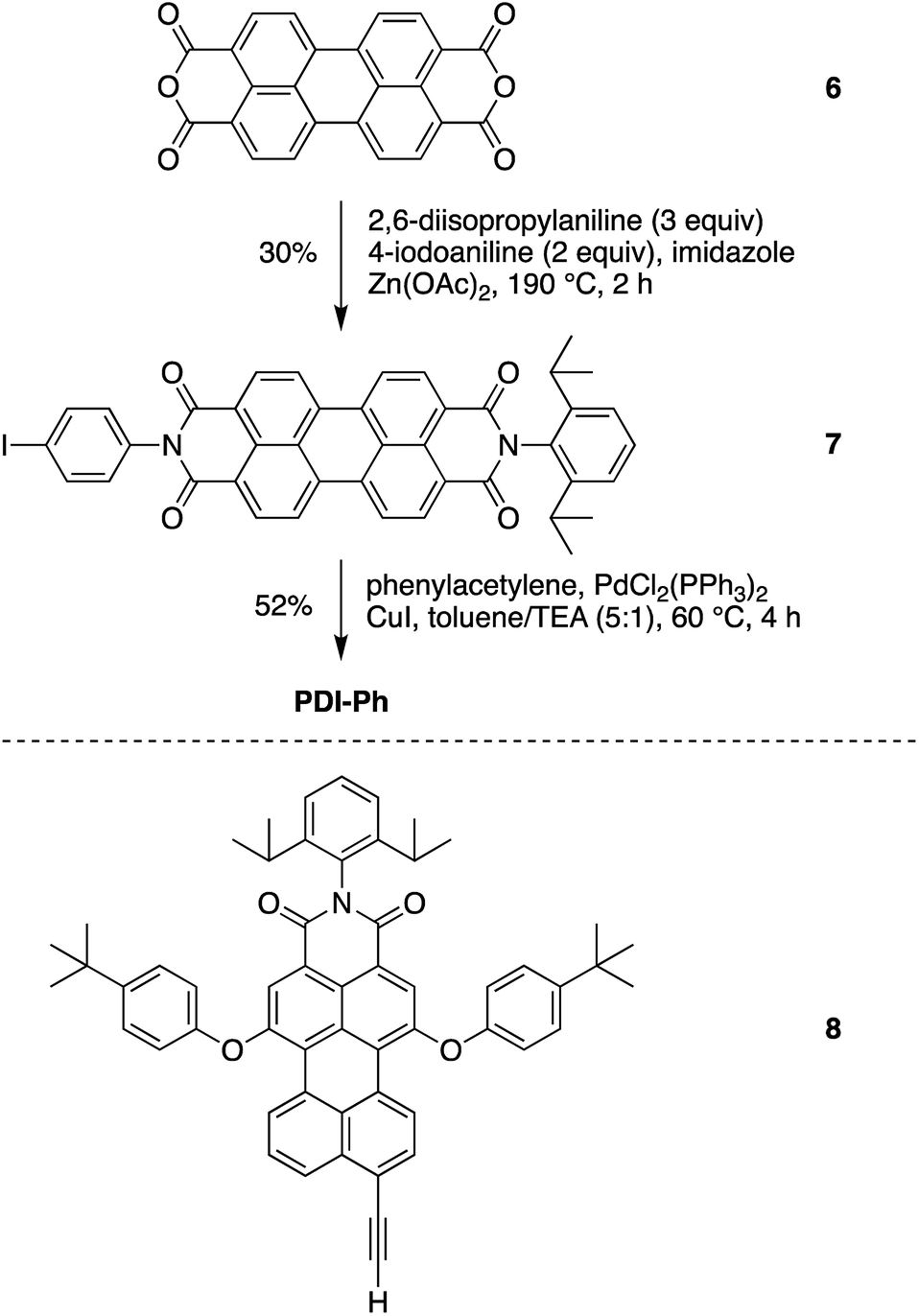 | ||
| Scheme 4 Synthesis of the perylene-diimide building block 7, and the benchmark PDI-Ph. | ||
 | ||
| Scheme 5 Preparation of triads (T-Ph, ZnT-Ph) and tetrads (C-T, T-PDI, ZnT-PDI). | ||
Phenylethynyl groups were installed at the meso-positions of 4-Br2/TMS/TIPS through a Sonogashira reaction to afford the porphyrin benchmark P-TMS/TIPS in 78% yield (Scheme 6). The TMS group in P-TMS/TIPS was thereafter removed with K2CO3 at room temperature, whereupon coupling of the resulting porphyrin and perylene-diimide 7 afforded the porphyrin–perylene-diimide dyad 9 (49% yield). Dyad 9 has limited solubility in many organic solvents (toluene, CH2Cl2, chloroform, THF, acetonitrile, methanol, or DMSO), but the solubility could be improved upon zinc chelation (not shown). Despite the meager solubility, dyad 9 in a saturated solution (∼1 mM) was deprotected with tetrabutylammonium fluoride (TBAF) and then coupled with the chlorin building block 5 in the presence of Pd2(dba)3/P(o-tol)3 to produce the chlorin–porphyrin–perylene-diimide triad C-P-PDI. Subsequent zinc chelation generated the triad ZnC-ZnP-PDI, wherein both the porphyrin and the chlorin macrocycles are zinc chelated.
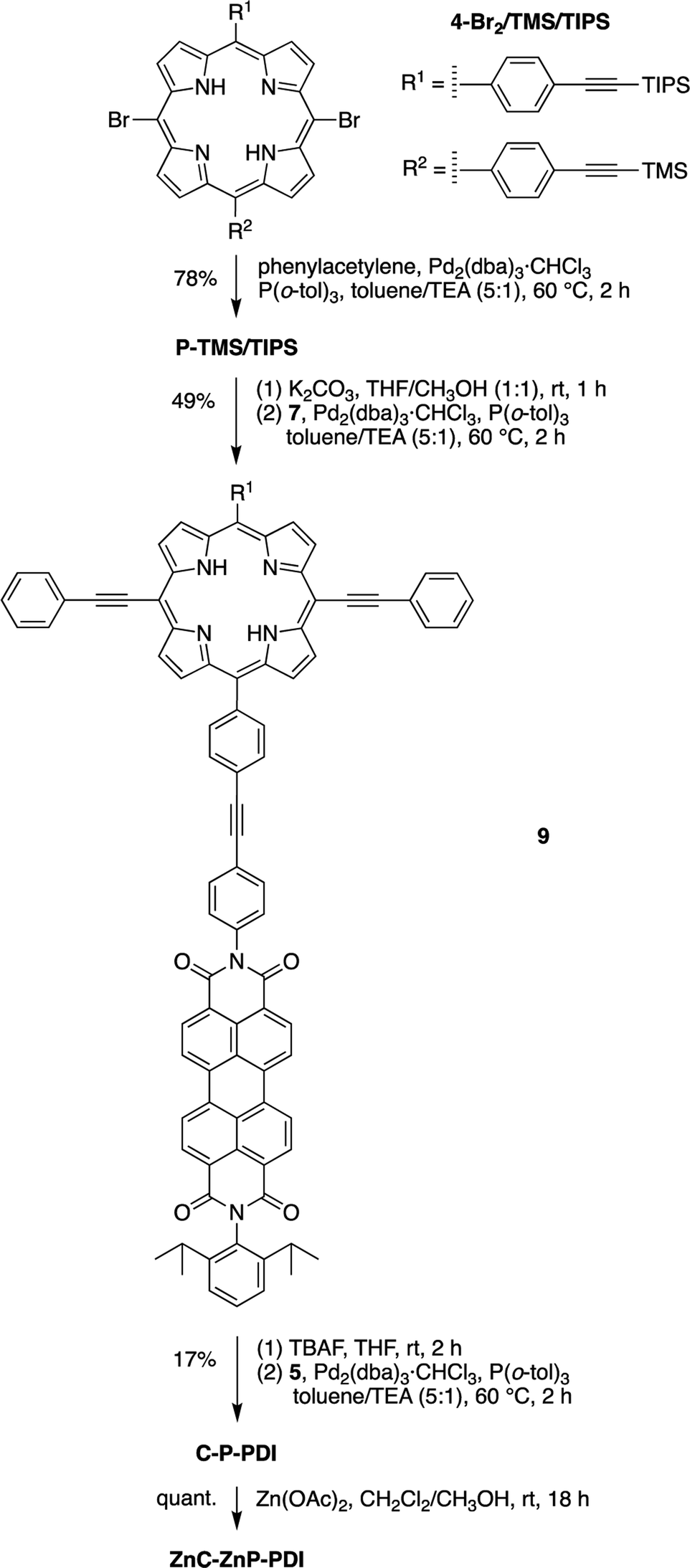 | ||
| Scheme 6 Synthesis of triads C-P-PDI and ZnC-ZnP-PDI from porphyrin 4-Br2/TMS/TIPS. | ||
The pentad C-T-PDI was synthesized similarly through the stepwise incorporation of various building blocks (Scheme 7). Perylene-monoimide 8, instead of phenylethynyl groups in the case of C-P-PDI, was installed on the meso-positions of the porphyrin 4-Br2/TMS/TIPS, generating the panchromatic triad T-TMS/TIPS. A succession of reactions – deprotecting with K2CO3, coupling with perylene-diimide 7, and again deprotecting with TBAF – afforded the tetrad T-H/PDI in 12% overall yield for the three steps. The final coupling entailed reaction of the tetrad with iodophenyl–chlorin 5 to complete the synthesis of the target pentad C-T-PDI in 27% yield. The pentad-forming reaction was monitored with analytical size exclusion chromatography (SEC), as has been done previously with multiporphyrin arrays (see ESI†).31,32 The pentad array and all benchmarks were also characterized by analytical SEC and MALDI-MS prior to any photophysical studies. On the basis of the analytical SEC traces, the purity of each array was estimated to be >99%, which is suitable for further photophysical characterization.
