
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of porous calcium phosphate microspheres with phosphate-containing molecules at room temperature for drug delivery and osteogenic differentiation
Jun-Feng Liu†
a,
Lu Wei†a,
Dilixiati Duolikuna,
Xiao-Dong Houa,
Feng Chen *ab,
Jun-Jian Liu*a and
Long-Po Zheng*ab
*ab,
Jun-Jian Liu*a and
Long-Po Zheng*ab
aDepartment of Orthopedics, Shanghai Tenth People's Hospital, Tongji University School of Medicine, Shanghai 200072, China. E-mail: fchen@tongji.edu.cn; liujunjian1968@126.com; trauma_zlp@163.com
bShanghai Bone Tumor Institution, Shanghai 200072, China
First published on 16th July 2018
Abstract
Calcium phosphate (CaP) has similar chemical properties to those of the inorganic component of human bone tissue, for potential application in drug delivery for the chemotherapy of osteosarcoma. In this work, CaP with a porous microsphere structure has been synthesized using fructose-1,6-bisphosphate (FBP) as the phosphorus source by a simple wet-chemical strategy at room temperature. The CaP porous microspheres, as an organic–inorganic hybrid nano-platform, exhibit good doxorubicin (Dox) loading capacity, and Dox-loading CaP, enhancing the in vitro chemotherapy of osteosarcoma cells. The CaP porous microspheres show high biocompatibility, and induce the osteogenic differentiation of MC3T3-E1. These results indicate that the CaP porous microspheres reported in this study are promising for application as an anti-osteosarcoma drug carrier and osteoinductive material for bone regeneration in the treatment of osteosarcoma.
Introduction
Osteosarcoma (OS) is the most common primary malignant bone tumor that mainly affects adolescents and young adults.1 The five-year survival rate of non-metastatic osteosarcoma patients has improved to approximately 70% with the application of neoadjuvant chemotherapy combined with surgical resection, but has hardly been further improved upon over the past three decades due to distal metastasis and chemotherapy resistance.2,3 Moreover, most of the chemotherapeutic agents may bring about severe side effects in normal tissues and organs due to their low selectivity and low sensitivity to the target tumor tissue.Nowadays, the development of nanotechnology provides approaches to overcome the chemotherapy resistance of osteosarcoma tissue and improve the efficiency of chemotherapy. Drug carriers with a porous structure may be favorable for the loading of anti-osteosarcoma drugs. Calcium phosphate (CaP) has similar chemical properties to those of the inorganic component of human bone tissue, displaying excellent biocompatibility, bioactivity, and osteoconductivity. CaP based biomaterials have been widely used in drug/gene delivery,4–8 and bone tissue engineering/repair.9,10 Therefore, CaP based biomaterials may be a potential candidate for delivering chemotherapeutic agents to improve the efficiency of osteosarcoma treatment.
Fructose-1,6-bisphosphate (FBP), a fructose sugar phosphorylated on carbons 1 and 6, lies within the glycolysis metabolic pathway of mammalian cells and regulates the activity of several enzymes involved in metabolism in the human body. In the 1980s, animal experiments and clinical studies showed that FBP has effects on myocardial infarction, tissue ischemia, hypoxic disease, and so on.11 Oral FBP calcium salt tablets are used for the adjuvant treatment of metabolic diseases, such as cerebral metabolic diseases, ossification diseases, skeletal muscle metabolic disorders, and diabetes. Meanwhile, FBP is thought to be a nontoxic natural biomolecule, which may affect the differentiation of mesenchymal stem cells.12 Increasing evidence has shown that mesenchymal stem cells in the tumor microenvironment play a pivotal role in the proliferation, metastasis and chemotherapy resistance of osteosarcoma. Thus, induction of the terminal differentiation of the mesenchymal stem cells has been estimated to be a promising therapeutic approach in the treatment of osteosarcoma.13–15
Herein, we have developed a facile wet-chemical strategy to prepare CaP porous microspheres using FBP molecules as the phosphorous source at room temperature. The CaP porous microspheres are an organic–inorganic hybrid system, which enhances chemotherapy efficiency in osteosarcoma treatment as a drug nanocarrier and promotes the osteogenic differentiation of MC3T3-E1. The results of the current study provide a potential strategy for the future treatment of osteosarcoma.
Experimental
Materials
Fructose-1,6-bisphosphate (FBP) disodium salt was purchased from Sangon Biotech (Shanghai) Co., Ltd. Doxorubicin hydrochloride (Dox·HCl) was purchased from Aladdin Chemistry Co., Ltd. The other chemicals used in the preparation of the samples were purchased from Sinopharm Chemical Reagent Co. MC3T3-E1 cells (293 [HEK-293] (Procell CL-0001)) were kindly provided by Procell Life Science & Technology Co., Ltd. The MG-63 and 143b cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). The alizarin red staining solution, von Kossa staining solution, DAPI staining solution, alkaline phosphatase assay kit and phalloidine staining solution were purchased from Wuhan Servicebio Technology Co., Ltd. The cell counting kit-8 (CCK8) was purchased from Dojindo, Japan. All chemicals were of analytical grade and used directly without being further purified.Preparation of the CaP microspheres
Typically, the CaP microspheres were prepared as follows: 0.550 g of Na3FBP was dissolved in 8 mL of deionized water, followed by the addition of 8 mL of an aqueous solution containing 0.185 g of CaCl2. Then, 1.7 mL of NaOH solution (1 M) was added slowly dropwise under magnetic stirring, and the resulting solution was stirred for another 2 h at room temperature. The product was collected by centrifugation, washed three times with deionized water, and freeze-dried to a powder.Control samples (HA nanorods) were prepared using a normal phosphate source as follows: 0.660 g CaCl2 was dissolved into 50 mL of deionized water, to which a 50 mL aqueous solution containing 4.020 g Na3PO4 was quickly added under magnetic stirring. The mixture was then stirred for 1 h in a water bath at a temperature of 80 °C. The control samples were collected by centrifugation, washed three times with deionized water, and freeze-dried to a powder.
Characterization
The as-prepared samples were characterized using powder X-ray diffraction (PXRD, Rigaku D/max 2550 V, Cu Kα radiation, l = 1.54178 Å), and Fourier transform infrared (FTIR) spectroscopy (FTIR-7600, Lambda Scientific, Australia). The morphology of the samples was characterized using scanning electron microscopy (SEM, Hitachi S-4800, Japan), and transmission electron microscopy (FEI TF20/2100F, USA). The doxorubicin (Dox) concentrations were analyzed using a UV-Vis spectrophotometer (UV-2300, Techcomp) at a wavelength of 232.6 nm. A specific surface area and pore size analyzer (V-sorb 2800P, Gold APP Instruments, China) was used to obtain the Brunauer–Emmett–Teller (BET) specific surface area and pore size distribution of the as-prepared samples.In vitro Dox loading and release experiments
The freeze-dried powder of CaP porous microspheres and HA nanorods (100 mg each) were respectively dispersed in 50 mL of water with a Dox concentration of 1 mg mL−1. After ultrasonic treatment for 10 min, the suspensions were shaken at a constant rate (140 rpm) in two sealed vessels at 37 °C for 16 h, followed by centrifugation and freeze drying to obtain the Dox-loaded CaP porous microspheres and HA nanorods. Then, the solution was centrifuged and the amount of Dox in the supernatant was measured by UV-Vis absorption at a wavelength of 232.6 nm. The Dox loading capacity is defined as (M0 − M1)/MS and the Dox loading efficiency is defined as (M0 − M1)/M0 × 100%, where M0 is the total mass of Dox, M1 is the mass of Dox in the supernatant and MS is the mass of the sample.For the Dox release assay, 40 mg of freeze-dried powder of Dox-loaded CaP porous microspheres and HA nanorods were dispersed into 40 mL of phosphate buffer saline solution (PBS, pH 7.4), and each solution was shaken at a constant rate at 37 °C. At predetermined time points, 1 mL of release medium was withdrawn to measure the concentration of the released Dox, and then 1 mL of fresh PBS was added.
