
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAg2O nanoparticle-catalyzed substrate-controlled regioselectivities: direct access to 3-ylidenephthalides and isocoumarins†
Sandeep Chaudhary *,
Bharti Rajesh K. Shyamlal,
Lalit Yadav,
Mohit K. Tiwari and
Krishan Kumar
*,
Bharti Rajesh K. Shyamlal,
Lalit Yadav,
Mohit K. Tiwari and
Krishan Kumar
Laboratory of Organic and Medicinal Chemistry, Department of Chemistry, Malaviya National Institute of Technology Jaipur, Jawaharlal Nehru Marg, Jaipur-302017, India. E-mail: schaudhary.chy@mnit.ac.in
First published on 26th June 2018
Abstract
Herein, we disclose the first example of an efficient, silver oxide nanoparticle-catalyzed, direct regioselective synthesis of 3-ylidenephthalides 11–16 and isocoumarins 17–20 via sonogashira type coupling followed by substrate-controlled 5-exo-dig or 6-endo-dig cyclization reaction, respectively. This one pot coupling involves reaction of substituted 2-halobenzoic acid with meta/para-substituted and ortho-substituted terminal alkynes, which proceeded in a regioselective manner resulting in the formation of 3-ylidenephthalides or isocoumarins, respectively, in excellent yields (up to 95%) with complete Z-selectivity. This protocol features relatively broad substrate scope, mild conditions, operational simplicity, and is favourable with aromatic/alicyclic terminal alkynes. The competition experiments and gram-scale synthesis further highlight the importance and versatility of the methodology. The proposed mechanistic pathways illustrate that the regioselectivity is substantially being controlled by the substituent(s) present on the acetylenic phenyl ring.
Introduction
Phthalide (isobenzofuran-1(3H)-one) is an important building block present in many natural products, pharmaceuticals, and biological materials, and shows a wide range of biological activities, such as anti-HIV, antidiabetic, antispasmodic, anti-allergic, antifungal, pesticidal, herbicidal, insecticidal, and anticancer activity.1a–d It has been used as an intermediate for the synthesis of various phthalide-based natural products and therapeutics.2 Similarly, an inverted δ-lactone moiety, i.e., isocoumarin (1H-2-benzopyran-1-one), also displays astounding bio-activities, namely antitumor, antiallergenic, antimicrobial, anti-inflammatory, antidiabetic, phytotoxic, anti-HIV and immunomodulatery responses.3,1bConsequently, a number of methods have been reported to construct 3-ylidenephthalides and isocoumarins via traditional methods as well as C–H bond functionalization.4 It has been reported that 3-ylidenephthalides and isocoumarins can be easily prepared, respectively, from metal-catalyzed 5-exo-dig and 6-endo-dig cyclization of 2-alkynylbenzoic acid/esters, which can be generated in situ from the Sonogashira-type coupling reaction of 2-halobenzoic acids with terminal alkynes.5 However, earlier methods suffer from low regioselectivities affording mixture of phthalides and isocoumarins.6a,b
The controlled regioselectivity is an important concept for the synthesis of pharmaceutically important molecules with definite regio- and stereo-selectivities. Pertaining to the present study that is the regioselective synthesis of 5-exo-dig and 6-endo-dig cyclization; only a few regioselective methods, i.e., Cu2+-NCS catalyzed (Scheme 1a),7 acid–base controlled,8 and temperature controlled synthesis (Scheme 1b),9 have been reported to synthesize 3-ylidenephthalides 2 and 7 as well as isocoumarins 6 via C–C and C–O bond formations. Recently, Rh-catalyzed oxidative coupling/annulation of benzoic acids with terminal alkynes has been reported to construct 3-ylidenephthalides 8 with complete Z-selectively via C–H bond activation concept (Scheme 1c).10
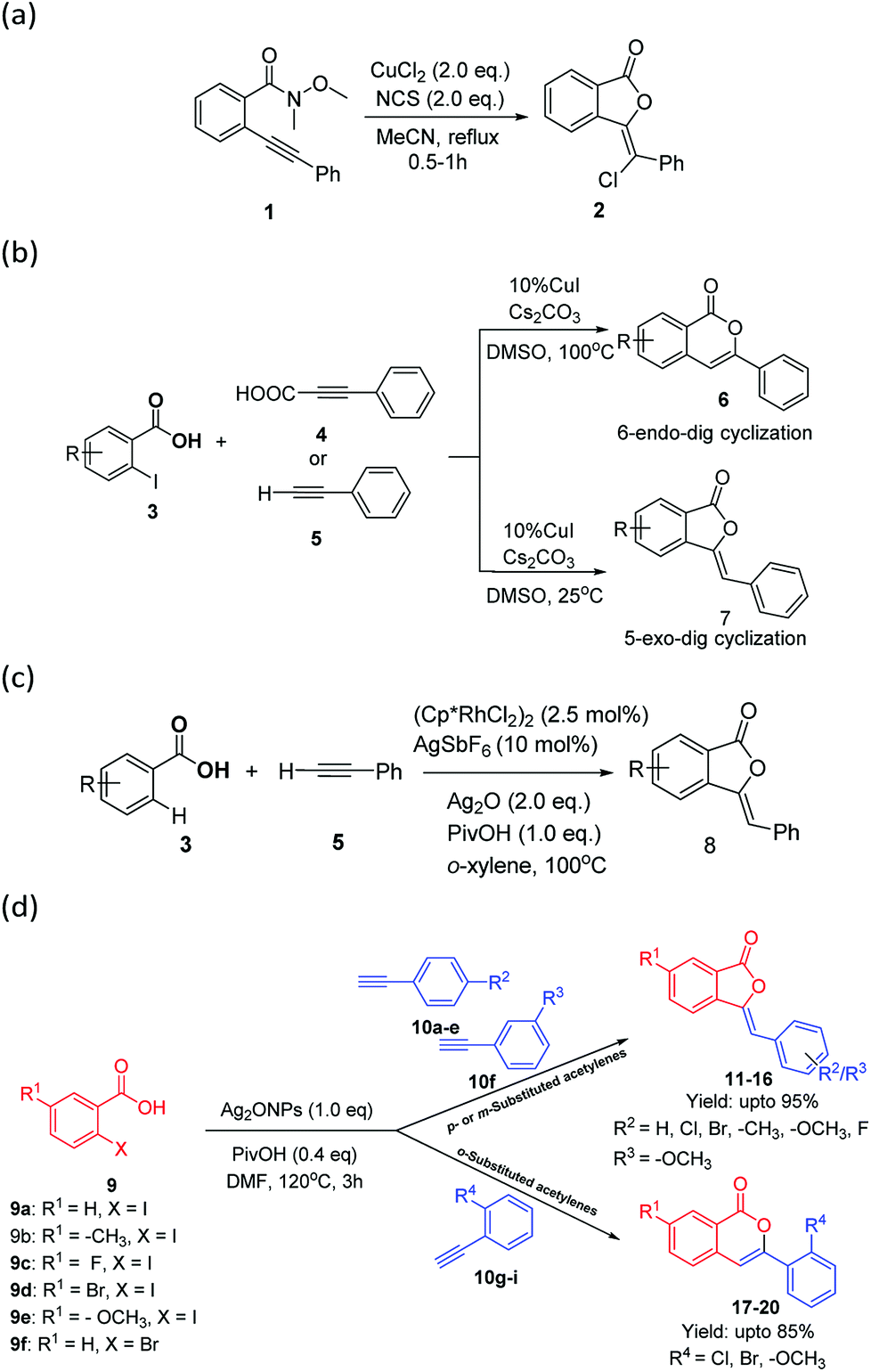 | ||
| Scheme 1 Present study versus previous representative approaches for the regioselective synthesis of 3-ylidenephthalides and isocoumarins. (a) Miyata et al., (b) Manian et al., (c) Liu et al., (d) present work. | ||