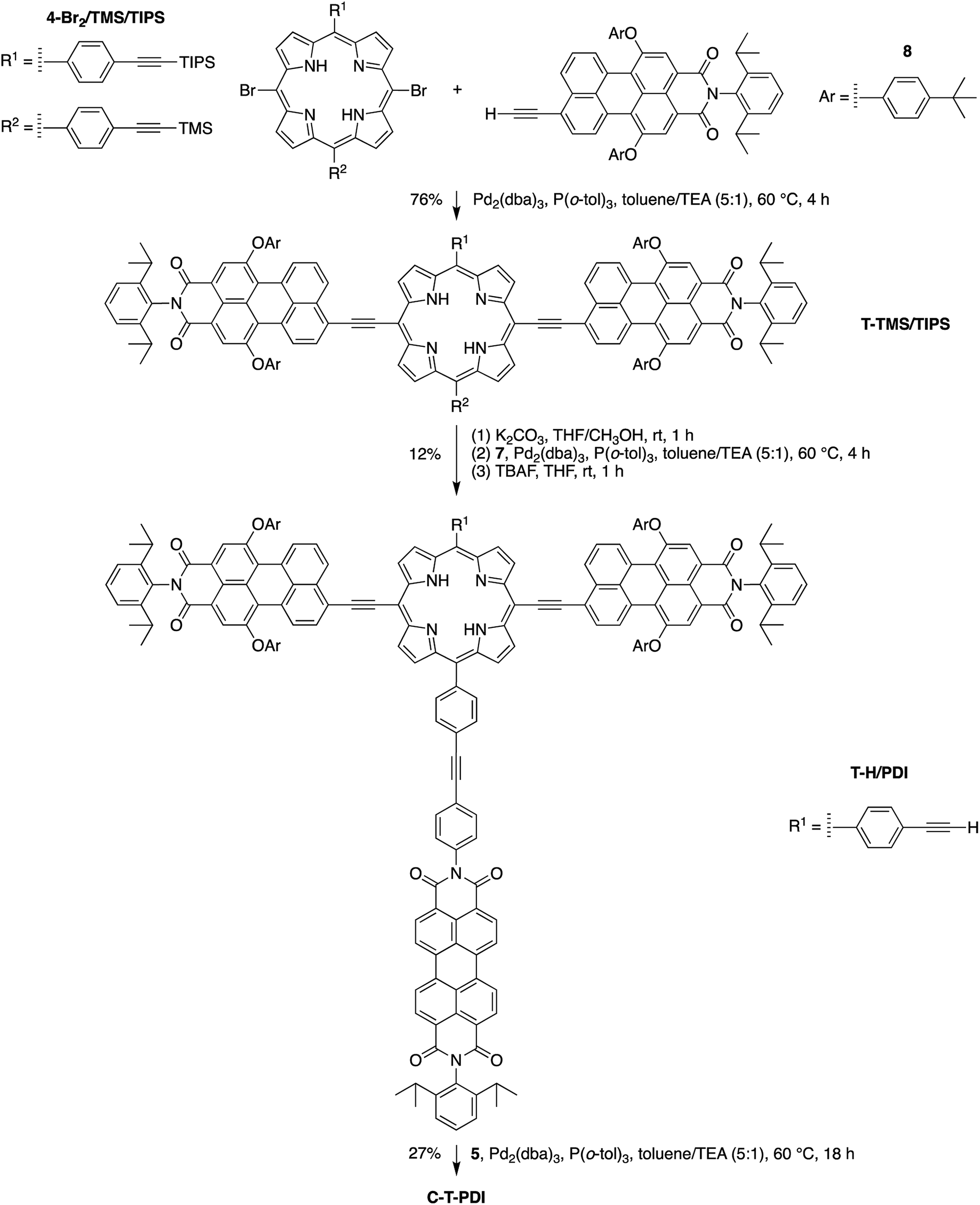 | ||
| Scheme 7 Synthesis of the target pentad C-T-PDI. | ||
Photophysical studies
000 M−1 cm−1) reflects a substantial shift of absorption strength from the typically intense porphyrin near-UV Soret feature(s) (typical peak ε of 350000–400000 M−1 cm−1) into the typically weak visible transitions (typical peak ε of 10000–20000 M−1 cm−1), accompanied by a bathochromic shift of features such as the S0 → S1 band along with the possible generation of new bands (e.g. at ∼650 nm). Corresponding differences can be seen for the corresponding zinc chelates—panchromatic triad ZnT-Ph (Fig. 3F) compared to porphyrin ZnP-H/TIPS (Fig. 4D). The substantial shift of intensity from the porphyrin near-UV Soret region into and across the visible-region features upon attachment of perylene-monoimides via direct ethyne linkers arises at least in part from a significant diminution in mixing of the key porphyrin one-electron excited-state configurations within the four-orbital model.28,30 Such a diminution in configuration interaction reduces the constructive configurational interference that affords the normal intense near-UV bands and the destructive interference that gives the typically weak visible bands within the Gouterman four-orbital model.61–63 A decrease in configuration interaction is further accentuated by the shifts in energies of the frontier molecular-orbital, which also contributes to the bathochromic shift in the S0 → S1 transition.
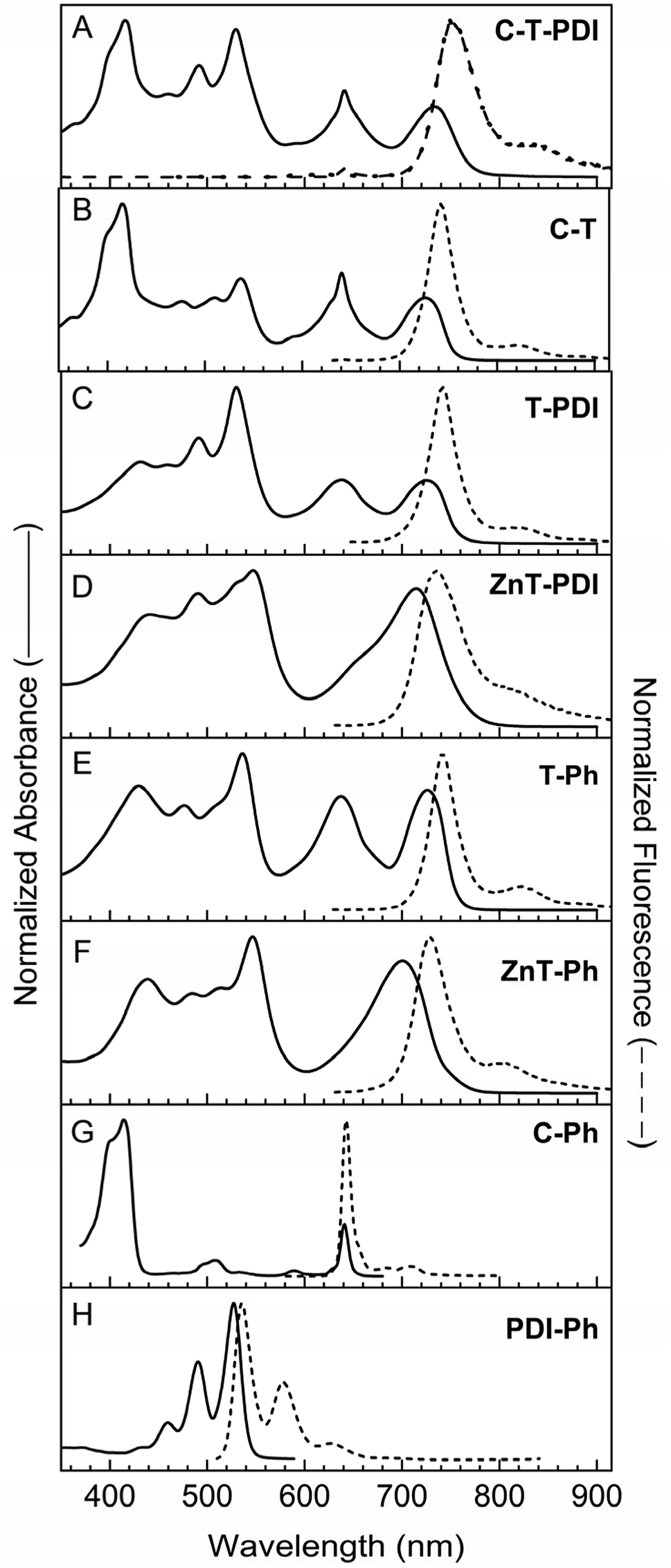 | ||
| Fig. 3 The absorption (solid) and fluorescence (dashed) spectra of pentad (A) containing a central panchromatic bis(perylene-monoimide)porphyrin triad (denoted T) and benchmark tetrads (B–D), triads (E and F) and monomers (G and H). In (A), fluorescence spectra for the pentad were obtained using primarily excitation of a nominal absorption band of the chlorin or perylenes, with essentially the same result. | ||
 | ||
| Fig. 4 The absorption (solid) and fluorescence (dashed) spectra of triads (A and B) containing a central porphyrin (denoted P) and benchmark monomers (C–F). | ||
| Compound | Abs λ1 (nm) | Abs λ2 (nm) | Abs λ3 (nm) | Abs λ4 (nm) | Abs λ5 (nm) | Abs λ6 (nm) | Abs λ6 (nm) | Flu λ1 (nm) | Flu λ2 (nm) | Abs–Flu (cm−1)b | Φf |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a All data were acquired in toluene at 298 K. The value in parenthesis following the absorption or fluorescence wavelength is the intensity relative to the maximum in the spectrum.b Shift between the S0 → S1 absorption and S1 → S0 fluorescence maxima.c Shoulder. | |||||||||||
| Panchromatic arrays | |||||||||||
| C-T-PDI | 398c (0.78) | 416 (1.0) | 492 (0.71) | 530 (0.95) | 641 (0.55) | 734 (0.45) | 753 (1.0) | 841 (0.23) | 342 | 0.16 | |
| C-T | 403c (0.84) | 415 (1.0) | 476 (0.38) | 537 (0.52) | 640 (0.56) | 726 (0.40) | 742 (1.0) | 824 (0.10) | 293 | 0.26 | |
| T-PDI | 432 (0.52) | 491 (0.77) | 530 (1.0) | 638 (0.41) | 725 (0.41) | 742 (1.0) | 829 (0.10) | 309 | 0.23 | ||
| ZnT-PDI | 441 (0.72) | 491 (0.85) | 547 (1.0) | 663c (0.46) | 714 (0.88) | 735 (1.0) | 816 (0.23) | 400 | 0.13 | ||
| T-Ph | 431 (0.79) | 476 (0.67) | 536 (1.0) | 637 (0.73) | 726 (0.77) | 741 (1.0) | 824 (0.15) | 275 | 0.30 | ||
| ZnT-Ph | 439 (0.71) | 485 (0.64) | 547 (1.0) | 662c 0.48) | 700 (0.85) | 728 (1.0) | 807 (0.20) | 547 | 0.22 | ||
|
|||||||||||
| Non-panchromatic arrays | |||||||||||
| C-P-PDI | 418 (0.45) | 445 (1.0) | 492 (0.19) | 528 (0.27) | 600 (0.14) | 641 (0.10) | 692 (0.66) | 698 (1.0) | 775 (0.18) | 118 | 0.17 |
| ZnC-ZnP-PDI | 413 (0.56) | 451 (1.0) | 490(0.15) | 528 (0.21) | 609 (0.15) | 641 (0.12) | 646 (1.0) | 706 (0.20) | 130 | 0.04 | |
|
|||||||||||
| Benchmark monomers | |||||||||||
| C-Ph | 401 (0.87) | 414 (1.0) | 496 (0.075) | 508 (0.10) | 589 (0.04) | 640 (0.29) | 641 (1.0) | 706 (0.09) | 30 | 0.32 | |
| PDI-Ph | 491 (0.61) | 521 (1.0) | 536 (1.0) | 578 (0.57) | 301 | 0.99 | |||||
| P-TMS/TIPS | 445 (1.0) | 600 (0.22) | 692 (0.10) | 695 (1.0) | 772 (0.19) | 77 | 0.16 | ||||
| ZnP-H/TIPS | 450 (1.0) | 587 (0.03) | 639 (0.11) | 647 (1.0) | 707 (0.20) | 201 | 0.10 | ||||
As expected, the absorption spectra of the pentad (C-T-PDI) and two tetrads (C-T and T-PDI) show panchromatic features similar to those of triad T-Ph (Fig. 3, panels A–C and E). In analogy, the spectrum of the tetrad containing a zinc porphyrin (ZnT-PDI) has similar panchromaticity as reference triad ZnT-Ph (Fig. 3, panels D and F). The spectra of the tetrads generally reflect the contributions of the corresponding triad (T-Ph or ZnT-Ph) and monomer (C-Ph or PDI-Ph), with some modest changes in relative intensities and peak positions. In turn, the spectrum of pentad C-T-PDI has features that correspond to those in the spectra of tetrad C-T and monomer PDI-Ph, or tetrad T-PDI and monomer C-Ph, again with some shifts in intensities and wavelengths. For example a slight bathochromic shift in the Soret band of the chlorin moiety (at 418 nm) is observed in the pentad compared to the tetrad C-T (2 nm) and chlorin benchmark C-Ph (4 nm). Broadening of this band is also observed for the pentad [full width at half maximum (FWHM) = 65 nm] compared to the tetrad C-T (40 nm) and the monomer C-Ph (33 nm).