Cell culture
MC3T3-E1 cells were maintained in α-modified Eagle's minimum essential medium (α-MEM), and MG-63 cells and 143b cells were maintained in Dulbecco's modified Eagle's medium (DMEM). These two types of medium were supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (100 U mL−1 penicillin and 100 μg mL−1 streptomycin). These three cells lines were maintained at 37 °C in a 5% CO2 atmosphere and detached with 0.25% trypsin-EDTA (Gibco) for subculture after reaching 70–80% confluence. Generally, cells were seeded into cell culture wells and incubated overnight for attachment before co-culture with biomaterials.Cell viability assay
The cell viability assay was assessed using a CCK8 kit. All materials were sterilized using ultraviolet irradiation for 30 min. After cell attachment, media containing different concentrations of sterilized materials were added into the wells and co-cultured with the cells for 24 h. Sample free culture medium was used as the blank control. After incubation, the medium was removed and the cells were washed twice with PBS. Then, 90 μL of culture medium and 10 μL of CCK-8 solution were added into each well and incubated at 37 °C for 3 h. The absorbance of the samples was measured at 450 nm with a spectrophotometric microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).Meanwhile, the long-time influence of the as-prepared CaPs materials on the viability of MC3T3-E1 cells was also studied. The sterilized materials were dipped in cell culture medium at certain concentrations and incubated at 37 °C for 24 h. Then, the supernatant of the medium was added into the wells and co-cultured with the cells for a designated time period. The culture medium containing no extract was used as the blank control. Then, the cell viability was measured by a CCK8 kit as described as above, and the data are shown in the form of a percentage compared with the blank control from the first day.
Osteogenic differentiation analysis
MC3T3-E1 cells were cultured in culture medium containing CaP microspheres or HA nanorods for 14 days. The medium was changed every two days. Alkaline phosphatase (ALP) activity assays and calcium deposition detection assays were performed at both day 7 and day 14. After culturing for 7 days and 14 days, the cell layers were washed with cold PBS and lysed in 100 μL cell lysis buffer for 20 min. The lysates were centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min at 4 °C. The following procedures were all conducted according to the manufacturer's protocol (Beyotime Biotechnology, Shanghai, China). Calcium deposition in the extracellular matrix was tested with alizarin red staining and von Kossa staining. Briefly, the cells were washed with PBS and fixed in 4% (v/v) paraformaldehyde for 15 min. Then, cells were stained with 0.2% alizarin red solution for 30 min or von Kossa staining solution for 2 h under ultraviolet radiation. After being washed with deionized water twice, cells were dried at room temperature for alizarin red staining.
000 rpm for 15 min at 4 °C. The following procedures were all conducted according to the manufacturer's protocol (Beyotime Biotechnology, Shanghai, China). Calcium deposition in the extracellular matrix was tested with alizarin red staining and von Kossa staining. Briefly, the cells were washed with PBS and fixed in 4% (v/v) paraformaldehyde for 15 min. Then, cells were stained with 0.2% alizarin red solution for 30 min or von Kossa staining solution for 2 h under ultraviolet radiation. After being washed with deionized water twice, cells were dried at room temperature for alizarin red staining.
Fluorescent staining
After being fixed in 4% (v/v) paraformaldehyde solution for 15 min, cells were stained with DAPI staining solution for 5 min or phalloidine staining solution for 2 h. Then, the cells were washed with PBS twice. The intracellular fluorescence signals were observed under a fluorescence microscope (Leica, DMI6000B).Statistical analysis
The data were analyzed with SPSS 24 (SPSS, Chicago, IL, USA) and presented as mean ± SD for the three independent experiments. The Student's t-test was used for calculating the significance between the different groups. A p value of < 0.05 indicates a significant difference.Results and discussion
Characterization of the calcium phosphate microspheres
SEM and TEM micrographs of the HA nanorods and CaP porous microspheres are shown in Fig. 1. The HA sample displays a nanorod structure, which was used as the control. The selected area electron diffraction (SAED) pattern of the HA nanorods indicates that the sample is crystalline, which is in good agreement with the PXRD results shown in Fig. 2A. As shown in Fig. 1C–F, the CaP sample displays a porous microsphere structure. The SAED pattern of the CaP porous microspheres indicates that the sample has low crystallinity, which is also in good agreement with the PXRD results. | ||
| Fig. 1 SEM and TEM micrographs of HA samples (A and B) and CaP porous microspheres (C–F). The inset in B shows the SAED pattern of the HA samples, and the inset in F shows the SAED pattern of the CaP porous microspheres. | ||
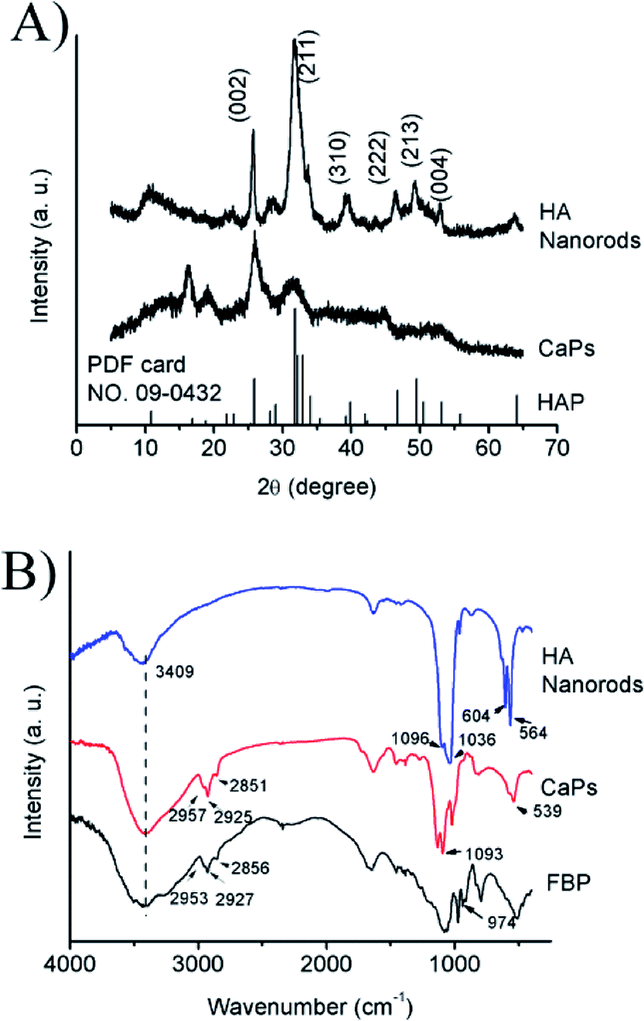 | ||
| Fig. 2 (A) PXRD patterns of the CaP porous microspheres and HA samples; (B) FTIR spectra of pure FBP, the CaP porous microspheres and HA nanorods. | ||
The PXRD pattern (Fig. 2A) of the HA control sample shows well-defined diffraction peaks, which can be well indexed to the different crystal planes of HA (Ca5(PO4)3(OH), JCPDS no. 09-0432). Meanwhile, the PXRD pattern of the CaP porous microspheres indicates low crystallinity, and shows several weak peaks, which can also be indexed to HA. Fig. 2B shows the FTIR spectra of pure FBP, the CaP porous microspheres and HA nanorods. The broad absorption band at around 3409 cm−1 is assigned to the absorbed water. The absorption peaks for the CaP porous microspheres at 2957, 2925 and 2851 cm−1 can be attributed to the symmetric and asymmetric stretching and bending vibrations of the C–H bonds of the FBP molecules. The intense absorption peaks at around 1093, 1020 and 539 cm−1 for the CaP porous microspheres can be attributed to PO43−. Meanwhile, in the spectrum of the HA nanorods, typical absorption peaks of PO43− were observed, with asymmetric and symmetric stretching modes found at around 1096, 1036, 604 and 564 cm−1. These results indicate that the samples of CaP porous microspheres are an organic–inorganic hybrid system.