In general, transition metal-catalyzed cross coupling reactions have been conveniently established for carrying out C–H bond activation via new systems that exhibit extremely high catalytic activity and/or unique reactivity. In recent years, the use of metal catalysts of Cu, Mn, Co, and Fe in C–H bond activation has gained significant attention over Pd and Ni catalysts. Since, Ag(I) salt have been reported to show exceptional potential to explicit tremendous affinity especially towards terminal alkynes11 and several Ag-catalyzed C–C coupling reaction have been well reported in the literature;12 we envisaged that if terminal alkynes with electron rich substituents at o-, m-, or p-position be chosen as starting material for the reaction with 2-iodobenzoic acid, then Ag(I) may coordinate simultaneously with triple bond as well as with electron-rich substituents (more likely with o- as compared to m- and p-position) e.g., Cl, Br, OMe etc. which could undergo 5-exo-dig or 6-endo-dig cyclisation in a regioselective manner (Fig. 1).
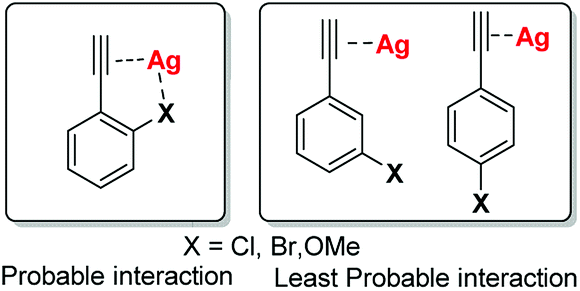 | ||
| Fig. 1 Structures depicting the probable interaction between Ag(I) compounds with terminal alkynes and electron rich substituents. | ||
To the best of our knowledge, there is no report on the silver-catalyzed substrate-controlled regioselectivities on 5-exo-dig or 6-endo-dig cyclization reaction of 2-iodobenzoic acid with o-, m-, or p-substituted terminal alkynes. Herein, we report an efficient, one-pot, silver oxide nanoparticle (Ag2ONPs) catalyzed substrate-controlled synthesis of 3-ylidenephthalides and isocoumarins in a regioselective manner upto 95% yields (Scheme 1d).
Results and discussions
In our endeavors in search for potent bioactive compounds;13 we were interested in the synthesis of 3-ylidenephthalides 11a and isocoumarins 11aa′ regioselectively (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Metal | Add. | Solvent | Temp. | Time (h) | Yield (%) |
| a Reaction conditions: 9a (1 eq.), 5 (1 eq.), Ag2ONPs (1 eq.), pivalic acid (0.4 eq.), 120 °C, 3 h.b Isocoumarin 11aa′ was also obtained in 5% yield in this reaction. | ||||||
| 1 | CuI | — | DMF | 80 °C | 3 | 10b |
| 2 | Ag2O | — | DMF | 80 °C | 3 | 45 |
| 3 | Ag2O | PivOH | DMF | 80 °C | 3 | 75 |
| 4 | AgOAc | PivOH | DMF | 80 °C | 3 | 43 |
| 5 | AgCl | PivOH | DMF | 80 °C | 3 | Nil |
| 6 | AgNO3 | PivOH | DMF | 80 °C | 3 | Nil |
| 7 | Ag2ONPs | PivOH | DMF | 80 °C | 3 | 83 |
| 8 | Ag2ONPs | AcOH | DMF | 80 °C | 3 | 46 |
| 9 | Ag2ONPs | PivOH | DMF | 80 °C | 24 | 59 |
| 10 | Ag2ONPs | PivOH | DMF | 100 °C | 3 | 90 |
| 11 | Ag2ONPs | PivOH | DMF | 120 °C | 3 | 95 |
| 12 | Ag2ONPs | PivOH | DMF | 130 °C | 3 | 88 |
| 13 | Ag2ONPs | PivOH | DMF | 120 °C | 1.5 | 88 |
| 14 | Ag2ONPs | PivOH | DMF | 120 °C | 2 | 88 |
| 15 | — | PivOH | DMF | 120 °C | 3 | No reaction |
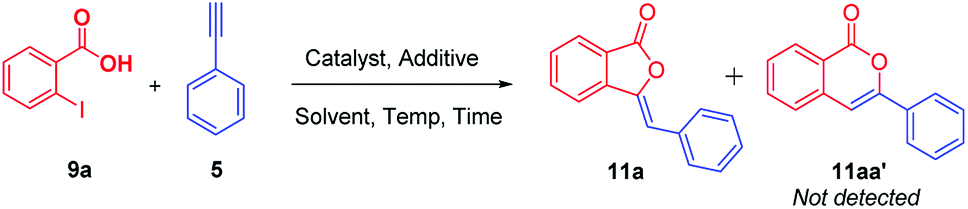
Consequently, we started our synthetic investigation via the reaction of 2-iodobenzoic acid 9a with phenylacetylene 5 as model reaction for the optimization study of the reaction. We initiated our investigation by carrying out the reaction using CuI in DMF which furnished the desired 3-ylidenephthalide 11a in 10% yield only (Table 1, entry 1). Since [Ag] salts have high affinity towards terminal alkynes; we used Ag2O to carry out our model reaction. To our surprise, we obtained only 11a in 45% yield (Table 1, entry 2) and 11aa′ was not detected at all. The structure of the desired compound was well characterized via 1H NMR, 13C NMR, FT-IR and HRMS analysis and it demonstrated complete Z-selectivity. Then, the same reaction has been carried out in the presence of pivalic acid as an additive which furnished 11a in 75% yield (Table 1, entry 3). Once realizing the unique nature of Ag2O; we also screened various other salts of silver, i.e., acetate, chloride, and nitrate. Among them, only silver acetate/pivalic acid combination furnished 11a in 43% yields (Table 1, entry 4–6). In addition, screening of metal oxides of Zn, Cu, Cd, Pb, Al, Cr, Ni, Mn, Fe, etc. were carried out. Either reaction does not occur at all or 11a was obtained in traces (see ESI†).