The band at ∼530 nm due to perylene-diimide absorption is essentially superimposable on that of the tetrad T-PDI and the monomeric PDI-Ph, except for a 6 nm hypsochromic shift for the former relative to the latter. For pentad C-T-PDI, the S0 → S1 feature at ∼741 nm due to the absorption of the panchromatic bis(perylene-monoimide)porphyrin moiety shows a batho-chromic shift and broadening (FWHM = 54 nm) compared with that of tetrad C-T (727 nm, FWHM = 46 nm), tetrad T-PDI (728 nm, FWHM = 71 nm), and triad T-Ph (725 nm, FWHM = 46 nm). This effect on the S0 → S1 energy and also in the relative intensity of the absorption band is likely due in part to substituent effects on the electronic properties of the central triad. In particular, T-Ph contains one phenyl ring and one ethynylphenyl group at the two meso-positions apart from the meso-positions that bear ethynyl linkers to the perylene-monoimides (Chart 3). In each tetrad the ethynylphenyl group is converted to a diphenylethyne linker to the chlorin or perylene-diimide (Chart 2). The pentad has two such diphenyl-ethyne groups, terminated with the chlorin or perylene-diimide (Chart 1). Thus, in progressing from the triad to pentad, the change in terminal moieties on the ethynylphenyl and phenyl groups propagates to the porphyrin and sufficiently alters the porphyrin molecular orbital energies and configuration interaction to affect the characteristics of the S0 → S1 transition.
Despite the above-noted differences in certain spectral characteristics, the generally similar spectral features of the pentad and the corresponding constituents indicate that ground-state electronic interaction between the panchromatic core and either the perylene-diimide or the chlorin (via a diphenylethyne linker) are relatively weak, but not zero. Thus, addition of the chlorin and perylene-diimide moieties does not impart a substantial electronic perturbation to the central triad. In contrast, replacing the perylene-monoimide-ethynyl units attached to the central porphyrin with phenylethynyl groups restores the strong Soret bands and effectively diminishes panchromaticity, affording the spectrum of the triad C-P-PDI as a linear combination of those from the three monomeric components (Fig. 4, panels A, C, E and F). The same is true for the triad containing a central zinc porphyrin (ZnC-ZnP-PDI) relative to its three components (Fig. 4, panels B, D–F).
There is a resemblance of the fluorescence spectra of the two tetrads (C-T and T-PDI) and that of the triad T-Ph, as well as a similar resemblance between the emission of the zinc-chelate tetrad ZnT-PDI and reference monomer (Fig. 3, panels D and F). Fluorescence emission of the pentad and tetrads are predominantly from the central panchromatic bis(perylene-monoimide)-porphyrin unit, regardless of excitation wavelength. This is shown in Fig. 4A for pentad C-T-PDI, for excitation wavelengths that should primarily excite the chlorin or perylenes. This finding is consistent with efficient energy transfer from either the chlorin (in C-T) or the perylene-diimide (in T-PDI) to the central bis(perylene-monoimide)porphyrin.
As noted above, prior work on arrays that contain one (or more) perylene-monoimide unit(s) coupled via an ethyne to a porphyrin are best treated as a supermolecule: excitation within any absorption band, including features in the green region that one would normally associate with the perylene, results in rapid internal conversion within the array to produce the tetrapyrrole-like S1 excited state, from which fluorescence occurs. Thus, it is expected that rapid energy transfer from the excited chlorin or excited perylene-diimide to the bis(perylene-monoimide)porphyrin unit in the pentad and tetrads will give rise to tetrapyrrole-like emission from the S1 excited state of that triad unit. This is observed for the pentad and tetrads, even if the (Förster) spectral overlap of the emission from the excited perylene-diimide unit (e.g., in C-T-PDI and T-PDI) would be the greatest with the green-region absorption of nominal perylene-monoimides of the central panchromatic bis(perylene-monoimide)porphyrin triad. Thus, observations here support the view that the arrays containing a bis(perylene-monoimide)porphyrin unit are efficient in absorbing light spanning the near-UV and into the near-IR and producing a discrete tetrapyrrole-like S1 state from which charge separation can ensue.
In comparison with the aforementioned arrays, the fluorescence of the non-panchromatic triad C-P-PDI is at a higher energy, because no strongly coupled perylene-monoimide pigments are integrated at the central porphyrin. However, the presence of the two meso-phenylethynyl groups on the central porphyrin causes sufficient bathochromic shifts in the S0 → S1 absorption and corresponding S1 → S0 fluorescence bands that the S1 excited state remains at a lower energy than the S1 of the chlorin and the perylene-diimide pigments (Fig. 4). The same is true for triad ZnC-ZnP-PDI, in which the central bis(phenylethynyl)porphyrin is a zinc chelate, which tends to hypsochromically shift the S0 ↔ S1 transitions compared to those of the free base analogue (Fig. 4F). Thus, the observation again of porphyrin fluorescence independent of excitation wavelength indicates that these triads afford efficient energy transfer from the two terminal chromophores to the central porphyrin, from which charge separation can occur.
The Φf of C-T (0.26) and T-PDI (0.23) are reduced from that for the benchmark panchromatic triad (0.30) by 13% and 23%, respectively, indicating modest yields of excited-state electron transfer. On the basis of the redox properties, the most likely product of hole/electron transfer would be C+-T− and T+-PDI− in the two tetrads, respectively. The Φf for pentad C-T-PDI (0.16) is reduced by 47% from the panchromatic triad benchmark, suggesting that the yield of hole/electron-transfer initiated in the excited triad (T*) in this case is roughly the sum of the yields in the two tetrads. These measurements do not identify the product or product ratio, but the results tend to suggest that both C+-T−-PDI and C-T+-PDI− form in the pentad. A subsequent ground-state hole/electron step would be needed for each intermediate to produce the full charge-separated state C+-T-PDI−.
Tetrad ZnT-PDI and benchmark panchromatic triad ZnT-Ph were prepared to test whether the more facile oxidization of a zinc chelate versus the free base form would enhance excited-state electron transfer in these types of systems. The Φf of ZnT-PDI (0.13) is reduced by 41% compared to benchmark panchromatic triad ZnT-Ph (0.22), indicating that the electron-transfer yield (to produce ZnT+-PDI−) is roughly doubled relative to the yield in the analogous tetrad T-PDI that contains the free base porphyrin in the panchromatic triad unit. The simplest interpretation is that stabilization of the electron-transfer product (ZnT+-PDI− versus T+-PDI−), together with the higher S1 excited-state energy of ZnT* versus T* (Fig. 3 and 4) places the process in the zincated array on a more favorable location on the Marcus rate versus free energy curve.
The analysis now turns to the triads C-P-PDI and ZnC-ZnP-PDI in which the central panchromatic triad of the above-mentioned pentad (and tetrad benchmarks) is replaced by a porphyrin, albeit one with two phenylethynyl and one ethynylphenyl substituents that renders the porphyrin lowest singlet excited state the S1 excited state of the entire array (vide supra). As noted above, the redox properties of analogous arrays suggest that the central porphyrin in these triads will be harder to oxidize than the panchromatic triad unit in C-T-PDI, T-PDI or ZnT-PDI. The Φf in triad C-P-PDI (0.17) is essentially the same as that in porphyrin benchmark P-TMS/TIPS (0.16) consistent with no excited-state electron transfer. On the other hand, the Φf in triad ZnC-ZnP-PDI (0.04) is reduced by 60% versus porphyrin benchmark ZnP-H/TIPS (0.10) indicating a substantial yield of excited-state electron transfer. Whether this reflects formation of ZnC+-P−-PDI or ZnC-P+-PDI− or some combination in the first step and whether this is followed by the ground-state process(es) that would produce the ZnC+-P-PDI− fully charge-separated array requires in-depth time-resolved absorption studies, which are beyond the scope of this current work. Regardless, the present studies show that making both the chlorin hole trap and the porphyrin excited-state hole/electron donor simultaneously more easily oxidized compared to the free base porphyrin analogue has a dramatic effect on the photodynamics of the arrays.
Conclusions and outlook
One target pentad and 11 benchmark compounds were prepared at the 10 mg scale. The two trans-A2BC dibromoporphyrin building blocks anchor the star-shaped architecture and were readily accessible through an established “2 + 2” condensation of dipyrromethanes followed by bromination. A modular strategy was employed for the construction of the arrays, in which several pigment building blocks (porphyrin, chlorin, perylene-diimide, and perylene-monoimide) were readily synthesized and then integrated into arrays via copper-free Pd-mediated Sonogashira coupling reactions. This building block approach opens new possibilities for next-generation design and synthesis of molecular systems for studies in the broad field of artificial photosynthesis. The availability of the trans-A2BC porphyrin building blocks in conjunction with successive Pd-mediated coupling reactions provides access to star-shaped architectures that are compositionally richer than those we have prepared previously for light-harvesting and charge separation.64 The inclusion of a panchromatic supermolecule constitutes a molecular entity that differs considerably from chromophores found in natural systems.The absorption spectrum of the pentad array shows panchromatic absorption from below 400 nm to above 700 nm due to the central bis(perylene-monoimide)porphyrin unit, complemented by absorption features due to the perylene-diimide and chlorin. The latter two constituents can efficiently transfer energy to the central unit and also serve as electron acceptor and hole trap, respectively, for charge separation that is initiated in the core triad. Two chlorin–porphyrin–perylene-diimide triads that are capable of charge separation but lack the panchromatic-absorbing core were also prepared as functional benchmarks. The results described herein provide the foundation for in-depth photophysical investigations of all the arrays.
Experimental section
General methods
All chemicals obtained commercially were used as received unless otherwise noted. Reagent-grade solvents (CH2Cl2, CHCl3, methanol, toluene, ethyl acetate) and HPLC-grade solvents (toluene, CH2Cl2, hexanes) were used as received. ACS-grade hexanes was distilled (with a rotary evaporator) and dried (over 4 Å molecular sieves) immediately before use. THF was freshly distilled from sodium/benzophenone ketyl and used immediately. Matrix-assisted laser-desorption ionization mass spectrometry (MALDI-MS) was performed with the matrix 1,4-bis(5-phenyl-2-oxaxol-2-yl)benzene (POPOP)65 unless noted otherwise. ESI-MS data are reported for the molecular ion or cationized molecular ion. Noncommercial compounds 1,57 3,56 5,60 Zn560 and 834 were prepared following literature procedures, whereas the known dipyrromethane 1-TMS58 was synthesized following a more recent method.57 All other compounds were used as received from commercial sources.Chromatography
A sequence of three-chromatography procedures31 was employed for purification of arrays following Pd-mediated coupling reactions. First, adsorption column chromatography (flash silica, Silicycle) was applied to remove catalysts and other materials. Second, preparative-scale SEC was applied. A glass column (o.d. 4.3 cm) was packed using BioRad Bio-Beads S-X1 (MWCO = 12000 Da) in HPLC-grade toluene with a length of 53 cm. The chromatography was performed with gravity flow (∼1 drop per s). Eluate was collected in 3 mL portions and analyzed with MALDI-MS and absorption spectrometry. The portion with the desired product was combined and concentrated. Third, adsorption column chromatography (flash silica, Silicycle) was performed with HPLC-grade CH2Cl2 and n-hexane unless noted otherwise.