In the reaction solution, there is an abundance of OH− ions. We deem that the FBP molecules could slowly release phosphate groups into the reaction solution through a hydrolysis effect. Then, the calcium ions will react with the free phosphate ions to form CaP (Scheme 1). Meanwhile, the FBP molecules have a strong interaction with Ca2+, and form a complex product. Therefore, the as-prepared CaP porous microspheres using FBP as an organic phosphorus source is an organic–inorganic hybrid system, containing CaP and FBP molecules and their derivatives.
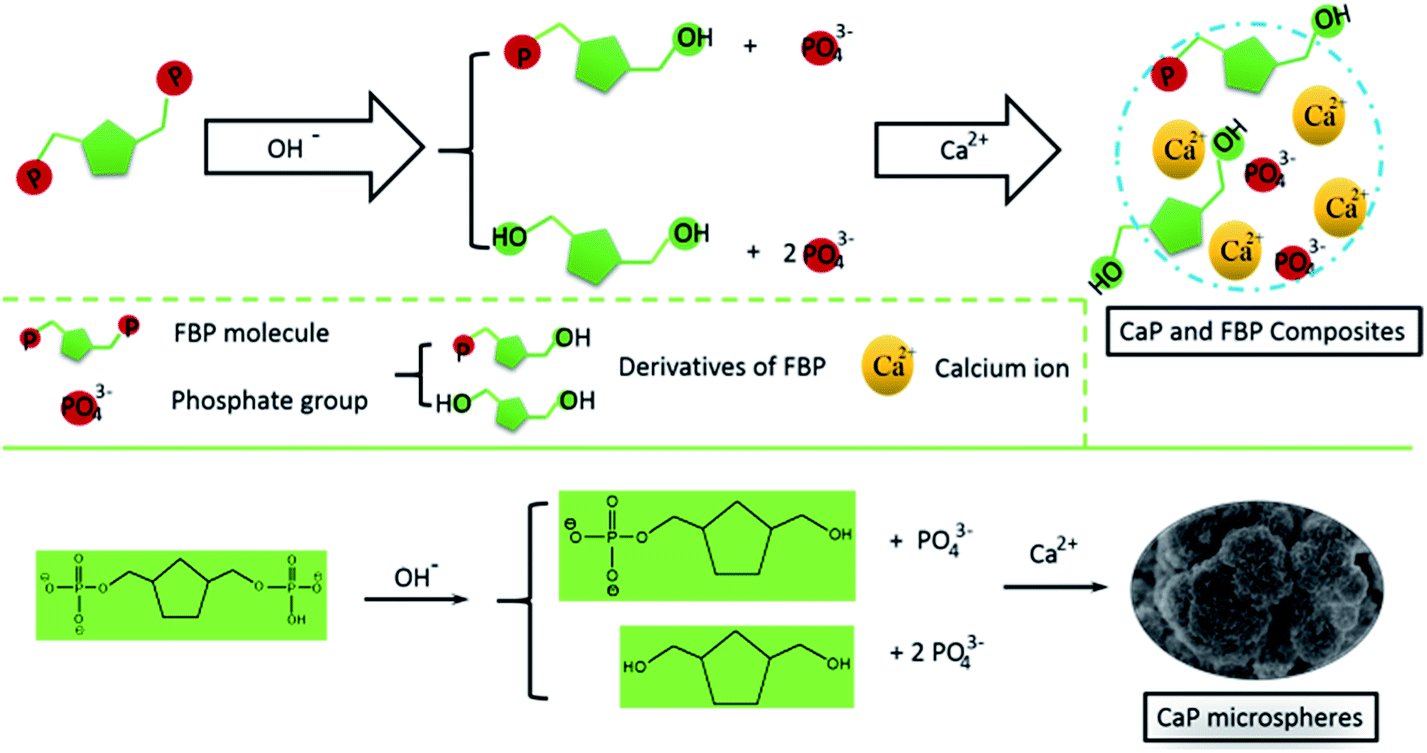 | ||
| Scheme 1 Schematic illustration of the strategy for the preparation of CaP porous microspheres using FBP disodium salt as a phosphorus source. | ||
Fig. 3 shows the N2 adsorption–desorption isotherm of the CaP porous microspheres synthesized using FBP molecules as the phosphorus source. According to the International Union of Pure and Applied Chemistry, it can be classified as a type-IV isotherm loop. The BET specific surface area is 167.1 m3 g−1, and the BJH desorption cumulative pore volume and the average pore size of the sample are 0.335132 cm3 g−1, and 29.4 nm, respectively. The porous structure of the CaP microspheres is suitable for drug loading. We chose the anti-tumor drug Dox as a typical drug model, and found that the Dox-loading capacity of CaP microspheres reaches 471.3 mg g−1. Meanwhile, the Dox-loading capacity of HA nanorods was found to be 197.8 mg g−1. The higher loading capacity of the as-prepared CaP microspheres could be explained by the porous structure and high surface area, which are beneficial for drug adsorption. The CaP porous microspheres may be used as an in situ drug release system for anti bone tumors and promoting bone regeneration. Therefore, the as-prepared CaP microspheres do not require a high Dox loading capacity for the in vivo studies, compared with the amount required for a drug delivery system in the circulatory system.
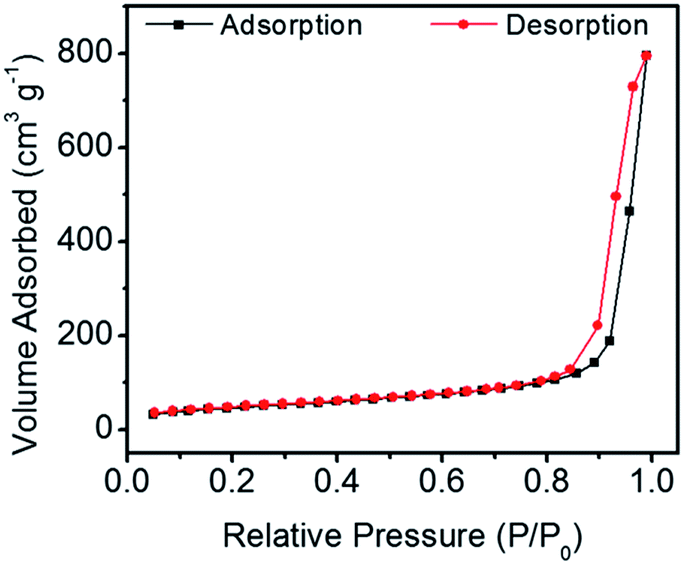 | ||
| Fig. 3 The N2 adsorption–desorption isotherm of CaP porous microspheres synthesized using FBP molecules as the phosphorus source. | ||
The release kinetics of the CaP porous microspheres were studied, and the release curve is shown in Fig. 4. Dox-loaded HA showed a fast release of Dox and reached an equilibrium release in a period of about 10 h. Meanwhile, as for the sample of Dox-loaded CaPs, the release of Dox increased dramatically within 6 h. Then, the concentration of released Dox gradually decreased after the first 6 hours. CaPs is an inorganic–organic composite with a porous structure. We suppose that the surface properties of CaPs were changed after being immersed in the PBS solution, which influenced the interaction between the CaPs and Dox molecules and further affected the release curves of Dox-loaded CaPs.