It has been known that nanoparticle leveraged C–H activation has been identified as an efficient sustainable approach for industrial applications by transforming unactivated C(sp3)–H and C(sp2)–H bonds present in feedstock into important bio-active molecules.14 We anticipated that silver oxide nanoparticles (Ag2ONPs), having larger surface area, may results in high yield of our desired products.15 Thus, the model reaction was carried out using Ag2ONPs/pivalic acid in DMF which, to our expectation, furnished the desired 11a in 83% yields (Table 1, entry 7). Decrease in catalytic loading of Ag2ONPs leads to decrease in yield of the reaction (see ESI†). The model reaction has been also carried out using Ag2ONPs/acetic acid in DMF which furnished 11a in only 46% yield (Table 1, entry 8). Extending the reaction for 24 h at the same temperature leads to decrease in reaction yield (Table 1, entry 9). Further examination of variation in temperature and time interval were carried out. Increasing the temperature from 80 °C to 100 °C furnished 11a in 90% yield (Table 1, entry 10). Interestingly, we further increase the temperature to 120 °C for 3 h which furnished the desired product 11a in excellent (95%) yield (Table 1, entry 11). Further increase in temperature or decrease in time interval do not have much beneficial effects on the yield of the reaction (Table 1, entry 12–14). While the reaction in other solvents such as DMSO, 1,4-dioxane, toluene, o-xylene, diminishes the catalytic activity; the reaction in PEG and water did not occur at all (see ESI†). Further, change in the nature of additives such as Na2CO3, Cs2CO3, NaOH, 1,10-phenanthroline etc. do not furnished the desired product 11a or 11aa′ (see ESI†). This infer us that our model reaction facilitates under acidic conditions. It has been also observed that no reaction occur when Ag2ONPs is not added to the optimized reaction conditions (Table 1, entry 15). Overall, Ag2ONPs/pivalic acid in DMF at 120 °C for 3 h was found to be the best optimized reaction conditions for this regioselective reaction.
To explore the substrate scope and generality of the present methodology, various substituted 2-iodobenzoic acids 9a–f were reacted with several o-/m-/p-substituted alkynes 10a–i under optimized reaction conditions. Thus, 9a–f were reacted with para-and meta-substituted alkynes 10a–f under optimized reaction conditions, which undergoes substrate-controlled 5-exo-dig cyclization to furnish 3-ylidenephthalides 11–16 upto 95% yields in a highly regioselective manner (Scheme 2).
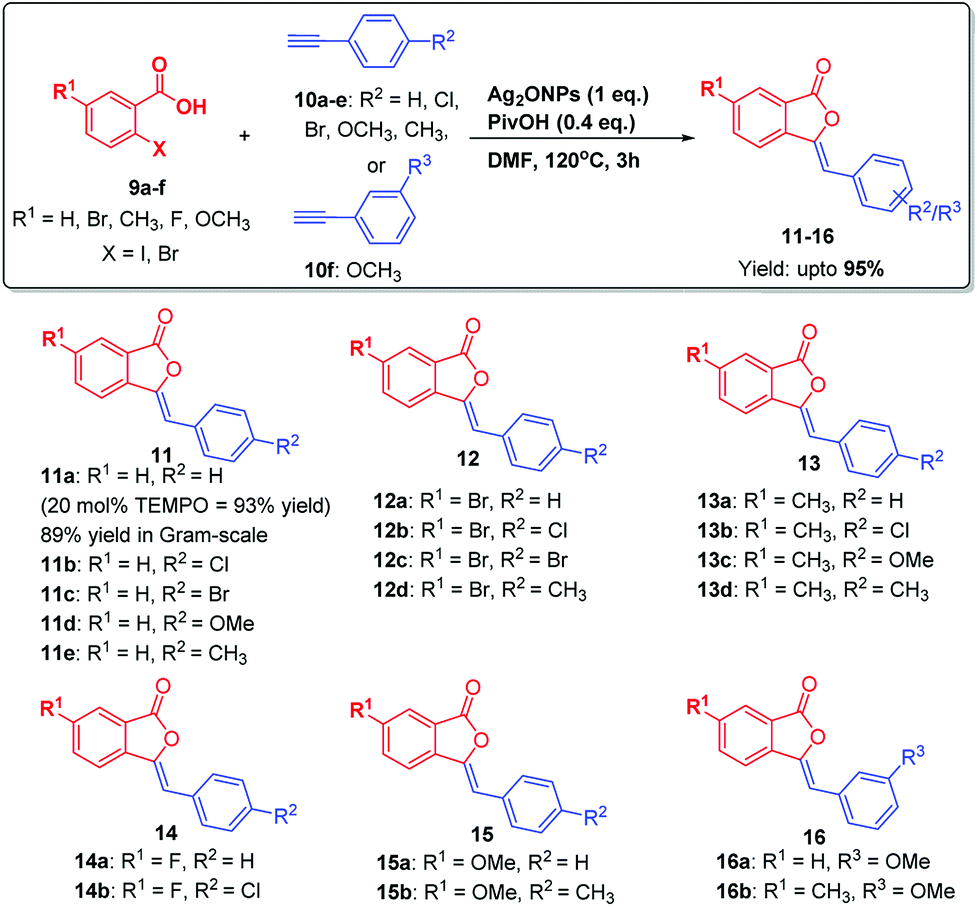 | ||
| Scheme 2 Substrate scope: synthesis of 3-ylidenephthalides 11–16. | ||
Then, 9a was reacted with 10g under optimized conditions. As envisioned in the case of ortho-substitutent in acetylenic phenyl ring, instead of getting 3-ylidenephthalides; we obtained only isocoumarin 17a in 42% yield. This encourage us to investigate this unusual observation in details to understand the cause of regioselectivity. Since chlorine having lone pair of electrons at ortho-position of the acetylenic phenyl ring displayed different behaviour, we analyzed other groups containing lone pair of electrons such as Br, OMe at ortho-position of phenyl acetylenes. Thus, substituted 2-iodobenzoic acid 9a–c were reacted with ortho-substituted alkynes 10g–j under optimized reaction conditions which furnished isocoumarins 17–20 upto 85% yields via substrate-controlled 6-endo-dig cyclization reaction (Scheme 3). These groups also furnished isocoumarins regioselectively.