Analytical-scale SEC (styrene–divinylbenzene copolymer columns) was performed to characterize the purity of the resulting samples. Analytical SEC was performed with 100 Å, 500 Å, and 1000 Å columns in series eluting with HPLC-grade toluene (flow rate = 1.0 mL min−1, 25 °C). Sample detection was achieved by absorption spectroscopy using a diode-array detector (λdet = 430, 476, 537, 638 nm).
General procedure for Pd-catalyzed coupling reactions
Toluene (or THF) and TEA were deaerated by bubbling with argon for 1 h. The solid starting materials, including Pd2(dba)3 and P(o-tol)3, were charged in a Schlenk flask. The flask was evacuated under high vacuum for 5 min, then purged with argon. The evacuation-purge procedure was repeated three times. The deaerated solvents were then added to the Schlenk flask via a syringe. The resulting mixture was again deaerated by three freeze-pump-thaw cycles. Then liquid reagents, if any, were added to the flask. The mixture was then heated to initiate reaction. The copper-free method has been described in detail.54,555-Phenyl-15-[4-(2-trimethylsilylethynyl)phenyl]porphyrinatozinc(II) (Zn2-TMS)
Following a reported method,56 a solution of diacyldipyrromethane 1 (42 mg, 0.15 mmol) in THF (0.50 mL) was treated with propylamine (0.18 g, 3.0 mmol) and stirred at room temperature for 1 h. The solution was concentrated to dryness. The resulting solid was dissolved in ethanol (15 mL) and treated with dipyrromethane 1-TMS (48 mg, 0.15 mmol) and Zn(OAc)2 (0.28 g, 1.5 mmol). The mixture was then refluxed open to the air for 18 h. Upon cooling to room temperature, the mixture was concentrated and chromatographed [silica gel, hexanes/CH2Cl2 (2:1)] to afford a purple solid (37 mg, 40%): 1H NMR (THF-d8, 400 MHz) δ 10.28 (s, 2H), 9.40 (d, J = 4.4 Hz, 4H), 9.02 (d, J = 4.4 Hz, 4H), 8.21–8.23 (m, 4H), 7.78–7.88 (m, 5H), 0.38 (s, 9H); 13C NMR (THF-d8, 100 MHz) δ 150.9, 150.6, 150.5, 144.9, 144.3, 135.6, 132.7, 132.3, 130.8, 128.1, 127.2, 123.2, 120.5, 119.2, 106.63, 106.56, 106.4, 95.2, 0.127; MALDI-MS obsd 620.7 [M+], ESI-MS obsd 621.1428 [(M + H)+], calcd 620.1375 (M = C37H28N4SiZn); λabs (CH2Cl2) 409, 536, 572 nm.
5-Phenyl-15-[4-(2-trimethylsilylethynyl)phenyl]porphyrin (2-TMS)
Following a reported method43 with slight modifications, a solution of zinc porphyrin Zn2-TMS (33 mg, 0.053 mmol) in CHCl3 (50 mL) was treated dropwise with trifluoroacetic acid (0.20 mL, 1.2 mmol). The solution was stirred at room temperature for 30 min. Then the reaction was quenched by the addition of saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried with Na2SO4, concentrated and chromatographed [silica gel, hexanes/CH2Cl2 (2:1)] to afford a purple solid (17 mg, 58%): 1H NMR (THF-d8, 300 MHz) δ 10.38 (s, 2H), 9.44 (m, 4H), 9.04 (m, 4H), 8.22–8.26 (m, 4H), 7.82–7.94 (m, 5H), 0.38 (s, 9H), −3.12 (br, 2H); 13C NMR (THF-d8, 100 MHz) δ 150.8, 150.6, 150.5, 148.1, 147.8, 146.3, 142.9, 142.4, 135.7, 132.8, 132.7, 132.6, 132.5, 131.6, 131.3, 131.2, 128.6, 127.8, 123.7, 120.0, 118.7, 106.2, 106.1, 95.6, 0.127; MALDI-MS obsd 558.4 [M+], ESI-MS obsd 559.2326 [(M + H)+], calcd 558.2240 (M = C37H30N4Si); λabs (CH2Cl2) 406, 503, 537, 575, 630 nm.
10,20-Dibromo-5-phenyl-15-[4-(2-trimethylsilylethynyl)phenyl]porphyrin (2-Br2/TMS)
Following a standard bromination method,59 a solution of porphyrin 2-TMS (12 mg, 0.021 mmol) in CHCl3 (6.8 mL) containing pyridine (9.0 μL) was cooled to 0 °C and treated with NBS (9.2 mg, 0.052 mmol). The mixture was stirred at 0 °C for 1 h. The reaction was quenched by the addition of saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried with Na2SO4, concentrated, and chromatographed [silica gel, hexanes/CH2Cl2 (4:1)] to afford a purple-red solid (11 mg, 74%): 1H NMR (THF-d8, 300 MHz) δ 9.62 (d, J = 4.5 Hz, 4H), 8.82 (d, J = 4.5 Hz, 4H), 8.12–8.16 (m, 4H), 7.78–7.89 (m, 5H), 0.38 (s, 9H), −2.74 (s, 2H); 13C NMR (THF-d8, 100 MHz) δ 142.6, 142.3, 135.4, 131.2, 129.0, 127.8, 124.2, 122.7, 121.5, 105.9, 104.2, 95.9, 0.089; MALDI-MS obsd 717.1 [(M + H)+], ESI-MS 715.0520 [(M + H)+], calcd 714.0405 (M = C37H28Br2N4Si); λabs (CH2Cl2) 422, 485, 520, 557, 661 nm.
5-(4-Iodophenyl)-15-[4-(2-trimethylsilylethynyl)phenyl]porphyrinatozinc(II) (Zn4-I/TMS)
Following a reported method,56 a solution of diformyldipyrromethane–tin complex 3 (0.40 g, 0.63 mmol) in propylamine (1.0 mL) was stirred at room temperature for 2 h and then concentrated to dryness. A solution of the resulting solid and Zn(OAc)2 (1.20 g, 6.3 mmol) in ethanol (63 mL) was refluxed open to the air for 17 h. The cooled mixture was passed through a pad of silica gel (CH2Cl2), concentrated, and chromatographed [silica gel, hexanes/ethyl acetate (4:1)] to afford a purple-red solid (73 mg, 15%): 1H NMR (THF-d8, 300 MHz) δ 10.29 (s, 2H), 9.42 (d, J = 4.4 Hz, 4H), 9.02 (dd, J = 5.6, 4.4 Hz, 4H), 8.22 (d, J = 8.0 Hz, 2H), 8.16 (d, J = 8.0 Hz, 2H), 8.02 (d, J = 8.0 Hz, 2H), 7.88 (d, J = 8.0 Hz, 2H), 0.38 (s, 9H); MALDI-MS obsd 748.0 [(M + H)+], ESI-MS obsd 747.0392 [(M + H)+], calcd 747.0414 (M = C37H27IN4SiZn); λabs (CH2Cl2) 411, 538, 572 nm.
5-[4-(2-Triisopropylsilylethynyl)phenyl]-15-[4-(2-trimethylsilylethynyl)phenyl]porphyrinatozinc(II) (Zn4-TMS/TIPS)
Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of Zn4-I/TMS (67 mg, 0.090 mmol), PdCl2(PPh3)2 (3.2 mg, 4.5 μmol), and CuI (1.7 mg, 1.0 μmol) in THF/TEA (9:1, 9.0 mL) was treated with TIPS-acetylene (0.100 mL, 0.45 mmol) under argon and then stirred at 60 °C for 4 h. The reaction was quenched by the addition of water. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, concentrated, and chromatographed [silica gel, hexanes/ethyl acetate (9:1)] to afford a red solid (58 mg, 80%): 1H NMR (THF-d8, 300 MHz) δ 10.29 (s, 2H), 9.42 (d, J = 4.5 Hz, 4H), 9.02 (dd, J = 7.2, 4.8 Hz, 4H), 8.22 (dd, J = 7.8, 1.8 Hz, 4H), 7.89 (dd, J = 9.9, 7.8 Hz, 4H), 1.50 (m, 3H), 1.10 (m, 18H), 0.37 (s, 9H); MALDI-MS obsd 802.4 [(M + H)+], ESI-MS obsd 801.2775 [(M + H)+], calcd 801.2782 (M = C48H48N4Si2Zn); λabs (CH2Cl2) 411, 538, 576 nm.
5,15-Dibromo-10-[4-(2-triisopropylsilylethynyl)phenyl]-20-[4-(2-trimethylsilylethynyl)phenyl]porphyrin (4-Br2/TMS/TIPS)
A solution of Zn4-TMS/TIPS (54 mg, 67 μmol) in CH2Cl2 (22 mL) containing pyridine (0.030 mL, 0.33 mmol) was treated with NBS (0.030 g, 0.17 mmol) at 0 °C for 1 h. The solution was allowed to warm to room temperature and then treated with saturated aqueous NH4Cl solution. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, and concentrated to dryness. A solution of the resulting solid in CH2Cl2 (40 mL) was treated dropwise with trifluoroacetic acid (0.26 mL, 3.4 mmol) and stirred at room temperature for 30 min. The reaction was quenched by the addition of saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, concentrated and chromatographed [silica gel, hexanes/CH2Cl2 (3:1)] to afford a red solid (16 mg, 27%): 1H NMR (THF-d8, 300 MHz) δ 9.64 (d, J = 5.1 Hz, 4H), 8.84–8.87 (m, 4H), 8.14 (d, J = 7.8 Hz, 4H), 7.90 (m, 4H), 1.49 (m, 3H), 1.29, (s, 18H), 0.37 (s, 9H), −2.74 (s, 2H); MALDI-MS obsd 896.6 [(M + H)+], ESI-MS obsd 895.1836 [(M + H)+], calcd 895.1857 (M = C48H48Br2N4Si2); λabs (CH2Cl2) 429, 524, 562, 605, 659 nm.
5-(4-Ethynylphenyl)-10,20-bis(2-phenylethynyl)-15-[4-(2-triisopropylsilylethynyl)phenyl]porphyrinatozinc(II) (ZnP-H/TIPS)
A solution of Zn4-TMS/TIPS (118 mg, 0.146 mmol, 1.0 equiv.) in CH2Cl2 (49 mL, [Zn4-TMS/TIPS] ∼3 mM) containing pyridine (65 μL, 0.81 mmol, 5.5 equiv.) was cooled to 0 °C under argon, and treated with NBS (65 mg, 0.37 mmol, 2.5 equiv.) at 0 °C for 1 h. The reaction was then quenched by the addition of saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2. The organic extract was dried, concentrated, and chromatographed (silica gel, hexanes/CH2Cl2 = 1:1) to afford a blue solid (140 mg): TLC (silica, hexanes/CH2Cl2 = 1:1) Rf = 0.28; MALDI-MS obsd 957.0 [(M + H)+]. Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of the resulting solid (140 mg, 0.146 mmol, 1.0 equiv.), phenylacetylene (35 μL, 0.32 mmol, 2.2 equiv.), Pd2(dba)3 (40 mg, 0.044 mmol, 0.30 equiv.), P(o-tol)3 (107 mg, 0.350 mmol, 2.4 equiv.), and CuI (1.4 mg, 7.3 μmol, 0.050 equiv.) in deaerated THF/TEA (5:1, 14.6 mL) was stirred at 60 °C for 17 h. The mixture was allowed to cool to room temperature, dried, and chromatographed (silica gel, hexanes/CH2Cl2 = 2:1 to 3:2) to afford a blue solid (72.6 mg): TLC (silica, hexanes/CH2Cl2 = 3:2) Rf = 0.30; MALDI-MS obsd 1001.9 [(M + H)+]. Then, a mixture of the resulting solid (72.6 mg) and K2CO3 (1.00 g, 7.24 mmol, 100 equiv.) in THF/CH3OH (1:1, 15 mL) was stirred at room temperature for 3 h. MALDI-MS analysis showed the completion of the reaction [obsd 929.8, [(M + H)+]]. The reaction was then quenched by the addition of water. The mixture was extracted with CH2Cl2. The organic extract was dried, concentrated, and chromatographed (silica gel, hexanes/CH2Cl2 = 3:2 to 1:1) to afford a blue solid (60.2 mg, 44% for three steps): 1H NMR (THF-d8, 400 MHz) δ 9.76 (d, J = 4.3 Hz, 4H), 8.86 (d, J = 4.7 Hz, 4H), 8.20–7.47 (m, 18H), 3.28 (s, 1H), 1.30–1.22 (m, 21H); MALDI-MS obsd 929.8 [(M + H)+], ESI-MS obsd 929.3038 [(M + H)+], calcd 929.3012 (M = C61H48N4SiZn).