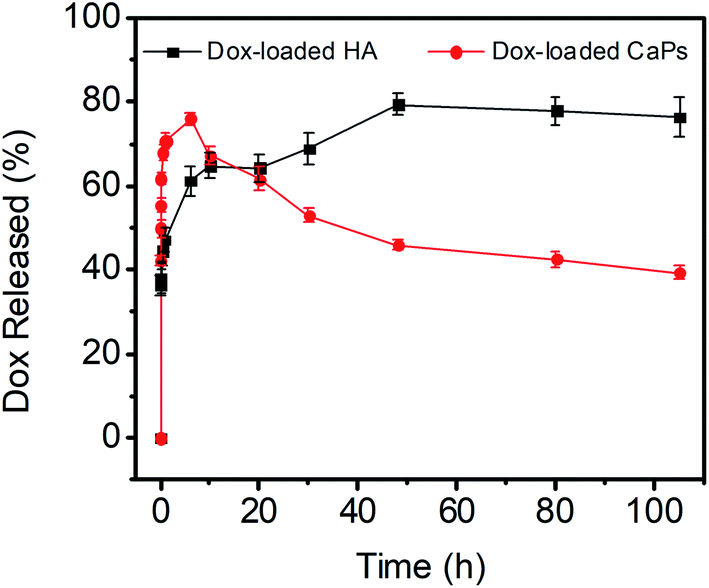 | ||
| Fig. 4 Dox release curves of the Dox-loaded CaPs and Dox-loaded HA in PBS solutions (pH 7.4, 37 °C). | ||
To study the stability of the CaP porous microspheres, the CaPs were immersed in PBS solution (pH 7.4) at 37 °C for 105 h. The SEM micrographs and PXRD pattern of the immersed CaPs are shown in Fig. 5. Compared with the fresh samples, the morphology of the CaPs immersed in the PBS solution for 105 h obviously changed, but a porous structure could still be identified. The PXRD pattern in Fig. 5C indicates that the crystal phase of CaPs immersed in the PBS solution for 105 h can be well indexed to the crystal planes of HA (Ca5(PO4)3(OH), JCPDS no. 09-0432), indicating that CaPs gradually transformed into a well crystalline HA phase in PBS solution.
 | ||
| Fig. 5 SEM micrographs (A and B) and PXRD pattern (C) of the CaP porous microspheres after being immersed in PBS solution (pH 7.4) at 37 °C for 105 h. | ||
Effect of the CaP porous microspheres on cell viability
When the MC3T3-E1 cells were directly cultured with CaP porous microspheres for 1 day, no significant difference in the cell viability was found, when compared with the HA nanorods or the blank control group (Fig. 6A). Meanwhile, when the MC3T3-E1 cells were cultured with an extract of CaP porous microspheres of different concentrations for 1 day, no significant difference in the cell viability was found, compared with the HA nanorods or the blank control group (Fig. 6B). Moreover, when the co-culturing time increased to 3 and 7 days, the extract of CaP porous microspheres showed a slight advance over the extract of HA nanorods in terms of cell viability. The results suggest that both the CaP porous microspheres and HA nanorods have good biocompatibility. The results can be well explained by the high biocompatibility of the CaP and FBP molecules, both of which are native to the human body.16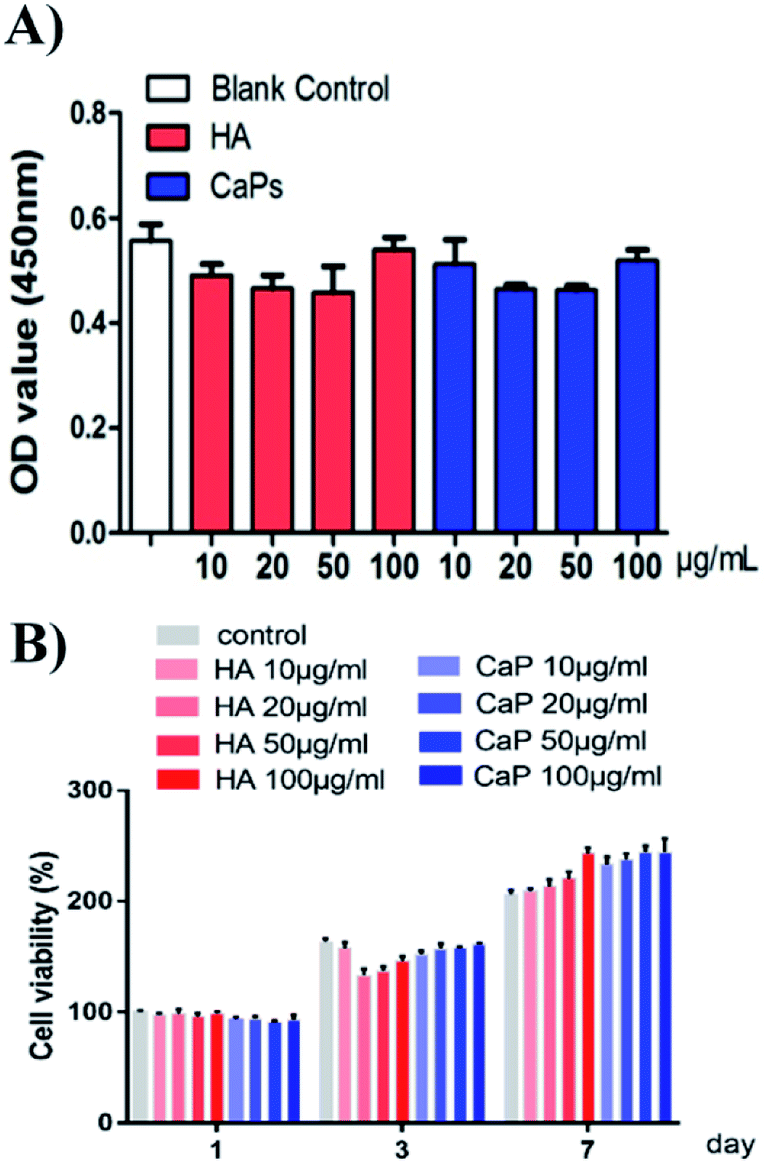 | ||
| Fig. 6 Cell viability studies of the HA nanorods and CaP porous microspheres. (A) Cell viability assay of MC3T3-E1 cells co-cultured with CaP porous microspheres or HA nanorods for 1 day. (B) Cell viability assay of MC3T3-E1 cells co-cultured with the extract solution of CaP porous microspheres or HA nanorods at different concentrations for 1, 3 and 7 days. | ||
Cellular toxicity of the Dox-loaded CaP porous microspheres on osteosarcoma cells
After loading Dox, we studied the cellular toxicity of the Dox-loaded CaP porous microspheres on osteosarcoma cells. The inhibition rate of the Dox-loaded CaP porous microspheres on the cell viability is significantly higher than that of the Dox-loaded HA nanorods, with evidence of their inhibitory effect on MG-63 cells (Fig. 7A) and 143b cells (Fig. 7B). In particular, a low concentration (5 μg mL−1) of Dox-loaded CaP porous microspheres showed a good inhibition ratio (77.26 ± 3.446%) in 143b cells after 1 day of co-culturing, much higher than that of Dox-loaded HA nanorods (9.37 ± 3.96%). Even at a high concentration (100 μg mL−1), the Dox-loaded HA nanorods showed a comparable inhibition ratio (80.22 ± 2.21%) to that of