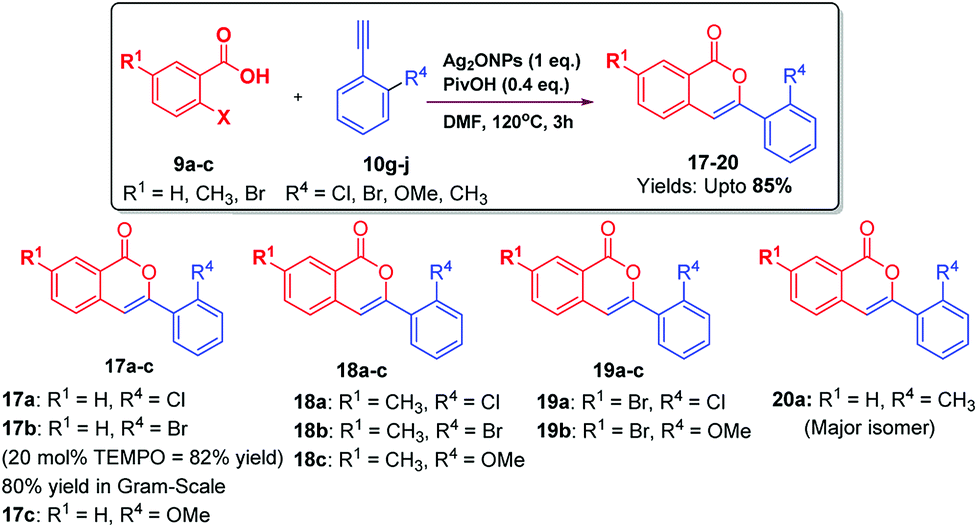 | ||
| Scheme 3 Substrate scope: synthesis of isocoumarins 17–20. | ||
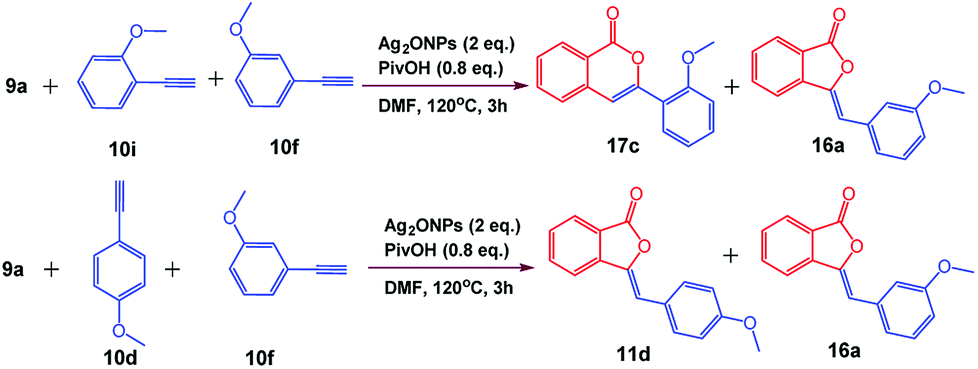
The generality of the reaction was demonstrated by carrying out gram-scale synthesis of 3-ylidinephthalides 11a as well as isocoumarins 17b which were obtained in 89% and 80% yields, respectively. In order to check the competition between 5-exo-dig and 6-endo-dig cyclization reaction, reaction of 9a with an equimolar ratio of 10f and 10i was carried out which furnished 17c/16a in 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This infer us that the 5-exo-dig and 6-endo-dig cyclization occurs in almost equal proportion under optimized reaction conditions. Furthermore, competition reaction between 9a with an equimolar ratio of 10d and 10f furnished 11d/16a in 1.2:1 ratio which indicates that m- and p-substituted phenylacetylenes undergoes 5-exo-dig cyclization in almost equal proportion (Scheme 5).
:1 ratio. This infer us that the 5-exo-dig and 6-endo-dig cyclization occurs in almost equal proportion under optimized reaction conditions. Furthermore, competition reaction between 9a with an equimolar ratio of 10d and 10f furnished 11d/16a in 1.2:1 ratio which indicates that m- and p-substituted phenylacetylenes undergoes 5-exo-dig cyclization in almost equal proportion (Scheme 5).
 | ||
| Scheme 4 Substrate scope: Synthesis of alicyclic 3-ylidenephthalides 21a–c. | ||
 | ||
| Scheme 5 Intermolecular competition experiments. | ||
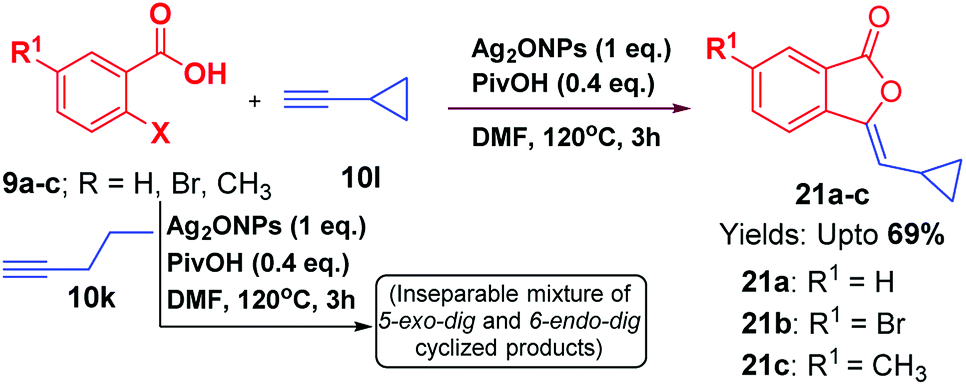
Like aromatic terminal alkynes, aliphatic terminal alkyne 10k was also attempted to perform this reaction under optimized reaction conditions; however, we obtained an inseparable mixture of 5-exo-dig and 6-endo-dig cyclized products. Hence, it may be inferred that the regioselectively of this reaction is favourable with aromatic alkynes as compared to aliphatic alkynes. In addition, when alicyclic acetylene 10l, was subjected to optimized reaction conditions; cyclopropyl-based 3-ylidenephthalides 21a–c were obtained favouring 5-exo-dig product regioselectively (Scheme 4).