10-Mesityl-18,18-dimethyl-5-(4-(2-phenylethynyl)phenyl)chlorin (C-Ph)
Following the general procedure for Pd-catalyzed coupling reactions described above, a solution of Zn5 (5.0 mg, 6.9 μmol) in THF/TEA (9:1, 0.70 mL) was treated with phenylacetylene (3.8 μL, 0.034 mmol) in the presence of PdCl2(PPh3)2 (0.70 mg, 1.0 μmol) and CuI (0.20 mg, 1.0 μmol) and stirred at 60 °C for 24 h. The reaction was quenched by the addition of water. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, and concentrated. A solution of the resulting sample in CH2Cl2 (4.1 mL) was treated dropwise with trifluoroacetic acid (0.027 mL) and stirred at room temperature for 30 min. The reaction was quenched by the addition of saturated aqueous NaHCO3 solution. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, concentrated, and chromatographed [silica gel, hexanes/CH2Cl2 (3:1)] to afford a green solid (3.7 mg, 84%): 1H NMR (CDCl3, 300 MHz) δ 8.93 (s, 1H), 8.86 (s, 1H), 8.83 (d, J = 4.5 Hz, 1H), 8.74–8.76 (m, 2H), 8.60 (d, J = 4.5 Hz, 1H), 8.43 (d, J = 4.5 Hz, 1H), 8.46 (d, J = 4.5 Hz, 1H), 8.08 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 8.1 Hz, 2H), 7.55–7.58 (m, 2H), 7.32–7.34 (m, 3H), 7.23 (s, 2H), 4.60 (s, 2H), 2.59 (s, 3H), 2.06 (s, 6H), 1.84 (s, 6H), −1.83 (s, 2H); MALDI-MS obsd 634.8 [M+], ESI-MS obsd 635.3169 [(M + H)+], calcd 634.3096 (M = C45H38N4); λabs (toluene) 414, 508, 589, 641 nm.
N-(4-Iodophenyl)-N′-(2,6-diisopropylphenyl)perylene-3,4,9,10-bis(dicarboximide) (7)
Following a reported procedure,37 a mixture of 6 (3.00 g, 7.64 mmol), 2,6-diisopropylaniline (4.3 mL, 23 mmol), 4-iodoaniline (3.35 g, 15 mmol), Zn(OAc)2 (1.05 g), and imidazole (22.5 g) was heated to 190 °C under argon. After 20 h, the mixture was allowed to cool to room temperature and then was treated with 60% ethanol (180 mL) for another 2 h. The suspension was then filtered. The filter cake was washed with 60% ethanol. The dry sample was suspended in 10% aqueous KOH solution and refluxed for 30 min. Upon cooling to room temperature, the mixture was filtered. The filter cake was washed with water and dried in an oven. The resulting solid was soaked in CH2Cl2 (100 mL), sonicated for 5 min, and filtered to remove insoluble perylene-diimide byproducts. The CH2Cl2 filtrate was concentrated and chromatographed [silica gel, hexanes/ethyl acetate (7:1 to 7:3)] to afford a red solid (1.52 g, 30%): 1H NMR (CDCl3, 300 MHz) δ 8.66–8.80 (m, 8H), 7.91 (dd, J = 6.6, 1.8 Hz, 2H), 7.51 (t, J = 7.5 Hz, 1H), 7.36 (d, J = 7.5 Hz, 2H), 7.12 (dd, J = 6.6, 1.8 Hz, 2H), 2.76 (quintet, J = 6.6 Hz, 2H), 1.18 (d, J = 6.6 Hz, 12H); MALDI-MS obsd 754.0 [(M + H)+], ESI-MS obsd 753.1235 [(M + H)+], calcd 752.1172 (M = C49H29IN2O4); λabs (CH2Cl2) 434, 459, 490, 527 nm.
N-[4-(2-Phenylethynyl)phenyl]-N′-(2,6-diisopropylphenyl)perylene-3,4,9,10-bis(dicarboximide) (PDI-Ph)
Following the general procedure for Pd-catalyzed coupling reactions described above, a solution of 7 (3.0 mg, 4.0 μmol) in toluene/TEA (5:1, 1.8 mL) was treated with phenylacetylene (2.2 μL, 0.020 mmol) in the presence of PdCl2(PPh3)2 (0.50 mg, 0.60 μmol) and CuI (0.10 mg, 0.60 μmol) and stirred at 60 °C for 3 h. Then the reaction was quenched by the addition of water. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, concentrated, and chromatographed [silica gel, hexanes/ethyl acetate (7:3)] to afford a red solid (1.5 mg, 52%): 1H NMR (CDCl3, 300 MHz) δ 8.69–8.81 (m, 8H), 7.73 (d, J = 8.7 Hz, 2H), 7.49–7.59 (m, 3H), 7.35–7.40 (m, 7H), 2.76 (pentad, J = 6.9 Hz, 2H), 1.19 (d, J = 6.9 Hz, 12H); MALDI-MS obsd 726.1 [M+], ESI-MS 727.2553 [(M + H+)], calcd 726.2519 (M = C50H34N2O4); λabs (toluene) 459, 491, 527 nm.
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-phenyl-20-(4-(2-trimethylsilylethynyl)phenyl)porphyrin (T-Ph)
Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of porphyrin 2-Br2/TMS (5.4 mg, 7.5 μmol), 8 (12 mg, 0.015 mmol), Pd2(dba)3 (2.1 mg, 2.2 μmol), and P(o-tol)3 (5.5 mg, 18 μmol) in toluene/TEA (5:1, 3.4 mL) was stirred at 60 °C for 4 h. The mixture was allowed to cool to room temperature and then was purified according to the three-chromatography method. The product was concentrated to afford a black solid (14 mg, 86%): 1H NMR (CDCl3, 300 MHz): δ 9.73 (d, J = 5.1 Hz, 4H); 9.27 (d, J = 7.2 Hz, 2H), 9.21 (d, J = 7.8 Hz, 2H), 9.02 (d, J = 8.1 Hz, 2H), 8.86 (d, J = 5.1 Hz, 2H), 8.80 (d, J = 5.1 Hz, 2H), 8.33 (s, 2H), 8.28 (s, 2H), 8.20–8.10 (m, 4H), 7.90 (d, J = 7.8 Hz, 2H), 7.79 (m, 3H), 7.67 (t, J = 7.8 Hz, 2H), 7.39–7.49 (m, 12H), 7.32 (d, J = 7.8 Hz, 4H), 7.08 (d, J = 9.0 Hz, 4H), 7.05 (d, J = 9.0 Hz, 4H), 2.77 (quintet, J = 6.6 Hz, 4H), 1.34 (s, 36H), 1.18 (d, J = 6.6 Hz, 24H), 0.089 (s, 9H), −1.90 (s, 2H); MALDI-MS obsd 2158.0 [(M + H)+], calcd 2156.9563 (M = C149H128N6O8Si); λabs (toluene) 429, 476, 537, 638, 726 nm.
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-phenyl-20-(4-(2-trimethylsilylethynyl)phenyl)porphyrinatozinc(II) (ZnT-Ph)
A solution of T-Ph (22 mg, 10 μmol) was treated with Zn(OAc)2·2H2O (44 mg, 0.20 mmol) in CH2Cl2/CH3OH (2:1, 3.0 mL) for 18 h. The resulting mixture was washed with water. The organic layer was dried and purified according to the three-chromatography method to afford a black solid (10 mg, 45%): 1H NMR (CDCl3, 300 MHz): δ 9.77 (m, 4H); 9.30–9.00 (m, 6H), 8.90–8.82 (m, 4H), 8.35 (s, 2H), 8.27 (s, 2H), 8.22–8.10 (m, 4H), 7.90–7.65 (m, 7H), 7.39–7.49 (m, 12H), 7.34 (d, J = 8.1 Hz, 4H), 7.08 (d, J = 8.7 Hz, 4H), 7.07 (d, J = 8.6 Hz, 4H), 2.77 (quintet, J = 6.5 Hz, 4H), 1.34 (s, 36H), 1.18 (d, J = 6.6 Hz, 24H), 0.090 (s, 9H); MALDI-MS obsd 2220.0 [(M + H)+], calcd 2219.8771 (M = C149H126N6O8SiZn).
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-(4-ethynylphenyl)-20-phenylporphyrin (T-Ph-H)
A solution of T-Ph (7.0 mg, 3.2 μmol) in toluene (3.0 mL) and methanol (3.0 mL) was treated with K2CO3 (44 mg, 0.32 mmol) and stirred at room temperature for 3 h. The reaction was quenched by the addition of water. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, and concentrated to afford a black solid (6.7 mg, quant.): 1H NMR (CDCl3, 300 MHz): δ 9.73 (d, J = 5.1 Hz, 4H); 9.27 (d, J = 7.2 Hz, 2H), 9.21 (d, J = 7.8 Hz, 2H), 9.02 (d, J = 8.1 Hz, 2H), 8.84 (d, J = 5.1 Hz, 2H), 8.80 (d, J = 5.1 Hz, 2H), 8.34 (s, 2H), 8.28 (s, 2H), 8.10–8.20 (m, 4H), 7.91 (d, J = 7.8 Hz, 2H), 7.79 (m, 3H), 7.67 (t, J = 7.8 Hz, 2H), 7.39–7.49 (m, 12H), 7.32–7.00 (m, 12H), 3.36 (s, 1H), 2.77 (quintet, J = 6.6 Hz, 4H), 1.34 (s, 36H), 1.18 (d, J = 6.6 Hz, 24H), −1.88 (s, 2H); MALDI-MS obsd 2085.7 [(M + H)+], calcd 2084.9168 (M = C146H120N6O8); λabs (toluene) 429, 476, 537, 638, 726 nm.5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-[4-(2-(4-(9,10-(N-(2,6-diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-20-phenylporphyrin (T-PDI)
Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of the triad T-Ph-H (34 mg, 16 μmol), iodophenyl-perylene-diimide 7 (12 mg, 16 μmol), Pd2(dba)3 (2.2 mg, 2.4 μmol), and P(o-tol)3 (5.4 mg, 18 μmol) in toluene/TEA (5:1, 4.0 mL) was stirred at 60 °C for 4 h. The mixture was allowed to cool to room temperature and then was chromatographed according to the three-chromatography method to afford a black solid (17 mg, 40%): 1H NMR (CDCl3, 400 MHz) δ 9.78 (d, J = 5.1 Hz, 4H); 9.30 (d, J = 7.2 Hz, 2H), 9.20 (d, J = 7.8 Hz, 2H), 9.01 (d, J = 8.1 Hz, 2H), 8.88 (d, J = 5.1 Hz, 2H), 8.80–8.60 (m, 10H), 8.34 (s, 2H), 8.28 (s, 2H), 8.10–8.20 (m, 4H), 7.94–7.90 (m, 4H), 7.79 (m, 3H), 7.67 (t, J = 7.8 Hz, 2H), 7.30–7.52 (m, 19H), 7.15–7.05 (m, 10H), 2.80–2.77 (m, 6H), 1.34 (s, 36H), 1.20–1.18 (m, 36H), −1.95 (s, 2H); MALDI-MS obsd 2711.0 [(M + H)+], calcd 2709.1217 (M = C188H148N8O12); λabs (toluene) 430, 463, 492, 530, 639, 727 nm.