the Dox-loaded CaP porous microspheres at a low concentration of 5 μg mL−1 (Fig. 7B). A similar anti-proliferation effect on MG 63 and 143b cells could be observed after treatment of the Dox-loaded CaP porous microspheres and HA nanorods for 2 days (Fig. 7C and D). On day two, the inhibition rate was high for both cell lines, even at low concentrations (5 μg mL−1). CaPs-Dox still performed the best in the inhibition of the two cell lines, which is in line with the data of the one day co-culture.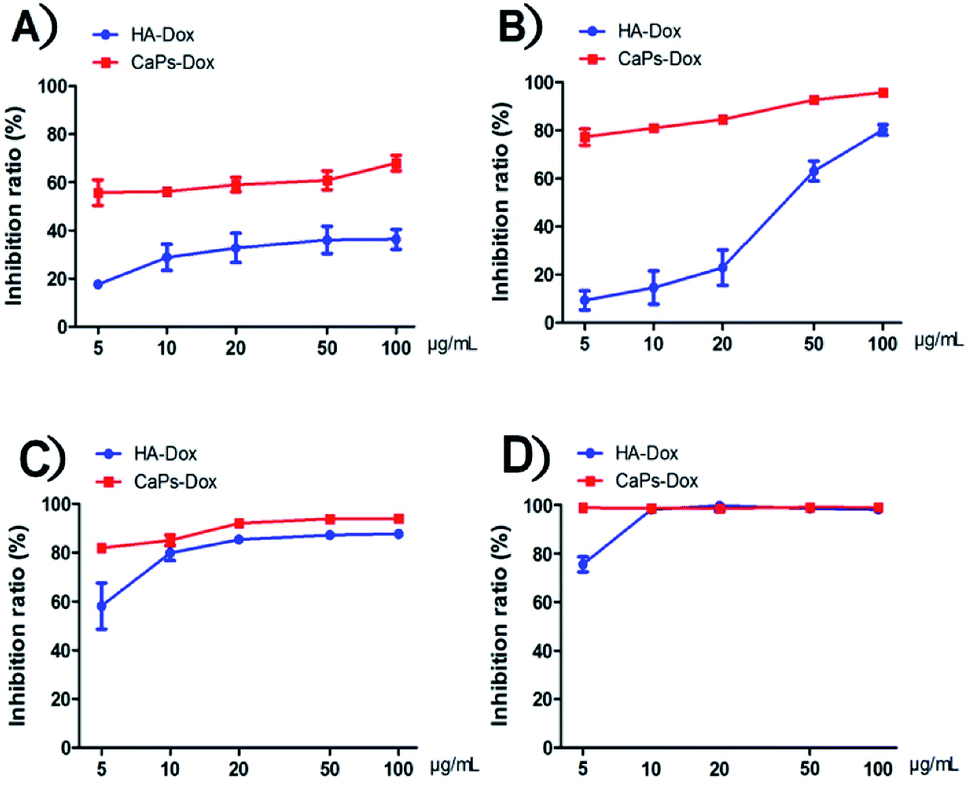 | ||
| Fig. 7 Dox-loaded CaP porous microspheres show a better inhibitory effect on cell proliferation than Dox-loaded HA nanorods on MG-63 cells (A) and 143b cells (B) co-cultured for 1 day at various concentrations. The inhibition rate of the MG-63 cells (C) and 143b cells (D) co-cultured with Dox-loaded CaP porous microspheres (CaPs-Dox) and Dox-loaded HA nanorods (HA-Dox) for two days at various concentrations. | ||
The intracellular accumulation of Dox was observed using a fluorescence microscope. Red fluorescence indicated Dox absorbed into the cells, while blue fluorescence indicated DAPI-labeled cell nuclei. The fewer cell nuclei in Fig. 8 indicates the presence of fewer cells after co-incubation of the Dox-loaded materials and osteosarcoma MG63 cells, which verified the anti-proliferative results shown in Fig. 7. As shown in Fig. 8, the Dox fluorescence intensity in Dox-loaded HA nanorod treated MG63 cells (Fig. 8B) was higher than that of the blank control group (Fig. 8A), after co-culturing for 24 h. Meanwhile, Dox-loaded CaP porous microsphere treated MG63 cells exhibited the highest Dox fluorescence intensity (Fig. 8C). In both Dox-loaded HA nanorods and Dox-loaded CaP porous microsphere treated cells, a higher concentration of Dox loaded materials induced a higher Dox fluorescence intensity (Fig. 8B and C). Above all, the CaP porous microspheres exhibited a high performance in the inhibition of the proliferation of osteosarcoma cells in vitro, which is in line with the results from the Dox loading experiment.
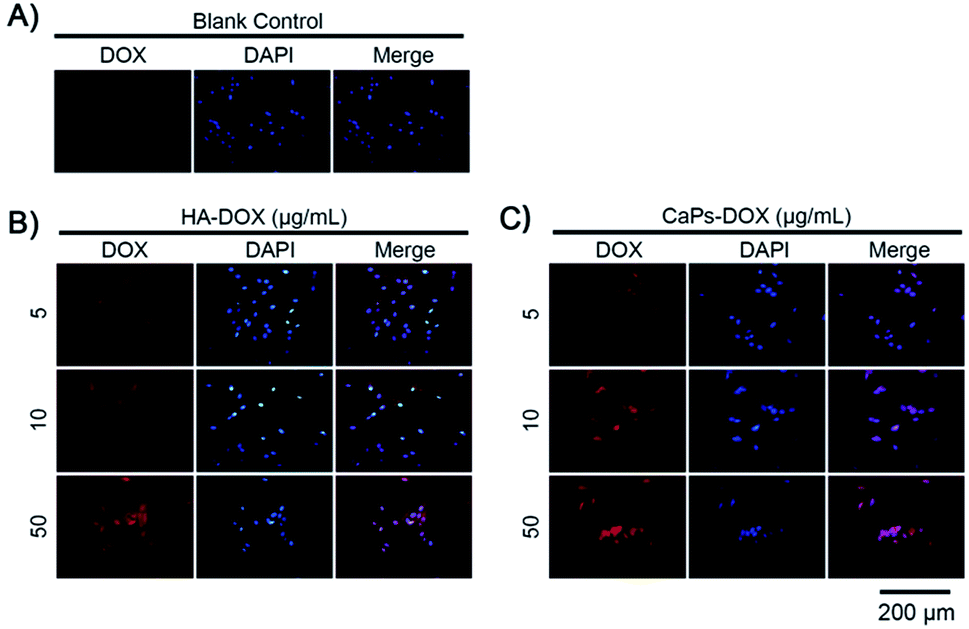 | ||
| Fig. 8 Increased Dox fluorescence intensity in MG-63 cells after being co-cultured with Dox-loaded HA nanorods (HA-Dox) and Dox-loaded CaP porous microspheres (CaPs-Dox) for 24 h. | ||
Phalloidine staining shows the microfiber within the cells, presenting the whole cell morphology. As is shown in Fig. 9, MG63 cells exhibited normal morphology in the blank control group. However, Dox-loaded material treated MG63 cells showed cellular atrophy and fewer numbers of cells, especially in the Dox-loaded CaP porous microsphere treated MG63 cells. Similar data were collected for the 143b cells (Fig. 10). The 143b cells exhibited normal morphology in the blank control group. However, Dox-loaded material treated 143b cells showed cellular atrophy and fewer numbers of cells, especially in the CaPs-Dox treated 143b cells. The worse cellular morphology and fewer cell numbers in the Dox-loaded CaP porous microsphere treated group represent the better Dox loading efficiency of the CaP porous microspheres than the HA nanorods.