In consideration of the unique features of the Ag2ONPs-catalyzed substrate-controlled regioselective 5-exo-dig or 6-endo-dig cyclization reaction, we were interested in delineating the tentative mechanism of this reaction. The evidence for the occurrence of the Sonogashira type C–H activation followed by oxycation-mediated cyclization was gathered by carrying out reactions in the presence of typical free-radical scavenger such as 2,2,6,6,-tetramethylpiperidine-N-oxyl (TEMPO, 20 mole%) under the same optimized conditions. The occurrence of 11a in 93% yields and 17b in 82% yields, respectively, indicates that the reaction involves cationic mechanism rather than free-radical process.
To study the mechanistic role of groups having lone pair of electrons at ortho-position of acetylenic phenyl ring; we were also interested to depict the role of group(s) without having lone pair of electrons at ortho-position. Thus, isocoumarin 20a was obtained as major isomer along with 3-ylidenephthalides (minor) via reaction of 9a with 10j which infer us that ortho-group having lone pair of electrons furnish isocoumarins exclusively; this regioselective methodology is also applicable with ortho-group without having lone pair of electrons. Presumably, the formation of 20a was due to the coordinated association of electron clouds of CH3 group with [Ag] complex which facilitates oxyanion attacks at C-2 position of acetylenic bond via 6-endo-dig cyclization more favourably than 5-exo-dig cyclization which generated 3-ylidenephthalides 20b as minor product (see ESI†).
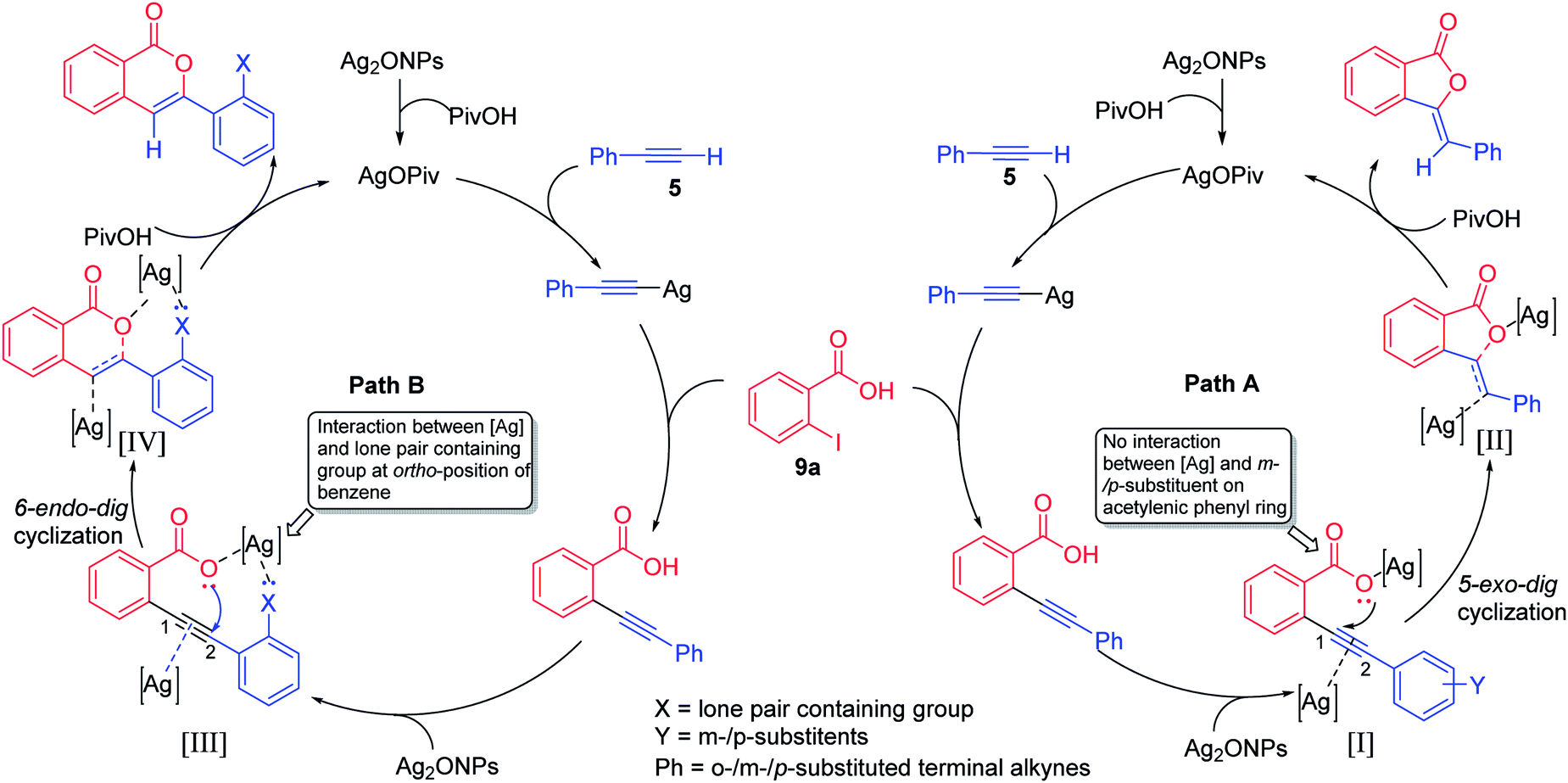
Thus, the tentative mechanism of this heterogeneous reaction may be proposed based on previous similar studies (Fig. 2).16 It involves deprotonation of 5 which furnishes the silver phenylacetylide in situ.17 Due to the ligand to metal charge transfer (LMCT),18 partial positive charge developed at the acetylene site which favour the attack of 9a and gives 2-(phenylethynyl)benzoic acid on which residual Ag2ONPs could coordinate with electron rich alkynyl bond as well as with the carboxylate oxyanion [I].19 In the case of m-/p-substituted terminal alkynes as in intermediate [I] (path A), no interaction of lone pair electrons or electron cloud between [Ag] and m-/p-substituent on acetylenic phenyl ring occurs and hence, carboxylate oxyanion undergoes attack at C-1 position of acetylenic bond via 5-exo-dig cyclization to give intermediate [II], which is further converted into 3-ylidenephthalides upon protonation with PivOH. However, in the case o-substituted terminal alkynes as in intermediate [III] (path B), there is significant electronic interaction between the [Ag] and the electronic lobe or lone pair of electrons containing group at o-substituted acetylenic phenyl ring. Consequently, oxyanion attacks at C-2 position of acetylenic bond via 6-endo-dig cyclization to give intermediate [IV], which is further converted into isocoumarins upon protonation with PivOH.