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-[4-(2-(4-(9,10-(N-(2,6-diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-20-phenylporphyrinatozinc(II) (ZnT-PDI)
A solution of T-PDI (0.020 g, 7.4 μmol, 1.0 equiv.) in CH2Cl2/CH3OH (2:1, 2.2 mL) was treated with Zn(OAc)2·2H2O (32 mg, 0.15 mmol, 20 equiv.) at room temperature for 22 h, then washed with water. The organic layer was dried, concentrated, and chromatographed by SEC (HPLC-grade toluene) followed by silica gel (CH2Cl2/CH3OH = 1:1 to 9:1) to afford a black solid (0.020 g, quant.): 1H NMR (CDCl3, 400 MHz) δ 9.78 (d, J = 5.1 Hz, 4H); 9.30 (d, J = 7.2 Hz, 2H), 9.20 (d, J = 7.8 Hz, 2H), 9.01 (d, J = 8.1 Hz, 2H), 8.88 (d, J = 5.1 Hz, 2H), 8.80–8.60 (m, 10H), 8.34 (s, 2H), 8.28 (s, 2H), 8.10–8.20 (m, 4H), 7.94–7.90 (m, 4H), 7.79 (m, 3H), 7.67 (t, J = 7.8 Hz, 2H), 7.30–7.52 (m, 19H), 7.15–7.05 (m, 10H), 2.80–2.77 (m, 6H), 1.34 (s, 36H), 1.20–1.18 (m, 36H); MALDI-MS obsd 2772.9 [(M + H)+], calcd 2771.0352 (M = C188H146N8O12Zn).
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-[4-(2-(4-(10-mesityl-18,18-dimethylchlorin-5-yl)phenyl)ethynyl)phenyl]-20-phenylporphyrin (C-T)
Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of the triad T-Ph-H (33 mg, 16 μmol), iodophenyl–chlorin 5 (11 mg, 16 μmol), Pd2(dba)3 (2.2 mg, 2.4 μmol), and P(o-tol)3 (5.4 mg, 18 μmol) in toluene/TEA (5:1, 4.0 mL) was stirred at 60 °C for 3 h. The mixture was allowed to cool to room temperature and then was chromatographed according to the three-chromatography method to afford a black solid (10 mg, 24%): 1H NMR (CDCl3, 400 MHz) δ 9.79–9.77 (m, 2H), 9.53–9.44 (m, 4H), 9.10 (d, J = 7.6 Hz, 2H), 8.98 (s, 1H), 8.92–8.85 (m, 4H), 8.78 (d, J = 4.0 Hz, 1H), 8.63 (d, J = 4.8 Hz, 1H), −1.89 (s, 2H), 8.54 (d, J = 4.4 Hz, 1H), 8.50 (m, 1H), 8.42 (d, J = 4.4 Hz, 1H), 8.36 (s, 1H), 8.35 (s, 1H), 8.32 (d, J = 8.0 Hz, 2H), 8.19–8.21 (m, 3H), 8.08 (d, J = 8.4 Hz, 1H), 8.01 (d, J = 8.4 Hz, 2H), 7.87–7.77 (m, 5H), 7.52–7.42 (m, 11H), 7.31 (d, J = 8.0 Hz, 6H), 7.13–7.10 (m, 8H), 4.65 (s, 2H), 2.74 (quintet, J = 6.8 Hz, 6H), 2.62 (s, 3H), 2.10 (s, 6H), 1.89 (s, 6H), 1.34 (s, 36H), 1.16 (d, J = 6.8 Hz, 36H), −1.81 (s, 2H), −1.90 (s, 2H); MALDI-MS obsd 2618.6 [(M + H)+], calcd 2617.1795 (M = C183H152N10O8); λabs (toluene) 416, 477, 511, 537, 641, 728 nm.
5,15-Bis(2-phenylethynyl)-10-[4-(2-trimethylsilylethynyl)phenyl]-20-[4-(2-triisopropylsilylethynyl)phenyl]porphyrin (P-TMS/TIPS)
Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of porphyrin 4-Br2/TMS/TIPS (47 mg, 52 μmol, 1.0 equiv.), phenylacetylene (13 mg, 0.13 mmol, 2.5 equiv.), Pd2(dba)3·CHCl3 (8.1 mg, 7.9 μmol, 0.15 equiv.), and P(o-tol)3 (19 mg, 62 μmol, 1.2 equiv.) in toluene/TEA (5:1, 10.2 mL) was stirred at 60 °C for 2 h. The mixture was then washed with water. The organic layer was dried, concentrated, and chromatographed (silica gel, hexanes/CH2Cl2, 2:1) to afford a blue solid (38 mg, 78%): TLC (silica, hexanes/CH2Cl2, 3:2) Rf = 0.54; 1H NMR (CDCl3, 300 MHz) δ 9.65 (d, J = 4.7 Hz, 4H), 8.78 (dd, J = 9.3, 4.8 Hz, 4H), 8.12 (d, J = 8.0 Hz, 4H), 8.02 (d, J = 6.6 Hz, 4H), 8.00–7.88 (m, 4H), 7.60–7.50 (m, 6H), 1.27–1.25 (m, 21H), 0.40 (s, 9H), −2.09 (s, 2H); ESI-MS obsd 939.4105 [(M + H)+], calcd 938.4200 (M = C64H58N4Si2).
5-[4-(2-(4-(9,10-(N-(2,6-Diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-10,20-bis(2-phenylethynyl)-15-[4-(2-triisopropylsilylethynyl)phenyl]porphyrin (9)
A mixture of P-TMS/TIPS (10 mg, 0.011 mmol, 1.0 equiv.) and K2CO3 (0.15 g, 1.1 mmol, 100 equiv.) in THF/CH3OH (1:1, 10 mL) was stirred at room temperature for 30 min. The reaction was then quenched by the addition of 2 M HCl solution (10 mL). The mixture was extracted with CH2Cl2. The organic extract was dried and concentrated to dryness. Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of the resulting sample, iodophenyl-perylene-diimide 7 (8.3 mg, 0.011 mmol, 1.0 equiv.), Pd2(dba)3·CHCl3 (1.7 mg, 1.6 μmol, 0.15 equiv.), and P(o-tol)3 (4.0 mg, 0.013 mmol, 1.2 equiv.) in toluene/TEA (5:1, 2.4 mL) was stirred at 60 °C for 2 h. The mixture was then allowed to cool to room temperature and passed through a pad of silica gel (CH2Cl2). The eluate was concentrated and chromatographed (silica gel, CH2Cl2) to afford a green solid (7.9 mg, 49%): TLC (silica, CH2Cl2) Rf = 0.22; Analytical SEC (toluene, 1.0 mL min−1) tR = 21.12 min; MALDI-MS obsd 1491 (M+), calcd 1490.5854 (M = C103H78N6O4Si); λabs (toluene) 445, 491, 528, 600, 692 nm. The compound was not characterized by NMR spectroscopy due to limited solubility in organic solvents.
5-[4-(2-(4-(9,10-(N-(2,6-Diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-15-[4-(2-(4-(10-mesityl-18,18-dimethylchlorin-5-yl)phenyl)ethynyl)phenyl]-10,20-bis(2-phenylethynyl)porphyrin (C-P-PDI)
A solution of 9 (6.5 mg, 0.0044 mmol, 1.0 equiv.) in THF (2.2 mL) was treated with 1 M TBAF solution in THF (8.7 μL, 0.0087 mmol, 2.0 equiv.) at room temperature for 2 h, then treated with water. The mixture was extracted with CH2Cl2. The organic extract was dried and concentrated to dryness. Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of the resulting sample, iodophenyl–chlorin 5 (2.9 mg, 0.044 mmol, 1.0 equiv.), Pd2(dba)3·CHCl3 (1.4 mg, 0.00132 mmol, 0.30 equiv.), and P(o-tol)3 (1.6 mg, 0.0053 mmol, 1.2 equiv.) in toluene/TEA (5:1, 1.2 mL) was stirred at 60 °C for 2 h. The mixture was then allowed to cool to room temperature. Purification according to the three-chromatography method afforded a green solid (1.4 mg, 17%): TLC (silica, CH2Cl2/CH3OH = 99:1) Rf = 0.52; 1H NMR (THF-d8, 400 MHz) δ 9.68 (dd, J = 11.8, 4.3 Hz, 4H), 8.87–8.86 (m, 4H), 8.68–8.64 (m, 2H), 8.58 (s, 2H), 8.54 (d, J = 4.5 Hz, 1H), 8.40–8.32 (m, 4H), 8.29–8.27 (m, 5H), 8.21–7.98 (m, 15H), 7.71–7.51 (m, 12H), 7.24 (s, 2H), 4.56 (s, 2H), 3.10–3.02 (m, 2H), 2.05 (s, 9H), 1.89 (s, 6H), 1.30–1.29 (m, 16H), −1.82 (s, 2H), −1.88 (s, 2H); MALDI-MS obsd 1867.6 [(M + H)+], calcd 1866.7143 (M = C131H90N10O4); λabs (toluene) 418, 446, 528, 600, 642, 692 nm.
5-[4-(2-(4-(9,10-(N-(2,6-Diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-15-[4-(2-(4-(10-mesityl-18,18-dimethylchlorinatozinc(II)-5-yl)phenyl)ethynyl)phenyl]-10,20-bis(2-phenylethynyl)porphyrinatozinc(II) (ZnC-ZnP-PDI)
A solution of C-P-PDI (1.0 mg, 0.54 μmol, 1.0 equiv.) in THF (1.0 mL) was treated with Zn(OAc)2 (2.3 mg, 0.011 mmol, 20 equiv.) at room temperature for 18 h. The sample was treated with water and extracted with CH2Cl2. The organic extract was dried and concentrated to afford a green solid (1.0 mg, quant.): TLC (silica, CH2Cl2/CH3OH = 99:1) Rf = 0.44; 1H NMR (THF-d8, 400 MHz) δ 9.65 (dd, J = 11.8, 4.3 Hz, 4H), 8.87–8.86 (m, 4H), 8.68–8.64 (m, 2H), 8.58 (s, 2H), 8.54 (d, J = 4.5 Hz, 1H), 8.40–8.36 (m, 4H), 8.29–8.27 (m, 5H), 8.21–7.98 (m, 15H), 7.71–7.41 (m, 12H), 7.24 (s, 2H), 4.53 (s, 2H), 3.06–3.02 (m, 2H), 2.05 (s, 9H), 1.89 (s, 6H), 1.30–1.29 (m, 16H); MALDI-MS obsd 1991.5 [(M + H)+], calcd 1990.5 (M = C131H86N10O4Zn2); λabs (toluene) 418, 446, 528, 600, 642, 692 nm.