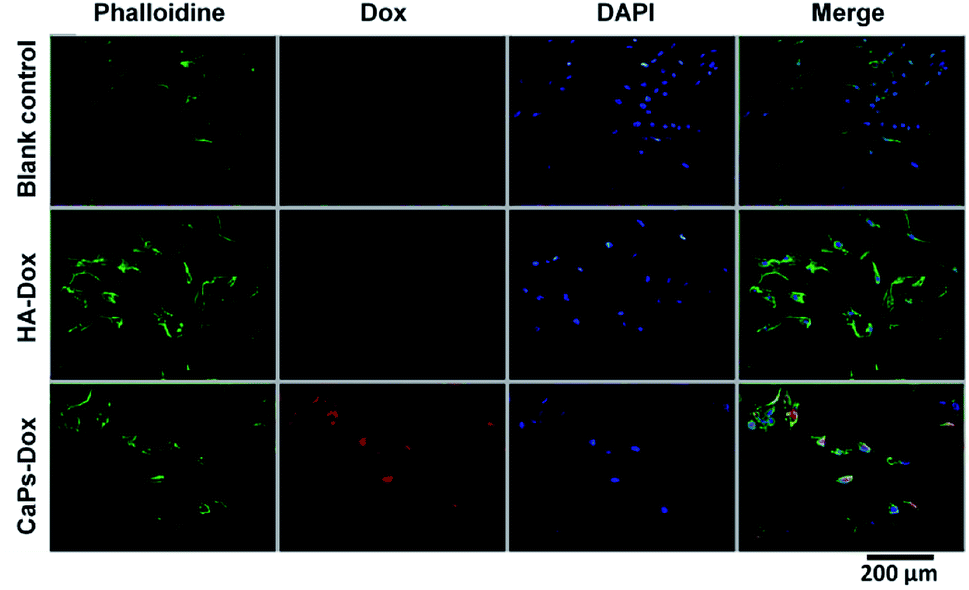 | ||
| Fig. 9 DAPI staining and phalloidine staining of MG-63 cells when co-cultured with Dox-loaded HA nanorods (HA-Dox) and Dox-loaded CaP porous microspheres (CaPs-Dox) for 24 h. | ||
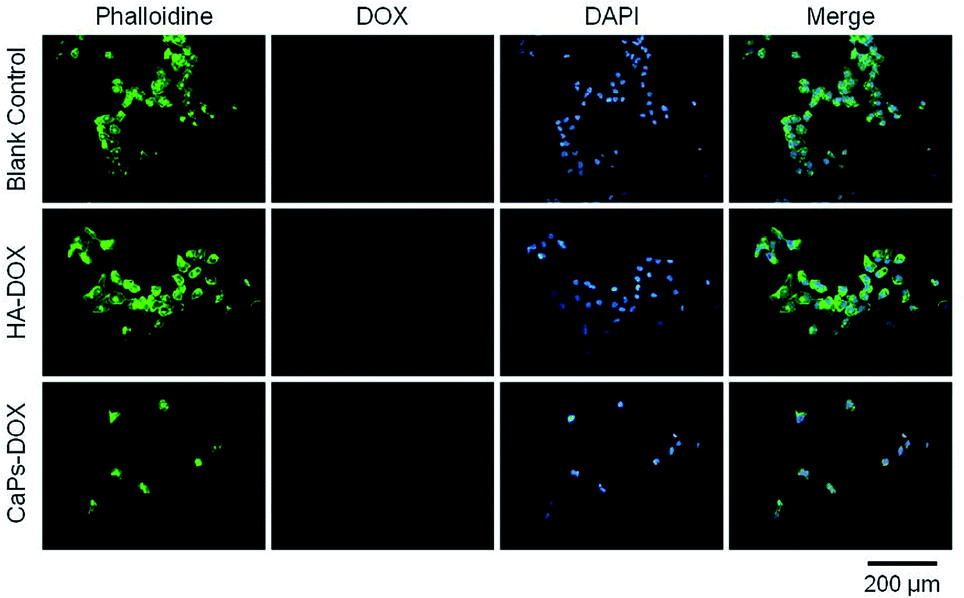 | ||
| Fig. 10 143b cells were co-cultured with CaP porous microspheres (CaPs-Dox) and HA-Dox for one day at a concentration of 5 μg mL−1. | ||
As a biocompatible material, CaP is widely used to develop various types of drug delivery systems that have superior functions in terms of biocompatibility, drug loading capability, cellular uptake efficiency and controlled drug release.6,17–23 In this study, we synthesized CaP porous microspheres using FBP biomolecules as an organic phosphorus source, which have enhanced drug loading capability compared with those previously reported using an inorganic phosphorus source.24,25 Therefore, the as-prepared CaP porous microspheres exhibit promise for potential application in the promotion of the chemotherapy efficiency of osteosarcoma, as a drug carrier with high biocompatibility.
Effect of the CaP porous microspheres on the ALP activity
As an early marker of osteogenesis, the ALP activity can be used to assess the osteogenic potential of MC3T3-E1 cells.26 Fig. 11 shows that the ALP activity displayed an increasing tendency from day 7 to day 14 for both the CaP porous microspheres and HA group. Moreover, MC3T3-E1 cells co-cultured with the CaP porous microspheres (10 μg mL−1) displayed higher ALP activity compared with HA nanorods at the same concentration on day 7 and day 14. Interestingly, on day 7, a high concentration (20 μg mL−1) of CaP porous microspheres induced higher ALP activity in the MC3T3-E1 cells, but on day 14, a high concentration (20 μg mL−1) of CaP porous microspheres induced lower ALP activity, which showed that the concentration of CaP porous microspheres did not show positive correlation with the ALP activity. Therefore, the ALP activity was found to be influenced by the type and concentration of the biomaterials, as well as the treatment time. Overall, a low concentration of CaP porous microspheres showed better osteogenic potential compared with the HA nanorods in promoting ALP activity.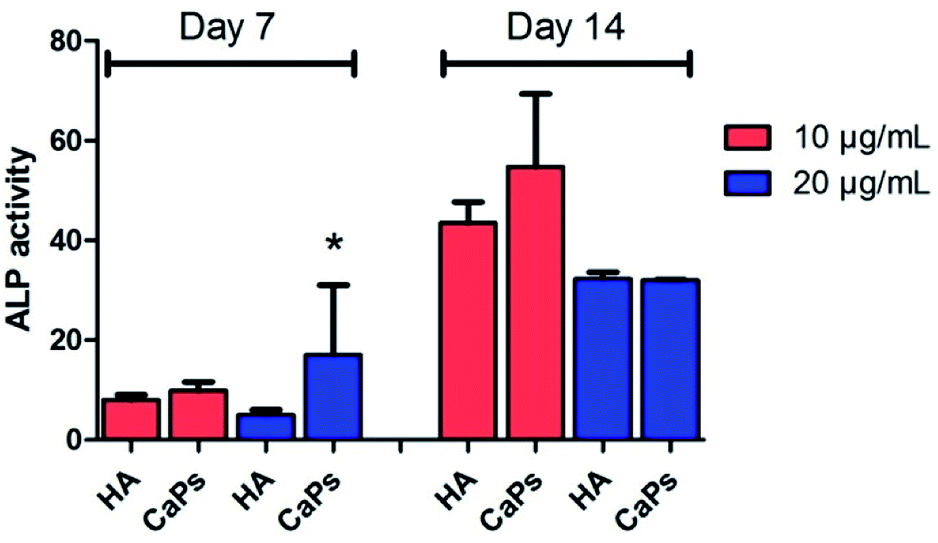 | ||
| Fig. 11 The CaP porous microspheres induced higher ALP activity than the HA nanorods at certain concentrations and co-culture times in the MC3T3-E1 cells. *Significant difference (p < 0.05) compared with the HA nanorods at the same concentration. | ||
Effect of CaPs on calcium nodule formation
Calcium nodule formation is the result of extracellular matrix mineralization, which reflects late stage osteogenic differentiation.26 The formed calcium nodules were stained as black stained dots or red stained dots by von Kossa staining and alizarin red staining, respectively (Fig. 12). As is shown in Fig. 12A, MC3T3-E1 cells co-cultured with CaP porous microspheres (10 μg mL−1) exhibit better mineralized nodule formation ability, compared with the HA nanorods (10 μg mL−1) at day 7 and day 14. Moreover, such time- and concentration-dependent promotion of the CaP porous microspheres in mineralized nodule formation was verified by the alizarin red staining (Fig. 12B). This result further demonstrated the superiority of CaP porous microspheres over HA nanorods in terms of osteogenic induction.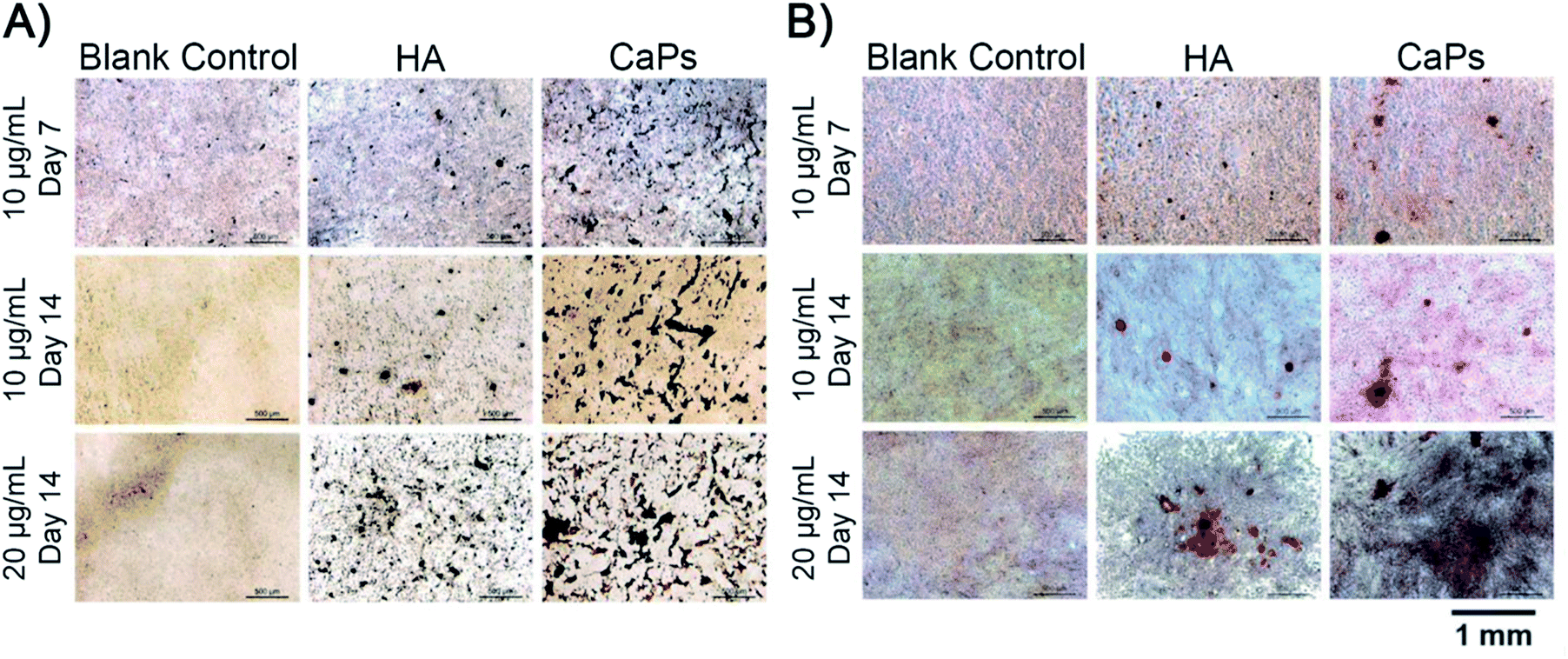 | ||
| Fig. 12 von Kossa staining (A) and alizarin red staining (B) of the calcium nodules of MC3T3-E1 cells co cultured with the CaP porous microspheres and HA nanorods. | ||
Previous studies suggest that mesenchymal stem cells (MSCs) are progenitor cells of osteosarcoma cells and that osteosarcoma originates from defects in the differentiation of MSCs.27–29 The osteogenic differentiation of MSCs can be divided into a variety of stages, including MSCs, committed osteoprogenitor, pro-osteoblast, early osteoblast, mature osteoblast and osteocyte.30 Defects in any of these stages can induce osteosarcoma. Moreover, MSCs promote tumor metastasis in a variety of cancer types.31–33 MSCs also interact with osteosarcoma cells to orchestrate the development of osteosarcoma.34–36 Qi et al.37 reported that exosomes derived from MSCs promote osteosarcoma MG-63 cell proliferation through a hedgehog signaling pathway. Taken together, MSCs play a pivotal role in osteosarcoma origination, proliferation, metastasis and chemoresistance. In this regard, facilitating the terminal differentiation of MSCs is predicted to be a potential therapeutic approach to inhibit osteosarcoma progression.