 | ||
| Fig. 2 Plausible mechanism | ||
Conclusions
For the first time, we have developed a highly efficient and straightforward route to access two bioactive moieties, 3-ylidinephthalides and isocoumarins, via Ag2O nanoparticle-catalyzed Sonogashira type coupling followed by substrate-controlled 5-exo-dig or 6-endo-dig cyclization in a highly regioselective manner, respectively, with complete Z-selectivity. This protocol features relatively broad substrate scope, mild conditions, operationally simple, and is more favorable with aromatic/alicyclic terminal alkynes as compared to aliphatic terminal alkynes. The intermolecular competition experiments and gram-scale synthesis further highlights the importance and versatility of the newly developed methodology. The proposed mechanistic pathways interprets that the regioselectivity is substantially being controlled by the substituent(s) present on the acetylenic phenyl ring.Experimental section
All glass apparatus were oven dried prior to use. High quality reagents were purchased from Sigma Aldrich (USA), TCI Chemicals (Tokyo, Japan) and Spectrochem (India) and were used without further purification. Laboratory grade commercial reagents and solvents were purified by standard procedures prior to use. Infrared spectra were recorded on a FT-IR Spectrum 2 (Perkin-Elmer) spectrophotometer. NMR spectra were obtained on a Jeol resonance ECS 400 MHz spectrometer (operating at 400 MHz respectively for 1H; 100 MHz respectively for 13C). The 1H-NMR (400 MHz) chemical shifts were measured relative to CDCl3 or DMSO-d6 as the internal reference (CDCl3: δ = 7.249 ppm; DMSO-d6: δ = 2.50 ppm). The 13C-NMR (100 MHz) chemical shifts were given using CDCl3 or DMSO-d6 as the internal standard (CDCl3: δ = 77.16 ppm; DMSO-d6: δ = 39.52 ppm). Tetramethylsilane (δ = 0.00 ppm) served as an internal standard in 1H-NMRand CDCl3 (δ 77.16 ppm) in 13C NMR. Chemical shifts are reported in parts per million. Splitting patterns are described as singlet (s), doublet (d), triplet (t) and multiplet (m). Electrospray emission mass spectrometry (ESI-MS) and high resolution mass spectra (HRMS) were obtained with a Gevo G-2 S Q-TOF (Waters). Melting points were taken in open capillaries on Complab melting point apparatus and are presented uncorrected. Unless otherwise noted, column chromatography was performed over Rankem silica gel (particle size: 100–200 mesh) procured from Rankem™ (India).General procedure (GP) for the synthesis of 3-ylidenephthalides 11–16
To a stirred mixture of substituted halo-aromatic carboxylic acid 9a–f (1.209 mmol, 1.0 eq.), terminal alkynes 10a–e/10f (1.209 mmol, 1.0 eq.), Ag2ONPs (1.209 mmol, 1.0 eq.), and PivOH (0.484 mmol, 0.4 eq.) in dry DMF (2 mL) at 120 °C for 3 h. The reaction mixture was then cooled to ambient temperature; diluted with ethyl acetate (10 mL), filtered through a celite pad and washed further with ethyl acetate (15 mL). The combined organic solvents were extracted with ethyl acetate (3 × 10 mL), washed with distilled water (2 × 20 mL) and then with brine solution (20 mL). The combined organic layer was dried over anhyd. Na2SO4 & concentrated under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) using 2% ethyl acetate: hexane as eluant to furnish 11–16.General procedure (GP) for the synthesis of isocoumarins 17–20
To a stirred mixture of substituted halo aromatic carboxylic acid 9a–c (1.209 mmol, 1.0 eq.), terminal alkynes 10g–j (1.209 mmol, 1.0 eq.), Ag2ONPs (1.209 mmol, 1.0 eq.), and PivOH (0.484 mmol, 0.4eq.) in dry DMF (2 mL) at 120 °C for 3 h. The reaction mixture was then cooled to ambient temperature; diluted with ethyl acetate (10 mL), filtered through a celite pad and washed further with ethyl acetate (15 mL). The combined organic solvents were extracted with ethyl acetate (3 × 10 mL), washed with distilled water (2 × 20 mL) and then with brine solution (20 mL). The combined organic layer was dried over anhyd. Na2SO4 & concentrated under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) using 2% ethyl acetate: hexane as eluant to furnish 17–20.General procedure for the reaction of 9a with 10k
To a stirred mixture of substituted 2-iodobenzoic acid 9a (1.209 mmol, 1.0 eq.), 1-pentyne 10k (1.209 mmol, 1.0 eq.), Ag2ONPs (1.209 mmol, 1.0 eq.), and PivOH (0.484 mmol, 0.4eq.) in dry DMF (2 mL) at 120 °C for 3 h. The reaction mixture was then cooled to ambient temperature; diluted with ethyl acetate (20 mL), filtered through a celite pad and washed further with ethyl acetate (25 mL). The combined organic solvents were extracted with ethyl acetate (3 × 20 mL), washed with distilled water (2 × 20 mL) and then with brine solution (20 mL). The combined organic layer was dried over anhyd. Na2SO4 & concentrated under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) using 2% ethyl acetate: hexane as eluant which furnished an inseparable mixture of 5-exo-dig and 6-endo-dig cyclized product in 72% yields.General procedures for the synthesis of alicyclic 3-ylidenephthalides 21a–c
To a stirred mixture of substituted halo-aromatic carboxylic acid 9a–c (1.209 mmol, 1.0 eq.), alicyclic terminal alkyne 10l (1.209 mmol, 1.0 eq.), Ag2ONPs (1.209 mmol, 1.0 eq.), and PivOH (0.484 mmol, 0.4 eq.) in dry DMF (2 mL) at 120 °C for 3 h. The reaction mixture was then cooled to ambient temperature; diluted with ethyl acetate (10 mL), filtered through a celite pad and washed further with ethyl acetate (15 mL). The combined organic solvents were extracted with ethyl acetate (3 × 10 mL), washed with distilled water (2 × 20 mL) and then with brine solution (20 mL). The combined organic layer was dried over anhyd. Na2SO4 & concentrated under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) using 2% ethyl acetate: hexane as eluant to furnish 21a–c in 61–69% yield range.Control experiments
:1).:1).TEMPO-mediated reaction in the case of 3-ylidenephthalides. To a stirred mixture of 2-iodo benzoic acid 9a (1.209 mmol, 1.0 eq.), phenylacetylene 5 (1.209 mmol, 1.0 eq.), Ag2ONPs (1.209 mmol, 1.0 eq.), PivOH (0.484 mmol, 0.4 eq.) and TEMPO (0.242 mmol, 0.2 eq.) in dry DMF (2 mL) at 120 °C for 3 h. The reaction mixture was then cooled to ambient temperature; diluted with ethyl acetate (20 mL), filtered through a celite pad and washed further with ethyl acetate (20 mL). The combined organic solvents were extracted with ethyl acetate (3 × 10 mL), washed with distilled water (2 × 20 mL) and then with brine solution (20 mL). The combined organic layer was dried over anhyd. Na2SO4 & concentrated under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) using 2% ethyl acetate: hexane as eluant which furnished 11a in 93% yield. This illustrates that the mechanism of the reaction do not occur via free-radical pathway.