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-[4-(2-trimethylsilylethynyl)phenyl]-20-[4-(2-triisopropylsilylethynyl)phenyl]porphyrin (T-TMS/TIPS)
Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of dibromoporphyrin 4-Br2/TMS/TIPS (14 mg, 16 μmol), ethynylperylene-monoimide 8 (26 mg, 32 μmol), Pd2(dba)3 (4.4 mg, 4.8 μmol), and P(o-tol)3 (12 mg, 38 μmol) in toluene/TEA (5:1, 6.4 mL) was stirred at 60 °C for 4 h. The mixture was allowed to cool to room temperature and then chromatographed according to the three-chromatography method to afford a black solid (28 mg, 76%): 1H NMR (CDCl3, 300 MHz), δ 9.78 (d, J = 4.8 Hz, 4H), 9.48 (dd, J = 7.2, 3.9 Hz, 2H), 9.11 (d, J = 7.8 Hz, 2H), 8.86 (dd, J = 7.8, 4.5 Hz, 4H), 8.37 (s, 2H), 8.36 (s, 2H), 8.34 (d, J = 8.4 Hz, 2H), 8.15 (d, J = 7.8 Hz, 4H), 7.92 (dd, J = 8.4, 3.0 Hz, 4H), 7.86 (d, J = 8.1 Hz, 2H), 7.73 (t, J = 5.7 Hz, 2H), 7.40–7.43 (m, 10H), 7.32 (s, 2H), 7.30 (s, 2H), 7.12 (dd, J = 9.0, 2.1 Hz, 8H), 2.75 (hept, J = 6.9 Hz, 4H), 2.04 (hept, J = 6.9 Hz, 3H), 1.36 (d, J = 1.8 Hz, 36H), 1.17 (d, J = 6.6 Hz, 24H), 0.99 (d, J = 6.9 Hz, 18H), 0.40 (s, 9H), −1.81 (s, 2H); MALDI-MS obsd 2337.2 [M+], calcd 2337.0897 (M = C160H148N6O8Si2); λabs (CH2Cl2) 423, 471, 531, 636, 725 nm.
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-[4-(2-(4-(9,10-(N-(2,6-diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-20-[4-(2-triisopropylsilylethynyl)phenyl]porphyrin (T-H/PDI)
A solution of T-TMS/TIPS (28 mg, 12 μmol) in THF/MeOH (3:1, 12 mL) was treated with K2CO3 (11 mg, 80 μmol) at room temperature for 1 h. The reaction was quenched by the addition of water. The mixture was extracted with CH2Cl2. The organic extract was washed with brine, dried, and concentrated to dryness. Following the general procedure for Pd-catalyzed coupling reactions described above, a mixture of the resulting solid, iodophenyl-perylene-diimide 7 (11 mg, 14 μmol), Pd2(dba)3 (1.6 mg, 1.8 μmol), and P(o-tol)3 (4.3 mg, 14 μmol) in toluene/TEA (5:1, 4.8 mL) was stirred at 60 °C for 4 h. The mixture was washed with water and extracted with CH2Cl2. The organic extract was washed with brine, dried, and concentrated to dryness. A solution of the resulting solid in THF (2.0 mL) was treated dropwise with 1.0 M TBAF solution in THF (7.8 μL, 7.8 μmol) and stirred for 1 h at room temperature. The reaction was quenched by the addition of saturated aqueous NaHCO3 solution. The mixture was extracted with ethyl acetate. The organic extract was washed with brine, dried, concentrated, and chromatographed according to the three-chromatography method to afford a black solid (11 mg, 40%): MALDI-MS obsd 2889.7 (M+), calcd 2889.2551 (M = C199H168N8O12Si); λabs (toluene) 431, 459, 491, 530, 641, 730 nm. The purity of the sample was estimated to be >95% according to analytical SEC. The sample was used directly in the next step without further characterization.
5,15-Bis[2-(3,4-(N-(2,6-diisopropylphenyl)iminodicarbonyl)-1,6-bis(4-tert-butylphenoxy)perylen-9-yl)ethynyl]-10-[4-(2-(4-(9,10-(N-(2,6-diisopropylphenyl)iminodicarbonyl)perylene-3,4-dicarboximido)phenyl)ethynyl)phenyl]-20-[4-(2-(4-(10-mesityl-18,18-dimethylchlorin-5-yl)phenyl)ethynyl)phenyl]porphyrin (C-T-PDI)
Following the general procedure for Pd-catalyzed coupling reactions described above, a solution of T-H/PDI (11 mg, 4.0 μmol), iodophenyl–chlorin 5 (4.0 mg, 6.0 μmol), Pd2(dba)3 (1.0 mg, 1.1 μmol), and P(o-tol)3 (1.5 mg, 4.8 μmol) in toluene/TEA (5:1, 1.6 mL) was stirred at 60 °C for 18 h. The mixture was chromatographed according to the three-chromatography method to afford a black solid (3.5 mg, 27%): 1H NMR (CDCl3, 700 MHz) δ 9.77–9.73 (m, 4H), 9.37–9.11 (m, 4H), 8.93–8.64 (m, 12H), 8.55–8.44 (m, 6H), 8.42–8.32 (m, 4H), 8.25–7.91 (m, 12H), 7.82 (d, J = 6.2 Hz, 4H), 7.52 (d, J = 7.7 Hz, 3H), 7.43–7.49 (m, 16H), 7.35–7.43 (m, 6H), 7.32 (d, J = 8.2 Hz, 4H),–1.91 (br, 1H), 7.04–7.13 (m, 6H), 4.66 (s, 2H), 2.76 (pent, J = 7.1 Hz, 6H), 2.64 (s, 3H), 2.01–2.09 (m, 6H), 1.36–1.38 (m, 36H), 1.18 (d, J = 5.8 Hz, 42H), −1.73 (br, 1H), −1.79 (br, 1H), −1.84 (br, 2H); MALDI-MS obsd 3265.8 [M+], calcd 3265.3844 (M = C227H180N12O12); λabs (toluene) 418, 494, 530, 641, 742 nm.
Photophysical properties
Static absorption (Shimadzu UV-1800) and emission (Horiba Nanolog) and other measurements were performed on dilute (μM), Ar-purged solutions in toluene at room temperature. Fluorescence quantum yields (Φf) utilized samples with A ≤ 0.1 at the excitation wavelength and were obtained by absolute measurements (Horiba QuantiPhi). Values for several arrays were also obtained by relative Φf measurements using meso-tetraphenylporphyrin in non-degassed toluene (Φf = 0.070)30 as a standard and gave the same values as the absolute measurements within experimental uncertainty (±5% of the reported value).Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by a grant from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, of the U. S. Department of Energy (DE-FG02-05ER15661). All mass spectrometry measurements were carried out in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University.References
- A. A. Krasnovsky, The Fragments of the Photosynthetic Electron Transfer Chain in Model Systems,, Biophys. J., 1972, 12, 749–763 CrossRef PubMed.
- G. Porter and M. D. Archer, In Vitro Photosynthesis, Interdiscip. Sci. Rev., 1976, 1, 119–143 CrossRef.
- D. Mauzerall, Electron-Transfer Reactions and Photoexcited Porphyrins, Brookhaven Symp. Biol., 1977, 28, 64–73 Search PubMed.
- D. Mauzerall, Photoinduced Electron Transfer at the Water-Lipid Bilayer Membrane, in Light-Induced Charge Separation in Biology and Chemistry, ed. H. Gerischer and J. J. Katz, Berlin: Dahlem Konferenzen, Verlag-Chemie, Weinheim, 1979, vol. 12, pp. 241–257 Search PubMed.
- A. Harriman and J. Barber, Photosynthetic Water-Splitting Process and Artificial Chemical Systems, in Photosynthesis in Relation to Model Systems. Topics in Photosynthesis, ed. J. Barber, Elsevier/North-Holland Biomedical Press, NY, 1979, vol. 3, ch. 8, pp. 243–280 Search PubMed.
- M. Calvin, Synthetic Chloroplasts, Energy Res., 1979, 3, 73–87 CrossRef.
- P. Cuendet and M. Grätzel, Artificial Photosynthetic Systems, Experientia, 1982, 38, 223–228 CrossRef.
- S. G. Boxer, Model Reactions in Photosynthesis, Biochim. Biophys. Acta, 1983, 726, 265–292 CrossRef.
- H. T. Tien, Planar Bilayer Lipid Membranes, Prog. Surf. Sci., 1985, 19, 169–274 CrossRef.
- M. R. Wasielewski and M. P. Niemczyk, Distance-Dependent Rates of Photoinduced Charge Separation and Dark Charge Recombination in Fixed-Distance Porphyrin–Quinone Molecules, in Porphyrins – Excited States and Dynamics, ACS Symposium Series, ed. M. Gouterman, P. M. Rentzepis and K. D. Straub, American Chemical Society, Washington, DC, 1986, vol. 321, pp. 154–165 Search PubMed.
- D. Gust and T. A. Moore, Mimicking Photosynthesis, Science, 1989, 244, 35–41 CrossRef PubMed.
- K. Maruyama and A. Osuka, A Chemical Approach Toward Photosynthetic Reaction Center,, Pure Appl. Chem., 1990, 62, 1511–1520 Search PubMed.
- P. D. Harvey, Recent Advances in Free and Metalated Multiporphyrin Assemblies and Arrays; a Photophysical Behavior and Energy Transfer Perspective, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2003, vol. 18, ch. 113, pp. 63–250 Search PubMed.
- T. S. Balaban, Tailoring Porphyrins and Chlorins for Self-Assembly in Biomimetic Artificial Antenna Systems, Acc. Chem. Res., 2005, 38, 612–623 CrossRef PubMed.
- L. Flamigni, Photoinduced Processes in Interlocked Structures Containing Porphyrins, J. Photochem. Photobiol., C, 2007, 8, 191–210 CrossRef.
- V. Balzani, A. Credi and M. Venturi, Photochemical Conversion of Solar Energy, ChemSusChem, 2008, 1, 26–58 CrossRef PubMed.
- M. R. Wasielewski, Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef PubMed.
- I. A. Maretina, Porphyrin–Ethynyl Arrays: Synthesis, Design, and Application, Russ. J. Gen. Chem., 2009, 79, 1544–1581 CrossRef.
- N. Aratani and A. Osuka, Synthetic Strategies toward Multiporphyrinic Architectures, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2010, vol. 1, ch. 1, pp. 1–132 Search PubMed.
- S. Fukuzumi, Artificial Photosynthetic Systems Composed of Porphyrins and Phthalocyanines, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2010, vol. 10, ch. 46, pp. 183–243 Search PubMed.
- P. D. Harvey, C. Stern and R. Guilard, Bio-Inspired Molecular Devices based on Systems Found in Photosynthetic Bacteria, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co., Singapore, 2011, vol. 11, ch. 49, pp. 1–179 Search PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Realizing Artificial Photosynthesis, Faraday Discuss., 2012, 155, 9–26 RSC.
- P. D. Frischmann, K. Mahata and F. Würthner, Powering the Future of Molecular Artificial Photosynthesis with Light-Harvesting Metallosupramolecular Dye Assemblies, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- M. E. El-Khouly, S. Fukuzumi and F. D′Souza, Photosynthetic Antenna–Reaction Center Mimicry by Using Boron Dipyrromethene Sensitizers, ChemPhysChem, 2014, 15, 30–47 CrossRef PubMed.
- A. Harriman, Artificial Light-Harvesting Arrays for Solar Energy Conversion, Chem. Commun., 2015, 51, 11745–11756 RSC.