In this study, we tested the osteoinductive potential of the as-prepared product using the murine pre-osteoblast cell line MC3T3-E1, which is widely used in the research of osteogenic differentiation. The results showed that the CaP porous microspheres could facilitate osteogenic differentiation of this pre-osteoblast cell line. Similar results have been reported using MSCs.16 Moreover, apart from alleviating the tumor progression caused by MSCs, facilitating the osteogenic differentiation of MSCs could promote calcium deposition and bone formation at the lesion sites, filling up the bone defects after the surgical resection of the osteosarcoma.4 This highlighted the therapeutic potential of CaP porous microspheres against osteosarcoma (Fig. 13).
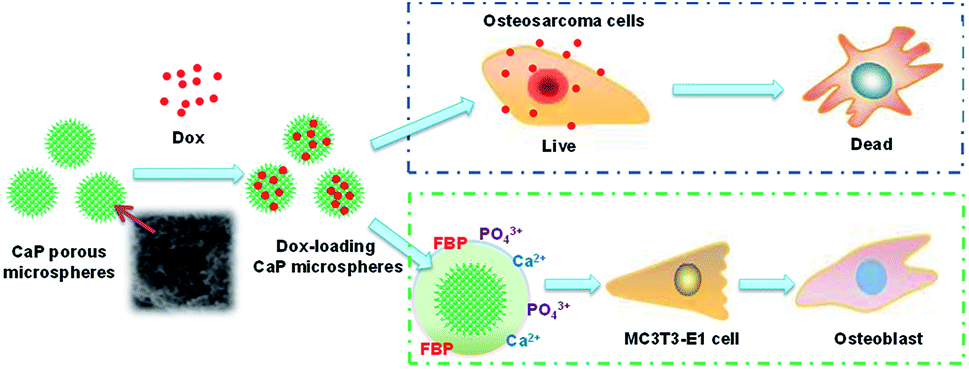 | ||
| Fig. 13 Schematic illustration of the functions of CaP porous microspheres in osteosarcoma treatment. CaP porous microspheres display dual functions, to deliver Dox to inhibit the proliferation of osteosarcoma cells and promote the osteogenic differentiation of MC3T3-E1. | ||
In addition, increasing evidence has been found that cancer is a metabolic disease.38,39 As an intermediate product of glycolysis, FBP not only shows cytoprotective effects in a variety of pathological conditions, such as ischemia, shock, inflammatory diseases and toxic lesions,40,41 it also shows anticancer effects in certain cancer types. FBP inhibits the proliferation of hepatocellular carcinoma cell lines in vitro through decreasing IL-8 levels, which is closely related to tumor progression and chemotherapeutic resistance and increasing the activity of pro-apoptotic proteins.42 In this perspective, the as-prepared CaP porous microspheres may exert a synergistic inhibitory effect on inflammation in the process of osteosarcoma progression, which further demonstrates the great therapeutic potential of CaP porous microspheres against osteosarcoma.
Conclusions
Herein, we have synthesized CaP porous microspheres using a facile wet-chemical strategy at room temperature, with phosphate-containing FBP molecules as the phosphorous source. The CaP porous microspheres are an organic–inorganic hybrid system. With a porous structure, the sample displayed a higher Dox-loading capability than the control sample of HA nanorods. The Dox-loaded CaP porous microspheres exhibit a high performance in the inhibition of the proliferation of osteosarcoma cells in vitro, and promote the enrichment of Dox in osteosarcoma cells. Meanwhile, the CaP porous microspheres are effective in inducing the osteogenic differentiation of the pre-osteoblast cell line MC3T3-E1. All of the results demonstrate that CaP porous microspheres are promising as a multifunctional drug carrier in osteosarcoma treatment. The chemical composition of CaP porous microspheres is CaP and FBP molecules, both of which are native to the human body. Therefore, CaP porous microspheres display high biocompatibility, which is significantly important in osteosarcoma treatment. The CaP porous microspheres reported in this study display potential for application in osteosarcoma related drug delivery and the promotion of osteogenic differentiation in subsequent bone regeneration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (31771081, 51472259, 81541055), the Science and Technology Commission of Shanghai (18ZR1445100) and the Fundamental Research Funds for the Central University (1501219171) is gratefully acknowledged.Notes and references
- D. J. Harrison, D. S. Geller, J. D. Gill, V. O. Lewis and R. Gorlick, Expert Rev. Anticancer Ther., 2018, 18, 39–50 CrossRef PubMed.
- B. A. Lindsey, J. E. Markel and E. S. Kleinerman, Rheumatol Ther, 2017, 4, 25–43 CrossRef PubMed.
- S. S. Bielack, S. Hecker-Nolting, C. Blattmann and L. Kager, F1000Research, 2016, 5, 2767 Search PubMed.
- Z. F. Zhou, T. W. Sun, F. Chen, D. Q. Zuo, H. S. Wang, Y. Q. Hua, Z. D. Cai and J. Tan, Biomaterials, 2017, 121, 1–14 CrossRef PubMed.
- K. D. Son and Y. J. Kim, Biomater. Res., 2017, 21, 13 CrossRef PubMed.
- U. Hess, S. Shahabi, L. Treccani, P. Streckbein, C. Heiss and K. Rezwan, Mater. Sci. Eng., C, 2017, 77, 427–435 CrossRef PubMed.
- Y. Tanzawa, H. Tsuchiya, T. Shirai, H. Nishida, K. Hayashi, A. Takeuchi, M. Kawahara and K. Tomita, J. Orthop. Sci., 2011, 16, 77–84 CrossRef PubMed.