TEMPO-mediated reaction in the case of isocoumarins. To a stirred mixture of 2-iodo benzoic acid 9a (1.209 mmol, 1.0 eq.), 2-bromo phenylacetylene 10h (1.209 mmol, 1.0 eq.), Ag2ONPs (1.209 mmol, 1.0 eq.), PivOH (0.484 mmol, 0.4 eq.) and TEMPO (0.242 mmol, 0.2 eq.) in dry DMF (2 mL) at 120 °C for 3 h. The reaction mixture was then cooled to ambient temperature; diluted with ethyl acetate (15 mL), filtered through a celite pad and washed further with ethyl acetate (20 mL). The combined organic solvents were extracted with ethyl acetate (3 × 10 mL), washed with distilled water (2 × 20 mL) and then with brine solution (20 mL). The combined organic layer was dried over anhyd. Na2SO4 & concentrated under reduced pressure. The crude product was purified by column chromatography over silica gel (100–200 mesh size) using 2% ethyl acetate: hexane as eluant which furnished 17b in 82% yield. This also illustrates that the mechanism of the reaction do not occur via free-radical pathway.
3-(o-Tolyl)-1H-isochromen-1-one (20a). See spectral data of 20a.
(Z)-3-(2-Methylbenzylidene)isobenzofuran-1(3H)-one (20b). This compound was prepared according to the GP and obtained in 2% EtOAc/hexane solution as light yellow liquid in 25% yield. IR (Neat, cm−1): 3067, 2923, 2863, 1727, 1639, 1481, 1331, 1228, 1050, 757, 687; 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 7.96 Hz, 1H), 7.74–7.70 (m, 1H), 7.53–7.46 (m, 3H), 7.36–7.32 (m, 1H), 7.28 (d, J = 7.4 Hz, 2H), 6.60 (s, 1H), 2.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.69, 155.67, 137.58, 136.87, 134.92, 132.85, 131.14, 129.80, 129.28, 128.33, 125.99, 120.42, 106.01, 20.87; HRMS (ESI/QTOF) m/z [M + H]+ calculated for [C16H12O2]+: 237.0910, found 237.0914.
Conflicts of interest
All the authors declare no conflict of interest.Acknowledgements
S. C. acknowledges CSIR, New Delhi for CSIR-EMR Grant [02 (0189)/14/EMR-II]; DST, New Delhi for DST-RFBR Indo-Russian Joint Research Project (INT/RUS/RFBR/P-169) and SERB, New Delhi for Fast Track young scientist scheme (Grant No. CS-037/2013). B. R. K.·S., L. Y., M. K. T. and K. K. thanks UGC, New Delhi; DST, New Delhi; MNIT, Jaipur and CSIR, New Delhi for providing financial assistance in the form of fellowships, respectively. Materials Research Centre (MRC), MNIT, Jaipur is gratefully acknowledged for providing analytical facilities.Notes and references
- (a) Y. Liu, Y. Yang, Y. Shi, X. Wang, L. Zhang, Y. Cheng and J. You, Organometallics, 2016, 35(10), 1350–1353 CrossRef; (b) R. K. Manian, F. M. Irudayanathan, J. H. Moon and S. Lee, Adv. Synth. Catal., 2013, 355, 3221–3230 CrossRef; (c) N. G. Kundu, M. Pal and B. Nandi, J. Chem. Soc., Perkin Trans. 1, 1998, 561–568 RSC; (d) N. Yu and M. Wang, Curr. Med. Chem., 2008, 15, 1350–1375 CrossRef PubMed.
- R. Karmakar, P. Pahari and D. Mal, Chem. Rev., 2014, 114(12), 6213–6284 CrossRef PubMed.
- R. G. Chary, G. R. Reddy, Y. S. S. Ganesh, K. V. Prasad, S. K. Pani Chandra, S. Mukherjee and M. Pal, RSC Adv., 2013, 3, 9641–9644 RSC.
- (a) S. L. Shapiro, K. Geiger and L. Freedman, J. Org. Chem., 1960, 25, 1860–1865 CrossRef; (b) H. Zimmer and R. D. Barry, J. Org. Chem., 1962, 27, 3710–3711 CrossRef; (c) P. A. Chopard, R. P. Hudson and R. J. G. Searle, Tetrahedron Lett., 1965, 6, 2357–2360 CrossRef; (d) P. G. Ciattini, G. Mastropietro, E. Morera and G. Ortar, Tetrahedron Lett., 1993, 34, 3763–3766 CrossRef; (e) R. C. Larock and T. R. Hightower, J. Org. Chem., 1993, 58, 5298–5300 CrossRef; (f) E.-I. Negishi, C. Coperet, T. Sugihara, I. Shimoyama, Y. Zhang, G. Wu and J. M. Tour, Tetrahedron, 1994, 50, 425–436 CrossRef; (g) J. Campora, C. M. Maya, P. Palma, E. Carmona, E. Gutierrez- Puebla and C. Ruiz, J. Am. Chem. Soc., 2003, 125, 1482–1483 CrossRef PubMed; (h) K. Y. Lee, J. M. Kim and J. N. Kim, Synlett, 2003, 0357–0360 Search PubMed; (i) J. Safari, H. Naeimi, A. A. Khakpour, R. S. Jondani and S. D. Khalili, J. Mol. Catal. A: Chem., 2007, 270, 236–240 CrossRef; (j) T. Satosh and M. Miura, Synthesis, 2010, 2010, 3395–3409 CrossRef; (k) D. Rambabu, G. P. Kumar, B. D. Kumar, R. Kapavarapu, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2013, 54, 2989–2995 CrossRef; (l) S. Dhara, R. Singha, M. Ghosh, A. Ahmed, Y. Nuree, A. Das and J. K. Ray, RSC Adv., 2014, 4, 42604–42607 RSC.
- (a) H. Sashida and A. Kawamukai, Synthesis, 1999, 1145–1148 CrossRef; (b) X. Li, A. R. Chianese, T. Vogel and R. H. Crabtree, Org. Lett., 2005, 7, 5437–5440 CrossRef PubMed; (c) E. Marchal, P. Uriac, B. Legouin, L. Toupet and P. Van deWeghe, Tetrahedron, 2007, 63, 9979–9990 CrossRef; (d) C. Kanazawa and M. Terada, Tetrahedron Lett., 2007, 48, 933–935 CrossRef; (e) H.-Y. Liao and C.-H. Cheng, J. Org. Chem., 1995, 60, 3711–3716 CrossRef; (f) V. Subramanian, V. R. Batchu, D. Barange and M. Pal, J. Org. Chem., 2005, 70, 4778–4783 CrossRef PubMed; (g) L. Zhou and H.-F. Jiang, Tetrahedron Lett., 2007, 48, 8449–8452 CrossRef.
- (a) M. Uchiyama, H. Ozawa, K. Takuma, Y. Matsumoto, M. Yonehara, K. Hiroya and T. Sakamoto, Org. Lett., 2006, 8, 5517–5520 CrossRef PubMed; (b) S. Inack-Ngi, R. Rahmani, L. Commeiras, G. Chouraqui, J. Thibonnet, A. DuchÞne, M. Abarbri and J. L. Parrain, Adv. Synth. Catal., 2009, 351, 779–788 CrossRef.
- M. Jithunsa, M. Ueda and O. Miyata, Org. Lett., 2011, 13(3), 518–521 CrossRef PubMed.
- M. Uchiyama, H. Ozawa, K. Takuma, Y. Mastumoto, M. Yonehara, K. Hiroya and T. Sakamoto, Org. Lett., 2006, 8(24), 5517–5520 CrossRef PubMed.
- R. K. Manian, F. M. Irudayanathan, J. H. Moon and S. Lee, Adv. Synth. Catal., 2013, 355, 3221–3230 CrossRef.
- Y. Liu, Y. Yang, Y. Shi, X. Wang, L. Zhang, Y. Cheng and J. You, Organometallics, 2016, 35(10), 1350–1353 CrossRef.
- H. Someya, H. Ohmiya, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10(5), 969–971 CrossRef PubMed.
- G. Fang and X. Bi, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- (a) D. K. Yadav, S. Kumar, Saloni, S. Misra, L. Yadav, M. Teli, P. Sharma, S. Chaudhary, N. Kumar, E. H. Choi and M. –H. Kim, Sci. Rep., 2018, 8, 4777, DOI:10.1038/s41598-018-22972-9; (b) V. Sharma, P. K. Jaiswal, M. Saran, D. K. Yadav, Saloni., M. Mathur, A. K. Swami, S. Misra, M.-H. Kim and S. Chaudhary, Front. Chem., 2018, 6(56), 1–17, DOI:10.3389/fchem.2018.00056; (c) P. K. Jaiswal, V. Sharma, S. Kumar, M. Mathur, A. K. Swami, D. K. Yadav and S. Chaudhary, Arch. Pharm., 2018, 1–17, DOI:10.1002/ardp.201700349; (d) A. Negi, N. Bhandari, B. R. K. Shyamlal and S. Chaudhary, Saudi Pharm. J., 2018, 26(4), 546–567 CrossRef PubMed; (e) V. Sharma, P. K. Jaiswal, D. K. Yadav, M. Saran, J. Prikhodko, M. Mathur, A. K. Swami, I. V. Mashevskaya and S. Chaudhary, Acta Chim. Slov., 2017, 67(4), 988–1004 CrossRef; (f) R. Sharma, L. Yadav, J. Lal, P. K. Jaiswal, M. Mathur, A. K. Swami and S. Chaudhary, Bioorg. Med. Chem. Lett., 2017, 27(18), 4393–4398 CrossRef PubMed; (g) P. K. Jaiswal, V. Sharma, J. Prikhodko, I. V. Mashevskaya and S. Chaudhary, Tetrahedron Lett., 2017, 58(22), 2077–2083 CrossRef; (h) L. Yadav, M. K. Tiwari, B. R. K. Shyamlal, M. Mathur, A. K. Swami, S. K. Puri, N. K. Naikade and S. Chaudhary, RSC Adv., 2016, 6, 23718–23725 RSC; (i) S. Chaudhary, N. K. Naikade, M. K. Tiwari, L. Yadav, B. R. K. Shyamlal and S. K. Puri, Bioorg. Med. Chem. Lett., 2016, 26(6), 1536–1541 CrossRef PubMed; (j) S. Chaudhary, V. Sharma, P. K. Jaiswal, A. N. Gaikwad, S. K. Sinha, S. K. Puri, A. Sharon, P. R. Maulik and V. Chaturvedi, Org. Lett., 2015, 17(20), 4948–4951 CrossRef PubMed.
- D. Pla and M. Gómez, ACS Catal., 2016, 6(6), 3537–3552 CrossRef.
- X. Zhou, Y. Lu, L. –L. Zhai, Y. Zhao, Q. Liu and W. –Y. Sun, RSC Adv., 2013, 3, 1732–1734 RSC.
- (a) E. Parker, N. Leconte, T. Godet and P. Belmont, Chem. Commun., 2011, 47, 343 RSC; (b) T. Godet, C. Vaxelaire, C. Michel, A. Milet and P. Belmont, Chem.–Eur. J., 2007, 13, 5632 CrossRef PubMed; (c) C. Michel, T. Godet, M.-L. Dheu-Andries, P. Belmont and A. Milet, J. Mol. Struct.: THEOCHEM, 2007, 811, 175 CrossRef; (d) A. S. K. Hashmi, A. M. Schuster, S. Gaillard, L. Cavallo, A. Poater and S. P. Nolan, Organometallics, 2011, 30, 6328 CrossRef; (e) V. H. L. Wong, A. J. P. White, T. S. Hor and K. K. Hii, Adv. Synth. Catal., 2015, 357, 3943 CrossRef.
- (a) 9a was reacted with silver phenylacetylide under optimized reaction conditions which furnished the desired 11a in 35% yield.; (b) Following a suggestion from one of the reviewers, the reusability of the silver oxide nanoparticle was checked. It was observed that the reaction diminished tremendously by using recovered residue..
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef.
- PivOAg was also utilized to underline the tentative mechanism. 9a was reacted with either 10a or 10g under optimized conditions (DMF as solvent at 120 °C for 3 h). The desired 3-ylidinephthalides 11a and isocoumarins 17a were obtained in 51% and 35% yield, respectively..
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of all synthesized compounds 11–16, 17–20, 21, 22a–c. See DOI: 10.1039/c8ra03926g |
| This journal is © The Royal Society of Chemistry 2018 |