- T. Tanaka and A. Osuka, Conjugated Porphyrin Arrays: Synthesis, Properties, and Applications for Functional Materials, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- M. Rudolf, S. V. Kirner and D. M. Guldi, A Multicomponent Molecular Approach to Artificial Photosynthesis – the Role of Fullerenes and Endohedral Metallofullerenes,, Chem. Soc. Rev., 2016, 45, 612–630 RSC.
- J. Wang, E. Yang, J. R. Diers, D. M. Niedzwiedzki, C. Kirmaier, D. F. Bocian, J. S. Lindsey and D. Holten, Distinct Photophysical and Electronic Characteristics of Strongly Coupled Dyads Containing a Perylene Accessory Pigment and a Porphyrin, Chlorin, or Bacteriochlorin, J. Phys. Chem. B, 2013, 117, 9288–9304 CrossRef PubMed.
- E. J. Alexy, J. M. Yuen, V. Chandrashaker, J. R. Diers, C. Kirmaier, D. F. Bocian, D. Holten and J. S. Lindsey, Panchromatic Absorbers for Solar Light-Harvesting, Chem. Commun., 2014, 50, 14512–14515 RSC.
- A. K. Mandal, J. R. Diers, D. M. Niedzwiedzki, G. Hu, R. Liu, E. J. Alexy, J. S. Lindsey, D. F. Bocian and D. Holten, Tailoring Panchromatic Absorption and Excited-State Dynamics of Tetrapyrrole–Chromophore (Bodipy, Rylene) Arrays. The Interplay of Orbital Mixing and Configuration Interaction,, J. Am. Chem. Soc., 2017, 139, 17547–17564 CrossRef PubMed.
- R. W. Wagner, T. E. Johnson and J. S. Lindsey, Soluble Synthetic Multiporphyrin Arrays. 1. Modular Design and Synthesis, J. Am. Chem. Soc., 1996, 118, 11166–11180 CrossRef.
- M. del Rosario Benites, T. E. Johnson, S. Weghorn, L. Yu, P. D. Rao, J. R. Diers, S. I. Yang, C. Kirmaier, D. F. Bocian, D. Holten and J. S. Lindsey, Synthesis and Properties of Weakly Coupled Dendrimeric Multiporphyrin Light-Harvesting Arrays and Hole-Storage Reservoirs, J. Mater. Chem., 2002, 12, 65–80 RSC.
- D. Holten, D. F. Bocian and J. S. Lindsey, Probing Electronic Communication in Covalently Linked Multiporphyrin Arrays. A Guide to the Rational Design of Molecular Photonic Devices,, Acc. Chem. Res., 2002, 35, 57–69 CrossRef PubMed.
- G. Hu, R. Liu, E. J. Alexy, A. K. Mandal, D. F. Bocian, D. Holten and J. S. Lindsey, Panchromatic Chromophore–Tetrapyrrole Light-Harvesting Arrays Constructed from Bodipy, Perylene, Terrylene, Porphyrin, Chlorin, and Bacteriochlorin Building Blocks, New J. Chem., 2016, 40, 8032–8052 RSC.
- J. S. Lindsey, De Novo Synthesis of Gem-Dialkyl Chlorophyll Analogues for Probing and Emulating our Green World, Chem. Rev., 2015, 115, 6534–6620 CrossRef PubMed.
- D. Gosztola, M. P. Niemczyk and M. R. Wasielewski, Picosecond Molecular Switch Based on Bidirectional Inhibition of Photoinduced Electron Transfer Using Photogenerated Electric Fields, J. Am. Chem. Soc., 1998, 120, 5118–5119 CrossRef.
- S. Prathapan, S. I. Yang, J. Seth, M. A. Miller, D. F. Bocian, D. Holten and J. S. Lindsey, Synthesis and Excited-State Photodynamics of Perylene-Porphyrin Dyads. 1. Parallel Energy and Charge Transfer via a Diphenylethyne Linker, J. Phys. Chem. B, 2001, 105, 8237–8248 CrossRef.
- E. Yang, J. Wang, J. R. Diers, D. M. Niedzwiedzki, C. Kirmaier, D. F. Bocian, J. S. Lindsey and D. Holten, Probing Electronic Communication for Efficient Light-Harvesting Functionality: Dyads Containing a Common Perylene and a Porphyrin, Chlorin, or Bacteriochlorin, J. Phys. Chem. B, 2014, 118, 1630–1647 CrossRef PubMed.
- J. Amanpour, G. Hu, E. J. Alexy, A. K. Mandal, H. S. Kang, J. M. Yuen, J. R. Diers, D. F. Bocian, J. S. Lindsey and D. Holten, Tuning the Electronic Structure and Properties of Perylene–Porphyrin–Perylene Panchromatic Absorbers, J. Phys. Chem. A, 2016, 120, 7434–7450 CrossRef PubMed.
- H. Langhals, Synthese von hochreinen Perylen-Fluoreszenzfarbstoffen in großen Mengen – gezielte Darstellung von Atrop-Isomeren, Chem. Ber., 1985, 118, 4641–4645 CrossRef.
- H. Langhals, S. Demmig and H. Huber, Rotational Barriers in Perylene Fluorescent Dyes, Spectrochim. Acta, Part A, 1988, 44, 1189–1193 CrossRef.
- H. Langhals, Cyclic Carboxylic Imide Structures as Structure Elements of High Stability. Novel Developments in Perylene Dye Chemistry, Heterocycles, 1995, 40, 477–500 CrossRef.
- C. Kirmaier, H.-E. Song, E. K. Yang, J. K. Schwartz, E. Hindin, J. R. Diers, R. S. Loewe, K.-y. Tomizaki, F. Chevalier, L. Ramos, R. R. Birge, D. F. Bocian, J. S. Lindsey and D. Holten, Excited-State Photodynamics of Perylene–Porphyrin Dyads 5. Tuning Light-Harvesting Characteristics via Perylene Substituents, Connection Motif, and 3-Dimensional Architecture, J. Phys. Chem. B, 2010, 114, 14249–14264 CrossRef PubMed.
- J. S. Lindsey, Synthetic Routes to meso-Patterned Porphyrins, Acc. Chem. Res., 2010, 43, 300–311 CrossRef PubMed.
- K.-y. Tomizaki, P. Thamyongkit, R. S. Loewe and J. S. Lindsey, Practical Synthesis of Perylene-Monoimide Building Blocks That Possess Features Appropriate for Use in Porphyrin-Based Light-Harvesting Arrays, Tetrahedron, 2003, 59, 1191–1207 CrossRef.
- D. P. Arnold, A. W. Johnson and M. Mahendran, Some Reactions of meso-Formyloctaethylporphyrin, J. Chem. Soc., Perkin Trans. 1, 1978, 366–370 RSC.
- H. L. Anderson, Meso-Alkynyl Porphyrins, Tetrahedron Lett., 1992, 33, 1101–1104 CrossRef.
- V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Highly Conjugated, Acetylenyl Bridged Porphyrins: New Models for Light-Harvesting Antenna Systems, Science, 1994, 264, 1105–1111 CrossRef PubMed.
- D. P. Arnold and L. J. Nitschinsk, Porphyrin Dimers Linked by Conjugated Butadiynes, Tetrahedron, 1992, 48, 8781–8792 CrossRef.
- D. P. Arnold, D. Manno, G. Micocci, A. Serra, A. Tepore and L. Valli, Porphyrin Dimers Linked by a Conjugated Alkyne Bridge: Novel Moieties for the Growth of Langmuir–Blodgett Films and Their Applications in Gas Sensors, Langmuir, 1997, 13, 5951–5956 CrossRef.
- D. P. Arnold and D. A. James, Dimers and Model Monomers of Nickel(II) Octaethylporphyrin Substituted by Conjugated Groups Comprising Combinations of Triple Bonds with Double Bonds and Arenes. 1. Synthesis and Electronic Spectra, J. Org. Chem., 1997, 62, 3460–3469 CrossRef.
- K. Susumu and M. J. Therien, Design of Diethynyl Porphyrin Derivatives with High Near Infrared Fluorescence Quantum Yields, J. Porphyrins Phthalocyanines, 2015, 19, 205–218 CrossRef.
- M. Rickhaus, A. V. Jentzsch, L. Tejerina, I. Grübner, M. Jirasek, T. D. W. Claridge and H. L. Anderson, Single-Acetylene Linked Porphyrin Nanorings, J. Am. Chem. Soc., 2017, 139, 16502–16505 CrossRef PubMed.
- R. W. Wagner, T. E. Johnson, F. Li and J. S. Lindsey, Synthesis of Ethyne-Linked or Butadiyne-Linked Porphyrin Arrays Using Mild, Copper-Free, Pd-Mediated Coupling Reactions, J. Org. Chem., 1995, 60, 5266–5273 CrossRef.
- R. W. Wagner, Y. Ciringh, C. Clausen and J. S. Lindsey, Investigation and Refinement of Palladium-Coupling Conditions for the Synthesis of Diarylethyne-Linked Multiporphyrin Arrays, Chem. Mater., 1999, 11, 2974–2983 CrossRef.
- M. Taniguchi, A. Balakumar, D. Fan, B. E. McDowell and J. S. Lindsey, Imine-Substituted Dipyrromethanes in the Synthesis of Porphyrins Bearing One or Two Meso Substituents, J. Porphyrins Phthalocyanines, 2005, 9, 554–574 CrossRef.
- J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, A Scalable Synthesis of Meso-Substituted Dipyrromethanes,, Org. Process Res. Dev., 2003, 7, 799–812 CrossRef.
- C.-H. Lee and J. S. Lindsey, One-Flask Synthesis of Meso-Substituted Dipyrromethanes and Their Application in the Synthesis of Trans-Substituted Porphyrin Building Blocks, Tetrahedron, 1994, 50, 11427–11440 CrossRef.
- I. Schmidt, J. Jiao, P. Thamyongkit, D. S. Sharada, D. F. Bocian and J. S. Lindsey, Investigation of Stepwise Covalent Synthesis on a Surface Yielding Porphyrin-Based Multicomponent Architectures, J. Org. Chem., 2006, 71, 3033–3050 CrossRef PubMed.
- R. Liu, M. Liu, D. Hood, C.-Y. Chen, C. J. MacNevin, D. Holten and J. S. Lindsey, Chlorophyll-Inspired Red-Region Fluorophores. Building Block Syntheses and Studies in Aqueous Media, Molecules, 2018, 23, 130 CrossRef PubMed.
- M. Gouterman, Study of the Effects of Substitution on the Absorption Spectra of Porphin, J. Chem. Phys., 1959, 30, 1139–1161 CrossRef.
- M. Gouterman, Spectra of Porphyrins, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef.
- M. Gouterman, Optical Spectra and Electronic Structure of Porphyrins and Related Rings, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, ch. 1, pp. 1–165 Search PubMed.
- D. Kuciauskas, P. A. Liddell, S. Lin, T. E. Johnson, S. J. Weghorn, J. S. Lindsey, A. L. Moore, T. A. Moore and D. Gust, An Artificial Photosynthetic Antenna-Reaction Center Complex, J. Am. Chem. Soc., 1999, 121, 8604–8614 CrossRef.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, Investigation of MALDI-TOF Mass Spectrometry of Diverse Synthetic Metalloporphyrins, Phthalocyanines, and Multiporphyrin Arrays, J. Porphyrins Phthalocyanines, 1999, 3, 283–291 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all new compounds; MALDI-MS data and analytical SEC traces for all arrays and benchmarks; and reaction monitoring by analytical SEC for C-T-PDI. See DOI: 10.1039/c8ra04052d |
| This journal is © The Royal Society of Chemistry 2018 |