- L. Jin, X. Zeng, M. Liu, Y. Deng and N. Y. He, Theranostics, 2014, 4, 240–255 CrossRef PubMed.
- D. R. Bienek and D. Skrtic, Biointerface Res. Appl. Chem., 2017, 7, 1989–1994 Search PubMed.
- A. M. A. Ambrosio, J. S. Sahota, Y. Khan and C. T. Laurencin, J. Biomed. Mater. Res., 2001, 58, 295–301 CrossRef PubMed.
- A. K. Markov, Ann Emerg Med, 1986, 15, 1470–1477 CrossRef PubMed.
- B. Ma, Q. Zhang, D. Wu, Y. L. Wang, Y. Y. Hu, Y. P. Cheng, Z. D. Yang, Y. Y. Zheng and H. J. Ying, Acta Pharmacol. Sin., 2012, 33, 479–489 CrossRef PubMed.
- Y. Zheng, G. Wang, R. Chen, Y. Hua and Z. Cai, Stem Cell Res. Ther., 2018, 9, 22 CrossRef PubMed.
- M. Cortini, S. Avnet and N. Baldini, Cancer Lett., 2017, 405, 90–99 CrossRef PubMed.
- H. K. Brown, M. Tellez-Gabriel and D. Heymann, Cancer Lett., 2017, 386, 189–195 CrossRef PubMed.
- C. Qi, D. Zhou, Y. J. Zhu, T. W. Sun, F. Chen and C. Q. Zhang, Mater. Sci. Eng., C, 2017, 77, 846–856 CrossRef PubMed.
- C. Wu, J. Xu, Y. Hao, Y. Zhao, Y. Qiu, J. Jiang, T. Yu, P. Ji and Y. Liu, Int. J. Nanomed., 2017, 12, 7979–7992 CrossRef PubMed.
- X. Wang, M. Zhang, L. Zhang, L. Li, S. Li, C. Wang, Z. Su, Y. Yuan and W. Pan, Chem.–Eur. J., 2017, 23, 6586–6595 CrossRef PubMed.
- H. Y. Nam, K. H. Min, D. E. Kim, J. R. Choi, H. J. Lee and S. C. Lee, Colloids Surf., B, 2017, 157, 215–222 CrossRef PubMed.
- S. Huang, C. Li and Q. Xiao, Nanoscale Res. Lett., 2017, 12, 275 CrossRef PubMed.
- Y. He, B. Zeng, S. Liang, M. Long and H. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 44402–44409 CrossRef PubMed.
- C. Qi, Y. J. Zhu, T. W. Sun, J. Wu and F. Chen, Chem.–Asian J., 2015, 10, 2503–2511 CrossRef PubMed.
- J. Tang, C. B. Howard, S. M. Mahler, K. J. Thurecht, L. Huang and Z. P. Xu, Nanoscale, 2018, 10, 4258–4266 RSC.
- W. Yu, T. W. Sun, C. Qi, Z. Ding, H. Zhao, F. Chen, D. Chen, Y. J. Zhu, Z. Shi and Y. He, ACS Appl. Mater. Interfaces, 2017, 9, 3306–3317 CrossRef PubMed.
- C. Qi, Y. J. Zhu and F. Chen, Chem.–Asian J., 2013, 8, 88–94 CrossRef PubMed.
- I. S. Kim, Y. M. Song, T. H. Cho, Y. D. Park, K. B. Lee, I. Noh, F. Weber and S. J. Hwang, Dev., Growth Differ., 2008, 50, 553–564 Search PubMed.
- R. C. Haydon, H. H. Luu and T. C. He, Clin. Orthop. Relat. Res., 2007, 454, 237–246 CrossRef PubMed.
- D. M. Thomas, S. A. Johnson, N. A. Sims, M. K. Trivett, J. L. Slavin, B. P. Rubin, P. Waring, G. A. McArthur, C. R. Walkley, A. J. Holloway, D. Diyagama, J. E. Grim, B. E. Clurman, D. D. Bowtell, J. S. Lee, G. M. Gutierrez, D. M. Piscopo, S. A. Carty and P. W. Hinds, J. Cell Biol., 2004, 167, 925–934 CrossRef PubMed.
- J. Y. Wang, P. K. Wu, P. C. Chen, C. W. Lee, W. M. Chen and S. C. Hung, Stem Cells Transl. Med., 2017, 6, 512–526 CrossRef PubMed.
- X. Luo, J. Chen, W. X. Song, N. Tang, J. Luo, Z. L. Deng, K. A. Sharff, G. He, Y. Bi, B. C. He, E. Bennett, J. Huang, Q. Kang, W. Jiang, Y. Su, G. H. Zhu, H. Yin, Y. He, Y. Wang, J. S. Souris, L. Chen, G. W. Zuo, A. G. Montag, R. R. Reid, R. C. Haydon, H. H. Luu and T. C. He, Lab. Invest., 2008, 88, 1264–1277 CrossRef PubMed.
- A. E. Karnoub, A. B. Dash, A. P. Vo, A. Sullivan, M. W. Brooks, G. W. Bell, A. L. Richardson, K. Polyak, R. Tubo and R. A. Weinberg, Nature, 2007, 449, 557–563 CrossRef PubMed.
- K. Shinagawa, Y. Kitadai, M. Tanaka, T. Sumida, M. Kodama, Y. Higashi, S. Tanaka, W. Yasui and K. Chayama, Int. J. Cancer, 2010, 127, 2323–2333 CrossRef PubMed.
- S. R. Baglio, T. Lagerweij, M. Perez-Lanzon, X. D. Ho, N. Leveille, S. A. Melo, A. M. Cleton-Jansen, E. S. Jordanova, L. Roncuzzi, M. Greco, M. A. J. van Eijndhoven, G. Grisendi, M. Dominici, R. Bonafede, S. M. Lougheed, T. D. de Gruijl, N. Zini, S. Cervo, A. Steffan, V. Canzonieri, A. Martson, K. Maasalu, S. Koks, T. Wurdinger, N. Baldini and D. M. Pegtel, Clin. Cancer Res., 2017, 23, 3721–3733 CrossRef PubMed.
- S. Avnet, G. Di Pompo, T. Chano, C. Errani, A. Ibrahim-Hashim, R. J. Gillies, D. M. Donati and N. Baldini, Int. J. Cancer, 2017, 140, 1331–1345 CrossRef PubMed.
- K. C. Vallabhaneni, M. Y. Hassler, A. Abraham, J. Whitt, Y. Y. Mo, A. Atfi and R. Pochampally, PLoS One, 2016, 11, e0166027 CrossRef PubMed.
- P. Avril, L. R. Le Nail, M. A. Brennan, P. Rosset, G. De Pinieux, P. Layrolle, D. Heymann, P. Perrot and V. Trichet, J Bone Oncol, 2016, 5, 5–14 CrossRef PubMed.
- J. Qi, Y. Zhou, Z. Jiao, X. Wang, Y. Zhao, Y. Li, H. Chen, L. Yang, H. Zhu and Y. Li, Cell. Physiol. Biochem., 2017, 42, 2242–2254 CrossRef PubMed.
- H. Li, J. Wang, H. Xu, R. Xing, Y. Pan, W. Li, J. Cui, H. Zhang and Y. Lu, Mol. Cancer, 2013, 12, 110 CrossRef PubMed.
- F. Ye, Y. Chen, L. Xia, J. Lian and S. Yang, Biochem. Biophys. Res. Commun., 2018, 497, 639–645 CrossRef PubMed.
- A. K. Markov, Ann Emerg Med, 1986, 15, 1470–1477 CrossRef PubMed.
- G. V. Bochi, V. D. Torbitz, L. P. Cargnin, M. B. Sangoi, R. C. Santos, P. Gomes and R. N. Moresco, Inflammation, 2012, 35, 1786–1792 CrossRef PubMed.
- A. Adah, H. Benghuzzi, M. Tucci, D. Huang, L. Franklin and F. Adah, Biomed. Sci. Instrum., 2006, 42, 120–125 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |